

Abstract
Alcoholism is a highly visible and prevalent issue in the United States. Although binge-drinking is assumed to be a college-age problem, older adults (ages 65+) consume binge amounts of alcohol and have alcohol use disorders (AUDs). Moreover, individuals with alcohol dependence in their youth often continue to drink as they age. As such, this study tested the hypothesis that the effects of alcohol on hippocampal microglia are exacerbated in aged versus younger rodents in two AUD models. Briefly, adult (2–3 months) and aged (15+ months) Sprague-Dawley rats were administered alcohol or control diet using the Majchrowicz model to study alcohol-induced neurodegeneration. To study the effects of non-dependent binge consumption on microglia, adolescent (6–8 weeks) and aged (18+ months) C57/BL6N were subjected to the Drinking in the Dark paradigm. Microglia number and densitometry were assessed using immunohistochemistry. Hippocampal subregional and model/species-specific effects of alcohol were observed, but overall, aging did not appear to increase the alcohol-induced microglia reactivity as measured by Iba-1 densitometry. However, analysis of microglial counts revealed a significant decrease in the number microglia cells in both the alcohol-induced neurodegeneration and DID model across age groups. In the dentate gyrus, the loss of microglia was exacerbated by aging, particularly in mice after DID, non-dependent model. Using qRT-PCR, the persistence of alcohol and aging effects was assessed following the DID model. Allograft Inflammatory Factor 1 mRNA was increased in both young and aged mice by alcohol exposure; however, only in the aged mice did the alcohol effect persist. Overall, these data imply that the microglial response to alcohol is complex with evidence of depressed numbers of microglia but also increased reactivity with advanced age.
1. Introduction
1.1. Alcohol use disorders: A variety of bad drinking habits
Although consuming alcohol is a socially accepted pastime, excessive alcohol consumption is a common theme of various problems seen in the United States and internationally (Schomerus et al., 2011). Problems associated with the misuse and abuse of alcohol affects the economy, social relationships, and health of the population in various nations (Schomerus et al., 2011). Alcohol abuse encompasses various forms of problematic drinking, including alcohol dependence, episodic binge drinking, and alcohol consumption among vulnerable populations (e.g., pregnant women and underage-adolescents). Drinking during pregnancy and adolescence is considered a health risk because of the extensive neurobiological development that occurs during these periods. It could reasonably be argued that the elderly also have a variety of neurobiological changes that make them vulnerable as well (Kelly et al., 2006). This article will focus on binge drinking in aged rodents using two distinct models that recapitulate either non-dependence or dependence.
Binge drinking is a pattern of alcohol consumption that results in blood alcohol concentrations (BACs) greater than 80 mg/dL (NIAAA, 2004). BACs greater than 80 mg/dL are often achieved after consuming 4 or 5 alcoholic beverages within 2 h for women or men, respectively (Fillmore & Jude, 2011). However, alcohol dependent individuals often achieve BACs that far exceed the binge limit and often drink to BACs that surpass 300 mg/dL (Afshar, Netzer, Salisbury-Afshar, Murthi, & Smith, 2016; Roffman & Stern, 2006; Urso, Gavaler, & Van Thiel, 1981). Drinking patterns (social vs binge), the history and pattern of alcohol use (chronic vs acute), and BACs significantly change the impact of ethanol. Likewise, the neurobiological consequences observed in chronic heavy alcohol exposure are often distinct from those associated with acute binge drinking (Cui et al., 2013; Sprow & Thiele, 2012). For example, binge drinking engages corticotropin-releasing factor (CRF) signaling during intoxication, but chronic heavy alcohol use results in depression of CRF during abstinence (Quadros, Macedo, Domingues, & Favoretto, 2016). Interestingly, it is hypothesized that maladaptations that occur during binge drinking increase the risk for AUD development and perpetuate excessive consumption (Koob & Volkow, 2010). Understanding the neurobiological mechanisms that are engaged during dependent and non-dependent alcohol consumption is necessary to develop treatments to alleviate alcohol misuse at various stages of an AUD.
Determining the unique consequences of binge drinking during alcohol dependence compared with episodic binging also has epidemiological implications. Reports suggest that approximately one in six adults in the United States participate in binge drinking episodes multiple times a month (CDC, 2013), but only about 4% of people are considered alcohol dependent (Grant et al., 2004). It is important to acknowledge that the new Diagnostics and Statistics Manual V has a different way of categorizing AUDs. This categorization does not specifically delineate dependence and abuse, but rather, the DSM V categorizes AUDs as mild, moderate, and severe (American Psychiatric, American Psychiatric, & Force, 2013). Despite the changes in nomenclature, the trend for a smaller number of individuals being “dependent” still persists (Grant et al., 2015, 2016). Current estimates suggest that only 3.4% would meet the diagnostic criteria for a current severe AUD but the lifetime prevalence is approximately 14% (Grant et al., 2015). Understanding the distinct effects of alcohol during both binge drinking and alcohol dependence warrants attention given the number of people that are affected by AUDs.
1.2. Alcohol consumption in aging
When people think about problems with alcohol, they often envision younger adults and quintessential Hollywood movies like House Party and American Pie, with excessive alcohol consumption at high school and college parties (Osberg, Billingsley, Eggert, & Insana, 2012). Yet, approximately 46 million Americans over the age of 26 consume alcohol in the form of binge drinking (Quality, 2015). Moreover, this pattern of drinking persists as people progress into their “senior” years. In fact, over 40% of Americans over the age of 65 consume alcohol and approximately 10% binge drink (Blazer & Wu, 2012). When in a retirement community or communal living environments, estimates of binge drinking can be almost seven times higher (Blazer & Wu, 2009; Sacco et al., 2015). Estimating dependence rates among aging populations can be more difficult due to underreporting, but epidemiologists believe that both the rates of dependence and binge drinking will increase as baby boomers, many of whom previously abused alcohol and other drugs, continue to age (Kuerbis, Sacco, Blazer, & Moore, 2014). While many AUD studies have focused on adolescence or early adulthood, the impact of alcohol in older adults has not received much attention. This is unfortunate given the added risks associated with alcohol use in individuals over 65, including increased sensitivity to the pharmacological effects of alcohol (Squeglia, Boissoneault, Van Skike, Nixon, & Matthews, 2014), adverse reactions due to drug-drug interactions (Immonen, Valvanne, & Pitkala, 2013), and most importantly for these studies, comorbid conditions that exacerbate alcohol’s detrimental effects on various organ systems including the CNS and immune response (Pfefferbaum et al., 1992; Pfefferbaum, Sullivan, Mathalon, & Lim, 1997).
1.3. Alcohol consumption and the neuroimmune system
A growing body of evidence suggests that the neuroimmune system undergoes neuroplastic changes induced by alcohol and that dysregulation of the neuroimmune system plays an influential role in addictive behaviors (Coller & Hutchinson, 2012; Crews, 2012; Crews, Zou, & Qin, 2011). In terms of alcoholism, the majority of this work has focused on how the neuroinflammatory response elicited by excessive alcohol exposure or consumption may mediate alcohol-related brain damage (ARBD) in dependent models (Crews & Vetreno, 2014). Clinical reports have suggested that alcohol induces a hyper-reactive neuroimmune response evident in the postmortem brains of alcoholics (Crews, Qin, Sheedy, Vetreno, & Zou, 2013; Crews & Vetreno, 2016; He & Crews, 2008). However, further probing with preclinical studies has elucidated that the alcohol-induced neuroimmune response is much more complicated. Our previous reports highlighted the importance of temporal factors in understanding these responses (Marshall et al., 2013; Marshall, Geil, & Nixon, 2016). We, and others, have shown that there are distinct neuroimmune profiles during intoxication and abstinence (Doremus-Fitzwater et al., 2014; Marshall et al., 2013; Walter & Crews, 2017). For example, the Deak laboratory has shown during intoxication that IL-6 is increased with concurrent IL-1 and TNF-α suppression; however, during withdrawal, IL-6 returns to normal concurrent with TNF and IL-1 upregulation (Doremus-Fitzwater et al., 2014; Doremus-Fitzwater, Gano, Paniccia, & Deak, 2015; Gano, Doremus-Fitzwater, & Deak, 2017). Various components of the neuroimmune system have been explored in dependence models including microglial activation (Marshall, Geil, et al., 2016; Marshall et al., 2013; McClain, Morris, Marshall, & Nixon, 2014), astrocyte dysregulation (Alfonso-Loeches, Pascual-Lucas, Blanco, Sanchez-Vera, & Guerri, 2010; Kelso, Liput, Eaves, & Nixon, 2011; Wilhelm et al., 2016), altered expression of pro-inflammatory molecules (Coleman, Zou, Qin, & Crews, 2018; Qin & Crews, 2012; Qin, Liu, Hong, & Crews, 2013), and loss of glial neurotrophic support (Davis, 2008; Joe et al., 2007). Indeed, the complexities of the neuroimmune response to alcohol necessitates studying it under multiple conditions including alcohol dependence and non-dependent binge drinking.
Neuroimmune responses, particularly cytokines, have also been linked to a variety of behavioral disorders independent of brain damage including, but not limited to, depression (Enache, Pariante, & Mondelli, 2019), attention deficit hyperactivity disorder (Dunn, Nigg, & Sullivan, 2019), and anxiety disorders (Hou & Baldwin, 2012). More importantly, these neuroimmune responses have been associated with drug abuse behaviors including alcoholism (Coller & Hutchinson, 2012). Our previous reports showed that even without ARBD, binge-like consumption can elicit changes in the neuroimmune response. More specifically, there was increased pro-inflammatory signaling molecules and astrocyte activation caused by the drinking in the dark paradigm (Grifasi et al., 2019; Marshall et al., 2016; Marshall, McKnight, Blose, Lysle, & Thiele, 2017). The Roberto lab has shown repeatedly that these changes in cytokines have direct implications for neurotransmission and can alter alcohol-GABAergic signaling (Bajo, Herman, et al., 2015; Bajo, Varodayan, et al., 2015; Suryanarayanan et al., 2016). Moreover, these biological changes in cytokines have behavioral implications. For example, administration of anti-inflammatory molecules, like anakinra, reduce binge-like consumption and ameliorate alcohol-induced motor impairment in rodents (Marshall, Casachahua, et al., 2016; Wu et al., 2011). Likewise, administration of phosphodiesterase-4 inhibitors reduced alcohol consumption in preclinical studies (Blednov, Benavidez, Black, & Harris, 2014; Hu et al., 2011), and even reduced alcohol cravings in a recent clinical trial (Ray et al., 2017). Because the neuroimmune system has the capacity to impact both the behavioral and biological aspects of AUDs, the studies presented here examined the effects of alcohol dependence and binge-like consumption on microglia.
1.4. Alcohol abuse, neuroimmune system, and aging
Although normal aging and alcohol abuse impact a variety of neurological functions, this manuscript focuses on their combined impact on the neuroimmune system. Alcohol abuse and aging are both independently associated with dysregulation of the neuroimmune system. For example, in alcohol-dependence, it has been hypothesized that a hyperactive neuroimmune response contributes to ARBD (Crews & Nixon, 2009; Crews & Vetreno, 2014). Likewise, in pathological aging, a hyperactive neuroimmune response has been tied to a variety of diseases including multiple sclerosis, Alzheimer’s disease, and Parkinson’s Disease (Glass, Saijo, Winner, Marchetto, & Gage, 2010; Morales, Guzman-Martinez, Cerda-Troncoso, Farias, & Maccioni, 2014; Perry & Holmes, 2014). In fact, even in normal aging there seems to be a steady increase in basal pro-inflammatory signaling within the CNS (Patterson, 2015). However, an alternate theory suggests that during aging there is an impaired neuroimmune response to events that would normally cause a reaction. This age-associated hypo-reactive response to immunomodulators could also exacerbate neurodegeneration because the brain has lost the ability to surmount a sufficient immune response and remove damaged cells and/or cellular debris (Streit & Xue, 2009; Wainwright et al., 2009). This phenomenon of increased basal activity combined with impaired reactivity has been termed “inflamm-aging” (Costantini, D’Angelo, & Reale, 2018; Deleidi, Jaggle, & Rubino, 2015).
Previous studies of alcohol effects on the neuroimmune response during aging have focused on cytokines and chemokines. For example, Kane and colleagues have shown increased pro-inflammatory responses in the cerebellum of aged mice following binge-like levels of alcohol exposure compared with younger mice (Kane et al., 2013), and a recent study by the Deak laboratory suggests that acute alcohol may exacerbate the pro-inflammatory cytokine milieu in the hippocampus of aged rats relative to young adults (Gano et al., 2017). These studies however, utilized gene expression approaches in tissue punches of the hippocampus, and therefore could not discriminate the specific contribution of cells such as microglia to the aging-alcohol interaction. Mounting an appropriate microglial response to injury elicits both a pro-inflammatory response necessary to remove damaged cells, but is subsequently followed by an anti-inflammatory and neurotrophic response to facilitate recovery (Frank-Cannon, Alto, McAlpine, & Tansey, 2009). The lack of, or an insufficient, neuroimmune response can be as detrimental to the neuronal milieu as a hyper-inflammatory response (Streit, 2005; Wainwright et al., 2009). Streit and colleagues suggest that a special subset of microglia, called dystrophic or senescent, are damaged and impair the ability of the brain to ameliorate neurodegenerative events (Streit, 2005; Streit, Braak, Xue, & Bechmann, 2009; Streit, Sammons, Kuhns, & Sparks, 2004; Tischer et al., 2016). Considering that studies have shown both a pro-inflammatory response (Crews & Nixon, 2009; Morales et al., 2014) and an impaired neuroimmune response in both aging and alcohol studies (Marshall, Geil, et al., 2016; Streit et al., 2009; Wilhelm et al., 2016), it is critical to consider the confluence of alcohol and aging on the neuroimmune response.
1.5. Microglia: A snapshot of neuroimmune responses
Microglia have often been called the “macrophages of the CNS” because of their phagocytic capacity and the fact that microglia share the same hematopoietic lineage as peripheral macrophages (Penfield, 1925; Saijo & Glass, 2012). The term microglia literally means “small glue” because microglia were originally thought to be supportive cells that simply held neurons together (Dermietzel & Spray, 1998). However, we now realize that microglia are much more dynamic and respond to minute changes within the CNS microenvironment (Harting, Jimenez, Adams, Mercer, & Cox, 2008; Lai & Todd, 2008). Microglia display heterogeneity in their morphology, cytokine secretions, and cell surface proteins when perturbed, allowing for tailored actions to respond effectively to a variety of pathogens, toxins, and/or damage (Carson et al., 2007; Michelucci, Heurtaux, Grandbarbe, Morga, & Heuschling, 2009). Following environmental changes, resting ramified microglia transform from a “bushy” morphology to an amoeboid shape. These morphological changes mean that the cell bodies of microglia physically increase in size as processes are retracted and the microglia become activated. Immunohistochemistry and densitometric analysis has often been used as a way of determining if microglia have been activated using a variety of markers (Hovens, Nyakas, & Schoemaker, 2014; Raivich et al., 1999). This article uses ionized calcium-binding adaptor protein-1, a microglial specific marker, in order to assess changes in microglial morphology and number (Ito et al., 1998; Ito, Tanaka, Suzuki, Dembo, & Fukuuchi, 2001). Likewise, Aif1, the gene that encodes Iba-1, was measured to see if alcohol and aging converge to alter mRNA concentrations (Postler, Rimner, Beschorner, Schluesener, & Meyermann, 2000). These markers were chosen as an initial assessment because Iba-1/Aif1 are constitutively expressed in microglia; whereas, many other microglia markers are only expressed after activation (Raivich et al., 1999). Upregulation of these markers is, however, consistent with increased microglial activation.
1.6. The susceptibility of the hippocampus to alcohol abuse and aging
The hippocampus has been chosen as the focus of this chapter because studies have repeatedly shown that the hippocampus is particularly vulnerable to alcohol-induced neurodegeneration (Liput, Hammell, Stinchcomb, & Nixon, 2013; Obernier, Bouldin, & Crews, 2002). These deficits are believed to contribute to the decline in cognition seen in human alcoholics (Bartels et al., 2007; Hunt, 1993). Moreover, our previous studies as well as others report that the hippocampal neuroimmune system, especially microglia and cytokines, are dysregulated by binge alcohol exposure (He & Crews, 2008; Marshall et al., 2013; Zahr, Luong, Sullivan, & Pfefferbaum, 2010). Characterizing neuroimmune dysregulation caused by alcohol within the hippocampus will further our understanding of how the neuroimmune system may contribute directly to neurotoxicity and ultimately ARBD.
Understanding the impact of alcohol on the hippocampus is particularly important in aging. Older individuals often have deficits in memory and recall. Moreover, damage to the hippocampus is one of the hallmarks of aging and Alzheimer’s Disease (Bartsch & Wulff, 2015; Patterson, 2015). Microglia may play an important role in both the neurodegeneration and memory deficits associated with pathological aging and alcohol abuse. For example, dysregulation of microglia is believed to be a mechanism of inhibited neurogenesis seen in both alcohol and aging (Crews & Nixon, 2009; Nixon & Morris, 2008; Wyss-Coray, 2016). Moreover cytokines and/or microglia can alter hippocampal long-term potentiation (Prieto, Tong, Smith, & Cotman, 2019; Raghuraman, Karthikeyan, Lik Wei, Dheen, & Sajikumar, 2019) and memory formation/recall. Alcohol abuse and aging are both independently associated with memory deficits and/or neurodegeneration in the hippocampus (see Matthews, Schneider, Kastner, Scaletty, & Szenay, 2019; Nunes, Kipp, Reitz, & Savage, 2019). Elucidating the microglial response within the hippocampus will highlight how alcohol and aging may coalesce to impact neuronal function and therein impair memory.
2. Methods
2.1. Animals
All experimental procedures for this study were approved by the North Carolina Central University and High Point University’s Institutional Animal Care and Use Committees. Adolescent (6–8 weeks; N = 14, 8 male and 6 female) and aged (18–20 months; N = 12, 8 male and 4 female) C57BL/6 N mice were housed in individual wire top cages in a vivarium with a reverse 12:12-h (7 AM–7 PM) dark-light cycle kept at approximately 22°C. Likewise, young adult Sprague-Dawley rats (2–3 months; N = 13, 7 male and 6 female) and aged Sprague-Dawley rats (15–18 months; N = 10, 6 male and 4 female) were housed individually in ventilated blue-line cages in a separate vivarium with identical temperature and lighting settings. All rodents were acquired from Charles River Laboratories (Raleigh, NC). The aged animals used herein were retired breeders gifted from Charles River colony, but the younger animals purchased were all sexually naive. In this study, we chose to combine female and male animals into single groups and that may have impacted some of our variables. Neuroimmune responses have previously been shown to be impacted by sex, including in alcohol studies (Maynard, Barton, Robinson, Wooden, & Leasure, 2018; Schwarz & Bilbo, 2012; Tronson & Collette, 2017). All rodents were housed in their individual experimental cages 1 week prior to the beginning of both experimental procedures to allow animals to acclimate to their new environment and single housing conditions. Rodents were given unlimited access to water and rodent chow (Teklad Diet® 2920X) (Marshall et al., 2015), unless otherwise stated.
2.2. DID paradigm
The DID paradigm is a well-validated model of alcohol consumption that simulates non-dependent voluntary binge consumption (Rhodes, Best, Belknap, Finn, & Crabbe, 2005; Thiele & Navarro, 2014). This model is useful in the investigation of problematic drinking prior to alcohol dependence and independent of ARBD (Marshall, Casachahua, et al., 2016; Sprow et al., 2015). The voluntary consumption design of the experiment has a high face validity for modeling the human condition (Thiele & Navarro, 2014). Moreover, the DID paradigm consistently produces alcohol drinking rodents with BACs well over the 80 mg/dL binge limit (Cox et al., 2013; Marshall et al., 2015). Over a period of four consecutive days, mice were given access to 20% (v/v) alcohol solution or tap water. Approximately 3 h into the dark cycle, regular cage water bottles were removed and replaced with the experimental or control solution, so that during the DID procedure, mice only had access to a single sipper tube with either ethanol or water. On the first 3 days, mice were given 2 h of access, but on the 4th day of consumption mice were given 4 h of access, similar to previous reports (Cox et al., 2013; Thiele & Navarro, 2014). Consumption was normalized to body weight and is expressed as g of alcohol/kg of body weight. All mice underwent three cycles of the 4-day DID paradigm, each of which were separated by 3 days. Three cycles of DID was chosen based on our previous studies showing that the changes in cytokines occurred after this period (Marshall et al., 2017; Marshall, Casachahua, et al., 2016). Immediately following the end of the last 4-day DID cycle, a collection of venous tail blood samples (about 60 μL per rodent) were acquired via tail nicks in order to measure BACs. Depending on the experiment, mice were euthanized immediately or 7 days following alcohol consumption.
2.3. Majchrowicz paradigm
The Majchrowicz model has been used extensively to recapitulate alcohol dependence (Faingold, 2008; Majchrowicz, 1975; Morris, Kelso, Liput, Marshall, & Nixon, 2010). Moreover, reports have shown that this model results in ARBD (Liput et al., 2013; Obernier, White, Swartzwelder, & Crews, 2002). In this AUD model, involuntary gavage of alcohol typically elicits dangerously high BACs above 300 mg/dL in rats (Morris et al., 2010; Obernier, Bouldin, et al., 2002), which is four to five times above the legal limit in most American states (Wagenaar, Maldonado-Molina, Ma, Tobler, & Komro, 2007). In this study, we modified the 4-day period electing to use 2-day procedure similar to previous reports (Hayes, Deeny, Shaner, & Nixon, 2013). We chose to limit the study to 2 days because it has previously been established that 2 days of ethanol administration in the Majchrowicz model produces gliosis and neurodegeneration (Hayes et al., 2013; Obernier, Bouldin, et al., 2002). Although the 4 day model has been used more extensively, both the two and 4 day Majchrowicz model have shown evidence of vimentin upregulation, increased Fluoro-Jade B+ cells, and BACs greater than 300 mg/dL (Hayes et al., 2013; Kelso et al., 2011; Liput et al., 2013; Morris et al., 2010). It is important to note that withdrawal, a hallmark in AUD dependence models, is not consistently observed after 2 days of alcohol exposure in the Majchrowicz model (Hayes et al., 2013; Obernier, Bouldin, et al., 2002), but the BACs (>300 mg/dL) achieved by gavage are reflective of the concentrations usually only observed in a select group of alcohol dependent individuals (Crabbe, Harris, & Koob, 2011; Majchrowicz, 1975; Roffman & Stern, 2006).
In this experiment, access to food but not water was removed; however, rats were administered a nutritionally complete diet with alcohol [25% (w/v) in Vanilla Ensure Plus®] or control diet (isocaloric dextrose in Vanilla Ensure Plus®). Solutions were given by intragastric gavage over a 2-day period every 8-h. As previously described (Morris et al., 2010), alcohol dosing was titrated based on the behavioral intoxication observations. For the first initial dose, all rats were given 5 g/kg of the alcohol solution. For all of the subsequent treatments, rats were dosed with alcohol solution calculated from their individual behavioral intoxication score, which was determined based on experimenter observations. The intoxication scores were quantified using a six-point scale, ranging of scores from 0 to 5 (see Table 1). All control animals were dosed based on the overall average dose of experimental animal every hour. All rats were euthanized immediately following their final gavage, and trunk bloods were collected for BAC analysis.
Table 1.
Intoxication scoring characteristics.
| Intoxication score | Behavioral attributes | Ethanol dose (g/kg) |
|---|---|---|
| 0 | Normal animal | 5 |
| 1 | Hypoactive, mildly ataxic | 4 |
| 2 | Ataxic, elevated abdomen | 3 |
| 3 | Ataxic, absence of abdominal elevation, delayed righting reflex | 2 |
| 4 | Loss of righting reflex, retain eye blink reflex | 1 |
| 5 | Loss of righting reflex, loss of eye blink reflex | 0 |
Rat intoxication was scored based on behavioral attributes to determine the necessary ethanol dose.
2.4. Blood alcohol concentration analysis
Trunk or tail blood samples were taken at the time of euthanasia to determine the BACs. Bloods were only collected during intoxication, so no samples were taken from mice who were euthanized a week after intoxication to avoid complications of restraint stress on neuroimmune gene expression (Walker, Nilsson, & Jones, 2013). Serum was separated from plasma by centrifugation. Using the EnzyChrom™ Ethanol Assay Kit (BioAssay Systems; Hayward, CA), serum samples were run in duplicates, and the BACs were determined by comparing the absorbance of samples to known concentration standards according to manufacturer instructions ( Jeong et al., 2008; Paris & Frye, 2009).
2.5. Immunohistochemistry
For immunohistochemistry studies, mice or rats were euthanized immediately following alcohol exposure by transcardial perfusion with 0.1 M phosphate buffered saline (PBS, pH 7.4), followed by 4% paraformaldehyde (PFA). Extracted brains were post-fixed for 2 h in 4% PFA, and then stored in cryopreserve solution (50% (v/v) 0.2 M phosphate buffer; 50% (v/v) ethylene glycol; 1% (w/v) polyvinylpyrrolidone). Preserved brains were sliced into coronal sections (40 μm) using a microtome (Compresstome VF-300; Precisionary Instruments). For the mice sectioning was done using a 1:4 collection method; whereas, a 1 in 12 series was used for rats. All sections were stored in cryopreserve at −20 °C until processed for immunohistochemistry. Similar to our previously published studies (Marshall et al., 2013; Marshall, Geil, et al., 2016; McClain et al., 2011), the antibody against ionized calcium binding adaptor 1 (Iba-1) binding protein was used because it is specific to microglia (Imai, Ibata, Ito, Ohsawa, & Kohsaka, 1996; Ito et al., 1998). Briefly, every 4th (mice) or 12th (rats) section was rinsed in a 0.1 M phosphate buffered saline solution (PBS; pH 7.4) prior to a 30-min incubation in 0.6% H2O2 to block endogen peroxidase activity. After additional PBS washes, non-specific binding was blocked using goat serum (3% goat serum/0.1% triton-X/PBS), and the tissue was incubated in the rabbit anti-Iba-1 primary antibody (1:1000; Wako, Cat #: 019-19741; Richmond, VA) for 48 h at 4 °C. A series of PBS washes removed the primary antibody before a goat anti-rabbit secondary (1:2000; Vector Labs, Cat #: AI-1000; Burlingame, CA) incubation for 2 h. To amplify and visualize the Iba-1 antibody, a conjugate of avidin-biotin-peroxidase complex (ABC elite kit, Vector Labs) and 3,3′-diaminobenzidine tetrahydrochloride (Polysciences; Warrington, PA) was formed. Processed sections were mounted onto glass slides and cover-slipped with SHUR/Mount™ (Triangle Biomedical Sciences; Durham, NC).
2.6. Iba-1 IHC quantification
Iba-1 was quantified using both densitometric analysis and cell counts to assess the effects of aging on alcohol-induced microglial changes. Images of the hippocampus from Iba-1 processed sections were captured using an Olympus DP73 attached to a Nikon Eclipse 80i microscope. To avoid experimental bias, images were coded prior to quantification. Subregions of the hippocampus [dentate gyrus (DG), cornu amonis 1 (CA1), and the cornu amonis 2 and 3 (CA2/3); between Bregma −1.06 and −2.70 mm for mice or −2.10 and −4.00 mm for rats] were traced separately for each image (Paxinos & Franklin, 2004; Paxinos & Watson, 2009). Immunoreactivity was measured with CellSens (Olympic; Tokyo, Japan) using experimenter-determined optical density thresholds (Grifasi et al., 2019; Waise et al., 2015). Immunoreactivity is expressed as percent area (immunoreactive positive area/total area of region of interest). Microglia were also assessed using automated cell counts in CellSens. By defining a minimum and maximum number of Iba-1+ pixels in a single microglia, automated cell counts can be used to estimate the number of microglia in a given region (Davis, Salinas-Navarro, Cordeiro, Moons, & De Groef, 2017; Gallego & de Gracia, 2016; Marshall, Geil, et al., 2016). Cell counts are reported as Iba-1+ cells/section.
2.7. Quantitative polymerase chain reaction
Quantitative polymerase chain reaction (qRT-PCR) was performed similarly to our previous report (Grifasi et al., 2019). Mice in this cohort were euthanized by rapid decapitation immediately or after 7 days of abstinence following three cycles of alcohol and compared to a single water group. The hippocampus was removed by microdissection and snap frozen. RNA was extracted from homogenized hippocampal tissue using trizol (200 μL; Invitrogen Corporation, Carlsbad, CA) according to the manufacturer’s instructions. RNA concentrations were normalized to 1.0 μg/μL following total mRNA quantification with a Qubit® 3.0 Fluorometer (Invitrogen). Samples were then reverse transcribed to cDNA using Maxima™ H Minus cDNA (ThermoScientific; Waltham MA). TaqMan® assays (ThermoScientific) were used to determine the relative mRNA expression of Aif-1 (Mm00479862_g1) compared with an internal control PPIA (Mm02342430_g1). Aif-1 was chosen as the mRNA of interest because AIF1 gene encodes for Iba-1. Aif-1 and Iba-1 are often used interchangeably to describe the same microglial specific 17-kDa-peptide (Ito et al., 1998; Postler et al., 2000). Measurements were compared to the water groups using the ΔΔct method and are expressed as the fold change, similar to previous reports (Grifasi et al., 2019; Kim, Lee, John, Maeda, & Smithies, 2002).
2.8. Statistical analysis
All data were analyzed and graphed using GraphPad Prism Version 7 (GraphPad Software, Inc., La Jolla, Ca). Two-way ANOVAs were used to assess voluntary consumption, gavage dose, immunoreactivity, and cell counts. Posthoc Bonferroni or Dunnet’s tests were only done if a significant interaction or main effect of alcohol was found. Each subregion of the hippocampus was considered independent and therefore run separately in all analyses. BACs and body weights were analyzed using t-tests. For behavioral scores, data points were averaged across time and analyzed using the Mann-Whitney test. All data are reported as the mean standard error of the mean and considered significantly different if P < 0.05, two-tailed.
3. Results
3.1. No differences in alcohol consumption with aging in DID model
In the DID paradigm, C57BL/6 N mice consumed similar amounts of alcohol despite differences in their age. For simplicity, the binge parameters of ethanol mice were collapsed across animals used for qRT-PCR and immunohistochemistry studies (Fig. 1A). A two-way ANOVA (age × time) of alcohol consumption indicated that there was no interaction between age and time [F(11,624) = 0.83, P = 0.62], nor were there main effects of either age [F(1,624) = 2.48, P = 0.12] or time [F(11,624) = 1.67, P = 0.08]. It is important to denote that these groups contained both male and female mice despite research indicating that female mice consume more ethanol (g/kg) than their male counterparts (Crabbe et al., 2009; Juarez & de Barrios Tomasi, 1999; Sneddon, White, & Radke, 2019) (see Fig. 1A). Likewise, a t-test indicated there were no differences in the BACs between the young (95% CI [72.34, 98.1 mg/dL]) and aged (95% CI [76.69, 108.3 mg/dL]) mice [t(32) = 0.75, P = 0.46]. BACs and consumption were similar despite the expected significant differences in weight between aged and young mice (see Table 2).
Fig. 1.
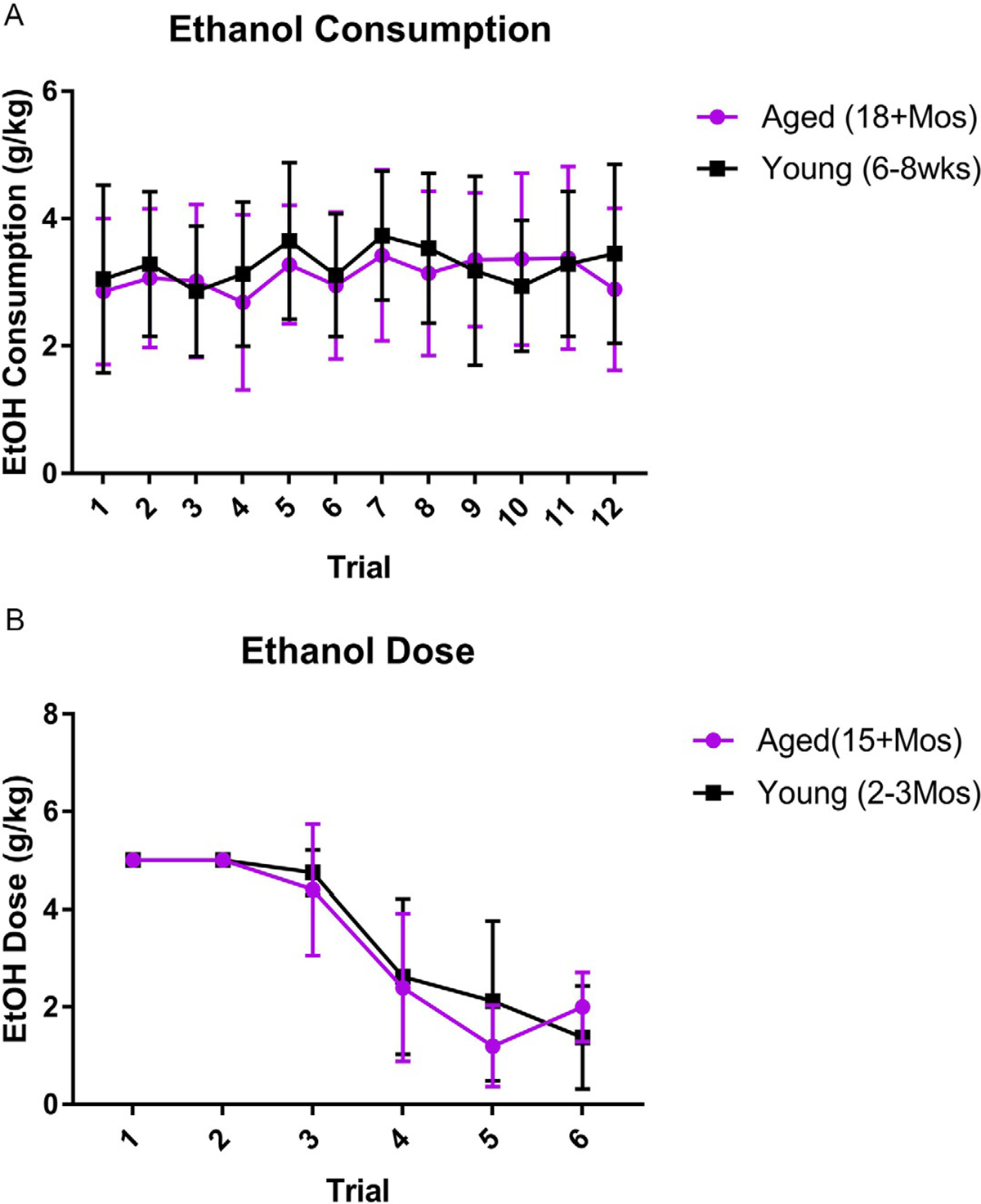
No differences in ethanol dose between aged and young animals: In both the mouse voluntary consumption of the DID model (A) and the rat intragastric gavage of the Majchrowicz model (B), there was no significant differences in the dose between the aged and young groups. In the Majchrowicz model (B), time significantly impacted dosing with a decrease as the model progressed and animals appeared more intoxicated.
Table 2.
Drinking in the dark ethanol animal characteristics.
| Age | Treatment | Study | N | Average BACs (mg/dL) | Average weights (g) |
|---|---|---|---|---|---|
| Young (6–8 weeks) | Ethanol | IHC | 8 | 120.5 ± 18.9 | 21.23 ± 0.68 |
| qRT-PCR | 20 | 85.2 ± 7.0a | 22.46 ± 0.57 | ||
| Aged (18+ months) | Ethanol | IHC | 8 | 115.9 ± 15.9 | 31.26 ± 1.72 |
| qRT-PCR | 16 | 101.6 ± 9.8a | 29.66 ± 1.07 |
BACs were collected from a smaller subset of animals to reduce the impact of restraint stress on mRNA measures.
No significant differences were seen in the BACs among young and aged groups despite a significant change in the body weights.
3.2. DID alcohol exposure significantly increases microglia reactivity
Microglia reactivity was assessed using immunohistochemistry and densitometric measurements of Iba-1 in the hippocampal subregions. A two-way ANOVA (age × treatment) of Iba-1 immunoreactivity indicated that there was an interaction [F(1,22) = 4.32, P = 0.049], as well as main effects of treatment [F(1,22) = 4.31, P = 0.049] and age [F(1,22) = 4.57, P = 0.044] in the DG. Bonferroni analysis showed that alcohol increased microglial reactivity in the young, but not aged animals. It also indicated that age alone increased Iba-1 density in the DG (see Fig. 2). In the CA1 field, a two-way ANOVA indicated no interaction [F(1,23) = 0.002, P = 0.97] or main effect of treatment [F(1,23) = 0.87, P = 0.36], but there was a main effect of age [F(1,23) = 6.47, P = 0.02], indicating that age caused an increase in Iba-1 immunoreactivity independent of alcohol. A similar effect was observed in the CA2/3 region. A two-way ANOVA indicated no interaction [F(1,23) = 0.48, P = 0.50] or main effect of treatment [F(1,23) = 0.19, P = 0.66], but there was again a main effect of age [F(1,23) = 21.37, P < 0.001], demonstrating that with age there is an increase in Iba-1 density in the hippocampus of mice.
Fig. 2.
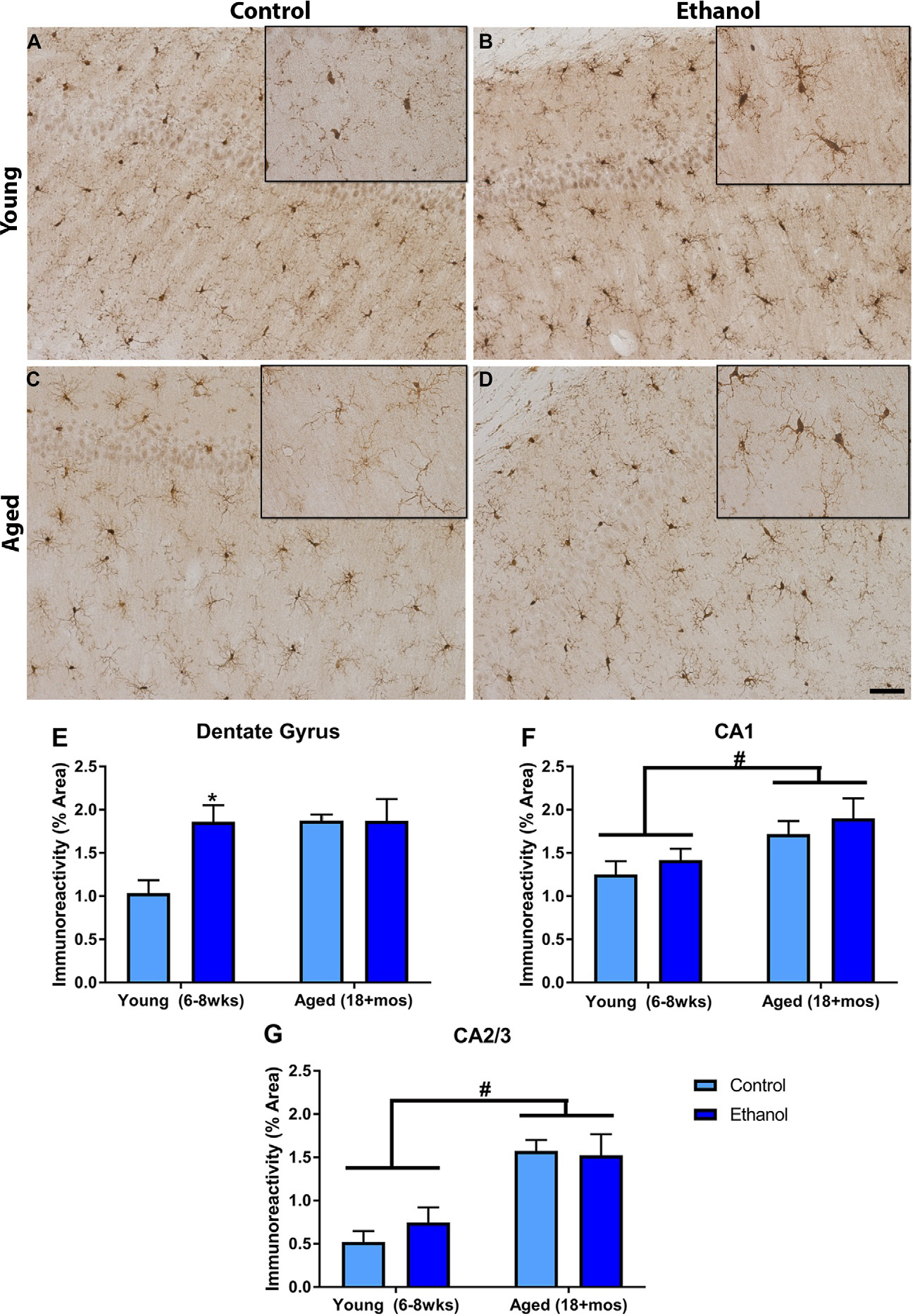
Different microglial responses to alcohol based on age in the mouse DID model: Iba-1 was ubiquitous in the hippocampus. Representative photomicrographs of the dentate gyrus show that Iba-1 was present in the young (A) and aged control animals (C), but that in the young animals ethanol increased the microglial response (B). However, ethanol’s effects on microglia were not present in the aged group (D). Quantification of the subregions indicated that ethanol only increased Iba-1 immunoreactivity in the DG of the young animals (E). In all of the hippocampal subregions, age significantly increased Iba-1 immunoreactivity (E–G). *P < 0.05 compared to control treatment; #P < 0.05 compared to age matched group. Scale Bar= 200 μm.
3.3. DID alcohol exposure significantly decreased microglia number
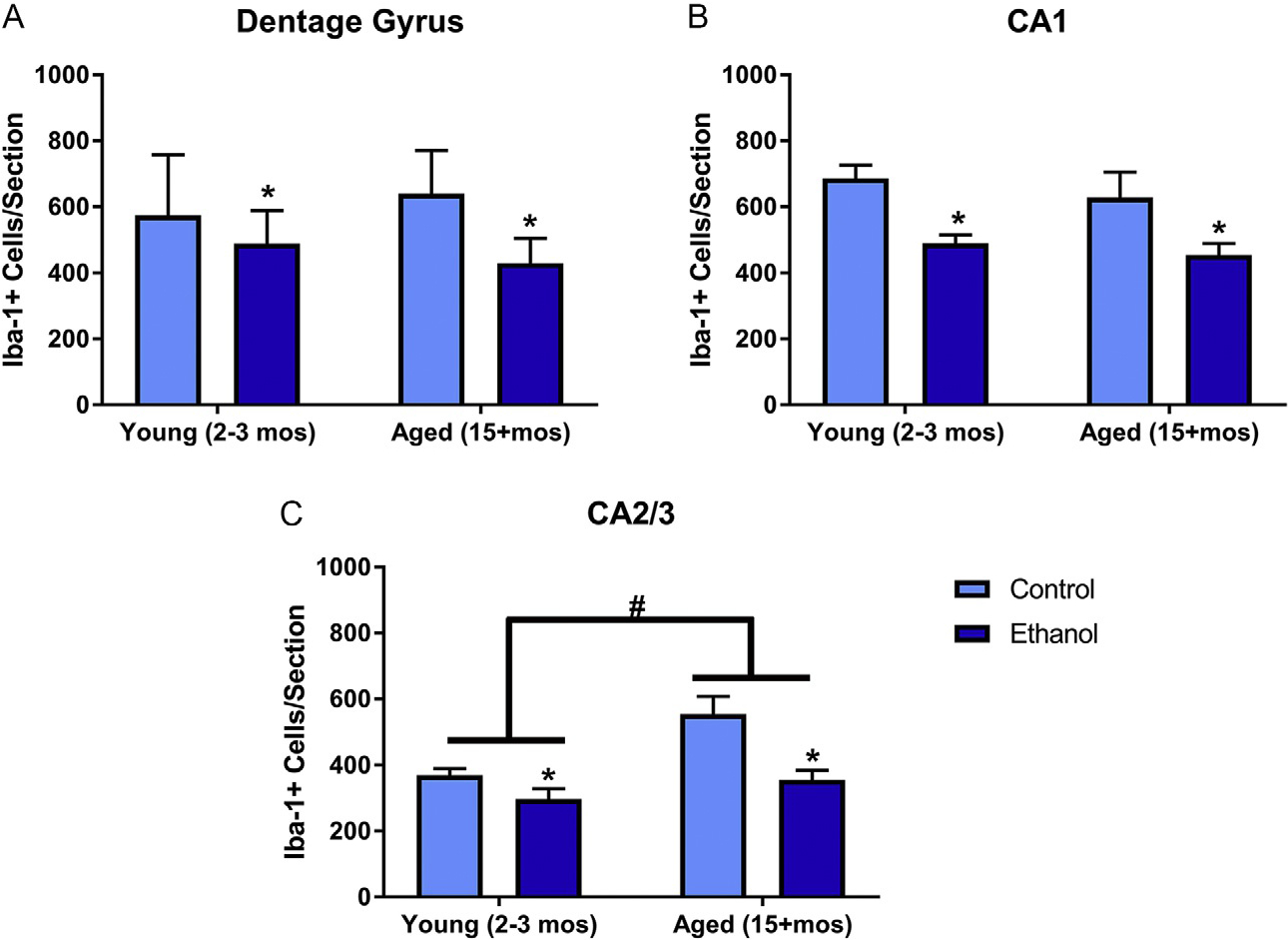
Cell counts were performed to determine if binge alcohol exposure altered the number of microglia following the DID procedure. A two-way ANOVA of Iba-1+ cell number in the DG revealed there was no interaction between treatment and age [F(1,23) = 0.14, P = 0.71]; however, there were main effects of both treatment [F(1,23) = 21.37, P < 0.001] and age [F(1,23) = 47.72, P < 0.0001]. Because both age and treatment had independent effects, the percent difference from control was analyzed to determine if aging exacerbated alcohol’s effects. A t-test indicated that the percent decrease in the number of microglia [t(14) = 2.3, P = 0.04] was greater in aged mice (μ = 60.8%±6), compared with young mice (μ = 40.2%±7). In the CA1 field, there was no interaction [F(1,23) = 0.13, P = 0.72] or a main effect of age [F(1,23) = 0.33, P = 0.57] on the number of microglia, but there was an overall main effect of treatment [F(1,23) = 8.72, P = 0.007], revealing an alcohol-induced decrease in microglial number. However, aging caused an increase in the number of microglia in the CA2/3. A two-way ANOVA indicated no interaction [F(1,23) = 0.43, P = 0.52] or main effect of treatment [F(1,23) = 0.83, P = 0.37], but there was a significant effect of age [F(1,23) = 9.57, P = 0.005] (see Fig. 3).
Fig. 3.
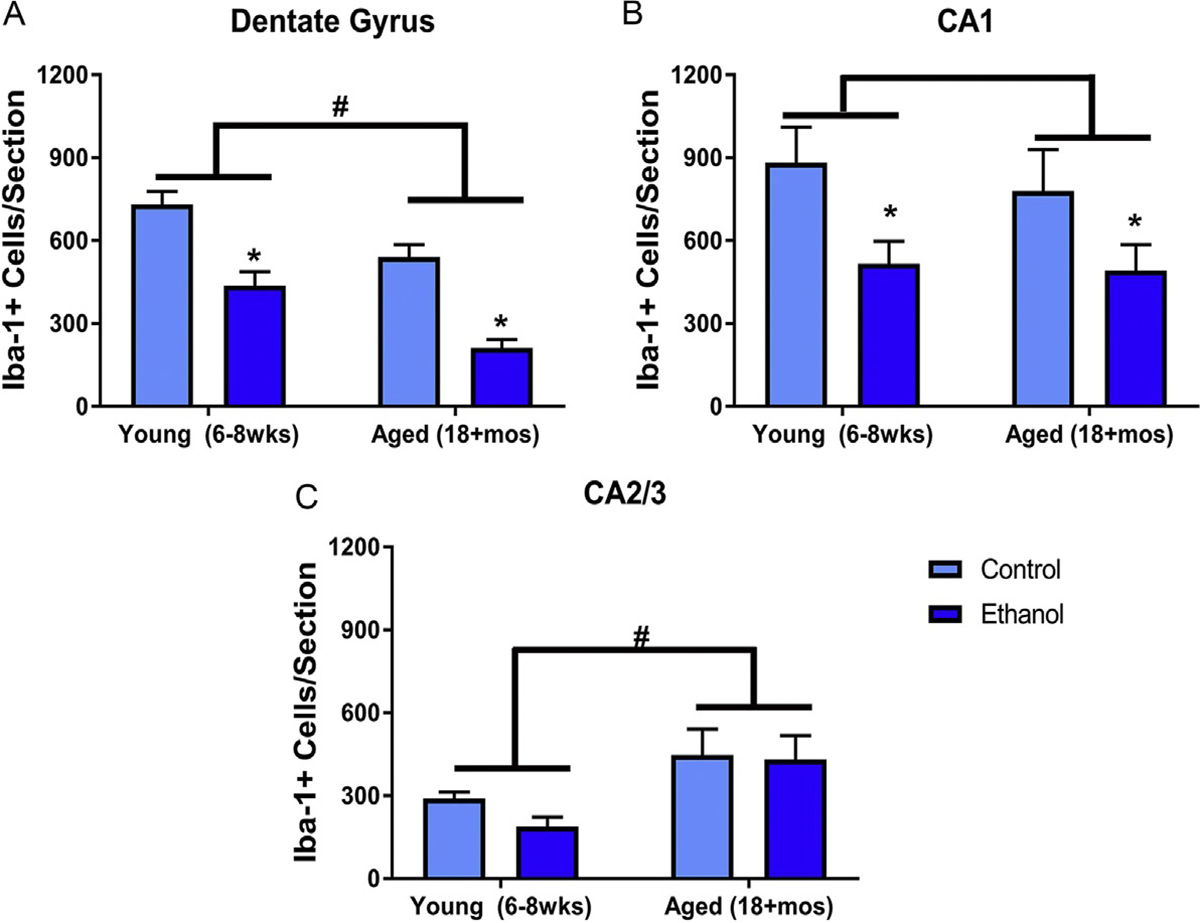
DID ethanol exposure decreases microglia number in mice: In the dentate gyrus (A) and CA1 (B), ethanol significantly decreased the number of microglia. However, in each of the hippocampal subregions, there was also an effect of aged on the number of microglia. In the dentate gyrus (A) and CA1 (B), age cause a loss of microglia, but age caused a significant increase in microglia of the CA2/3 fields (C). *P < 0.05 compared to control treatment; #P < 0.05 compared to age matched group.
3.4. Persistent microglia gene expression changes in aging following DID alcohol exposure
A two-way ANOVA of Aif-1 gene expression indicated there was no interaction between age and alcohol exposure [F(2,45) = 2.40, P = 0.10] or a main effect of age [F(1,45) = 1.77, P = 0.19], but there was a main effect of alcohol [F(2,45) = 8.56, P < 0.001]. Posthoc Dunnett’s tests, comparing alcohol groups to the water control group, revealed that in the young C57BL/6 N mice there was a significant increase in Aif-1 expression during intoxication, but in the aged group the increase in Aif-1 expression was seen both during intoxication and persisted after 7 days of abstinence (see Fig. 4).
Fig. 4.
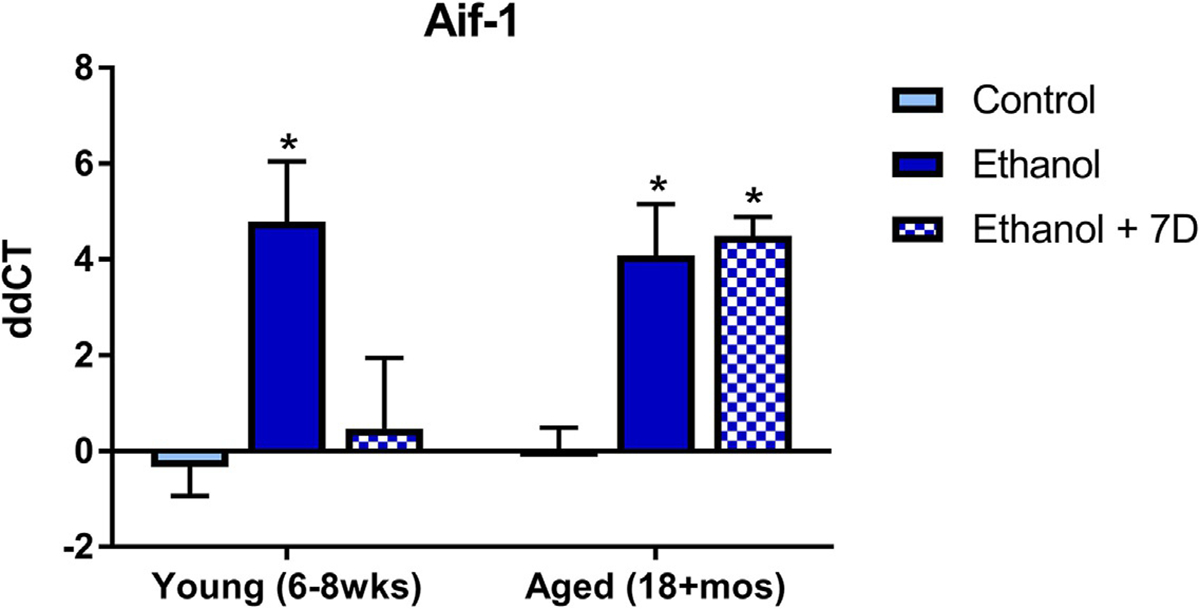
Age causes persistent upregulation of alcohol-induced microglial gene changes in mice exposed to the DID model: Ethanol significantly increases Aif-1, but after 7 days of abstinence this abates in the young animals but remains elevated in the aged ethanol group. *P < 0.05 compared to control treatment.
3.5. No age effects on intoxication scores or alcohol metabolism in Majchrowicz model
There were no effects of age on the perceived intoxication of the rats according to a Mann-Whitney test [U(5,7) = 15.5, P = 0.81]. Because aged rats had greater mass, the total volume of alcohol administered was significantly different than that given to young rats [t(10) = 4.2, P = 0.002], but the dose (g/kg) was consistent between age groups, as it is based on the intoxication score (see Fig. 1). A two-way ANOVA (age × treatment duration) of alcohol dose indicated that there was no interaction [F(5,66) = 0.78, P = 0.57] or main effect of age [F(1,66) = 0.38, P = 0.54], but there was an expected effect of treatment duration [F(5,66) = 31.95, P < 0.0001] (Morris et al., 2010). There was also no statistical difference in the BACs of the different aged groups [t(10) = 0.58, P = 0.57]. Both aged (95% CI [312.8, 387.5 mg/dL]) and young (95% CI [233.4, 392.6 mg/dL]) rats had BACs that were approximately four times the legal limit (see Table 3 for additional details).
Table 3.
Majchrowicz model ethanol animal characteristics.
| Age | Treatment | N | Average dose (g/kg) | Average BACs (mg/dL) | Average weights (g) |
|---|---|---|---|---|---|
| Young (2–3 mos) | Ethanol | 7 | 3.5 ± 0.2 | 313.0 ± 28.67 | 242.87 ± 20.09 |
| Aged (15+ mos) | Ethanol | 5 | 3.4 ± 0.2 | 350.1 ± 13.44 | 464.60 ± 62.70 |
There were no significant differences in the doses or in the BACs of aged and young rats used in the Majchrowicz model.
3.6. Majchrowicz alcohol exposure has age specific effects on microglia reactivity
Following the modified Majchrowicz model, Iba-1 densitometry was assessed in the hippocampus. A two-way ANOVA found that in the DG there was an interaction between age and treatment [F(1,19) = 5.60, P = 0.029] but no main effects of either age [F(1,19) = 3.58, P = 0.07] or treatment [F(1,19) = 0.004, P = 0.94]. Bonferroni tests revealed a significant increase in Iba-1 density in the young alcohol group compared with controls, but this alcohol-induced difference was not observed in the aged group. In the CA1, a similar effect was observed. A two-way ANOVA showed a significant interaction [F(1,19) = 8.40, P = 0.01], but no main effect of either age [F(1,19) = 2.9, P = 0.10] or treatment [F(1,19) = 0.17, P = 0.68]. Bonferroni tests indicated that only in the young animals did alcohol significantly increase the Iba-1 immunoreactivity. Finally in the CA2/3 region, there was no effect of either alcohol [F(1,19) = 2.63, P = 0.12] or age [F(1,19) = 3.02, P = 0.09 nor a significant interaction [F(1,19) = 2.85, P = 0.11] between the two factors according to a two-way ANOVA (see Fig. 5).
Fig. 5.
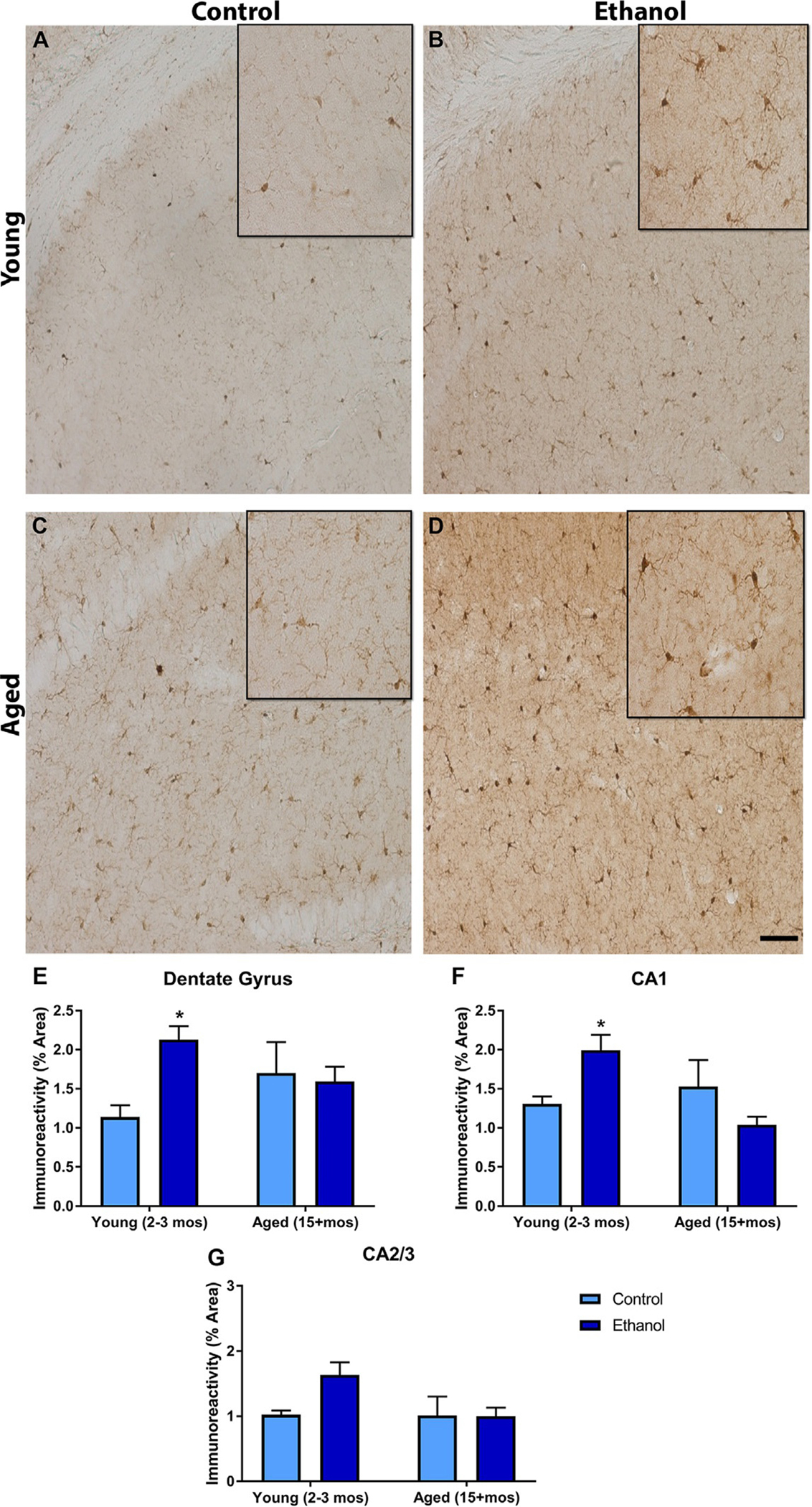
Different microglial responses to alcohol based on age in the modified Majchrowicz Rat model: Iba-1 was expressed throughout the hippocampus. Representative photomicrographs of the dentate gyrus show that Iba-1 was present in the young (A) and aged control animals (C), but that in the young animals ethanol increased the microglial response (B). However, ethanol’s effects on microglia were not present in the aged group (D). Quantification of the subregions indicated that ethanol only increased Iba-1 immunoreactivity in the DG (E) and CA1 (F) of the young animals. No effect of ethanol or age was observed on Iba-1 immunoreactivity in the CA2/3 region (G). *P < 0.05 compared to control treatment. Scale bar= 150 μm.
3.7. Majchrowicz alcohol exposure decreases microglia number
Cell counts were done to further assess Iba-1 in rats. A two-way ANOVA showed that in the DG there was a main effect of alcohol [F(1,19) = 7.28, P = 0.01], but not age [F(1,19) = 0.01, P = 0.93] nor any interaction [F(1,19) = 1.28, P = 0.27] between the two factors. Bonferroni tests showed that alcohol significantly reduced the number of microglia in the DG of both aged and young animals. In the CA1, a two-way ANOVA revealed that there was a main effect of alcohol [F(1,19) = 16.91, P < 0.001], but no effect of age [F(1,19) = 1.06, P = 0.32], nor an interaction [F(1,19) = 0.07, P = 0.80]. Alcohol significantly reduced the number of CA1 microglia for both aged and young rats according to Bonferroni tests. In the CA2/3, a two-way ANOVA indicated there were main effects of both age [F(1,19) = 11.94, P = 0.003] and alcohol [F(1,19) = 14.99, P = 0.001], but no significant interaction between those factors [F(1,19) = 3.25, P = 0.08]. Although age appeared to increase the number of microglia in the CA2/3, Bonferroni tests concluded that alcohol caused a decrease in microglia number in both the aged and young groups (see Fig. 6).
Fig. 6.
Binge drinking-like exposure decreases microglia number in the modified Majchrowicz Rat model: In the dentate gyrus (A), CA1 (B), and CA2/3 fields (C) ethanol significantly decreased the number of microglia among both aged and young animals. However, age significant increased microglia of the CA2/3 fields (C). *P < 0.05 compared to control treatment; #P < 0.05 compared to age matched group.
4. Discussion
Excessive alcohol use has been repeatedly shown to alter a variety of neuroimmune responses including astrocyte activation (Alfonso-Loeches et al., 2010; Kelso et al., 2011), blood-brain barrier integrity (Abdul Muneer, Alikunju, Szlachetka, & Haorah, 2012; Haorah, Knipe, Gorantla, Zheng, & Persidsky, 2007), microglial activation (Crews, Walter, Coleman, & Vetreno, 2017; Marshall et al., 2013), and cytokine production (Doremus-Fitzwater et al., 2014; Sanchez-Alavez et al., 2019). Moreover, human alcoholics have been shown to have an upregulation of a variety of pro-inflammatory glial and cytokine markers (Coleman et al., 2018; Crews et al., 2011, 2017). However, a deeper probing of the literature shows that these responses differ depending on the type of alcohol exposure (chronic versus acute), as well as the period within or after the alcohol exposure (intoxication, withdrawal, or abstinence) (Doremus-Fitzwater et al., 2014; Marshall et al., 2013; Walter & Crews, 2017). Likewise, aging seems to include multiple signs of microglial activation including evidence of excessive inflammation (Block & Hong, 2005; Norden, Muccigrosso, & Godbout, 2015) and senescent, dystrophic microglia (Streit et al., 2004, 2009). Considering the variable neuroimmune response observed in both aging and AUDs, the current studies sought to determine the interaction between aging and binge drinking on microglia using models of both dependence and non-dependence. It is important to denote that these models used two different species to compare dependent and non-dependent binge drinking, which makes a direct comparison difficult. However, the current results provide a snapshot into the microglial response under two separate AUD conditions. The major findings of this report are: (1) the effects of alcohol on microglia are exacerbated by aging, (2) alcohol had disparate effects on microglia in mice after the DID and in rats after the Majchrowicz- binge drinking models, and (3) the effects are not likely to be mediated by pharmacokinetic differences based on age.
4.1. Changes in microglial immunoreactivity: Alcohol and aging effects
Microglial activation is often assessed using the density of cell specific markers like Iba-1, OX-42, ED-1, or OX-6 (Marshall et al., 2013; Raivich et al., 1999). After the DID paradigm, our results indicated that alcohol increased Iba-1 immunoreactivity in the DG in only the young animals. Our results suggest that perhaps the DG is more susceptible to the effects of alcohol compared with the other subregions of the hippocampus. This susceptibility could be due to differences in the density of microglia within the hippocampus. Our results indicate that Iba-1 immunoreactivity between the CA1 and DG are greater than the CA2/3 region even in young control animals. Similarly, others have reported that the density of microglia within the dorsal DG and CA1 are higher than the CA2/3 regions ( Jinno, Fleischer, Eckel, Schmidt, & Kosaka, 2007). One of the unique characteristic of the DG from other hippocampal subregions is its neurogenic niche. We know that excessive alcohol consumption in the DID model can reduce neurogenesis (Belmer, Patkar, Lanoue, & Bartlett, 2018), and that in other pathologies the microglia of the DG play a critical role in neurogenesis (Ekdahl, Kokaia, & Lindvall, 2009; Morrens, Van Den Broeck, & Kempermann, 2012; Sierra et al., 2010). The microglial response can be critical in the proliferation of progenitor cells as well as the survival, migration, and differentiation of new born cells (Ekdahl et al., 2009). It is not possible with the current work to tie changes in microglia to neurogenic changes, but it represents a future avenue of work. At minimum, we see that the DG seems to be more susceptible to the influence binge-like alcohol consumption on microglia in this DID model.
In the modified Majchrowicz procedure with dependent-like BACs, binge-like alcohol exposure resulted in an increase in the Iba-1 immunoreactivity in the young animals in both the DG and the CA1 during intoxication. This finding aligns with other reports showing that the 4-day Majchrowicz model induces microglial activation during intoxication (Marshall et al., 2013; Marshall, Geil, et al., 2016; Zhao et al., 2013). Unlike the DID model, the changes in microglia extended beyond the DG and include the CA1 region. This could be an example of more extensive changes in the CNS caused by the higher BACs of this model (Ende et al., 2006); however, future studies should directly compare dependence and non-dependence in the same animal model. Again, it is also possible that the effects observed here may be due to a difference in rat and mice neuroimmune responses independent of higher BACs or alcohol-dependence.
Importantly, in both the Majchrowicz and DID model, alcohol failed to significantly impact Iba-1 immunoreactivity in the aged animals. In the current studies, there was a main effect of aging on Iba-1 immunoreactivity in mice regardless of the alcohol effects. Our data suggest that there is an increase in microglia activation throughout the subregions of the hippocampus in the aged mice. A similar trend for an aging effect was seen in the rat hippocampus, but failed to reach statistical significance. The lack of an exacerbation of alcohol’s effects on microglia density during aging was surprising. Many have postulated that aging is associated with microglial priming, meaning that the microglial response would be exacerbated by other modulators (e.g., binge-alcohol) to produce more pro-inflammatory cytokines and/or increase phagocytic factors (Dilger & Johnson, 2008; Norden et al., 2015; Perry & Holmes, 2014). Our results did not indicate an exacerbation of Iba-1 immunoreactivity by alcohol in aged mice or rats. These findings concur with a previous study that found no age-related increases in microglia reactivity following lipopolysaccharide administration (Hart, Wyttenbach, Perry, & Teeling, 2012). However, it is not possible to completely rule out the priming theory without a more thorough classification of microglial activation. These studies only used Iba-1 as a marker of microglia reactivity, but it is possible that the microglia after alcohol exposure in the aged groups have other indicators of more pro-inflammatory microglial activation, or that the morphology of microglia may have changed as a result of alcohol exposure. In the future, markers like OX-6 or ED-1 should be used to assess whether microglia are progressing to a more proinflammatory state. Using only Iba-1 is a limitation of the current study, but it does provide a basic snapshot of the changes to microglia reactivity following binge alcohol in aged rodents.
4.2. Changes in microglia number: Alcohol and aging effects
Iba-1 immunohistochemistry was also assessed to determine whether binge drinking differentially affects microglia number as a function of age. Following the DID paradigm, we found that alcohol decreased the number of microglial cells in the DG in both aged and young mice. The extent of the decrease in microglia seems to be greater in the aged population, but this was difficult to tease apart as both age and alcohol had independent effects. Our t-test of the percent difference indicated that there was an exacerbated cell loss following alcohol consumption in aged mice. However, in the CA1, there was only a main effect of alcohol, suggesting that there was no additive effect of aging on the decrease in microglial cell number. Similar to the immunoreactivity data, it appears that the DG may be more sensitive to alcohol’s effects on microglia compared with the other portions of the hippocampus. Interestingly, in the CA2/3 regions we found that aging alone resulted in an increase in the number of microglia in mice regardless of treatment. Together these results indicate region-specific effects of alcohol consumption and aging in non-dependent subjects. However, in our dependent model, there was a main effect of alcohol on microglia number in all of the hippocampal subregions. A reduction in the overall number of microglia in the hippocampus following the Majchrowicz model has previously been reported (Marshall, Geil, et al., 2016), but this study failed to find an exacerbation in cell loss from aging.
Microglial cell loss could have various implications. For example, in a recent study, the Crews lab showed that depletion of microglia ameliorated withdrawal-induced pro-inflammatory genes and actually led to increased production of anti-inflammatory genes (Walter & Crews, 2017). The loss of microglia after alcohol has the potential to be beneficial; however, in other models of neurodegeneration, significant losses in microglial function actually exacerbated cell loss (Lalancette-Hebert, Gowing, Simard, Weng, & Kriz, 2007; Wainwright et al., 2009). The timing of microglial activation (e.g., acute vs prolonged; immediate vs delayed) is critical in whether microglia are beneficial or detrimental within a specific cellular microenvironment (DiSabato, Quan, & Godbout, 2016; Kriz, 2006). Whether the current loss in the number of microglia caused by alcohol is protective or damaging remains to be determined. Moreover, the implications of microglial dysregulation may be more negative in aging populations with the increase in senescent microglia and the loss of other support mechanisms mediated by microglia (Streit, 2006; Streit et al., 2009). Alcohol-induced microglial cell loss may be a component of the machinery leading to neurodegeneration in human alcoholics (Sullivan, Marsh, Mathalon, Lim, & Pfefferbaum, 1995). Future studies should elucidate the functional implication of the reduction in the number of microglia and whether there is direct toxicity to these cells.
4.3. Age-associated prolonged increases in Aif-1
Alcohol caused an upregulation in the Aif-1 gene in both age and young mice within the hippocampus following DID exposure. This effect may be specific to the hippocampus or to Aif-1 as others have not found significant changes in a conglomerate of microglial genes in the prefrontal cortex (Osterndorff-Kahanek, Ponomarev, Blednov, & Harris, 2013). More importantly, these findings indicate, that unlike in young mice, the increase in Aif-1 mRNA persists during abstinence in aged mice. This finding suggests that with age the effects of non-dependent binge drinking may have more permanence. Unfortunately, it was not possible to test this in the Majchrowicz dependence model, but the persistence in gene changes after DID exposure in mice is particularly concerning considering the myriad of neuroimmunomodulatory events that are present in aging.
4.4. Age exacerbation of microglia is not caused by metabolic differences
In both the DID and Majchrowicz model, the BACs among aged and young animals were similar, suggesting that the additional effects of aging on microglia observed were not due to differences in BACs. Older people are thought to be more sensitive to the behavioral effects of alcohol (Nixon, Prather, & Lewis, 2014; Squeglia et al., 2014), but we did not find that age impacted either voluntary consumption in the DID model or the observed intoxication in the Majchrowicz model. It is probable that if the Majchrowicz model was extended beyond 2 days a difference in intoxication between young and aged animals may have been observed— similar to that seen between adolescents and adults (Morris et al., 2010). Others have reported similar findings that the differences in observed behavior between age groups are not merely due to metabolic changes (Hoffman et al., 2019; Wood, Armbrecht, & Wise, 1982). This is important because it suggests that there is a mechanism that underlies the change in microglial response that goes beyond metabolic differences and the alcohol concentration that reaches the brain. The unknown mechanism represents a novel point of intervention for ameliorating the influence of aging on ethanol’s effects on microglia and the neuroimmune response.
5. Conclusion
This study is a small step toward understanding the consequences of binge drinking on the neuroimmune system as a function of advanced age. Microglia are generally some of the first responders to perturbation within the CNS. The differential effects that were observed based on age during binge-alcohol exposure may represent a prelude to increased brain damage and susceptibility to neurodegenerative diseases. This is particularly true given the persisting effects of alcohol that were observed in the non-dependent model (DID). Moreover, in many of the hippocampal subregions there was a lack of alcohol-induced microglial reactivity in the aged group. The age-induced priming aspect of microglia would normally manifest as a greater pro-inflammatory response in the presence of other immunomodulators. The fact that alcohol does not elicit such a response could mean that the brain has lost a critical first responder to a toxic event. To further understand whether aging truly primes or exacerbates alcohol’s effects, future studies should use more definitive markers of microglial phenotypes to assess whether they become more pro-inflammatory in nature.
Intriguingly, in both models, we found that alcohol seemed to decrease microglia. An additive effect of aging on alcohol-related decrease in microglia number was only observed in particular hippocampal subregions and only in the DID model; however, the functional aspects of losing microglia may be very different in an aged versus young binge drinkers. This is particularly true as it is believed that aged microglia begin to have a decline in their neuroprotective function and therefore a loss of microglia caused by binge drinking may be more detrimental as individuals age (Streit et al., 2009). Future studies are necessary to determine the functional implications of the microglial loss caused by alcohol. As the nation continues to age, it will be critical to explore how alcohol abuse and misuse impacts on the CNS vary with age. This is especially true of microglia and the neuroimmune system because of the diversity of responses possible as well as the pervasive nature of their influence on biological and behavioral consequences.
Acknowledgments
The authors thank Scot McIntosh and Christopher Trevisani for technical assistance. This research was supported by the NIAAA (U54 AA019765) and generous startup funds from the High Point University Fred Wilson School of Pharmacy.
References
- Abdul Muneer PM, Alikunju S, Szlachetka AM, & Haorah J (2012). The mechanisms of cerebral vascular dysfunction and neuroinflammation by MMP-mediated degradation of VEGFR-2 in alcohol ingestion. Arteriosclerosis, Thrombosis, and Vascular Biology, 32(5), 1167–1177. ATVBAHA.112.247668 [pii] 10.1161/ATVBAHA.112.247668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afshar M, Netzer G, Salisbury-Afshar E, Murthi S, & Smith GS (2016). Injured patients with very high blood alcohol concentrations. Injury, 47(1), 83–88. 10.1016/j.injury.2015.10.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, & Guerri C (2010). Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. The Journal of Neuroscience, 30(24), 8285–8295. 30/24/8285 [pii] 10.1523/JNEUROSCI.0976-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. (2013). Diagnostic and statistical manual of mental disorders (5th ed.). 10.1176/appi.books.9780890425596. [DOI]
- Bajo M, Herman MA, Varodayan FP, Oleata CS, Madamba SG, Harris RA, et al. (2015). Role of the IL-1 receptor antagonist in ethanol-induced regulation of GABAergic transmission in the central amygdala. Brain, Behavior, and Immunity, 45, 189–197. 10.1016/j.bbi.2014.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajo M, Varodayan FP, Madamba SG, Robert AJ, Casal LM, Oleata CS, et al. (2015). IL-1 interacts with ethanol effects on GABAergic transmission in the mouse central amygdala. Frontiers in Pharmacology, 6, 49. 10.3389/fphar.2015.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels C, Kunert HJ, Stawicki S, Kroner-Herwig B, Ehrenreich H, & Krampe H (2007). Recovery of hippocampus-related functions in chronic alcoholics during monitored long-term abstinence. Alcohol and Alcoholism, 42(2), 92–102. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17179161. [DOI] [PubMed] [Google Scholar]
- Bartsch T, & Wulff P (2015). The hippocampus in aging and disease: From plasticity to vulnerability. Neuroscience, 309, 1–16. 10.1016/j.neuroscience.2015.07.084. [DOI] [PubMed] [Google Scholar]
- Belmer A, Patkar OL, Lanoue V, & Bartlett SE (2018). 5-HT1A receptor-dependent modulation of emotional and neurogenic deficits elicited by prolonged consumption of alcohol. Scientific Reports, 8(1), 2099. 10.1038/s41598-018-20504-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazer DG, & Wu LT (2009). The epidemiology of at-risk and binge drinking among middle-aged and elderly community adults: National Survey on Drug Use and Health. The American Journal of Psychiatry, 166(10), 1162–1169. 10.1176/appi.ajp.2009.09010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazer DG, & Wu LT (2012). Patterns of tobacco use and tobacco-related psychiatric morbidity and substance use among middle-aged and older adults in the United States. Aging & Mental Health, 16(3), 296–304. 10.1080/13607863.2011.615739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Benavidez JM, Black M, & Harris RA (2014). Inhibition of phosphodiesterase 4 reduces ethanol intake and preference in C57BL/6J mice. Frontiers in Neuroscience, 8, 129. 10.3389/fnins.2014.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, & Hong JS (2005). Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Progress in Neurobiology, 76(2), 77–98. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16081203. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Bilousova TV, Puntambekar SS, Melchior B, Doose JM, & Ethell IM (2007). A rose by any other name? The potential consequences of microglial heterogeneity during CNS health and disease. Neurotherapeutics, 4(4), 571–579. S1933-7213(07)00137-7 [pii] 10.1016/j.nurt.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center for Behavioral Health Statistics and Quality. (2015). Behavioral health trends in the United States: Results from the 2014 National Survey on Drug Use and Health. HHS Publication No. SMA. Retrieved from http://www.samhsa.gov/data/.
- Centers for Disease Control and Prevention (CDC). (2013). Vital signs: Binge drinking prevalence, frequency, and intensity among adults—United States, 2010. MMWR. Morbidity and Mortality Weekly Report, 61(1), 14–19. doi:mm6101a4 [pii]. [PubMed] [Google Scholar]
- Coleman LG Jr., Zou J, Qin L, & Crews FT (2018). HMGB1/IL-1beta complexes regulate neuroimmune responses in alcoholism. Brain, Behavior, and Immunity, 72, 61–77. 10.1016/j.bbi.2017.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller JK, & Hutchinson MR (2012). Implications of central immune signaling caused by drugs of abuse: Mechanisms, mediators and new therapeutic approaches for prediction and treatment of drug dependence. Pharmacology & Therapeutics, 134(2), 219–245. 10.1016/j.pharmthera.2012.01.008. [DOI] [PubMed] [Google Scholar]
- Costantini E, D’Angelo C, & Reale M (2018). The role of immunosenescence in neurodegenerative diseases. Mediators of Inflammation, 2018, 6039171. 10.1155/2018/6039171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox BR, Olney JJ, Lowery-Gionta EG, Sprow GM, Rinker JA, Navarro M, et al. (2013). Repeated cycles of binge-like ethanol (EtOH)-drinking in male C57BL/6J mice augments subsequent voluntary EtOH intake but not other dependence-like phenotypes. Alcoholism, Clinical and Experimental Research, 37(10), 1688–1695. 10.1111/acer.12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC, Harris RA, & Koob GF (2011). Preclinical studies of alcohol binge drinking. Annals of the New York Academy of Sciences, 1216, 24–40. 10.1111/j.1749-6632.2010.05895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC, Metten P, Rhodes JS, Yu CH, Brown LL, Phillips TJ, et al. (2009). A line of mice selected for high blood ethanol concentrations shows drinking in the dark to intoxication. Biological Psychiatry, 65(8), 662–670. 10.1016/j.biopsych.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT (2012). Immune function genes, genetics, and the neurobiology of addiction. Alcohol Research: Current Reviews , 34(3), 355–361. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=23134052. [PMC free article] [PubMed] [Google Scholar]
- Crews FT, & Nixon K (2009). Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol and Alcoholism, 44(2), 115–127. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18940959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Qin L, Sheedy D, Vetreno RP, & Zou J (2013). High mobility group box 1/toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biological Psychiatry, 73(7), 602–612. 10.1016/j.biopsych.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, & Vetreno RP (2014). Neuroimmune basis of alcoholic brain damage. International Review of Neurobiology, 118, 315–357. 10.1016/B978-0-12-801284-0.00010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, & Vetreno RP (2016). Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology, 233(9), 1543–1557. 10.1007/s00213-015-3906-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Walter TJ, Coleman LG Jr., & Vetreno RP (2017). Toll-like receptor signaling and stages of addiction. Psychopharmacology, 234(9–10), 1483–1498. 10.1007/s00213-017-4560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Zou J, & Qin L (2011). Induction of innate immune genes in brain create the neurobiology of addiction. Brain, Behavior, and Immunity, 25(Suppl. 1), S4–S12. S0889-1591(11)00071-7 [pii] 10.1016/j.bbi.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C, Noronha A, Morikawa H, Alvarez VA, Stuber GD, Szumlinski KK, et al. (2013). New insights on neurobiological mechanisms underlying alcohol addiction. Neuropharmacology, 67, 223–232. 10.1016/j.neuropharm.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI (2008). Ethanol-BDNF interactions: Still more questions than answers. Pharmacology & Therapeutics, 118(1), 36–57. 10.1016/j.pharmthera.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BM, Salinas-Navarro M, Cordeiro MF, Moons L, & De Groef L (2017). Characterizing microglia activation: A spatial statistics approach to maximize information extraction. Scientific Reports, 7(1), 1576. 10.1038/s41598-017-01747-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleidi M, Jaggle M, & Rubino G (2015). Immune aging, dysmetabolism, and inflammation in neurological diseases. Frontiers in Neuroscience, 9, 172. 10.3389/fnins.2015.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermietzel R, & Spray DC (1998). From neuro-glue (‘Nervenkitt’) to glia: A prologue. Glia, 24(1), 1–7. . [pii]. [DOI] [PubMed] [Google Scholar]
- Dilger RN, & Johnson RW (2008). Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. Journal of Leukocyte Biology, 84(4), 932–939. jlb.0208108 [pii] 10.1189/jlb.0208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiSabato DJ, Quan N, & Godbout JP (2016). Neuroinflammation: The devil is in the details. Journal of Neurochemistry, 139(Suppl. 2), 136–153. 10.1111/jnc.13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doremus-Fitzwater TL, Buck HM, Bordner K, Richey L, Jones ME, & Deak T (2014). Intoxication- and withdrawal-dependent expression of central and peripheral cytokines following initial ethanol exposure. Alcoholism, Clinical and Experimental Research, 38(8), 2186–2198. 10.1111/acer.12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doremus-Fitzwater TL, Gano A, Paniccia JE, & Deak T (2015). Male adolescent rats display blunted cytokine responses in the CNS after acute ethanol or lipopolysaccharide exposure. Physiology & Behavior, 148, 131–144. 10.1016/j.physbeh.2015.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GA, Nigg JT, & Sullivan EL (2019). Neuroinflammation as a risk factor for attention deficit hyperactivity disorder. Pharmacology, Biochemistry, and Behavior, 182, 22–34. 10.1016/j.pbb.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl CT, Kokaia Z, & Lindvall O (2009). Brain inflammation and adult neurogenesis: The dual role of microglia. Neuroscience, 158(3), 1021–1029. S0306-4522(08)00971-8 [pii] 10.1016/j.neuroscience.2008.06.052. [DOI] [PubMed] [Google Scholar]
- Enache D, Pariante CM, & Mondelli V (2019). Markers of central inflammation in major depressive disorder: A systematic review and meta-analysis of studies examining cerebrospinal fluid, positron emission tomography and post-mortem brain tissue. Brain, Behavior, and Immunity, 81, 24–40. 10.1016/j.bbi.2019.06.015. [DOI] [PubMed] [Google Scholar]
- Ende G, Walter S, Welzel H, Demirakca T, Wokrina T, Ruf M, et al. (2006). Alcohol consumption significantly influences the MR signal of frontal choline-containing compounds. NeuroImage, 32(2), 740–746. 10.1016/j.neuroimage.2006.03.049. [DOI] [PubMed] [Google Scholar]
- Faingold CL (2008). The Majchrowicz binge alcohol protocol: An intubation technique to study alcohol dependence in rats. Current Protocols in Neuroscience, 44, 9.28.1–9.28.12. 10.1002/0471142301.ns0928s44. Chapter 9, Unit 9 28. [DOI] [PubMed] [Google Scholar]
- Fillmore MT, & Jude R (2011). Defining “binge” drinking as five drinks per occasion or drinking to a .08% BAC: Which is more sensitive to risk? The American Journal on Addictions, 20(5), 468–475. 10.1111/j.1521-0391.2011.00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank-Cannon TC, Alto LT, McAlpine FE, & Tansey MG (2009). Does neuroinflammation fan the flame in neurodegenerative diseases? Molecular Neurodegeneration, 4, 47. 1750-1326-4-47 [pii] 10.1186/1750-1326-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego BI, & de Gracia P (2016). Automatic counting of microglial cell activation and its applications. Neural Regeneration Research, 11(8), 1212–1215. 10.4103/1673-5374.189166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gano A, Doremus-Fitzwater TL, & Deak T (2017). A cross-sectional comparison of ethanol-related cytokine expression in the hippocampus of young and aged Fischer 344 rats. Neurobiology of Aging, 54, 40–53. 10.1016/j.neurobiolaging.2017.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, & Gage FH (2010). Mechanisms underlying inflammation in neurodegeneration. Cell, 140(6), 918–934. 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BF, Dawson DA, Stinson FS, Chou SP, Dufour MC, & Pickering RP (2004). The 12-month prevalence and trends in DSM-IV alcohol abuse and dependence: United States, 1991–1992 and 2001–2002. Drug and Alcohol Dependence, 74(3), 223–234. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15194200. [DOI] [PubMed] [Google Scholar]
- Grant BF, Goldstein RB, Saha TD, Chou SP, Jung J, Zhang H, et al. (2015). Epidemiology of DSM-5 alcohol use disorder: Results from the National Epidemiologic Survey on alcohol and related conditions III. JAMA Psychiatry, 72(8), 757–766. 10.1001/jamapsychiatry.2015.0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BF, Saha TD, Ruan WJ, Goldstein RB, Chou SP, Jung J, et al. (2016). Epidemiology of DSM-5 drug use disorder: Results from the National Epidemiologic Survey on alcohol and related conditions-III. JAMA Psychiatry, 73(1), 39–47. 10.1001/jamapsychiatry.2015.2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifasi IR, McIntosh SE, Thomas RD, Lysle DT, Thiele TE, & Marshall SA (2019). Characterization of the hippocampal neuroimmune response to binge-like ethanol consumption in the drinking in the dark model. Neuroimmunomodulation, 26(1), 19–32. 10.1159/000495210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haorah J, Knipe B, Gorantla S, Zheng J, & Persidsky Y (2007). Alcohol-induced blood-brain barrier dysfunction is mediated via inositol 1,4,5-triphosphate receptor (IP3R)-gated intracellular calcium release. Journal of Neurochemistry, 100(2), 324–336. JNC4245 [pii]. 10.1111/j.1471-4159.2006.04245.x. [DOI] [PubMed] [Google Scholar]
- Hart AD, Wyttenbach A, Perry VH, & Teeling JL (2012). Age related changes in microglial phenotype vary between CNS regions: Grey versus white matter differences. Brain, Behavior, and Immunity, 26(5), 754–765. 10.1016/j.bbi.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harting MT, Jimenez F, Adams SD, Mercer DW, & Cox CS Jr. (2008). Acute, regional inflammatory response after traumatic brain injury: Implications for cellular therapy. Surgery, 144(5), 803–813. S0039-6060(08)00356-5 [pii] 10.1016/j.surg.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes DM, Deeny MA, Shaner CA, & Nixon K (2013). Determining the threshold for alcohol-induced brain damage: New evidence with gliosis markers. Alcoholism, Clinical and Experimental Research, 37(3), 425–434. 10.1111/j.1530-0277.2012.01955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, & Crews FT (2008). Increased MCP-1 and microglia in various regions of the human alcoholic brain. Experimental Neurology, 210(2), 349–358. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18190912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman JL, Faccidomo S, Kim M, Taylor SM, Agoglia AE, May AM, et al. (2019). Alcohol drinking exacerbates neural and behavioral pathology in the 3xTg-AD mouse model of Alzheimer’s disease. In Deak T & Savage LM (Eds.), International Review of Neurobiology. Vol. 148 (pp. 169–230). Academic Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou R, & Baldwin DS (2012). A neuroimmunological perspective on anxiety disorders. Human Psychopharmacology, 27(1), 6–14. 10.1002/hup.1259. [DOI] [PubMed] [Google Scholar]
- Hovens I, Nyakas C, & Schoemaker RG (2014). A novel method for evaluating microglial activation using ionized calcium-binding adaptor protein-1 staining: Cell body to cell size ratio. Neuroimmunology and Neuroinflammation, 1, 82–88. Retrieved from https://nnjournal.net/article/view/79/511. [Google Scholar]
- Hu W, Lu T, Chen A, Huang Y, Hansen R, Chandler LJ, et al. (2011). Inhibition of phosphodiesterase-4 decreases ethanol intake in mice. Psychopharmacology, 218(2), 331–339. 10.1007/s00213-011-2290-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt WA (1993). Are binge drinkers more at risk of developing brain damage? Alcohol, 10(6), 559–561. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=8123218. [DOI] [PubMed] [Google Scholar]
- Imai Y, Ibata I, Ito D, Ohsawa K, & Kohsaka S (1996). A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochemical and Biophysical Research Communications, 224(3), 855–862. S0006-291X(96)91112-2 [pii] 10.1006/bbrc.1996.1112. [DOI] [PubMed] [Google Scholar]
- Immonen S, Valvanne J, & Pitkala KH (2013). The prevalence of potential alcohol-drug interactions in older adults. Scandinavian Journal of Primary Health Care, 31(2), 73–78. 10.3109/02813432.2013.788272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, & Kohsaka S (1998). Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Research Molecular Brain Research, 57(1), 1–9. doi:S0169328X98000400 [pii]. [DOI] [PubMed] [Google Scholar]
- Ito D, Tanaka K, Suzuki S, Dembo T, & Fukuuchi Y (2001). Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke, 32(5), 1208–1215. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11340235. [DOI] [PubMed] [Google Scholar]
- Jeong WI, Osei-Hyiaman D, Park O, Liu J, Batkai S, Mukhopadhyay P, et al. (2008). Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metabolism, 7(3), 227–235. 10.1016/j.cmet.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Jinno S, Fleischer F, Eckel S, Schmidt V, & Kosaka T (2007). Spatial arrangement of microglia in the mouse hippocampus: A stereological study in comparison with astrocytes. Glia, 55(13), 1334–1347. 10.1002/glia.20552. [DOI] [PubMed] [Google Scholar]
- Joe KH, Kim YK, Kim TS, Roh SW, Choi SW, Kim YB, et al. (2007). Decreased plasma brain-derived neurotrophic factor levels in patients with alcohol dependence. Alcoholism, Clinical and Experimental Research, 31(11), 1833–1838. ACER507 [pii] 10.1111/j.1530-0277.2007.00507.x. [DOI] [PubMed] [Google Scholar]
- Juarez J, & de Barrios Tomasi E (1999). Sex differences in alcohol drinking patterns during forced and voluntary consumption in rats. Alcohol, 19(1), 15–22. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/10487383. [DOI] [PubMed] [Google Scholar]
- Kane CJ, Phelan KD, Douglas JC, Wagoner G, Johnson JW, Xu J, et al. (2013). Effects of ethanol on immune response in the brain: Region-specific changes in aged mice. Journal of Neuroinflammation, 10, 66. 10.1186/1742-2094-10-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly KM, Nadon NL, Morrison JH, Thibault O, Barnes CA, & Blalock EM (2006). The neurobiology of aging. Epilepsy Research, 68(Suppl. 1), S5–20. 10.1016/j.eplepsyres.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Kelso ML, Liput DJ, Eaves DW, & Nixon K (2011). Upregulated vimentin suggests new areas of neurodegeneration in a model of an alcohol use disorder. Neuroscience, 197, 381–393. 10.1016/j.neuroscience.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Lee G, John SW, Maeda N, & Smithies O (2002). Molecular phenotyping for analyzing subtle genetic effects in mice: Application to an angiotensinogen gene titration. Proceedings of the National Academy of Sciences of the United States of America, 99(7), 4602–4607. 10.1073/pnas.072083799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, & Volkow ND (2010). Neurocircuitry of addiction. Neuropsychopharmacology, 35(1), 217–238. 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriz J (2006). Inflammation in ischemic brain injury: Timing is important. Critical Reviews in Neurobiology, 18(1–2), 145–157. 2b9060795f4edc81,5c2b39a3421e698b [pii]. [DOI] [PubMed] [Google Scholar]
- Kuerbis A, Sacco P, Blazer DG, & Moore AA (2014). Substance abuse among older adults. Clinics in Geriatric Medicine, 30(3), 629–654. 10.1016/j.cger.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AY, & Todd KG (2008). Differential regulation of trophic and proinflammatory microglial effectors is dependent on severity of neuronal injury. Glia, 56(3), 259–270. 10.1002/glia.20610. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Gowing G, Simard A, Weng YC, & Kriz J (2007). Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. The Journal of Neuroscience, 27(10), 2596–2605. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17344397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liput DJ, Hammell DC, Stinchcomb AL, & Nixon K (2013). Transdermal delivery of cannabidiol attenuates binge alcohol-induced neurodegeneration in a rodent model of an alcohol use disorder. Pharmacology, Biochemistry, and Behavior, 111, 120–127. 10.1016/j.pbb.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majchrowicz E (1975). Induction of physical dependence upon ethanol and the associated behavioral changes in rats. Psychopharmacologia, 43(3), 245–254. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=1237914. [DOI] [PubMed] [Google Scholar]
- Marshall SA, Casachahua JD, Rinker JA, Blose AK, Lysle DT, & Thiele TE (2016). IL-1 receptor signaling in the basolateral amygdala modulates binge-like ethanol consumption in male C57BL/6J mice. Brain, Behavior, and Immunity, 51, 258–267. 10.1016/j.bbi.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SA, Geil CR, & Nixon K (2016). Prior binge ethanol exposure potentiates the microglial response in a model of alcohol-induced neurodegeneration. Brain Sciences, 6(2), 16. 10.3390/brainsci6020016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SA, McClain JA, Kelso ML, Hopkins DM, Pauly JR, & Nixon K (2013). Microglial activation is not equivalent to neuroinflammation in alcohol-induced neurodegeneration: The importance of microglia phenotype. Neurobiology of Disease, 54, 239–251. 10.1016/j.nbd.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SA, McKnight KH, Blose AK, Lysle DT, & Thiele TE (2017). Modulation of binge-like ethanol consumption by IL-10 signaling in the basolateral amygdala. Journal of Neuroimmune Pharmacology, 12(2), 249–259. 10.1007/s11481-016-9709-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SA, Rinker JA, Harrison LK, Fletcher CA, Herfel TM, & Thiele TE (2015). Assessment of the effects of 6 standard rodent diets on binge-like and voluntary ethanol consumption in male C57BL/6J mice. Alcoholism, Clinical and Experimental Research, 39(8), 1406–1416. 10.1111/acer.12773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews DB, Schneider A, Kastner A, Scaletty S, & Szenay R (2019). I can’t drink what I used to: The interaction between ethanol and the aging brain. In Deak T & Savage LM (Eds.), International Review of Neurobiology. Vol. 148 (pp. 79–99). Academic Press. [DOI] [PubMed] [Google Scholar]
- Maynard ME, Barton EA, Robinson CR, Wooden JI, & Leasure JL (2018). Sex differences in hippocampal damage, cognitive impairment, and trophic factor expression in an animal model of an alcohol use disorder. Brain Structure & Function, 223(1), 195–210. 10.1007/s00429-017-1482-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain JA, Morris SA, Deeny MA, Marshall SA, Hayes DM, Kiser ZM, et al. (2011). Adolescent binge alcohol exposure induces long-lasting partial activation of microglia. Brain, Behavior, and Immunity, 25(Suppl. 1), S120–S128. 10.1016/j.bbi.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain JA, Morris SA, Marshall SA, & Nixon K (2014). Ectopic hippocampal neurogenesis in adolescent male rats following alcohol dependence. Addiction Biology, 19(4), 687–699. 10.1111/adb.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelucci A, Heurtaux T, Grandbarbe L, Morga E, & Heuschling P (2009). Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. Journal of Neuroimmunology, 210(1–2), 3–12. 10.1016/j.jneuroim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Morales I, Guzman-Martinez L, Cerda-Troncoso C, Farias GA, & Maccioni RB (2014). Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Frontiers in Cellular Neuroscience, 8, 112. 10.3389/fncel.2014.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrens J, Van Den Broeck W, & Kempermann G (2012). Glial cells in adult neurogenesis. Glia, 60(2), 159–174. 10.1002/glia.21247. [DOI] [PubMed] [Google Scholar]
- Morris SA, Kelso ML, Liput DJ, Marshall SA, & Nixon K (2010). Similar withdrawal severity in adolescents and adults in a rat model of alcohol dependence. Alcohol, 44(1), 89–98. 10.1016/j.alcohol.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIAAA. (2004). NIAAA council approves definition of binge drinking. National Institute on Alcohol Abuse and Alcoholism Newsletter. [Google Scholar]
- Nixon K, & Morris SA (2008). Neural stem cells: A potential mechanism in alcoholic neurodegeneration and recovery. In Sher L (Ed.), Research on the neurobiology of alcohol use disorders. New York: Nova Science Publishers. [Google Scholar]
- Nixon SJ, Prather R, & Lewis B (2014). Sex differences in alcohol-related neurobehavioral consequences. Handbook of Clinical Neurology, 125, 253–272. 10.1016/B978-0-444-62619-6.00016-1. [DOI] [PubMed] [Google Scholar]
- Norden DM, Muccigrosso MM, & Godbout JP (2015). Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology, 96(Pt. A), 29–41. 10.1016/j.neuropharm.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes PT, Kipp BT, Reitz NL, & Savage LM (2019). Aging with alcohol-related brain damage: Critical brain circuits associated with cognitive dysfunction. In Deak T & Savage LM (Eds.), International Review of Neurobiology. Vol. 148 (pp. 101–168). Academic Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obernier JA, Bouldin TW, & Crews FT (2002). Binge ethanol exposure in adult rats causes necrotic cell death. Alcoholism, Clinical and Experimental Research, 26(4), 547–557. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11981132. [PubMed] [Google Scholar]
- Obernier JA, White AM, Swartzwelder HS, & Crews FT (2002). Cognitive deficits and CNS damage after a 4-day binge ethanol exposure in rats. Pharmacology, Biochemistry, and Behavior, 72(3), 521–532. doi:S0091305702007153 [pii]. [DOI] [PubMed] [Google Scholar]
- Osberg TM, Billingsley K, Eggert M, & Insana M (2012). From animal house to old school: A multiple mediation analysis of the association between college drinking movie exposure and freshman drinking and its consequences. Addictive Behaviors, 37(8), 922–930. 10.1016/j.addbeh.2012.03.030. [DOI] [PubMed] [Google Scholar]
- Osterndorff-Kahanek E, Ponomarev I, Blednov YA, & Harris RA (2013). Gene expression in brain and liver produced by three different regimens of alcohol consumption in mice: Comparison with immune activation. PLoS One, 8(3). e59870. 10.1371/journal.pone.0059870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris JJ, & Frye CA (2009). Alcohol dose-dependently enhances 3 -Androstanediol formation in frontal cortex of male rats concomitant with aggression. The Open Neuropsychopharmacology Journal, 2, 1–11. Retrieved from https://benthamopen.com/contents/pdf/TONEUROPPJ/TONEUROPPJ-2-1.pdf. [Google Scholar]
- Patterson SL (2015). Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1beta, BDNF and synaptic plasticity. Neuropharmacology, 96(Pt. A), 11–18. 10.1016/j.neuropharm.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, & Franklin KBJ (2004). The mouse brain in stereotaxic coordinates. Compact (2nd ed.). Amsterdam; Boston: Elsevier Academic Press. [Google Scholar]
- Paxinos G, & Watson C (2009). In Paxinos G & Watson C (Eds.), The rat brain in stereotaxic coordinates. (6th ed.). London: Elsevier Academic. Compact. [Google Scholar]
- Penfield W (1925). Microglia and the process of phagocytosis in gliomas. The American Journal of Pathology, 1(1), 77–90. 15. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19969634. [PMC free article] [PubMed] [Google Scholar]
- Perry VH, & Holmes C (2014). Microglial priming in neurodegenerative disease. Nature Reviews Neurology, 10(4), 217–224. 10.1038/nrneurol.2014.38. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Lim KO, Zipursky RB, Mathalon DH, Rosenbloom MJ, Lane B, et al. (1992). Brain gray and white matter volume loss accelerates with aging in chronic alcoholics: A quantitative MRI study. Alcoholism, Clinical and Experimental Research, 16(6), 1078–1089. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=1471762. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, Mathalon DH, & Lim KO (1997). Frontal lobe volume loss observed with magnetic resonance imaging in older chronic alcoholics. Alcoholism, Clinical and Experimental Research, 21(3), 521–529. [DOI] [PubMed] [Google Scholar]
- Postler E, Rimner A, Beschorner R, Schluesener HJ, & Meyermann R (2000). Allograft-inflammatory-factor-1 is upregulated in microglial cells in human cerebral infarctions. Journal of Neuroimmunology, 104(1), 85–91. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/10683518. [DOI] [PubMed] [Google Scholar]
- Prieto GA, Tong L, Smith ED, & Cotman CW (2019). TNFalpha and IL-1beta but not IL-18 suppresses hippocampal long-term potentiation directly at the synapse. Neurochemical Research, 44(1), 49–60. 10.1007/s11064-018-2517-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, & Crews FT (2012). Chronic ethanol increases systemic TLR3 agonist-induced neuroinflammation and neurodegeneration. Journal of Neuroinflammation, 9, 130. 1742–2094-9–130 [pii] 10.1186/1742-2094-9-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Liu Y, Hong JS, & Crews FT (2013). NADPH oxidase and aging drive microglial activation, oxidative stress, and dopaminergic neurodegeneration following systemic LPS administration. Glia, 61(6), 855–868. 10.1002/glia.22479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadros IM, Macedo GC, Domingues LP, & Favoretto CA (2016). An update on CRF mechanisms underlying alcohol use disorders and dependence. Frontiers in endocrinology, 7, 134. 10.3389/fendo.2016.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman R, Karthikeyan A, Lik Wei W, Dheen TS, & Sajikumar S (2019). Activation of microglia in acute hippocampal slices affects activity-dependent long-term potentiation and synaptic tagging and capture in area CA1. Neurobiology of Learning and Memory, 163, 107039. 10.1016/j.nlm.2019.107039. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Kloss CU, Werner A, Jones LL, & Kreutzberg GW (1999). Neuroglial activation repertoire in the injured brain: Graded response, molecular mechanisms and cues to physiological function. Brain Research. Brain Research Reviews, 30(1), 77–105. doi:S0165017399000077 [pii]. [DOI] [PubMed] [Google Scholar]
- Ray LA, Bujarski S, Shoptaw S, Roche DJ, Heinzerling K, & Miotto K (2017). Development of the neuroimmune modulator Ibudilast for the treatment of alcoholism: A randomized, placebo-controlled, human laboratory trial. Neuropsychopharmacology, 42(9), 1776–1788. 10.1038/npp.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes JS, Best K, Belknap JK, Finn DA, & Crabbe JC (2005). Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiology & Behavior, 84(1), 53–63. 10.1016/j.physbeh.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Roffman JL, & Stern TA (2006). Alcohol withdrawal in the setting of elevated blood alcohol levels. Primary care companion to the Journal of clinical psychiatry, 8(3), 170–173. 10.4088/pcc.v08n0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco P, Burruss K, Smith CA, Kuerbis A, Harrington D, Moore AA, et al. (2015). Drinking behavior among older adults at a continuing care retirement community: Affective and motivational influences. Aging & Mental Health, 19(3), 279–289. 10.1080/13607863.2014.933307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, & Glass CK (2012). Microglial cell origin and phenotypes in health and disease. Nature Reviews. Immunology, 11(11), 775–787. nri3086 [pii] 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- Sanchez-Alavez M, Nguyen W, Mori S, Wills DN, Otero D, Ehlers CL, et al. (2019). Time course of microglia activation and brain and blood cytokine/chemokine levels following chronic ethanol exposure and protracted withdrawal in rats. Alcohol, 76, 37–45. 10.1016/j.alcohol.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schomerus G, Lucht M, Holzinger A, Matschinger H, Carta MG, & Angermeyer MC (2011). The stigma of alcohol dependence compared with other mental disorders: A review of population studies. Alcohol and Alcoholism: international journal of the Medical Council on Alcoholism, 46(2), 105–112. agq089 [pii] 10.1093/alcalc/agq089. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, & Bilbo SD (2012). Sex, glia, and development: Interactions in health and disease. Hormones and Behavior, 62(3), 243–253. 10.1016/j.yhbeh.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, et al. (2010). Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell, 7(4), 483–495. S1934–5909(10)00437–6 [pii] 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneddon EA, White RD, & Radke AK (2019). Sex differences in binge-like and aversion-resistant alcohol drinking in C57BL/6J mice. Alcoholism, Clinical and Experimental Research, 43(2), 243–249. 10.1111/acer.13923. [DOI] [PubMed] [Google Scholar]
- Sprow GM, Rinker JA, Lowery-Gointa EG, Sparrow AM, Navarro M, & Thiele TE (2015). Lateral hypothalamic melanocortin receptor signaling modulates binge-like ethanol drinking in C57BL/6J mice. Addiction Biology, 21(4), 835–846. 10.1111/adb.12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprow GM, & Thiele TE (2012). The neurobiology of binge-like ethanol drinking: Evidence from rodent models. Physiology & Behavior, 106(3), 325–331. 10.1016/j.physbeh.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squeglia LM, Boissoneault J, Van Skike CE, Nixon SJ, & Matthews DB (2014). Age-related effects of alcohol from adolescent, adult, and aged populations using human and animal models. Alcoholism, Clinical and Experimental Research, 38(10), 2509–2516. 10.1111/acer.12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ (2005). Microglia and neuroprotection: Implications for Alzheimer’s disease. Brain Research Brain Research Reviews, 48(2), 234–239. S0165-0173(04)00190-0 [pii] 10.1016/j.brainresrev.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Streit WJ (2006). Microglial senescence: Does the brain’s immune system have an expiration date? Trends in Neurosciences, 29(9), 506–510. S0166-2236(06)00144-5 [pii] 10.1016/j.tins.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Braak H, Xue QS, & Bechmann I (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathologica, 118(4), 475–485. 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Sammons NW, Kuhns AJ, & Sparks DL (2004). Dystrophic microglia in the aging human brain. Glia, 45(2), 208–212. 10.1002/glia.10319. [DOI] [PubMed] [Google Scholar]
- Streit WJ, & Xue QS (2009). Life and death of microglia. Journal of Neuroimmune Pharmacology, 4(4), 371–379. 10.1007/s11481-009-9163-5. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Marsh L, Mathalon DH, Lim KO, & Pfefferbaum A (1995). Anterior hippocampal volume deficits in nonamnesic, aging chronic alcoholics. Alcoholism, Clinical and Experimental Research, 19(1), 110–122. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7771636. [DOI] [PubMed] [Google Scholar]
- Suryanarayanan A, Carter JM, Landin JD, Morrow AL, Werner DF, & Spigelman I (2016). Role of interleukin-10 (IL-10) in regulation of GABAergic transmission and acute response to ethanol. Neuropharmacology, 107, 181–188. 10.1016/j.neuropharm.2016.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele TE, & Navarro M (2014). “Drinking in the dark” (DID) procedures: A model of binge-like ethanol drinking in non-dependent mice. Alcohol, 48(3), 235–241. 10.1016/j.alcohol.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischer J, Krueger M, Mueller W, Staszewski O, Prinz M, Streit WJ, et al. (2016). Inhomogeneous distribution of Iba-1 characterizes microglial pathology in Alzheimer’s disease. Glia, 64(9), 1562–1572. 10.1002/glia.23024. [DOI] [PubMed] [Google Scholar]
- Tronson NC, & Collette KM (2017). (Putative) sex differences in neuroimmune modulation of memory. Journal of Neuroscience Research, 95(1–2), 472–486. 10.1002/jnr.23921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urso T, Gavaler JS, & Van Thiel DH (1981). Blood ethanol levels in sober alcohol users seen in an emergency room. Life Sciences, 28, 1053–1056. [DOI] [PubMed] [Google Scholar]
- Wagenaar AC, Maldonado-Molina MM, Ma L, Tobler AL, & Komro KA (2007). Effects of legal BAC limits on fatal crash involvement: Analyses of 28 states from 1976 through 2002. Journal of Safety Research, 38(5), 493–499. 10.1016/j.jsr.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Wainwright DA, Xin J, Mesnard NA, Beahrs TR, Politis CM, Sanders VM, et al. (2009). Exacerbation of facial motoneuron loss after facial nerve axotomy in CCR3-deficient mice. ASN Neuro, 1(5), e00024. AN20090017 [pii] 10.1042/AN20090017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waise TMZ, Toshinai K, Naznin F, NamKoong C, Md Moin AS, Sakoda H, et al. (2015). One-day high-fat diet induces inflammation in the nodose ganglion and hypothalamus of mice. Biochemical and Biophysical Research Communications, 464(4), 1157–1162. 10.1016/j.bbrc.2015.07.097. [DOI] [PubMed] [Google Scholar]
- Walker FR, Nilsson M, & Jones K (2013). Acute and chronic stress-induced disturbances of microglial plasticity, phenotype and function. Current Drug Targets, 14(11), 1262–1276. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/24020974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter TJ, & Crews FT (2017). Microglial depletion alters the brain neuroimmune response to acute binge ethanol withdrawal. Journal of Neuroinflammation, 14(1), 86. 10.1186/s12974-017-0856-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm CJ, Hashimoto JG, Roberts ML, Bloom SH, Andrew MR, & Wiren KM (2016). Astrocyte dysfunction induced by alcohol in females but not males. Brain Pathology, 26(4), 433–451. 10.1111/bpa.12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood WG, Armbrecht HJ, & Wise RW (1982). Ethanol intoxication and withdrawal among three age groups of C57BL/6NNIA mice. Pharmacology, Biochemistry, and Behavior, 17(5), 1037–1041. 10.1016/0091-3057(82)90490-7. [DOI] [PubMed] [Google Scholar]
- Wu Y, Lousberg EL, Moldenhauer LM, Hayball JD, Robertson SA, Coller JK, et al. (2011). Attenuation of microglial and IL-1 signaling protects mice from acute alcohol-induced sedation and/or motor impairment. Brain, Behavior, and Immunity, 25(Suppl. 1), S155–S164. 10.1016/j.bbi.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T (2016). Ageing, neurodegeneration and brain rejuvenation. Nature, 539(7628), 180–186. 10.1038/nature20411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr NM, Luong R, Sullivan EV, & Pfefferbaum A (2010). Measurement of serum, liver, and brain cytokine induction, thiamine levels, and hepatopathology in rats exposed to a 4-day alcohol binge protocol. Alcoholism, Clinical and Experimental Research, 34(11), 1858–1870. ACER1274 [pii]. 10.1111/j.1530-0277.2010.01274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YN, Wang F, Fan YX, Ping GF, Yang JY, & Wu CF (2013). Activated microglia are implicated in cognitive deficits, neuronal death, and successful recovery following intermittent ethanol exposure. Behavioural Brain Research, 236(1), 270–282. S0166-4328(12)00578-5 [pii]. 10.1016/j.bbr.2012.08.052. [DOI] [PubMed] [Google Scholar]