

Abstract
Recent structural studies have shown that the carboxyl-terminus of many TRP channels, including TRPC3, are folded into a horizontal rib helix that is connected to the vertical pole helix, which play roles in inter-structural interactions and multimerization. In a previous work we identified I807 located in the pole helix with a role in regulation of TRPC3 by STIM1 (Lee K.P. et. al., 2014, Liu H. et al, 2022). To further determine the role of the pole helix in TRPC3 function, here we identified key hydrophobic residues in the pole helix that form tight tunnel-like structure and used mutations to probe their role in TRPC3 regulation by Ca2+ and Calmodulin. Our findings suggest that the hydrophobic starch formed by the I807-L818 residues has several roles, it modulates gating of TRPC3 by Ca2+, affects channel selectivity and the channel Ca2+ permeability. Mutations of I807, I811, L814 and L818 all attenuated the Ca2+-dependent inactivation (CDI) of TRPC3, with I807 having the most prominent effect. The extent of modulation of the CDI depended on the degree of hydrophobicity of I807. Moreover, the TRPC3(I807S) mutant showed altered channel monovalent ion selectivity and increased Ca2+ permeability, without affecting the channel permeability to Mg2+ and Ba2+ and without changing the pore diameter. The CDI of TRPC3 was reduced by an inactive calmodulin mutant and by a pharmacological inhibitor of calmodulin, which was eliminated by the I807S mutation. Notably, deletion of STIM1 caused similar alteration of TRPC3 properties. Taken together, these findings reveal a role of the pole helix in CDI, in addition to its potential role in channel multimerization that required gating of TRPC3 by STIM1. Since all TRPC and most TRP channels have pole helix structures, our findings raise the possibility that the pole helix may have similar roles in all the TRP family.
Keywords: canonical transient receptor potential channel, coiled-coil domain, Calcium-dependent inactivation, calmodulin, STIM1
Graphical abstract
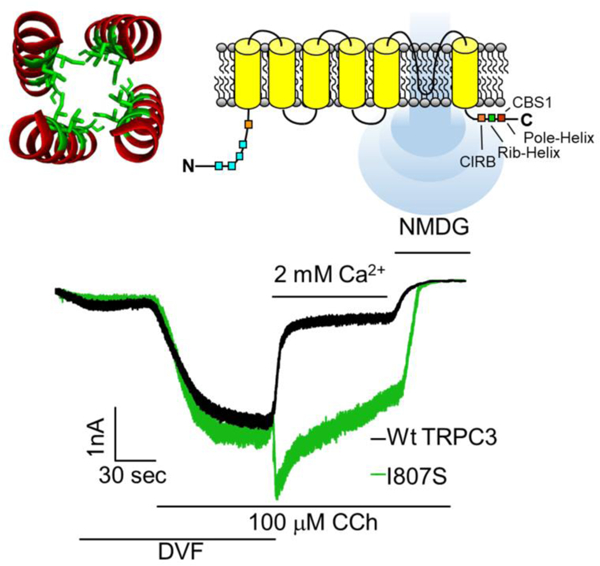
1. Introduction
The super family of TRP channels functions as Ca2+ permeable cation channels that mediate numerous physiological functions [1–3]. The canonical TRP (TRPC) channels are a subfamily of TRP channels that in human consists of 6 members. TRPC channels are grouped into TRPC1/4/5 and TRPC3/6/7 subgroups based on their homology [3]. All TRPC channels are activated in response to stimulation of Gq/11 protein coupled receptors and mediate significant portion of the receptor mediated Ca2+ influx [3]. Excessive Ca2+ influx is highly toxic to cells and an important mechanism guarding against Ca2+ toxicity is inhibition of TRP channels by Ca2+ itself, including TRPC channels [3]. Therefore, understanding regulation of TRPC channels by Ca2+ is of paramount physiological significance. In the present work we selected the canonical transient receptor potential channel 3 (TRPC3) as a model to better understand regulation of TRP channels by Ca2+. TRPC3 is expressed and plays key roles in the nervous, endothelial, smooth muscle and secretory gland tissues [4–8]. Change in TRPC3 functional expression or mutations that modify gating of TRPC3 are associated with various diseases. For example, TRPC3 over-expression has been reported in human ovarian adenocarcinoma tissue [9] and overactivity of TRPC3 in moonwalker mice result in abnormal Purkinje cell development and cerebellar ataxia [10]. In secretory epithelia over activation of TRPC3 contribute to acute pancreatitis [7, 8].
TRPC3 is activated by receptor stimulation and is regulated by STIM1 in response to ER Ca2+ store depletion. Receptor-mediated activation starts by hydrolysis of PI(4,5)P2 to generate diacylglycerol (DAG), which directly activates TRPC3 [11], likely by interacting with lipid site 2 in the Cryo-EM structure of TRPC3 [12–14], in which DAG was identified as the lipid in the structures of TRPC5 [15, 16] and TRPC6 [17]. DAG interaction with lipid 2 site also induce a sensitized channel state [18]. In addition to activation by DAG, TRPC3 requires activation by STIM1 in response to store depletion [19, 20]. STIM1 appears to directly activate TRPC3 by interacting with the channel [20] and by targeting TRPC3 to PI(4,5)P2-rich domains at the ER/PM junctions [21]. Overactivation of TRPC3 is prevented by the cytosolic Ca2+-binding protein, calmodulin (CaM). When bound to Ca2+, CaM competes with IP3 receptors (IP3Rs) for binding to an overlapping region on TRPC3 to inactivate TRPC3 currents [22]. This region was termed the CaM/IP3Rs binding (CIRB) domain, which extends from residue 761 to 785, spanning a flexible region and the horizontal helix coiled-coil domain (rib helix) of TRPC3 [23]. In this manner, CaM can provide a negative feedback mechanism to control cytoplasmic Ca2+ levels ([Ca2+]i) by inactivating TRPC3 when bound to Ca2+ entering the cell via TRPC3 or released from the ER. However, the molecular mechanism and structural requirements by which CaM inhibits TRPC3 has not yet been fully elucidated.
TRPC3 has six transmembrane domains, of which 5 and 6 form the pore region and 1–4 resembles a voltage sensor domain and assemble into a functional tetrameric ion channel [12, 14]. The Cryo-EM structures show estimated pore diameter of <5.8 Å with close state of <1.0 Å [12–14, 24]. The cytoplasmic N-terminus (NT) consists of four pairs of ankyrin repeat domains (ARDs) that are followed by helical linker domains. The carboxyl terminus (CT) consists of a TRP box, an unsolved disordered region and terminates with two helical coiled-coil domains (CCDs), a horizontal rib helix and a connected vertical pole helix. The flexible disordered region between the TRP box and the rib helix appears to participate in multiple interactions and may regulates the gating of TRPC3 [13, 24]. The rib and pole helices form multiple interactions with the NT ARDs, and the pole helix was suggested to play a role in subunits multimerization [13, 25]. The four pole helices interact to form a central tunnel under the cytoplasmic exit of the pore domain. The functional role of the tunnel is not known and was examined at the present work.
Early structures suggested that the acidic E630 acts as a selective Ca2+ filter [12, 13, 24]. The same authors recently reported Cryo-EM structures of TRPC3 and TRPC6 in the presence and in the absence (EGTA+EDTA) of Ca2+ that revealed compact structure of the pole helices in the presence of Ca2+ and loose structure in the absence of Ca2+ [14]. The compact structure contained three potential Ca2+ bindings sites (CBS), with CBS1 located between ankyrin repeat 2 and the pole helix and mutation of D798A at this site reduce inhibition of TRPC3 by intracellular Ca2+ ([Ca2+]i). CBS2 was in a negatively charged region at the top of the pole helix, but mutation analysis suggested no role in regulation of TRPC3 by Ca2+. Finally, CBS3 was located within the voltage sensor-like domain that may function to activate rather than inhibit TRPC3 [14]. When bound to the CBS1 in each of the 4 subunits, the Ca2+ ions surround the tunnel form by the pole helices. Communication between CBS1 and the tunnel formed by the pole helices in channel gating and regulation by Ca2+ have not been thoroughly investigated. In previous studies we reported that TRPC3 NT and CT CCDs are involved in channel gating. The studies revealed that mutation of the hydrophobic I807 (I807S) that partially disrupts the pole helix is involved in the regulation of TRPC3 by STIM1 [20, 21] and participates in multimerization of TRPC3 with TRPC1 [26, 27]. In the present study we took advantage of these findings to examine the role of the hydrophobic tunnel formed by the pole helix to examine its role in TRPC3 gating by Ca2+, likely through influencing the function of CBS1.
2. Materials and Method
2.1. Cell culture and transfection
Human embryonic kidney 293 (HEK293) cells and STIM1−/− HEK293 cells were cultured in 12-well cell culture plates at 37 °C and 5% CO2 in Dulbecco’s modified Eagle medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with antibiotic-antimycotic reagent (Life Technologies, Grand Island, NY, USA) and 10% fetal bovine serum (Gibco). Cells were transiently transfected using Lipofectamine 2000 Transfection Reagent (Life Technologies), as recommended by the manufacturer.
2.2. Constructs and mutagenesis
The human TRPC3 transcript variant 2 (NM_003305) cDNA, tagged with yellow fluorescent protein, was inserted into pcDNA3.1 (pcDNA3.1-hTRPC3-YFP) as detailed before [20, 21]. The mutagenic primers were designed to contain unmodified nucleotide sequences flanking the desired mutation site. The primers used to generate the various mutants are listed in supplementary table 1. PCR was performed using the BioFACT™ Lamp Pfu DNA Polymerase kit (Biofact, Daejeon, South Korea) with a Bio-Rad T100 Thermocycler (Bio-Rad Laboratories, Hercules, CA, USA), according to the protocol provided by the manufacturer.
2.3. Current measurement
The patch pipettes were pulled from thin-wall filament glass capillaries GC 150TF-7.5 (Harvard Apparatus, Holliston, MA, USA) to a resistance of 3–4 MΩ using a vertical pipette puller PC-10 (Narishige Group Products, Tokyo, Japan). An inverted microscope (Nikon ECLIPSE Ti, Tokyo, Japan) was used to identify positively transfected cells based on their yellow fluorescence when illuminated at 514 nm. Whole-cell voltage-clamp experiments were performed at room temperature using an Axopatch 200 B capacitor-feedback patch-clamp amplifier connected to a Digidata-1440A Digitizer (Molecular Devices). Currents were elicited by applying linear voltage-ramp protocols ranging from −100 to +100 mV from a holding potential at −60 mV. The rectification score was determined from the relative change in the inward current at −80mV compared to the outward current at +80mV. The percentage current remaining after exposure of the cells to at 2 mM external Ca2+ for 30–60 seconds was used to estimate the inhibition by Ca2+, by calculating the fraction of current at −60mV remaining in the presence of 2 mM external calcium compared with the current in the fully activated state under divalent-free conditions (DVF).
2.4. Solutions and materials
sThe standard divalent free extracellular solution contained (in mM): NaCl 140, KCl 5, 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic a (HEPES) 10, and Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA)0.5 (pH7.4 using NaOH). The extracellular solution used for response for external calcium contained (in mM): NaCl 140, KCl 5, HEPES 10, MgCl2 1 and CaCl2 X (pH7.4 using NaOH). CaCl2 concentration was adjusted as indicated in the Figures. External solution used for pore size measurements contained (in mM): HEPES 10, EDTA 10 and Cation Salt 150 (pH7.4 using HCl or NMDG). Cation salts used were NaCl, methylamine, dimethyl amine, trimethyl amine and (1-Deoxy-1-(methylamino)-D-glucitol, Meglumine) NMDG. External saline used for permeability measurements contained (in mM): XCl 150, MgCl2 1, HEPES 10 and Glucose 10 (7.4 pH with XOH) (X being Li, Na, Rb and Cs). All saline solutions were within 290–320 mosM. The standard intracellular solution contained (in mM): CsCl 140, MgCl2 3, ATP 1, CaCl2 1.5 (to clamp free Ca2+ at 70 nM), HEPES 10 and EGTA 5 titrated to pH 7.2 with CsOH unless specified. Calmidazolium chloride, carbamoylcholine chloride (CCh), methyl ammonium, dimethyl ammonium, trimethylammonium, tetramethylammonium, N-methyl-d-glucamine, and 1-oleoyl-2-acetyl-sn-glycerol were purchased from Sigma–Aldrich (Seoul, Korea).
2.5. Structural analysis
The Marcoil 1.0 coiled-coil software predicted the CCD based on a hidden Markov model [28]. The first CCD was predicted to span between residues E240 and E284, and the second CCD spans between Q777 and L836 with an initial coiled-coil probability of 11.9% at K789. The probability increases along with the predicted CCD domain, with the peak probability values (p) greater than 89% as calculated from L804 to the end of the protein sequence. The hydrophobic line in the pole helix in the TRPC3 structure (PDB; 6CUD) was visualized by PyMOL 2.2 (The PyMOL Molecular Graphics System)
2.6. Statistical Analysis
Statistical significance of the data was calculated using one-way analysis of variance with Bonferroni correction (Origin Pro 8.1, Northampton, Massachusetts, USA) when comparing three or more dataset groups, and the unpaired two-tailed Student’s t-test was used when comparing two datasets. The linear fit, Y = mX + c, was used to fit the data for a linear relationship. Data are expressed as mean ± SEM; statistical significance is shown as *p < 0.05, **p < 0.01, and ***p < 0.001.
3. Results
3.1. Disrupting hydrophobic interactions within the pole helix tunnel hinders Ca2+ dependent inactivation of TRPC3
Previous studies on the regulation of TRPC3 by Ca2+ used activation of TRPC3 by OAG [29] and the measurement of Ca2+-dependent inactivation (CDI) associated with the structural studies relied on spontaneous activity of TRPC3 [14]. To examine the physiological role of the regulation of TRPC3 by Ca2+, we activated TRPC3 by stimulation of the muscarinic M3 receptors. To demonstrate regulation of TRPC3 by Ca2+, HEK cells expressing TRPC3 and the M3Rs were stimulated with 100 μM CCh while the cells were perfused with divalent ions free solution (Fig. 1). The stimulation resulted in a large inward Na+ current that was promptly and markedly inhibited upon addition of 2 mM Ca2+ to the bath perfusate (Fig. 1A). This form of CDI was proposed to result from interaction of cytoplasmic Ca2+ ([Ca2+]i) with Ca2+ binding site 1 (CBS1) of the three TRPC3 CBS, based on measurement of spontaneous activity of TRPC3 [14]. We extended these findings to show similar CDI for the receptor-activated TRPC3 (n = 8). Disrupting the pole helix by mutating the hydrophilic amino acid I807 (I807S) modified the inhibition by Ca2+, causing rapid transient increase in the current and significantly reduced inactivation rate and extent. Interestingly, buffering the cytosol with 5 mM EGTA and even 10 mM BAPTA failed to prevent the CDI (small residual current after addition of 2 mM external Ca2+) of TRPC3, although BAPTA further reduced CDI of I807S, suggesting that even the I807S mutants retained some CDI that requires Ca2+ at the mouth of the channel (Fig. 1B).
Fig 1. Hydrophobic stretch of the pole helix controls Ca2+-dependent inactivation of TRPC3.
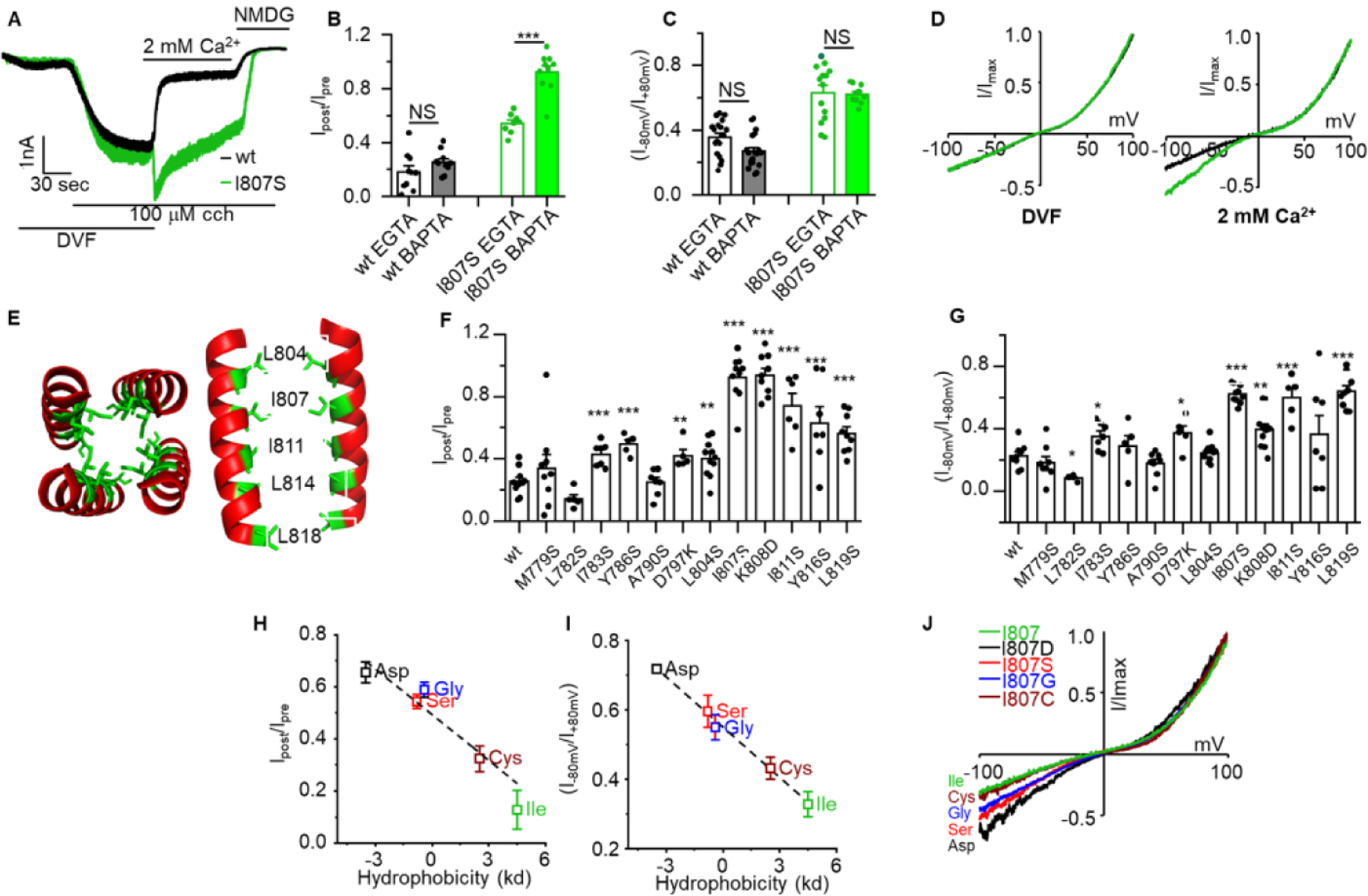
Panel (A): HEK cells transfected with TRPC3 (black trace) or TRPC3(I807S) (green trace) in divalent free (DVF) solution were stimulated with 100 μM carbachol (CCH). When maximum current was attained, the cells were exposed to media containing 2 mM Ca2+. (B): The percentage of residual current after 10 seconds exposure to external Ca2+ was determined in pipette solutions containing 5 mM EGTA or 10 mM BAPTA. (C): Representative IV curves recorded with DVF media (left) and in the presence of 2 mM Ca2+ (right). (D): shown is the channel rectification score determined as the ratio of current at −80mV/+80mV for TRPC3 (black) and TRPC3(I807S) (green). (E): Structure of the human TRPC3 pole helix (PDB 6CUD) visualized with hydrophobic side chains in the pole helix interface between two subunits. (F): Summary of residual current after exposure to 2 mM external Ca2+ of the indicated TRPC3 pole helix mutants. (G): Summary of rectification score of the indicated the same TRPC3 mutants as in F. (H): Correlation of hydrophobicity with residual current measured with the indicated I807 mutants (I): Correlation of hydrophobicity with current rectification measured with the indicated I807 mutants. (J): Representative IV curves of the indicated I807 mutants. Results are presented as mean ± SEM with ∗p < 0.05 and ∗∗∗p < 0.001.
Summary of the current density of TRPC3 and TRPC3(I807S) under the various conditions (Supplementary Fig. S1) showed that external Ca2+ and the I807S mutation mainly affected the inward current, with minimal effect on the outward current. A trend of small reduction in outward current was the same for TRPC3 and TRPC3(I807S), but a did not reach statistical significance. However, the I897S mutation affected channel rectification defined as the ratio of the currents at −80mV/+80mV, and the shape of the IV curve only when Ca2+ was present in the extracellular perfusate (Fig. 1C). Notably, the rectification score was 0.36 ± 0.03 for TRPC3 and 0.63 ± 0.046 (p < 0.001) for TRPC3(I807S) in the presence of 2 mM extracellular Ca2+ (Fig 1D). Buffering [Ca2+]i had no effect on TRPC3 and TRPC3(I807S) rectification or the increased rectification by TRPC3(I807S) (Fig. 1C), suggesting that the rectification may involve one of the additional CBS that mediates inhibition of TRPC3 by Ca2+ that cannot be accessed by cytoplasmic BAPTA.
I807 is located at the top of the pole helix and is part of a hydrophobic tunnel at the bottom of the pore presumed ionic exit site [14]. To analyze the role of the pole helix in gating of TRPC3 by Ca2+, first we truncated the residues below I807. Truncation of the pole helix below L804 resulted in impaired TRPC3 current (supplementary Fig. S2). In fact, truncation of only the last 8 residues of the pole helix was sufficient to inhibit channel conductance [21], highlighting the importance of the pole helix to channel function. In another approach, we substituted the hydrophobic amino acids at the pole helix with hydrophilic amino acids that are predicted to disrupt the helical structure. The region at the pole helix (red) in which the hydrophobic residues (green) in each subunit appose each other is shown in Fig. 1E. Mutations of residues upstream of I807 had minimal effect on CDI, as determined by the residual current after inhibition by 2 mM external Ca2+ (Fig. 1F). The same assay showed that CDI gradually decreased along the pole helix from its peak at I807 and down to L819 (Fig. 1F). Mutation of the same residues also showed the highest rectification score (Fig. 1G). Therefore, to further explore the importance of the pole helix hydrophobicity we substituted I807 with amino acids with a gradual change in hydrophobicity and compared their CDI. Fig. 1H shows linear relationship between the pole helix hydrophobicity and CDI and Fig. 1I shows that the rectification score of the I807 mutants followed a linear relationship with reduced hydrophobicity (R2 = 0.997, with intercept 0.55 ± 0.006, slope – 0.048 ± 0.002, n = 7–12). Example I/Vs are shown in Fig. 1J.
3.2. TRPC3-mediated calcium current is required for calcium-depended inhibition
In principle, the CDI triggered by addition of extracellular Ca2+ can be caused either by interaction of Ca2+ with an extracellular site or by Ca2+ entering the cells through TRPC3. To distinguish between the two possibilities, we took advantage of the Ca2+-impermeable TRPC3(E630Q) mutant [30]. Figs. 2A and 2B show that TRPC3(E630Q) lacks CDI to an extent comparable to that of TRPC3(I807S), which is also apparent in the I/Vs in Figs. 2C and 2D. Similarly, the TRPC3(E630Q) mutation increased the rectification score to a level similar to that obtained for TRPC3(I807S) (Figs. 2E). To provide further evidence that the CDI of TRPC3 was due to inward Ca2+ current, we characterized the properties of a TRPC3(E630Q/I807S) double mutant. Analyzing the current within the first seconds of addition of external Ca2+ revealed the I807S mutation increased the relative Ca2+/Na+ permeability (calculated as in [31]) of both TRPC3 and TRPC3(E630Q) (Supplementary Fig. S3). Yet, TRPC3(E630Q/I807S) still lacked CDI and displayed rectification score comparable to TRPC3(I807S) (Figs. 2A-E). Finally, to study the specific role of Ca2+ in the CDI, we replaced extracellular Ca2+ with Ba2+. Addition of 2 mM external Ba2+ failed to inactivate TRPC3 and TRPC3(I807S) (time course in Fig. 2F and averages in Fig. 2G). The I/Vs in Fig. 2H for Ca2+ and 2I for Ba2+ and the rectification scores in Fig. 2J further show that CDI is specific for Ca2+ and cannot be mediated by Ba2+ that is often used as a Ca2+ surrogate with Ca2+ channels. When taken together these findings demonstrated that the inactivation of TRPC3 is specifically mediated by Ca2+ influx through TRPC3. However, the question remains as to whether the lack of CDI by the pole helix TRPC3 mutants is due to either the lack of Ca2+ permeability or altered sensitivity of the mutants to Ca2+. This is addressed below.
Fig 2. Ca2+ influx through TRPC3 is required for Ca2+-dependent inactivation of TRPC3.
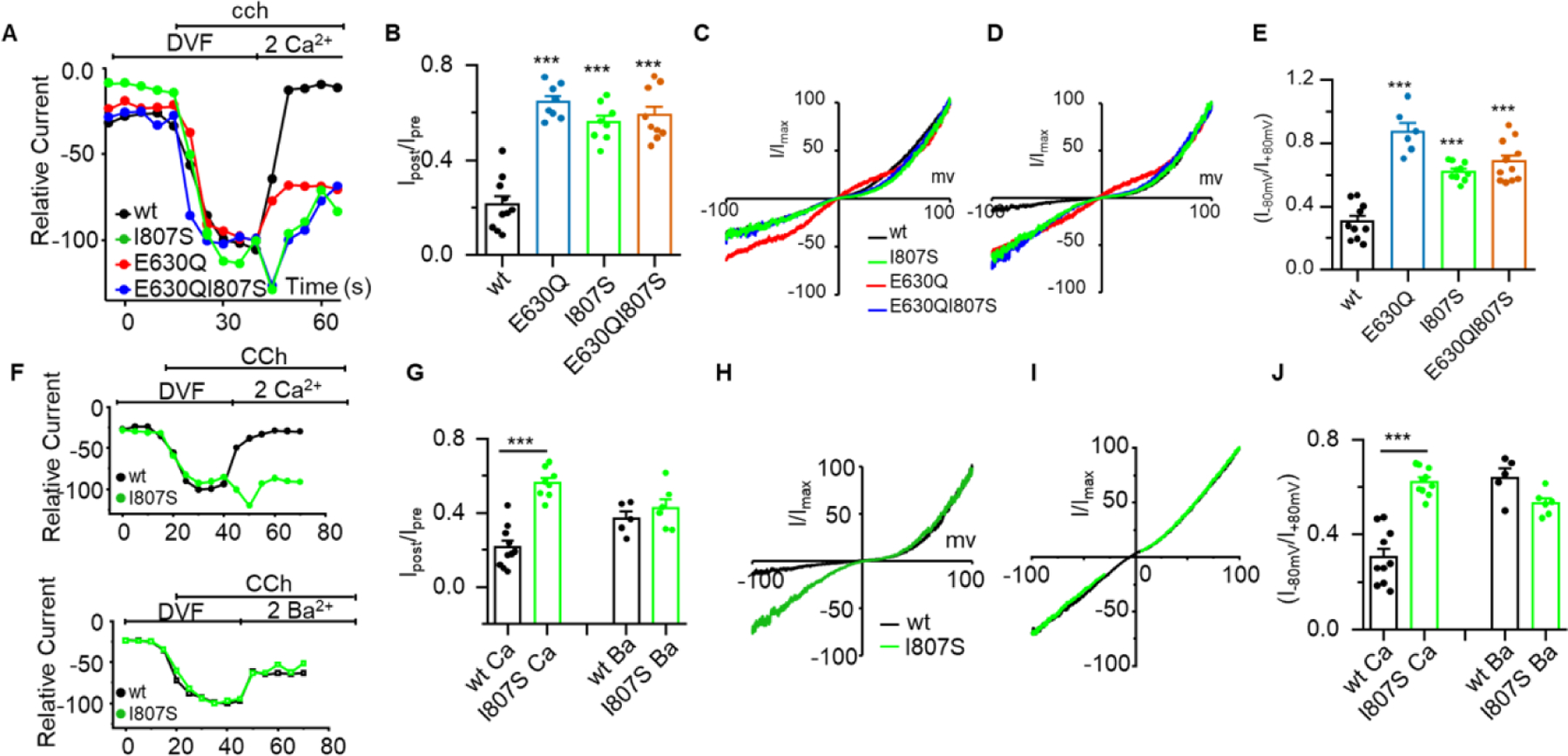
Panel (A): Time course of the inward current measured with TRPC3 (black), TRPC3(I807S) (green), the Ca2+ impermeable TRPC3(E630Q) (red) and the double mutant TRPC3(E630Q/I807S) (blue). Note restoration of Ca2+ influx to TRPC3(E630Q) by the I807S mutation (green and blue traces and I/V in D). (B): Residual current after exposure to 2 mM external Ca2+. (C, D): Example IV curves recorder in DVF solution (C) and in the presence of 2 mM Ca2+ (D). (E): rectification score of the indicated mutants. (F): Time course of TRPC3 and TRPC3(I807S) currents measured in DVF solution. Where indicated the cells were exposed to 2 mM Ca2+ (top traces) or 2 mM Ba2+ (lower traces). (G): Summary of experiments as in F showing the effect of Ca2+ and Ba2+ on the residual current. (H, I) Example I/Vs measured with Ca2+ (H) and Ba2+ (I). (J): The rectification score measured in DVF solutions and solutions containing 2 mM Ca2+ or 2 mM Ba2+ measured with TRPC3 and TRPC3(I807S). Results are presented as mean ± SEM. ∗p < 0.05, ∗∗ p < 0.01, and ∗∗∗p < 0.001.
3.3. Mutations in the pole helix increase the Ca2+ permeability of TRPC3.
To address the question if TRPC3(I807S) mediates Ca2+ influx, first we measured Ca2+ influx by selectively stimulating TRPC3 and TRPC3(I807S) with the channel ligand GSK that activates the channel like DAG [32, 33]. Figs. 3A and 3B show that 10 μM GSK increased [Ca2+]i in cells transfected with TRPC3 and TRPC3(I807S) and TRPC3(I807S) was significantly more permeable to Ca2+ than TRPC3, suggesting that the reduced TRPC3(I807S) CDI was not due to reduced Ca2+ influx.
Fig 3. Ca2+ influx by TRPC3(I807S) and effect of the I807 and E630 mutants on TRPC3 selectivity and pore size.
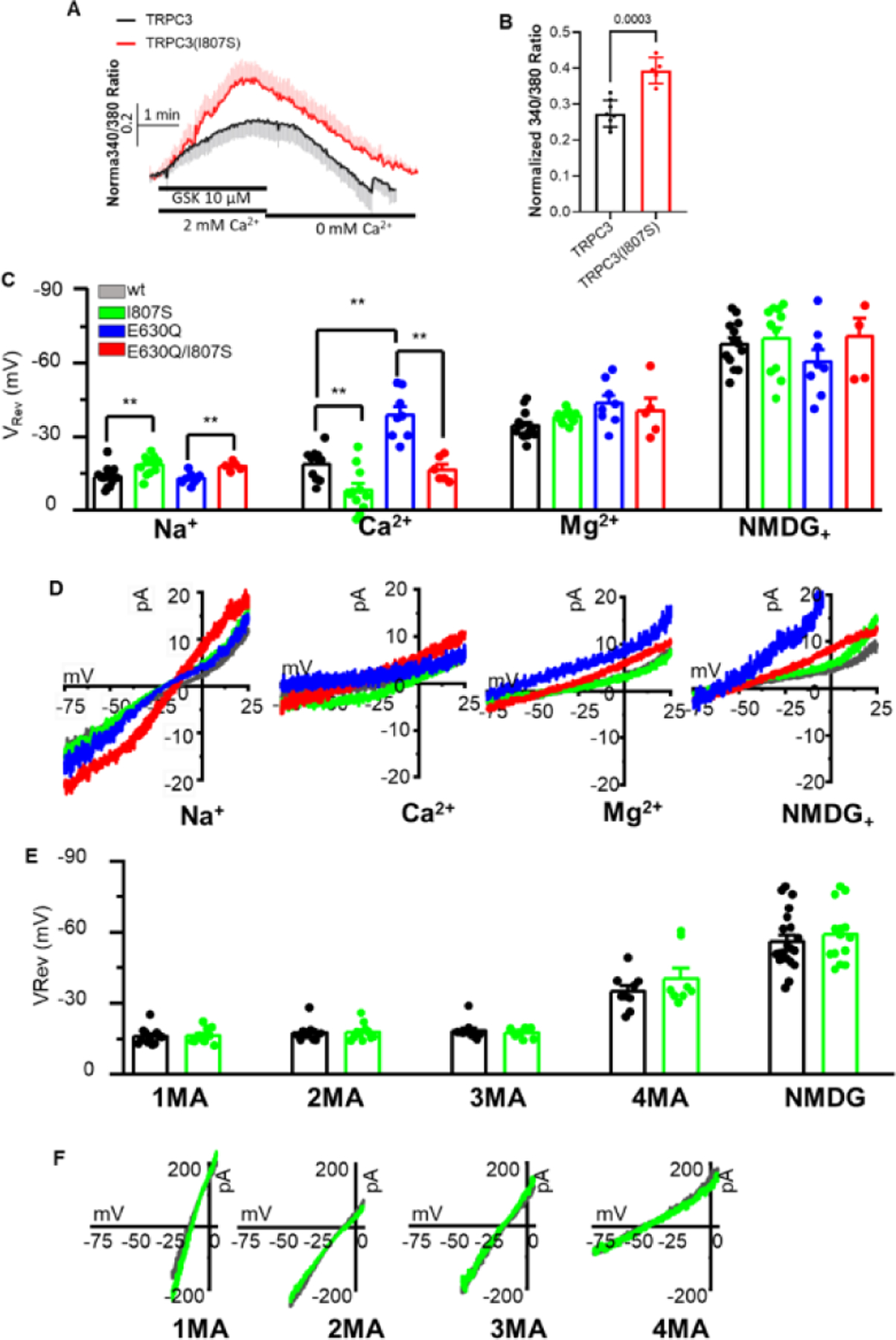
Panel (A, B): Ca2+ influx was measured in HEK cells transfected with TRPC3 (black) or TRPC3(I807S) (red) and stimulated with 10 μM GSK in Ca2+ containing solution and then perfused with Ca2+ free solution (A). The peak currents are shown in (B). (C): The reversal potentials were measured in solution when the only permeable extracellular ions were Na+, Ca2+, Mg2+ or NMDG with the indicated TRPC3 variants. (D): Magnified portion of the I/V curves measured with the indicated mutants and external ions. (E): TRPC3 and TRPC3(I807S) currents were measured with the indicated methylated ammonium ions. The results are presented as mean ± SEM. ∗p < 0.05, ∗∗ p < 0.01, and ∗∗∗p < 0.001
Second, we determined the selectivity and ionic permeability of TRPC3 and the mutants and how they affect divalent ions permeability. The divalent ion permeability of TRPC3, TRPC3(I807S), TRPC3(E630Q) and TRPC3(E630Q/I807S) in the presence Na+, Ca2+ and Mg2+ as the only permeable cations are shown in Figs. 3C and 3D. As we reported recently [21], TRPC3(I807S) showed reduced Na permeability that was maintained by TRPC3(E630Q/I807S), as shown by the more negative reversal potentials. On the other hand, TRPC3(I807S) had higher Ca2+ permeability than TRPC3 and prominently increased the Ca2+ permeability of the Ca2+ impermeable TRPC3(E630Q) mutant. The reversal potentials measured by addition of external Mg2+ and by replacing external Na+ with NMDG shifted to a more negative value, and was the same for all constructs, indicating that all TRPC3 variants showed low permeability for Mg2+ and NMDG. Hence, the finding that the I807S mutation markedly shifted the reversal potential of TRPC3 towards a positive potential when Ca2+ was the only permeable external ion, indicate that disruption of the pole helix increased TRPC3 Ca2+ permeability, consistent with the increased [Ca2+]i when TRPC3(I807S) is activated by GSK (Fig. 3A, B).
The surprising finding of increased Ca2+ permeability due to disruption of hydrophobic interactions within the pole helix led us to investigate whether the mutants alter the TRPC3 pore diameter. To this end, we compared the permeation of TRPC3 and TRPC3(I807S) to methylated ammonium ions of increased size, mono, di, tri, and tetra methyl ammonium. Both TRPC3 and TRPC3(I807S) displayed similar reversal potentials to the methyl ammonium ions (Figs. 3E and 3F), indicating that the increased TRPC3(I807S) Ca2+ permeability is not due to increased pore diameter. Together, the Ca2+ influx and current analysis suggest that the loss of CDI phenotype of the pole helix mutants (Figs. 1) is due to disruption of Ca2+ binding by TRPC3. This conclusion is in agreement with a recent report that the absence of Ca2+ and mutations in CBS1 that prevent CDI disrupt the compactness of the pole helix [14].
3.4. Ca/CaM contributes to CDI of TRPC3
The TRPC3 rib helix forms part of the CIRB domain that was shown to interact with calmodulin (CaM) to inactivate TRPC3 [22, 34]. Recent structural analysis of drosophila TRP channel C terminal domains identified similar CaM binding site at the rib helix and a site at the pole helix [35]. The TRPC6 CIRB domain, a close relative of TRPC3, interacts with CaM in a Ca2+-dependent manner to affect TRPC6 activity [36]. It is therefore possible that binding of CaM to the CIRB domain and rib helix affects the structure of the pole helix to regulate CDI. To test this hypothesis, we evaluated the effect of a CaM inhibitor calmidazolium chloride (CMZ) and of the dominant negative CaM EF hand mutant (CaMEF) on TRPC3 function. Figs. 4A-E show the effect of CaM and of the CaMEF mutant and Figs. 4F-J show the effect of CMZ on TRPC3 function. Transfection of CaM had no effect the CDI of TRPC3 or TRPC3(I807S), the time course of activation, residual currents, I/V or rectification scores. This is likely because TRPC3 is saturated with endogenous CaM, which in HEK cells can range from 6–10 μM [37]. By contract, transfection of the CaMEF mutant increased the residual current of TRPC3 after exposure to 2 mM external Ca2+ while CaMEF had no effect on the residual current of TRPC3(I807S), suggesting that TRPC3(I807S) has reduced sensitive to or lost interaction with Ca/CaM (Fig. 4B). In line with the CaMEF findings, Figs. 4F-J show that incubating the cells with 1 μM CMZ hindered the CDI of TRPC3 to a level measured with TRPC3(I807S) by increasing the residual TRPC3 current, while having no effect on the residual current of TRPC3(I807S) (Fig. 4G). Similarly, CMZ increased TRPC3 rectification without affecting the rectification of TRPC3(I807S) (Fig. 4J). These findings suggest functional coupling between the pole helix and the CIRB domain that affect Ca2+ regulation of TRPC3.
Fig 4. Effect of Calmodulin, the Calmodulin EF hand mutant and calmidazolium on Ca2+-dependent inactivation of TRPC3 and TRPC3(I807S) currents.
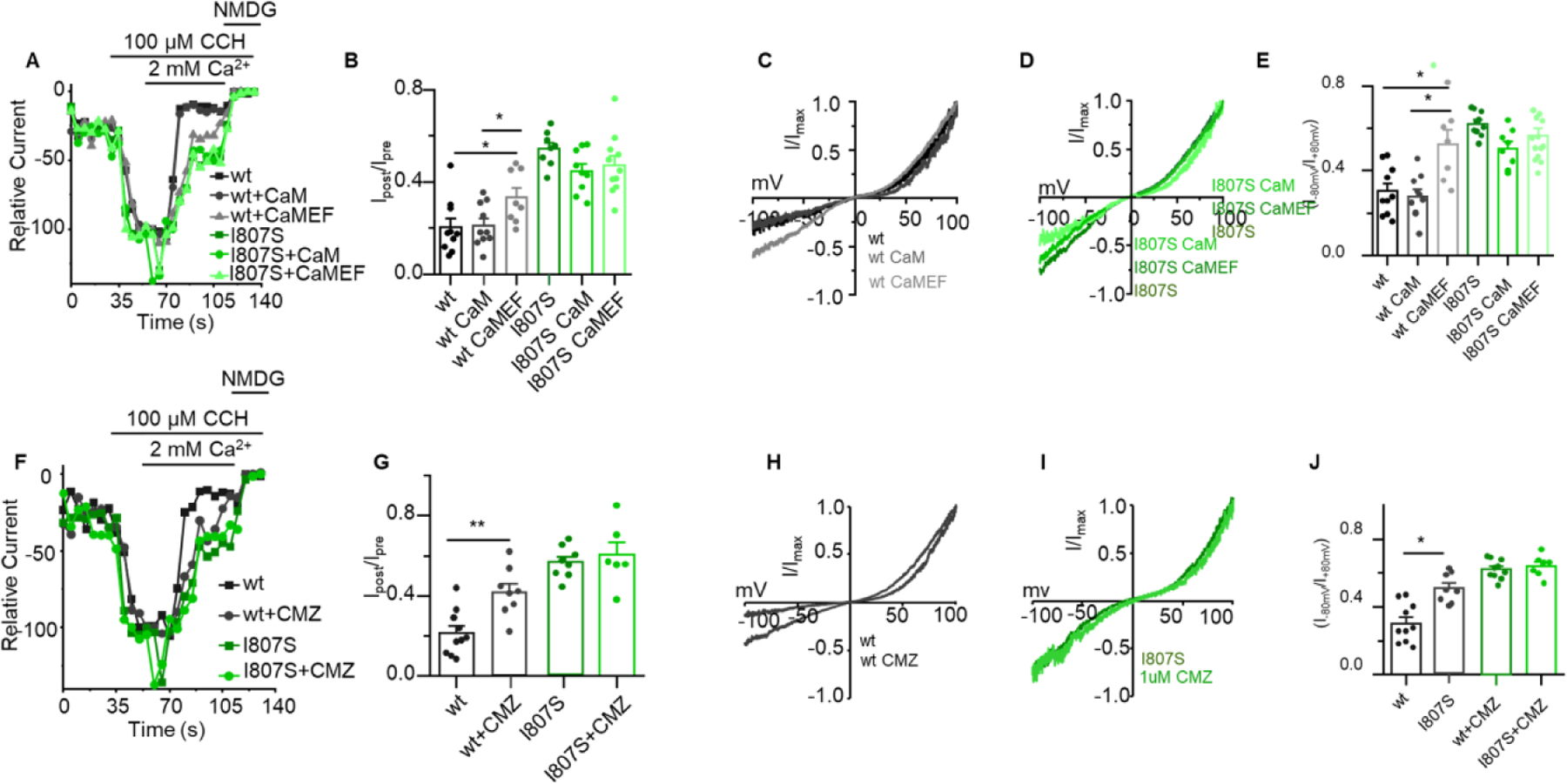
Panel (A): Time course of TRPC3 (black traces) and TRPC3(I807S) (green traces) expressed alone or together with calmodulin (CaM) or the calmodulin EF mutant (CaMEF) measured in DVF solution and where indicated exposed to 2 mM Ca2+. (B): Summary of residual current after exposure to 2 mM external Ca2+ measured as in A. (C, D): Example I/Vs for TRPC3 (C) and TRPC3(I807S) (D). (E): Rectification score for the indicated conditions. (F): Time course of TRPC3 and TRPC3(I807S) current in control cells and cells treated with 1 μM calmidazolium for 5 min and stimulated by 100 μM CCh. (G): Summary of residual current after exposure to 2 mM external Ca2+ measured as in F. (H, I): Example I/Vs for TRPC3 (H) and TRPC3(I807S) (I). (J): Rectification score for the indicated conditions. Results are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001
3.5. Localization at the STIM1-formed ER/PM junction is required for CDI
TRPC channels, including TRPC3, interacts with and are regulated by STIM1 [19, 20, 38]. Localization of TRPC3 in STIM1-formed ER/PM junction is required for regulation of TRPC3 by lipids and modulates TRPC3 ionic selectivity [21]. Therefore, we asked whether CDI of TRPC3 is affected by STIM1. This was tested by expressing TRPC3 and TRPC3(I807S) in STIM1−/− cells and measuring the effect of external Ca2+ on their activity (Fig. 5). To be able to compare TRPC3 behavior in the presence and absence of STIM1, the cells were maximally stimulated with 100 μM CCh since STIM1 affect the apparent Michaelis-Menton constant (Km) but not the maximal velocity (Vmax) for stimulation with CCh [21]. The behavior of TRPC3 and TRPC3(I807S) were similar in STIM1−/− cells, with the addition of 2 mM external Ca2+ causing an initial increase in the current (Fig. 5A), although the increased current was lower with TRPC3 (Fig. 5B) and prominently and similarly reduced CDI (Fig. 5C). At present, the reason for the reduced increased current with TRPC3 compared to TRPC3(I807S) upon addition of external Ca2+ is not known. Among the possibilities can be faster TRPC3 inactivation of better access of Ca2+ to TRPC3 CBS1.
Fig 5. Localization in the STIM1-formed junctions in required for CDI.
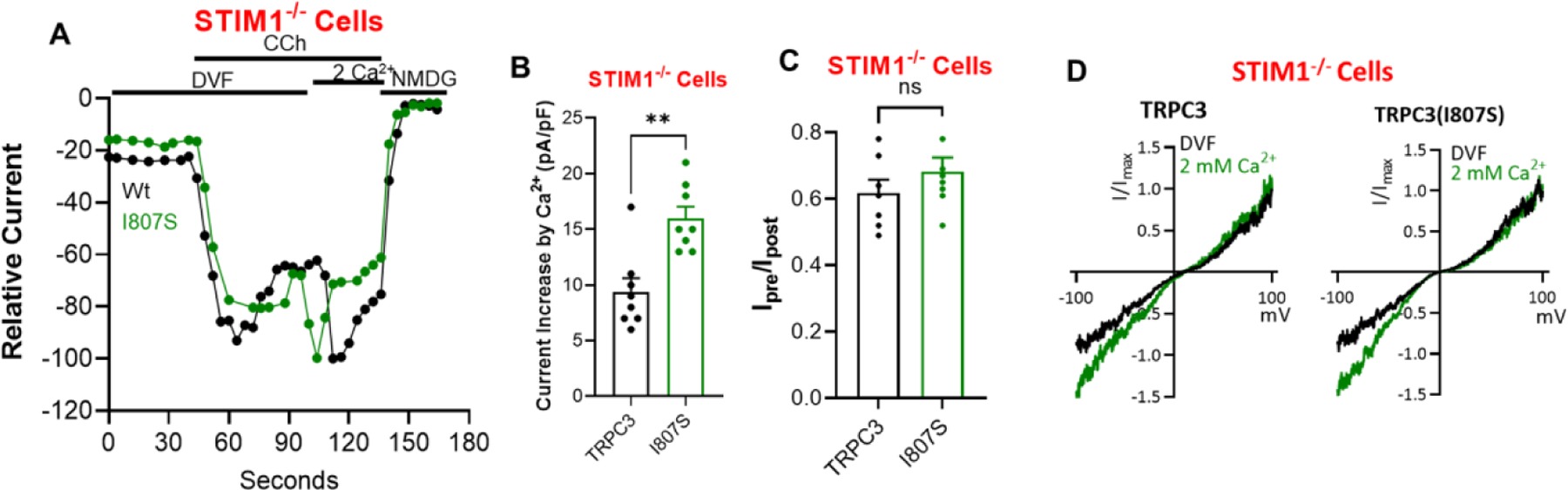
TRPC3 (black) and TRPC3(I807S) (green) were transfected in STIM1−/− cells and current was measured in response to stimulation with 100 μM CCh in DFM and in the presence of 2 mM external Ca2+, as indicated. Panel (A) is the time course, Panel (B) the maximal increase increased current in pA/pF measured on addition of Ca2+, panel (C) is the residual current after exposure to 2 mM external Ca2+ and panel (D) are example I/V.
4. Discussion
Excess [Ca2+]i is highly toxic and many cellular mechanisms have been evolved to ensure that [Ca2+]i will be maintained within physiologically beneficial levels. One of the key mechanism involves the acute limit of the period of the [Ca2+]i increase. This is achieved by Ca2+ clearance by the SERCA and PMCA pumps that are Ca2+-activated transporters [39]. However, most of the Ca2+ toxicity is due to excessive Ca2+ influx from the virtually unlimited source of extracellular Ca2+ that enters the cells through Ca2+ permeable channels [40]. An effective mechanism in controlling Ca2+ influx channels is their inhibition by Ca2+. This is well documented for voltage gated Ca2+ channels [41], the Orai channels [42] and the TRP channels [3]. However, the molecular details of the inhibition and its regulation by channel domains are not fully understood. As for the TRPC channels, a major strid was made recently by obtaining the structure of TRPC3 and TRPC6 in the presence and absence of Ca2+ [14]. The Ca2+ bound structure is compact and with 3 Ca2+ ions bound to three separate sites CBS1–3. However, binding of Ca2+ to CBS1 appears to be the main site regulating inhibition of TRPC3 by Ca2+ [14].
A major issue not addressed before is how binding of Ca2+ to TRPC channels CBS1 is modulated by the channel domains and by localization of the channels at the ER/PM junctions. A unique structure found in all TRP channels is the connected pole and rib helices [43]. The pole helices of the four protomers are assembled into a tunnel just under the presumed cytoplasmic ion exit site. The inner portion of the protomers interact with each other, as shown in Figure 1, to form a hydrophobic domain close to CBS1 while the rib helices together with an unstructured domain form the site at which IP3 receptors and CaM compete for interaction. This compact rib-pole structure is held together by Ca2+ binding to TRPC3 and is disrupted when Ca2+ is removed from all three sites [14], although the contribution of each of the three CBSs to the structure is not known at present. In the present studies, we examined the role of the pole helix and its hydrophobic nature in inhibition of TRPC3 by Ca2+. TRPC3 is directly activated by DAG and is regulated by STIM1 [23, 26] and by STIM1-dependent localization at the ER/PM junctions [21]. In previous studies we have shown that the carboxyl terminus CCD that includes the pole helix is required for interaction of TRPC3 with TRPC1 and subsequent activation by STIM1 [26]. In the present studies we extended these findings to show that pole helix modulates the CDI of TRPC3 and regulate channel ion selectivity and gating. Moreover, regulation by CDI and by CaM requires localization of TRPC3 in the STIM1 formed ER/PM junctions.
The pole helix has an essential role in TRPC3 function since deletion of residues below L804 (Fig. S2) or even the last 8 residues [21] resulted in inactive channel. Moreover, the pole helix controls channel gating and selectivity. It limits the Ca2+ permeability of TRPC3 and confer higher Na+/Ca2+ selectivity (Figs. 3, 5). The effect of the pole helix of channel selectivity is independent of Ca2+ influx, and thus intracellular Ca2+, since it takes place, and it is similar with Ca2+ permeable and impermeable TRPC3 (Fig. S3). Finally, the pole helix is essential for a robust CDI of TRPC3, as shown by the marked reduced CDI upon disruption of pole helix hydrophobicity (Fig. 1). Together, these effects limit Ca2+ influx into cells with activated TRPC3 to guard against Ca2+ toxicity.
The CDI of TRPC3 is mediated by Ca2+ binding to CBS1 [14]. The present findings show that CDI is modulated by the pole helix and by Ca2+/CaM. A surprising finding is that even 10 mM cytoplasmic BAPTA could not prevent the strong CDI, which suggests close vicinity of CBS1 to the channel pore and tight binding of Ca2+ to CBS1. Nevertheless, CDI appears to be modulated by Ca2+/CaM that was shown to interact with the TRPC3 [22, 34] and TRPC6 [36] CIRB domains (Fig. 4) located in the unstructured loop between the rib helix and TRP box. How CaM exert its effect needs further examination. Measurement of TRPC3 current in inside-out patches showed that the current was inhibited by Ca2+ and the inhibition was not affected by CaM or the CaM mutant [14]. It is possible that regulation by CaM was lost when the patches were isolated, or the regulation required cell stimulation as was used in the present studies. Indeed, when rhabdomeric drosophila TRP was activated by light, CaM affected the current measured at low external Ca2+, although not during large Ca2+ influx [35]. CaM interacted with two sites in the C terminus of drosophila TRP, with the CaM N lob bound to an upstream site and the CaM C lob bound to a site formed partially by the pole helix. The site at the pole helix is highly conserved among the drosophila and mammalian TRPs and includes the hydrophobic stretch examined in the present studies [35]. Indeed, the I807S mutation that disrupts this site also eliminated regulation by CaM (Figure 4). The somewhat variable observation on regulation of TRP channels by CaM that appears to depend on the channel isoform studied and the method used and thus required further analysis.
The hydrophobic interface within the pole helix has several roles in channel function. Disruption of the hydrophobicity even by mutation of a single residue, I807, change TRPC3 ionic selectivity (Fig. S3), Ca2+ permeability (Fig. 3), rectification (Fig. 1C, D) and inhibition by Ca2+ that requires Ca2+ binding to CBS1 (Fig. 1A, B). These findings indicate that the hydrophobic interface within the pole helix is required for the pole helix to provide an anchor that allows the transduction of conformational movement of the rib helix and the disordered flexible region to the channel pore. The flexible disordered region appears important for channel function and likely for polymodal regulation of TRPC3 by intracellular signals and transfer of conformational changes. Accordingly, truncations and extension of the flexible loop by introduction of glycines increased and decreased channel function when activated by GSK [24]. Clearly additional structural and functional studies are required to fully understand the conformational effect of the pole and rib helices in channel gating. The present findings highlight the multiple roles of these structures in TRPC3 function. The presence of similar rib and pole helices that appears to exist in different conformation suggest that our findings are relevant to other members of the TRP channels family.
Regulation of TRPC3 by lipids, channel selectivity [21] and CDI (Fig. 5), all required localization of TRPC3 in STIM1-formed ER/PM junctions. Similar regulatory mechanism by Ca2+ at the STIM1-formed ER/PM junction may be common to all TRP channels that contain pole-rib helices structure to limit Ca2+ influx through the channels. Interestingly, Ca2+-dependent inhibition of Orai1 that is mediated by the SARAF protein also required localization at the ER/PM junctions [44]. Ca2+-dependent inactivation of voltage-gated Ca2+ channel take place in junctional domains [45]. Thus, disruption of the junctions should lead to uncontrolled Ca2+ influx and Ca2+ toxicity. It should be informative to explore further the loss of junctional control of CDI as part of the pathology of cellular Ca2+ toxicity and cell death.
Supplementary Material
Highlights.
Hydrophobic interaction in the pole helix in TRPC3 is important for TRPC channel function.
Mutation interrupting Pole helix hydrophobic interaction attenuates calcium dependent inactivation.
TRPC3 has calcium dependent feedback regulation either by direct biding of calcium to CBSs and Calcium/Calmodulin dependent regulation to CIRB.
5. Acknowledgements
We thank Spencer Leibow (NIH/NIDCR) for careful reading of the manuscript and Dr. Mohamed Trebak (University of Pittsburgh) for providing the STIM1−/− cells. This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (2019R1F1A1060643) and by intramural grant NIH/NIDCR DE000735–13.
Abbreviations
- ARDs
ankyrin repeat domains
- BAPTA
1,2-bis(2-aminophenoxy)ethane-N,N,N,N-tetraacetic acid
- CaM
calmodulin
- CIRB
CaM/IP3R binding
- CCD
coiled-coil domain
- CMZ
calmidazolium chloride
- CT
carboxyl terminus
- DAG
diacylglycerol
- ER
endoplasmic reticulum
- HEK293
human embryonic kidney 293
- HH
horizontal helix
- NT
N-terminal
- STIM1
stromal interaction molecule 1
- TRPC
canonical transient receptor potential channel
- VH
vertical helix
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT author statement
Tharaka Wijerathne: Conceptualization, Methodology, investigation, analysis, writing-original draft. Wei-Yin Lin.: investigation, data Curation, Writing-original draft. Akila Cooray: writing - review and editing. Shmuel Muallem: conceptualization, data curation, writing-review and editing. Kyu Pil Lee: Conceptualization, investigation, Writing- Reviewing and Editing,
References
- [1].Li X, Fine M, TRP Channel: The structural era, Cell Calcium, 87 (2020) 102191. [DOI] [PubMed] [Google Scholar]
- [2].Goretzki B, Guhl C, Tebbe F, Harder JM, Hellmich UA, Unstructural Biology of TRP Ion Channels: The Role of Intrinsically Disordered Regions in Channel Function and Regulation, J Mol Biol, 433 (2021) 166931. [DOI] [PubMed] [Google Scholar]
- [3].Wang H, Cheng X, Tian J, Xiao Y, Tian T, Xu F, Hong X, Zhu MX, TRPC channels: Structure, function, regulation and recent advances in small molecular probes, Pharmacol Ther, 209 (2020) 107497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Um KB, Hahn S, Kim SW, Lee YJ, Birnbaumer L, Kim HJ, Park MK, TRPC3 and NALCN channels drive pacemaking in substantia nigra dopaminergic neurons, Elife, 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kassahun Gebremeskel A, Wijerathne TD, Kim JH, Kim MJ, Seo CS, Shin HK, Lee KP, Psoralea corylifolia extract induces vasodilation in rat arteries through both endothelium-dependent and -independent mechanisms involving inhibition of TRPC3 channel activity and elaboration of prostaglandin, Pharm Biol, 55 (2017) 2136–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cozart MA, Phelan KD, Wu H, Mu S, Birnbaumer L, Rusch NJ, Zheng F, Vascular smooth muscle TRPC3 channels facilitate the inverse hemodynamic response during status epilepticus, Sci Rep, 10 (2020) 812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kim MS, Hong JH, Li Q, Shin DM, Abramowitz J, Birnbaumer L, Muallem S, Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis, Gastroenterology, 137 (2009) 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kim MS, Lee KP, Yang D, Shin DM, Abramowitz J, Kiyonaka S, Birnbaumer L, Mori Y, Muallem S, Genetic and pharmacologic inhibition of the Ca2+ influx channel TRPC3 protects secretory epithelia from Ca2+-dependent toxicity, Gastroenterology, 140 (2011) 2107–2115, 2115 e2101–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zeng B, Yuan C, Yang X, Atkin SL, Xu SZ, TRPC channels and their splice variants are essential for promoting human ovarian cancer cell proliferation and tumorigenesis, Curr Cancer Drug Targets, 13 (2013) 103–116. [PubMed] [Google Scholar]
- [10].Becker EB, Oliver PL, Glitsch MD, Banks GT, Achilli F, Hardy A, Nolan PM, Fisher EM, Davies KE, A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice, Proceedings of the National Academy of Sciences of the United States of America, 106 (2009) 6706–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G, Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol, Nature, 397 (1999) 259–263. [DOI] [PubMed] [Google Scholar]
- [12].Fan C, Choi W, Sun W, Du J, Lu W, Structure of the human lipid-gated cation channel TRPC3, Elife, 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tang Q, Guo W, Zheng L, Wu JX, Liu M, Zhou X, Zhang X, Chen L, Structure of the receptor-activated human TRPC6 and TRPC3 ion channels, Cell Res, 28 (2018) 746–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Guo W, Tang Q, Wei M, Kang Y, Wu JX, Chen L, Structural mechanism of human TRPC3 and TRPC6 channel regulation by their intracellular calcium-binding sites, Neuron, 110 (2022) 1023–1035 e1025. [DOI] [PubMed] [Google Scholar]
- [15].Duan J, Li J, Chen GL, Ge Y, Liu J, Xie K, Peng X, Zhou W, Zhong J, Zhang Y, Xu J, Xue C, Liang B, Zhu L, Liu W, Zhang C, Tian XL, Wang J, Clapham DE, Zeng B, Li Z, Zhang J, Cryo-EM structure of TRPC5 at 2.8-A resolution reveals unique and conserved structural elements essential for channel function, Sci Adv, 5 (2019) eaaw7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Song K, Wei M, Guo W, Quan L, Kang Y, Wu JX, Chen L, Structural basis for human TRPC5 channel inhibition by two distinct inhibitors, Elife, 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bai Y, Yu X, Chen H, Horne D, White R, Wu X, Lee P, Gu Y, Ghimire-Rijal S, Lin DC, Huang X, Structural basis for pharmacological modulation of the TRPC6 channel, Elife, 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Erkan-Candag H, Clarke A, Tiapko O, Gsell MA, Stockner T, Groschner K, Diacylglycerols interact with the L2 lipidation site in TRPC3 to induce a sensitized channel state, EMBO Rep, 23 (2022) e54276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bodnar D, Chung WY, Yang D, Hong JH, Jha A, Muallem S, STIM-TRP Pathways and Microdomain Organization: Ca(2+) Influx Channels: The Orai-STIM1-TRPC Complexes, Adv Exp Med Biol, 993 (2017) 139–157. [DOI] [PubMed] [Google Scholar]
- [20].Lee KP, Choi S, Hong JH, Ahuja M, Graham S, Ma R, So I, Shin DM, Muallem S, Yuan JP, Molecular determinants mediating gating of Transient Receptor Potential Canonical (TRPC) channels by stromal interaction molecule 1 (STIM1), J Biol Chem, 289 (2014) 6372–6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu H, Lin WY, Leibow SR, Morateck AJ, Ahuja M, Muallem S, TRPC3 channel gating by lipids requires localization at the ER/PM junctions defined by STIM1, J Cell Biol, 221 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang Z, Tang J, Tikunova S, Johnson JD, Chen Z, Qin N, Dietrich A, Stefani E, Birnbaumer L, Zhu MX, Activation of Trp3 by inositol 1,4,5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain, Proc Natl Acad Sci U S A, 98 (2001) 3168–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wedel BJ, Vazquez G, McKay RR, St JBG, Putney JW Jr., A calmodulin/inositol 1,4,5-trisphosphate (IP3) receptor-binding region targets TRPC3 to the plasma membrane in a calmodulin/IP3 receptor-independent process, J Biol Chem, 278 (2003) 25758–25765. [DOI] [PubMed] [Google Scholar]
- [24].Sierra-Valdez F, Azumaya CM, Romero LO, Nakagawa T, Cordero-Morales JF, Structure-function analyses of the ion channel TRPC3 reveal that its cytoplasmic domain allosterically modulates channel gating, J Biol Chem, 293 (2018) 16102–16114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lepage PK, Lussier MP, Barajas-Martinez H, Bousquet SM, Blanchard AP, Francoeur N, Dumaine R, Boulay G, Identification of two domains involved in the assembly of transient receptor potential canonical channels, The Journal of biological chemistry, 281 (2006) 30356–30364. [DOI] [PubMed] [Google Scholar]
- [26].Lee KP, Choi S, Hong JH, Ahuja M, Graham S, Ma R, So I, Shin DM, Muallem S, Yuan JP, Molecular determinants mediating gating of Transient Receptor Potential Canonical (TRPC) channels by stromal interaction molecule 1 (STIM1), The Journal of biological chemistry, 289 (2014) 6372–6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lee KP, Yuan JP, Hong JH, So I, Worley PF, Muallem S, An endoplasmic reticulum/plasma membrane junction: STIM1/Orai1/TRPCs, FEBS letters, 584 (2010) 2022–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Delorenzi M, Speed T, An HMM model for coiled-coil domains and a comparison with PSSM-based predictions, Bioinformatics, 18 (2002) 617–625. [DOI] [PubMed] [Google Scholar]
- [29].Lintschinger B, Balzer-Geldsetzer M, Baskaran T, Graier WF, Romanin C, Zhu MX, Groschner K, Coassembly of Trp1 and Trp3 proteins generates diacylglycerol- and Ca2+-sensitive cation channels, J Biol Chem, 275 (2000) 27799–27805. [DOI] [PubMed] [Google Scholar]
- [30].Lichtenegger M, Stockner T, Poteser M, Schleifer H, Platzer D, Romanin C, Groschner K, A novel homology model of TRPC3 reveals allosteric coupling between gate and selectivity filter, Cell calcium, 54 (2013) 175–185. [DOI] [PubMed] [Google Scholar]
- [31].Owsianik G, Talavera K, Voets T, Nilius B, Permeation and selectivity of TRP channels, Annu Rev Physiol, 68 (2006) 685–717. [DOI] [PubMed] [Google Scholar]
- [32].Lichtenegger M, Tiapko O, Svobodova B, Stockner T, Glasnov TN, Schreibmayer W, Platzer D, de la Cruz GG, Krenn S, Schober R, Shrestha N, Schindl R, Romanin C, Groschner K, An optically controlled probe identifies lipid-gating fenestrations within the TRPC3 channel, Nat Chem Biol, 14 (2018) 396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Svobodova B, Lichtenegger M, Platzer D, Di Giuro CML, de la Cruz GG, Glasnov T, Schreibmayer W, Groschner K, A single point mutation in the TRPC3 lipid-recognition window generates supersensitivity to benzimidazole channel activators, Cell Calcium, 79 (2019) 27–34. [DOI] [PubMed] [Google Scholar]
- [34].Boulay G, Brown DM, Qin N, Jiang M, Dietrich A, Zhu MX, Chen Z, Birnbaumer M, Mikoshiba K, Birnbaumer L, Modulation of Ca(2+) entry by polypeptides of the inositol 1,4, 5-trisphosphate receptor (IP3R) that bind transient receptor potential (TRP): evidence for roles of TRP and IP3R in store depletion-activated Ca(2+) entry, Proc Natl Acad Sci U S A, 96 (1999) 14955–14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chen W, Shen Z, Asteriti S, Chen Z, Ye F, Sun Z, Wan J, Montell C, Hardie RC, Liu W, Zhang M, Calmodulin binds to Drosophila TRP with an unexpected mode, Structure, 29 (2021) 330–344 e334. [DOI] [PubMed] [Google Scholar]
- [36].Polat OK, Uno M, Maruyama T, Tran HN, Imamura K, Wong CF, Sakaguchi R, Ariyoshi M, Itsuki K, Ichikawa J, Morii T, Shirakawa M, Inoue R, Asanuma K, Reiser J, Tochio H, Mori Y, Mori MX, Contribution of Coiled-Coil Assembly to Ca(2+)/Calmodulin-Dependent Inactivation of TRPC6 Channel and its Impacts on FSGS-Associated Phenotypes, J Am Soc Nephrol, 30 (2019) 1587–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Black DJ, Tran QK, Persechini A, Monitoring the total available calmodulin concentration in intact cells over the physiological range in free Ca2+, Cell Calcium, 35 (2004) 415–425. [DOI] [PubMed] [Google Scholar]
- [38].Zeng W, Yuan JP, Kim MS, Choi YJ, Huang GN, Worley PF, Muallem S, STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction, Mol Cell, 32 (2008) 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ahuja M, Chung WY, Lin WY, McNally BA, Muallem S, Ca(2+) Signaling in Exocrine Cells, Cold Spring Harb Perspect Biol, 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu H, Kabrah A, Ahuja M, Muallem S, CRAC channels in secretory epithelial cell function and disease, Cell Calcium, 78 (2019) 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ames JB, L-Type Ca(2+) Channel Regulation by Calmodulin and CaBP1, Biomolecules, 11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Prakriya M, Lewis RS, Store-Operated Calcium Channels, Physiol Rev, 95 (2015) 1383–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Fine M, Li X, Dang S, Structural insights into group II TRP channels, Cell Calcium, 86 (2020) 102107. [DOI] [PubMed] [Google Scholar]
- [44].Maleth J, Choi S, Muallem S, Ahuja M, Translocation between PI(4,5)P2-poor and PI(4,5)P2-rich microdomains during store depletion determines STIM1 conformation and Orai1 gating, Nat Commun, 5 (2014) 5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Flucher BE, Campiglio M, STAC proteins: The missing link in skeletal muscle EC coupling and new regulators of calcium channel function, Biochim Biophys Acta Mol Cell Res, 1866 (2019) 1101–1110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.