

Abstract
The regulation of coordination configurations of single-atom sites is highly desirable to boost the catalytic performances of SA catalysts. Here, we demonstrate a versatile complexation-deposition strategy for the synthesis of 13 kinds of dual-metal SA site pairs with uniform and exclusive coordination configurations. The preparation is specifically exemplified by the fabrication of Cu and Co single-atom pairs with the co-existence of N and P heteroatoms through etching and pyrolysis of a pre-synthesized metal–organic framework template. Systematic characterizations reveal the uniform and exclusive coordinative configuration of Cu and Co SA sites in CuN4/CoN3P1 and CuN4/CoN2P2, over which the electrons are unsymmetrically distributed. Impressively, the CuN4/CoN2P2 site pairs exhibit significantly enhanced catalytic activity and selectivity in the synthesis of a variety of natural flavonoids in comparison with the CuN4/CoN3P1 and CuN4/CoN4 counterparts. Theoretical calculation results suggest that the unsymmetrical electron distribution over the CuN4/CoN2P2 sites could facilitate the adsorption and disassociation of oxygen molecules via reducing the energy barriers of the generation of the key intermediates and thus kinetically accelerate the oxidative-coupling reaction process.
Keywords: metal−organic frameworks, single-atom, heterogeneous catalysis, domino synthesis, flavone
Single-atom catalysts attract continuously increasing interest owing to their highest atom utilization and well-defined active sites. In most cases, the SA sites are in a metal–N4 coordination, and the highly symmetrical structure is demonstrated to be beneficial to achieving high catalytic selectivity.1−4 However, the symmetric electron distribution is not favorable for the adsorption and activation of reactants.5−10 To break through this limitation, efforts have been devoted to modulating the coordination configuration of SA sites through directly altering the non-metal coordinating atoms with heteroatoms or immobilizing them into a second shell.11−16 Normally, the heteroatom precursor is introduced via adsorption, space confinement, or ion exchanging to enable a homogeneous dispersion,17−19 but the weak interaction usually results in high randomness of the yielded coordination heteroatoms. Besides, a little part of the metal and non-metal species tend to aggregate in the subsequent thermal treatment, which may unwillingly lead to an uneven composition and eventually unsatisfied catalytic performance (especially selectivity).
Recently, considerable achievements have been made in fabricating uniform SA sites with unsymmetrical coordination configurations (e.g., N–S and N–P co-coordinating ones).20−22 The electron polarization atop the metal center leads to an attractive catalytic performance in terms of both activity and selectivity. Nevertheless, the high homogeneity of SA sites still causes analogous electronic properties throughout the catalyst interfaces. To further boost the catalytic performance, one effective way is to introduce another kind of atomic site to construct a primary-assistant site pair, which could redistribute electrons over the hetero-SA pairs by synergistic interactions.23−27 However, to date, there is still a lack of an effective synthesis strategy to integrate two different SA sites with uniform and designable coordination configurations.
Herein, we report a novel complexation-deposition strategy for the synthesis of 13 kinds of SA pairs with N, P co-coordination configurations. As a proof of concept, a Cu-based metal–organic framework (MOF) is employed as both the source of Cu–N4 site and the template for the formation of a Co-NP complexation layer on the external surface. After the inside-out migration process, the N-coordinated Cu complexes derived from MOFs are uniformly deposited on the internal surfaces with Co-NP complexation, which undergoes subsequent thermal treatment to yield Cu–N4/Co-NP pairs. Systematical characterizations indicate the uniform and exclusive coordination configuration for each kind of SA sites between which the synergistic interactions lead to significant electron re-distribution. In addition, the metal–NP also features a designable electron distribution due to the modifiable coordination (i.e., N3P1 and N2P2). The higher P content strengthens the electron polarization due to the electron transfer effect, which facilitates oxygen adsorption and disassociation and eventually leads to a boosted catalytic performance in the general synthesis of natural flavonoids.
Results and Discussion
Synthesis of Cu-Complex@MP-Co Precursors
In this work, aiming to demonstrate the material preparation details, interpret the interactions between two SA sites, and verify the feasibility in flavone synthesis, the fabrication of Cu and a Co composite is exemplified. First, the monodispersed Cu-ZIF crystals (Figures S1a,b) with a rhombic dodecahedral morphology were assembled (Figure 1a).28 Powder X-ray diffraction (XRD) patterns (Figure S1c) of Cu-ZIF match well with the reported and simulated ZIF-8, suggesting their highly analogous crystallization. The as-prepared Cu-ZIF was subsequently immerged into an aqueous solution containing melamine, Co2+, and phytic acid to form a melamine-Co-phytic acid (MP-Co) layer along the external surfaces of Cu-ZIF. Under acidic conditions, the solid Cu-ZIF gradually decomposes into a Cu-complex, which partially anchors onto the internal surface of the MP-Co layer. Consequently, hollow dodecahedrons composed of the Cu-complex and MP-Co layer are obtained (Figure S2).
Figure 1.

(a) Schematic illustration of the synthesis of CuN4/CoN2P2@NPC. (b,c) SEM. (d) TEM. (e,f1) HAADF-STEM. (f2–f7) Corresponding elemental mapping images of the Cu-complex@MP-Co. (g–i) HAADF-STEM. (j) AC HAADF-STEM. (k1) ABF-STE.M (k2–k7) Corresponding elemental mapping images of the CuN4/CoN2P2@NPC.
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Figure 1b–d) indicate that the as-synthesized Cu-complex@MP-Co has a well-defined rhombic dodecahedral morphology and uniform particle size similar to the parent Cu-ZIF. The high-angle annular dark-field scanning TEM (HAADF-STEM) image (Figure 1e) clearly verifies the even internal cavity (ca. 600 nm) and shell thickness (ca. 30 nm). Elemental mappings (Figure 1f) of Co, Cu, P, O, N, and C elements reveal their homogeneous distribution without obvious aggregations. No characteristic diffraction peaks of pristine Cu-ZIF are detected in the XRD patterns of Cu-complex@MP-Co and the Cu-complex@MP counterpart (Figure S3a), suggesting the complete decomposition of Cu-ZIF after the etching-coating process. Moreover, the etching-coating treatments also lead to the generation of a small amount of micro- and mesopores in Cu-complex@MP-Co (Figure S3c,d).
Synthesis of CuN4/CoN2P2@NPC Nano-Dodecahedrons
An annealing treatment was then carried out at 700 °C (Figure S3b) to convert Cu-complex@MP-Co into CuN4/CoN2P2@NPC (NPC denotes N, P co-doped carbon). The thin dodecahedron shell (<30 nm) could minimize metal aggregations during the pyrolysis. HAADF-STEM (Figure 1g–i) and annular bright field scanning TEM (Figure 1k1) images reveal the hollow dodecahedral morphology and concavo-convex external surfaces of CuN4/CoN2P2@NPC. The spherical aberration-corrected HAADF-STEM (AC HAADF-STEM) image (Figure 1j) clearly discloses the atomic Co and Cu sites with homogeneous distribution of the elements (Figure 1k2–k7). The contents of Cu and Co in CuN4/CoN2P2@NPC are 1.6 and 1.1 wt %, respectively (Table S1). The unique morphology of CuN4/CoN2P2@NPC endows it with a small amount of micro- and mesopores (smaller than 25 nm) (Figure S4c). The wide pore size distribution (larger than 25 nm) might be related to the stacking holes among the monodispersed dodecahedrons. The XRD patterns (Figure S4a) of CuN4/CoN2P2@NPC show a broad diffraction peak assigned to graphite carbons without any remarkable signal of metal aggregation, consistent with the TEM results.
X-ray photoelectron spectroscopy (XPS) and X-ray absorption fine structure (XAFS) measurements (Figures 2, S5, S6, Tables S2, and S3) were conducted to investigate the chemical states and coordination configurations of Cu and Co components in the as-synthesized samples. As shown in Figure S7a, the C 1s XPS spectra are fitting well with three peaks at the binding energies of 284.6, 285.3, and 287.6 eV, assigned to C–C/C=C, C–N/C–P, and O=C–N, respectively. The high-resolution N 1s spectra of CuN4/CoN2P2@NPC confirm the presence of pyridinic-N (398.5 eV), metal–N (399.4 eV), graphitic-N (401.1 eV), and oxide-N (403.8 eV) species (Figure S7b). The P 2p spectra display three peaks at 132.1, 133.9, and 129.7 eV, which corresponds to P–C, P–O, and Co–P species (Figure S7c).7,11,21
Figure 2.

XANES spectra at the (a) Cu K-edge and (k) Co K-edge. FT k3-weighted EXAFS spectra at the (b) Cu K-edge and (l) Co K-edge. (c, m) Corresponding EXAFS fitting curves in R space. Inset of (c): model of the CuN4 site; inset of (m): model of the CoN4 site, CoN3P1 site, and CoN2P2 site, respectively. Cu (orange), Co (purple), N (blue), P (pink), C (gray). (d–j,n–t) WT for the k3-weighted EXAFS signals.
As the Co species could simultaneously coordinate with the −NH2 group in melamine and −OH in phytic acid during the synthesis, the coordination configurations of the atomic Co sites could be modulated through tuning the relative content of melamine and phytic acid (for details, see the Experimental Section). Without the addition of melamine, Co is coordinated to four N atoms rather than P atoms, indicating the decisive role of melamine in the generation of MP-Co aggregates and consequently the introduction of P into the coordination configurations. Note the high difficulty for the Co ion to coordinate with three or four −OH of the phytic acid due to its topology and steric effect; only CuN4/CoN2P2@NPC, CuN4/CoN3P1@NPC, and CuN4/CoN4@NPC were fabricated in this work. As expected, the characterization results (Figures S4–S9) suggest that atomic Cu and Co sites are deposited throughout CuN4/CoN3P1@NPC and CuN4/CoN4@NPC with analogous C, N, and P contents in comparison with CuN4/CoN2P2@NPC.
The Cu K-edge X-ray absorption near-edge structure (XANES) spectra (Figure 2a) indicate that the absorption edge positions of Cu in CuN4/CoN2P2@NPC, CuN4/CoN3P1@NPC, and CuN4/CoN4@NPC are close to that of the CuO reference, suggesting their oxidized valence states. Specifically, the valence state of Cu in CuN4/CoN2P2@NPC is slightly higher than those of CuN4/CoN3P1@NPC and CuN4/CoN4@NPC (inset of Figure 2a), which may be related to the strengthened electron accumulation over Co due to the higher content of P atoms in the coordination configuration. A significant peak at ca. 1.50 Å is detected in the Fourier-transformed (FT) k3-weighted extended XAFS (EXAFS) spectra (Figure 2b) of CuN4/CoN2P2@NPC, CuN4/CoN3P1@NPC, and CuN4/CoN4@NPC, implying the dominant Cu–N coordination. The high similarity of the characteristic peaks among these samples suggests that the introduction of P barely affects the Cu–N configuration. No apparent peaks could be observed at around 2.24 Å, revealing a negligible amount of Cu–Cu bonds. The fitting EXAFS spectra (Figure 2c) uncover that Cu coordinates to the N4 site in CuN4/CoN2P2@NPC, CuN4/CoN3P1@NPC, and CuN4/CoN4@NPC with a mean Cu–N bond length of ca. 1.5 Å. Considering the negligible amount of Cu–P bonds, we infer the coordination configuration of the Cu SA site is CuN4.
The Co K-edge XANES spectra (Figure 2k) show that CuN4/CoN2P2@NPC, CuN4/CoN3P1@NPC, and CuN4/CoN4@NPC have a similar near-edge absorption to CoO, indicating that the valence states of Co are about +2. The FT k3-weighted EXAFS spectra (Figure 2l) of Co in the three samples indicate one main peak at 1.50 Å without the obvious peak of the Co–Co bond at 2.18 Å, suggesting that Co species exist in the form of isolated single atoms. The fitting EXAFS spectra (Figure 2m) reveal that the homogeneous coordination configurations of Co–N2P2, Co–N3P1, and Co–N4 feature similar Co–N and Co–P bond lengths of ca. 1.5 Å. Additionally, the wavelet transform (WT) contour plots of both Cu and Co show the maximum intensity at around 4–4.5 Å–1 (Figure 2d–j,n–t), confirming the presence of uniform Cu–N4 and Co–N2P2/Co–N3P1/Co–N4 coordination. The above results suggest that the Cu and Co species in CuN4/CoN2P2@NPC, CuN4/CoN3P1@NPC and CuN4/CoN4@NPC should dominantly coexist as hetero-SA centers (i.e., Cu–N4 and Co–N2P2/Co–N3P1/Co–N4).
Considering the homogeneous distribution of Cu and Co sites throughout the catalyst, we infer that each small region consists of adjacent Cu and Co pairs. Density functional theory (DFT) calculations were then conducted to investigate the impacts of the coordination environment on the electronic properties of the materials (Figure S10). The representative coordination structures of CuN4/CoN2P2@NPC (Figure S10a) and CuN4/CoN3P1@NPC (Figure S10e) were selected from the potential structures after the stability evaluation. For CuN4/CoN4@NPC, a classical structure (Figure S10g) was selected. To begin with, the charge distribution over these models is simulated (Figure 3a,e,i). Both Cu and Co tend to lose electrons from d orbitals to form chemical bonds with N and/or P atoms, as is verified by electron accumulations inside the metal–N(P) bonds. Lone pair electrons of the N atom (specifically pyridinic N) are transformed into the pz orbital and contribute to the strong metal–N interactions. On the other hand, because of the relatively smaller electronegativity, P atoms generally lose electrons to form Co–P bonds and result in significant electron-accumulation (yellow area) around Co atoms. In this regard, the increased number of P atoms in the coordination configuration leads to a strengthened electron accumulation around Co, which is in the order of CuN4/CoN2P2@NPC > CuN4/CoN3P1@NPC > CuN4/CoN4@NPC (Figure 3a,e,i). This tendency is quantitively verified by Bader charge analysis (Table S4) with the positive charge carried by Co atom in CuN4/CoN2P2@NPC (0.373 |e|), CuN4/CoN3P1@NPC (0.555 |e|), and CuN4/CoN4@NPC (0.849 |e|). Accordingly, the charge distribution over the adjacent CuN4 site is also affected due to the different numbers of P atoms. The CuN4 site in CuN4/CoN2P2@NPC, CuN4/CoN3P1@NPC, and CuN4/CoN4@NPC is negatively charged with −4.270, −3.730, and −3.015 |e|, respectively. The strengthened electron accumulation after the introduction of P is also demonstrated by the calculated density-of-states (DOS, Figure 3b,f,j).
Figure 3.

Top view of the differential charge densities of (a) CuN4/CoN2P2@NPC, (e) CuN4/CoN3P1@NPC, and (i) CuN4/CoN4@NPC; yellow and cyan isosurfaces with an isosurface level of 0.0055 e/a03 represent electron accumulation and depletion areas, respectively. Local DOSs of (b) CuN4/CoN2P2@NPC, (f) CuN4/CoN3P1@NPC, and (j) CuN4/CoN4@NPC. PDOSs of Co 3d (dz2, dxz, and dyz) and O 2p (pz, px, and py) in (c) CuN4/CoN2P2@NPC, (g) CuN4/CoN3P1@NPC, and (k) CuN4/CoN4@NPC. Orbital interactions between O* and the Co site (dz2–pz, dxz–px, and dyz–py) in (d) CuN4/CoN2P2@NPC, (h) CuN4/CoN3P1@NPC, and (l) CuN4/CoN4@NPC; the schematic illustrations are extracted from the corresponding DOS. (m) CI-NEB results of O2 disassociation. (n) Top and side views of charge difference plots for O2 adsorption on the samples; yellow and cyan isosurfaces with an isosurface level of 0.003 e/a03 represent electron accumulation and depletion areas, respectively.
Synthesis Versatility
In order to evaluate the application scope of the proposed strategy, a series of dual-metal single atoms with controllable P coordination configurations (Figures S11 and S12) were also fabricated. Characterization results clearly reveal the atomic distribution of metal sites throughout the hollow dodecahedrons, demonstrating the high versatility of this synthesis strategy.
Domino Synthesis of Flavones
Flavones and derivatives are an important category of natural products that are widely utilized as bioactive anti-inflammatory and anti-oxidant compounds in specific diseases including cancer and cardiovascular and cerebrovascular ones.29−36 Natural flavones are commonly collected from green plants, while the unsatisfied purity, cumbersome extraction process, and even limited supplement shackle their applications. Therefore, establishing an effective catalytic system for the synthesis of natural flavones offers a promising alternative to the conventional biological extraction protocol. Currently, the homogeneous chemical synthesis methods such as the Baker–Venkataraman rearrangement and Kostanecki–Robinson and Allan–Robinson reactions not only require harsh reaction conditions, dangerous co-catalysts, and difficult-to-separate and costly catalysts but also face considerable challenges in the synthesis of flavone derivatives with tunable functional groups from simple substrates.
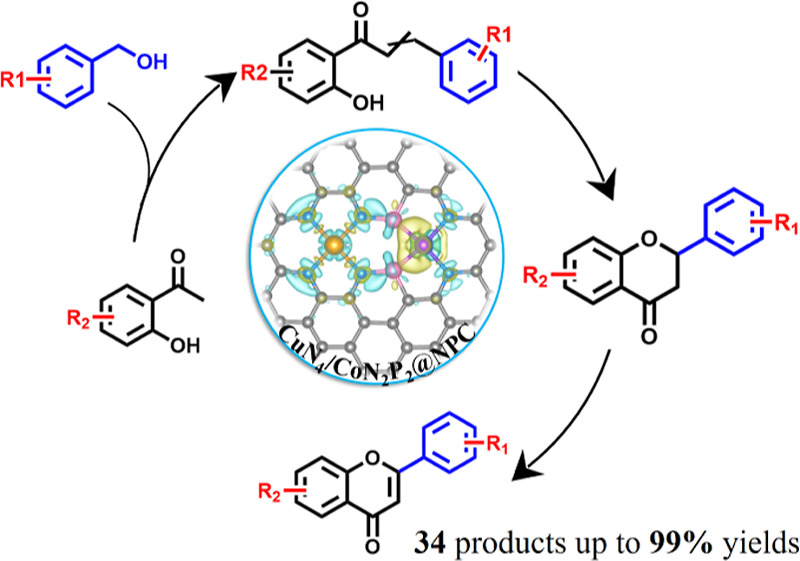
In this work, the combination of CuN4 and CoN2P2 sites achieves remarkable electron redistribution and an up-shifted DOS of the catalyst, therefore offering high potentials to simultaneously facilitate the activation and transformation of different reactants. To elucidate the impact of synergistic interactions between Cu and Co sites, we have rationally designed a domino reaction route starting from easily available substrates (substituted aromatic alcohols and ketones) for the preparation of various flavones. Taking flavone synthesis as an example, the first step is oxidative-coupling of benzyl alcohol and 2′-hydroxyacetophenone to 2′-hydroxychalcone, followed by the intramolecular cyclization to a flavanone, which undergoes the final oxidative dehydrogenation to yield a flavone (Figure 4b). In comparison with the conventional catalytic systems, this catalytic system features not only promoted efficiency but also considerable atom economy and environmental benign. Moreover, it also provides broad opportunities in the direct production of flavone derivatives from easily available reactants.
Figure 4.

(a) Oxidative coupling-dehydrogenation cascade reaction of benzyl alcohol and 2′-hydroxyacetophenone over different catalysts (for details, see Table S5). (b) Proposed reaction routes. (c, d) Free energy diagram. (e) Atomistic structures of the initial state, transition state, and final state for the reaction pathways over CuN4/CoN2P2@NPC, CuN4/CoN3P1@NPC (Figure S17), and CuN4/CoN4@NPC (Figure S18). The gray, blue, pink, orange, purple, red, and white balls represent C, N, P, Cu, Co, O, and H atoms, respectively. “TS” denotes a transition state.
Typically, the domino reaction was carried out at 120 °C and 2 bar of O2 (Figure 4a and Table S5), and a typical gas chromatography–mass spectrometry spectrum of the reaction solution is shown in Figure S13. The yields of products (e.g., flavone and flavanone) were also confirmed by nuclear magnetic resonance (NMR, Figures S14 and S15). The reaction did not produce flavones in the absence of either catalyst (Table S5, entry 1) or O2 (Table S5, entry 2). As expected, CuN4/CoN2P2@NPC, CuN4/CoN3P1@NPC, and CuN4/CoN4@NPC successfully trigger the reaction, all of which give a complete conversion of benzyl alcohol (Table S5, entries 3–5). However, their product distributions show remarkable differences, and CuN4/CoN2P2@NPC affords the highest flavone yield (>99%). Different Cu- or Co-based counterparts composed of Cu, Co nanoparticles and single-metal single-atom active sites are also employed as catalysts in this transformation (Table S5, entries 6–9). The unsatisfied conversions (<36%) and flavone yields (<3%) suggest the pivotal importance of Co/Cu dual single-atom sites in achieving high catalytic activity and selectivity.
Reaction Mechanism Investigations
To gain a theoretical insight into the reaction mechanism and disclose the underlying reasons for the excellent catalytic performance of CuN4/CoN2P2@NPC, DFT simulations were performed to identify the intrinsic reaction energy barriers. In the domino reaction, one of the most important steps is to activate molecular O2 to form O* free radicals. Thus, the activation and disassociation of O2 were investigated in detail (Figures 3, S16, and Table S6). Initially, the O2 molecule is adsorbed on the Co atom of the catalysts, and the adsorption energies on CuN4/CoN2P2@NPC, CuN4/CoN3P1@NPC, and CuN4/CoN4@NPC are −1.41, −1.27, and −0.82 eV with the corresponding O–O bond lengths of 1.31, 1.30, and 1.29 Å, respectively (Table S6). As compared with the bond length (1.23 Å) of the free O2 molecule, the increased O–O bond lengths suggest the activation of the adsorbed O2.
Detailed interactions between molecular orbitals of O2 and d orbitals of the Co center in the catalysts were investigated by projected DOS (PDOS). After O2 adsorption on the CoN4 site, the px and pz orbitals of O2 overlap with the dx and dz2 orbitals of the CoN4 site to form π (−7.5 to −5 eV) and π* (−2–2 eV) bonds, implying that the adsorbed O2 molecule is activated by CoN4. In terms of the CoN3P1 and CoN2P2 sites, an increased number of P atoms within the coordination configuration results in strengthened interactions originating from the Co site to the adsorbed O2 molecule, as is clearly verified by the enhanced contribution of the dx and dz2 orbitals of the Co site to the PDOS. The orbital hybridization results indicate that the increased P atom number within the coordination configuration is beneficial for O2 activation, and the O2 activation capability is in the order of CuN4/CoN2P2@NPC > CuN4/CoN3P1@NPC > CuN4/CoN4@NPC.
The subsequent O2 disassociation configurations were also examined (Figure S16). For CuN4/CoN4@NPC, the most stable O2 disassociation configuration is where one O atom locates on the top of the Co center and the other one moves to the bridge site of a C–C bond. For CuN4/CoN3P1@NPC and CuN4/CoN2P2@NPC, one O atom locates on the top of Co and the other moves to the bridge site of a P–C bond. The corresponding O2 disassociation energies are 0.85, 0.48, and 0.04 eV, respectively (Table S6). According to the disassociation energy barriers, it is apparent that CuN4/CoN2P2@NPC is the most active catalyst for O2 splitting.
Then, the transition state and activation energy for O2 disassociation were studied by the climbing image nudged elastic band (CI-NEB). The O2 activation energies are 3.21, 1.69, and 0.88 eV on CuN4/CoN4@NPC, CuN4/CoN3P1@NPC, and CuN4/CoN2P2@NPC, respectively (Figure 3m and Table S6). Noteworthily, a new O2 splitting configuration takes place on CuN4/CoN2P2@NPC, more stable than those of the previously reported catalysts. The optimized configuration (Figures 3n and S16) exhibits an extremely low O2 splitting energy of −0.89 eV, in which one O atom is bonded to P and Co and the other one locates at the Co center.
Finally, the charge redistribution after O2 adsorption was examined by charge difference plots (Figure 3d,h,l).37−39 The O–O bond of O2 is weakened after adsorption with electron depletion clearly observed. On the other hand, electrons around O and Co atoms are redistributed to form Co–O bonds, in which the Co atom of the catalyst tends to lose electrons. The Co atom in CuN4/CoN2P2@NPC transfers the most electrons to the adsorbed O2 molecule for its activation and splitting, outperforming the CuN4/CoN3P1@NPC and CuN4/CoN4@NPC counterparts.
To interpret the coordination configuration virtues of CuN4/CoN2P2@NPC, the free energy diagram of the domino reaction was calculated (Figures 4c–e, S17, and S18). The whole reaction process was divided into 14 elementary steps. First, one 2′-hydroxyacetophenone molecule was exothermically adsorbed on the Co atom of the catalysts via the −CH3 group to form (R1). Subsequently, the adsorbed 2′-hydroxyacetophenone lost an H atom from −CH3 to form an enolate anion (R2), and the energy barriers over CuN4/CoN2P2@NPC, CuN4/CoN3P1@NPC, and CuN4/CoN4@NPC were 1.77, 1.68, and 1.84 eV, respectively. Afterward, benzyl alcohol participated in the reaction with slight energy fluctuations and formed (R3), overcoming a 1.72 eV energy barrier over CuN4/CoN2P2@NPC (1.70 and 1.86 eV for CuN4/CoN3P1@NPC and CuN4/CoN4@NPC, respectively). Then, the enolate anion reacted with another substrate to generate a C–C single bond via a nucleophilic addition reaction, and the resultant hydroxyaldehyde adduct anion underwent a protonated process to produce β-hydroxyketone (R4). After such a complex process, the system energy changed a lot to 1.55 eV over CuN4/CoN2P2@NPC (1.24 and 1.23 eV over CuN4/CoN3P1@NPC and CuN4/CoN4@NPC, respectively). Further, the active β-hydroxyketone was endothermically dehydrated into 2′-hydroxychalcone (R5) as a stable conjugate structure. The energy barriers over CuN4/CoN2P2@NPC, CuN4/CoN3P1@NPC, and CuN4/CoN4@NPC were 4.01, 4.76, and 5.19 eV, respectively.
The obtained 2′-hydroxychalcone molecule was adsorbed onto the Co sites through the C=C group (R6) and went through the TS-1(R7) to form (R8), overcoming an energy barrier of 6.61 eV over CuN4/CoN2P2@NPC (7.56 and 7.73 eV for CuN4/CoN3P1@NPC and CuN4/CoN4@NPC, respectively). Then, the adsorbed 2′-hydroxychalcone molecule underwent an intramolecular cyclization reaction via TS-2(R9) to form flavanone (R10), overcoming energy barriers of 4.54 eV (over CuN4/CoN2P2@NPC), 5.09 eV (over CuN4/CoN3P1@NPC), and 5.02 eV (over CuN4/CoN4@NPC). Afterward, the disassociated O atom took part in the reaction (R11) with a negative energy over CuN4/CoN2P2@NPC (−0.58 eV) and CuN4/CoN3P1@NPC (−0.71 eV) and a positive energy of 1.36 eV over CuN4/CoN4@NPC. Flavanone was finally transformed into flavone via two dehydrogenation steps. The (R11) was first transformed into (R13) through TS-3(R12), during which an H atom of the intermediate was transferred to the adsorbed disassociated O, overcoming energy barriers of 0.79 eV (CuN4/CoN2P2@NPC), 1.66 eV (CuN4/CoN3P1@NPC), and 2.74 eV (CuN4/CoN4@NPC), respectively. Then, another H atom of (R13) was transferred to produce (R14) (*flavone). This step exothermically proceeded over CuN4/CoN2P2@NPC (−2.28 eV), CuN4/CoN3P1@NPC (−2.43 eV), and CuN4/CoN4@NPC (−2.59 eV). CuN4/CoN2P2@NPC outperformed the other two catalysts in reducing the reaction energy barriers of most elementary steps, which is believed to be attributable to the strengthened electron redistribution over Cu and Co sites (i.e., enhanced electron deficiency over the Co site and accordingly electron accumulation over the adjacent CuN4 site).
The overall reaction mechanism investigations indicate that regulated coordination configurations of the dual-metal SA catalyst had a significant impact on the catalytic performance. Based on the control experiments and DFT simulations, the CuN4/CoN2P2@NPC with suitable N and P content maximizes the electron redistribution throughout the catalyst, hence simultaneously facilitating O2 activation and consecutive catalysis for the preparation of flavone and derivatives.
Substrate Scope and Stability
To investigate the scope of the flavones, various substituted benzyl alcohols and 2′-hydroxyacetophenone with different functional groups (e.g., −CH3, −OCH3, −OH, and −NH2) were employed as substrates (Figure S19). As expected, CuN4/CoN2P2@NPC is also highly active for these transformations, affording the corresponding flavones in 81–99% yields. As we know, these flavone products possess unexpected biological activities owing to the presence of various functional groups. For instance, chrysin features a wide range of pharmacological activities and is also able to effectively inhibit HIV activation in a latent infection model. Acacetin shows effective anti-cancer activity through inducing cancer cell cycle arrest, apoptosis, and autophagy.
Finally, the stability and reusability of the CuN4/CoN2P2@NPC catalyst were examined. After the reaction, the catalyst was easily isolated from the solution by centrifugation and directly reused after washing and drying. No obvious loss in catalytic efficiency was observed within at least six cycles (Figure S20a), implying the reliable recyclability of the CuN4 and CoN2P2 sites under the investigated conditions. Moreover, the structural properties and chemical composition of the used CuN4/CoN2P2@NPC were well preserved after recycling, and the metal species were still atomically dispersed throughout the support without obvious aggregations (Figures S21 and S22). Furthermore, the hot filtration experiment (Figure S20b) demonstrates once again the negligible metal leaching during the reaction, which not only confirms the heterogeneous nature of this reaction but also further verifies the stability of the CuN4/CoN2P2@NPC catalyst.
Conclusions
In summary, we have demonstrated a versatile and highly efficient complexation-deposition strategy for the construction of 13 kinds of dual-metal single-atom site pairs with adjustable contents of N and P coordination atoms. The synthesis takes the advantages of atomic distribution of metal nodes in the MOF and strong interactions (Coulomb force) between the metal and heteroatoms. Taking the as-prepared CuN4/CoN3P1@NPC and CuN4/CoN2P2@NPC as examples, Cu and Co single-atom sites feature a homogeneous distribution and exclusive coordination configurations (CuN4, CoN3P1, or CoN2P2), and the CoN3P1 or CoN2P2 induces remarkable charge redistribution. Remarkably, CuN4/CoN2P2@NPC exhibits excellent catalytic performances in the domino synthesis of flavone and 33 kinds of derivatives from aromatic aldehydes and ketones. The DFT and control experiments indicate that the electron redistributions atop single-atom site pairs are beneficial for the adsorption and disassociation of oxygen molecules and hence eventually boost the entire domino reaction. To the best of our knowledge, this work represents the first example of the general synthesis of dual-metal single-atom composites with co-existing N and P coordination atoms and adjustable contents and therefore might open a new avenue for the development of advanced single-atom catalysts with tunable electron properties for high-efficiency catalysis applications.
Experimental Section
Synthesis of Cu-ZIF and Co-ZIF
In a typical synthesis, 2-MI (13.6 mmol) was dissolved in 5 mL of deionized water. After stirring for 5 min at room temperature, a 5 mL aqueous solution containing Zn(OAc)2·2H2O (1.36 mmol) and M(OAc)2 (0.136 mmol, M = Cu or Co) was added to the above solution under vigorous stirring. After stirring for 1 min at room temperature, the mixed solution was left undisturbed and aged for 5 h. Then, the product was collected by centrifugation and washed with methanol three times to obtain Cu-ZIF and Co-ZIF, respectively.
Synthesis of Hollow Cu-Complex@MP-Co Precursors and CuN4/CoN2P2@NPC Nano-Dodecahedrons
In a typical synthesis, the as-synthesized Cu-ZIF dodecahedrons (0.05 g) were dispersed in an aqueous solution of melamine (0.016 M, 50 mL) by sonication to form a homogeneous colloid. Then, a 1 mL aqueous solution containing Co(NO3)2·6H2O (0.005 g) was added to it dropwise. After stirring for 10 min at room temperature, phytic acid (0.065 g) was added to the above mixture under vigorous stirring. After stirring for 30 min at room temperature, the product was collected by centrifugation and washed with deionized water and ethanol three times, followed by drying at 50 °C to obtain Cu-complex@MP-Co. Typically, the as-prepared Cu-complex@MP-Co was placed in a tubular furnace, which was heated to 600 °C using a ramp rate of 1 °C min–1 and kept for 120 min and then heated to 700 °C using a ramp rate of 2 °C min–1 under an Ar atmosphere. The obtained black powders were recognized as CuN4/CoN2P2@NPC nano-dodecahedrons.
Other Experimental Details
Other experimental details are provided in the supplementary Experimental Section in the Supporting Information.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (21825802, 21908068, 22138003), the State Key Laboratory of Pulp and Paper Engineering (2022C04, 2022ZD05), the Fundamental Research Funds for the Central Universities (2022ZYGXZR108), the Natural Science Foundation of Guangdong Province (2017A030312005, 2020A1515010376), and the Science and Technology Program of Qingyuan City (2021YFJH01002).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.2c00582.
Experimental section, additional characterizations of MOFs and their derivatives, hetero SAs and counterparts, control experiments, catalytic results, and reaction mechanism (PDF)
Author Contributions
CRediT: Xin Zhao conceptualization, data curation, formal analysis, investigation, methodology, writing-original draft; Ruiqi Fang conceptualization, formal analysis, funding acquisition, investigation, project administration, writing-review & editing; Fengliang Wang formal analysis; Xiang-Peng Kong formal analysis; Yingwei Li conceptualization, funding acquisition, project administration, supervision, writing-review & editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Lang R.; Du X.; Huang Y.; Jiang X.; Zhang Q.; Guo Y.; Liu K.; Qiao B.; Wang A.; Zhang T. Single-atom catalysts based on the metal-oxide interaction. Chem. Rev. 2020, 120, 11986–12043. 10.1021/acs.chemrev.0c00797. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Zhou H.; Zhu X.; Qu Y.; Xiong C.; Xue Z.; Zhang Q.; Liu X.; Zhou F.; Mou X.; Wang W.; Chen M.; Xiong Y.; Lin X.; Lin Y.; Chen W.; Wang H.-J.; Jiang Z.; Zheng L.; Yao T.; Dong J.; Wei S.; Huang W.; Gu L.; Luo J.; Li Y.; Wu Y. Simultaneous oxidative and reductive reactions in one system by atomic design. Nat. Catal. 2021, 4, 134–143. 10.1038/s41929-020-00563-0. [DOI] [Google Scholar]
- Lin L.; Yu Q.; Peng M.; Li A.; et al. Atomically dispersed Ni/α-MoC catalyst for hydrogen production from methanol/water. J. Am. Chem. Soc. 2021, 143, 309–317. 10.1021/jacs.0c10776. [DOI] [PubMed] [Google Scholar]
- Shan J.; Ye C.; Jiang Y.; Jaroniec M.; Zheng Y.; Qiao S.-Z. Metal-metal interactions in correlated single-atom catalysts. Sci. Adv. 2022, 8, eabo0762 10.1126/sciadv.abo0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang H.; Zhou X.; Dong J.; Li A.; Zhao X.; Liu Q.; Lin Y.; Pei J.; Li Z.; Jiang Z.; Zhou D.; Zheng L.; Wang Y.; Zhou J.; Yang Z.; Cao R.; Sarangi R.; Sun T.; Yang X.; Zheng X.; Yan W.; Zhuang Z.; Li J.; Chen W.; Wang D.; Zhang J.; Li Y. Engineering unsymmetrically coordinated Cu-S1N3 single atom sites with enhanced oxygen reduction activity. Nat. Commun. 2020, 11, 3049–3059. 10.1038/s41467-020-16848-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy N.; Tylus U.; Jia Q.; Mukerjee S. Activity descriptor identification for oxygen reduction on nonprecious electrocatalysts: linking surface science to coordination chemistry. J. Am. Chem. Soc. 2013, 135, 15443–15449. 10.1021/ja405149m. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Gao R.; Ji S.; Li H.; Tang K.; Jiang P.; Hu H.; Zhang Z.; Hao H.; Qu Q.; Liang X.; Chen W.; Dong J.; Wang D.; Li Y. Atomic-level modulation of electronic density at cobalt single-atom sites derived from metal-organic frameworks: enhanced oxygen reduction performance. Angew. Chem., Int. Ed. 2021, 60, 3212–3221. 10.1002/anie.202012798. [DOI] [PubMed] [Google Scholar]
- Cao R.; Thapa R.; Kim H.; Xu X.; Gyu Kim M.; Li Q.; Park N.; Liu M.; Cho J. Promotion of oxygen reduction by a bio-inspired tethered iron phthalocyanine carbon nanotube-based catalyst. Nat. Commun. 2013, 4, 2076–2082. 10.1038/ncomms3076. [DOI] [PubMed] [Google Scholar]
- Chen K.; Liu K.; An P.; Li H.; Lin Y.; Hu J.; Jia C.; Fu J.; Li H.; Liu H.; Lin Z.; Li W.; Li J.; Lu Y. R.; Chan T. S.; Zhang N.; Liu M. Iron phthalocyanine with coordination induced electronic localization to boost oxygen reduction reaction. Nat. Commun. 2020, 11, 4173–4180. 10.1038/s41467-020-18062-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmieri L.; Monteverde Videla A.; Ocón P.; Specchia S. Kinetics of oxygen electroreduction on Me-N-C (Me = Fe, Co, Cu) catalysts in acidic medium: insights on the effect of the transition metal. J. Phys. Chem. C 2017, 121, 17796–17817. 10.1021/acs.jpcc.7b02455. [DOI] [Google Scholar]
- Jin H.; Li P.; Cui P.; Shi J.; Zhou W.; Yu X.; Song W.; Cao C. Unprecedentedly high activity and selectivity for hydrogenation of nitroarenes with single atomic Co1-N3P1 sites. Nat. Commun. 2022, 13, 723–731. 10.1038/s41467-022-28367-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia C.; Tan X.; Zhao Y.; Ren W.; Li Y.; Su Z.; Smith S. C.; Zhao C. Sulfur-dopant-promoted electroreduction of CO2 over coordinatively unsaturated Ni-N2 moieties. Angew. Chem., Int. Ed. 2021, 60, 23342–23348. 10.1002/anie.202109373. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Ji S.; Guo C.; Li H.; Dong J.; Guo P.; Wang D.; Li Y.; Toste F. D. A heterogeneous iridium single-atom-site catalyst for highly regioselective carbenoid O-H bond insertion. Nat. Catal. 2021, 4, 523–531. 10.1038/s41929-021-00637-7. [DOI] [Google Scholar]
- Chen Z.; Niu H.; Ding J.; Liu H.; Chen P.-H.; Lu Y.-H. Y.-R.; Lu W.; Zuo L.; Han Y.; Guo S.-F.; Hung Y.; Zhai Y. Unraveling the origin of sulfur-doped Fe-N-C single-atom catalyst for enhanced oxygen reduction activity: effect of iron spin-state tuning. Angew. Chem., Int. Ed. 2021, 60, 25404–25410. 10.1002/anie.202110243. [DOI] [PubMed] [Google Scholar]
- Jung E.; Shin H.; Lee B. H.; Efremov V.; Lee S.; Lee H. S.; Kim J.; Hooch Antink W.; Park S.; Lee K. S.; Cho S. P.; Yoo J. S.; Sung Y. E.; Hyeon T. Atomic-level tuning of Co-N-C catalyst for high-performance electrochemical H2O2 production. Nat. Mater. 2020, 19, 436–442. 10.1038/s41563-019-0571-5. [DOI] [PubMed] [Google Scholar]
- Wei X.; Zheng D.; Zhao M.; Chen H.; Fan X.; Gao B.; Gu L.; Guo Y.; Qin J.; Wei J.; Zhao Y.; Zhang G. Cross-linked polyphosphazene hollow nanosphere-derived N/P-doped porous carbon with single nonprecious metal atoms for the oxygen reduction reaction. Angew. Chem., Int. Ed. 2020, 59, 14639–14646. 10.1002/anie.202006175. [DOI] [PubMed] [Google Scholar]
- Hou C.-C.; Wang Y.; Zou L.; Wang M.; Liu H.; Liu Z.; Wang H.-F.; Li C.; Xu Q. A gas-steamed MOF route to P-doped open carbon cages with enhanced Zn-ion energy storage capability and ultrastability. Adv. Mater. 2021, 33, 2101698. 10.1002/adma.202101698. [DOI] [PubMed] [Google Scholar]
- Qin J.; Liu H.; Zou P.; Zhang R.; Wang C.; Xin H. L. Altering ligand fields in single-atom sites through second-shell anion modulation boosts the oxygen reduction reaction. J. Am. Chem. Soc. 2022, 144, 2197–2207. 10.1021/jacs.1c11331. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Zhao Y.; Gan J.; Xu J.; Wang Y.; Lv H.; Fang S.; Wang Z.; Deng Z.; Wang X.; Liu P.; Guo W.; Mao B.; Wang H.; Yao T.; Hong X.; Wei S.; Duan X.; Luo J.; Wu Y. Cation-exchange induced precise regulation of single copper site triggers room-temperature oxidation of benzene. J. Am. Chem. Soc. 2020, 142, 12643–12650. 10.1021/jacs.0c03415. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Zhao Y.; Chen C.; Huang Y.-C.; Dong C.-L.; Chen C.-J.; Liu R.-S.; Wang C.; Yan K.; Li Y.; Wang G. Tuning the coordination environment in single-atom catalysts to achieve highly efficient oxygen reduction reactions. J. Am. Chem. Soc. 2019, 141, 20118–20126. 10.1021/jacs.9b09352. [DOI] [PubMed] [Google Scholar]
- Yuan K.; Lützenkirchen-Hecht D.; Li L.; Shuai L.; Li Y.; Cao R.; Qiu M.; Zhuang X.; Leung M. K. H.; Chen Y.; Scherf U. Boosting oxygen reduction of single iron active sites via geometric and electronic engineering: nitrogen and phosphorus dual coordination. J. Am. Chem. Soc. 2020, 142, 2404–2412. 10.1021/jacs.9b11852. [DOI] [PubMed] [Google Scholar]
- Chen D.; Zhang L.-H.; Du J.; Wang H.; Guo J.; Zhan J.; Li F.; Yu F. A tandem strategy for enhancing electrochemical CO2 reduction activity of single-atom Cu-S1N3 catalysts via integration with Cu nanoclusters. Angew. Chem., Int. Ed. 2021, 60, 24022–24027. 10.1002/anie.202109579. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Wang F.; Kong X.-P.; Fang R.; Li Y. Dual-metal hetero-single-atoms with different coordination for efficient synergistic catalysis. J. Am. Chem. Soc. 2021, 143, 16068–16077. 10.1021/jacs.1c06349. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Fischer J. M. T. A.; Jia Y.; Yan X.; Xu W.; Wang X.; Chen J.; Yang D.; Liu H.; Zhuang L.; Hankel M.; Searles D. J.; Huang K.; Feng S.; Brown C. L.; Yao X. Coordination of atomic Co-Pt coupling species at carbon defects as active sites for oxygen reduction reaction. J. Am. Chem. Soc. 2018, 140, 10757–10763. 10.1021/jacs.8b04647. [DOI] [PubMed] [Google Scholar]
- Zhu Z.; Yin H.; Wang Y.; Chuang C.-H.; Xing L.; Dong M.; Lu Y.-R.; Casillas-Garcia G.; Zheng Y.; Chen S.; Dou Y.; Liu P.; Cheng Q.; Zhao H. Coexisting single-atomic Fe and Ni sites on hierarchically ordered porous carbon as a highly efficient ORR electrocatalyst. Adv. Mater. 2020, 32, 2004670. 10.1002/adma.202004670. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Xiao M.; Ren D.; Gao R.; Liu X.; Zhang Z.; Luo D.; Xing W.; Su D.; Yu A.; Chen Z. Quasi-covalently coupled Ni-Cu atomic pair for synergistic electroreduction of CO2. J. Am. Chem. Soc. 2022, 144, 9661–9671. 10.1021/jacs.2c00937. [DOI] [PubMed] [Google Scholar]
- Liu C.; Li T.; Dai X.; Zhao J.; He D.; Li G.; Wang B.; Cui X. Catalytic activity enhancement on alcohol dehydrogenation via directing reaction pathways from single- to double-atom catalysis. J. Am. Chem. Soc. 2022, 144, 4913–4924. 10.1021/jacs.1c12705. [DOI] [PubMed] [Google Scholar]
- Lu X. F.; Yu L.; Zhang J.; Lou X. W. Ultrafine dual-phased carbide nanocrystals confined in porous nitrogen-doped carbon dodecahedrons for efficient hydrogen evolution reaction. Adv. Mater. 2019, 31, 1900699. 10.1002/adma.201900699. [DOI] [PubMed] [Google Scholar]
- Ellis G. P.Chromenes, Chromanones, and Chromones; Wiley: New York, 1977. [Google Scholar]
- Biddle M. M.; Lin M.; Scheidt K. A. Catalytic enantioselective synthesis of flavanones and chromanones. J. Am. Chem. Soc. 2007, 129, 3830–3831. 10.1021/ja070394v. [DOI] [PubMed] [Google Scholar]
- Wang L.; Liu X.; Dong Z.; Fu X.; Feng X. Asymmetric intramolecular Oxa-Michael addition of activated α, β-unsaturated ketones catalyzed by a chiral N,N’-dioxide nickel(II) complex: highly enantioselective synthesis of flavanones. Angew. Chem., Int. Ed. 2008, 47, 8670–8673. 10.1002/anie.200803326. [DOI] [PubMed] [Google Scholar]
- Chen J.; Chen J.; Lang F.; Zhang X.; Cun L.; Zhu J.; Deng J.; Liao J. A C2-symmetric chiral bis-sulfoxide ligand in a rhodium-catalyzed reaction: asymmetric 1,4-addition of sodium tetraarylborates to chromenones. J. Am. Chem. Soc. 2010, 132, 4552–4553. 10.1021/ja1005477. [DOI] [PubMed] [Google Scholar]
- Diao T.; Stahl S. S. Synthesis of cyclic enones via direct palladium-catalyzed aerobic dehydrogenation of ketones. J. Am. Chem. Soc. 2011, 133, 14566–14569. 10.1021/ja206575j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan G.; Yang X.; Ma L.; Rao Y. Pd-catalyzed C-H oxygenation with TFA/TFAA: expedient access to oxygen-containing heterocycles and late-stage drug modification. Angew. Chem., Int. Ed. 2012, 51, 13070–13074. 10.1002/anie.201207458. [DOI] [PubMed] [Google Scholar]
- Zhao D.; Beiring B.; Glorius F. Ruthenium-NHC-catalyzed asymmetric hydrogenation of flavones and chromones: general access to enantiomerically enriched flavanones, flavanols, chromanones, and chromanols. Angew. Chem., Int. Ed. 2013, 52, 8454–8458. 10.1002/anie.201302573. [DOI] [PubMed] [Google Scholar]
- Yatabe T.; Jin X.; Yamaguchi K.; Mizuno N. Gold nanoparticles supported on a layered double hydroxide as efficient catalysts for the one-pot synthesis of flavones. Angew. Chem., Int. Ed. 2015, 54, 13302. 10.1002/anie.201507134. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Gao G.; Li Y.; Chu W.; Wang L. W. Transition-metal single atoms in nitrogen-doped graphenes as efficient active centers for water splitting: A theoretical study. Phys. Chem. Chem. Phys. 2019, 21, 3024–3032. 10.1039/c8cp06755d. [DOI] [PubMed] [Google Scholar]
- Yao Y.; Xu Z.; Cheng F.; Li W.; Cui P.; Xu G.; Xu S.; Wang P.; Sheng G.; Yan Y.; Yu Z.; Yan S.; Chen Z.; Zou Z. Unlocking the potential of graphene for water oxidation using an orbital hybridization strategy. Energy Environ. Sci. 2018, 11, 407–416. 10.1039/c7ee02972a. [DOI] [Google Scholar]
- Huang Q.; Wang B.; Ye S.; Liu H.; Chi H.; Liu X.; Fan H.; Li M.; Ding C.; Li Z.; Li C. Relation between water oxidation activity and coordination environment of C,N-coordinated mononuclear Co catalyst. ACS Catal. 2022, 12, 491–496. 10.1021/acscatal.1c04644. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.