

Abstract
With the advent of next-generation sequencing (NGS), identifying and better understanding genetic mutations in cancer pathways has become more feasible. A mutation now commonly reported in NGS panels is the SETD2 gene (H3K36 trimethyltransferase). However, its contributions to colorectal cancer (CRC) are not well-described. In this study we describe the clinicopathologic characteristics of SETD2-mutated CRC, determine common mutation sites on the SETD2 gene, and correlate these mutations with the loss of H3K36 trimethylation and the aberrant expression of beta-catenin. By searching pathology reports at our institution which included the 161-gene NGS panel from 2019–2021, we identify 24 individuals with SETD2-mutated CRC. All samples were evaluated for microsatellite status, H3K36 trimethylation, and beta-catenin via immunohistochemistry. In this cohort of 24 SETD2-mutated CRC individuals {a median age of 62.4 years (IQR: 49.1–73.6)}, Ten (41.7%) patients presented at American Joint Committee on Cancer (AJCC) tumor stage II, seven (29.2%) at stage III, six (25%) at stage IV and one (4.2%) at stage I. Most tumors studied were adenocarcinomas with no further specification (22, 92%) and most tumors were microsatellite stable (18, 82.5%). 33 mutation locations were represented by 24 patients, with one patient having six mutations in the SETD2 gene and two patients having three mutations. The dominant mutation type is missense mutations (N = 29, 87.9%), and no mutation hotspots were found. Only two samples lost trimethylation of Histone H3K36, both from individuals with multiple SETD2 mutations and aberrant nuclear beta-catenin expression. SETD2-mutated CRC is similar in clinical and histologic presentation to other commonly-reported CRC. SETD2 mutations were missense dominant, showed no hotspots, and multiple mutations are likely necessary for loss of H3K36 trimethylation. These results warrant further study on determining a role of SETD2 -Histone H3K36 pathway in CRC.
Keywords: SETD2, colorectal cancer, next-generation sequencing, H3K36
1. Introduction
Colorectal cancer (CRC) is the third most common and second most deadly cancer worldwide.1,2 Though previously considered a malignancy primarily affecting high-income countries, incidence and deaths in low- and middle-income countries are increasing.3,4 As global life-expectancy increases in the coming decades, global colorectal cancer incidence, which is positively correlated with age, is expected to continue to rise.5 Primary colorectal cancer is a result of the well-described adenoma to carcinoma sequence, first reported by Vogelstein and Fearon.6 This transformation involves sequential mutations driving tumorigenesis in colorectal cancer, now serving as a model for other solid tumor malignancies. Further studies are however still needed to better understand this tumorigenesis process to ameliorate the global burden of colorectal cancer.
With the advent of Sanger and now next-generation sequencing (NGS), identifying and better understanding genetic mutations in cancer pathways has become more feasible.7 NGS has become routine practice in academic centers in the U.S., enabling molecular testing of more colorectal cancer samples. In addition to exploring common mutations and their potential role in diagnostic and therapeutic approaches,8,9 rare genes may be studied with NGS as well to better understand the heterogeneity of the tumor microenvironment.
SETD2 is a histone methyltransferase that is responsible for the trimethylation of histone H3K36 and downstream cellular processes such as transcriptional regulation and DNA damage repair.10,11 As a member of the nuclear receptor SET domain-containing (NSD) family, SETD2 and its dysregulation have been implicated in the development of solid organ cancers such as liver, pancreatic, lung, breast, and renal cancer.11–14 In colorectal carcinoma, SETD2 was shown to modulate the Wnt signaling pathway in a mouse model, with SETD2 loss promoting tumor progression.15 However, SETD2 has only clinically been implicated in colorectal cancer in case reports. 16–19
Though the role of SETD2 in colorectal cancer has been postulated in previous reports, a larger case series has not been described. The purpose of this study is to first describe the clinicopathologic characteristics of SETD2-mutated CRC, determine common mutation sites on the SETD2 gene, and finally correlate these mutations with the loss of trimethylation of H3K36 and the aberrant expression of beta-catenin using the first and largest cohort to date.
2. Methods
2.1. Patient cohort and clinical data
We searched all pathology reports beginning with the adoption of the 161-gene NGS panel at our institution in 2019 and ending in 2021, and 24 individual cases of colorectal carcinoma were identified with SETD2 mutations. All these samples had concurrent mismatch repair (MMR) immunohistochemical staining. Clinical and demographic data for these 24 individuals was abstracted using chart review from the electronic health record. This study, and specifically both the database search and chart review of clinical. And demographic characteristics, was approved by the Institutional Review Board at the Northwestern University Feinberg School of Medicine.
2.2. Immunohistochemistry
First, all tumors were evaluated for microsatellite status and beta-catenin. This was done via immunohistochemistry (IHC) and is standard for tumors processed at our institution. The IHC staining method for microsatellite status and beta-catenin is Clinical Laboratory Improvement Amendments–approved and was completed with an automated immunostainer (Leica-Bond III; Leica Biosystems, Buffalo Grove, IL) and Bond Refine PolymerTM biotin-free 3,3′-diaminobenzidine detection kit. Antibodies for MLH1 (mouse anti-human antibody clone ES05, Leica, Buffalo Grove, IL), MSH2 (mouse anti-human antibody clone G219-1129, Cell-Marque, Rocklin, CA), MSH6 (mouse anti-human clone 44, Cell-Marque, Rocklin, CA), and PMS2 (mouse anti-human antibody clone MRQ-28, Cell-Marque, Rocklin, CA), and Beta-Catenin (mouse anti-human antibody clone 17C2; Leica, Buffalo Grove, IL) were used to determine nuclear microsatellite expression, which was deemed positive signal intensity in more than 1% of cells within a tumor sample, and to determine aberrant nuclear and cytoplasm beta-catenin expression (normally it shows an intensely membranous staining in the cells). Samples in which there was loss of mismatch repair protein’s expression were deemed microsatellite unstable, as per clinical protocol.
Histone H3 was stained as follows. Formalin-fixed and paraffin embedded colorectal carcinoma samples were stained with H3K36me3 PA5-96118 at concentration 1:200 and incubated at 4°C overnight. Subsequently, biotinylated horse anti-rabbit IgG secondary antibody was added and incubated in diaminobenzidine tetrahydrochloride for three minutes. Finally, high-pressure heat-mediated antigen retrieval was performed with citrate buffer pH 6 before commencing with IHC staining protocol. Staining was assessed in a binary fashion as total loss of methylation or retention of methylation.
2.3. Protein Modeling
Because the crystal structure of the full SETD2 protein is not known, estimation of protein structure was done using an online, publicly available version of AlphaFold. AlphaFold has been shown to accurately model protein structures.20,21
2.4. Next-Generation Sequencing
A full description of the NGS method used by our institution is described in our supplementary methods.
2.5. Data Analysis
All statistical analysis was completed using Stata, Version 15 (StataCorp, College Station, TX). Standard summary statistics of clinical and demographic information were reported with either frequencies and percentages or medians with interquartile ranges. Mutation sites and co-mutations were reported in frequencies and percentages. Statistical significance was defined as P < 0.05.
3. Results
3.1. Full Cohort
The cohort comprised of 24 individuals with a median age of 62.4 years (IQR: 49.1–73.6) with five patients (20.8%) being under age 50. The cohort was majority female (N = 13, 54% female, white (17, 70.8%) and non-Hispanic (21, 87.5%). Tumors were located equally in the proximal colon (12, 50%) and distal colon (12, 50%). The median tumor size was 5.9cm (4–8) and most tumors were low grade (16, 66.7%). Ten (41.7%) patients presented at American Joint Committee on Cancer (AJCC) tumor stage II, seven (29.2%) at stage III, and one (4.2%) at. Stage I. Six (25%) individuals presented with metastatic disease at AJCC stage IV (Full TNM information in Table 1). Almost all of the tumors studied were adenocarcinomas with no further specification (92%) and most tumors were microsatellite stable (82.5%) (Table 1).
Table 1 –
Clinicopathologic Characteristics of SETD2-Mutated Colorectal Carcinoma
| Total Number | 24 | 100% |
|---|---|---|
| Age | ||
| Median, IQR | 62.4 | [49.1–73.6] |
| Sex | ||
| Male | 11 | 46% |
| Female | 13 | 54% |
| Race | ||
| White | 17 | 71% |
| Black | 5 | 21% |
| Asian | 1 | 4% |
| Declined | 1 | 4% |
| Ethnicity | ||
| Non-Hispanic | 21 | 88% |
| Hispanic | 1 | 4% |
| Declined | 1 | 4% |
| Tumor Location | ||
| Proximal Colon | 12 | 50% |
| Cecum | 3 | 13% |
| Ascending | 6 | 25% |
| Transverse | 3 | 13% |
| Distal Colon | 12 | 50% |
| Descending | 2 | 8% |
| Sigmoid | 5 | 21% |
| Rectum | 5 | 21% |
| Tumor Stage | ||
| I | 1 | 4% |
| II | 10 | 42% |
| III | 7 | 29% |
| IV | 6 | 25% |
| Size | ||
| Median, IQR | 5.9 | [4–8] |
| Tumor Grade | ||
| Unknown | 2 | 8% |
| Low/Mod | 16 | 67% |
| High | 6 | 25% |
| Tumor Stage | ||
| Unknown | 3 | 13% |
| pT1 | 1 | 4% |
| pT2 | 3 | 13% |
| pT3 | 11 | 46% |
| pT4 | 6 | 25% |
| Lymph Node | ||
| pNX | 4 | 17% |
| pN0 | 9 | 38% |
| pN1 | 4 | 17% |
| pN2 | 7 | 29% |
| Metastases | ||
| pMX | 15 | 63% |
| pM0 | 3 | 13% |
| pM1 | 6 | 25% |
| Histology Subtypes | ||
| Adenocarcinoma, NOS | 22 | 92% |
| Mucinous | 2 | 8% |
| Microsatellite Status | ||
| Stable | 20 | 83% |
| Unstable | 4 | 17% |
3.2. Mutation Types and Sites and Histone H3K36 Trimethylation
Thirty-three mutation locations were represented by 24 patients, with one patient having six mutations in the SETD2 gene and two patients having three mutations. Patients with multiple mutations all were microsatellite stable. The dominant mutation type is missense mutations (N = 29, 87.9%), while nonsense (2, 6.1%) and star site (2, 6.1%) mutations were less common. There were also no mutation hotspots, with no mutation sites occurring in more than two (6.1%) patients (Table 2). Only two mutations were in regions of the SETD2 protein with known crystal structure, with p.D1616Y appearing to be in the core of the protein, and p.T2421A appearing to be on the surface of the protein. The rest of the mutations occurred in regions that have only been modeled in silico20,21 (Figure 1).
Table 2 –
SETD2 Mutation Types and Mutation Sites
| Mutation Type and Location | N | % |
|---|---|---|
| Missense Mutations | 29 | 87.9% |
| p.T592K | 2 | 6.1% |
| p.T928R | 2 | 6.1% |
| p.A2350T¤ | 1 | 3.0% |
| p.C714R‡ | 1 | 3.0% |
| p.D1616Y‡ | 1 | 3.0% |
| p.D350N¤ | 1 | 3.0% |
| p.I635V | 1 | 3.0% |
| p.K1074N | 1 | 3.0% |
| p.L1357I‡ | 1 | 3.0% |
| p.L600V | 1 | 3.0% |
| p.M2369R | 1 | 3.0% |
| p.N2058S | 1 | 3.0% |
| p.P193L | 1 | 3.0% |
| p.Q2030† | 1 | 3.0% |
| p.R1879H | 1 | 3.0% |
| p.R402Q | 1 | 3.0% |
| p.S1001C† | 1 | 3.0% |
| p.S1076C† | 1 | 3.0% |
| p.S1098F† | 1 | 3.0% |
| p.S1888I | 1 | 3.0% |
| p.S803C† | 1 | 3.0% |
| p.T1033A | 1 | 3.0% |
| p.T1077A | 1 | 3.0% |
| p.T1233A | 1 | 3.0% |
| p.T2421A | 1 | 3.0% |
| p.V2229A | 1 | 3.0% |
| p.Y389H | 1 | 3.0% |
| Nonsense Mutation | 2 | 6.1% |
| p.S512*† | 1 | 3.0% |
| p.E2215*¤ | 1 | 3.0% |
| Translation Start Site | 2 | 6.1% |
Three patients had multiple mutations. Mutations labeled
came from one patient, those labeled with
came from a second patient, and those labeled with
came from a third patient.
Figure 1: AlphaFold Modeling of the SETD2 Protein.

Figure 1 shows the AlphaFold predicted structure of the SETD2 protein. Orange lines represent unknown areas of the protein and blue/yellow areas represent regions with. A known crystal structure. Only two mutations, p.D1616Y and p.T2421A are within known regions of the protein. The former is estimated to be within the core of the protein while the latter is predicted to be towards. The surface of the protein, based on predicted structure and folding.
Additionally, we stained all samples for Histone H3K36 trimethylation. Two samples lost H3K36 trimethylation – one sample was from an individual with six SETD2 mutations and the second was from an individual with three SETD2 mutations (Figure 2A). All other samples retained H3K36 trimethylation (Figure 2B).
Figure 2: Trimethylation of H3K36 and aberrant expression of Beta-catenin.
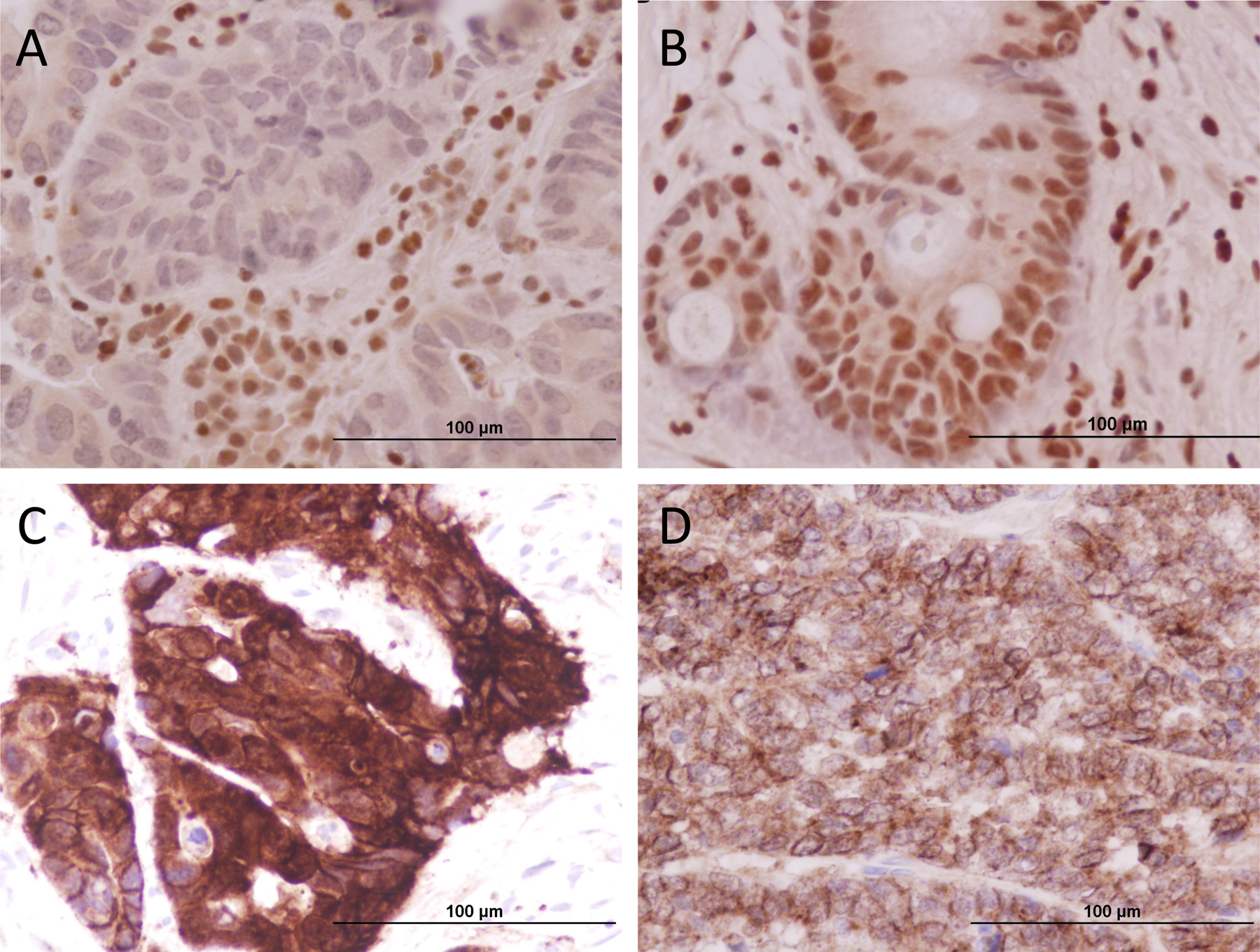
Figure 2A shows a loss of H3K36 trimethylation. This was found in 2 individuals with multiple SETD2 mutations. Figure 2B shows retained H3K36 trimethylation. All individuals with single mutations retained H3K36 trimethylation and one individual with multiple mutations. This may suggest that multiple SETD2 mutations are required for full loss of trimethylation of H3K36. Figure 2C shows aberrant cytoplasmic/nuclear beta-catenin expression in SETD2-mutated CRC with the loss of H3K36 trimethylation. Figure 2D shows dominant membranous beta-catenin expression.
Beta-catenin immunostaining showed 87.5% (21/24) CRC samples had intense cytoplasmic/nuclear expression of beta-catenin (Fig.2C) and only three CRC retained membranous staining pattern (Fig.2D). Two samples with the loss of H3K36 trimethylation exhibited aberrant cytoplasmic/nuclear beta-catenin expression (Fig.2D).
3.3. Co-Occurring Mutations
All 24 SETD2-mutated CRC had at least two other co-occurring mutations and no patients had only SETD2 mutations. The most common co-occurring mutation was in the TP53 gene (N = 14, 58.3%), followed by KRAS (12, 50%), ATM (9, 37.5%), BRCA2 (8, 33.3%), and ARID1A (8, 33.3%). Only five patients had SETD2 mutations without either a p53 or a KRAS mutation, and seven patients had all three mutations. Interestingly, all five patients without either a p53 or a KRAS mutation had aberrant cytoplasmic/nuclear beta-catenin expression detected immunohistochemically. A total of 89 genes were represented in the co-occurring mutations. 17 patients had less than ten total mutations, while seven had more than ten total mutations. Full information can be found in Table 3.
Table 3 –
Mutations Co-Occurring with SETD2
| Gene | N | % | Gene | N | % | Gene | N | % |
|---|---|---|---|---|---|---|---|---|
| TP53 | 14 | 58.3% | PIK3R1 | 3 | 12.5% | CTNNB1 | 1 | 4.2% |
| KRAS | 12 | 50.0% | RB1 | 3 | 12.5% | ERBB2 | 1 | 4.2% |
| ATM | 9 | 37.5% | RET | 3 | 12.5% | ESR1 | 1 | 4.2% |
| BRCA2 | 8 | 33.3% | SMAD4 | 3 | 12.5% | EZH2 | 1 | 4.2% |
| ARID1A | 8 | 33.3% | SMARCB1 | 3 | 12.5% | FANCA | 1 | 4.2% |
| ATR | 6 | 25.0% | TSC1 | 3 | 12.5% | FANCD2 | 1 | 4.2% |
| BRCA1 | 5 | 20.8% | TSC2 | 3 | 12.5% | FGF19 | 1 | 4.2% |
| CREBBP | 5 | 20.8% | AR | 2 | 8.3% | FGF3 | 1 | 4.2% |
| POLE | 5 | 20.8% | BAP1 | 2 | 8.3% | FGFR2 | 1 | 4.2% |
| SMARCA4 | 5 | 20.8% | DDR2 | 2 | 8.3% | FLT3 | 1 | 4.2% |
| CDK12 | 4 | 16.7% | EGFR | 2 | 8.3% | JAK1 | 1 | 4.2% |
| CDKN2B | 4 | 16.7% | FANCI | 2 | 8.3% | JAK3 | 1 | 4.2% |
| FBXW7 | 4 | 16.7% | MLH1 | 2 | 8.3% | KNSTRN | 1 | 4.2% |
| MSH6 | 4 | 16.7% | MRE11A | 2 | 8.3% | MAP2K1 | 1 | 4.2% |
| NF1 | 4 | 16.7% | MTOR | 2 | 8.3% | MAP2K4 | 1 | 4.2% |
| NOTCH1 | 4 | 16.7% | NBN | 2 | 8.3% | MDM2 | 1 | 4.2% |
| PMS2 | 4 | 16.7% | NTRK1 | 2 | 8.3% | MRE11A | 1 | 4.2% |
| PTEN | 4 | 16.7% | NTRK3 | 2 | 8.3% | MSH2 | 1 | 4.2% |
| RAD50 | 4 | 16.7% | PDGFRA | 2 | 8.3% | MYC | 1 | 4.2% |
| RNF43 | 4 | 16.7% | PIK3CB | 2 | 8.3% | MYCL | 1 | 4.2% |
| SLX4 | 4 | 16.7% | PTCH1 | 2 | 8.3% | MYCN | 1 | 4.2% |
| ATRX | 3 | 12.5% | ROS1 | 2 | 8.3% | NF2 | 1 | 4.2% |
| BRAF | 3 | 12.5% | STK11 | 2 | 8.3% | PPARG | 1 | 4.2% |
| ERBB3 | 3 | 12.5% | AKT2 | 1 | 4.2% | PPP2R1A | 1 | 4.2% |
| KIT | 3 | 12.5% | AKT3 | 1 | 4.2% | RAD51 | 1 | 4.2% |
| NOTCH2 | 3 | 12.5% | AXL | 1 | 4.2% | RAD51B | 1 | 4.2% |
| NOTCH3 | 3 | 12.5% | CCND3 | 1 | 4.2% | RAD51C | 1 | 4.2% |
| PALB2 | 3 | 12.5% | CDK6 | 1 | 4.2% | RAD51D | 1 | 4.2% |
| PDGFRB | 3 | 12.5% | CHEK1 | 1 | 4.2% | RHEB | 1 | 4.2% |
| PIK3CA | 3 | 12.5% | CSF1R | 1 | 4.2% | |||
4. Discussion
Although SETD2 mutations within colorectal cancer have been described, they are limited to case reports. This is the largest study to date to describe the clinicopathologic characteristics of SETD2 mutated CRC, determine common mutation sites on the SETD2 gene, and correlate these mutations with the loss of trimethylation of H3K36. We found that, within our sample, SETD2 mutated CRC presents in the seventh decade of life, affects the proximal and distal colon with equal frequency, is more likely to be AJCC Stage III or IV, and is likely to be microsatellite stable. Additionally, we found that most mutations are missense mutations and that there are no “hostpot” mutation sites within the SETD2 gene. Finally, we demonstrate that multiple mutations within the SETD2 gene may be required to lose trimethylation of H3K36, although the clinical correlates of this finding are unclear.
There is limited literature regarding the clinical characteristics of SETD2-mutated CRC. The age of diagnosis in our sample is similar to that reported in the general population, although the rate of CRC onset under age 50 is increased in our sample. 22,23 Additionally, we report 25% of patients presented with metastatic disease, which is also similar to that reported in the general population.22,23 Although we have a limited sample, we report the largest cohort to date for SETD2-mutated CRC and hope this study may serve as a foundation for future research. The increasing adoption of NGS nationally will facilitate larger studies, which are warranted to fully elucidate the clinical presentation of SETD2-mutated CRC. Further, as NGS adoption to test all tumor samples is relatively recent, analysis of SETD2 as related to prognosis, treatment efficacy, and survival will be possible and is necessary for understanding SETD2 mutation as related to clinical outcomes.
We are also one of the first studies to attempt to identify hotspot mutation sited within the SETD2 gene. The majority of mutations in our sample were missense mutations. However, only two sites were mutated in multiple individuals, with both of these sites being mutated in two patients each. This suggests there are no mutation hotspots within the SETD2 gene in SETD2-mutated CRC. It is possible that the mutations we identified may be in specific, functionally crucial portions of the SETD2 protein. However, the full crystal structure of the SETD2 protein has not been fully identified, with only a small region definitively mapped and the rest only modeled in silico. Additionally, it appears one of the mutations in the known regions of the protein was in the core, while another was closer to the protein surface. Research investigating the structure and function of the SETD2 protein is warranted in order to fully understand how mutations may lead to initial dysplasia with progression to invasive carcinoma. Additionally, we investigated how different SETD2 mutations affected histone H3K36 trimethylation – as methylation of histone H3 modulated DNA repair, we reasoned that loss of this trimethylation may represent total loss of expression of functional SETD2 protein. We found that no individuals with single SETD2 mutations had tumor samples with loss of H3K36 trimethylation. However, two of the three patients with multiple mutations did show loss of H3K36 trimethylation. Both of these patients had AJCC Stage II (T3N0Mx) CRC. Interestingly, all patients were microsatellite stable, and so SETD2 mutation status may not be associated with MMR status. Although a small sample, this may suggest that multiple SETD2 mutations are required for functional protein loss. As our full cohort is small, and only three patients (12.5%) within our full sample had multiple SETD2 mutations, future, larger studies are warranted to confirm this observation and to correlate functional loss with clinical presentation.
Another important finding of our study was identifying common co-occurring mutations with SETD2. First, we demonstrated that no patients had only SETD2 mutations, and all had other co-mutations, potentially indicating that SETD2 mutations by themselves are not sufficient for producing neoplasia but may modify existing oncogenic pathways. We found that TP53 and KRAS were the most commonly co-mutated genes, with only five patients having mutations in neither gene. As TP53 and KRAS are crucially implicated in the canonical APC adenoma-carcinoma sequence, this may suggest that SETD2 mutations modulate oncogenic risk for precancerous lesions in that pathway. In fact, SETD2 is known to interact with p53 by activating p53-mediated double-strand break DNA repair, regulating downstream p53 targets such as HDM2, and enhancing p53 stability.24–27 As such, co-occurring p53 and SETD2 mutation may further derange physiologic DNA repair and increase oncogenic risk. Additionally, in the ten individuals without p53 co-mutations, there is a high ATM mutation (7, 70%). SETD2 is also known to interact with ATM-mediated DNA repair, with functional ATM kinase and SETD2 regulating double-strand break repair.25,28,29 These findings, taken together, suggest that the significance of SETD2 may be as a key modulator of various DNA damage repair pathways which, in the presence of other co-mutations involved in DNA repair, may enhance oncogenic potential.30 Alternatively, SETD2 mutation, and thus defective DNA repair, may facilitate the mutation of other genes known to more directly cause oncogenesis. Further basic and translational research is warranted to understand how SETD2 mutations are implicated in these pathways, and future clinical research is warranted to correlate the presence of SETD2 mutations in these pathways with clinical outcomes.
One limitation is that no APC gene in the NGS panel utilized at our institution, and so the true prevalence of SETD2-APC co-mutation can only be inferred but not known directly. Thus, we analyzed the aberrant beta-catenin expression immunohistochemically. We found that aberrant cytoplasmic/nuclear beta-catenin expression is the most common alteration in our cohort patients. Interestingly, all five SETD2 mutant CRC without either a p53 or a KRAS mutation had aberrant beta-catenin expression, indicating Co-operation with WNT pathway to driving carcinogenesis is another pathologic feature of SETD2 mutant CRC.
This study has limitations. The first and most significant limitation is our small sample size. NGS has only recently been adopted and performed on all CRC samples at our institution. Additionally, SETD2 mutation is fairly uncommon, and these factors combine to result in a sample size of 24 cases. The small sample size makes it difficult to draw conclusions regarding clinical presentation of SETD2-mutated CRC, particularly its stage at presentation. Larger studies would provide more reliable characterization of clinical presentation. However, although clinical conclusions may be obscured by the small sample size, molecular characterization of SETD2 mutations, namely missense dominance, lack of hotspots and low rates of H3 loss, can be generalized with more reliability. The sample size does affect our ability to study how functional H3 loss impact clinical outcomes, and this is a promising area of future investigation. This small sample size limits our analysis of subgroups of individuals with different patterns of co-mutations, as well. Finally, this study was done at a tertiary referral center in a large urban area, which may limit generalizability, especially given the widespread prevalence of CRC in various geographic areas and across demographics.
In conclusion, there is a dearth of literature regarding SETD2-mutated CRC. This study is one of the first and largest to describe the clinicopathologic characteristics of SETD2-mutated CRC, identify mutation sites within the SETD2 gene, and correlate SETD2 mutations with total loss of H3K36 trimethylation. We found that the clinical presentation of SETD2-mutated CRC is similar to that reported for CRC in the general population, although it may be more common in the proximal colon. SETD2-mutated CRC is also likely to be found in the context of co-occurring p53 mutations and aberrant beta-catenin expression, and the two proteins are known to interact to modulate DNA repair. Finally, multiple SETD2 mutations appear to be required for total loss of H3K36 trimethylation, although future studies are required to confirm this observation. The increasing adoption of utilizing NGS on all tumor samples and thus the growing detection of SETD2-mutated CRC will facilitate future study of the significance of SETD2 mutation in CRC.
Supplementary Material
HIGHLIGHTS.
SETD2 tumors have similar characteristics to CRC in the general population, although may have a predilection for the proximal colon
SETD2 mutations are missense dominant, show no hotspots, and multiple mutations are seemingly required for total loss of histone H3K36 trimethylation
SETD2 mutations do not occur in isolation, and thus their significance may be further derangement of DNA repair mechanisms that furthers the potential for carcinogenesis by known oncogenic mutations
Funding Statement:
NIH R01 DK10776, CA172431, and CA164041 to Dr. Guang-Yu Yang; and NIH R35GM146979 and the ACS Research Scholar Grant (RSG-22-039-01-DMC) to Dr. Lu Wang.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COI: The authors declare no conflicts of interest for this article.
This article is not associated with a clinical trial.
References
- 1.Ranasinghe R, Mathai M, Zulli A. A synopsis of modern - day colorectal cancer: Where we stand. Biochim Biophys Acta Rev Cancer 2022;1877(2):188699. [DOI] [PubMed] [Google Scholar]
- 2.Organization WH. Cancer: The problem. https://www.who.int/news-room/fact-sheets/detail/cancer. Published 2022. Updated 3 February 2022. Accessed 19 April 2022.
- 3.Hossain MS, Karuniawati H, Jairoun AA, et al. Colorectal Cancer: A Review of Carcinogenesis, Global Epidemiology, Current Challenges, Risk Factors, Preventive and Treatment Strategies. Cancers (Basel) 2022;14(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021;71(3):209–249. [DOI] [PubMed] [Google Scholar]
- 5.Papamichael D, Audisio RA, Glimelius B, et al. Treatment of colorectal cancer in older patients: International Society of Geriatric Oncology (SIOG) consensus recommendations 2013. Ann Oncol 2015;26(3):463–476. [DOI] [PubMed] [Google Scholar]
- 6.Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med 1988;319(9):525–532. [DOI] [PubMed] [Google Scholar]
- 7.Del Vecchio F, Mastroiaco V, Di Marco A, et al. Next-generation sequencing: recent applications to the analysis of colorectal cancer. J Transl Med 2017;15(1):246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benson AB, Venook AP, Al-Hawary MM, et al. NCCN Guidelines Insights: Colon Cancer, Version 2.2018. J Natl Compr Canc Netw 2018;16(4):359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee MKC, Loree JM. Current and emerging biomarkers in metastatic colorectal cancer. Curr Oncol 2019;26(Suppl 1):S7–S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fahey CC, Davis IJ. SETting the Stage for Cancer Development: SETD2 and the Consequences of Lost Methylation. Cold Spring Harb Perspect Med 2017;7(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao C, Fan T, Tian H, et al. H3K36 trimethylation-mediated biological functions in cancer. Clin Epigenetics 2021;13(1):199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu M, Hu M, Zhang Q, Lai J, Liu X. SETD2, an epigenetic tumor suppressor: a focused review on GI tumor. Front Biosci (Landmark Ed) 2020;25(4):781–797. [DOI] [PubMed] [Google Scholar]
- 13.Lam UTF, Chen ES. Molecular mechanisms in governing genomic stability and tumor suppression by the SETD2 H3K36 methyltransferase. Int J Biochem Cell Biol 2022;144:106155. [DOI] [PubMed] [Google Scholar]
- 14.Chen R, Zhao WQ, Fang C, Yang X, Ji M. Histone methyltransferase SETD2: a potential tumor suppressor in solid cancers. J Cancer 2020;11(11):3349–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan H, Li N, Fu D, et al. Histone methyltransferase SETD2 modulates alternative splicing to inhibit intestinal tumorigenesis. J Clin Invest 2017;127(9):3375–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi YJ, Oh HR, Choi MR, et al. Frameshift mutation of a histone methylation-related gene SETD1B and its regional heterogeneity in gastric and colorectal cancers with high microsatellite instability. Human Pathology 2014;45(8):1674–1681. [DOI] [PubMed] [Google Scholar]
- 17.Chakrabarty S, Varghese VK, Sahu P, et al. Targeted sequencing-based analyses of candidate gene variants in ulcerative colitis-associated colorectal neoplasia. British Journal of Cancer 2017;117(1):136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu M, Rao H, Liu J, et al. The histone methyltransferase SETD2 modulates oxidative stress to attenuate experimental colitis. Redox Biology 2021;43:102004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puccini A, Lenz HJ, Marshall JL, et al. Impact of Patient Age on Molecular Alterations of Left‐Sided Colorectal Tumors. Oncologist 2019;24(3):319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varadi M, Anyango S, Deshpande M, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research 2022;50(D1):D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021;596(7873):583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Virostko J, Capasso A, Yankeelov TE, Goodgame B. Recent trends in the age at diagnosis of colorectal cancer in the US National Cancer Data Base, 2004–2015. Cancer 2019;125(21):3828–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA: A Cancer Journal for Clinicians 2022;72(1):7–33. [DOI] [PubMed] [Google Scholar]
- 24.Chen R, Zhao W-Q, Fang C, Yang X, Ji M. Histone methyltransferase SETD2: a potential tumor suppressor in solid cancers. J Cancer 2020;11(11):3349–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carvalho S, Vítor AC, Sridhara SC, et al. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife 2014;3:e02482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al Sarakbi W, Sasi W, Jiang WG, Roberts T, Newbold RF, Mokbel K. The mRNA expression of SETD2 in human breast cancer: correlation with clinico-pathological parameters. BMC Cancer 2009;9:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie P, Tian C, An L, et al. Histone methyltransferase protein SETD2 interacts with p53 and selectively regulates its downstream genes. Cell Signal 2008;20(9):1671–1678. [DOI] [PubMed] [Google Scholar]
- 28.Ji Z, Sheng Y, Miao J, et al. The histone methyltransferase Setd2 is indispensable for V(D)J recombination. Nature Communications 2019;10(1):3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chu SH, Chabon JR, Matovina CN, et al. Loss of H3K36 Methyltransferase SETD2 Impairs V(D)J Recombination during Lymphoid Development. iScience 2020;23(3):100941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stone L SETD2 affects DNA replication. Nature Reviews Urology 2015;12(4):183–183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.