

Abstract
An immunosuppressive tumour microenvironment is a major obstacle in the control of pancreatic and other solid cancers1–3. Agonists of the stimulator of interferon genes (STING) protein trigger inflammatory innate immune responses to potentially overcome tumour immunosuppression4. Although these agonists hold promise as potential cancer therapies5, tumour resistance to STING monotherapy has emerged in clinical trials and the mechanism(s) are unclear5–7. Here we show that the administration of five distinct STING agonists, including cGAMP, results in an expansion of human and mouse interleukin (IL)-35 regulatory B lymphocytes in pancreatic cancer. Mechanistically, cGAMP drives expression of IL-35 by B cells in an IRF3-dependent but type I interferon-independent manner. In several preclinical cancer models, the loss of STING signalling in B cells increases tumour control. Furthermore, anti-IL-35 blockade or genetic ablation of IL-35 in B cells also reduces tumour growth. Unexpectedly, the STING–IL-35 axis in B cells reduces proliferation of natural killer (NK) cells and attenuates the NK-driven anti-tumour response. These findings reveal an intrinsic barrier to systemic STING agonist monotherapy and provide a combinatorial strategy to overcome immunosuppression in tumours.
Tumours referred to as being cold, characterized by an immunosuppressive tumour microenvironment, pose a major obstacle to immunotherapy. These tumours include pancreatic cancer, which has a five-year survivalrate of only 10% and few treatment options2. Numerous preclinical studies have shown that activation of the cGAS–STING pathway in innate cells by STING agonists can elicit antigen-specific adaptive immune responses in a manner that is dependent on type I interferon4,5. In mice with pancreatic and other types of tumour, the intratumoral injection of STING agonist represents the most efficacious route8, leading to enhanced tumour-intrinsic IFN-I5, elevated CXCR3-dependent anti-tumour immunity9 and increased survival10. However, in clinical trials, STING agonist monotherapy has encountered profound tumour resistance5,7. Emerging studies suggest that STING may promote immune evasion and metastasis through increased PD-L1 expression11, T cell death12 and/or immunoinhibitory molecules including indoleamine 2,3-dioxygenase13. However, the precise mechanisms contributing to the lack of efficacy of STING monotherapy remain ill-defined5,7.
Although the role of T cells in tumour immunity has been extensively studied, a detailed understanding of B cell function is still emerging14–18. B cells induce tumour immunity through tertiary lymphoid structure formation16,17, antibody production and antigen presentation19,20. In contrast, B regulatory (Breg) cells have immunosuppressive properties and contribute to immunological tolerance of cancers14,15,18. Here, we show the unexpected impact of STING activation on the expansion of regulatory B cells which then diminish NK cell-mediated anti-tumour immunity.
STING activation enhances B cells
To delineate the in vivo response of pancreatic ductal adenocarcinoma (PDAC) to systemic STING agonist exposure, we injected wild-type (WT) mice with mouse PDAC KPC4662 cells derived from LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx1Cre/+ (KPC) mice21. After tumours were palpable, mice were given cGAMP (10μg/mL) intravenously (i.v.) every three days (Fig. 1a) and tumours were collected on days 11, 17 and 21 post-tumour implantation. Administration of cGAMP had no effect on tumour growth (Fig. 1b, Extended Data Fig. 1a) or CD45+ cell number in the tumours or spleens (Extended Data Fig. 1b, c). However, it increased the percent of CD19+ B cells among the CD45+ cells and the absolute number of B cells in the tumours by day 21 (Fig. 1c, Extended Data Fig. 1d). In contrast, cGAMP did not alter the percent or number of splenic CD19+ cells (Extended Data Fig. 1e), CD11b+ or CD8+ cells, and slightly decreased intratumoral CD4+ cells (Extended Data Fig. 1f).
Fig. 1 |. Inhibition of STING signaling in B cells improves tumor growth control in multiple models.
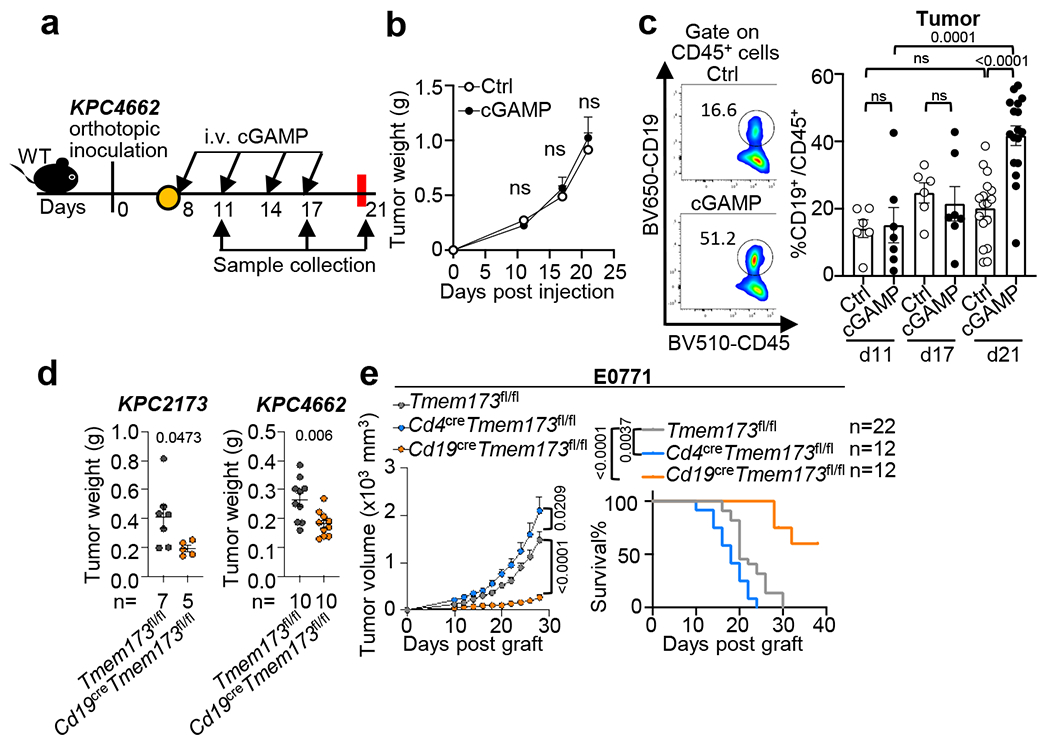
a, Workflow of the orthotopic pancreatic tumor model and treatment. WT mice were injected with KPC4662 tumor cells on Day 0 (d0) and i.v. injected with cGAMP or PBS at indicated timepoints. Tumors were harvested as indicated. b, Tumor weights at indicated timepoints (n=6 for control(Ctrl) at d11 and d17, n=7 for Ctrl at d21, n=7 for cGAMP-treated groups at d11 and d17, n=8 cGAMP-treated group at d21). c, Representative scatterplots (left) and frequency (right) of intratumoral CD19+ B cells among CD45+ cells (n=6 for Ctrl d11 and d17, n=7 for both cGAMP-treated groups d11 and d17, n=17 for both Ctrl and cGAMP-treated groups at d21). d, Tumor weights at d21 from Cd19cre Tmem173fl/fl or Tmem173fl/fl mice bearing KPC2173 (left) or KPC4662 (right) tumors (KPC2173, n=5 Cd19cre Tmem173fl/fl mice and n=7 Tmem173fl/fl; KPC4662, n=10 Cd19cre Tmem173fl/fl and Tmem173fl/fl mice). e, Tumor growth (left) and survival curves (right) in E0771-bearing Cd19cre Tmem173fl/fl, Cd4cre Tmem173fl/fl or Tmem173fl/fl mice. Data represent two (b-d) or three (e) independent experiments with symbols for individual mice and mean ± s.e.m. (d) shows a representative experiment. Statistical significance was determined by two-way ANOVA (b), unpaired two-tailed Student’s t tests (c-d), unpaired two-tailed Student’s t test for area under the curve (AUC) and log-rank (Mantel-Cox) test (e). P values indicated; NS, not significant.
To test the role of STING in B cells, we compared the growth of two orthotopic PDAC tumour lines (KPC2173 and KPC4662) in control mice (Tmem173fl/fl) vs. mice with B cell-specific deletion of the Sting1, also known as Tmem173 gene (CD19creTmem173fl/fl). In both models, tumour burden was reduced in Cd19creTmem173fl/fl mice compared to controls (Fig. 1d). Similar results were obtained for the triple-negative breast cancer cell line E0771 (Fig. 1e), the lung carcinoma (LLC) cell line and the B16-ova melanoma cell line(Extended Data Fig. 1g). In contrast, E0771 and LLC tumours showed accelerated growth in mice with T cell-specific STING deletion (Cd4creTmem173fl/fl) (Fig. 1e, Extended Data Fig. 1g), agreeing with a previous report22. In KPC tumour cells, STING appeared to be inactive as cGAMP did not change the expression of Ifnb1/Ifna4 transcripts, IFN-β protein or cell viability (Extended Data Fig. 1h–j).To explore the effect of cGAMP on B cells, we performed RNA sequencing (RNAseq) on B cells from WT and Tmem173 deficient mice (Tmem173−/−) stimulated using anti-IgM with or without cGAMP. STING activation was verified by increased phospho (p)-IRF3 and p-p65 in WT controls but not in Tmem173−/− B cells23 (Extended Data Fig. 2a). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of differentially expressed genes (DEGs) from pairwise comparisons of cGAMP-treated vs. untreated WT B cells and of cGAMP-treated WT vs. Tmem173−/− B cells was conducted. Genes downstream of cytokine-cytokine receptor activation and JAK-STAT signaling pathways were affected by cGAMP as expected (Extended Data Fig. 2b). Gene set enrichment analysis (GSEA) showed that the top 10 gene sets altered by cGAMP in B cells from WT vs. Tmem173−/− mice, included IFN response and inflammatory response genes (Extended Data Fig. 2c, d). Surprisingly, Il10 and Ebi3 mRNA were upregulated by cGAMP in WT but not in Tmem173−/− B cells (Fig. 2a, Extended Data Fig. 2e). EBI3 is a subunit of the immunosuppressive cytokines IL-27 and IL-35, members of the IL-12 family. In B cells, other transcripts of IL-12 family members24 such as the IL-35 subunit, p3525, were unaffected by cGAMP, whereas the expression of IL27, IL23a and IL12b were nearly undetectable (Extended Data Fig. 2f). These results were confirmed by real-time PCR, which showed that anti-IgM plus cGAMP increased the expression of Ebi3, Il10, Ifna and Ifnb1 in WT, but not in Tmem173−/− B cells, whereas p35 mRNA remained unchanged (Fig. 2b, Extended Data Fig. 2g).
Fig. 2 |. STING activation contributes to the expansion of IL-35+ and IL-10+ Breg cells.
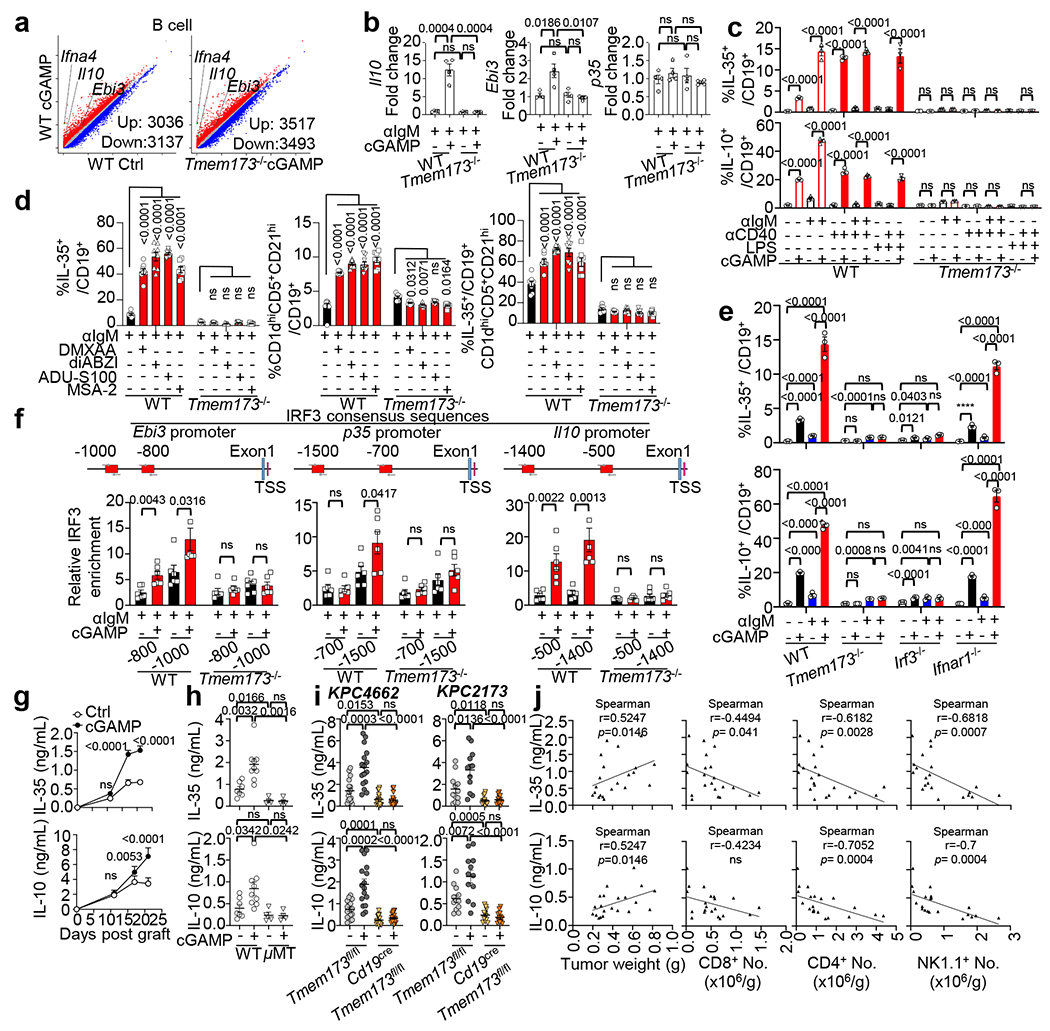
a, Scatterplots of DEGs in cGAMP-treated vs. untreated WT (left) or cGAMP-treated Tmem173−/− B cells (right) (data adjusted P<0.05; red, upregulated genes; blue, downregulated genes). b, Realtime PCR for Il10, Ebi3 and p35 mRNA in B cells; n=4 per group. c, Percent of IL-35+ or IL-10+ in CD19+ splenic B cells by flow cytometry. d, Percent of IL-35+ B cells (left), Breg cells (middle) and IL-35+ cells among Breg cells (right) stimulated by different STING agonists in WT and Tmem173−/− splenic B cells. e, IL-35+ (top) and IL-10+ (bottom) cells among CD19+ B cells from indicated mice. f, Relative enrichment of IRF3 by ChIP analysis on Ebi3 (left), p35 (middle) and IL-10 (right) promoters in anti-IgM+/−cGAMP treated B cells from WT or Tmem173−/− mice. IRF3 consensus sequences are shown as red rectangles. g, IL-35 (top) and IL-10 (bottom) levels in KPC4662 tumors from samples in Fig. 1a (n=11 per Ctrl group, n=12 per cGAMP group). h, IL-35 (top) and IL-10 (bottom) levels in tumour homogenates at d21 (n=7 WT Ctrl, n=9 WT cGAMP-treated group, n=4 μMT Ctrl and μMT cGAMP-treated groups). i, IL-35 (top) and IL-10 (bottom) levels in tumors from cGAMP-treated KPC4662 (left, n=16) or KPC2173 bearing mice (right, n=12) at d21. j, Correlation of tumor IL-35 (top) and IL-10 (bottom) protein with tumour weights(left) and the number of intratumoral CD8 (column 2), CD4 (column 3) and NK1.1 (right) cells (n = 21). Data represent three independent experiments for all panels except for (j) (two experiments); mean ± s.e.m. Statistical significance was determined by unpaired two-tailed Student’s t tests (b, f, h, i), Spearman correlation (j) and two-way ANOVA (c, d, e, g). P values indicated; NS, not significant .
STING induces B regulatory cells
To confirm cGAMP-induced IL-10 and IL-35, purified splenic B cells from WT and Tmem173−/− mice were treated with anti-CD40, anti-IgM or LPS, with or without cGAMP (Fig. 2c). anti-IgM treatment increased EBI3+p35+ (IL-35+) and IL-10+ cells, while anti-CD40 increased IL-35+ cells over baseline. cGAMP significantly augmented the frequency of IL-35 and IL-10 in WT but not in Tmem173−/− B cells (Fig. 2c) and the effect was dose-dependent (Extended Data Fig. 2h). These CD19+IL-35+ cells were CD1dhiCD5+CD21hi B cells (Extended Data Fig. 2j), a phenotype characteristic of murine Breg cells25,26,27,28. Compared to anti-IgM alone, cGAMP plus anti-IgM further increased the IL-10 production and the frequency of CD19+CD1dhiCD5+CD21hi Breg (Extended Data Fig. 2i, left and middle panel). Additionally, cGAMP treatment increased the frequency of IL-35+ cells among this Breg population (Extended Data Fig. 2i, right panel).
We next tested other STING agonists, DMXAA, diABZI, ADU-S100 and MSA-2, and all increased the frequency of IL-35+ and IL-10+ Breg cells (Fig. 2d, Extended Data Fig. 2k). As observed for cGAMP, DMXAA and diABZI (30μg) did not reduce tumour growth (Extended Data Fig. 2l) but increased the percent of CD19+IL-35+ cells in tumours and spleens, and increased EBI3 and p35 levels in splenic CD19+ B cells (Extended Data Fig. 2l). The frequency of IL-35+ or IL-10+ CD19+ cells was also increased by cGAMP in intratumoral B cells (Extended Data Fig. 2m).
STING-IRF3 mediates Breg cytokines
To delineate the source of B cells that infiltrated PDAC, splenic and peritoneal B cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) and cell trace violet (CTV) respectively, followed by adoptive transfer into B cell-deficient, PDAC-bearing mice treated with cGAMP. The majority of proliferating B cells recovered from tumours were CFSE+ (Extended Data Fig. 2n), indicating a splenic-origin.
To delineate the STING pathways that induced IL-35 and IL-10 expression in B cells, splenic B cells from WT, Tmem173−/−, Irf3−/− or Ifnar1−/− mice were sorted by magnetic-activated cell sorting (MACS), treated with anti-IgM and/or cGAMP ex vivo and analyzed by flow cytometry. Frequencies of IL-35+ and IL-10+ B cells in response to cGAMP or cGAMP plus anti-IgM were significantly diminished in Tmem173−/− and Irf3−/− but not in Ifnar1−/− B cells (Fig. 2e). Next, we used chromatin immuno-precipitation assays (ChIP) to test for IRF3 binding to the promoter regions (Fig. 2f), and found enhanced binding of IRF3 to the promoters of Ebi3, p35 and Il10 in WT but not in Tmem173−/− B cells (Fig. 2f). Thus, the upregulation of IL-35 and IL-10 expression in CD19+ B cells is mediated by an interferon-independent but STING-IRF3-dependent mechanism.
We next examined the source of IL-35 and IL-10 in PDAC. Both cytokines were increased in tumour homogenates as tumours grew and cGAMP significantly elevated the expression of these cytokines in tumours (Fig. 2g) and spleens (Extended Data Fig. 2o). Intratumoral IL-35 and IL-10 were nearly abrogated in B cell-deficient μMT mice, indicating a B cell-origin for these cytokines (Fig. 2h). IL-35 and IL-10 remained at baseline in Cd19creTmem173fl/fl PDAC-bearing mice treated with cGAMP (Fig. 2i, Extended Data Fig. 2p), indicating that the STING pathway in B cells is required for increased IL-35 and IL-10. To analyze the impact of these immunosuppressive cytokines on tumours, we showed that increased IL-35 and IL-10 in KPC4662 tumour homogenates positively correlated with tumour weight, and negatively correlated with tumour-infiltrating CD8+, CD4+ and NK1.1+ effectors (Fig. 2j), implicating their contribution to the establishment of an immunosuppressive milleu.
As IL-35 and IL-10 are also expressed by T regulatory cells (Treg)15,29,30, we explored whether cGAMP enhanced their expression in T cells. T and B cells were MACS-sorted from the spleens of PDAC-bearing and non-tumour-bearing WT mice. B cells were treated with anti-IgM with or without cGAMP and T cells were treated with anti-CD3 and anti-CD28 with or without cGAMP, revealing cGAMP increased IL-35+ and IL-10+ tumour-associated B cells but not IL-35+ or IL-10+ T cells (Extended Data Fig. 3a, b). To assess the role of Treg-derived IL-35, we orthotopically engrafted KPC4662 tumours into mice bearing Treg-specific deletion of Ebi3 (Foxp3creEbi3fl/fl) or control Ebi3fl/fl animals. cGAMP treatment caused no difference in tumour weight in these two groups, thus Ebi3 deficiency in Treg cells did not affect tumour growth in mice treated with cGAMP (Extended Data Fig. 3c).
IL-35+ B cells subdue NK cell responses
We next investigated the effect of cGAMP treatment in mice with B cell-specific deficiency in Ebi3, p35 or Il10, designated as B(Ebi3−/−)(Cd19creEbi3fl/fl), B(p35−/−) and B(Il10−/−), respectively (Extended Data Fig. 4a, b)28. These strains were orthotopically injected with KPC cells and treated with PBS or cGAMP (Fig. 3a,b). Treatment with cGAMP reduced tumour growth compared to PBS in B(Ebi3−/−)and B(p35−/−) mice but not control B(Ebi3+/−) and B(WT) or B(Il10−/−) mice (Fig. 3c, d). Conversely, the treatment increased tumour-infiltrating CD19+IL-35+ cells in B(Ebi3+/−), B(WT) and B(Il10−/−) but not B(Ebi3−/−) and B(p35−/−) mice (Supplementary Fig. 1, Fig. 3e). While we previously showed that the lack of Ebi3 and p35 in B cells increased total CD45+CD8+ cells in tumour-bearing mice28, cGAMP did not further augment this population (Fig. 3f) but instead increased the percent of CD8+ cells expressing granzyme (GzmB) or TNF (Fig. 3g, h). Furthermore, B cell-deletion of Ebi3 and p35 modestly increased intratumoral NK1.1+ cells, but cGAMP significantly increased NK1.1+ cells (Fig. 3i, j). GzmB+ NK cells were increased in cGAMP-treated B(Ebi3−/−) and B(p35−/−) mice compared to controls (B(Ebi3+/−), B(WT)) and B(Il10−/−) mice, suggesting that IL-35 but not IL-10 reduced GzmB+ NK cells (Fig. 3k). In contrast, total CD45+ cells (Extended Data Fig. 4c), IL-35 or IL-10 expressing CD4+ T cells (Extended Data Fig. 4d, e), cells other than T and B cells were not increased (Extended Data Fig. 4f, g). As previously reported28, B(p35−/−) and B(Ebi3−/−) mice had fewer CD4+Foxp3+ Treg cells and increased intratumoral CD4+ IFNγ+ and CD4+TNF+ T cells compared to controls, but these populations were not affected by cGAMP (Extended Data Fig.4h–j). Thus, the notable consequence of B cell-specific IL-35 expression is reduced frequency of NK cells.
Fig. 3 |. IL-35 ablation in B cells synergizes with STING agonist to modulate NK cell activity.
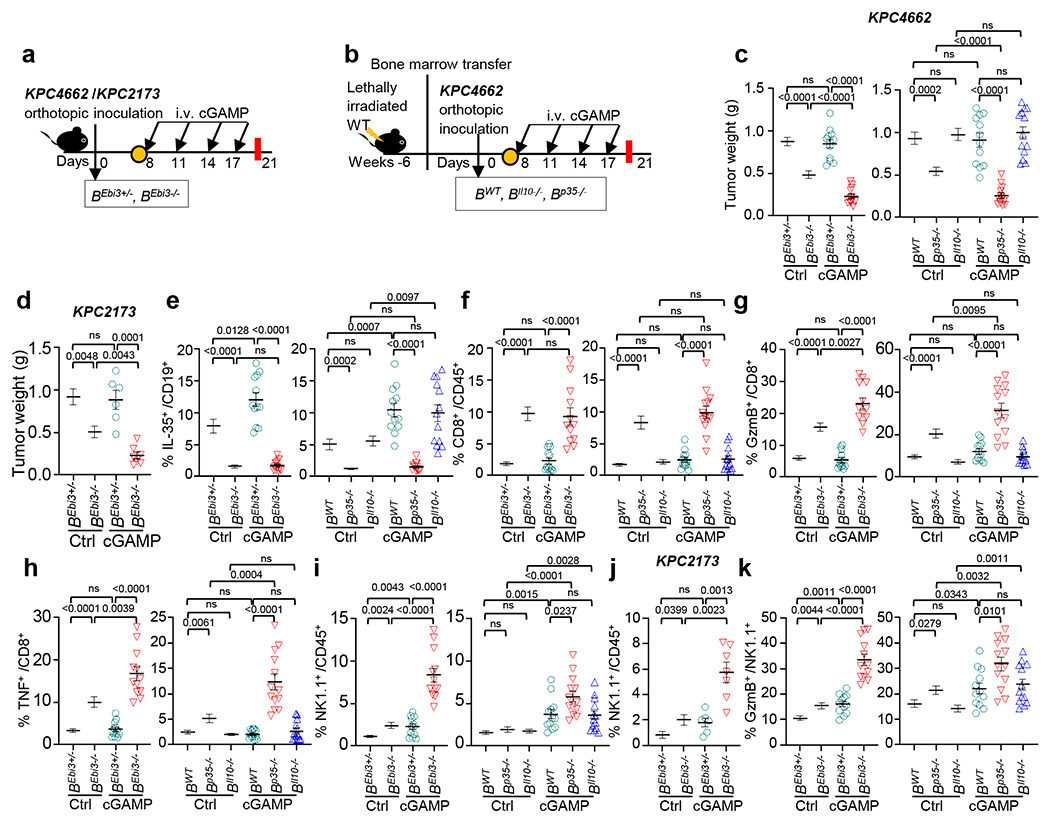
a, Workflow of the pancreatic tumor modeling and treatment. B(Ebi3−/−) mice and B(Ebi3+/−) mice were orthotopically injected with KPC4662 or KPC2173 tumor cells and given cGAMP. b, Bone marrow reconstituted chimeric mice with B cell-specific deficiency in Il10 or p35 were orthotopically injected with KPC4662 tumor cells and treated with cGAMP (i.v.) every three days beginning on d8, with tumors harvested on d21. c, Weights of KPC4662 tumors from cGAMP− and PBS-treated B(Ebi3+/−) and B(Ebi3−/−) mice (left) and B(WT), B(Il10−/−) and B(p35−/−) chimeric mice (right) harvested on d21 (n=11 for Ctrl B(Ebi3+/−) and Ctrl B(Ebi3−/−), n=12 for cGAMP-treated B(Ebi3+/−) and cGAMP-treated B(Ebi3−/−), n=12 for Ctrl and cGAMP-treated B(WT), n=13 for Ctrl and cGAMP-treated B(Il10−/−), n=13 for Ctrl and cGAMP-treated B(p35−/−)). d, Tumor weights of cGAMP− or PBS-treated B(Ebi3+/−) and B(Ebi3−/−) mice bearing KPC2173 tumor on d21 (n=6 Ctrl B(Ebi3+/−), Ctrl B(Ebi3−/−) and cGAMP-treated B(Ebi3+/−); n=7, cGAMP-treated B(Ebi3−/−)). e, Frequency of tumoral CD19+IL-35+ B cells from (c). f, Frequency of tumoral CD45+CD8+ T cells from (c). g, Frequency of tumoral CD8+ T cells expressing GzmB from (c). h, Frequency of tumoral TNF+CD8+ T cells from (c). i, Frequency of tumoral CD45+NK1.1+ cells from (c). j, Frequency of tumoral CD45+NK1.1+ cells from (d). k, Frequency of NK1.1+ cells expressing GzmB from (c). Data represent a combination of three independent experiments for all panels except for (d, j – two experiments), with each symbol representing an individual animal and mean ± s.e.m. Statistical significance was determined by unpaired two-tailed Student’s t tests. P values indicated; NS, not significant.
We next evaluated if cGAMP combined with IL-35 or IL-10 neutralizing antibodies could control the growth of PDAC, Lewis lung cancer (LLC) or B16-ova melanoma (Fig. 4a–e and Extended Data Fig. 5). Anti-IL-35 combined with cGAMP was most effective in reducing tumour burden and/or prolonging survival. Treatment with anti-IL-35 but not anti-IL-10, reduced the number of intratumoral CD19+IL-35+ B cells compared to the control group as expected (Extended Data Fig. 6a). The combination of cGAMP plus anti-IL-35 did not change the number of total CD45+ leukocytes (Fig. 4f), but significantly increased infiltrating NK1.1+ cells and NK1.1+GzmB+ over single treatment (Fig. 4g, Extended Data Fig. 6b). Treatment with anti-IL-35 alone increased CD8+ cells and CD8+TNF+ cells in the tumours, while the addition of cGAMP augmented this increase (Fig. 4h, Extended Data Fig. 6c). By contrast, cGAMP plus anti-IL-35 had no additive effect over anti-IL-35 alone on CD8+GzmB+, CD8+IFNγ+, CD4+CD25−, CD4+TNF+, CD4+IFNγ+ or CD4+Foxp3+ Treg cells (Extended Data Fig. 6d–h). Histologically, cGAMP plus anti-IL-35-treated mice showed increased cell death (caspase-3+) among keratin+ cells and more tumour infiltrating NK1.1+ cells (Extended Data Fig. 7a–c). STING activation is reported to reduce fibrosis31,32, a prevalent feature of PDAC and we confimed that cGAMP reduced expression of fibrosis markers including collagen deposition, Acta2 and Col3a1 mRNA regardless of inclusion of anti-IL-35 (Extended Data Fig. 7a, d, e).
Fig. 4 |. Systemic anti-IL-35 synergizes with STING agonist to potentiate NK cell efficacy.
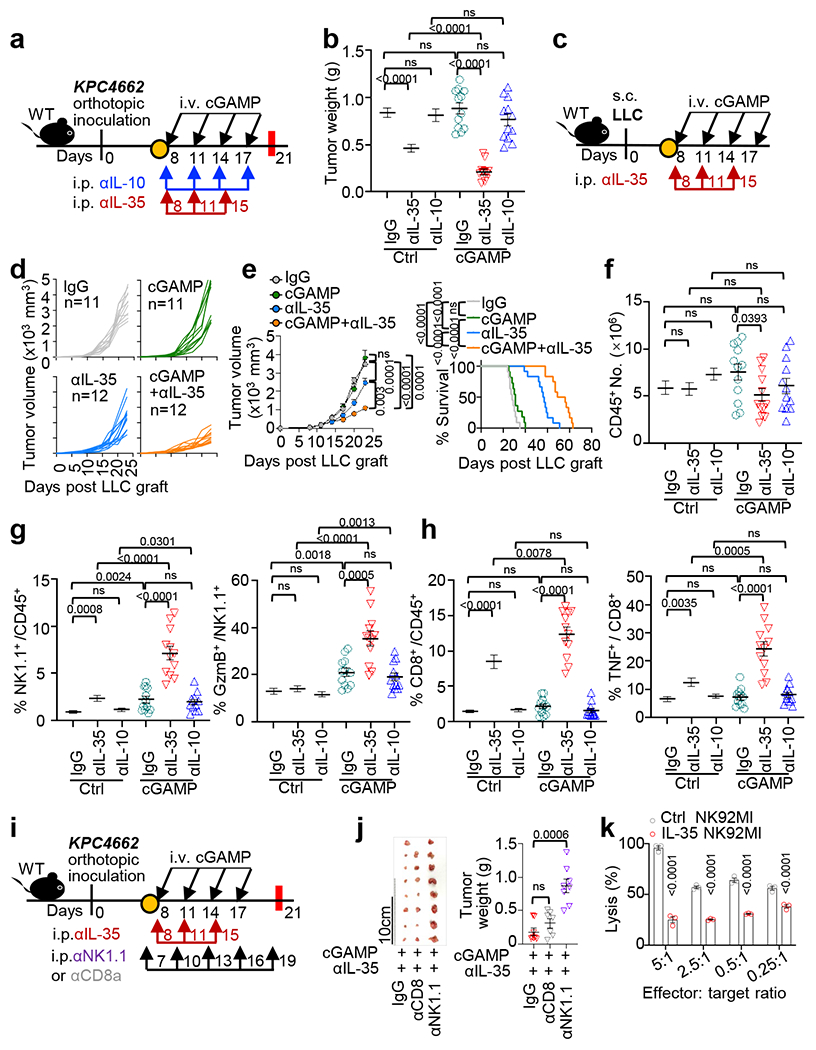
a, Workflow of the pancreatic tumor modeling with cGAMP+anti-IL-35 or anti-IL-10 treatments. b, Tumor weights from (a) collected at d21 (n=11 for Ctrl IgG, n=12 for anti-IL-10 and anti-IL-35-treated groups with or without cGAMP). c, Schematic workflow of the LLC tumor modeling and treatment with cGAMP and/or anti-IL-35. Tumor growth (d) and survival (e) in the LLC mouse model from (c) (n=11 for IgG and cGAMP groups, n=12 for anti-IL-35 and dual therapy groups). f, Number of total intratumoral CD45+ leukocytes in mice from (b). g, Frequency of tumoral CD45+NK1.1+ cells (left) and GzmB+NK1.1+ cells (right) from (b). h, Frequency of tumoral CD45+CD8+ cells (left) and TNF+CD8+ T cells (right) from (b). i, Schematic workflow of the pancreatic tumor modeling with cGAMP and anti-IL-35 plus either anti-CD8 or anti-NK1.1 depleting antibody treatments. j, Tumor images (middle) and weights (right) at d21 in mice from (i) (n=8 for IgG and anti-NK1.1 groups, n=7 for anti-CD8 group). k, Effect of IL-35 on NK cell-mediated lysis of K562 cells assessed by the lactate dehydrogenase (LDH) cell death assay (n=3 per group). Data represent a combination of three independent replicates (b, d-h) or a representative experiment (j, k), with mean ± s.e.m. Statistical significance was determined by unpaired two-tailed Student’s t tests (b, f-h, j, k), unpaired two-tailed Student’s t tests for AUC and Log-rank (Mantel-Cox) test (e). P values indicated; NS, not significant.
As the combination of cGAMP and anti-IL-35 increased both size of NK1.1+ and CD8+ subpopulations in orthotopic PDAC, we tested the relevance of these cell types in tumour containment by depleting NK or CD8+ T cells (Fig. 4i). The efficiency and specificity of antibody-mediated cell depletion was validated by flow cytometry (Extended Data Fig. 8a). Tumour reduction following cGAMP plus anti-IL-35 treatment was abrogated when NK1.1+ cells, but not CD8+ T cells, were depleted (Fig. 4j). This indicates that the anti-tumour effect of the dual therapy is mediated by NK cells. To further explore this link between IL-35 and NK cells, we incubated the NK cell line NK92 MI with IL-35 in vitro, and found that IL-35 treatment reduced NK cytotoxicity against K562, a typical NK target cell (Fig. 4k). To explore possible mechanisms of reduced NK cytotoxicity, we measured the expression of TRAIL, a key mediator of NK killing, and showed that IL-35 reduced TRAIL expression in NK cells (Extended Data Fig. 8b). Finally, anti-TRAIL neutralizing antibodies abolished tumour reduction following dual therapy (Extended Data Fig. 8c–e).
To further understand how intratumoral NK cells are affected by cGAMP and anti-IL-35 treatment in tumours, we sorted NK cells from established KPC tumours in mice treated with cGAMP and/or anti-IL-35 at day 21 post-tumour cell injection, extracted RNA from NK cells and performed RNAseq analysis. Principal component analysis (PCA) of RNAseq data distinguished NK cells treated with single or combination treatments (Fig. 5a). Gene set enrichment analysis (GSEA) identified top 20 hallmark pathways altered by a combination of cGAMP and anti-IL-35 treatment (Extended Data Fig. 9a). Among the affected hallmark pathways, known STING associated pathways such as TNFA signaling via NF-kB; IL6, JAK, STAT3 signaling and Interferon alpha response were increased (Extended Data Fig. 9a, b). The PI3K/AKT/MTOR signaling and glycolysis pathways were also increased. The former is known to drive general cell proliferation and the development, proliferation and effector function of NK cells33,34 , whereas the latter is required for NK cell cytotoxicity5,35,36 (Fig. 5b, Extended Data Fig. 9a). Additionally, the cell cycle associated “Mitotic Spindle” gene set was enriched in the combination treatment group (Fig. 5b, Extended Data Fig. 9c).
Fig. 5 |. STING agonist therapy suppresses NK cell-mediated anti-tumor responses via IL-35.
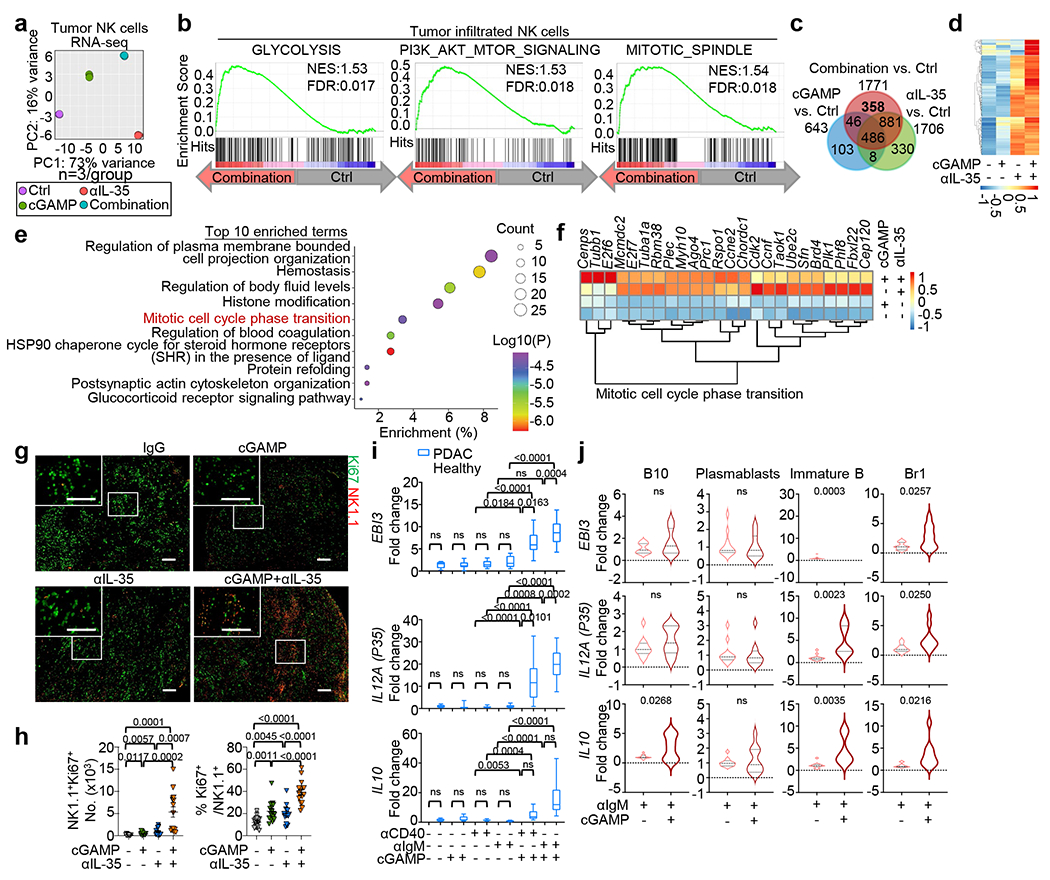
a, PCA of RNAseq results from intratumoral NK cells from Ctrl and mice(n=3 per group) treated with cGAMP, anti-IL-35 or a combination of cGAMP+anti-IL-35. b, Enrichment of “glycolysis”, “Pi3k, Akt, mTOR signaling” and “mitotic spindle” pathways in tumoral NK cells from cGAMP+anti-IL-35 vs. Ctrl groups by GSEA as noted in Extended Data Fig. 9a. c, Venn diagram of upregulated DEGs in combination cGAMP+anti-IL-35, cGAMP or anti-IL-35-treated groups vs. control. d, Heatmap showing n=358 upregulated genes noted in (c) in the Venn diagram. e, Top 10 enriched pathway terms in intratumoral NK cells from dual cGAMP+anti-IL-35-treated group in (d). f, Heatmap showing upregulated Mitotic cell cycle phase transition genes in intratumoral NK cells as noted in (e). g, Representative microscopy images of tumor sections from indicated groups as described in Fig. 4a. NK1.1+ cell (red) and Ki67+ marker (green) stainings are shown. (Scale bars, 100 μm.) h, Quantification of the data in (g); n=15 per group. i, Realtime-PCR of EBI3, IL12A and IL10 mRNA in human B cells collected from pancreatic cancer patients and healthy donors (n=15, each group) stimulated with anti-IgM +/− cGAMP. HPRT was used as the internal control. The box plots show minima, maxima and median, displaying the 10-90 percentile at the whiskers. j, EBI3, IL12A and IL10 transcript levels in B10, plasmablasts, immature B cells and Br1 cells sorted from peripheral blood of pancreatic cancer patients and healthy donors (n=10 each) and stimulated with anti-IgM +/− cGAMP. For RNAseq, each group has n=3 biological repetitions. Two experiments were combined (h) with mean ± s.e.m. Statistical significance was determined by unpaired two-tailed Student’s t tests (h-j). P values indicated; NS, not significant.
To distinguish the effects of combination versus single treatments, we noted that the combination treatment vs. control group upregulated 358 (Fig. 5c, d) and downregulated 610 DEGs (Extended Data Fig. 9d), which were unchanged in either the cGAMP or anti-IL-35 vs. control groups. The top 10 enriched pathways of upregulated DEGs included “mitotic cell cycle phase transition” (Fig. 5e). Genes that were not significantly altered by single treatment but were increased by the combination treatment included Tubb137, which promotes cell proliferation (Fig. 5f). To directly test whether the combination treatment altered NK cell proliferation, we show that the combination treatment resulted in robust NK cell proliferation as shown by increased total number and percentage of NK1.1+Ki67+ cells in the tumour (Fig. 5g, quantitated in Fig. 5h).
To investigate the link between STING and immunosuppressive cytokines in human pancreatic tumours, we examined the pancreatic adenocarcinoma dataset from The Cancer Genome Atlas (TCGA-PAAD)38. Correlation analysis revealed that human EBI3, IL12A (p35) and IL10 expression positively correlated with TMEM173 transcripts (Extended Data Fig. 10a). We directly evaluated the effect of cGAMP treatment in human PBMC-derived B cells from pancreatic cancer patients and healthy volunteers. cGAMP treatment enhanced EBI3, IL12A and IL10 expression in activated human B cells from healthy controls. EBI3 and IL12A but not IL10 were further elevated in B cells from pancreatic cancer patients compared to healthy controls (Fig. 5i). To identify the human B cell subsets that produce IL-35 cytokines in response to cGAMP, we analyzed B10 cells, plasmablasts, immature B cells and Br1 cells (Supplementary Fig. 2), all have been previously reported as immunosuppressive in auto-immune diseases28,39,40. Treatment with cGAMP plus either anti-IgM (Fig. 5j) or anti-CD40 (Extended Data Fig. 10b) selectively enhanced EBI3 and IL12A expression in immature B cells and Br1 cells. Finally, the Breg signature21,28 correlated positively with the interferon-stimulated gene (ISG) signature41, but negatively with activated NK cell signature42 in the TCGA-PAAD (pancreatic adenocarcinoma) (Extended Data Fig. 10c) and the LUAD (lung adenocarcinoma) databases (Extended Data Fig. 10d). Collectively, these data indicate that cGAMP induces immunosuppressive IL-35 preferentially in tumour-bearing patients over healthy controls.
In summary, our findings show that resistance to systemic treatment with STING agonist is attributable partly to the expansion of immunosuppressive B cells that impede NK cells. Whereas others have shown that the STING-triggered IFN response may be critical for anti-tumour NK cell function43, our work provides a previously unappreciated counter scenario wherein the systemic delivery of STING agonist suppresses NK cell-mediated anti-tumour responses through B cell-derived IL-35 (Extended Data Fig. 10e). This is pertinent as systemic delivery of STING agonist exhibits less efficacy than intratumoral delivery of STING agonists8. In viral infection, NK cell-dependent cytotoxicity during infection is STING-dependent44,45 and EBI3 induced IL-10 in NK cells leads to diminished anti-viral CD8 T cell activity46. However, the importance of EBI3 in the regulation of NK cells has not been previously reported in cancer nor have B cells been implicated as major regulators of NK cell during either infection or cancer. This report reveals a new immunoregulatory circuit in cancer where STING activates B regulatory function by inducing IL-35, which then subdues NK cell responses. This negative circuit can be relieved by blocking B cell-specific IL-35 during cGAMP treatment and represents a stratregy to control tumours.
Methods
Mice
All mouse protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill. Animals were maintained in a specific pathogen-free facility (12-hour light/dark cycle, temperature: 21–23 °C, humidity: 30–70%,). All mice, male or female, were used at 6–12 weeks of age, matching age and sex, randomly grouping for experiments. C57BL/6J or wildtype, WT (stock #000664), μMT (stock #002288), Tmem173−/− (stock #017537), Ifnar1−/−(stock #028288), Il10−/− (stock #002251) and p35−/− (stock #002692) strains were from the Jackson Laboratory (Bar Harbor, ME). Irf3−/− mice were provided by T. Taniguchi (University of Tokyo, Tokyo, Japan) and obtained from J. K. Whitmire (University of North Carolina, Chapel Hill, NC). Ebi3Tom.L/L and Foxp3Cre-YFPEbi3Tom.L/L (Treg(Ebi3−/−))mice were obtained from D. Vignali (University of Pittsburgh, Pittsburgh, PA)47–51. Cd19CreEbi3fl/fl (B(Ebi3−/−)) mice were generated by crossing Cd19Cre mice to Ebi3fl/fl mice for two generations to obtain homozygosity at the Ebi3 locus28 with resulting progeny lacking expression of IL-35 in B cells. Cd19CreTmem173fl/fl mice or Cd4CreTmem173fl/fl were generated by crossing Cd19Cre or Cd4Cre mice to Tmem173fl/fl mice (University of Washington School of Medicine, Seattle, WA) with resulting progeny lacking expression of STING in B or T cells. Littermates with WT allele or lacking Cre were used as controls.
Cell lines
The mouse PDAC cell lines KPC4662 and KPC2173 were derived from primary pancreatic tumor isolated from C57BL/6J LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx1Cre/+ (KPC) mice (R. H. Vonderheide, University of Pennsylvania, Philadelphia, PA)21. KPC, LLC, E0771 and B16-ova cells were cultured in complete DMEM (#11995065, Thermo Fisher) containing 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin-streptomycin (PS) (#15140122, Thermo Fisher). Cells were used at <16 passages and confirmed to be mycoplasma- and endotoxin-free. NK-92 MI NK cells (#CRL-2408, ATCC) were cultured in MEMα (#12561056, Thermo Fisher) media containing 10% FBS, 1% PS, 2mM L-glutamine (#25030081, Thermo Fisher), 0.1mM 2-mercaptoethanol, 10mM HEPES (#15630106, Thermo Fisher) and 0.1mM non-essential amino acids (#11140050, Thermo Fisher). The erythroleukemic cell line K562 was cultured in complete RPMI-1640 media. All cells were maintained at 37°C and 5% CO2.
Orthotopic surgery
For orthotopic intrapancreatic injection of KPC cells, mice were anesthetized using ketamine (100mg/kg)/xylazine (10mg/kg; Med-Vet International) cocktail. The depth of anesthesia was confirmed by verifying absence of toe pinch response. An left flank incision was made and KPC cells were injected (28-gauge needle) into the tail of the pancreas. The wound was closed in two layers, with running 5-0 Vicryl RAPIDE sutures (Ethicon) for the body wall and 5-0 PROLENE sutures (Ethicon) for the skin. All animals were given buprenorphine (0.1mg/kg; Med-Vet International) via subcutaneous (s.c.) injection to alleviate pain once after orthotopic surgery.
Generation of B(p35−/−), B(Il10−/−) and control B(WT) bone marrow chimeric mice
B(p35−/−) and B(Il10−/−) mice with deficiencies in p35 and Il10 restricted to B cells were obtained by reconstituting lethally irradiated recipient mice with a mixture of bone marrow cells from B cell deficient μMT mice (80%) and p35−/−, Il10−/− or WT mice (20%) (Extended Data Fig. 4a, b). Six weeks later, chimeras were orthotopically inoculated with tumor cells. For validation, B(WT), B(p35−/−)and B(Il10−/−) chimeras were assessed for deletion of p35 and Il10 genes restricted to B cells. Splenic CD19+ B and CD3+ T cells were purified from B(p35−/−), B(Il10−/−) and corresponding B(WT) chimeras by flow sorting. All sorted populations and remaining non-T/B cells were lysed with genomic DNA extracted using the DNAeasy kit (Qiagen) per manufacturer instructions. The amount of genomic DNA present in these samples was normalized using Actb as the standard, to determine the presence of WT p35 and Il10 alleles in cell subsets.
Tumour growth and treatments
KPC cells (75,000) in 25μL ice-cold PBS mixed with 25μL matrigel (354234, Corning) were injected into the pancreas. Mice received 10μg cGAMP or PBS by intravenous (i.v.) injection, starting on day 8 post tumor cell injection, and repeated every 3 days for a total of 4 times. For dynamic timepoint experiments, tumors were harvested on days 11, 17 and 21. For breast cancer studies, 1x106 E0771 cells were orthotopically inoculated. For lung cancer and melanoma studies, 5x105 LLC or B16-ova cells respectively were s.c. injected into the right flank of mice. B16-ova tumor-bearing mice were vaccinated with 50μg poly(I:C)-ova on day 6. Tumor growth and survival were monitored. For antibody blockade studies in vivo, mice were intraperitoneally (i.p.) injected with 200μg anti-IL-35 (clone V1.4C4.22, Shenandoah) twice a week for a total of 3 times starting on day 8 post tumor injection, 200μg anti-IL-10 (clone JES5-2A5, BioXcell) every three days for four times or IgG isotype control starting on day 8 post tumor injection. For studying the effects of combination therapy of anti-IL-35 with STING agonist and further depleting CD8+ T or NK1.1+ cells, tumor-bearing mice were treated as described above and received either 200μg of anti-CD8 (clone YTS 169.4, BioXcell), anti-NK (clone PK136, BioXcell) depleting antibody or IgG isotype control every 3 days, starting on day 7. Tumors were harvested on day 21. For mechanisms of NK cytotoxicity, 50μg of TRAIL neutralizing antibodies (clone N2B2, Thermo Fisher) were injected intratumorally (i.t.) every three days starting on day 7. Tumor growth and survival were monitored52.
Lymphocyte isolation
Single-cell suspensions were prepared from isolated tumors and spleens. Spleens were mechanically disrupted, re-suspended in 1% FBS/PBS and processed following RBC lysis (#00-4300-54, Thermo Fisher). For isolation of tumor-infiltrating lymphocytes, tumor tissue was minced into 1 to 2 mm pieces and digested with collagenase IV (1.25mg/mL; #LS004188, Worthington), 0.1% soybean trypsin inhibitor (#T9128, Sigma), hyaluronidase (1mg/mL; #LS002592, Worthington) and DNase I (100 mg/mL; #LS002007, Worthington) in DMEM for 30 min at 37°C. Cell suspensions were passed through a 70μm cell strainer (Falcon) and resuspended in 2% FBS DMEM media. Lymphocytes were isolated from processed tumor tissues by Ficoll density gradient centrifugation. For some experiments, isolation of total CD45+ leukocytes by magnetic-activated cell sorting (#130-052-301, MACS Miltenyi Biotec) was performed on the leukocyte-enriched fraction according to manufacturer’s protocol, with purity of >90% achieved. Cells were collected in complete RPMI media containing 10% FCS with 1% PS.
Intracellular cytokine staining
For ex vivo stimulation, 106 single cells from tumors or spleens were incubated with PMA (50ng/mL; #P8139, Sigma) and ionomycin (200ng/mL; #I-0634, Sigma) in the presence of Golgistop Brefeldin A (1X, BioLegend) in complete RPMI medium for 4 hr at 37°C. Cells were washed and blocked with anti-CD16/CD32 (#101320, Biolegend, 0.1mg/100,000 cells) for 5 min on ice. After washing, cells were stained with labeled cell surface antibodies on ice for 30 min in FACS buffer (PBS with 1% FBS and 0.05% sodium azide). Viability was assessed using Fixable Viability Ghost Dye Red 780 (#13-0865, Tonbo). After staining, cells were washed, fixed and permeabilized using Cytofix/cytoperm buffer (#554714, BD Biosciences) for 15 min at 4°C in the dark. Intracellular staining was performed using fluorophore-conjugated cytokine antibodies for 1 hr at 4°C in the dark. After intracellular staining, cells were washed and resuspended in FACS buffer for acquisition by flow cytometer via FACSDiva (BD, version 8.0.1). Intracellular staining for Foxp3 was performed using a Foxp3 staining kit (#00-5523, eBioscience). Samples were acquired on Attune NxT Cytometer (Thermo Fisher) or LSRII Fortessa (BD Biosciences) with data analyzed using FlowJo version 10.6.2 (BD Biosciences). For human B cells, human BD Fc Block (#422301, Biolegend, 0.1mg/100,000 cells,) was applied for blocking. All antibodies and reagents are listed in Supplementary Table 1.
Immunoblotting
A million cells were lysed on ice for 30 min using RIPA lysis and extraction buffer (Thermo Fisher), supplemented with protease inhibitor cocktail (Roche). The lysate was centrifuged at 16,000×g at 4°C for 10 min. Clarified supernatants were loaded into 4–12% NuPAGE Bis-Tris gels (Thermo Fisher), transferred to PVDF membranes (Bio-Rad) and blocked for 1 hr with 5% non-fat milk in TBST buffer. Membranes were incubated with primary antibodies in blocking buffer overnight at 4°C. After washing five times, membranes were incubated with secondary antibodies for 1 hr at room temperature (RT). Proteins were detected using Femto Chemiluminescent reagent (Thermo Scientific). Antibodies are listed in Supplementary Table 1.
Enzyme-linked immunosorbent assay (ELISA)
Tumor and spleen homogenates were prepared using extraction buffer (100 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, 0.5% Sodium deoxycholate, Phosphatase inhibitor cocktail, Protease inhibitor cocktail, PMSF). Samples were incubated at 4°C for 1 hr on the orbital shaker and supernatants were collected by centrifugation at 9000×rpm for 10 min at 4°C. All samples were stored at −80°C. The concentrations of IL-35 and IL-10 were measured using LEGEND MAX™ Mouse IL-35 Heterodimer ELISA Kit (#440507) and ELISA MAX™ Standard Set Mouse IL-10 kit (#431414) according to manufacturer’s protocols (Biolegend).
Realtime PCR analysis for gene expression
RNA was prepared from treated cells using RNeasy Mini Kit (#74106, Qiagen) or TRIzol extraction (#15596026, Thermo Fisher). Complementary DNA was generated using a High Capacity cDNA-RT Kit (Invitrogen). Quantitative PCR analyses were performed using universal SYBR green super-mix reagent (Bio-Rad) and QuantStudio 6 Flex real-time PCR system (Thermo Fisher). Results were normalized to Actb expression. Gene expression was determined by the ΔΔCt method (2-ΔΔCt). Primer sequences are listed in Supplementary Table 2.
Cell stimulation assays
Mouse splenic B or T cells were sorted using negative selection (#130-090-862 and #130-095-130, Miltenyi Biotec) from spleens of tumor-bearing or healthy mice. Total B cells were seeded in 96-well flat-bottom plates and stimulated with 5μg/mL anti-IgM. T cells were seeded and stimulated with 5μg/mL anti-CD3 and 2μg/mL anti-CD28. Cells were treated simultaneously with or without 5μg/mL cGAMP. For experiments in Fig. 2c, B cells were stimulated by 5μg/mL anti-IgM, 2μg/mL anti-CD40 or 2μg/mL LPS with or without 5μg/mL cGAMP for 24 hr. For experiments in Extended Data Fig. 2h–j, B cells were stimulated by 5μg/mL anti-IgM with increasing doses of cGAMP (0, 1, 5, 10μg/mL) for 24 hr. For experiments in Fig. 2d and Extended Data Fig. 2k, B cells were stimulated by 5μg/mL anti-IgM with 5μg/mL DMXAA53, ADU-S10054, diABZI55 or MSA-256 for 24 hr. Cells were processed for flow cytometry. For RNA evaluation, mouse B cells were first stimulated using 5μg/mL anti-IgM and cGAMP for 12 hr and then processed for analysis.
Primary NK cell treatment
Tumoral NK cells were sorted from KPC bearing mice by FACS cell sorter. NK cells (3x105) were seeded in 24-well plates and stimulated with 100ng/mL mouse IL-35 (#CHI-MF-11135, Chimerigen Labs). Cells were analyzed by FACS staining.
MTT viability assay
For the MTT assay, 103 KPC4662 cells were seeded, stimulated with cGAMP for 24 hr and 250μg/mL MTT was added 4 hr before the end point. The plate was centrifuged and supernatant was discarded. DMSO (150μL) was added to dissolve the pellets. The absorbance was read at OD590nm.
NK cell-mediated cytotoxicity assay
After incubation with 100ng/mL IL-35 for 48 hr, NK-92 MI cells were subjected to Ficoll density gradient centrifugation. Viable cells were washed and counted to add target K562 and effector NK-92 MI cells in effector/target ratios of 5:1, 2.5:1, 0.5:1 and 0.25:1 in 96-well U-bottom plates. After 3 hr of co-culture, Lactate Dehydrogenase (LDH) assay was performed according to manufacturer’s instructions (#11644793001, SigmaAldrich).
Human samples
The study was carried out in accordance with The University of North Carolina at Chapel Hill School of Medicine (Chapel Hill, NC) and Washington University School of Medicine (St. Louis, MO) guidelines and was approved by institutional review board ethics committees at respective institutions. The study was conducted in accordance to ethical standards as in the Declaration of Helsinki. Informed consent was obtained from the patients and healthy donors before blood donation. The PDAC patient blood samples were collected at Washington University School of Medicine. Blood samples were collected from 15 patients with pancreatic cancer and 15 healthy controls. PBMCs were obtained by separating whole blood samples via density gradient centrifugation (Lymphoprep, Axis-shield). Briefly, collected blood samples in yellow top ACD tubes (~20mL each) were diluted with an equal volume of 0.9% NaCl. Carefully, 20mL of diluted blood were layered on top of 10mL Lymphoprep in a 50mL centrifuge tube to avoid mixing blood and separation fluid and then, centrifuged at 800×g for 20 min at 18°C. Afterward, the mononuclear cells were removed from the sample/medium interface using a pasteur pipette. The cells were collected by centrifugation at 250×g for 5 min and washed three times with RPMI-1640 media. Human CD19+ B cells of pancreatic cancer patients and healthy donors were sorted from PBMCs and stimulated using 5μg/mL anti-IgM or 2μg/mL anti-CD40 with or without 5μg/mL cGAMP for 24 hr. Cells were collected for qPCR assessment of mRNA expression.
Chromatin immunoprecipitation assays
ChIP assays were performed as described previously57. Briefly, splenic B cells isolated from WT and Tmem173−/− mice were stimulated with anti-IgM and cGAMP for 24 hr. After activation, cells were harvested and cross-linked with 37% (W/V) formaldehyde at a final concentration of 1.42% for 15 min at RT. Formaldehyde quenching was done with 125 mM glycine for 5 min at RT. Cell lysis was performed using immunoprecipitation (IP) buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.5, 5 mM EDTA, 0.5% V/V NP-40 and 1% V/V Triton X-100, 0.1 M PMSF, phosphatase inhibitor). Chromatin was sheared into 500-1000 base pair fragments with four rounds of 15 sec sonication pulses with a 2 min rest between each round using a Diagenode Bioruptor. Sheared chromatin was then subjected to IP with IRF3 (Biolegend; 12A4A35; 1:50 diluted) or isotype control antibody (Biolegend; 27-35), followed by overnight incubation with rotation. DNA-protein complexes were immunoprecipitated using protein A-agarose beads (Millipore:Sigma, 17-5280-01) at 4°C for 1 hr with rotation, washed with IP buffer without inhibitors and subjected to DNA isolation, performed using 10% (W/V) chelex-100 (Bio-Rad; 142-1253) slurry followed by precipitation with 70% ethanol. Purified DNA was used to perform real-time PCR with SYBR green master-mix in 10 μL reaction volumes (2.5 μL DNA template, 0.3 μL of 10 μM primer pair, 5 μL master-mix and 2.2 μL PCR grade water). Relative occupancy of the immunoprecipitated factor at the target locus was estimated by using 2^(CTcontrol - CTsample) methods. Relative enrichment of upstream binding at transcriptional start site (TSS) are scaled to ChIP with control isotype antibody and input. The primers are listed in Supplementary Table 2.
RNA sequencing (RNAseq)
For RNAseq analysis, total RNA was extracted from splenic B cells or sorted tumoral NK cells using the RNeasy Mini Kit (Qiagen, 74106). RNAseq was performed by Novogene Corporation (Shenzhen, Guangdong, US). A total amount of 1 μg RNA per sample was used to generate sequencing libraries using NEBNext® UltraTM RNA Library Prep Kit from Illumina® (New England Biolabs, Inc., NEB, MA) according to manufacturer’s recommendations. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. First-strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase (RNase H-). Second strand cDNA synthesis was performed using DNA Polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of 3’ ends of DNA fragments, NEBNext Adaptor with hairpin loop structures were ligated for hybridization. To select cDNA fragments of preferable lengths of 150-200base pairs, the library fragments were purified with AMPure XP system (Beckman Coulter, Beverly, MA). Then 3 μL USER Enzyme (NEB, MA) was incubated with size-selected, adaptor-ligated cDNA at 37°C for 15 min followed by 5 min at 95°C. PCR was performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers and Index (X) Primers. Finally, PCR products were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2100 system. Clean data were obtained by removing reads from raw data containing adapter or poly-N sequences and reads with low quality using fastp58 with default parameters. All the downstream analyses were based on clean data with high quality. Paired-end clean reads were aligned to the GRCm38/mm10 genome assembly of the Mus Musculus reference genome using the Spliced Transcripts Alignment to a Reference (STAR59) software. FeatureCounts60 were used to calculate read counts for each gene based on the aligned files. The count matrix for all samples was used to perform differential expression analysis between two conditions or groups (three biological replicates per condition) using DESeq261 R package. The resulting P-values were adjusted using the Benjamini and Hochberg’s approach for controlling the False Discovery Rate (FDR). Genes with an adjusted P < 0.05 found by DESeq2 were assigned as differentially expressed. To obtain gene expression value matrix for each sample, we also used FeatureCounts to convert the reads counts to RPKM (reads per kilobase of exon model per million mapped reads) values. We used R package pheatmap (https://CRAN.R-project.org/package=pheatmap) to generate heatmaps based on this RPKM matrix. KEGG pathway enrichment analysis for DEGs was performed by the R package clusterProfiler62.
Gene Set Enrichment Analysis (GSEA)
GSEA was performed using the GSEA 4.1.0 Java desktop application downloaded from GSEA (https://www.gsea-msigdb.org/gsea/index.jsp). For this study, the hallmark gene sets (MSigDB version7.2) and Kyoto Encyclopedia of Genes and Genomes gene set collections (MSigDB version 7.2) were used in the analysis (http://www.gseamsigdb.org/gsea/downloads_archive.jsp). GSEA was performed using the gene_set permutation type with the number of permutations set at 1000. We used FDR cutoff of P < 0.05 to identify significantly enriched gene sets.
The Cancer Genome Atlas analyses
Gene expression and clinical data for Pancreatic adenocarcinoma (PAAD) were downloaded from TCGA38. For signatures, differential expression analysis was conducted using DESeq261. The Breg gene signature28 value was calculated for each sample with cancer types of PAAD or LUAD using the geometric mean. The ISG gene signature41 was calculated in the same way (geometric mean for each sample with cancer types PAAD or LUAD). The activated NK cell signature was from CIBERSORT(version 1.05)42. Spearman correlations were used to compare the Breg gene signature with the ISG gene signature or the activated NK cell signature and plotted using ggplot263.
Immunohistochemistry and microscopy
Tumor blocks were fixed with 10% formalin overnight, transferred into 70% ethanol, followed by paraffin embedding and cutting into 5 μM sections. Prior to immunostaining, tissues were deparaffinized, rehydrated and subjected to antigen retrieval (abcam, #ab93678). The vendor’s instructions for Sirius red staining (abcam, #ab150681) were followed. The sections were blocked with 10% goat serum. The anti-Ki67 (thermofisher, #11-5698-82, 1:200), anti-Keratin (Sigma-Aldrich, #MABT329, 1:200), anti-NK1.1 (biolegend, #108747, 1:200) or anti-Cleaved Caspase-3 (cell signaling, #9661, 1:400) antibodies were used to stain sections. Tissue sections were incubated with primary antibodies for 2 hr at RT. Fluorescently labelled secondary antibodies were incubated for 30 min at RT. Images were acquired with a Keyence BZ-X710 microscope and analysed in a blinded fashion with image J software for automated counting.
Statistical analyses
Experiments were independently repeated two to five times. For in vivo experiments, mice were assigned to groups randomly and maintained mixed among cages when possible. Data were analyzed using Prism version 9.0 software (GraphPad) and presented as mean values ± s.e.m. Unpaired two-tailed Student’s t tests, one-way ANOVA, two-way ANOVA and Log-rank (Mantel Cox) test were used, where P < 0.05 was considered statistically significant.
Extended Data
Extended Data Fig. 1 |. Effect of the cGAS-STING pathway on immune cell profiles and tumour growth.
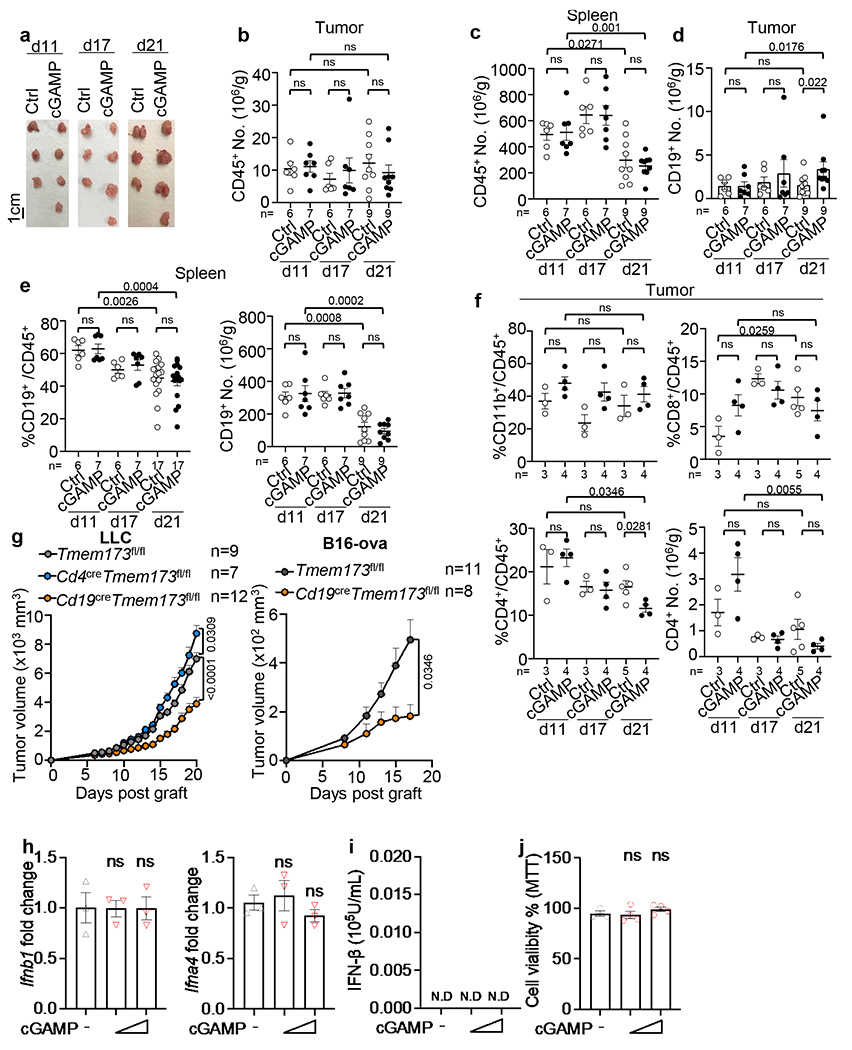
a, Representative images of mouse tumours from Fig. 1a. b, Number of tumoral CD45+ leukocytes from mice as treated in Fig. 1a (n=6 for Ctrl at d11 and d17, n=7 for cGAMP-treated groups at d11 and d17, n=9 for groups at d21). c, Number of splenic CD45+ leukocytes from mice in b. d, Number of tumoral CD19+ B cells in cGAMP or PBS groups from mice in e in b. e, Frequency (left) and number (right) of splenic CD19+ B cells among CD45+ cells from mice as treated in Fig. 1a (n=6 for Ctrl at d11 and d17, n=7 for cGAMP-treated groups at d11 and d17, n=17 (left) or 9 (right) for d21 groups). f, Frequency of tumoral CD11b+ and CD8+ cells among CD45+ cells (top); frequency and cell number of tumoral CD45+CD4+ cells (bottom). g, Left, LLC tumour growth in Cd19cre Tmem173fl/fl (n=12), Cd4cre Tmem173fl/fl (n=7) or Tmem173fl/fl (n=9) mice. Right, B16-ova cells were s.c. injected in Cd19cre Tmem173fl/fl (n=8) or Tmem173fl/fl (n=11) mice. Poly(I:C)-ova vaccine was administered at d6. Tumour growth was monitored. h, Realtime PCR revealed Ifnb1 and Ifna4 mRNA levels in KPC4662 at 12 hr post cGAMP treatment (n=3 per group). i, IFN-β in the KPC4662 cell culture supernatants at 24 hr post cGAMP treatment as detected by ELISA. j, Cell viability percentage of KPC4662 at 24 hr post cGAMP treatment as detected by MTT assay (n=3 for Ctrl, n=4 for cGAMP-treated group). Data are the combination of two independent experiments (Extended Data Fig. 1b–e) or a representative experiment (Extended Data Fig. 1a, f–j). Mean ± s.e.m. is shown, with each symbol representing an individual animal. Unpaired two-tailed Student’s t tests was performed for statistical analysis. P values indicated; ns, not significant.
Extended Data Fig. 2 |. Molecular profiles of cGAMP-treated B cells and the effect of STING agonists on B cells.
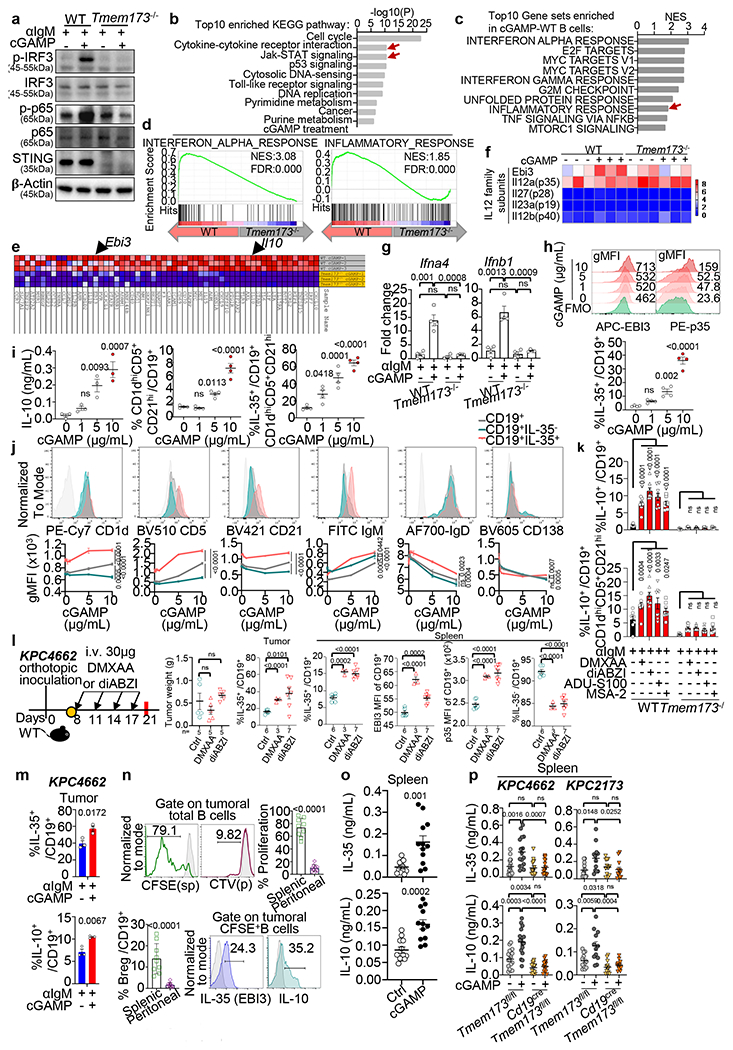
a, Immunoblot analysis of STING pathway components and β-actin in WT and Tmem173−/− B cells stimulated with anti-IgM +/− cGAMP. b, KEGG enrichment of upregulated DEGs in cGAMP-treated WT B cells vs. untreated WT B cells and cGAMP-stimulated Tmem173−/− B cells. c, GSEA analysis of RNAseq data. Normalized Enrichment Score (NES) plot of top10 gene sets enriched in the cGAMP-treated WT B cells. d, GSEA of RNAseq data shows enrichment of “interferon alpha response” (left) and “inflammatory response” pathways (right). e, Heat map of “inflammatory response pathway” genes in cGAMP-treated WT and Tmem173−/− B cells identified increased Ebi3 and Il10 expression (arrowheads). f, Heat map of IL-12 family subunit genes in cGAMP vs. PBS-treated WT and Tmem173−/− B cells. g, Real-time PCR of Ifna4 and Ifnb1 in B cells (n=4 per group) with Actb as the internal control. h, Representative histograms of EBI3 (top, left) and p35 (top, right) induced by cGAMP and analyzed by flow cytometry. Frequency of IL-35+ among CD19+ B cells treated with anti-IgM +/− cGAMP (bottom); n=4 per group. Green, fluorescence minus one control (FMO). i, Left, IL-10 in anti-IgM/cGAMP-treated B cell supernatants. CD1dhiCD5+CD21hi Bregs among CD19+ B cells (middle). IL-35+ cells among CD1dhiCD5+CD21hi Bregs (right); n=4 per group. j, Representative histograms (top) and gMFI plots (bottom) showing abundance of CD19+IL-35+ (pink), CD19+IL-35− (green) and total live CD19+ (dark gray) splenic B cells (top) in response to cGAMP. Light gray shadow represents FMO (n=4 per group). k, Frequency of IL-10+ (top) among live CD19+ B cells and IL-10+ cells among CD1dhiCD5+CD21hi Bregs (bottom) treated with four different STING agonists. WT and Tmem173−/− splenic B cells were studied as in Fig. 2d. l, Orthotopic pancreatic tumour model experimental schematic, tumour weight and frequency of tumoral and splenic CD19+IL-35+ B cells, EBI3 and p35 MFI, CD19+IL-35− B cell frequency of splenic CD19+ B cells in KPC4662 bearing mice treated with DMXAA or diABZI. m, IL-35+ (top) and IL-10+ (bottom) cells among CD19+ B cells sorted from KPC4662 tumours after anti-IgM +/− cGAMP. n, Representative histograms (top, left) and quantification (top, right) of proliferating intratumoral CFSE+ (splenic) or Cell Trace Violet (CTV, peritoneal) labelled B cells. The frequency of Breg cells (bottom, left) and expression of immunosuppressive IL-35 (EBI3) and IL-10 cytokines (bottom, right) by CFSE+ splenic or CTV+ peritoneal Breg cells. n=9 per group. o, IL-35 (top) and IL-10 (bottom) levels in spleens from samples described in Fig. 1a (n=12 per group). p, IL-35 (top) and IL-10 (bottom) levels in spleens from cGAMP-treated KPC4662 (left) (n=16) or KPC2173 (right) (n=12) bearing mice at d21. Representative of three (Extended Data Fig. 2g–k) or two (Extended Data Fig. 2a, l–m) independent experiments or combined two experiments (Extended Data Fig. 2n–p) is shown. For RNAseq, n=3 replicates/group. Mean ± s.e.m. with each symbol representing an individual animal. Unpaired two-tailed Student’s t tests (Extended Data Fig. 2g, l–p), unpaired two-tailed Student’s t tests for AUC (Extended Data Fig. 2j), one-way ANOVA (Extended Data Fig. 2h, i) and two-way ANOVA (Extended Data Fig. 2k) were performed. P values indicated; ns, not significant.
Extended Data Fig. 3 |. Differential effects of cGAMP on IL-35 and IL-10 induction are compared in B and T cells.
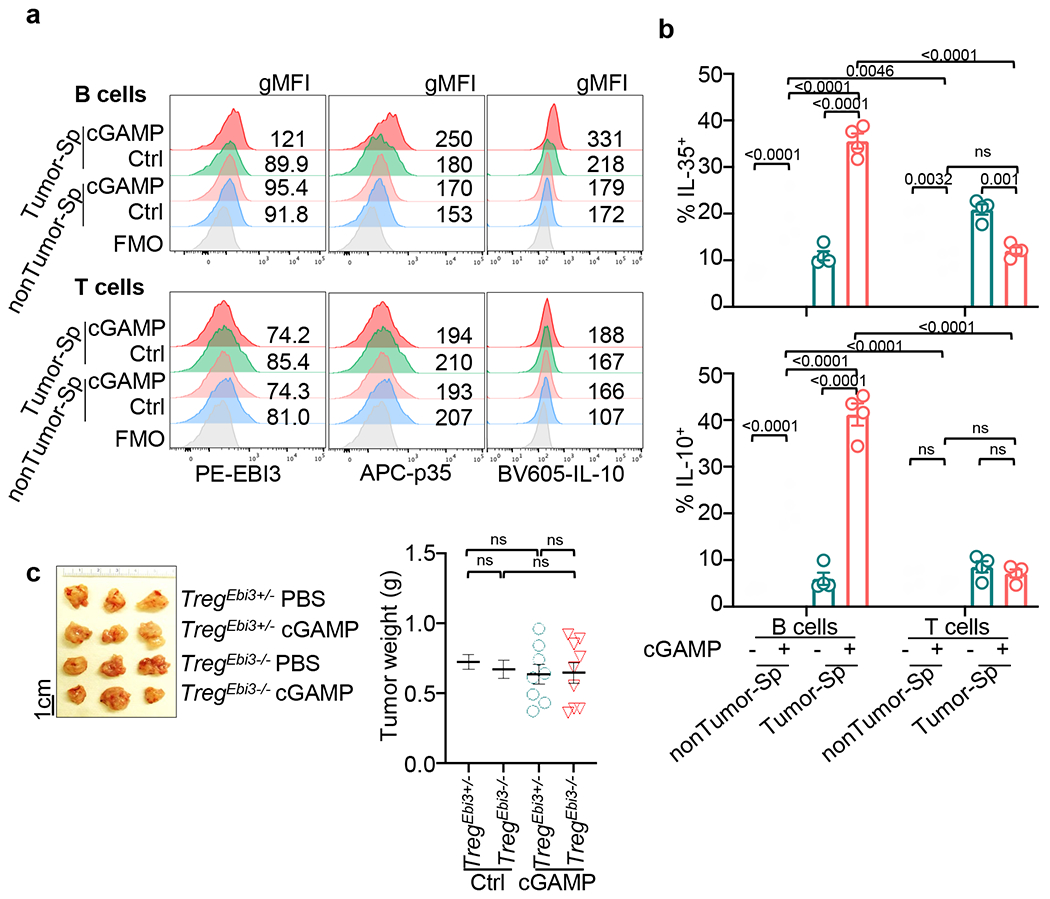
a, Representative histograms of EBI3 (left), p35 (middle) and IL-10 (right) with geometric mean fluorescence intensity (gMFI) are shown. B and T cells were sorted from spleens of KPC4662 tumour-bearing or non-tumour bearing mice. B cells were stimulated with anti-IgM. T cells were stimulated with anti-CD3 and anti-CD28. Cells were treated with or without cGAMP and analyzed by flow cytometry analysis at 24 hr post treatment. b, Quantification of the percentages of IL-35+ (top) and IL-10+ (bottom) cells in live CD19+ B cells and CD3+ T cells (n=4, representative of two independent experiments). c, Treg(Ebi3+/−) and Treg(Ebi3−/−) mice orthotopically inoculated with KPC4662 cells and treated with cGAMP. Representative images of tumours (left) and tumour weights (right) at d21 are shown (n=8 Ctrl PBS, n=8 Ctrl cGAMP, n=9 Treg(Ebi3−/−) PBS, n=9 Treg(Ebi3−/−) cGAMP). Representative of two replicate studies (Extended Data Fig. 3a–c) are shown, with each symbol representing an individual animal and mean ± s.e.m. denoted. Statistical tests of two-way ANOVA (Extended Data Fig. 3b) and unpaired two-tailed Student’s t tests (Extended Data Fig. 3c) were performed. P values indicated; ns, not significant.
Extended Data Fig. 4 |. Immune profiling of pancreatic tumour-bearing B cell-specific IL-35 or IL-10 ablation mice.
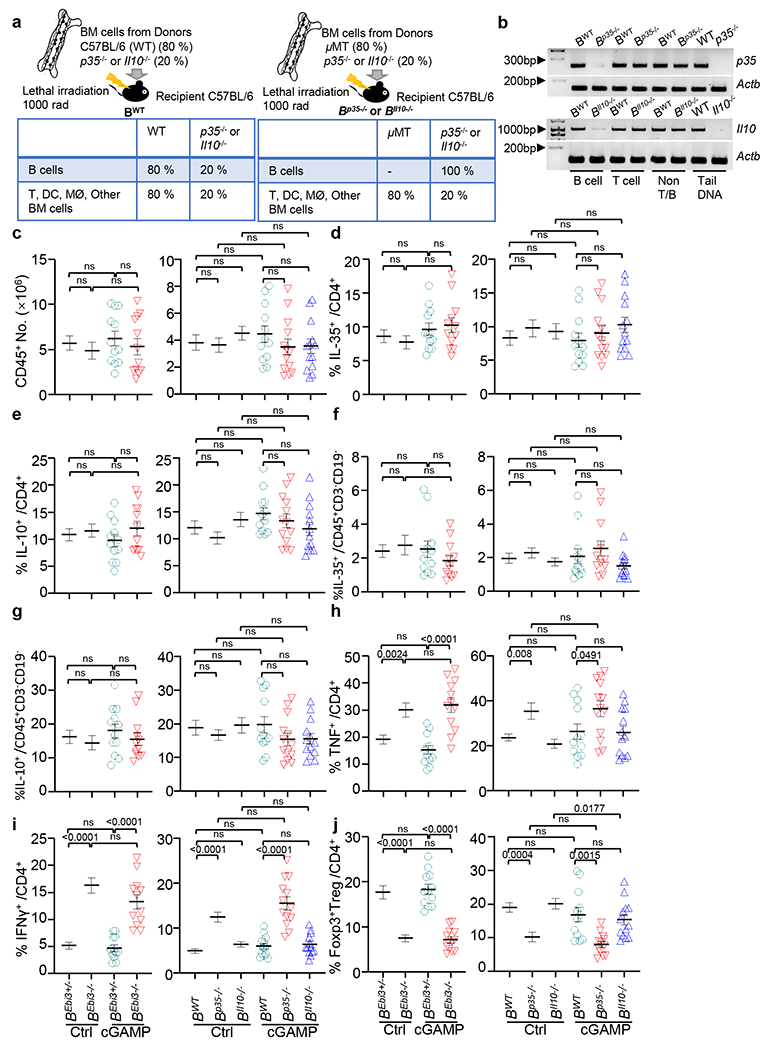
a, Mixed bone marrow chimeras containing B cell–specific deletion of p35 (B(p35−/−)) or Il10 (B(Il10−/−)) with WT (B(WT)) controls were generated (top) with expected cell phenotypes shown (bottom). b, Validation of loss of p35 and Il10 in B cells from B(p35−/−) and B(Il10−/−) mice. Genomic DNA was isolated from CD19+ B cells, CD3+ T cells and CD19−, CD3− cells (non-T or B cells). Genomic DNA samples from tails of WT, p35−/− and Il10−/− mice were used as controls. Actb was used as the internal control. c, Number of total tumoral CD45+ leukocytes from KPC4662 bearing cGAMP− and control PBS-treated mice as in Fig. 3c. d, Frequency of tumoral IL-35 producing CD4+ T cells from mice as in Fig. 3c. e, Frequency of tumoral IL-10 producing CD4+ T cells from mice as in Fig. 3c. f, Frequency of tumoral IL-35 producing non-T, B cells from mice as in Fig. 3c. g, Frequency of tumoral IL-10 producing non-T, B cells from mice as in Fig. 3c. h, Frequency of tumoral TNF secreting CD4+ T cells from mice as in Fig. 3c. i, Frequency of tumoral IFNγ secreting CD4+ T cells from mice as in Fig. 3c. j, Frequency of tumoral CD4+Foxp3+ Treg cells from mice as in Fig. 3c. Samples analyzed in Extended Data Fig. 4c–j are described in Fig. 3a,b. Representative experiments (Extended Data Fig. 4b) or a combination of three independent experiments (Extended Data Fig. 4c–j) are shown, with each symbol representing an individual animal and mean ± s.e.m. denoted. Unpaired two-tailed Student’s t tests were performed (Extended Data Fig. 4c–j) for statistical analysis. P values indicated; ns, not significant.
Extended Data Fig. 5 |. Anti-IL-35 synergizes with cGAMP to restrain tumour growth and enhance overall survival in the B16-ova melanoma mouse model.
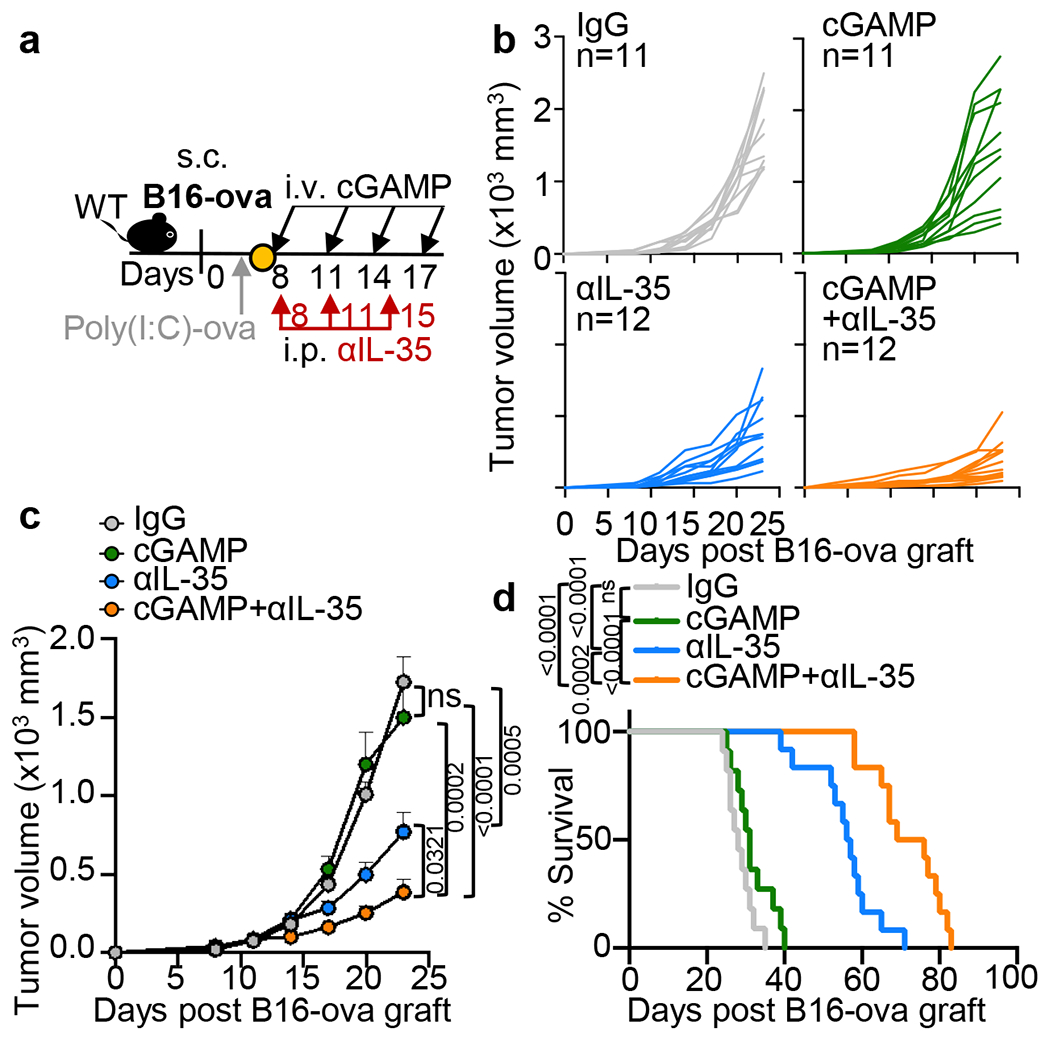
a, B16-ova tumour-bearing mice were vaccinated with poly(I:C)-ova on d6, followed by i.v. administration with cGAMP (n=11), i.p. injection of anti-IL-35 (n=12) or IgG (n=11) or cGAMP+anti-IL-35(n=12) on indicated timepoints. Mice were monitored for tumour growth (b, c) and survival (d). A combination of three independent experiments is shown. Mean ± s.e.m. is shown. Unpaired two-tailed Student’s t tests and Log-rank (Mantel-Cox) test were performed for statistical analyses. P values indicated; ns, not significant.
Extended Data Fig. 6 |. Immune cells of combination anti-IL-35- plus cGAMP-treated tumours are examined.
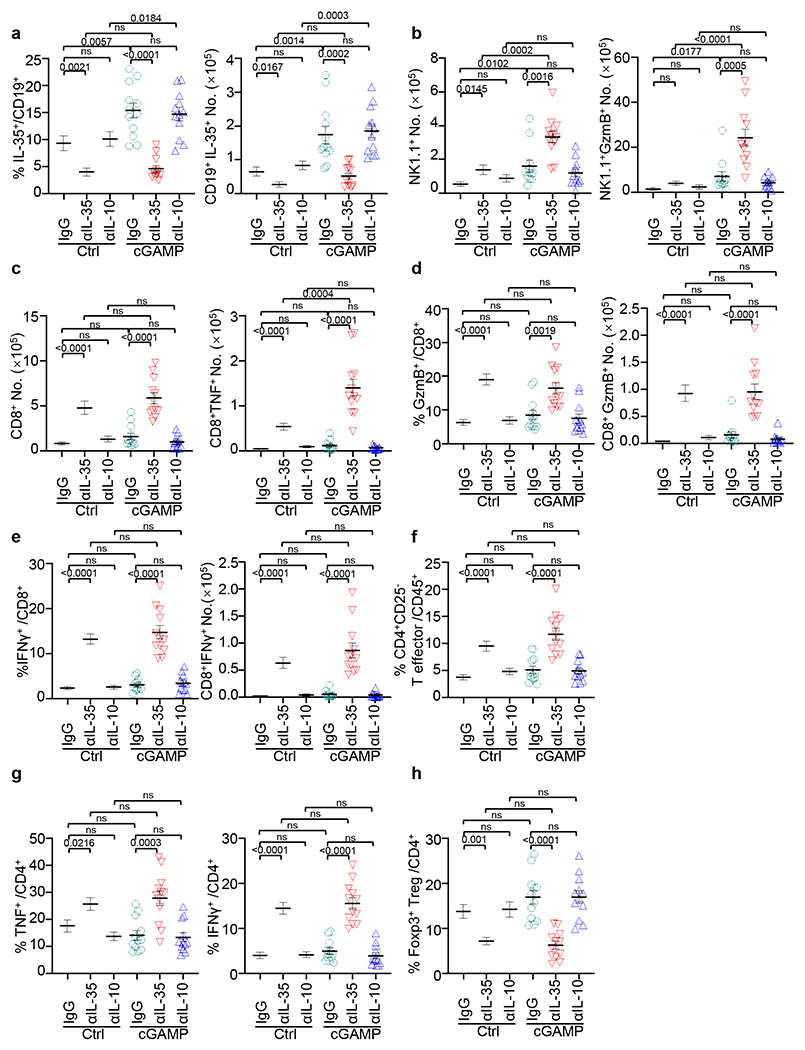
All samples analyzed were obtained from KPC4662 tumour-bearing mice that were injected with cGAMP or PBS control in combination with anti-IL-10, anti-IL-35 or control IgG as depicted in Fig. 4a. a, Frequency (left) and number (right) of tumoral IL-35 producing CD19+ B cells in Fig. 4b. b, Number of tumoral CD45+NK1.1+ cells (left) and NK1.1+ cells expressing GzmB (right) as noted in Fig. 4b. c, Number of tumoral CD45+CD8+ cells (left) and CD8+ T cells expressing TNF (right) as noted in Fig. 4b. d, Frequency (left) and number (right) of tumoral CD8+ T cells expressing GzmB from mice as in Fig. 4b. e, Frequency (left) and number (right) of tumoral CD8+ T cells expressing IFNγ from mice as in Fig. 4b. f, Frequency of tumoral CD45+CD4+CD25− T effector cells from mice as in Fig. 4b. g, Frequency of tumoral CD4+ T cells expressing TNF (left) or IFNγ (right) from mice as in Fig. 4b. h, Frequency of tumoral CD4+Foxp3+ Treg cells from mice as in Fig. 4b. A combination of three experiments (Extended Data Fig. 6a–h) are shown. Mean ± s.e.m. is shown. Unpaired two-tailed Student’s t tests were performed for statistical analysis. P values indicated; ns, not significant.
Extended Data Fig. 7 |. Immune profiles of combination cGAMP+anti-IL-35 treated tumours are examined.
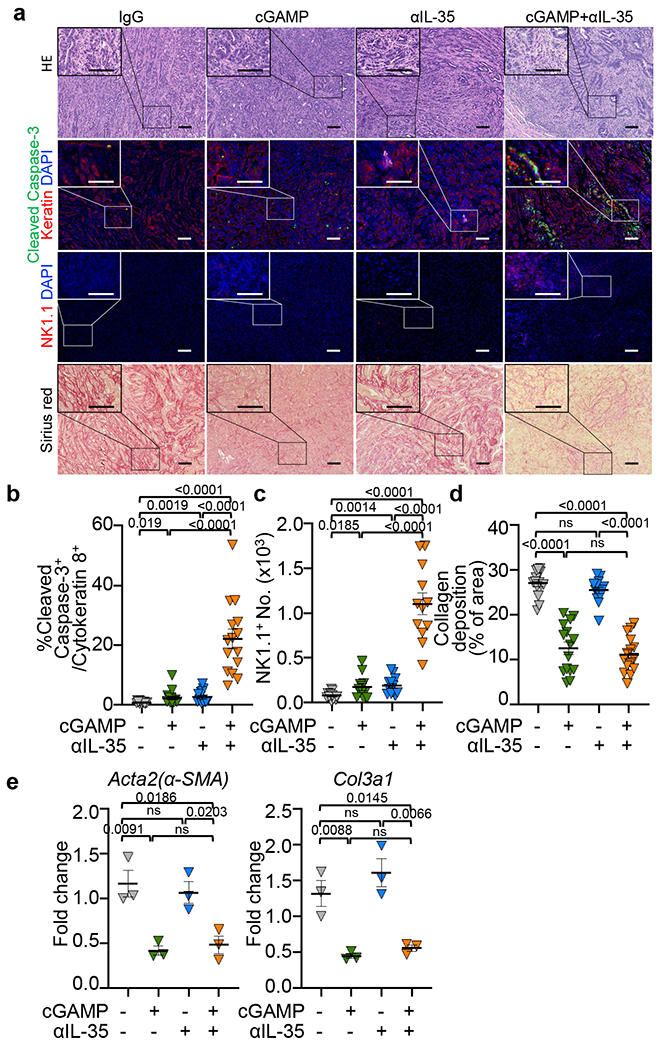
All samples analyzed were obtained from KPC4662 tumour-bearing mice that were injected with cGAMP or PBS in combination with anti-IL-35 or control IgG as depicted in Fig. 4a. a, H&E, immunohistochemistry with the indicated markers and Sirius red staining of sections from groups as described in Fig. 4a (Scale bars, 100 μm.). b, The frequency of cleaved caspase-3+ in cytokeratin 8+ cells as shown in (a), n=15 per group. c, The number of NK1.1+ cells as shown in (a), n=12 per group. d, The quantification of collagen deposition depicted by Sirius red staining in sections as shown in (a), n=15 per group. e, Real-time PCR results revealed the expression of fibrosis markers Acta2 (αSMA) and Col3a1 in tumour homogenates of indicated groups; n=3 per group. Actb was used as the internal control. A combination of two (Extended Data Fig. 7a–d) or representative of two independent experiments (Extended Data Fig. 7e) are shown. Mean ± s.e.m. is shown, Unpaired two-tailed Student’s t tests was performed for statistical analysis. P values indicated; ns, not significant.
Extended Data Fig. 8 |. Dual therapy of cGAMP plus anti-IL-35 depends on TRAIL mediated NK cytotoxity.
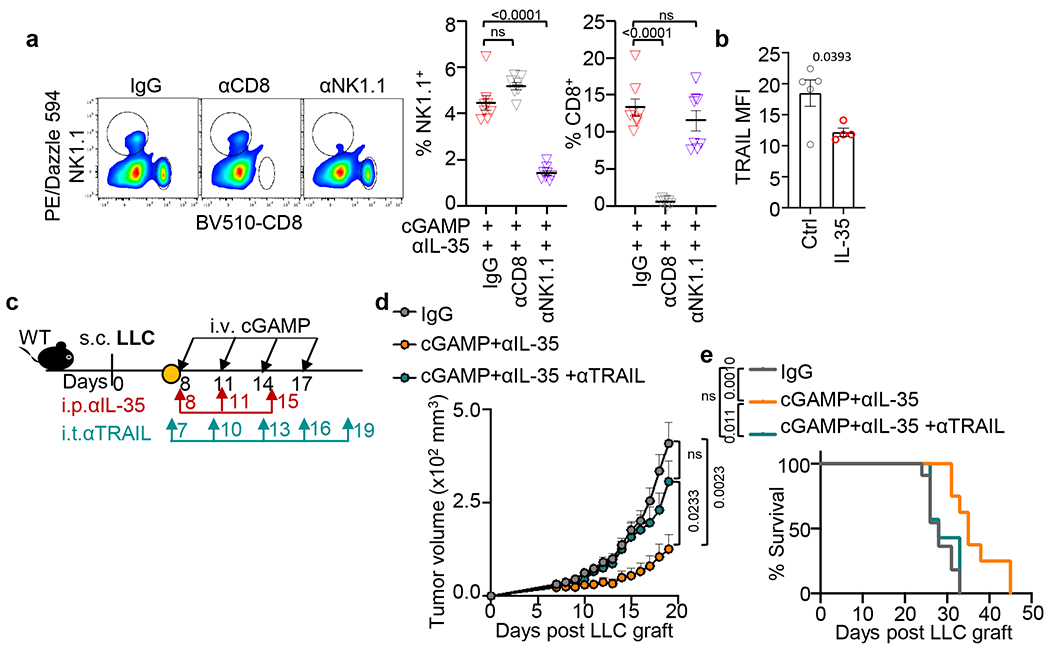
a, The efficiency of specific antibody depletion of CD8+ or NK1.1+ cells in spleens from treated mice (n=8 for IgG and anti-NK1.1 groups, n=7 for anti-CD8 group). b, Flow cytometric analysis of TRAIL, shown as MFI, on tumoral NK1.1+ cells in response to IL-35 with IL-15 stimulation for 24 hr (n=5 for Ctrl, n=4 for IL-35-stimulated group). c, Experimental schematic: LLC bearing mice received cGAMP (i.v.) and anti-IL-35 (i.p.) with anti-TRAIL (i.t.). d, Tumour growth of mice described in (c). e, Survival curve of mice described in (c). A representative of two independent experiments is shown for all panels. Mean ± s.e.m. is shown. Unpaired two-tailed Student’s t tests (Extended Data Fig. 8a, b), unpaired two-tailed Student’s t tests for AUC and Log-rank (Mantel-Cox) test (Extended Data Fig. 8d, e) were performed for statistical analysis. P values indicated; ns, not significant.
Extended Data Fig. 9 |. RNAseq analysis of tumoral NK cells from KPC tumour-bearing mice under mono or dual therapy of cGAMP+anti-IL-35.
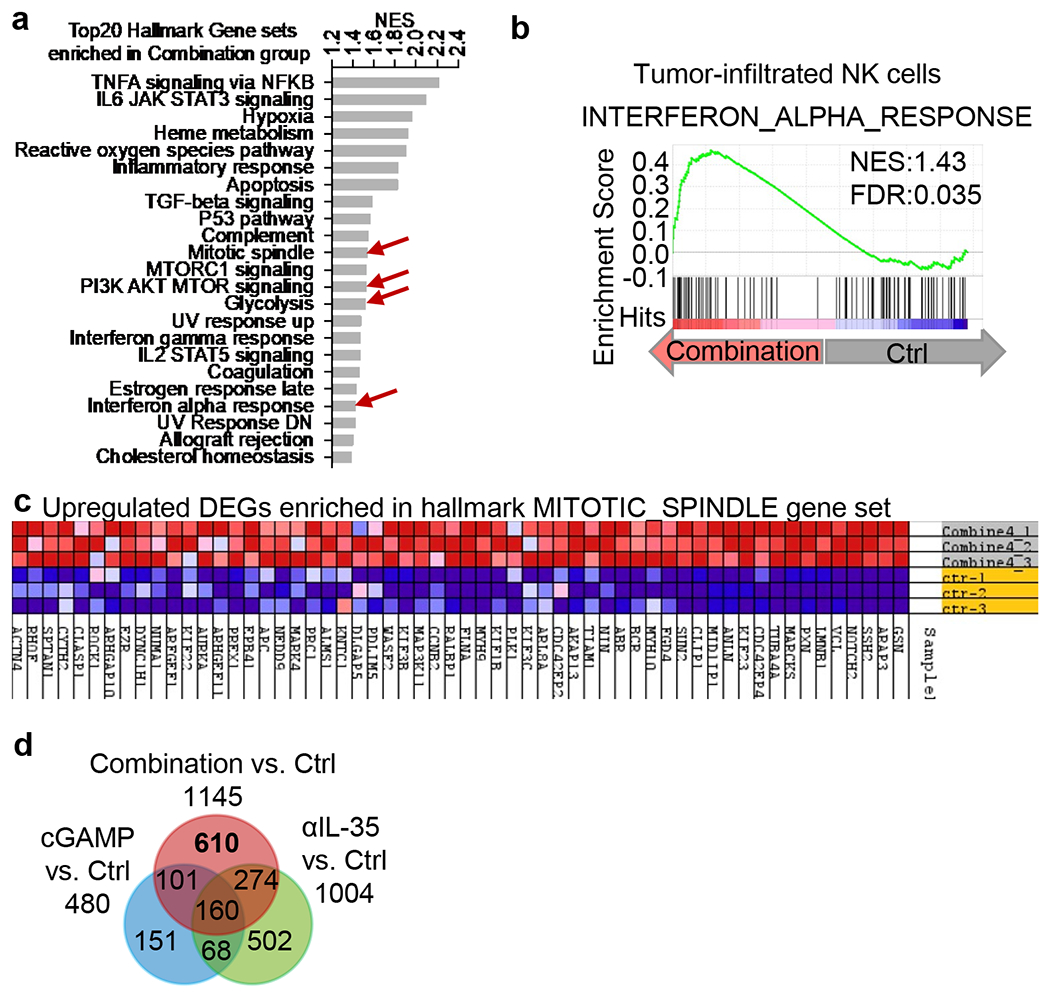
a, GSEA analysis of RNAseq of tumoral NK cells. Normalized Enrichment Score (NES) plot of top 20 hallmark signature gene sets enriched in combination cGAMP+anti-IL-35 vs. Ctrl treated NK cells. b, GSEA showing the enrichment of “interferon alpha response” in tumoral NK cells isolated from mice treated with cGAMP+anti-IL-35 vs. Ctrl NK cells as noted in (a). c, Heatmap of “mitotic spindle pathway” genes in NK cells isolated from animals treated with cGAMP+anti-IL-35 vs untreated Ctrl NK cells. d, Venn diagram showing down-regulated DEGs in combination cGAMP+anti-IL-35, cGAMP or anti-IL-35 vs. Ctrl groups.
Extended Data Fig. 10 |. Human pancreatic cancer Breg signatures are associated with STING signaling or activated NK cell signature.
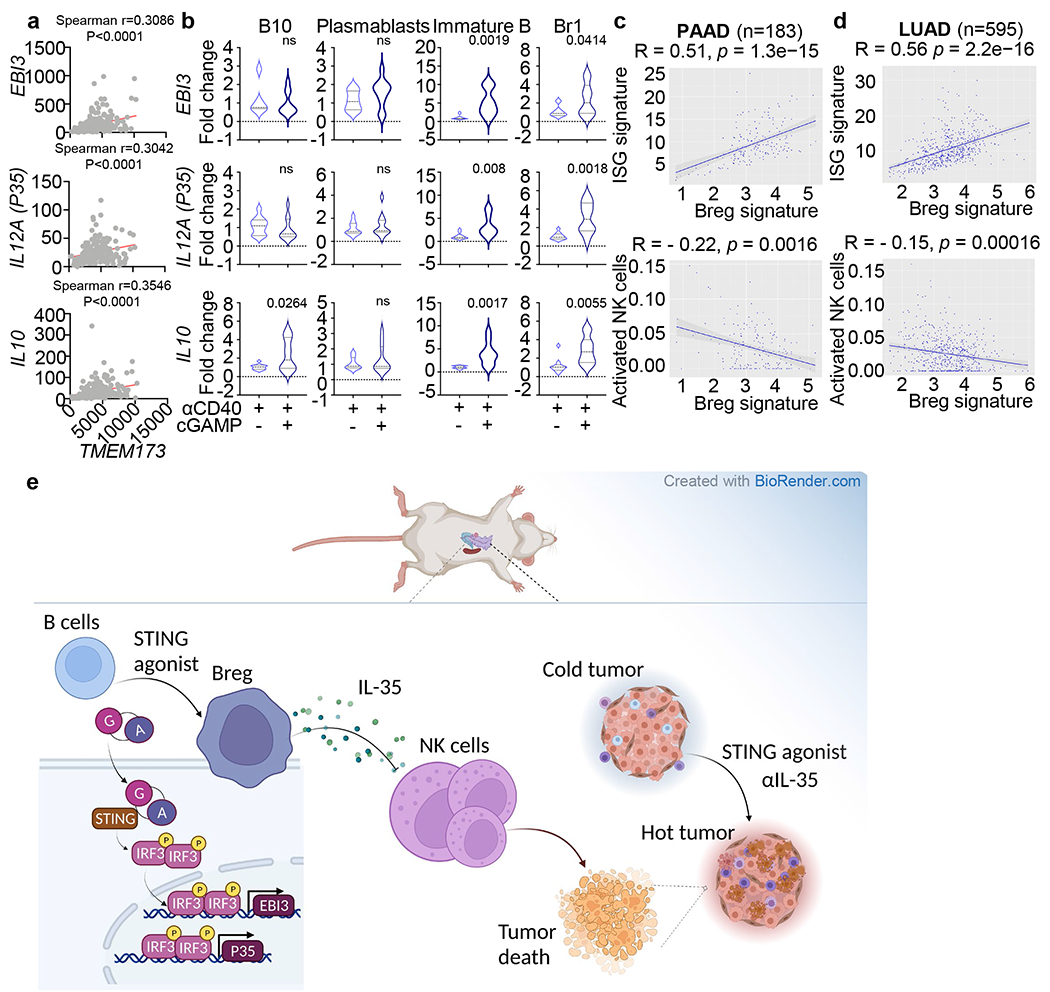
a, Correlations of EBI3 (top), IL12A (P35) (middle) or IL10 (bottom) with TMEM173 expression in TCGA-PAAD patient samples (n=183). b, Real-time PCR assessment of EBI3 (top), IL12A (middle) and IL10 (bottom) transcript levels in B10 cells, plasmablasts, immature B cells and Br1 cells sorted from pancreatic cancer patients and healthy donors (n=10) and stimulated with anti-CD40 +/− cGAMP. HPRT was used as the internal control. c, Correlation of Breg signature to ISG signature or activated NK cell index in TCGA PAAD (n=183) patient samples. d, Correlation of Breg signature to ISG signature or activated NK cell index in TCGA LUAD (n=595) patient samples. e, Model illustrating the effect of STING agonist on IL-35 production by Breg cells through an IRF3-dependent pathway, which suppresses NK cell function in controlling pancreatic cancer. The graphical illustration was made using BioRender (https://biorender.com/). Spearman correlation (Extended Data Fig. 10a, c, d) and Unpaired two-tailed Student’s t tests (Extended Data Fig. 10b) were performed. The shaded gray region indicates the 95% confidence bands of the fit, as shown in Extended Data Fig. 10c, d. P values indicated; ns, not significant.
Supplementary Material
Acknowledgments
The following funding sources are acknowledged: National Institutes of Health/National Cancer Institute Grants (R35-CA232109 and RO1-AI029564 to J.P-Y.T.; U19-AI067798 to J.P.-Y.T. and W.J.B.; R37-CA230786 to Y.P.-G.; 1P50CA196510-01A1 to Y.P.-G., Ryan Fields and Willian Hawkins; Developmental grants from the University Cancer Research Fund at the University of North Carolina at Chapel Hill to J.P.-YT and Y.P.-G.; North Carolina Biotechnology grant to J.P.-Y.T; Institutional Research Training Grant T32NHLBI7106-39 to B.M.J. and National Institute of Health AID Research Grants AI100625 to M.G.Jr. We received help from Ryan Fields and William Hawkins at the Washington University School of Medicine with providing human samples funded by the Washington University Specialized Program of Research Excellence. This work utilized the computational resources of the NIH HPC Biowulf cluster ([http://hpc.nih.gov]). We thank Ramiro Diz, Ayrianna Woody and Janet Dow (UNC-CH flow cytometry facility funded in part by P30 CA016086 Cancer Center Core Support Grant) for flow cytometry support.
Footnotes
Conflict of Interest
The authors have declared that no conflict of interest exists.
Data availability
All the databases used in the study were publicly accessible. The hallmark gene sets (MSigDB version7.2) and Kyoto Encyclopedia of Genes and Genomes gene set collections (MSigDB version 7.2) were used in the analysis (http://www.gseamsigdb.org/gsea/downloads_archive.jsp). The datasets of The Cancer Genome Atlas are available at https://portal.gdc.cancer.gov/. Raw and processed RNAseq data used in the study are available at the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) under repository accession number GSE199343 (B cells) and GSE211386 (NK cells). All other data supporting the findings of this study are available from corresponding authors upon reasonable request.
References
- 1.Evans RA et al. Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI insight 1, doi: 10.1172/jci.insight.88328 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hegde PS & Chen DS Top 10 Challenges in Cancer Immunotherapy. Immunity 52, 17–35, doi: 10.1016/j.immuni.2019.12.011 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Lutz ER et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer immunology research 2, 616–631, doi: 10.1158/2326-6066.Cir-14-0027 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng N et al. A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1-insensitive models of triple-negative breast cancer. JCI insight 3, doi: 10.1172/jci.insight.120638 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amouzegar A, Chelvanambi M, Filderman JN, Storkus WJ & Luke JJ STING Agonists as Cancer Therapeutics. Cancers (Basel) 13, doi: 10.3390/cancers13112695 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meric-Bernstam F et al. Phase I Dose-Escalation Trial of MIW815 (ADU-S100), an Intratumoral STING Agonist, in Patients with Advanced/Metastatic Solid Tumors or Lymphomas. Clinical cancer research : an official journal of the American Association for Cancer Research 28, 677–688, doi: 10.1158/1078-0432.Ccr-21-1963 (2022). [DOI] [PubMed] [Google Scholar]
- 7.Gogoi H, Mansouri S & Jin L The Age of Cyclic Dinucleotide Vaccine Adjuvants. Vaccines (Basel) 8, doi: 10.3390/vaccines8030453 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun X et al. Amplifying STING activation by cyclic dinucleotide-manganese particles for local and systemic cancer metalloimmunotherapy. Nature nanotechnology 16, 1260–1270, doi: 10.1038/s41565-021-00962-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vonderhaar EP et al. STING Activated Tumor-Intrinsic Type I Interferon Signaling Promotes CXCR3 Dependent Antitumor Immunity in Pancreatic Cancer. Cell Mol Gastroenterol Hepatol 12, 41–58, doi: 10.1016/j.jcmgh.2021.01.018 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jing W et al. STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models. J Immunother Cancer 7, 115, doi: 10.1186/s40425-019-0573-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghaffari A et al. STING agonist therapy in combination with PD-1 immune checkpoint blockade enhances response to carboplatin chemotherapy in high-grade serous ovarian cancer. Br J Cancer 119, 440–449, doi: 10.1038/s41416-018-0188-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu J, Dobbs N, Yang K & Yan N Interferon-Independent Activities of Mammalian STING Mediate Antiviral Response and Tumor Immune Evasion. Immunity 53, 115–126.e115, doi: 10.1016/j.immuni.2020.06.009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lemos H et al. Overcoming resistance to STING agonist therapy to incite durable protective antitumor immunity. J Immunother Cancer 8, doi: 10.1136/jitc-2020-001182 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang B et al. B cell-derived GABA elicits IL-10(+) macrophages to limit anti-tumour immunity. Nature 599, 471–476, doi: 10.1038/s41586-021-04082-1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mirlekar B, Michaud D, Searcy R, Greene K & Pylayeva-Gupta Y IL35 Hinders Endogenous Antitumor T-cell Immunity and Responsiveness to Immunotherapy in Pancreatic Cancer. Cancer immunology research 6, 1014–1024, doi: 10.1158/2326-6066.Cir-17-0710 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Helmink BA et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555, doi: 10.1038/s41586-019-1922-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meylan M et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity 55, 527–541.e525, doi: 10.1016/j.immuni.2022.02.001 (2022). [DOI] [PubMed] [Google Scholar]
- 18.Pylayeva-Gupta Y et al. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer discovery 6, 247–255, doi: 10.1158/2159-8290.Cd-15-0843 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong S et al. B Cells Are the Dominant Antigen-Presenting Cells that Activate Naive CD4(+) T Cells upon Immunization with a Virus-Derived Nanoparticle Antigen. Immunity 49, 695–708.e694, doi: 10.1016/j.immuni.2018.08.012 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Cui C et al. Neoantigen-driven B cell and CD4 T follicular helper cell collaboration promotes anti-tumor CD8 T cell responses. Cell 184, 6101–6118.e6113, doi: 10.1016/j.cell.2021.11.007 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bayne LJ et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer cell 21, 822–835, doi: 10.1016/j.ccr.2012.04.025 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li W et al. cGAS-STING-mediated DNA sensing maintains CD8(+) T cell stemness and promotes antitumor T cell therapy. Science translational medicine 12, doi: 10.1126/scitranslmed.aay9013 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Barber GN STING: infection, inflammation and cancer. Nature reviews. Immunology 15, 760–770, doi: 10.1038/nri3921 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Floss DM et al. IL-6/IL-12 Cytokine Receptor Shuffling of Extra- and Intracellular Domains Reveals Canonical STAT Activation via Synthetic IL-35 and IL-39 Signaling. Scientific Reports 7, 15172, doi: 10.1038/s41598-017-15173-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen P et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 507, 366–370, doi: 10.1038/nature12979 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Radomir L et al. The survival and function of IL-10-producing regulatory B cells are negatively controlled by SLAMF5. Nat Commun 12, 1893, doi: 10.1038/s41467-021-22230-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS & Bhan AK Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity 16, 219–230, doi: 10.1016/s1074-7613(02)00274-1 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Mirlekar B et al. B cell-Derived IL35 Drives STAT3-Dependent CD8(+) T-cell Exclusion in Pancreatic Cancer. Cancer immunology research 8, 292–308, doi: 10.1158/2326-6066.Cir-19-0349 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collison LW et al. IL-35-mediated induction of a potent regulatory T cell population. Nature immunology 11, 1093–1101, doi: 10.1038/ni.1952 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sawant DV et al. Adaptive plasticity of IL-10(+) and IL-35(+) T(reg) cells cooperatively promotes tumor T cell exhaustion. Nature immunology 20, 724–735, doi: 10.1038/s41590-019-0346-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y et al. STING signaling activation inhibits HBV replication and attenuates the severity of liver injury and HBV-induced fibrosis. Cellular & molecular immunology 19, 92–107, doi: 10.1038/s41423-021-00801-w (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Q, Manohar M, Wei Y, Pandol SJ & Habtezion A STING signalling protects against chronic pancreatitis by modulating Th17 response. Gut 68, 1827–1837, doi: 10.1136/gutjnl-2018-317098 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marçais A et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nature immunology 15, 749–757, doi: 10.1038/ni.2936 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nandagopal N, Ali AK, Komal AK & Lee SH The Critical Role of IL-15-PI3K-mTOR Pathway in Natural Killer Cell Effector Functions. Front Immunol 5, 187, doi: 10.3389/fimmu.2014.00187 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Z et al. Glycolysis and Oxidative Phosphorylation Play Critical Roles in Natural Killer Cell Receptor-Mediated Natural Killer Cell Functions. Front Immunol 11, 202, doi: 10.3389/fimmu.2020.00202 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheppard S et al. Lactate dehydrogenase A-dependent aerobic glycolysis promotes natural killer cell anti-viral and anti-tumor function. Cell reports 35, 109210, doi: 10.1016/j.celrep.2021.109210 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsumura T et al. TUBB1 dysfunction in inherited thrombocytopenia causes genome instability. Br J Haematol 185, 888–902, doi: 10.1111/bjh.15835 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Weinstein JN et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45, 1113–1120, doi: 10.1038/ng.2764 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blair PA et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity 32, 129–140, doi: 10.1016/j.immuni.2009.11.009 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Flores-Borja F et al. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Science translational medicine 5, 173ra123, doi: 10.1126/scitranslmed.3005407 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Liu H et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nature medicine 25, 95–102, doi: 10.1038/s41591-018-0302-5 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Newman AM et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 37, 773–782, doi: 10.1038/s41587-019-0114-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marcus A et al. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 49, 754–763 e754, doi: 10.1016/j.immuni.2018.09.016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mujal AM, Delconte RB & Sun JC Natural Killer Cells: From Innate to Adaptive Features. Annual review of immunology 39, 417–447, doi: 10.1146/annurev-immunol-101819-074948 (2021). [DOI] [PubMed] [Google Scholar]
- 45.Piersma SJ et al. Virus infection is controlled by hematopoietic and stromal cell sensing of murine cytomegalovirus through STING. eLife 9, doi: 10.7554/eLife.56882 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jensen H, Chen SY, Folkersen L, Nolan GP & Lanier LL EBI3 regulates the NK cell response to mouse cytomegalovirus infection. Proc Natl Acad Sci U S A 114, 1625–1630, doi: 10.1073/pnas.1700231114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turnis ME et al. Interleukin-35 Limits Anti-Tumor Immunity. Immunity 44, 316–329, doi: 10.1016/j.immuni.2016.01.013 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fontenot JD, Gavin MA & Rudensky AY Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nature immunology 4, 330–336, doi: 10.1038/ni904 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Kim JM, Rasmussen JP & Rudensky AY Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nature immunology 8, 191–197, doi: 10.1038/ni1428 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Rubtsov YP et al. Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671, doi: 10.1126/science.1191996 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rubtsov YP et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558, doi: 10.1016/j.immuni.2008.02.017 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Cardoso Alves L et al. Non-apoptotic TRAIL function modulates NK cell activity during viral infection. EMBO Rep 21, e48789, doi: 10.15252/embr.201948789 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conlon J et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol 190, 5216–5225, doi: 10.4049/jimmunol.1300097 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramanjulu JM et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 564, 439–443, doi: 10.1038/s41586-018-0705-y (2018). [DOI] [PubMed] [Google Scholar]
- 55.Corrales L et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell reports 11, 1018–1030, doi: 10.1016/j.celrep.2015.04.031 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pan BS et al. An orally available non-nucleotide STING agonist with antitumor activity. Science 369, eaba6098 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Nelson JD, Denisenko O & Bomsztyk K Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1, 179–185, doi: 10.1038/nprot.2006.27 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Chen S, Zhou Y, Chen Y & Gu J fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, doi: 10.1093/bioinformatics/bty560 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930, doi: 10.1093/bioinformatics/btt656 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Wang L, Feng Z, Wang X, Wang X & Zhang X DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138, doi: 10.1093/bioinformatics/btp612 (2010). [DOI] [PubMed] [Google Scholar]
- 62.Yu G, Wang LG, Han Y & He QY clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287, doi: 10.1089/omi.2011.0118 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wickham H ggplot2: Elegant Graphics for Data Analysis. (Springer-Verlag, 2009). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the databases used in the study were publicly accessible. The hallmark gene sets (MSigDB version7.2) and Kyoto Encyclopedia of Genes and Genomes gene set collections (MSigDB version 7.2) were used in the analysis (http://www.gseamsigdb.org/gsea/downloads_archive.jsp). The datasets of The Cancer Genome Atlas are available at https://portal.gdc.cancer.gov/. Raw and processed RNAseq data used in the study are available at the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) under repository accession number GSE199343 (B cells) and GSE211386 (NK cells). All other data supporting the findings of this study are available from corresponding authors upon reasonable request.