

Abstract
Major advances have been made to improve the sensitivity of mass analyzers, spectral quality, and speed of data processing enabling more comprehensive proteome discovery and quantitation. While focus has recently begun shifting toward robust proteomics sample preparation efforts, a high-throughput proteomics sample preparation is still lacking. We report the development of a highly automated universal 384-well plate sample preparation platform with high reproducibility and adaptability for extraction of proteins from cells within a culture plate. Digestion efficiency was excellent in comparison to a commercial digest peptide standard with minimal sample loss while improving sample preparation throughput by 20- to 40-fold (the entire process from plated cells to clean peptides is complete in ~300 min). Analysis of six human cell types, including two primary cell samples, identified and quantified ~4000 proteins for each sample in a single high-performance liquid chromatography (HPLC)–tandem mass spectrometry injection with only 100 – 10,000 cells, thus demonstrating universality of the platform. The selected protein was further quantified using a developed HPLC-multiple reaction monitoring method for HeLa digests with a heavy labeled internal standard peptide spiked in. Excellent linearity was achieved across different cell numbers indicating a potential for target protein quantitation in clinical research.
Graphical Abstract
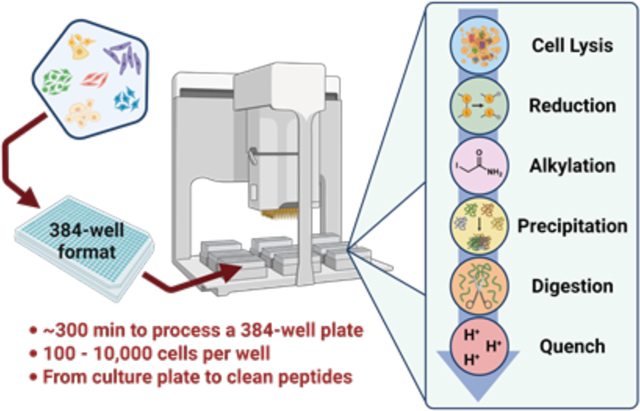
Introduction
Proteomics has become an essential tool to address critical biological questions through identification and characterization of the total protein content within a biological system1,2. Discovery of distinct protein biomarkers for specific disease targets can aid in the development of therapeutic treatments. A proteomics-based high-throughput compound screening approach that applies mass spectrometry analysis is a promising approach as it can provide more comprehensive information than other methods, which improves the probability of success for drug development efforts. Unfortunately, proteomics-based compound screening is still in the early stages of development due to sample preparation for HPLC-MS/MS analysis being primarily a manual or at its best semiautomated process with low to medium throughput.
Most of the advancements in proteomics have focused on improvements to the sensitivity of mass analyzers, speed of data processing, and data quality for discovery and quantitation3–6. However, emphasis has begun to shift toward robust proteomics sample preparation in order to produce thousands of samples required for large-scale discovery experiments and screens and the establishment of statistical significance7. The pursuit of otherwise unfeasible proteomics drug screens, especially in dosing regimens, could be realistic with high-throughput processing of cells to yield the requisite peptides. Mass spectrometry-based proteomics sample preparation methods are vastly complex due to the variety of options in each of the steps (i.e., multiple lysis strategies, reducing and alkylating agents, digestion enzymes, and cleanup methods), as well as the inherent technical challenges in their adaptability to high-throughput automation. In the past decade, a diverse array of strategies has emerged to circumvent these problems with all achieving medium-throughput, most notably a 96-well plate format7,8. Additionally, the strategies employ varying levels of automation, which includes semiautomation with either all steps automated except centrifugation and plate movements8–11 or selected liquid transfers being automated,12 or manual preparation in high-throughput labware without use of laboratory automation13–21. While various approaches provide unique advantages such as time saving, protein coverage, ease of use, and reduced cost, which are all important considerations depending on the desired goal, the highest level of throughput remains at the 96-well plate format with limited automation. One aspect that is noticeably not addressed in any of the protocols is treatment and processing of the cell samples directly from the culture plate. By utilizing the culture plate to perform the method operations rather than an initial cell scratch and transfer, we can not only increase efficiency of sample preparation but also reduce cell loss and limit cellular damage. This is extremely important in protocols involving a treatment of the cells with compounds for high-throughput screening to observe protein biomarker modulation for discovery of potential drugs.
With the goal of increasing efficiency and throughput for mass spectrometry-based proteomics analysis, here, we developed a 384-well sample preparation platform utilizing a liquid handling system outfitted with a small transfer head for disposable tips. An in-solution digest strategy was employed to take cells directly from the 384-well plate where the cells are cultured through to clean peptides for HPLC-MS/MS analysis. This universal method is performed on a high-throughput platform that incorporates comprehensive automation and has proven to be highly reproducible and adaptable with application to six different human cell types.
Materials and Methods
Reagents and Solvents.
Tris(2-carboxyethyl)phosphine (TCEP), protease inhibitor cocktail (PIC), iodoacetamide (IAA), MS-grade water, ammonium bicarbonate, and MS-grade acetonitrile (MeCN) were obtained from Millipore Sigma (Burlington, MA, U.S.A.). Trypsin and the Promega Rapid Digestion-Trypsin kit were obtained from Promega (Madison, WI, U.S.A.). Nonidet P-40 (NP40) cell lysis buffer, formic acid (FA), phosphate-buffered saline (PBS) and HeLa commercial digest were obtained from Thermo Fisher Scientific (Waltham, MA, U.S.A.). RapiGest was obtained from Waters Corporation (Milford, MA, U.S.A.). A full reagent and solvent list containing vendors and catalog numbers can be found in Table S1.
High-Throughput Sample Handling.
The automated portion of the sample preparation workflow was executed on an Agilent Bravo liquid handling platform equipped with a 384ST liquid handling head using disposable 70 μL tips, a Peltier thermal station, and an orbital shaker (Agilent Technologies, Santa Clara, CA, U.S.A.). The deck layout is shown in Figure 1A,B and includes the following labeled plates along with their respective deck positions: pos 1, wash station; pos 2, vacuum station; pos 3, tips; pos 4, analyte; pos 5, empty; pos 6, cells; pos 7, MeCN/waste/FA; pos 8, lysis-denaturation-reduction (LyDeR) rapid buffer; pos 9, IAA/trypsin. Since the protocol initially spanned 2 days due to overnight digestion, the procedure was divided into two automation steps (i.e., digestion and FA quenching) in VWorks Automation Control software (Agilent Technologies, Santa Clara, CA, U.S.A.). All Agilent Bravo liquid handling parameters can be found in Figure S1B.
Figure 1. Experimental design for the 384-well-based proteomics sample preparation platform.

Agilent Bravo automated liquid handling system (A) and deck layout indicating location of cells, reagents, and consumables (B); iodoacetamide (IAA), formic acid (FA), lysis-denaturation-reduction (LyDeR) rapid buffer, a combination buffer stock solution containing NP40 (cell lysis buffer), protease inhibitor cocktail (PIC), RapiGest (denaturant), and TCEP (reductant). (C) Sample preparation schematic from plated cells to clean peptides.
The protocol includes six key steps, which are (i) addition of LyDeR buffer to the wells of the cultured cell plate followed by incubation at 4 °C for 1 h then 40 °C for 30 min, (ii) transfer of the protein solution in LyDeR buffer to a new plate after centrifugation, (iii) addition of IAA followed by incubation at room temperature in the absence of light for 20 min, (iv) addition of MeCN at 4 °C to precipitate the protein followed by centrifugation and removal of MeCN, (v) addition of trypsin for digestion at 37 °C overnight, and (vi) addition of FA to quench the reaction prior to HPLC-MS/MS analysis. Detailed experimental procedures are provided in the Supporting Information (S5–S7).
NanoLC-MS/MS.
All discovery proteomics HPLC-MS/MS analyses were performed using an UltiMate 3000-nano LC system coupled to an Orbitrap Fusion Lumos Tribrid mass spectrometer equipped with a Nanospray Flex ion source (Thermo Fisher Scientific, Waltham, MA, U.S.A.). Peptides were loaded onto the trap column (Acclaim PepMap 100 C18, 75 μm × 2 cm, particle size of 3 μm, 100 Å) and separated with an analytical column (Acclaim PepMap RSLC, 75 μm × 50 cm, 2 μm, particle size of 100 Å) (Thermo Fisher Scientific, Waltham, MA, U.S.A.) using a 120 min method (~90 min gradient). Detailed conditions are provided in the Supporting Information (S7 and S8).
HPLC-Multiple Reaction Monitoring (MRM) Analysis.
All targeted proteomics MRM HPLC-MS/MS analyses were performed using an Agilent 1290 Infinity II LC system equipped with a high-speed pump, a multicolumn thermostat, and a multisampler, which was coupled to a 6470 Triple Quad LC/MS with a DUAL AJS ESI source (Agilent Technologies, Santa Clara, CA, U.S.A.). An ACQUITY UPLC BEH C18 (2.1 mm × 150 mm, 1.7 μm, particle size of 130 Å) column (Waters Corporation, Milford, MA, U.S.A.) was used for analysis. The peptides were loaded and separated over a 10 min gradient using the detailed conditions shown in the Supporting Information (S8) and Table S4.
Mass Spectrometry Data Search and Analysis.
Proteome Discoverer software suite (v2.2; Thermo Fisher Scientific, Waltham, MA, U.S.A.) with the Sequest algorithm was used for peptide identification and quantitation. The MS raw data were searched against a Swiss-Prot Human database (version January 2019, reviewed database) consisting of 20,350 entries using the following parameters: a precursor ion mass tolerance of 10 ppm and a fragment ion mass tolerance of 0.6 Da. Peptides were searched using fully tryptic cleavage constraints, and up to two internal cleavage sites were allowed for tryptic digestion. Fixed modifications consisted of carbamidomethylation of cysteine. Variable modifications considered were oxidation of methionine residues and N-terminal protein acetylation. Peptide identification false discovery rates (FDR) were limited to a maximum of 0.01 using identifications from a concatenated database from nondecoy and decoy databases. Label-free quantification analysis used the “Precursor Ions Quantifier” node from Proteome Discoverer and was normalized by the total peptide amount. The output from Proteome Discoverer was used to generate radar plots, violin plots, and heat maps using R (v3.4.2) and the following packages: “ggplot2” and “grid.” The mass spectrometric raw data files and associated search results of the HeLa cells and six human cell lines have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository22 with the dataset identifier PXD019733 (Figures 1 and 5) and PXD023374 (Figures 2 – 4).
Figure 5. Cell typing with the 384-well proteomics platform.

(A) Bar graph summarizing the total protein and peptide count for 5000 cells/sample for six cell types with three biological replicates. (B) Violin plots of the coefficient of variation (CV) for abundance for the 384-well plate platform with six cell types. (C) PCA analysis for six cell types using protein quantitation information. (D) Heat map showing biological pathways enriched in the six cell types. Each column is a cell type, and each row is a GO biological process. The heat map is colored by the significance of the enrichment p-value with red indicating that a pathway is significant for a cell type and blue is not significant. For each cell type, a few of the most significant pathways are given to demonstrate specificity and importance.
Figure 2. Method evaluation using HeLa cells.

Dot plot graphs summarizing total protein (A) and peptide (B) count for commercial HeLa digest and the 384-well plate platform (two technical replicates for (A), five biological replicates for (B)). (C) Ranking of HeLa proteins identified by the 384-well plate platform, normalized intensity (five biological replicates). (D) Protein overlap analysis for the 384-well plate platform from 500 to 10,000 cells across three biological replicates. Violin plots of the coefficient of variation (CV) for abundance for the 384-well plate platform at both protein (E) and peptide (F) level across five biological replicates at each cell count.
Figure 4. Universal applicability demonstrated with various human cell types.

Dot plot graphs summarizing the total protein and peptide count for the 384-well plate platform application on different cell types: HepG2 (A), HEK (B), primary ADPKD and primary NeoKerat (C), and AC16 (D) with 3–5 biological replicates for each cell count.
Pathway Analysis.
For each protein, the expression level differences between one cell type and the other five cell types were determined using the Student’s t-test. The p-values measuring the statistical significance were used for subsequent gene set enrichment analysis (GSEA)23. GSEA was performed for each cell type using Gene Ontology (GO) biological process (BP) terms24. Briefly, a Fisher’s exact test was performed at different protein level p-value cutoffs, and the cutoff that produced the most significant Fisher’s exact p-value was recorded25. At this optimal cutoff, the protein labels were randomly permutated 100 times and the Fisher’s exact p-value was recalculated. The number of times that the random p-value was smaller than the optimal p-value was counted to determine the bootstrap p-value. The GO BP terms with a bootstrap p-value of <0.01 were kept for further analysis. Heat maps and PCA plots were generated using TIBCO Spotfire version 7.11.1.
Results and Discussion
Method Design and Development.
For bottom-up proteomics, protein extraction and digestion are critical steps that often directly impact HPLC-MS/MS data quality for protein identification and quantitation. Despite this, there is a lack of efficient automated and high-throughput sample preparation methods capable of reproducibly processing hundreds of samples per day, which slows the pace of large-scale proteomics analysis for clinical samples and drug screening. In the development of a high-throughput sample preparation platform, we intended to leverage a 384-sample liquid handling system to automate the liquid transfer steps (Figure 1A,B). As such, it was necessary to adapt and optimize an in-solution digestion protocol, which was feasible for miniaturization (Figure 1C). The developed protocol includes six key steps (detailed above) to process plated cells from lysis to clean peptides. To minimize liquid handling manipulations and thus reduce sample loss, we prepared the in-house LyDeR rapid buffer mixture, which was added to the cell plates during the initial step of the method. Cell lysis was completed by incubating the cells at 4 °C with NP40 cell lysis buffer26 and assisted by RapiGest27 for solubilizing, unfolding and denaturing proteins. The incubation temperature was increased to 40 °C allowing TCEP to reduce the protein by breaking disulfide bonds. Subsequent alkylation, protein precipitation, trypsin digestion, and formic acid (FA) quenching produced clean peptide samples ready for HPLC-MS/MS analysis.
The miniaturization to a 384-well format was reliant on our ability to optimize each transfer event for a minimal amount of liquid remaining in the wells while maintaining reproducibility. To ensure that the maximum amount of the analyte was transferred in each step, careful consideration was given to having the lowest possible aspiration height (AH) of the pipette tips. The residual volume (RV) left within each well is dependent on numerous factors including the type of plate, solvent used, and the aspiration height (Figure S1A). The RV is typically less than the dead volume (DV) as over-aspiration occurs, which is advantageous when the goal is to remove the maximum amount of the solvent while trying not to remove cellular debris at the bottom of the well. It is important to note that the surface coating within the wells of some plates can significantly affect the RV due to competition between the surface tension of the liquid and the attractive forces of the plate surface. Furthermore, the shape of the well was observed to alter the RV. As such, it is essential to standardize values for each type of labware used as the recovery of every microliter is vital when working with such small volumes. For the deck locations (Figure 1B), the 384-liquid handling head was trained to our specific calibration routine.
During the calibration, aspiration, dispensing, and mixing parameters were determined empirically (Figure S1B). The aspiration height was initially set to either 0.1 or 0.2 mm for each liquid transfer event on the Bravo system and a preaspirate volume of air was incorporated as we recognized that it aided in complete dispensing of the liquid from the pipette tip. Additionally, a postaspirate volume of air ensured that the liquid did not inadvertently leak from the pipette during operation. In general, a pre and postaspirate of 2 μL was selected for the addition or removal of every reagent or solvent except acetonitrile (MeCN) while postaspirates were omitted for mixing cycles to prevent air bubbles. An aspiration speed of 10 μL/s was used except for liquid transfer events involving recovery or removal of the supernatant, which operated at 2 μL/s to ensure that the pelleted cellular debris or protein was not disturbed. A uniform dispensing speed of 10 μL/s was found to be sufficient, but the dispensing height depended on the liquid height within the wells of the specific transfer steps to prevent a distribution of liquid droplets on the well walls.
Method Evaluation Using HeLa Cells.
HeLa cells were cultured in 384-well plates with various seeding densities ranging from 500 to 10K cells/well, and subsequent washing with cold PBS prior to proteomics sample preparation. Utilizing the aforementioned method (Figure 1C), the HeLa cells were lysed, the proteins were extracted and digested, and the isolated peptides were subjected to HPLC-MS/MS analysis for protein identification and label-free-based quantitation. The quality of the method was assessed by preparing different concentrations of a commercial HeLa digest standard followed by direct HPLC-MS/MS analysis in duplicate with the following final estimated peptide amounts loaded onto the column: 31.25, 62.5, 125, 250, 500, and 1000 ng. A comprehensive database search was performed for total peptide and protein identification whose results are shown in Figure 2A. Approximately 3900 proteins (average of two runs, ~10,000 peptides) were identified for the 31.25 ng injection of total digested peptides with a gradual increase to ~4700 proteins (23,000 peptides) as the injection amount was progressively increased to 250 ng and leveled off at higher concentrations. For HeLa cell samples processed by the 384-well plate platform, the samples with multiple different cell numbers (five biological replicates) were analyzed by HPLC-MS/MS using the same method with the protein and peptide identification results shown in Figure 2B and Table S5. Satisfactorily, a similar trend to the commercial HeLa digests was observed where protein and peptide identification increased relative to the cell number. While a similar number of total proteins (3,700 proteins) were identified at 500 cells as compared to the 31.25 ng commercial digest sample, the total peptides (~15,000 peptides) increased more than 50%. These results correlate, as 500 HeLa cells correspond to ~95 ng of total proteins (micro-bicinchoninic acid (BCA) results and the literature reported 100 – 200 pg protein per cell)28,29. After transferring and digestion, around 40% of the sample within each of the 384 wells, equating to ~38 ng, was injected into the HPLC system. The comparable protein identification and 50% greater peptide identification as related to traditional methods demonstrated that the 384-well sample preparation steps where the cells are processed directly from the culture plate caused minimal protein and peptide loss, which is in part due to bypassing otherwise necessary cell scratch and transfer steps.
The dynamic range of the identified protein is plotted in Figure 2C, representing around six orders of the protein range. The protein identification reproducibility was evaluated for 500 cells up to 10K cells, shown in Figure 2D, with >85% of the proteins being identified in all three biological replicates. Additionally, well-to-well variability of the method was assessed by calculating the coefficient of variation (CV) of each individual protein or peptide for the HeLa digestion at each cell number and the datasets were visualized in violin plots with five biological replicates (Figure 2E,F). The HeLa digestion samples resulted in median CVs of 14.05 to 25.70% for protein abundance and 16.33 to 24.17% for peptide abundance, showing an excellent reproducibility for label-free-based protein quantitation.
Reproducibility Assessment for Intraplate, Interplate and Interuser.
The intraplate, interplate, and interuser variability was assessed by comparing the results from three 384-well plates containing HeLa cells subjected to the developed protocol where two plates were run by one user (i.e., plate 1 and plate 2) and one plate was run by a different user (i.e., plate 3). Similar protein and peptide identification and abundance CVs were achieved for the intraplate and interuser plate (Figures S1C,D and S2A–D). The logarithm of the protein or peptide abundance values for two replicates can be plotted, and the Pearson’s correlation coefficient (R) value was obtained from a linear regression model to evaluate reproducibility (Figure 3). The linear regression model was obtained with all combinations of samples between 500 and 10,000 cells in five replicates plotting the associated R values on heat maps to compare the intraplate, interplate, and interuser variability (Figure 3A–C and Figures S3A–D and S4A–F). Furthermore, when the comparisons are constrained to only between equivalent cell count replicates, there is an increase in protein and peptide abundance values. These results align with our hypothesis that intraplate sample preparation would have the greatest reproducibility followed by interplate and lastly interuser, but the differences were quite small, especially between interplate and interuser.
Figure 3. Intraplate, interpolate, and interuser reproducibility and method adaptability.

Pearson correlation coefficient (R) associated with a color and size scale of 0.6–1.0 and plotted on several heat maps comparing intraplate (A) and interplate (B) variability both at the protein level for HeLa cells. (C) Scatter plot of a linear regression model with each data point representing the abundance of an individual protein in two separate biological samples. (D) Violin plots of the coefficient of vVariation (CV; five biological replicates) for abundance for the 384-well plate platform using a Rapid Digest kit. (E) Radar plots of total confident protein identifications from four separate digestions of HeLa cells: plates 1 and 2 (individual plates performed by user 1), plate 3 (an individual plate performed by user 2), and plate 4 (adapted Rapid Digest protocol).
Method Adaptability.
To assess the adaptability of the workflow to other proteomics protocols, we performed the highly automated 384-well digestion on HeLa cells using the commercially available Rapid Digestion-Trypsin kit (Table S1). The kit is reported to complete proteomics digestion in as fast as 1 h and a maximum of 3 h for more difficult digestions such as disulfide-rich or membrane proteins. Besides the purported time savings gained by reducing the digest from overnight to 1–3 h, the Rapid Digestion-Trypsin kit was an attractive means to assess robustness of the sample preparation platform with changes in reagents and conditions for enabling expedient integration with other novel workflows. The rapid digestion protocol utilizes a Rapid Digest buffer that replaces ammonium bicarbonate for trypsin resuspension and a Rapid Trypsin Gold reagent substituting for the sequencing grade modified trypsin. An additional modification to the rapid digestion protocol was an increase in digestion temperature from 37 to 70 °C, which caused heterogeneous heating microtiter plates and warping of the plates that resulted in greater variability. The use of a custom copper thermal conducting block and enclosure provided homogeneous heating across the plate with minimized warping. The general observed trend and total number of protein and peptide identifications from 500 to 10,000 cells (Figure S1E), as well as protein and peptide abundance CVs (Figure 3D and Figure S2E) were found to be similar for the modified rapid digestion protocol as compared to the traditional overnight digestion with multiple replicates (Figure 3E and Figure S5A). Moreover, variability among several proteomics metrics (Figure S5B) such as the average Sequest score, average sequence coverage, average q-value, average posterior error probability (PEP) score, and average number of peptide spectral matches (PSMs) was used to assess reproducibility of replicate plates within the adapted protocol. The results displayed in Figure S5B illustrate a similarity of values for each of the metrics, which indicates that there are no significant macro-changes between datasets despite variation at the individual protein and peptide levels. Taken all together, this not only validates that the Rapid Digestion-Trypsin kit delivers comparable results ~10-fold faster than a traditional overnight digestion but also demonstrates that the sample preparation method is adaptable to other in-solution digestion strategies enabling wider applicability.
Universal Application with Six Human Cell Types.
The ability of a protocol to achieve a sufficient yield of confident protein and peptide identifications independent of the cell type is an indication of the method’s universality. Therefore, we applied the proteomics sample preparation workflow to six different human cell types: HeLa cervical epithelial cells (HeLa), AC16 cardiomyocytes (AC16), human embryonic kidney 293 cells (HEK), HepG2 hepatocytes (HepG2), primary autosomal dominant polycystic kidney disease epithelial cells (primary ADPKD), and primary neonatal keratinocytes (primary NeoKerat) (Figure S6). While five cell types underwent digestion and analysis with our standard cell counts of 500 to 10K cells/well, we were able to expand the range for AC16 to include 100, 200, and 400 cells due to its large cell size. The results show that the protocol went smoothly across all cell types with confident protein and peptide identifications being recorded for each cell type for 3–5 biological replicates (Figures 4A–D). Since HEK and HepG2 are highly proliferated cells, ~4000 proteins were identified even with a cell count of 500 whereas the less proliferated primary ADPKD and NeoKerat cells, which contain less protein overall within each cell, only achieved around 2000–2500 identified proteins at the same cell count. Additionally, the primary cells did not adhere well to the culture plate, and a portion may have been washed away lowering the amount of recoverable proteins. The number of identifications at higher cell counts either marginally increased, were maintained, or decreased depending on the cell type. The notable exception to this was the primary ADPKD and NeoKerat cells where their rate of protein and peptide identifications was slower to decrease and the curve plateau was reached at a higher cell number. However, a plateau curve was achieved by the 5K cell count for all cell types possibly due to detection saturation with the nano-HPLC-MS/MS method using current columns and gradient settings.
Cell Typing with Proteomics.
Protein and peptide identifications rely on statistical scoring of peptides which itself is largely dependent on the other peptides present in additional samples when searched against an online database together. As such, three biological replicates from each cell type at a 5K cell count were pooled and re-searched to enhance protein and peptide identifications. In these searches, averages of 3645 to 4708 proteins and 23,712 to 32,474 peptides were identified (Figure 5A and Table S5). The reproducibility of the six cell type digestions for just the 5K cells/well replicates resulted in CVs of 14.52 to 38.76% for protein abundance, shown in Figure 5B. The principal component analysis (PCA) results (Figure 5C), which are based on protein identification and quantitation analysis, distinguished well all six cell lines. The detected proteins from the different cell lines having label-free quantitation values (normalized MS1 intensity) were analyzed using bioinformatics tools to perform enriched pathway and function analysis. As shown in the enriched pathway heat map (Figure 5D), the clustered pathways of each cell line indicate that these up/downregulated proteins can be accordingly assigned to biological processes with most of the pathways matching to the identity or origin of the cell line. For example, fatty acid modulation and cholesterol biosynthesis are typical biological processes in hepatocytes30. In our study, HepG2 cells are derived from hepatocellular carcinoma, and the pathways listed in the corresponding green box (Figure 5D and Table S5) are related to lipid and cholesterol metabolism, which are very important functions in HepG2 cells. Keratinocytes have the ability to produce the complete repertoire of proinflammatory cytokines and recruit immunocompetent cells31. The secretion of chemokines and cytokines from damaged keratinocytes is essential for the recruitment of monocytes and subsequent downstream events of the inflammatory response32. Migration of monocytes from blood to the injured or infected skin through the endothelium (also called extravasation) is a crucial event in early inflammation33. In the primary neonatal keratinocytes, several pathways are related to acute inflammation, such as regulation of cytokines (e.g., cellular response to interleukin-12) and activation of inflammatory cells (e.g., monocyte extravasation). As expected, these significantly enriched inflammation pathways in proteomics resulted in matches to characteristics of keratinocytes.
MRM Method Development for Protein Quantitation.
In order to validate protocol feasibility for use in biomarker validation and drug compound screening, faster HPLC-MRM methods were developed (Table S4) for selected peptides derived from the protein HSPD1 (P10809, 60 kDa heat shock protein, mitochondrial), which is involved in positive regulation of interleukin-10 production. These two peptides LSDGVAVLK and VGEVIVTK, along with C13 and N15 double-labeled heavy peptides as internal standards, (IS, 200 fmol spiked in each sample) were used to conduct the protein quantitation analysis of the HPLC-MRM method. Unlike the 90 min HPLC-MS analysis established for our discovery proteomics workflow, the MRM analysis was performed on a triple quadrupole mass spectrometer with a run time of 10 min providing a higher throughput (>100 samples/day) that can be leveraged against proteome coverage. The HPLC-MRM quantitation results from three biological replicates of HeLa cells digested from 500 to 10K cells, using the high-throughput preparation platform (Figure S7), showed excellent linearity (R2=0.9753 and R2=0.9569 for LSDGVAVLK and VGEVIVTK, respectively) and CV demonstrating the success of the protein quantitation analysis.
Conclusions
Current available proteomics assays do not go far enough in balancing sensitivity, comprehensiveness, and throughput to make biomarker discovery and drug screening practical, often sacrificing throughput for the benefit of the other considerations. Here, we describe a highly automated high-throughput 384-well plate proteomics sample preparation platform, utilizing mammalian cells directly within the cultured plate for processing of peptides, which could overcome the throughput issue. This platform allows for targeted proteomics-based biomarker discovery and drug screening to finally be conducted in a dose response manner that is practical for the modern lab. Additionally, treatment of the cells with compounds of interest is better achieved when performed directly within the cultured plates. Presently, our HPLC-MS/MS method has a lower limit of 100 – 500 cells for identification of over 2000 proteins in a single run with a variety of cell types. While the protocol has yet to be tested at the single cell level and is currently not applicable to suspension cells, successful implementation of the Rapid Digestion-Trypsin protocol bolstered our confidence in the flexibility of the platform. The entire process from plated cells to clean peptides is complete in ~300 min but increases in throughput could be made with improvements to application strategies and incorporating protocols, which offer advancements in reagent technologies. Application of the method to six different cell types demonstrated its universality in protein digestion and cell typing along with suggesting a broader feasibility for use with varied sample sources including clinical samples, blood, saliva, and other fluids. By coupling the expedient sample preparation platform with the fast HPLC-MRM method, which exhibits an excellent quantitation curve from 500 to 10K cells, an overall advancement in throughput can be achieved. Implementation of multiplexing LC-MS technologies has the potential to provide an additional fourfold increase in throughput equating to >500 samples per day, and subsequent optimization of the gradient could improve throughput even further to nearly 1000 samples per day. In summary, the advancement in high-throughput proteomics sample preparation reported here is a step closer to realizing a fully automated proteomics screening platform that enables an efficient large-scale characterization of diverse biological signatures across a wide array of conditions and parameters.
Supplementary Material
Acknowledgements
We thank Eric Wallgren for creating the heating block conductor, Maya Gosztyla for training on the Bravo, Dr. Ilyas Singec for instrument support, and the University of Maryland School of Medicine for providing the ADPKD renal cells. The TOC graphic, Figure S6, and parts of Figure 1 were created with BioRender.com. This work was supported by the Intramural Research Program of the National Center for Advancing Translational Sciences, National Institutes of Health.
Footnotes
The authors declare no competing financial interest.
Supporting Information
Supplemental methods; full list of reagents, solvents, labware, instruments, and accessories including the vendors and catalog numbers; peptide sequence and MRM information; method development and evaluation using HeLa cells; protein and peptide intraplate reproducibility by CVs; protein reproducibility of intraplate and interuser replicates; peptide reproducibility of intraplate, interpolate, and interuser replicates; reproducibility of peptide abundance between different plates, users, and protocols; flowchart of universal application with six human cell types; HPLC-MRM data for two selected peptides (60 kDa mitochondrial heat shock protein) with C13 and N15 double-labeled internal standards (IS) from 500 to 10K cells (PDF)
Protein identification information and pathway analysis (XLSX)
References
- (1).Li J; Van Vranken JG; Pontano Vaites L; Schweppe DK; Huttlin EL; Etienne C; Nandhikonda P; Viner R; Robitaille AM; Thompson AH; Kuhn K; Pike I; Bomgarden RD; Rogers JC; Gygi SP; Paulo JA Nat. Methods 2020, 17, 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Meier F; Geyer PE; Virreira Winter S; Cox J; Mann M Nat. Methods 2018, 15, 440–448. [DOI] [PubMed] [Google Scholar]
- (3).Amon S; Meier-Abt F; Gillet LC; Dimitrieva S; Theocharides APA; Manz MG; Aebersold R Mol. Cell Proteomics 2019, 18, 1454–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Pappireddi N; Martin L; Wuhr M Chembiochem 2019, 20, 1210–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Li H; Han J; Pan J; Liu T; Parker CE; Borchers CH J. Mass Spectrom. 2017, 52, 319–341. [DOI] [PubMed] [Google Scholar]
- (6).Altelaar AF; Munoz J; Heck AJ Nat. Rev. Genet. 2013, 14, 35–48. [DOI] [PubMed] [Google Scholar]
- (7).Muller T; Kalxdorf M; Longuespee R; Kazdal DN; Stenzinger A; Krijgsveld J Mol. Syst. Biol. 2020, 16, e9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chen Y; Guenther JM; Gin JW; Chan LJG; Costello Z; Ogorzalek TL; Tran HM; Blake-Hedges JM; Keasling JD; Adams PD; Garcia Martin H; Hillson NJ; Petzold CJ J. Proteome Res. 2019, 18, 3752–3761. [DOI] [PubMed] [Google Scholar]
- (9).Dayon L; Nunez Galindo A; Corthesy J; Cominetti O; Kussmann MJ Proteome Res. 2014, 13, 3837–3845. [DOI] [PubMed] [Google Scholar]
- (10).Lee J; Kim H; Sohn A; Yeo I; Kim Y J. Proteome Res. 2019, 18, 2337–2345. [DOI] [PubMed] [Google Scholar]
- (11).Liang Y; Acor H; McCown MA; Nwosu AJ; Boekweg H; Axtell NB; Truong T; Cong Y; Payne SH; Kelly RT Anal. Chem. 2021, 93, 1658–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ruelcke JE; Loo D; Hill MM J. Proteomics 2016, 149, 3–6. [DOI] [PubMed] [Google Scholar]
- (13).Humphrey SJ; Karayel O; James DE; Mann M Nat. Protoc. 2018, 13, 1897–1916. [DOI] [PubMed] [Google Scholar]
- (14).Humphrey SJ; Azimifar SB; Mann M Nat. Biotechnol. 2015, 33, 990–995. [DOI] [PubMed] [Google Scholar]
- (15).Switzar L; van Angeren J; Pinkse M; Kool J; Niessen WM Proteomics 2013, 13, 2980–2983. [DOI] [PubMed] [Google Scholar]
- (16).Fu Q; Kowalski MP; Mastali M; Parker SJ; Sobhani K; van den Broek I; Hunter CL; Van Eyk JE J. Proteome Res. 2018, 17, 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kulak N; Pichler G; Paron I; Nagaraj N; Mann M Nat. Methods 2014, 11, 319–324. [DOI] [PubMed] [Google Scholar]
- (18).Berger ST; Ahmed S; Muntel J; Cuevas Polo N; Bachur R; Kentsis A; Steen J; Steen H Mol. Cel. Proteomics 2015, 14, 2814–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Geyer PE; Kulak NA; Pichler G; Holdt LM; Teupser D; Mann M Cell Syst. 2016, 2, 185–195. [DOI] [PubMed] [Google Scholar]
- (20).Yu Y; Bekele S; Pieper R J. Proteomics 2017, 166, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yu Y; Suh MJ; Sikorski P; Kwon K; Nelson KE; Pieper R Anal. Chem. 2014, 86, 5470–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Vizcaino JA; Csordas A; Del-Toro N; Dianes JA; Griss J; Lavidas I; Mayer G; Perez-Riverol Y; Reisinger F; Ternent T; Xu QW; Wang R; Hermjakob H Nucleic Acids Res. 2016, 44, 11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Subramanian A; Tamayo P; Mootha VK; Mukherjee S; Ebert BL; Gillette MA; Paulovich A; Pomeroy SL; Golub TR; Lander ES; Mesirov JP Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ashburner M; Ball CA; Blake JA; Botstein D; Butler H; Cherry JM; Davis AP; Dolinski K; Dwight SS; Eppig JT; Harris MA; Hill DP; Issel-Tarver L; Kasarskis A; Lewis S; Matese JC; Richardson JE; Ringwald M; Rubin GM; Sherlock G Nat. Genet. 2000, 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Simillion C; Liechti R; Lischer HE; Ioannidis V; Bruggmann R BMC Bioinformatics 2017, 18, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Peach M; Marsh N; Miskiewicz EI; MacPhee DJ Methods Mol. Biol. 2015, 1312, 49–60. [DOI] [PubMed] [Google Scholar]
- (27).Huang HZ; Nichols A; Liu D Anal. Chem. 2009, 81, 1686–1692. [DOI] [PubMed] [Google Scholar]
- (28).Volpe P; Eremenko-Volpe T Eur. J. Biochem. 1970, 12, 195–200. [DOI] [PubMed] [Google Scholar]
- (29).Finka A; Goloubinoff P Cell Stress Chaperones 2013, 18, 591–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Javitt NB FASEB J. 1990, 4, 161–168. [DOI] [PubMed] [Google Scholar]
- (31).Barker JN; Mitra RS; Griffiths CE; Dixit VM; Nickoloff BJ Lancet 1991, 337, 211–214. [DOI] [PubMed] [Google Scholar]
- (32).Ishibashi Y; Sugita T; Nishikawa A FEMS Immunol. Med. Microbiol. 2006, 48, 400–409. [DOI] [PubMed] [Google Scholar]
- (33).Olingy CE; San Emeterio CL; Ogle ME; Krieger JR; Bruce AC; Pfau DD; Jordan BT; Peirce SM; Botchwey EA Sci. Rep. 2017, 7, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.