

Abstract
Cyclin-dependent kinase-like 5 (CDKL5) deficiency disorder (CDD) is a developmental and epileptic encephalopathy with infantile-onset epilepsy. Most individuals with CDD develop refractory epilepsy with multiple seizure types. Management of seizures in CDD remains challenging for clinicians given the highly refractory nature of seizures and the limited number of disease-specific studies that offer a high level of evidence. Epileptic spasms are the most common seizure type in CDD and are more often refractory to standard first-line treatment compared to spasms of other etiologies. In other seizure types, effectiveness of anti-seizure medications is limited and wanes over time. Ketogenic diet and palliative surgical treatments both have mixed results in observational studies. When treating refractory seizures in CDD, we recommend carefully balancing seizure control and treatment related side effects to optimize each individual’s overall quality of life. There have been clinical trials for medications targeting epilepsy in CDD, and additional investigational small molecules, gene therapy, and other disease-modifying therapies are in development for CDD.
1. Introduction
Cyclin-dependent kinase-like 5 (CDKL5) deficiency disorder (CDD) is a developmental and epileptic encephalopathy (DEE) with infantile-onset epilepsy, global developmental delay with subsequent intellectual and motor disabilities, and cortical visual impairment as major features [1-4]. The first clinical report of CDD in 2003 described two girls with infantile spasms, hypsarrhythmia, and global developmental delay due to balanced translocation on Xp22.3 causing a breakage of CDKL5 [5]. Initially identified as the early-onset seizure variant of Rett syndrome, the spectrum and phenotypes of CDD that have emerged proved it to be an independent disorder [3, 4, 6] garnering a specific International Classification of Disease Clinical Modification (ICD-10-CM) code: G40.42. CDD, an X-linked disease, is more prevalent in girls, with a female-to-male ratio of approximately 4:1 [6, 7]. Overall, boys have more severe developmental impairment and lower quality of life compared to girls, though there is variability due to a high rate of somatic mosaicism in males [8-10].
Epilepsy in CDD has been described in three stages (Figure 1), starting with early infantile-onset generalized tonic or tonic-clonic seizures, with variable pharmaco-responsiveness [11, 12]. Median age of seizure onset has been reported from 4 weeks to 2 months; 90% of individuals with CDD present with seizures by 3 months of age [6, 11-13]. Early infantile electroencephalograms (EEG) may be normal or accompanied by a slow background with or without epileptiform activities [11]. In contrast to other severe early infantile-onset epileptic encephalopathies, CDD is rarely associated with a burst-suppression pattern on EEG [11]. In this first phase, a subset of individuals experience a transient period of seizure freedom, known as a honeymoon period [11]. There is wide variability in reports of a honeymoon period, defined as seizure freedom lasting at least 1 month (median duration 4-6 months), described in14% to 100% of individuals (38% when pooled across four studies), with median onset of 20-24 months [11-14]. In the second stage, there is relapse of previous seizures and onset of epileptic spasms [11]. While epileptic spasms are the most common seizure type in CDD, only about half of those with spasms have hypsarrhythmia captured on EEG at any point [12]. In the third and final stage, spasms may continue or evolve to other seizure types, and most individuals with CDD continue to have generalized, or mixed focal and generalized seizures [7, 11]. In children and adults, EEGs have shown multifocal and/or generalized interictal epileptiform discharges, including pseudoperiodic epileptiform discharges, focal or generalized slowing, and at times high amplitude background [7, 11]. Other common types of seizures include generalized tonic, generalized tonic-clonic, generalized myoclonic, and focal seizures, and less commonly clonic, atonic, and absence seizures [6, 12, 15]. Unique sequential seizures with multiple distinct motor and non-motor phases in a single seizure have been reported, including hypermotor-tonic-spasm and similar variations [12, 16], hyperkinetic-spasm [17], tonic-clonic-spasm [18], and tonic-spasm-myoclonic [19]. Three case reports document reflex seizures triggered by diaper change, noise, light, or water immersion [20-22]. Sudden unexpected death in epilepsy (SUDEP) has also been reported, and while the frequency of SUDEP in CDD is not established, the highly refractory nature of epilepsy in CDD puts many individuals with CDD at high risk for SUDEP [23-25].
Figure 1. Three stages of epilepsy in CDD:

During the initial stage, the EEG may be normal, and hypsarrhythmia may not be present at the onset of spasms. In later stages, hypsarrhythmia and generalized slowing with multifocal epileptiform discharges are common. A portion of individuals may experience a honeymoon period. Epileptic encephalopathy and refractory generalized or mixed epilepsy are common in later stages, and a subset of individuals are diagnosed with West syndrome or Lennox-Gastaut Syndrome. Sequential seizures with multiple motor phases are common in all three stages [7, 11, 13].
(Legend: CDD = CDKL5 deficiency disorder. EEG = electroencephalogram.)
Our goal is to discuss a treatment approach in the context of this highly refractory DEE. We synthesize the current observational and clinical trial data on the treatment of epilepsy in CDD, and given the limited amount of studies with high level of evidence, also aim to provide expert opinions from the CDKL5 Centers of Excellence. We conclude with a review of emerging therapies in development.
2. Treatment approach for epilepsy in CDD
In DEEs, epileptiform activity may interfere with normal brain function and negatively impact an individual’s development above what may be attributable to the underlying condition [26]. However, in certain DEEs with underlying genetic causes, including CDD, whether improving seizure control or epileptiform activities leads to better development has been debatable [13, 27, 28]. A multi-center retrospective study in 2016 of individuals with CDD found no improvement in cognitive function despite significant reduction of seizures [27]. While an association between better functional abilities and fewer seizures in individuals with CDD has been observed, whether better function results from improved seizure control or is an epiphenomenon of a less severe genotype remains to be answered [13].
When approaching epilepsy management in CDD, it is critical to consider seizures in the context of each individual’s broader neurodevelopmental disorder and each family’s priorities for quality of life. Seizure control in and of itself is not necessarily the highest priority for optimizing quality of life [9, 29], and we must consider to what extent additional attempts to minimize seizures may benefit individuals with CDD compared to the risk of side effects from medications or non-medication approaches to treatment.
2.1. Impact of seizure burden on quality of life
Families and caregivers of individuals with CDD have identified seizures as the second most burdensome symptom, after global developmental delay, and as an important factor in determining quality of life [29, 30]. Seizures in CDD are prevalent and frequency is high, with approximately two-thirds of individuals having daily seizures and nearly one-third experiencing five or more seizures per day [6, 13]. Frequent seizures can lead to a prolonged recovery period and somnolence, limiting communication, interaction, mobility, and participation in support services [30]. Those with reflex seizures and hypersensitivity to stimuli can experience a negative impact on their behavioral and emotional wellbeing [30]. Factors such as post-ictal sedation, need for rescue medication, and frequency of seizure-related emergency room visits or hospitalization may all play a role in how much seizures negatively influence an individual’s quality of life. Having more than five seizures per day and a high hospital admission rate was associated with lower quality of life scores [9].
2.2. Benefits and side effects of anti-seizure medications (ASMs)
Nearly three-quarters of individuals with CDD take two or more ASMs simultaneously [9, 13]. In a 2021 retrospective report with data on 168 individuals with CDD, the median number of ASMs prescribed per individual over their lifetime was six (range: 0 to 18) [15]. ASMs are the most commonly used medications in CDD. While some do help decrease the seizure burden, others may provide only transient or partial benefit for seizures and cause a negative impact on cognition and motor function [15, 27, 29]. Families have reported that sedating effects from ASMs, causing lethargy and decreased engagement in daily activities, negatively impact quality of life and in certain instances outweigh the positive effects associated with seizure control [9, 29, 30]. Treatment with monotherapy compared to polytherapy with three or more medications was associated with higher quality of life score [9].
2.3. Balance of seizure control and quality of life
A 2021 study assessing quality of life in children with CDD, based on questionnaires completed by 129 families registered with the International CDKL5 Disorder Database, found that functional impairment, including lack of ability to sit, use hands, and communicate, had the greatest adverse impact on quality of life [9]. At the 2019 U.S. Patient-Focused Drug Development Meeting for CDD, caregivers ranked the following in the top-three most desirable targets for new CDD treatment development: developmental milestones (76%, 35/46), improved language abilities (54%, 25/46), and improved social communication (35%, 16/46) [29]. Only 30% (14/46) rated reduced seizures in the top-three priorities [29]. These results highlight aspects to consider when using available treatments for epilepsy.
In the CDKL5 Centers of Excellence, we tend to be cautious about the high burden of medication side effects. Brief seizures that do not impact alertness and interaction may be better tolerated than escalating doses of ASMs that may cause sedation, impair cognitive function, and worsen hypotonia and dysphagia. Withdrawing medications when seizures are active can be challenging but may be necessary to avoid increasing polypharmacy. Simplifying the ASM regimen may minimize complex drug-drug interactions affecting pharmacokinetics and absorption; rarely does simplifying a regimen worsen seizure control more than transiently in our experience. Thus, the art of epilepsy management in CDD, as in other severe refractory epileptic encephalopathies, necessitates a careful balancing act to optimize each individual’s ability to interact with family and others.
3. Treatment for CDD-associated infantile spasms
Epileptic spasms are present at any point in time in 82% of individuals with CDD, and as the initial seizure type in 23% [12]. In addition, more than one-third of individuals with CDD report seizures with multiple phases that include epileptic spasms, often combined with tonic seizures [12]. Epileptic spasms in this population are highly refractory and may continue into childhood and adulthood [7, 12]. In one early case series, only three of eight individuals with CDD and epileptic spasms responded to high dose corticosteroids [11]. Data presented at the 2020 American Neurological Association meeting from three CDKL5 Centers of Excellence showed that response to first-line pharmacologic treatments for infantile spasms in individuals with CDD is low when compared to infantile spasms of other etiologies from the National Infantile Spasms Consortium (NISC) database [31, 32] (Table 1). A sustained response at 3 months was rare in the CDD cohort [31]. Ketogenic diet for refractory spasms initially had similar response rate between both cohorts at approximately 20%, but at 3 months, the response rate in individuals with CDD was 17% (2/12), compared to 38% (15/40) in the comparison group [31]. Future research should focus on an alternative first-line therapy specific for epileptic spasms in CDD.
Table 1.
Treatment response of infantile spasms in CDD is worse for standard first-line medications compared to a non-CDD population from the NISC database [32].
| Treatment | CDD Treated % (N) |
NISC Treated % (N) |
CDD 14-day response % (N) |
NISC 14-day response % (N) |
CDD 1-month response % (N) |
NISC 1-month response % (N) |
CDD 3-month response % (N) |
NISC 3-month response % (N) |
|---|---|---|---|---|---|---|---|---|
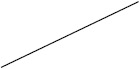
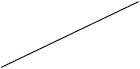
| ACTH | 38 (17/45) | 60 (225/376) | 24 (4/17) | 63 (138/219) |
|
|
0 (0/8) | 59 (128/217) |
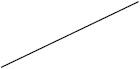
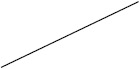
| Prednisolone | 40 (17/43) | 30 (111/376) | 12 (2/17) | 53 (51/97) |
|
|
0 (0/6) | 54 (45/84) |
| Vigabatrin | 67 (30/45) | 53 (197/375) | 27 (7/26) | 42 (78/184) |
|
|
11 (2/19) | 42 (78/184) |
| Ketogenic diet | 53 (24/45) | 14 (51/376) |
|
|
20 (4/20) | 19 (5/27) | 17 (2/12) | 38 (15/40) |
Individuals included in the analysis had infantile spasms onset between 2 months to 2 years of age. Exclusion criteria were tuberous sclerosis complex, trisomy 21, and unknown etiology with normal development. The CDD cohort showed poorer response to all first-line treatments. Early response of the CDD cohort to ketogenic diet for refractory spasms was similar to that of the non-CDD group, but response rate was lower in the CDD cohort at 3 months [31]*. (Legend: ACTH = adrenocorticotropic hormone. CDD = CDKL5 deficiency disorder cohort. NISC = non-CDD cohort from National Infantile Spasms Consortium. * = presented at the 2020 annual American Neurological Association meeting.)
4. Anti-seizure medications in CDKL5 Deficiency Disorder
4.1. Observational studies of currently available medications
There are four large studies that investigated benefits of different ASMs approved by the U.S. Food and Drug Administration (FDA) [14, 15, 27, 33] (Table 2). Three of them are multi-center retrospective observational studies with effectiveness defined as a greater than 50% seizure reduction at variable time points [14, 15, 27], and the other is a questionnaire based study that asked caregivers what they considered to be “the most effective medication” [33]. Levetiracetam was one of the top-five most used ASMs in all studies, and topiramate, phenobarbital, valproic acid, and vigabatrin were among the top-five most used ASMs in three of the four studies [14, 15, 27, 33]. Clobazam, lamotrigine, valproic acid, and vigabatrin were each in the top-five most effective ASMs in at least three of the four studies, none of which differentiated responses by seizure types [14, 15, 27, 33]. This excludes ASMs used in fewer than ten individuals per study, as well as adrenocorticotropin hormone (ACTH) and corticosteroid which are discussed separately. Two studies looked into percentage of responders to ASMs longitudinally and demonstrated a decline in effectiveness of ASMs over the course of 2 to 9 months [15, 27].
Table 2.
Summary of ASMs used for CDD-related epilepsy based on data from four large observational studies: Clobazam, lamotrigine, valproic acid, and vigabatrin were more commonly in the top-five most effective ASMs, but response rate declines over time.
| Study Type (Level of Evidence) |
Seizure Reduction Criteria |
ACTH / Cortico- steroid % (N) * |
CBZ / OXC % (N) |
CLB % (N) |
LTG % (N) |
LVT % (N) |
PB % (N) |
RFM % (N) |
TPM % (N) |
VGB % (N) |
VPA % (N) |
ZNS %(N) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Retros Obs (4) [27] | >50% at 3 months | 19 (5/26) | 10 (3/29) | 24 (4/17) | 22 (5/23) | 16 (5/31) | 8 (2/26) | 8 (1/13) | 16 (5/31) | 32 (8/25) | 21 (7/34) | 18 (2/11) |
| >50% at 12 months | 0 (0/26) | 7 (2/29) | 0 (0/17) | 9 (2/23) | 0 (0/31) | 8 (2/26) | 0 (0/13) | 3 (1/31) | 4 (1/25) | 9 (3/34) | 0 (0/18) | |
| Caregiver Survey (4) [33] | “Effective” not defined | 11 (1/9) | 0 (0/7) | 43 (6/14) | 15 (2/13) | 11 (3/27) | 0 (0/21) | 0 (0/1) | 7 (1/15) | 52 (12/23) | 19 (5/27) | 29 (2/7) |
| Retros Obs (4) [14] | ≥50% | 41 (9/22)** | 0 (0/21) | 0 (0/22) | 5 (1/21) | 0 (0/21) | 0 (0/19) | 0 (0/5) | 0 (0/20) | 50 (1/2) | 7 (2/28) | 0 (0/23) |
| Retros Obs (4) [15] | ≥50% for 2 weeks | 38 (15/40) | 19 (5/26) | 48 (12/25) | 40 (6/15) | 14 (8/56) | 37 (13/35) | 47 (7/15) | 26 (10/38) | 56 (15/27) | 36 (9/25) | 0 (0/13) |
| ≥50% for 3 months | 0 (0/40) | 8 (2/26) | 36 (9/25) | 13 (2/15) | 5 (3/56) | 9 (3/35) | 27 (4/15) | 13 (5/38) | 33 (9/27) | 28 (7/25) | 0 (0/13) |
Levetiracetam was commonly used, but clobazam, lamotrigine, vigabatrin, and valproic acid were most consistently effective for seizure reduction. Response rate to ASMs declined over time when reported longitudinally. Bold text indicates top-five most effective medications, excluding ACTH and corticosteroid, each with minimum ten or more sample size, from each study. Level of evidence is sourced from Oxford Centre for Evidence-Based Medicine (http://www.cebm.net/index.aspx?o=5653).
(Legend: ACTH = adrenocorticotropic hormone. ASMs = Anti-seizure medications. CBZ/OXC = carbamazepine or oxcarbazepine. CDD = CDKL5 deficiency disorder. CLB = clobazam. LTG = lamotrigine. LVT = levetiracetam. PB = phenobarbital. Retros Obs = retrospective observational study. RFN = rufinamide. TPM = topiramate. VGB = vigabatrin. VPA = valproic acid. ZNS = zonisamide.
= Response was not differentiated by seizure types (spasms versus other seizure types).
= ACTH effects were reported to be short-lived.)
One small case series reported a more than 90% decrease in seizure frequency in five individuals with a combined use of vigabatrin and zonisamide [18]. There is mixed data on use of sodium channel blockers, ranging from seizure exacerbation to seizure freedom lasting 3 years or longer [14, 15, 27, 29, 34]. In one multi-center retrospective observational study in 2021, six of nineteen individuals with CDD reported a greater than 50% reduction in seizure frequency at 6 months with sodium channel blockers [34]. Caution may be advised in using sodium channel blockers in infants, however, given their potential risk of inducing infantile spasms in high-risk populations including those with CDD [35]. Levetiracetam and cannabidiol have also been anecdotally reported to exacerbate the seizure burden in CDD in certain situations [14, 15, 27, 29, 36].
4.2. Cannabidiol
Following randomized double-blinded placebo-controlled trials, Epidiolex® (purified cannabidiol) was approved by the FDA in 2018 for Dravet syndrome and Lennox-Gastaut syndrome [37, 38]. Use of Epidiolex® and other non-FDA approved cannabidiol derivatives has gained popularity in CDD, with 20% of individuals reporting current use and 36% reporting use at some point in their treatment course [9, 15]. The perceived benefit of cannabidiol was also significant, with 54% (38/70) of caregivers reporting sustained benefit in seizure control and another 16% (11/70) reporting some temporary benefit, according to an analysis of caregivers’ questionnaires [36]. When used as an adjuvant treatment to clobazam, Epidiolex® reportedly granted one individual with CDD total seizure freedom at 8 weeks [39]. Other observational and retrospective studies, however, show variable results (Table 3). In open label studies of Epidiolex® including individuals with CDD [40, 41], disease-specific long-term outcomes were variable with the primary long-term extension study reporting only one of five individuals with CDD who remained in the study having a greater than 50% reduction in sustained motor seizures at 24 months [41]. An overlapping study of individuals with four genetic conditions reported 53% (9/17) individuals with CDD had a ≥50% reduction in motor seizures at 48 weeks [42]. A 2021 retrospective report from the CDKL5 Centers of Excellence that evaluated seizure response to Epidiolex® in individuals with CDD reported a ≥50% reduction in at least one seizure type in 29% (4/14) and 21% (3/14) at 2 weeks and 3 months, respectively [15]. Currently, there is not enough evidence for Epidiolex® to conclude whether it is more effective in certain seizure types.
Table 3.
Summary of observational and clinical trial data for cannabidiol and investigational medications for CDD-related epilepsy: cannabidiol results were highly variable, ganaxolone showed benefit in a phase III trial and is now FDA approved, and fenfluramine showed promise in a phase II trial.
| Treatment | Study Type (Level of Evidence) |
Seizure Outcome in CDD |
|---|---|---|
| Cannabidiol | Caregiver Survey (4) [36] |
"Improved" in 54% (38/70) “Temporary improved” in 16% (11/70) |
| Cannabidiol (Epidiolex®) | Open-label Pros (4)* in refractory epilepsy [41] |
>50% reduction in motor** seizures in 20% (1/5) at 24 months |
| Cannabidiol (Epidiolex®) | Open-label Pros (4) in genetic syndromes [42] | 41% (median, n=11) reduction of convulsive*** seizures at week 12 60% (median, n=10) reduction of convulsive*** seizures at week 48 ≥50% reduction of convulsive*** seizures in 41% (7/17) at week 12 ≥50% reduction of convulsive*** seizures in 53% (9/17) at week 48 |
| Cannabidiol (Epidiolex®) | Retros Obs (4) [15] | ≥50% reduction in at least one seizure type for 2 weeks in 29% (4/14) ≥50% reduction in at least one seizure type for 3 months in 21% (3/14) |
| Ganaxolone | RCT (2) (Phase III, 2020) [46] |
32% (median, n=50) reduction in motor seizures at 17 weeks (versus 4% in placebo, p=0.002) |
| Fenfluramine | Open-label Pros (4) (Phase II, 2021) [67] | 90% (median, n=5) reduction in generalized tonic-clonic seizures 55% (medina, n=2) reduction in tonic seizures 71% (n=1) reduction in myoclonic seizures |
| Soticlestat | Open-label Pros (4) (Phase II, 2021) [74] **** | 14% (median, n=12) reduction in motor seizures |
| Ataluren | RCT (2) (Phase III, 2021) [75] | No improvement at 12 weeks |
Data on cannabidiol were mixed, possibly due to different outcome measures used in different studies. Ganaxolone was approved by the FDA for CDD-related epilepsy in individuals 2 years or older in March 2022. Fenfluramine phase II results were promising, and phase III trial is in process. Level of evidence is sourced from Oxford Centre for Evidence-Based Medicine (http://www.cebm.net/index.aspx?o=5653).
(Legend: CDD = CDKL5 deficiency disorder. FDA = the U.S. Food and Drug Administration. Open-label Pros = open-label prospective study. Retros Obs = retrospective observational study. RTC = randomized, placebo-controlled, double-blinded study.
= long-term follow-up study from a short-term efficacy analysis of Epidiolex® in intractable childhood onset epilepsy including eight individuals with CDD [40].
= motor seizures included tonic, clonic, atonic, and myoclonic-absence seizures.
= convulsive seizures were defined as countable motor seizures lasting > 3 seconds.
= presented at the 2021 annual American Academy of Neurology meeting.)
4.3. Ganaxolone
At the time of this writing, ganaxolone is the only ASM approved specifically for CDD by the FDA. Ganaxolone is a synthetic analog of endogenous allopregnanolone, a progesterone-derived neurosteroid [43]. Neurosteroids are allosteric modulators of gamma-aminobutyric acid (GABA)-A receptors with a binding site distinct from that of benzodiazepines [43]. Both allopregnanolone and ganaloxone showed early promising results for the treatment of status epilepticus and focal seizures, but ultimately failed to show efficacy [43-45]. In CDD, the results of a randomized, placebo-controlled, double-blinded phase III trial (NCT03572933) showed a statistically significant reduction in bilateral tonic, generalized tonic-clonic, bilateral clonic, focal to bilateral tonic-clonic, and atonic seizures with 17-week use of ganaxolone (median 32% vs 4%) [46]. The percentage of individuals with a ≥50% reduction in seizures was also higher for ganaxolone (25% versus 10%), although this did not reach statistical significance [46]. Clinical global impression scales also tended to be better with ganaxolone without reaching statistical significance [46]. Adverse events and serious adverse events occurred at a similar rate in the treatment and placebo groups (88% versus 86% for adverse events, and 12% versus 10% for serious adverse events), with common side effects including somnolence, fever, and upper respiratory tract infection [46]. An expanded access program for ganaxolone in CDD (NCT04678479) was subsequently established for compassionate use. Ganaxolone was approved by the FDA for CDD-related epilepsy in individuals 2 years or older in March 2022 and is expected to be available in the second half of 2022.
4.4. Immune-modulating therapies
There is a lack of clear evidence to recommend immune-modulating or anti-inflammatory therapy in pediatric epilepsy, with certain exceptions such as infantile spasms and autoimmune-associated epilepsy [47-49]. Rare case reports describe successful reduction in seizures with corticosteroid in genetic epilepsy including in Angelman syndrome [50]. In CDD, three of the four large aforementioned studies in section 4.1 reported either ACTH or corticosteroid as among the top-five most used ASMs [14, 15, 27, 33]. These reports, however, did not differentiate whether the observed effects were on epileptic spasms or on other seizure types, and details on duration or titration schedule of these therapies are limited [14, 15, 27, 33]. Regardless, benefits of ACTH or corticosteroid on reducing seizure burden appeared to be transient at best [14, 15, 27, 33]. Two observational studies with longitudinal data both showed a decline in response rate to zero over 2 to 9 months after the initial response rate of 19 and 38% [15, 27]. In one of the two studies, however, the authors noted that discontinuation of medication, as is typical for corticosteroid or ACTH after a short course, would place the medication in the non-responder category [27]. The third observational study specifically reported that the response to ACTH was transient, and in the questionnaire-based study, merely 11% (1/9) of parents and caregivers considered corticosteroid as “the most effective medication” [14, 33]. Only in one small case series was high-dose prednisolone noted to be particularly effective, with a greater than 75% reduction of seizures in five of eight individuals with CDD who were diagnosed with either West syndrome or Lennox-Gastaut syndrome [51].
Literature on the use of intravenous immunoglobulin (IVIG) is limited to one case series presented at the 2017 annual American Epilepsy Society meeting [52]. Of the three individuals with CDD who were treated with corticosteroid followed by IVIG, all three had a reported reduction in both seizure frequency and duration with corticosteroid, and one became free of motor seizures during the eight month course of IVIG [52]. Overall, while ACTH, corticosteroid, or IVIG could be considered in CDD in select situations at the clinician’s judgement, more evidence is needed to decide on their indication, dose, and duration of therapy.
5. Diet Therapy
Ketogenic diet provides a non-pharmacologic approach to seizure management. Studies have estimated current use of ketogenic diet in approximately one-fifth to one-third of individuals with CDD, with more than half of individuals with CDD reporting use of ketogenic diet at some point [9, 15, 30, 53]. Ketogenic diet is often initiated after 1 year of age, with one study showing median age of diet initiation of 2 years and another study reporting median time between seizure onset and diet initiation as 4 years [15, 53]. Literature on benefits of ketogenic diet in CDD shows mixed results (Table 4). Three large studies (total N = 171) have reported at least 50% response rate of either a ≥50% reduction in seizure frequency or perceived benefits from caregivers, along with variable benefits on behavior, cognition, and quality of life [15, 33, 53]. On the other hand, five smaller studies (total N = 47) showed poorer outcomes [14, 27, 54-56]. Overall, more studies on ketogenic diet, including its use in infants and longitudinal follow-up, are warranted.
Table 4.
Observational studies on effectiveness of ketogenic diet in CDD demonstrate mixed results but more benefits in a larger case series and caregiver surveys.
| Study Type (Level of Evidence) |
Outcome |
|---|---|
| Caregiver Survey (4) [53] | Positive perceived effects on seizure characteristics in 59% (61/104) Positive behavioral changes in 25% (26/104) Worsening seizures in 8% (8/104) |
| Retros Obs (4) [15] | ≥50% reduction in seizure frequency in 50% (22/44) Subjective cognitive benefit in 25% (11/44) Worsening seizures in 11% (6/54) |
| Caregiver Survey (4) [33] | “Effective” for seizure control in 52% (12/23) Fewer anti-seizure medications in 36% (5/14) Fewer rescue medications in 33% (6/18) Improved quality of life in 53% (10/19) |
| Retros Obs (4) [27] | ≥50% reduction of seizure in 17% (2/12) at 3 months ≥50% reduction of seizure in 8% (1/12) at 12 months |
| Retros Obs (4) [55] | “Effective” for seizure control in 10% (1/10) |
| Retros Obs (4) [54] | ≥90% reduction of seizure in 0% (0/10) |
| Retros Obs (4) [14] | ≥50% reduction of seizure in 10% (1/10) |
| Retros Obs (4) [56] | “Significant reduction” in seizure frequency and duration in 20% (1/5) |
Results are mixed, but larger studies support the use of ketogenic diet for seizure control. Level of evidence is sourced from Oxford Centre for Evidence-Based Medicine (http://www.cebm.net/index.aspx?o=5653).
(Legend: CDD: CDKL5 deficiency disorder, Retros Obs = retrospective observational study.)
6. Surgical Treatments
Data on surgical intervention in CDD are sparse. Resective surgeries in genetic epilepsy are more effective in those with focal epilepsies related to variants in the phosphoinositide 3-kinase (PI3K) pathway and Gap Activity Toward Rag 1 (GATOR1) complexes [57]. Focal resective surgeries or hemispherectomies in CDD would be expected to be palliative at best and are, in general, not recommended. One individual with CDD had persistent seizures after a functional hemispherectomy performed prior to the genetic diagnosis [15].
Palliative surgeries including vagus nerve stimulator (VNS) and corpus callosotomy have been performed, likely with a bias towards those with more severe epilepsy. A study on quality of life in individuals with CDD showed that a quarter of individuals with CDD had a VNS placed [9]. Three studies (total N = 64, not accounting for possible overlap) reported overall reduction in seizure burden by 63-75% with VNS in individuals with CDD [15, 33, 58] (Table 5). Benefits of VNS in reducing seizure burden related to CDD have also been reported individually [1, 59-61]. On the other hand, the 2021 multi-center retrospective study from Japan reported three individuals with CDD with VNS implantation and another three individuals with CDD who underwent corpus callosotomy, none of whom achieved a ≥50% reduction in seizure frequency [14]. There are reports of at least short-term benefits of corpus callosotomy for seizure control in six individuals, including a greater than 50% reduction in at least four individuals, one of whom also had a VNS [15, 51, 55].
Table 5.
VNS demonstrates improvements in three of four retrospective case series and caregiver surveys for CDD-related epilepsy
| Study Type (Level of Evidence) |
Outcome |
|---|---|
| Caregiver Survey (4) [58] | Improvement in seizure activity in 69% (25/36) - reduction in duration in 72% (18/25), - reduction in frequency in 68% (17/25), - reduction in intensity in 60% (15/25) |
| Retros Obs (4) [15] | Improvement in seizure activity in 63% (10/16) - reduction in seizure duration in 12% (2/16) - reduction in seizure frequency in 43% (7/16) - reduction in seizure intensity in 18% (3/16) |
| Caregiver Survey (4) [33] | “Effective” for seizure control in 75% (9/12) Fewer anti-seizure medications in 17% (2/12) after 1 year Fewer rescue medications in 17% (2/12) after 1 year Improved quality of life in 75% (9/12) after 1 year |
| Retros Obs (4) [14] | ≥50% reduction of seizure in 0% (0/3) |
VNS may be effective for seizure reduction in select refractory cases. Level of evidence is sourced from Oxford Centre for Evidence-Based Medicine (http://www.cebm.net/index.aspx?o=5653).
(Legend: CDD = CDKL5 deficiency disorder, Retros Obs = retrospective observational study, VNS = vagal nerve stimulator.)
The use of deep brain stimulation or responsive neurostimulation for individuals with CDD has not been reported thus far. A preclinical study conducted in Cdkl5+/− mice found that chronic forniceal deep brain stimulation rescued hippocampal memory deficits, restored synaptic plasticity, and relieved feedforward inhibition, raising the possibility that deep brain stimulation could be used in the future to enhance the function of neural circuitry in individuals with CDD [62].
7. Investigational Drugs
7.1. Fenfluramine
Fenfluramine is a serotonergic agent originally developed as an appetite suppressant in combination with phentermine but was withdrawn from the market in 1997 due to complications related to valvular heart disease [63]. Since then, fenfluramine has reemerged as an effective ASM for its significant reduction of convulsive seizures in Dravet syndrome [64] as well as drop seizures in Lennox-Gastaut syndrome, presented at the 2020 annual American Epilepsy Society meeting [65]. The FDA subsequently approved fenfluramine under a risk evaluation and mitigation strategy for Dravet syndrome in June 2020 and for Lennox-Gastaut syndrome in March 2022. In CDD, a phase II open-label trial investigated the benefits of fenfluramine for convulsive seizures, defined as tonic-clonic, tonic, atonic, clonic, or focal motor seizures lasting ≥3 seconds [66]. In six individuals with four or more convulsive seizures per month, a 14-week course of fenfluramine showed 90% median reduction (n=5) for tonic-clonic seizures, 55% median reduction (n=2) for tonic seizures, and 71% reduction (n=1) for myoclonic seizures [66]. Clinically meaningful improvement and improvement in quality of life were reported as secondary outcome measures [66]. Echocardiograms performed at 6 weeks did not reveal signs of valvular heart disease [66]. Safety profile related to this potential adverse event has been also monitored in trials in Dravet syndrome, showing only trace, also known as physiologic, mitral or aortic regurgitation [64, 67, 68]. The phase III trial of fenfluramine for Lennox-Gastaut syndrome, presented at the 2020 annual American Epilepsy Society meeting, also reported no cases of valvular heart disease [65]. Overall, the degree of seizure reduction in these data is notable, but the efficacy and safety profile of this treatment in CDD will be best established through the phase III double-blinded, placebo-controlled, clinical trial in process (NCT05064878).
7.2. Soticlestat
Soticlestat (TAK-935 / OV-935), a cholesterol 24-hydroxylase inhibitor largely expressed in neurons, converts cholesterol to 24(S)-hydroxycholesterol for excretion [69]. 24(S)-hydroxycholesterol has various functions including being a direct positive allosteric modulator of N-methyl-D-aspartate (NMDA) signaling [69-71]. Thus, soticlestat provides a novel mechanism of action as a potential ASM. A phase 1b/2a randomized double-blinded, placebo-controlled trial followed by an open-label study showed a 36% reduction in bilateral motor seizure frequency in individuals with DEE at the end of the open-label phase [72]. Common adverse events included dysarthria, lethargy, upper respiratory infection, fatigue, and headache; treatment-related emergent or serious adverse events included gait disturbance, lethargy, asthenia, and seizure clusters [72]. A subsequent phase II open-label study in individuals with CDD and 15q duplication syndrome, presented at the 2021 annual American Academy of Neurology meeting, showed a 14% decrease in median (n=12) seizure frequency in CDD [73]. Eleven of twelve families and 8 of 12 clinicians reported improvement in the global impression of change scale, a secondary outcome measure [73]. No treatment-related serious adverse events occurred [73]. An open-label extension phase II trial to study long-term safety and tolerability (NCT03635073) is ongoing, and additional trials in other epileptic encephalopathies are also in recruitment (NCT04940624 and NCT04938427).
8. Disease-modifying and other targeted therapies in development
Disease-modifying therapies for CDD are in development. A randomized placebo-controlled phase II trial of ataluren, a drug that promotes a premature stop codon read-through, failed to show improvement in seizure frequency or quality of life in 15 individuals with CDD or Dravet syndrome [74]. Protein replacement therapy and virus-mediated gene transfer have been investigated in animal models of CDD, with promising results [75, 76]. CDKL5 gene delivery via an AAV vector demonstrated partial improvement in subsets of testing parameters representative of autistic behavior and motor coordination in Cdkl5 knockout (KO) mice [75]. Systemic infusion of a TAT-CDKL5 fusion protein rescued various neuroanatomical and behavioral defects in Cdkl5-null mice, including breathing pattern and visual responses, and intracerebrocortical infusion of TAT-CDKL5 restored hippocampal development, hippocampus-dependent memory, and breathing pattern [76]. In a conditional rescue mouse model, restoration of Cdkl5 after the early stages of brain development ameliorated a majority of CDD-related loss-of-function behavioral impairments (including hyperactivity, anxiety-related phenotypes, autistic-like behaviors, motor impairments, and learning and memory deficits) and aberrant NMDA receptor signaling, suggesting the potential for a broad therapeutic window for disease reversal in CDD [77].
Other possible pharmacologic approaches for treatment of CDD have been studied in mouse models. Sertraline, a selective serotonin reuptake inhibitor, has been studied in Cdkl5 KO female mice with promising effects, including normalization of locomotion, stereotypic and autistic-like features, and spatial memory [78]. Memantine, a non-competitive NMDA receptor antagonist, significantly mitigated behavioral deficits in Dlx-cKO mice, a model with selective ablation of CDKL5 expression in forebrain GABAergic neurons, and selectively ameliorated autistic-like features in a novel mouse CDD model bearing a known pathogenic nonsense variant, p.R59X [79]. Treatment with a combined glycogen synthase kinase 3-beta (GSK3-beta) / histone deacetylase (HDAC) inhibitor restored synapse development, neuronal survival, and microglia over-activation, and improved motor and cognitive abilities of Cdkl5 KO mice [80]. Another study suggested that neuroinflammatory processes contribute to the pathogenesis of CDD and showed that treatment with luteolin, a natural anti-inflammatory flavonoid, recovered microglia alterations and neuronal survival and maturation in Cdkl5 KO mice [81].
To date, CDD-related phenotypes in mice are mostly reproducible, including several cardinal elements of the human disease such as learning and memory impairments, motor deficits, and autistic-like behaviors [77]. However, although animal models of CDD have shown evidence of hyperexcitability, spontaneous seizures have been exhibited only in aged female heterozygous Cdkl5 mutant mice as epileptic spasms [82]. Additional research will thus be needed prior to considering a human trial phase for any of these compounds, although each sheds additional light on the underlying biology of the disorder.
9. Limitations and future direction
The majority of available literature related to epilepsy management in CDD are retrospective studies, surveys, case series, and case reports. These lack systematically collected quantifiable objective measurements and a pre-treatment period to establish baseline seizure burden for comparison post-treatment. In addition, while it is important to select ASMs based on seizure types, observational studies in CDD have largely not been able to differentiate treatment responses by specific seizure types. This limitation poses a particular challenge in interpreting treatment data of ACTH and corticosteroid for their use in epileptic spasms and other seizure types. Thus, caution is needed when interpreting these data for clinical use.
As emphasized throughout the manuscript, seizure reduction is only a part of many factors that contribute to the overall quality of life in individuals with CDD. Yet, most clinical trials in epilepsy rely on reduction in seizure count as their primary outcome measure, and there is a need for other objective quantifiable measures that reflect other aspects of quality of life in individuals with CDD. The ongoing clinical trial readiness project for CDD, funded through the National Institute of Neurological Disorders and Stroke (NINDS, 1U01NS114312-01A1), as well as the observational study in CDKL5 deficiency disorder (CANDID) are important steps toward defining these outcome measures for CDD. The FDA approval for ganaxolone as a CDD specific medication highlights the enthusiasm and hope in the CDKL5 community for better therapies. Additional clinical trials in small molecules, gene therapy, and other disease-modifying therapies will be expected in the coming years, which we anticipate will change the treatment approach to CDD.
Key points.
Anti-seizure medications tend to lose effectiveness with time in individuals with CDD.
Balance treatment side effects with caution in CDD, focusing on quality of life.
Clinical trials or expanded access programs are ongoing for CDD, and disease modifying therapies are in development.
Funding
This review was supported by the National Institute of Neurologic Disorders and Stroke (K23 NS107646-04, principal investigator HEO) and by philanthropic donation for CDD research to support WH’s fellowship in Epilepsy Genetics.
Footnotes
Conflicts of interest/Competing interests
WH declares that he has no conflicts of interest.
IH declares that she has no conflicts of interest.
EPK is on the scientific advisory board of Marinus Pharmaceuticals.
JLW has funding from the International Foundation for CDKL5 Research and is a site principal investigator for Marinus Pharmaceuticals.
SD has consulted for Upsher-Smith, Biomarin, Neurogene, Marinus, Tysha, and Ovid Therapeutics. He has funding from the National Institutes of Health, the International Foundation for CDKL5 Research, Project 8P and Mila's Miracle Foundation. He also serves on the advisory board for the non-profit foundations SLC6A1 Connect, Ring14 USA and FamilieSCN2A.
EDM is a site principal investigator for trials for Stoke Therapeutics, GW Pharmaceuticals, Zogenix Pharmaceuticals, Acadia Pharmaceuticals, and Marinus Pharmaceuticals. He was previously consultant for Cilpa Pharmaceuticals and Stoke Therapeutics. He has research support from the National Institutes of Health, the Eagles Autism Foundation, the Penn Orphan Disease center, RettSyndrome.org, the International Foundation for CDKL5 Research, and the LouLou Foundation.
HEO reports consulting for Marinus pharmaceuticals, Ovid Therapeutics, Zogenix, and Takeda Pharmaceuticals related to CDKL5 deficiency disorder. She has clinical research funding from the International Foundation for CDKL5 Research and from the LouLou Foundation/Orphan Disease Center at the University of Pennsylvania.
References
- 1.Tao J, Van Esch H, Hagedorn-Greiwe M, Hoffmann K, Moser B, Raynaud M, et al. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet. 2004. Dec;75(6):1149–54. DOI: 10.1086/426460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weaving LS, Christodoulou J, Williamson SL, Friend KL, McKenzie OL, Archer H, et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet. 2004. Dec;75(6):1079–93. DOI: 10.1086/426462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bahi-Buisson N, Nectoux J, Rosas-Vargas H, Milh M, Boddaert N, Girard B, et al. Key clinical features to identify girls with CDKL5 mutations. Brain. 2008. Oct;131(Pt 10):2647–61. DOI: 10.1093/brain/awn197 [DOI] [PubMed] [Google Scholar]
- 4.Fehr S, Wilson M, Downs J, Williams S, Murgia A, Sartori S, et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur J Hum Genet. 2013. Mar;21(3):266–73. DOI: 10.1038/ejhg.2012.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalscheuer VM, Tao J, Donnelly A, Hollway G, Schwinger E, Kubart S, et al. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am J Hum Genet. 2003. Jun;72(6):1401–11. DOI: 10.1086/375538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cutri-French C, Armstrong D, Saby J, Gorman C, Lane J, Fu C, et al. Comparison of Core Features in Four Developmental Encephalopathies in the Rett Natural History Study. Ann Neurol. 2020. Aug;88(2):396–406. DOI: 10.1002/ana.25797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson HE, Demarest ST, Pestana-Knight EM, Swanson LC, Iqbal S, Lal D, et al. Cyclin-Dependent Kinase-Like 5 Deficiency Disorder: Clinical Review. Pediatr Neurol. 2019. Aug;97:18–25. DOI: 10.1016/j.pediatrneurol.2019.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fehr S, Leonard H, Ho G, Williams S, de Klerk N, Forbes D, et al. There is variability in the attainment of developmental milestones in the CDKL5 disorder. Journal of Neurodevelopmental Disorders. 2015. 2015/January/05;7(1):2. DOI: 10.1186/1866-1955-7-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leonard H, Junaid M, Wong K, Demarest S, Downs J. Exploring quality of life in individuals with a severe developmental and epileptic encephalopathy, CDKL5 Deficiency Disorder. Epilepsy Res. 2021. Jan;169:106521. DOI: 10.1016/j.eplepsyres.2020.106521 [DOI] [PubMed] [Google Scholar]
- 10.Stosser MB, Lindy AS, Butler E, Retterer K, Piccirillo-Stosser CM, Richard G, et al. High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genetics in Medicine. 2018. 2018/April/01;20(4):403–10. DOI: 10.1038/gim.2017.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bahi-Buisson N, Kaminska A, Boddaert N, Rio M, Afenjar A, Gerard M, et al. The three stages of epilepsy in patients with CDKL5 mutations. Epilepsia. 2008. Jun;49(6):1027–37. DOI: 10.1111/j.1528-1167.2007.01520.x [DOI] [PubMed] [Google Scholar]
- 12.Demarest ST, Olson HE, Moss A, Pestana-Knight E, Zhang X, Parikh S, et al. CDKL5 deficiency disorder: Relationship between genotype, epilepsy, cortical visual impairment, and development. Epilepsia. 2019;60(8):1733–42. DOI: 10.1111/epi.16285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fehr S, Wong K, Chin R, Williams S, de Klerk N, Forbes D, et al. Seizure variables and their relationship to genotype and functional abilities in the CDKL5 disorder. Neurology. 2016;87(21):2206–13. DOI: 10.1212/wnl.0000000000003352 [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi Y, Tohyama J, Takahashi Y, Goto T, Haginoya K, Inoue T, et al. Clinical manifestations and epilepsy treatment in Japanese patients with pathogenic CDKL5 variants. Brain Dev. 2021. Apr;43(4):505–14. DOI: 10.1016/j.braindev.2020.12.006 [DOI] [PubMed] [Google Scholar]
- 15.Olson HE, Daniels CI, Haviland I, Swanson LC, Greene CA, Denny AMM, et al. Current neurologic treatment and emerging therapies in CDKL5 deficiency disorder. Journal of Neurodevelopmental Disorders. 2021. 2021/September/16;13(1):40. DOI: 10.1186/s11689-021-09384-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein KM, Yendle SC, Harvey AS, Antony JH, Wallace G, Bienvenu T, et al. A distinctive seizure type in patients with CDKL5 mutations: Hypermotor-tonic-spasms sequence. Neurology. 2011;76(16):1436–8. DOI: 10.1212/WNL.0b013e3182166e58 [DOI] [PubMed] [Google Scholar]
- 17.Grosso S, Brogna A, Bazzotti S, Renieri A, Morgese G, Balestri P. Seizures and electroencephalographic findings in CDKL5 mutations: case report and review. Brain Dev. 2007. May;29(4):239–42. DOI: 10.1016/j.braindev.2006.09.001 [DOI] [PubMed] [Google Scholar]
- 18.Melikishvili G, Epitashvili N, Tabatadze N, Chikvinidze G, Dulac O, Bienvenu T, et al. New insights in phenomenology and treatment of epilepsy in CDKL5 encephalopathy. Epilepsy Behav. 2019. May;94:308–11. DOI: 10.1016/j.yebeh.2019.02.013 [DOI] [PubMed] [Google Scholar]
- 19.Melani F, Mei D, Pisano T, Savasta S, Franzoni E, Ferrari AR, et al. CDKL5 gene-related epileptic encephalopathy: electroclinical findings in the first year of life. Dev Med Child Neurol. 2011. Apr;53(4):354–60. DOI: 10.1111/j.1469-8749.2010.03889.x [DOI] [PubMed] [Google Scholar]
- 20.Peikes T, Hartley JN, Mhanni AA, Greenberg CR, Appendino JP. Reflex Seizures in a Patient with CDKL5 Deficiency Disorder. Can J Neurol Sci. 2019. Jul;46(4):482–5. DOI: 10.1017/cjn.2019.29 [DOI] [PubMed] [Google Scholar]
- 21.Solazzi R, Fiorini E, Parrini E, Darra F, Dalla Bernardina B, Cantalupo G. Diaper changing-induced reflex seizures in CDKL5-related epilepsy. Epileptic Disord. 2018. Oct 1;20(5):428–33. DOI: 10.1684/epd.2018.0999 [DOI] [PubMed] [Google Scholar]
- 22.Saitsu H, Osaka H, Nishiyama K, Tsurusaki Y, Doi H, Miyake N, et al. A girl with early-onset epileptic encephalopathy associated with microdeletion involving CDKL5. Brain Dev. 2012. May;34(5):364–7. DOI: 10.1016/j.braindev.2011.07.004 [DOI] [PubMed] [Google Scholar]
- 23.Harden C, Tomson T, Gloss D, Buchhalter J, Cross JH, Donner E, et al. Practice guideline summary: Sudden unexpected death in epilepsy incidence rates and risk factors. Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the American Epilepsy Society. 2017;88(17):1674–80. DOI: 10.1212/wnl.0000000000003685 [DOI] [PubMed] [Google Scholar]
- 24.Paine SM, Munot P, Carmichael J, Das K, Weber MA, Prabhakar P, et al. The neuropathological consequences of CDKL5 mutation. Neuropathol Appl Neurobiol. 2012. Dec;38(7):744–7. DOI: 10.1111/j.1365-2990.2012.01292.x [DOI] [PubMed] [Google Scholar]
- 25.Bagnall RD, Crompton DE, Petrovski S, Lam L, Cutmore C, Garry SI, et al. Exome-based analysis of cardiac arrhythmia, respiratory control, and epilepsy genes in sudden unexpected death in epilepsy. Ann Neurol. 2016. Apr;79(4):522–34. DOI: 10.1002/ana.24596 [DOI] [PubMed] [Google Scholar]
- 26.Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017. Apr;58(4):512–21. DOI: 10.1111/epi.13709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Müller A, Helbig I, Jansen C, Bast T, Guerrini R, Jähn J, et al. Retrospective evaluation of low long-term efficacy of antiepileptic drugs and ketogenic diet in 39 patients with CDKL5-related epilepsy. Eur J Paediatr Neurol. 2016. Jan;20(1):147–51. DOI: 10.1016/j.ejpn.2015.09.001 [DOI] [PubMed] [Google Scholar]
- 28.Berg AT, Mahida S, Poduri A. KCNQ2-DEE: developmental or epileptic encephalopathy? Ann Clin Transl Neurol. 2021. Mar;8(3):666–76. DOI: 10.1002/acn3.51316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mingorance A, Jaksha A, Smart T, Sherriff L, Valentine J. The Voice of the Patient Report: CDKL5 Deficiency Disorder (CDD). https://www.cdkl5.com/pfdd/: Loulou Foundation; International Foundation for CDKL5 Research; 2019. [Google Scholar]
- 30.Tangarorang J, Leonard H, Epstein A, Downs J. A framework for understanding quality of life domains in individuals with the CDKL5 deficiency disorder. Am J Med Genet A. 2019. Feb;179(2):249–56. DOI: 10.1002/ajmg.a.61012 [DOI] [PubMed] [Google Scholar]
- 31.Olson H, Demarest S, Pestana-Knight E, Daniels C, Greene CA, DeLeo M, et al. Infantile Spasms In CDKL5 Deficiency Disorder Respond Poorly To First Line Treatments. 145th Annual Meeting of the American Neurological Association; 2020 October 4-9, 2020; Virtual; 2020. [Google Scholar]
- 32.Knupp KG, Coryell J, Nickels KC, Ryan N, Leister E, Loddenkemper T, et al. Response to treatment in a prospective national infantile spasms cohort. Ann Neurol. 2016. Mar;79(3):475–84. DOI: 10.1002/ana.24594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amin S, Majumdar A, Mallick A, Patel J, Scatchard R, Partridge C, et al. Caregiver's perception of epilepsy treatment, quality of life and comorbidities in an international cohort of CDKL5 patients. HIPPOKRATIA. 2017;21(3):130–5. [PMC free article] [PubMed] [Google Scholar]
- 34.Aledo-Serrano A, Gomez-Iglesias P, Toledano R, Garcia-Penas JJ, Garcia-Morales I, Anciones C, et al. Sodium channel blockers for the treatment of epilepsy in CDKL5 deficiency disorder: Findings from a multicenter cohort. Epilepsy Behav. 2021. May;118:107946. DOI: 10.1016/j.yebeh.2021.107946 [DOI] [PubMed] [Google Scholar]
- 35.Hussain SA, Heesch J, Weng J, Rajaraman RR, Numis AL, Sankar R. Potential induction of epileptic spasms by nonselective voltage-gated sodium channel blockade: Interaction with etiology. Epilepsy & Behavior. 2021. 2021/February/01/;115:107624. DOI: 10.1016/j.yebeh.2020.107624 [DOI] [PubMed] [Google Scholar]
- 36.Dale T, Downs J, Wong K, Leonard H. The perceived effects of cannabis products in the management of seizures in CDKL5 Deficiency Disorder. Epilepsy Behav. 2021. Sep;122:108152. DOI: 10.1016/j.yebeh.2021.108152 [DOI] [PubMed] [Google Scholar]
- 37.Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, et al. Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome. N Engl J Med. 2017. May 25;376(21):2011–20. DOI: 10.1056/NEJMoa1611618 [DOI] [PubMed] [Google Scholar]
- 38.Devinsky O, Patel AD, Cross JH, Villanueva V, Wirrell EC, Privitera M, et al. Effect of Cannabidiol on Drop Seizures in the Lennox-Gastaut Syndrome. N Engl J Med. 2018. May 17;378(20):1888–97. DOI: 10.1056/NEJMoa1714631 [DOI] [PubMed] [Google Scholar]
- 39.Geffrey AL, Pollack SF, Bruno PL, Thiele EA. Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia. 2015. Aug;56(8):1246–51. DOI: 10.1111/epi.13060 [DOI] [PubMed] [Google Scholar]
- 40.Devinsky O, Marsh E, Friedman D, Thiele E, Laux L, Sullivan J, et al. Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. The Lancet Neurology. 2016;15(3):270–8. DOI: 10.1016/s1474-4422(15)00379-8 [DOI] [PubMed] [Google Scholar]
- 41.Sands TT, Rahdari S, Oldham MS, Caminha Nunes E, Tilton N, Cilio MR. Long-Term Safety, Tolerability, and Efficacy of Cannabidiol in Children with Refractory Epilepsy: Results from an Expanded Access Program in the US. CNS Drugs. 2019. Jan;33(1):47–60. DOI: 10.1007/s40263-018-0589-2 [DOI] [PubMed] [Google Scholar]
- 42.Devinsky O, Verducci C, Thiele EA, Laux LC, Patel AD, Filloux F, et al. Open-label use of highly purified CBD (Epidiolex(R)) in patients with CDKL5 deficiency disorder and Aicardi, Dup15q, and Doose syndromes. Epilepsy Behav. 2018. Sep;86:131–7. DOI: 10.1016/j.yebeh.2018.05.013 [DOI] [PubMed] [Google Scholar]
- 43.Nohria V, Giller E. Ganaxolone. Neurotherapeutics. 2007;4(1):102–5. DOI: 10.1016/j.nurt.2006.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rossetti AO. Place of neurosteroids in the treatment of status epilepticus. Epilepsia. 2018. Oct;59 Suppl 2:216–9. DOI: 10.1111/epi.14481 [DOI] [PubMed] [Google Scholar]
- 45.Sperling MR, Klein P, Tsai J. Randomized, double-blind, placebo-controlled phase 2 study of ganaxolone as add-on therapy in adults with uncontrolled partial-onset seizures. Epilepsia. 2017. Apr;58(4):558–64. DOI: 10.1111/epi.13705 [DOI] [PubMed] [Google Scholar]
- 46.Pestana-Knight EM, Amin S, Bahi-Buisson N, Benke TA, Cross JH, Demarest ST, et al. Safety and efficacy of ganaxolone in patients with CDKL5 deficiency disorder: results from the double-blind phase of a randomised, placebo-controlled, phase 3 trial. The Lancet Neurology. 2022;21:417–27. [DOI] [PubMed] [Google Scholar]
- 47.Grinspan ZM, Knupp KG, Patel AD, Yozawitz EG, Wusthoff CJ, Wirrell E, et al. Comparative Effectiveness of Initial Treatment for Infantile Spasms in a Contemporary US Cohort. Neurology. 2021. Jul 15;97(12):e1217–28. DOI: 10.1212/wnl.0000000000012511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Demarest ST, Shellhaas RA, Gaillard WD, Keator C, Nickels KC, Hussain SA, et al. The impact of hypsarrhythmia on infantile spasms treatment response: Observational cohort study from the National Infantile Spasms Consortium. Epilepsia. 2017;58(12):2098–103. DOI: 10.1111/epi.13937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suleiman J, Brilot F, Lang B, Vincent A, Dale RC. Autoimmune epilepsy in children: case series and proposed guidelines for identification. Epilepsia. 2013. Jun;54(6):1036–45. DOI: 10.1111/epi.12142 [DOI] [PubMed] [Google Scholar]
- 50.Forrest KM, Young H, Dale RC, Gill DS. Benefit of corticosteroid therapy in Angelman syndrome. J Child Neurol. 2009. Aug;24(8):952–8. DOI: 10.1177/0883073808331344 [DOI] [PubMed] [Google Scholar]
- 51.Na JH, Shin S, Yang D, Kim B, Kim HD, Kim S, et al. Targeted gene panel sequencing in early infantile onset developmental and epileptic encephalopathy. Brain Dev. 2020. Jun;42(6):438–48. DOI: 10.1016/j.braindev.2020.02.004 [DOI] [PubMed] [Google Scholar]
- 52.Pestana-Knight EM, Naduvil AM, Sumit P, Zeft A. Clinical response to Oral prednisolone and IVIG in children with CDKL5 mutation. American Epilepsy Society Annual Meeting 2017: Washington, DC; 2017; 2017. [Google Scholar]
- 53.Lim Z, Wong K, Olson HE, Bergin AM, Downs J, Leonard H. Use of the ketogenic diet to manage refractory epilepsy in CDKL5 disorder: Experience of >100 patients. Epilepsia. 2017. Aug;58(8):1415–22. DOI: 10.1111/epi.13813 [DOI] [PubMed] [Google Scholar]
- 54.Ko A, Jung DE, Kim SH, Kang HC, Lee JS, Lee ST, et al. The Efficacy of Ketogenic Diet for Specific Genetic Mutation in Developmental and Epileptic Encephalopathy. Front Neurol. 2018;9:530. DOI: 10.3389/fneur.2018.00530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ko A, Youn SE, Kim SH, Lee JS, Kim S, Choi JR, et al. Targeted gene panel and genotype-phenotype correlation in children with developmental and epileptic encephalopathy. Epilepsy Res. 2018. Mar;141:48–55. DOI: 10.1016/j.eplepsyres.2018.02.003 [DOI] [PubMed] [Google Scholar]
- 56.Siri B, Varesio C, Freri E, Darra F, Gana S, Mei D, et al. CDKL5 deficiency disorder in males: Five new variants and review of the literature. Eur J Paediatr Neurol. 2021. Jul;33:9–20. DOI: 10.1016/j.ejpn.2021.04.007 [DOI] [PubMed] [Google Scholar]
- 57.Stevelink R, Sanders MW, Tuinman MP, Brilstra EH, Koeleman BP, Jansen FE, et al. Epilepsy surgery for patients with genetic refractory epilepsy: a systematic review. Epileptic Disord. 2018. Apr 1;20(2):99–115. DOI: 10.1684/epd.2018.0959 [DOI] [PubMed] [Google Scholar]
- 58.Lim Z, Wong K, Downs J, Bebbington K, Demarest S, Leonard H. Vagus nerve stimulation for the treatment of refractory epilepsy in the CDKL5 Deficiency Disorder. Epilepsy Res. 2018. Oct;146:36–40. DOI: 10.1016/j.eplepsyres.2018.07.013 [DOI] [PubMed] [Google Scholar]
- 59.Evans JC, Archer HL, Colley JP, Ravn K, Nielsen JB, Kerr A, et al. Early onset seizures and Rett-like features associated with mutations in CDKL5. Eur J Hum Genet. 2005. Oct;13(10):1113–20. DOI: 10.1038/sj.ejhg.5201451 [DOI] [PubMed] [Google Scholar]
- 60.Baba S, Sugawara Y, Moriyama K, Inaji M, Maehara T, Yamamoto T, et al. Amelioration of intractable epilepsy by adjunct vagus nerve stimulation therapy in a girl with a CDKL5 mutation. Brain Dev. 2017. Apr;39(4):341–4. DOI: 10.1016/j.braindev.2016.10.007 [DOI] [PubMed] [Google Scholar]
- 61.White R, Ho G, Schmidt S, Scheffer IE, Fischer A, Yendle SC, et al. Cyclin-Dependent Kinase-Like 5 (CDKL5) Mutation Screening in Rett Syndrome and Related Disorders. Twin Research and Human Genetics. 2010;13(2):168–78. DOI: 10.1375/twin.13.2.168 [DOI] [PubMed] [Google Scholar]
- 62.Hao S, Wang Q, Tang B, Wu Z, Yang T, Tang J. CDKL5 Deficiency Augments Inhibitory Input into the Dentate Gyrus That Can Be Reversed by Deep Brain Stimulation. J Neurosci. 2021. Oct 27;41(43):9031–46. DOI: 10.1523/JNEUROSCI.1010-21.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD, et al. Valvular Heart Disease Associated with Fenfluramine–Phentermine. New England Journal of Medicine. 1997;337(9):581–8. DOI: 10.1056/nejm199708283370901 [DOI] [PubMed] [Google Scholar]
- 64.Lagae L, Sullivan J, Knupp K, Laux L, Polster T, Nikanorova M, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double-blind, placebo-controlled trial. The Lancet. 2019;394(10216):2243–54. DOI: 10.1016/s0140-6736(19)32500-0 [DOI] [PubMed] [Google Scholar]
- 65.Knupp KG, Scheffer IE, Ceulemans B, Sullivan J, Nickels KC, Miller I, et al. Efficacy and safety of FINTEPLA (fenfluramine) for the treatment of seizures associated with Lennox-Gastaut syndrome: a randomized, double-blind, placebo-controlled clinical trial. American Epilepsy Society Annual Meeting 2020: Seattle, Washington; 2020; 2020. [Google Scholar]
- 66.Devinsky O, King L, Schwartz D, Conway E, Price D. Effect of fenfluramine on convulsive seizures in CDKL5 deficiency disorder. Epilepsia. 2021. Jul;62(7):e98–e102. DOI: 10.1111/epi.16923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lai WW, Galer BS, Wong PC, Farfel G, Pringsheim M, Keane MG, et al. Cardiovascular safety of fenfluramine in the treatment of Dravet syndrome: Analysis of an ongoing long-term open-label safety extension study. Epilepsia. 2020;61(11):2386–95. DOI: 10.1111/epi.16638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nabbout R, Mistry A, Zuberi S, Villeneuve N, Gil-Nagel A, Sanchez-Carpintero R, et al. Fenfluramine for Treatment-Resistant Seizures in Patients With Dravet Syndrome Receiving Stiripentol-Inclusive Regimens: A Randomized Clinical Trial. JAMA Neurology. 2020;77(3):300–8. DOI: 10.1001/jamaneurol.2019.4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nishi T, Kondo S, Miyamoto M, Watanabe S, Hasegawa S, Kondo S, et al. Soticlestat, a novel cholesterol 24-hydroxylase inhibitor shows a therapeutic potential for neural hyperexcitation in mice. Sci Rep. 2020. Oct 13;10(1):17081. DOI: 10.1038/s41598-020-74036-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Noguchi N, Saito Y, Urano Y. Diverse functions of 24(S)-hydroxycholesterol in the brain. Biochem Biophys Res Commun. 2014. Apr 11;446(3):692–6. DOI: 10.1016/j.bbrc.2014.02.010 [DOI] [PubMed] [Google Scholar]
- 71.Paul SM, Doherty JJ, Robichaud AJ, Belfort GM, Chow BY, Hammond RS, et al. The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-D-aspartate receptors. J Neurosci. 2013. Oct 30;33(44):17290–300. DOI: 10.1523/JNEUROSCI.2619-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Halford JJ, Sperling MR, Arkilo D, Asgharnejad M, Zinger C, Xu R, et al. A phase 1b/2a study of soticlestat as adjunctive therapy in participants with developmental and/or epileptic encephalopathies. Epilepsy Res. 2021. Aug;174:106646. DOI: 10.1016/j.eplepsyres.2021.106646 [DOI] [PubMed] [Google Scholar]
- 73.Demarest S, Jeste S, Agarwal N, Arkilo D, Forgacs PB, Asgharnejad M, et al. Efficacy, Safety and Tolerability of Soticlestat (TAK-935/OV935) as an Adjunctive Therapy in Patients with 15q Duplication Syndrome (Dup15q) or Cyclin-Dependent Kinase-Like 5 Deficiency Disorder (CDD) in a Signal-Finding Phase 2 Study (ARCADE) (4096). Neurology. 2021;96(15 Supplement):4096. [Google Scholar]
- 74.Devinsky O, King L, Bluvstein J, Friedman D. Ataluren for drug-resistant epilepsy in nonsense variant-mediated Dravet syndrome and CDKL5 deficiency disorder. Ann Clin Transl Neurol. 2021. Mar;8(3):639–44. DOI: 10.1002/acn3.51306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao Y, Irvine EE, Eleftheriadou I, Naranjo CJ, Hearn-Yeates F, Bosch L, et al. Gene replacement ameliorates deficits in mouse and human models of cyclin-dependent kinase-like 5 disorder. Brain. 2020. Mar 1;143(3):811–32. DOI: 10.1093/brain/awaa028 [DOI] [PubMed] [Google Scholar]
- 76.Trazzi S, De Franceschi M, Fuchs C, Bastianini S, Viggiano R, Lupori L, et al. CDKL5 protein substitution therapy rescues neurological phenotypes of a mouse model of CDKL5 disorder. Hum Mol Genet. 2018. May 1;27(9):1572–92. DOI: 10.1093/hmg/ddy064 [DOI] [PubMed] [Google Scholar]
- 77.Terzic B, Davatolhagh MF, Ho Y, Tang S, Liu Y-T, Xia Z, et al. Temporal manipulation of Cdkl5 reveals essential postdevelopmental functions and reversible CDKL5 deficiency disorder–related deficits. Journal of Clinical Investigation. 2021;131(20). DOI: 10.1172/jci143655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fuchs C, Gennaccaro L, Ren E, Galvani G, Trazzi S, Medici G, et al. Pharmacotherapy with sertraline rescues brain development and behavior in a mouse model of CDKL5 deficiency disorder. Neuropharmacology. 2020. May 1;167:107746. DOI: 10.1016/j.neuropharm.2019.107746 [DOI] [PubMed] [Google Scholar]
- 79.Tang S, Terzic B, Wang IJ, Sarmiento N, Sizov K, Cui Y, et al. Altered NMDAR signaling underlies autistic-like features in mouse models of CDKL5 deficiency disorder. Nat Commun. 2019. Jun 14;10(1):2655. DOI: 10.1038/s41467-019-10689-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Loi M, Gennaccaro L, Fuchs C, Trazzi S, Medici G, Galvani G, et al. Treatment with a GSK-3beta/HDAC Dual Inhibitor Restores Neuronal Survival and Maturation in an In Vitro and In Vivo Model of CDKL5 Deficiency Disorder. Int J Mol Sci. 2021. May 31;22(11). DOI: 10.3390/ijms22115950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Galvani G, Mottolese N, Gennaccaro L, Loi M, Medici G, Tassinari M, et al. Inhibition of microglia overactivation restores neuronal survival in a mouse model of CDKL5 deficiency disorder. J Neuroinflammation. 2021. Jul 8;18(1):155. DOI: 10.1186/s12974-021-02204-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mulcahey PJ, Tang S, Takano H, White A, Davila Portillo DR, Kane OM, et al. Aged heterozygous Cdkl5 mutant mice exhibit spontaneous epileptic spasms. Exp Neurol. 2020. Oct;332:113388. DOI: 10.1016/j.expneurol.2020.113388 [DOI] [PMC free article] [PubMed] [Google Scholar]