

Abstract
Oncolytic viruses (OVs) provide novel and promising therapeutic options for patients with cancers resistant to traditional therapies. Natural or genetically modified OVs are multifaceted tumor killers. They directly lyse tumor cells while sparing normal cells, and indirectly potentiate anti-tumor immunity by releasing antigens and activating inflammatory responses in the tumor microenvironment (TME). However, some limitations, such as penetration of OVs into tumors and the hosts’ antiviral immune response, are impeding the application of oncolytic virotherapy’s broad translation into the clinic. If these challenges can be overcome, combination therapies, such as OVs plus immune checkpoint blockades (ICBs), chimeric antigen receptor (CAR) T cells, or CAR natural killer (NK) cells, will provide powerful therapeutic platforms in the clinic.
Keywords: oncolytic virus, oncolytic virotherapy, immunotherapy, CAR-T, CAR-NK, combination therapy
Overview of oncolytic viruses for cancer therapy
Viruses have been used as experimental agents to induce cell death and/or dysfunction for more than a hundred years. In the 1990s, researchers stated a hypothesis: could viruses be used to kill tumor cells specifically? Therefore, oncolytic viruses (OVs) (see Glossary) have been identified. OVs are anti-tumor agents that selectively replicate in tumor cells and are thought to induce lysis as well as an immunogenic tumor cell death. The main difference between OV therapy and gene therapy is that the vectors used in the latter are engineered to lack the ability to replicate in infected cells. There are two main groups of OVs: naturally existing viruses and genetically modified viruses. Naturally existing OVs, including measles virus (MV), Newcastle disease virus (NDV), and reovirus, have been used for research studies and clinical trials in their native forms. To develop genetically modified OVs, some virulent genes are removed to attenuate the virus and/or target transgenes are inserted into the viral genome to promote anti-tumor immunity. In 2015, the United States Food and Drug Administration (U.S. FDA) approved the first OV (T-VEC) to treat patients with metastatic melanoma. T-VEC is a genetically engineered oncolytic herpes simplex virus type 1 (oHSV-1), which is a milestone in the field of oncolytic virotherapy [1]. In 2021, Japan approved the world’s first OV against malignant glioma (DELYTACT, oHSV-1 with G47Δ) [2].
The use of OVs as single-agent therapy or in combination with other strategies is supported by both preclinical and clinical studies. Therapeutic outcomes depend on the balance between a host’s antiviral response and an OV-induced anti-tumor immunity, but the biological mechanisms affecting that balance remain unclear. Here, we describe the unique characteristics of OVs, the therapeutic potentials of natural and genetically engineered OVs, and efficient administration routes for OV delivery. We also discuss the anti-tumor activities and challenges that are coming to light as OVs develop into a new class of immunotherapy drugs. Moreover, we highlight how combinations of OVs with other immunotherapies might overcome barriers to successful cancer treatment. The review aims to not only elucidate the current status of OV therapy but also to guide the design of more powerful therapeutic OVs for treating cancer patients.
Unique characteristics of oncolytic viruses
Viruses are small particles that can replicate in host cells, inducing host inflammation. The two main types are DNA and RNA viruses. DNA viruses generally have a large genome that can be edited to encode transgenes to increase therapeutic activity modulating the immune system or other mechanisms of action without impairing viral replication [3]. RNA viruses have a much smaller genome than DNA viruses, and can therefore cross the blood-brain barrier (BBB) to target tumor cells in the central nervous system (CNS) [4]. However, their small genomes limit their ability to encode large transgenes. Additionally, RNA viruses have more genetic instability, with much higher mutation rates than DNA viruses [5].
During the past decade, natural or engineered OVs from DNA and RNA viruses have been used experimentally, and some have been brought into the clinic (Box 1). We surveyed the current landscape of OV clinical trials from 2012 to 2022 and summarized them in Table S1 (see the supplemental information).
Box 1. The diversity and characteristics of OVs.
Adenovirus (Ads)
Ads are non-enveloped viruses with a single linear double-stranded DNA (dsDNA) genome (~30–40 kb). Oncolytic Ads (oAds) were some of the earliest OVs used in preclinical studies and the most commonly tested in clinical trials (Figure I). Intracerebral injection of an E1B-attenuated oAd (ONYX-015) was used to treat glioma patients [6]. Recently, intratumoral (IT) infusion of an oAd (DNX-2401) followed by radiotherapy in pediatric patients with newly diagnosed diffuse intrinsic pontine glioma (DIPG) showed immune responses and tumor reduction (Table S1, NCT03178032) [7].
Herpes simplex virus (HSV)
HSV, especially HSV-1, is one of the most widely explored dsDNA viruses with the benefits of (a) infecting most malignant cell types and replicating quickly; (b) a large genome (over 150 kb) with non-essential parts for replication, providing space for adding engineered transgenes without limiting the virus’s packaging efficiency; (c) easy access for modification and the flexibility to insert multiple transgenes [8]. G207, an oHSV with deleted ICP34.5 and ICP6, successfully converted immunologically “cold” tumors to “hot” tumors in 12 patients (NCT02457845). The median overall survival (OS) of the 12 patients who received G207 by IT injection alone or in combination with radiation was 12.2 months, compared with a historical median overall survival of 5.6 months [9]. 50 glioma patients were safely treated (IT) with rQNestin34.5v.2 (NCT03152318). Compared with HSV-1, HSV-2 has several unique features that can be exploited to engineer oncolytic agents [10].
Vaccinia virus (VACV)
VACV is an enveloped dsDNA poxvirus (~190 kb) with a natural selectivity to tumors and the potential for systemic administration. JX-594, a Wyeth VACV strain-derived OV, includes a thymidine kinase (TK) gene deletion to enable more selective replication in tumors and a human GM-CSF gene insertion to induce a systemic anti-tumor immune response. It is well tolerated and increases survival in patients with colorectal cancer after intravenous (IV) injection (NCT01469611) [11]. Prostvac, a neoantigen-based therapeutic vaccine consisting of a recombinant vaccinia vector (prime) and a recombinant fowlpox vector (boost), was safe in phase I/II trials when injected subcutaneously into prostate cancer patients (NCT02933255) and led to improvement of OS from 16.6 to 25.1 months [12].
Myxoma virus (MYXV)
MYXV is a dsDNA poxvirus responsible for rabbit myxomatosis (~161.8 kb, 171 genes). Like oHSV and VACV, multiple transgenes can be inserted into the big genome. The tropism of MYXV for tumor cells differs from that of many other viruses, as MYXV attaches to cell surface and fuses its envelope with the cell membrane to enter cells without a specific receptor. MYXV was developed as an OV for several tumor types with the potential to be in the clinic [13–15].
Measles virus (MV)
MV is a single-stranded, negative-sense, enveloped single-stranded (ss) RNA paramyxovirus (~16kb). MV interacts with host cells through three receptors: SLAM/CD150, CD46, and Nectin-4. Its favorable safety profile with no dose-limiting toxicities gives it more potential to be developed as an OV agent. In phase I/II trials (NCT02068794), MV-NIS, an oncolytic Edmonston strain-derived MV carrying the human sodium iodide symporter (NIS, Na+/I− symporter), triggered cellular immunity against drug-resistant ovarian cancer and was safe in patients treated intraperitoneally [16]. MV-NIS was administered (IV) to 32 patients with recurrent or refractory multiple myeloma, with one achieving a complete response and others showing a response (NCT00450814) [17].
Other viruses
NDV is an avian enveloped, negative ssRNA paramyxovirus virus (~15 kb). Parvovirus is a small non-enveloped ssDNA virus (~5 kb). Vesicular stomatitis virus (VSV) is an enveloped, small negative ssRNA virus (~11 kb) with substantial genome plasticity. Reovirus is a dsRNA virus (~23.5 kb). All these are being developed as oncolytic viral agents [18].
Figure I. Oncolytic viral backbones under clinical investigation from 2012 to 2022.
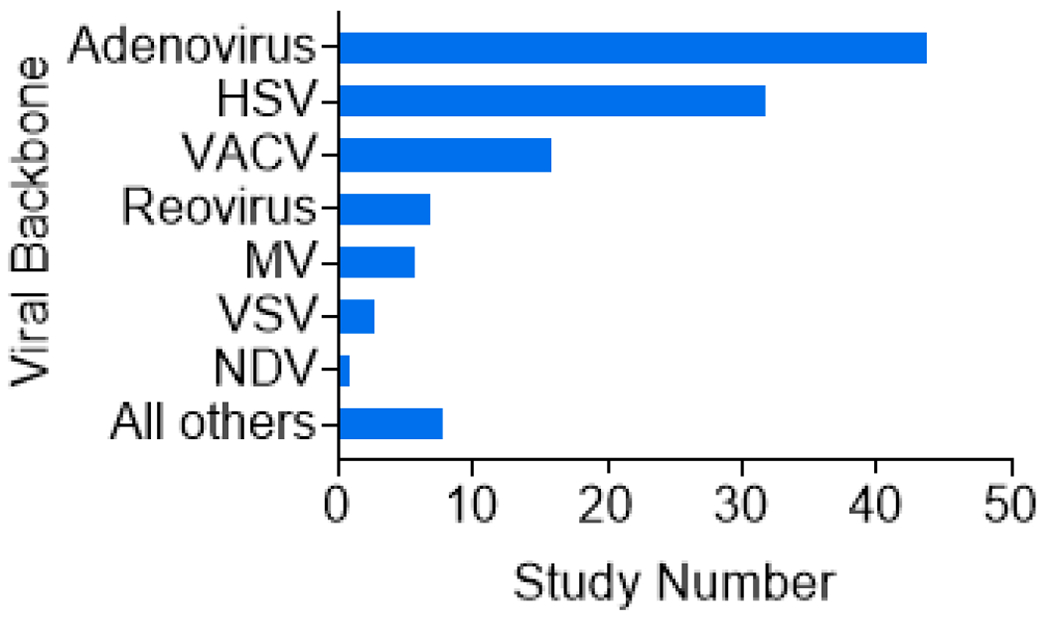
Characterization of oncolytic viruses used in clinical trials. Types of OVs reported in clinical trials from 2012 to 2022 as determined by searching ClinicalTrials.gov with the key words: Oncolytic virus, Not recruiting (N/R), Active not recruiting (A/NR), Recruiting (R), and Completed (C). Among the results, adenovirus is the most dominant viral backbone (n = 44) and HSV is second (n = 32). Vaccinia virus (VACV) (n = 16) and several other viruses are shown for comparison.
Mechanisms of action of oncolytic virotherapy
Direct oncolytic activity, i.e., lysis of tumor cells directly, is the initial mechanism by which OVs kill tumors. However, OVs also can exert their effects by indirectly inducing systemic anti-tumor immunity. OV-infected tumor cells release tumor antigens and activate inflammatory responses that can counteract tumor-induced immune suppression and evasion.
Tumor-selective infection, replication, and oncolysis
Several factors drive tumor selectivity in oncolytic virotherapy. The first is entry into cells via virus-specific receptors. Expression of SLAM/CD150 and CD46 receptors on tumors mediates oncolytic MV-specific recognition [19]. Modification or mutation of viral surface proteins to recognize tumor-specific cellular receptors enhances OVs’ ability to enter tumor cells [18]. Retargeting OVs to tumor-associated receptors including epidermal growth factor receptor (EGFR) facilitates efficient OV infection and consequently improved viral replication.
Second, tumor cells divide more rapidly and have a higher metabolism than normal quiescent cells. Therefore, it is advantageous for OVs to use tumor cells’ genetic machinery to replicate. Engineering OVs by deleting non-essential viral virulence genes and inserting tumor-specific promoters or combining microRNAs enhanced OV replication in tumors [20,21].
Third, tumor cells have a dysfunctional immune response. Deficiencies in antiviral type I interferon (IFN) signaling blocks viral clearance. However, healthy cells, including immune and non-immune cells, produce IFN elements [e.g., Janus kinase (JAK) / signal transducer and activator of transcription (STAT)] that induce a programmed transcriptional pathway, limiting viral replication [22,23]. Therefore, OVs do not damage healthy cells (Figure 1). There is considerable controversy about the antiviral role of IFN, as some patient-derived tumors/xenografts with mutations in the IFN pathway sometimes show considerable resistance to OV infection [24]. Identifying the mutated genes and understanding their roles may shed light on this observation.
Figure 1. Oncolytic viruses replicate selectively in tumor cells.
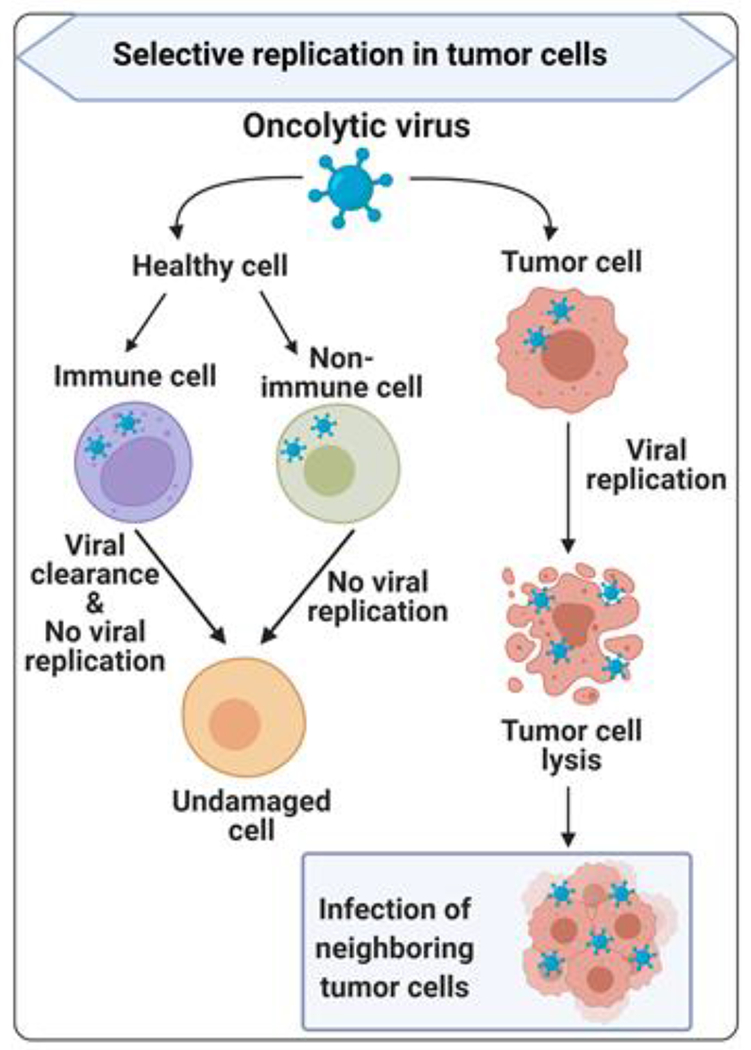
Oncolytic viruses (OVs) can specifically infect dysfunctional tumor cells and replicate until the tumor cells lyse and newborn viruses are released to infect neighboring tumor cells. In healthy cells, including immune cells and non-immune cells, OVs have low or no replication due to antiviral type I interferon (IFN) signaling and other mechanisms. OVs can be cleared by immune cells, and do not damage healthy cells.
After tumor cell lysis, OVs spread to the surrounding area, amplifying oncolysis. The OV-selective lytic potential depends on the type of virus, dose, natural or induced viral tropism, and a tumor cell’s susceptibility to various forms of cell death (e.g., apoptosis, necrosis, pyroptosis, and autophagy) [25]. However, over-enhanced oncolytic potential may increase the risk of off-target and unexpected toxic effects in normal healthy cells [26]. Moreover, expression of virally encoded transgenes may wane, as OV-infected tumor cells will be killed, leading to reduce OV reproduction to some extent. This action results in temporary debulking rather than long-lasting responses [27].
Induction of systemic anti-tumor response
Following viral replication, immunogenic cell death of tumor cells induced by OVs— characterized by the release of danger-associated molecular pattern signals (DAMPs) and tumor-associated antigens (TAAs)—can directly eliminate viable tumors and set the stage for systemic immune responses. Induction of systemic innate and tumor-specific adaptive anti-tumor responses further eradicates tumor cells (Figure 2).
Figure 2. Enhancement of immune responses by oncolytic viruses.
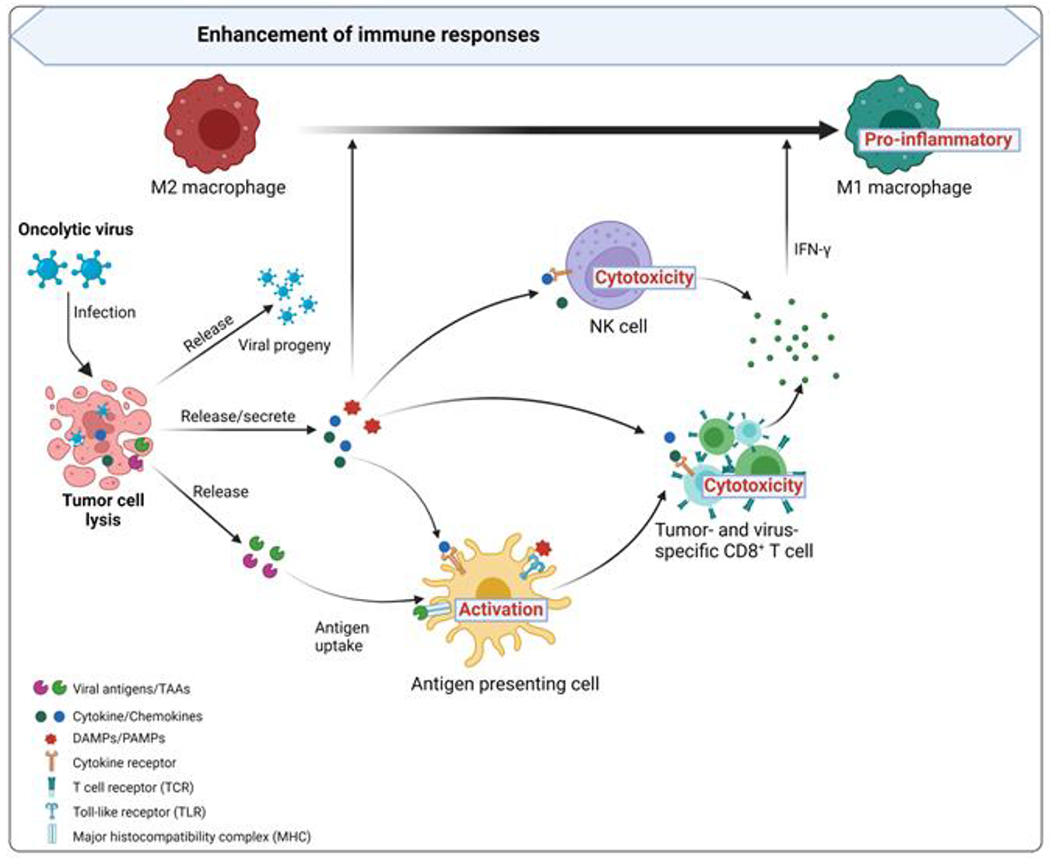
After infection with an oncolytic virus, tumor cells initiate an antiviral response by releasing antiviral cytokines (especially IFNs) that promote the maturation of antigen-presenting cells (APCs) such as dendritic cells (DCs) and that also stimulate CD8+ T cells and NK cells. After a tumor cell is lysed, viral progeny, DAMPs (including host cells proteins), PAMPs (viral particles), and TAAs (tumor-associated antigens) including neoantigens are released. The viral progeny infects more tumor cells. DAMPs and PAMPs stimulate the immune system by activating receptors, including TLRs. TAAs and neoantigens are taken up by APCs, activating antigen-/virus-specific CD8+ T cell responses, and thereby creating an immune-stimulatory environment. This change prompts tumor-supportive M2 macrophages to change to pro-inflammatory M1 phenotypes
Activation of the innate immune system might be both a hindrance and helper to OVs. It reduces viral persistence but is required to generate memory immune cells. Following oncolytic cell death, infected tumor cells release TAAs that promote an adaptive immune response to eliminate tumors at distant sites that are not injected or exposed to the virus [28]. Also, they can release cytokines [such as type I IFNs, tumor necrosis factor-α (TNFα), and interleukin-12 (IL-12)], cellular DAMPs, and viral pathogen-associated molecular patterns (PAMPs) to promote the maturation of antigen-presenting cells (APCs), thus activating antigen- and/or virus-specific CD8+ T cell responses. Once activated, naive CD8+ T cells become cytotoxic effector cells and traffic to tumor sites where they mediate anti-tumor immunity [28].
Type I IFNs and DAMPs directly or indirectly activate natural killer (NK) cells and macrophages. Activated cytotoxic NK cells kill OV-infected tumors by releasing cytolytic components, triggering FAS–FAS ligand signaling, and expressing IFNγ and TNFα [29,30]. These cytokines polarize tumor-supportive M2 macrophages towards pro-inflammatory M1 phenotypes and recruit more immune cells into the TME, presenting antigens to T-cells, which, in turn, produce more IFNs and TNFα to amplify the initial innate anti-tumor response [31–33].
Upon recognizing OV infection, dendritic cells (DCs) are also activated and change to a mature state. By secreting cytokines and chemokines, these DCs induce innate immune responses by NK cells and macrophages. Activated DCs also prime antigen-specific T cells to induce adaptive immunity by presenting viral antigens in MHC molecules along with costimulatory molecules and inflammatory cytokines, thus overcoming tumor-associated immunosuppression [34].
Remodeling the immunologically “cold” tumor microenvironment into “hot”
The tumor microenvironment (TME) is a complex niche for the development of cancerous and noncancerous cell components, and it can be characterized as immunologically “cold” or “hot” depending on pro-inflammatory cytokine production and immune cell infiltration levels. A “cold” TME is typified by few inflammatory immune cells, an immunosuppressive TME, a poor prognosis, and inadequate response to immunotherapy. In contrast, a “hot” TME associates with higher response rates with activated immune cells [35–37]. OV-infected tumors were more visible to the immune system for recognition and attack. OV-based immunotherapy converts immunogenic “cold” tumors to “hot” by killing tumor cells, releasing antigens, and recruiting innate effector cells to tumor sites [38] (Figure 3). However, in rare cases, OV-based immunotherapy can also produce too much inflammation and lead to adverse immunological events, such as autoimmune disease, which may damage tissues or organs. This is exemplified by vitiligo in two of twelve melanoma patients receiving a VACV expressing B7.1 [39].
Figure 3. Oncolytic viruses remodel the tumor immune microenvironment from “cold” to “hot.”.
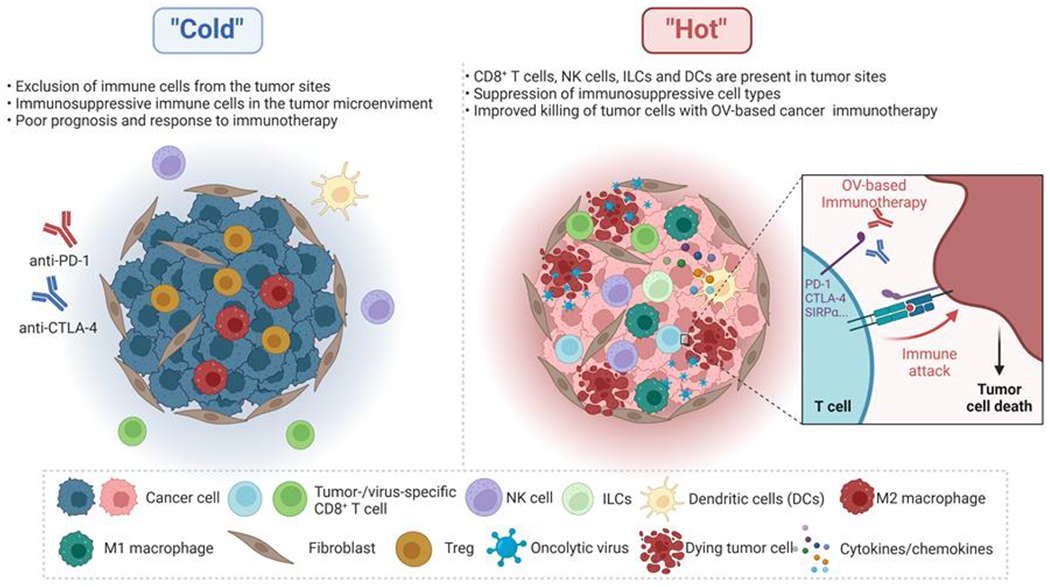
OV-based cancer immunotherapy can remodel the tumor microenvironment (TME). A “cold” TME often has high infiltration of immunosuppressive cells, including Treg cells and M2-polarized macrophages (M2 macrophage) from the tumor site, a poor prognosis, and inadequate response to immunotherapy. In contrast, a “hot” TME associates with higher response rates with more activated immune cells (e.g., CD8+ T cells, NK cells, innate lymphoid cells (ILCs), DCs, and M1-polarized macrophages (M1 macrophages)) to immunotherapy. OV infection can enhance the infiltration and activity of immune cells, including innate and adaptive immune cells, within the TME. At the same time, these therapeutic viruses reduce populations of immunosuppressive cell types and shift immune cells toward an anti-tumor phenotype, thereby overcoming immune suppression within the TME. Activated DCs or macrophages can generate a broadened repertoire of tumor neoantigen-specific T cells whose effector function can be augmented by immune checkpoint blockade (e.g., anti-PD-1 and anti-CTLA4 antibody) in their killing of tumor cells locally and systemically. Activation of anti-tumor immunity by oncolytic virotherapy is often accompanied by the production of various pro-inflammatory cytokines, which further helps generate a “hot” TME.
Therapy that combines OVs with immune checkpoint blockades (ICBs) has achieved impressive clinical responses. In a phase Ib clinical trial of T-VEC combined with an anti-PD-1 antibody (pembrolizumab), patients with metastatic melanoma showed a 62% and 33% overall and complete response, respectively (Table S1, NCT04068181) [40]. Unfortunately, a subsequent randomized phase III trial (NCT02263508) in melanoma patients was stopped because of clinical futility [41]. However, the two trials used different treatment protocols. In the phase Ib trial, T-VEC injections started five weeks before pembrolizumab, whereas the two treatments were administered simultaneously in the phase III trial. Hence, in the phase III trial, the virus lacked time to heat up the tumors. Therefore, choosing the best time points for administering agents in OV combination therapy appears to be critical.
Strategies for engineering innovative oncolytic viruses
As OVs are live viral particles, developing them into anti-cancer drugs has to consider attenuating viral pathogenesis, specifically targeting tumor cells, and promoting tumor cell death.
Generation of attenuated OVs
Most clinically relevant OVs utilize attenuated vectors or naturally existing less virulent variants of viruses to prevent acute and long-term toxicities. T-VEC is an engineered attenuated HSV-1-based virus. HSV-1 is known to cause neurovirulence and latent infection due to the viral gene product ICP34.5, which prevents the type I IFN response and antagonizes the protein kinase R signaling pathway within non-dividing cells [42]. As ICP34.5 is deleted in T-VEC, the virus is unable to grow within neurons or mediate latent infection. H101 is an E1B-deleted Ad that was approved in China for patients with nasopharyngeal tumors [43]. In a randomized phase III trial with 160 participants with advanced squamous cell carcinomas of the head and neck or esophagus, patients who were treated with cisplatin/5-FU and H101 had a 78.8% response rate compared to the 39.6% rate in the cisplatin/5-FU-only cohort [44]. Oncolytic NDV-PV701 treating glioblastoma (GBM) patient is another example of naturally existing less virulent variant of a particular virus [45].
Arming OVs with a transgene(s)
The ability to encode foreign transgenes makes OV an attractive vector for clinical development. The adenoviral genome is rather easily modified, and transgenes of up to 10 kb can be inserted without disrupting the virus’s ability to infect. The HSV genome provides plenty of space (~25 kb) for transgene insertion. Therefore, many oAds and oHSVs have been engineered to express TAAs [e.g., human melanoma-associated antigen A3 (MAGE-A3) / carcinoembryonic antigen (CEA)], immune-activating molecules (e.g., CD40L/4-1BBL), and immune anti-inhibitory molecules [e.g., single-chain variable fragment (scFv) against programmed cell death 1 (PD-1) / cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) / CD47] against various cancers [46–49].
Our group armed oHSVs with a full-length antibody against CD47 to target GBM and ovarian cancer [48,49]. CD47, an important immune checkpoint, collaborates with the signal regulatory protein alpha (SIRPα), thereby inhibiting innate immune cell phagocytosis. oHSVs armed with different engineered versions of anti-CD47 antibodies disrupted the interaction between CD47 and SIRPα, allowing for enhanced phagocytosis of tumor cells by macrophages and reducing tumor burden. A combination therapy—mouse OV-αCD47 and anti-PD-L1 mouse antibody—significantly prolonged survival compared with the corresponding single agents in immunocompetent mice bearing ID8 ovarian tumors [49].
Arming OVs with a cytokine(s)/chemokine(s)
Cytokines are a large group of soluble proteins, peptides, or glycoproteins that regulate the innate and adaptive immune system by controlling proliferation, differentiation, survival, and effector functions. Chemokines, the largest subfamily of cytokines, can act as chemoattractants for immune cells. However, systemic administration of super-physiological doses of cytokine(s)/chemokine(s) is often toxic in the clinic.
In contrast, cytokine or chemokine-expressing OVs are safer because both viral load and cytokine concentration can be controlled by local injection. OVs armed with cytokines or chemokines have shown potential anti-tumor properties in preclinical studies and clinical trials. oHSV armed with IL-12 remarkably increased IL-12 in the TME and induced infiltration of effector T cells, NK cells, and APCs into tumors, enhancing anti-tumor efficacy [50,51]. The safety of the oHSV-IL-12 as monotherapy or combination therapy was demonstrated preclinically [32,52]. OVs engineered with human IL-12 are being investigated in clinics. IL-23, an IL-12 cytokine family member, plays many roles in cancer immunity. VACV armed with IL-23 modulated the TME and exerted potent anti-tumor immunity in an animal model [53]. IL-15-armed OVs were safe and effective in diverse syngeneic murine tumor models [54]. Intravenous (IV) OV treatment with a fusion protein of IL-15 and IL-15Rα enhanced infiltration of CD8+ T and NK cells in vivo [55]. Our group demonstrated that an oHSV-1 expressing human IL-15/IL-15Rα sushi domain fusion protein (OV-IL15C) promoted cellular cytotoxicity in GBM and improved the trafficking and survival of NK and CD8+ T cells both in vitro and in vivo and led to a better survival when combined with chimeric antigen receptor (CAR)-NK cells [56]. OVs engineered with GM-CSF has been widely applied for cancer immunotherapies in the clinic. Like T-VEC, JX-594, a VACV encoding the human GM-CSF gene, is being used to treat patients with advanced hepatocellular carcinoma [11]. Although OV armed with a cytokine(s) is effective, it has a limitation as the cytokine can activate immune cells to clear the virus and thus restrict OV function. It is difficult to balance antiviral and anti-tumor immune responses. Some cytokines attract more attention than others (e.g., GM-CSF and IL-12 versus IL-23). However, clinical data are needed to determine what cytokines are better than others in terms of efficacy and safety.
Arming OVs with a bi-specific/tri-specific T cell engager (BiTE/TriTE) molecule
Bi-specific T cell engager (BiTE) is a recombinant bi-specific protein engager consisting of two scFvs recognizing different proteins. One scFv binds to a cell-surface molecule on T cells, and the other binds to an antigen on malignant cells. Arming OVs with a BiTE overcomes a particular shortcoming of BiTE molecules, which have an extremely short half-life in serum. When incorporated into a replicating OV, a BiTE achieves the potential for long-lasting tumor-selective expression. Studies with xenograft mouse models have revealed that OVs armed with a BiTE induce immune-mediated tumor destruction through increased oncolysis and activation of cytotoxic T cells. An oncolytic VACV targeting CD3 and the tumor cell-surface antigen EphA2 was the first BiTE-armed OV tested—in a murine xenograft model of human lung cancer [57]. Other BiTE-armed oncolytic Ads and MVs are under development [58–60].
Tri-specific T cell engager (TriTE), the next generation of T cell engager constructs that consists of three domains (such as CD3 × dual tumor antigens or tumor antigen × CD3/CD28). Dual antigens can decrease propensity for immune escape by antigen loss and reduce on-target off-tumor side effects because they improve tumor selectivity. Natural killer group 2D (NKG2D), a potent activating receptor, engages NK, CD8+, NKT, and γδ T cells, can be a better choice than traditional CD3 [61,62]. Given the hallmark of tumor cells—antigenic heterogeneity—choosing the best antigens for OV development is still challenging.
Administration routes for delivering oncolytic viruses
An appropriate administration route for delivering OVs is critical to the success of OV-based cancer immunotherapy. Local injection of OVs into a single tumor site, such as IT administration, can directly target a tumor’s location. However, it has limited application for metastatic tumors. Systemic injection, e.g., IV administration, is logistically simple and effective against metastatic lesions, but OVs are quickly cleared in the circulatory system before reaching tumors. Cell carriers, including transformed tumor cells, immune cells, and stem cells, have been adopted as systemic delivery vehicles for OVs [63]. Intraperitoneal (IP), subcutaneous (SC), intracranial (IC), intramuscular (IM), intranasal, intravesical (IVE), aerosol, and inhalation administration are all being used preclinically and clinically (Box 2). In the preclinical setting, selecting a delivery method depends primarily on a research project’s aim. Safety must take precedence in the clinical setting.
Box 2. Established and emerging methods for delivering OVs.
Intratumoral delivery
IT, the current most common choice for OV administration in clinical trials (Figure I), facilitates direct delivery of infectious viral particles into injected tumor sites, thereby bypassing systemic dilution in the blood volume. FDA-approved T-VEC is being administered by IT injection to patients with metastatic skin melanoma [64]. At the preclinical level, many GBM-targeted OVs generated by our group and others have shown good anti-tumor activity following IT delivery into the brain [65,66]. However, disadvantages of IT delivery include the low response in uninjected distant metastases (i.e., the abscopal effect) and safety challenges when multiple doses are needed.
Intravenous delivery
IV, the second most common choice for OV administration in clinical trials (Figure I), enables OVs to access multiple metastatic sites regardless of their location. IV infusion of JX-594 selectively infected, replicated, and expressed transgene products in tumor tissues in patients with advanced solid metastatic tumors [67]. However, IV delivery of OVs into brain tumors may be less effective than IT delivery due to the BBB. Moreover, OVs delivered by IV injection (except for those not derived from human viruses, such as MYXV) may be cleared very quickly by pre-existing and induced neutralizing antiviral antibodies and other immune responses, thus limiting effective OV delivery to tumor sites.
Cell carrier delivery
In this method, selected cells are infected with OVs ex vivo and then administered for cancer treatment. The selected cells should be permissive to OV infection and should release the virus efficiently when contacting tumor cells. Selected cells can protect OVs from clearance in the circulation and allow them to traffic to the established tumor sites.
Irradiated transformed leukemia or myeloma cells might be better vehicles than transformed solid tumor cells as they circulate much more easily due to their small size [68]. Immune cells such as T lymphocytes, DCs, and monocytes/macrophages are effective cell carriers because they can circulate systemically and recognize tumor cells specifically [69]. Stem cells, including mesenchymal stem cells, neural stem cells, and adipose-derived stem cells, have intrinsic tumor-homing ability, making them the most promising cell carriers for OV delivery [70–72]. However, some limitations, such as lysis of the carrier cells by the virus before reaching tumor cells and their transduction/infection efficacy and safety, need to be considered [73].
Other delivery routes
IP delivery is ideal for abdominal tumors, as it is relatively easy to administer and requires few special techniques [74]. SC delivery is the typical route used in animal models [75]. IC or intraventricular delivery is frequently used to target brain tumors, as OVs release high concentrations of viruses locally [76,77]. IM delivery of oAd-MAGEA3 has been used to treat patients with MAGE-A3-positive solid tumors with potential anti-tumor responses [78]. Intranasal delivery of OV is a promising platform for glioma therapy [79]. IVE delivery of OV is a novel treatment for high-risk bladder cancer as it exposes tumor sites to high concentrations of viruses [80]. Inhalation and aerosols provide potentially noninvasive methods for delivering OVs, especially for lung tumors or metastatic liver cancer, which commonly metastasizes to lung [81,82].
Figure I. Administration routes for OVs in clinical trials.
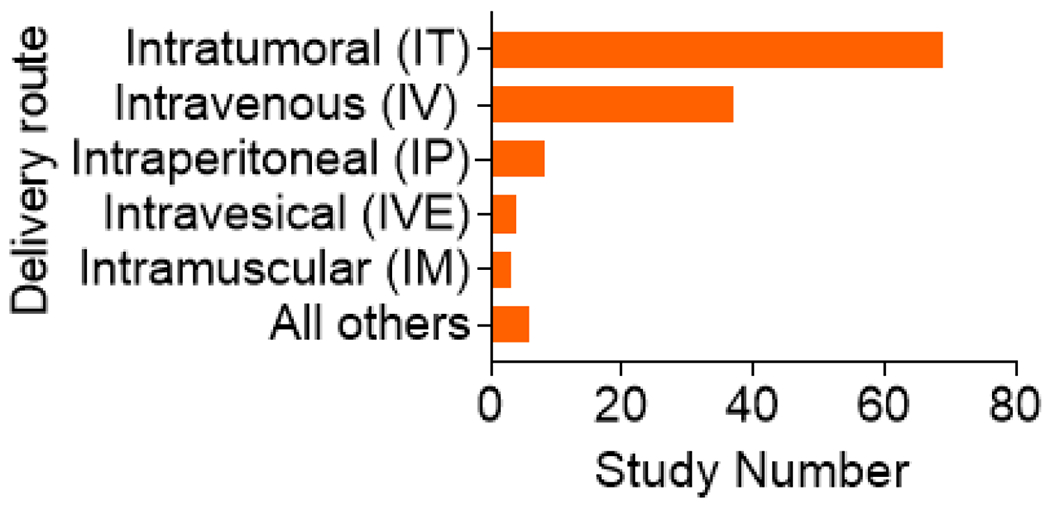
Delivery routes for OVs used in clinical trials from 2012 to 2022 as documented at ClinicalTrials.gov. IT was the most dominant route (n = 69), and IV was second (n = 37).
Current challenges and solutions to oncolytic virotherapy
Despite OVs’ anti-tumor potential, there are still some unique barriers for developing OVs into a new class of anti-cancer drugs, including inadequate OV penetration and spread, a host’s antiviral immunity, patient selection, and low or moderate efficacy when they are used as single agents. Thus, more practical studies are required.
OV penetration and spread
Physical barriers pose a big challenge to the penetration and spread of OVs as they require the virus to pass through endothelial layers to reach tumor sites. Extracellular matrix (ECM) is now known to actively contribute to tumor maintenance by interacting with tumor cells and cellular components in the TME. This complex of secreted proteins and proteoglycans from neoplastic and normal stromal cells is a major obstacle to OV transmission. OVs must pass through the ECM to enter tumor cells, lyse them, replicate, and spread to bystander tumor cells. Therefore, modulating the ECM is necessary for enhancing OV efficacy.
Engineered OVs, such as an oAd expressing relaxin (YDC002) and an oHSV armed with matrix metalloproteinase (MMP9), significantly improved penetration, spread, and persistence, leading to selective degradation of aberrant ECM and effective induction of tumor cell apoptosis [83,84]. Using viral fusion proteins to modify OVs is another approach. oHSV incorporated with human CDH1 (encoding E-cadherin) increased viral entry into GBM cells with cell membrane fusion and improved virus production, augmenting viral spread [77].
Host antiviral immunity
OV-infected cancer cells process and present virus-specific antigens on their surface, facilitating their recognition and destruction by antiviral immune cells such as T cells. The immunological events (e.g., PAMPs and viral antigens) that are provoked by OV-infected tumor cells facilitate the recruitment of effector immune cells against tumors [85]. The boosted antiviral immunity may benefit OV-induced anti-tumor immunity.
However, OVs can be cleared from the host’s immune system before they reach tumor sites. Our group demonstrated that NK cells impeded GBM virotherapy by preferentially lysing oHSV-infected tumor cells. These NK cells were recruited into the TME within 2 hours of viral infection. They recognized and cleared virally infected cells through the natural cytotoxicity receptors NKp30 and NKp46 [86]. Hence, initial suppression of immune cell recruitment or inhibition of inflammatory cell pathways could counteract immune cells’ tendency to clear viruses. For example, histone deacetylase inhibitors (HDACis) impaired NK cell function against oHSV-infected GBM cells by inhibiting STAT5/T-BET signaling, thereby augmenting the anti-tumor efficacy of oncolytic virotherapy [87]. HDACis also potentiated VSV replication and oncolysis in prostate cancer cells by stimulating NF-κB-dependent autophagy. Administering transforming growth factor β (TGF-β) prior to oHSV injection inhibited the innate immune response and improved anti-tumor activity in a GBM mouse model [88].
Patient selection for OV treatment
Most people, including cancer patients, have natural circulating neutralization antibodies against HSV, which may block the infection and expansion of oHSV. To overcome this obstacle, it is crucial in OV-based immunotherapy to select an appropriate OV administration method. In one rat study, pre-existing anti-HSV-1 antibodies significantly reduced but did not completely abolish the efficacy of gene transfer to brain tumors by a HSV-1 vector. In a phase III trial, however, circulating anti-HSV-1 antibodies failed to prevent the cell-to-cell spread of T-VEC after IT administration to patients with advanced melanoma [89]. IV injection of human EGFR2 (HER2) retargeted oHSV armed with IL-12 and GM-CSF, in combination with an anti-PD1 antibody, reduced lung tumor nodule formation in both HSV-naive and HSV-immune mice [90].
However, different tumor types, stages, and their heterogeneities pose a challenge in selecting appropriate patients for OV therapy. As many patients in clinical trials have already received numerous cycles of conventional therapy, their immune system has been disrupted, and tumor cells have been radically altered. Therefore, it is critical to identify tumor-specific biomarkers that might predict which patients are suitable for OV treatment.
Low or moderate efficacy of OV treatment as a single agent
OV administration as a single agent can reduce biosafety issues, complexity, and costs because only one therapeutic is required to be manufactured and administered. However, OVs must persist long enough to exert maximal therapeutic effects and induce sufficient oncolysis to generate long-lasting adaptive anti-tumor immunity. Unfortunately, a single OV administration may only have low or moderate efficacy because the host immune system may clear it from the circulation, the virus may express the transgene only briefly, and/or viral spread may be limited. Viral particles were cleared quickly (over 80% clearance within 3 days) from the brain of GBM mice treated with oHSV [91] or oAd [92]. oAd showed limited efficacy and poor persistence, especially in target tissues, in the clinic [93].
To prolong viral persistence, multiple OV injections and a higher initial dose can help, but they may cause safety concerns. Alternatively, OVs can be placed within carrier cells and trafficked to tumor sites to avoid clearance. However, transfection efficiency and lysis of the carrier cells are challenges. One possible solution is to combine OVs with TGF-β or low-dose chemotherapy to transiently suppress early immune responses to prolong viral persistence and improve OV delivery to tumor sites [94]. This approach requires careful consideration of the balance between antiviral response and anti-tumor immunity. Nevertheless, a safe combination of an OV with various immunotherapy strategies is likely required to improve the low or moderate efficacy of OV treatment.
Combination strategies with oncolytic virotherapy
As well as providing a promising platform for cancer therapy by itself, OV-based cancer immunotherapy offers an attractive opportunity for combination with other cancer therapies. This is especially true because, as discussed above, the efficacy of OV as a single agent has not yet been optimized for maximum anti-tumor effectiveness. Thus, OV treatments are now considered potent partners for immunotherapies. This section focuses mainly on combinations of OVs with other forms of cancer therapy (Figure 4).
Figure 4. Oncolytic viruses as the foundation of combination therapy for cancer.
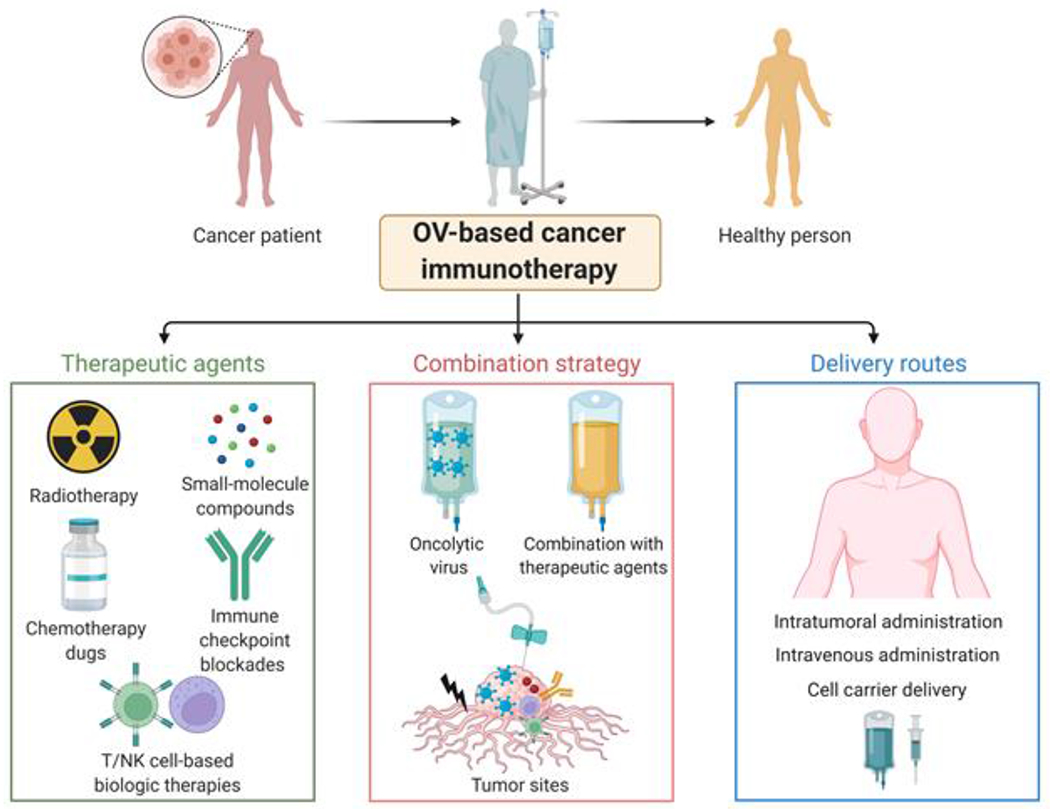
Conventional treatment strategies, such as radiotherapy, chemotherapeutic drugs, as well as small-molecule compounds and, more recently, immune checkpoint blockade and cell-based immunotherapy (CAR T/NK cell-based therapies) have been used to treat cancer. OVs provide a foundation and promising therapeutic platform for cancer treatment when combined with these conventional therapies and biologic therapies. Administration routes for delivering oncolytic viruses commonly include intratumoral injection, intravenous injection, and cell carriers.
Radiotherapy with OVs
OVs can be used as radiosensitizers. NIS (Na+/I− symporter) is an intrinsic plasma membrane protein that drives cellular uptake of radionuclides (e.g., 131I) [95]. Combined with radionuclide therapy, NIS-armed oncolytic VSV showed high IT viral replication, resulting in enhanced tumor regression and prolonged survival in either subcutaneous or orthotropic mouse tumor models [96]. Recently, oAd DNX-2401 plus radiotherapy has provided the rationale for combination immunotherapy in patients with newly diagnosed DIPG. Among 12 glioma patients receiving a single IT infusion of viral particles of DNX-2401, 11 patients received subsequent radiotherapy at a median dose of 54 Gy. Nine of 12 patients had enhanced T cell activity and reduced or stabilized tumor size [7].
Chemotherapy with OVs
Combining chemotherapy with OVs provides an alternative for cancer immunotherapy, as chemotherapy can enhance oncolytic virotherapy by enabling OVs to evade antiviral immune responses. Cyclophosphamide (CPA), a chemotherapeutic alkylating agent used as an immunosuppressive drug, in combination with oncolytic virotherapy induced apoptotic cell death of tumor cells and affected humoral and cellular mediators of both the innate and adaptive immune responses. CPA has shown global immunosuppressive function, thus potentiating viral oncolysis and improving the anti-tumor efficacy of HSV, Ad, MV, reovirus, and VACV [97,98]. CPA plus GM-CSF-armed-oAd boosted anti-tumor immunity by inhibiting regulatory T cells and inducing T helper type 1 immunity to counteract the immunosuppressive TME [99]. Additionally, OVs can be used as adjuvants for chemotherapy, particularly for in the setting of drug-resistant tumors. Although chemotherapy can enhance OV therapy by preventing OV elimination due to antiviral immunity, it may also diminish OV therapeutic efficacy. For example, low-dose CPA removed immunosuppressive cells (e.g., Treg cells) to improve vaccine-induced adaptive antitumor immune responses while promoting the antiviral immune response to clear the virus early [97]. However, high-dose CPA enhanced viral oncolytic capacity by widespread immunosuppression of innate and adaptive antiviral immune responses, but also completely abrogated antitumor immune responses [100]. Temozolomide, a current standard of care for GBM, adversely affected oHSV immunovirotherapy [101]. These results illustrate that the clinic’s combination of chemotherapy and OV therapy should be carefully considered.
Targeted small molecular agents plus OVs
Various classes of small molecular compounds have been used to inhibit antiviral signaling pathways, and they have the potential to enhance OV replication or even address OV resistance in tumor cells. The FDA-approved drug ruxolitinib, a specific JAK-1/2 inhibitor, enhanced the activity of numerous OVs by countering antiviral JAK/STAT signaling and no toxicities were observed in various preclinical studies [102]. The stimulator of interferon gene (STING) signaling pathway is a critical natural immune pathway against viral infection, resulting in tumor immune evasion [103,104]. Therefore, oncolytic virotherapy may benefit from STING deficiency or dysfunction when it incorporates specific inhibitors of STING.
Immune checkpoint blockade with OVs
Immune checkpoints are regulators of the immune system and are crucial for self-tolerance, which prevents the immune system from attacking cells indiscriminately. However, some cancers can protect themselves from attack by stimulating immune checkpoint targets. ICB therapy interrupts immunosuppressive tumor signals and normalizes anti-tumor immune responses by targeting checkpoint receptors [e.g., PD-1, CTLA-4, lymphocyte activation gene-3 (LAG-3), and T cell immunoglobulin and ITIM domain (TIGIT)] or ligands (e.g., PD-L1) [105]. Although combining OVs and ICB may result in more robust clearance of the viruses, it can achieve a more immunogenic TME and enhances anti-tumor efficacy.
Multiple ongoing clinical trials are combining OVs with ICBs, but PD-1/PD-L1 and CTLA-4 plus OV combination therapies have advanced the furthest. T-VEC combined with an anti-PD-1 antibody (pembrolizumab) produced encouraging anti-tumor outcomes in a phase Ib trial in patients with stages IIB and IV melanoma. It altered the TME, increased the infiltration of cytotoxic CD8+ T cells into tumor sites, and elevated IFNγ levels [40]. Although it was stopped because of clinical futility in a phase III trial, this combination therapy may warrant further clinical testing if the best treatment time points can be determined [41]. Another phase Ib trial (NCT01740297) that combined T-VEC with an anti-CTLA-4 antibody (ipilimumab) showed enhanced anti-tumor efficacy and produced tolerable safety profiles in patients with stage IIIB-IV melanoma [106]. In a subsequent phase II study, this combination therapy obtained a significantly higher objective response rate (39%) compared to that achieved with ipilimumab alone (18%) [107].
Adoptive T cell or CAR-T cell therapy with OVs
Adoptive T-cell transfer (ACT) therapy has contributed significantly to cancer treatment. ACT therapy benefits from being combined with OVs, particularly for solid tumor treatment, due to extended T cell persistence and greater anti-tumor response mediated by OVs armed with cytokine(s) or chemokine(s). IT administration of an oAd armed with hGM-CSF and combined with ovalbumin-specific CD8+ T-cells (OT-I) that had been activated ex vivo had increased the number of endogenous CD8+ T cells, resulting in the rejection of B16.F10 tumor re-challenge [108]. OVs armed with CXCL9-11 or CCL5 also increased T cell infiltration and persistence in the TME [109,110]. TAA-engineered OVs act as vaccines to stimulate T cells, resulting in maximum antigen-targeted responses [111,112]. Moreover, pretreating established tumors with OVs can reprogram the TME from immunologically “cold” to “hot,” improving the recruitment and effector functions of T cells.
Tumor-infiltrating lymphocytes (TILs) can be readily expanded in vitro (cell number up to 1011) and selected for recognition of tumors. A combination of OVs and TILs with effector functions is being investigated for enhanced anti-tumor activity in ovarian and pancreatic carcinoma mouse models [113,114]. Careful choice of the CAR target can achieve better tumor selectivity and reduce CAR targets, has shown promising results in multiple mouse cancer models. Park et al. designed an oncolytic VACV that delivered CD19, a naturally occurring antigen, to tumor cells, which could then be targeted with CD19-specific CAR-T cells. The dead tumor cells released additional virus copies, propagating CD19 expression to adjacent tumor cells and thus inducing more effective anti-tumor responses [115]. Oncolytic VSV or reovirus combined with dual-specific CAR-T cells potentiated CAR-T cell efficacy in subcutaneous melanoma and intracranial glioma mouse models. The OVs stimulated memory CAR-T cells with a specific viral T cell receptor, thereby enhancing T cell proliferation and CAR-directed anti-tumor function [116]. However, CAR-T cell therapy has produced certain serious adverse events, such as cytokine release syndrome (CRS). Thus, safety profiles need to be considered when using CAR-T cells as monotherapy or in combination with oncolytic virotherapy.
Adoptive NK or CAR-NK cell therapy with OVs
NK cells are a critical component of the innate immune system. Compared with T cells, NK cells offer multifaceted advantages for tumor immunotherapy. They do not require human leukocyte antigen (HLA) matching and produce no Graft vs. Host Disease (GvHD) [117,118]. Also, unlike adoptive T cell therapy, which to date has been autologous or HLA matched, it is possible to use the same batch of unmatched, allogeneic NK cells to treat the same type of cancer in different patients. The “off-the-shelf’ characteristic of NK cells makes them more suitable for commercial development. We successfully used umbilical cord blood (UCB) to derive NK cells expressing PD-L1 and secreting IL-15 (COH06). We demonstrated that COH06 killed more tumor cells with or without a PD-L1 inhibitor (atezolizumab), and that secreted IL-15 allowed the NK cells to live longer. Our group is treating non-small cell lung cancer patients for a phase I trial with these engineered NK cells (NCT05334329).
To enhance NK cell anti-tumor effects, OVs can be modified to express NK-stimulating cytokines such as IL-12, IL-15, and IL-18. Such modified OVs combined with NK cells significantly inhibited tumor cell growth in an NK-dependent manner because the OVs released potent cytokines into the TME [119]. However, combining a chemokine with a matched receptor can improve NK cell homing and therapeutic efficacy. Indeed, a combination of a CCL5-modified oncolytic VACV with CCR5-overexpressing NK cells was more effective than the corresponding single agents in a colon cancer model [120].
As with CAR-T cell therapy, NK cells can be engineered to express CARs, so they specifically target tumor cells. CAR-NK cells—which can be derived from UCB, induced pluripotent stem cells (iPSCs), peripheral blood mononuclear cells (PBMCs), or NK cell lines such as NK-92—can be manufactured and stored as “off-the-shelf’ products [121]. In a phase I-II trial (NCT03056339) of 11 patients with relapsed or refractory CD19-positive cancers, UCB-derived CAR-NK cells expanded and persisted for >12 months without neurotoxicity, CRS, or GvHD [122]. In a breast cancer metastatic CNS tumor model, combining IT administration of oHSV with CAR-NK-92 cells targeting EGFR improved anti-tumor efficacy and prolonged survival [123]. We demonstrated a synergistic anti-tumor effect when OV-IL15C was combined with “off-the-shelf’ EGFR-CAR-NK cells derived from PBMCs in an orthotopic GBM mouse model [56].
Concluding remarks
Here, we reviewed OVs that mediate immunotherapy in cancer treatment, either as single agents or when combined with other strategies. Survival outcomes in experimental models are generally improved when OVs are combined with ICBs, chemotherapy drugs, or other strategies. At the clinical stage, OVs are increasingly recognized as promising therapeutic tools against various advanced tumors and thus far, combination therapies have not produced additional observable toxicity.
Increased understanding of the molecular interactions between the immune system and viral pathogens and tumor cells will help us design better anti-tumor OVs. Also, it is important to select the most suitable OV vector for a given immunotherapeutic role. For example, viruses such as HSV, Ads, and VACV, which replicate more slowly and therefore can sustain transgene production, can reliably deliver immune-modulatory agents into the TME, changing a “cold” immunosuppressive TME into a “hot” inflammatory TME [124]. Additional research is needed to minimize antiviral activity but maximize anti-tumor activity as OV progresses (see Outstanding questions). This in turn should aid in the design of more effective and safer combination therapies.
Outstanding Questions.
To optimize therapeutic impact, how can we determine the best OV for each tissue and each type of cancer within a tissue?
How can we protect OV-induced anti-tumor benefits from antiviral immunity to obtain better anti-tumor activity?
How can we choose the best administration approach for each OV to maintain therapeutic potency against cancers while preventing the innate and/or adaptive immune system from eliminating the OV?
How can we improve constructs and reduce barriers such as inadequate viral persistence to ensure OV therapeutic efficacy?
Which payloads can be best used as OV generation targets to turn a “cold” tumor into a “hot” tumor?
Which host biomarkers could predict clinical responses or help select appropriate patients for OV?
How can we determine the best combination strategy among multi-modality treatments with OVs? When using a combination therapy, what study design should be used to determine the sequence of treatments and the time points at which they should be given?
How can we improve safety profiles when OVs are delivered systemically to treat metastatic tumors?
For those cancers with no current curative regimen, can we use experimental OV-based cancer immunotherapy at an earlier stage of the disease rather than assessing toxicity and evidence of anti-tumor activity in patients with end-stage disease?
In summary, OV-based virotherapy is an emerging form of cancer immunotherapy. Challenges to its success include, but are not limited to, inadequate OV persistence, limited viral replication and spread in the TME, low therapeutic efficacy as a monotherapy, and safety profiles of systemic treatment. Accordingly, an improved understanding of the interactions among OVs, immune cells, tumor cells, and other components of the TME may contribute to developing more innovative OVs and ultimately better clinical outcomes in patients.
Supplementary Material
Highlights.
Natural or genetically modified oncolytic viruses (OVs) are promising anti-tumor agents for specifically targeting tumor cells without harming normal cells.
OV-based oncolysis and OV-induced antiviral immune responses play an important role in remodeling the tumor microenvironment (TME) from immunologically “cold” to “hot.”
As single agents or combined with other strategies, OVs can enhance cancer immunotherapy.
OV persistence, replication, spread, systemic delivery, lack of therapeutic biomarkers, and moderate efficacy as single agents are among the main challenges in the field. If these problems can be solved, OV will most likely enhance clinical outcomes for cancer patients substantially.
Acknowledgments
This work was supported by grants from the National Institutes of Health (CA210087, CA265095, and CA163205 to M.A.C.; NS106170, AI129582, CA247550, CA264512, CA266457 and CA223400 to J.Y.) and the Leukemia and Lymphoma Society (1364-19 to J.Y.). The authors regret that it was not possible to include many interesting studies in the field due to limited space. Images were created with BioRender.com and GraphPad Prism 9.
Glossary
- Bi-specific T cell engager (BiTE)
an artificial fusion protein comprising two different single-chain variable fragments (scFvs)—one targets a molecule expressed on the surface of immune cells and the other targets tumor-specific antigen(s), which induces the activation and cytotoxicity of T cells.
- Chimeric antigen receptor (CAR)
A special and artificial receptor designed to express in immune cells (e.g., T or NK cells) and bind to certain proteins on cancer cells. This helps the T cells specifically spot and kill cancer cells.
- Cytokine release syndrome (CRS)
a large, rapid release of cytokines into the blood from immune cells affected by the immunotherapy. It occurs when the immune system responds to immunotherapy drugs more aggressively than it should. CRS symptoms include fever, headache, body ache, rapid heartbeat, nausea, fatigue, low blood pressure, and trouble breathing.
- Cytotoxic T lymphocyte-associated antigen-4 (CTLA-4)
an immune checkpoint receptor that is expressed on T cells, which suppresses T cell effector function.
- Damage-associated molecular patterns (DAMPs)
endogenous molecules, such as extracellular matrix, intracellular proteins (e.g., high-mobility group box 1 [HMGB1] and heat shock proteins), and non-proteins (e.g., ATP and DNA), released from damaged or dying cells due to cell death or tissue injury, promoting pathological inflammatory responses.
- Graft vs. Host Disease (GvHD)
a systemic disorder that occurs when the graft’s (transplanted cells from the donor) immune cells recognize the host (recipient that received the transplant) as foreign and attack the host’s cells.
- Immune checkpoint blockade (ICB)
A molecule inhibiting immune checkpoint molecules (e.g., PD-1 and CTLA-4) from binding their partner proteins such as PD-L1 (for PD-1), CD80, and CD86 (for CTLA-4), allowing immune cells to recognize and react against tumor cell antigens.
- Oncolytic viruses (OVs)
naturally existing or genetically modified viruses that can selectively replicate in tumor cells and then kill them without harming the healthy cells.
- Pathogen-associated molecular patterns (PAMPs)
small molecules (e.g., bacterial lipopolysaccharides (LPSs) and endotoxins) derived from bacteria or viruses that evoke an inflammatory reaction, triggering innate immune responses and protecting the host from infection.
- Programmed cell death protein-1 (PD-1)
an immune checkpoint receptor expressed on T cells’ cell surface, which inhibits T cell immune response.
- Tri-specific T cell engager (TriTE)
a next-generation specific T cell engager consisting of three domains (such as CD3 × dual tumor antigens or tumor antigen × CD3/CD28), promoting tumor cell binding and T cell anti-tumor response.
- Tumor-associated antigens (TAAs)
antigen molecules aberrantly and highly expressed on tumor cells, which at times can be recognized by the adaptive immune system. TAAs are useful tumor markers in identifying tumor cells with diagnostic tests and are potential candidates for use in cancer therapy in the form of antibodies, CARs, etc.
- Tumor microenvironment (TME)
a complex niche resulting from a tumor that often includes a large variety of cell types, of both cancerous and noncancerous origins.
- Type I interferon (IFN)
a type of cytokines that mediate inflammation, immunoregulation, tumor cell recognition, and anti-tumor or antiviral immunity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
E.A.C. is currently (within last year) an advisor to Amacathera, Bionaut Labs, Candel Therapeutics Inc., Genenta, inc., Insightec, Inc., DNAtrix Inc, Seneca Therapeutics, Synthetic Biologics. He has equity options in Bionaut Laboratories, DNAtrix, Immunomic Therapeutics, Seneca Therapeutics, Synthetic Biologics, Ternalys Therapeutics. He is co-founder and on the Board of Directors of Ternalys Therapeutics. E.A.C, M.A.C., and J.Y. have oncolytic virus patents awarded or pending. M.A.C. and J.Y are co-founders of CytoImmune Therapeutics. None of the remaining authors have any interests to declare.
References
- 1.Poh A (2016) First Oncolytic Viral Therapy for Melanoma. Cancer Discov 6, 6. [DOI] [PubMed] [Google Scholar]
- 2.Todo T et al. (2022) Intratumoral oncolytic herpes virus G47Δ for residual or recurrent glioblastoma: a phase 2 trial. Nat Med. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaufman HL et al. (2015) Oncolytic viruses: a new class of immunotherapy drugs. Nature reviews Drug discovery 14, 642–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haseley A et al. (2009) Advances in oncolytic virus therapy for glioma. Recent Patents on CNS Drug Discovery (Discontinued) 4, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tremaglio CZ et al. (2021) Chapter 2 - Genetic instability of RNA viruses. In Genome Stability (Second Edition) (Kovalchuk I and Kovalchuk O, eds), pp. 23–38, Academic Press [Google Scholar]
- 6.Chiocca EA et al. (2004) A Phase I Open-Label, Dose-Escalation, Multi-Institutional Trial of Injection with an E1B-Attenuated Adenovirus, ONYX-015, into the Peritumoral Region of Recurrent Malignant Gliomas, in the Adjuvant Setting. Molecular Therapy 10, 958–966. [DOI] [PubMed] [Google Scholar]
- 7.Gállego Pérez-Larraya J et al. (2022) Oncolytic DNX-2401 Virus for Pediatric Diffuse Intrinsic Pontine Glioma. New England Journal of Medicine 386, 2471–2481. [DOI] [PubMed] [Google Scholar]
- 8.Nakashima H and Chiocca EA (2014) Modification of HSV-1 to an oncolytic virus. Methods Mol Biol 1144, 117–127. [DOI] [PubMed] [Google Scholar]
- 9.Friedman GK et al. (2021) Oncolytic HSV-1 G207 Immunovirotherapyfor Pediatric High-Grade Gliomas. New England Journal of Medicine 384, 1613–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu X et al. (2006) Effective treatment of pancreatic cancer xenografts with a conditionally replicating virus derived from type 2 herpes simplex virus. Clin Cancer Res 12, 3152–3157. [DOI] [PubMed] [Google Scholar]
- 11.Park SH et al. (2015) Phase 1b Trial of Biweekly Intravenous Pexa-Vec (JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus in Colorectal Cancer. Molecular Therapy 23, 1532–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jochems C et al. (2014) A combination trial of vaccine plus ipilimumab in metastatic castration-resistant prostate cancer patients: immune correlates. Cancer Immunol Immunother 63, 407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pisklakova A et al. (2016) M011L-deficient oncolytic myxoma virus induces apoptosis in brain tumor-initiating cells and enhances survival in a novel immunocompetent mouse model of glioblastoma. Neuro Oncol 18, 1088–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nounamo B et al. (2017) Myxoma Virus Optimizes Cisplatin for the Treatment of Ovarian Cancer In Vitro and in a Syngeneic Murine Dissemination Model. Mol Ther Oncolytics 6, 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villa NY et al. (2016) Ex vivo virotherapy with myxoma virus does not impair hematopoietic stem and progenitor cells. Cytotherapy 18, 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galanis E et al. (2015) Oncolytic measles virus expressing the sodium iodide symporter to treat drug-resistant ovarian cancer. Cancer Res 75, 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dispenzieri A et al. (2017) Phase I trial of systemic administration of Edmonston strain of measles virus genetically engineered to express the sodium iodide symporter in patients with recurrent or refractory multiple myeloma. Leukemia 31, 2791–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiocca EA (2002) Oncolytic viruses. Nat Rev Cancer 2, 938–950. 10.1038/nrc948 [DOI] [PubMed] [Google Scholar]
- 19.Msaouel P et al. (2013) Oncolytic measles virus strains as novel anticancer agents. Expert Opln Biol Ther 13, 483–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kambara H et al. (2005) An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res 65, 2832–2839. [DOI] [PubMed] [Google Scholar]
- 21.Mazzacurati L et al. (2015) Use of miRNA response sequences to block off-target replication and increase the safety of an unattenuated, glioblastoma-targeted oncolytic HSV. Mol Ther 23, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia T et al. (2016) Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res 76, 6747–6759. [DOI] [PubMed] [Google Scholar]
- 23.Xia T et al. (2016) Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep 14, 282–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lipatova AV et al. (2021) Multi-Omics Analysis of Glioblastoma Cells’ Sensitivity to Oncolytic Viruses. Cancers (Basel) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seymour LW and Fisher KD (2016) Oncolytic viruses: finally delivering. British Journal of Cancer 114, 357–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buijs PR et al. (2015) Oncolytic viruses: From bench to bedside with a focus on safety. Hum Vaccin Immunother 11, 1573–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez-Quintanilla J et al. (2019) Oncolytic viruses: overcoming translational challenges. J Clin Invest 129, 1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaufman HL et al. (2015) Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 14, 642–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sathaiah M et al. (2012) Oncolytic poxvirus armed with Fas ligand leads to induction of cellular Fas receptor and selective viral replication in FasR-negative cancer. Cancer Gene Ther 19, 192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaddy DF and Lyles DS (2007) Oncolytic vesicular stomatitis virus induces apoptosis via signaling through PKR, Fas, and Daxx. J Virol 81, 2792–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paul S and Lal G (2017) The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front Immunol 8, 1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saha D et al. (2017) Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 32, 253–267.e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H et al. (2022) Oncolytic adenoviruses synergistically enhance anti-PD-L1 and anti-CTLA-4 immunotherapy by modulating the tumour microenvironment in a 4T1 orthotopic mouse model. Cancer Gene Ther 29, 456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma J et al. (2020) Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis 11, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Z et al. (2022) ILC1s control leukemia stem cell fate and limit development of AML. Nat Immunol 23, 718–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kansler ER et al. (2022) Cytotoxic innate lymphoid cells sense cancer cell-expressed interleukin-15 to suppress human and murine malignancies. Nat Immunol 23, 904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Serafini N et al. (2022) Trained ILC3 responses promote intestinal defense. Science 375, 859–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melcher A et al. (2021) Oncolytic virotherapy as immunotherapy. Science 374, 1325–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaufman HL et al. (2005) Targeting the local tumor microenvironment with vaccinia virus expressing B7.1 for the treatment of melanoma. J Clin Invest 115, 1903–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ribas A et al. (2017) Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 170, 1109–1119.e1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dimitriou F et al. (2022) Double Trouble: Immunotherapy Doublets in Melanoma—Approved and Novel Combinations to Optimize Treatment in Advanced Melanoma. American Society of Clinical Oncology Educational Book, 1–22. [DOI] [PubMed] [Google Scholar]
- 42.Conry RM et al. (2018) Talimogene laherparepvec: First in class oncolytic virotherapy. Hum Vaccin Immunother 14, 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang M (2018) Oncorine, the World First Oncolytic Virus Medicine and its Update in China. Curr Cancer Drug Targets 18, 171–176. [DOI] [PubMed] [Google Scholar]
- 44.Xia ZJ et al. (2004) [Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus]. Ai Zheng 23, 1666–1670 [PubMed] [Google Scholar]
- 45.Pecora AL et al. (2002) Phase I trial of intravenous administration of PV701, an oncolytic virus, in patients with advanced solid cancers. J Clin Oncol 20, 2251–2266. [DOI] [PubMed] [Google Scholar]
- 46.Liu W et al. (2022) Oncolytic adenovirus-mediated intratumoral expression of TRAIL and CD40L enhances immunotherapy by modulating the tumor microenvironment in immunocompetent mouse models. Cancer Lett 535, 215661. [DOI] [PubMed] [Google Scholar]
- 47.Passaro C et al. (2019) Arming an Oncolytic Herpes Simplex Virus Type 1 with a Single-chain Fragment Variable Antibody against PD-1 for Experimental Glioblastoma Therapy. Clin Cancer Res 25, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu B et al. (2021) An oncolytic virus expressing a full-length antibody enhances antitumor innate immune response to glioblastoma. Nature Communications 12, 5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tian L et al. (2022) Targeting Fc Receptor-Mediated Effects and the “Don’t Eat Me” Signal with an Oncolytic Virus Expressing an Anti-CD47 Antibody to Treat Metastatic Ovarian Cancer. Clin Cancer Res 28, 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hellums EK et al. (2005) Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro Oncol 7, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parker JN et al. (2000) Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci U S A 97, 2208–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saha D et al. (2018) Combinatorial Effects of VEGFR Kinase Inhibitor Axitinib and Oncolytic Virotherapy in Mouse and Human Glioblastoma Stem-Like Cell Models. Clin Cancer Res 24, 3409–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen L et al. (2021) Intratumoral expression of interleukin 23 variants using oncolytic vaccinia virus elicit potent antitumor effects on multiple tumor models via tumor microenvironment modulation. Theranostics 11, 6668–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pol JG et al. (2020) Cytokines in oncolytic virotherapy. Cytokine Growth Factor Rev 56, 4–27. [DOI] [PubMed] [Google Scholar]
- 55.Kowalsky SJ et al. (2018) Superagonist IL-15-Armed Oncolytic Virus Elicits Potent Antitumor Immunity and Therapy That Are Enhanced with PD-1 Blockade. Mol Ther 26, 2476–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma R et al. (2021) An Oncolytic Virus Expressing IL15/IL15Rα Combined with Off-the-Shelf EGFR-CAR NK Cells Targets Glioblastoma. Cancer Research 81, 3635–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu F et al. (2014) T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol Ther 22, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fajardo CA et al. (2017) Oncolytic adenoviral delivery of an EGFR-targeting T-cell engager improves antitumor efficacy. Cancer research 77, 2052–2063 [DOI] [PubMed] [Google Scholar]
- 59.Speck T et al. (2018) Targeted BiTE expression by an oncolytic vector augments therapeutic efficacy against solid tumors. Clinical Cancer Research 24, 2128–2137 [DOI] [PubMed] [Google Scholar]
- 60.Yu F et al. (2017) A T-cell engager-armed oncolytic vaccinia virus to target the tumor stroma. Cancer Translational Medicine 3. [Google Scholar]
- 61.Chan WK et al. (2018) A CS1-NKG2D Bispecific Antibody Collectively Activates Cytolytic Immune Cells against Multiple Myeloma. Cancer Immunol Res 6, 776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ravirala D et al. (2021) Co-delivery of novel bispecific and trispecific engagers by an amplicon vector augments the therapeutic effect of an HSV-based oncolytic virotherapy. J Immunother Cancer 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reale A et al. (2021) Giving Oncolytic Viruses a Free Ride: Carrier Cells for Oncolytic Virotherapy. Pharmaceutics 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Andtbacka RH et al. (2016) Patterns of clinical response with talimogene laherparepvec (T-VEC) in patients with melanoma treated in the OPTiM phase III clinical trial. Annals of surgical oncology 23, 4169–4177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Otani Y et al. (2020) Oncolytic HSV-Infected Glioma Cells Activate NOTCH in Adjacent Tumor Cells Sensitizing Tumors to Gamma Secretase Inhibition. Clin Cancer Res 26, 2381–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Han J et al. (2015) TGFβ Treatment Enhances Glioblastoma Virotherapy by Inhibiting the Innate Immune Response. Cancer Res 75, 5273–5282. 10.1158/0008-5472.Can-15-0894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Breitbach CJ et al. (2011) Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 477, 99–102. [DOI] [PubMed] [Google Scholar]
- 68.Möller C et al. (2003) Expression and function of chemokine receptors in human multiple myeloma. Leukemia 17, 203–210. [DOI] [PubMed] [Google Scholar]
- 69.Muthana M et al. (2013) Macrophage delivery of an oncolytic virus abolishes tumor regrowth and metastasis after chemotherapy or irradiation. Cancer research 73, 490–495 [DOI] [PubMed] [Google Scholar]
- 70.Studeny M et al. (2004) Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. Journal of the National Cancer Institute 96, 1593–1603 [DOI] [PubMed] [Google Scholar]
- 71.Aboody KS et al. (2000) Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proceedings of the National Academy of Sciences 97, 12846–12851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ahmed AU et al. (2013) A preclinical evaluation of neural stem cell–based cell carrier for targeted antiglioma oncolytic virotherapy. Journal of the National Cancer Institute 105, 968–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nakashima H and Chiocca EA (2014) Switching a Replication-Defective Adenoviral Vector into a Replication-Competent, Oncolytic Adenovirus. Journal of Virology 88, 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sugawara K et al. (2020) Efficacy of a Third-Generation Oncolytic Herpes Virus G47Δ in Advanced Stage Models of Human Gastric Cancer. Molecular Therapy-Oncolytics 17, 205–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Phuong LK et al. (2003) Use of a Vaccine Strain of Measles Virus Genetically Engineered to Produce Carcinoembryonic Antigen as a Novel Therapeutic Agent against Glioblastoma Multiforme. Cancer Research 63, 2462–2469 [PubMed] [Google Scholar]
- 76.Ma R et al. (2021) An oncolytic virus expressing IL-15/1L-15Rα combined with off-the-shelf EGFR-CAR NK cells targets glioblastoma. Cancer Res. 10.1158/0008-5472.Can-21-0035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu B et al. (2018) An oncolytic herpesvirus expressing E-cadherin improves survival in mouse models of glioblastoma. Nat Biotechnol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jonker DJ et al. (2017). Phase I study of oncolytic virus (OV) MG1 maraba/MAGE-A3 (MG1MA3), with and without transgenic MAGE-A3 adenovirus vaccine (AdMA3) in incurable advanced/metastatic MAGE-A3-expressing solid tumours: CCTG IND. 214. American Society of Clinical Oncology [Google Scholar]
- 79.Dey M et al. (2016) Intranasal oncolytic virotherapy with CXCR4-enhanced stem cells extends survival in mouse model of glioma. Stem Cell Reports 7, 471–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Simpson GR et al. (2012) Combination of a fusogenic glycoprotein, pro-drug activation and oncolytic HSV as an intravesical therapy for superficial bladder cancer. British Journal of Cancer 106, 496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Low N et al. (2015) A randomized, controlled trial of an aerosolized vaccine against measles. New England Journal of Medicine 372, 1519–1529 [DOI] [PubMed] [Google Scholar]
- 82.Agarkhedkar S et al. (2014) Safety and immunogenicity of dry powder measles vaccine administered by inhalation: a randomized controlled Phase I clinical trial. Vaccine 32, 6791–6797 [DOI] [PubMed] [Google Scholar]
- 83.Jung KH et al. (2017) Oncolytic adenovirus expressing relaxin (YDC002) enhances therapeutic efficacy of gemcitabine against pancreatic cancer. Cancer Lett 396, 155–166. [DOI] [PubMed] [Google Scholar]
- 84.Sette P et al. (2019) GBM-Targeted oHSV Armed with Matrix Metalloproteinase 9 Enhances Anti-tumor Activity and Animal Survival. Mol Ther Oncolytics 15, 214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gujar S et al. (2018) Antitumor Benefits of Antiviral Immunity: An Underappreciated Aspect of Oncolytic Virotherapies. Trends in Immunology 39, 209–221. [DOI] [PubMed] [Google Scholar]
- 86.Alvarez-Breckenridge CA et al. (2012) NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nature medicine 18, 1827–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Alvarez-Breckenridge CA et al. (2012) The histone deacetylase inhibitor valproic acid lessens NK cell action against oncolytic virus-infected glioblastoma cells by inhibition of STAT5/T-BET signaling and generation of gamma interferon. J Virol 86, 4566–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Han J et al. (2015) TGFβ Treatment Enhances Glioblastoma Virotherapy by Inhibiting the Innate Immune Response. Cancer Research 75, 5273–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Harrington KJ et al. (2015) Clinical development of talimogene laherparepvec (T-VEC): a modified herpes simplex virus type-1–derived oncolytic immunotherapy. Expert Review of Anticancer Therapy 15, 1389–1403. [DOI] [PubMed] [Google Scholar]
- 90.De Lucia M et al. (2020) Retargeted and Multi-cytokine-Armed Herpes Virus Is a Potent Cancer Endovaccine for Local and Systemic Anti-tumor Treatment. Molecular Therapy - Oncolytics 19, 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fulci G et al. (2006) Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci U S A 103, 12873–12878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lamfers ML et al. (2006) Cyclophosphamide increases transgene expression mediated by an oncolytic adenovirus in glioma-bearing mice monitored by bioluminescence imaging. Mol Ther 14, 779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jung B-K et al. (2017) A hydrogel matrix prolongs persistence and promotes specific localization of an oncolytic adenovirus in a tumor by restricting nonspecific shedding and an antiviral immune response. Biomaterials 147, 26–38. [DOI] [PubMed] [Google Scholar]
- 94.Groeneveldt C et al. (2020) Immunotherapeutic Potential of TGF-β Inhibition and Oncolytic Viruses. Trends Immunol 41, 406–420. [DOI] [PubMed] [Google Scholar]
- 95.Hingorani M et al. (2010) The biology of the sodium iodide symporter and its potential for targeted gene delivery. Curr Cancer Drug Targets 10, 242–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lau L et al. (2015) DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350, 568–571. [DOI] [PubMed] [Google Scholar]
- 97.Ikeda K et al. (1999) Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat Med 5, 881–887. [DOI] [PubMed] [Google Scholar]
- 98.Qiao J et al. (2008) Cyclophosphamide facilitates antitumor efficacy against subcutaneous tumors following intravenous delivery of reovirus. Clin Cancer Res 14, 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cerullo V et al. (2011) Immunological effects of low-dose cyclophosphamide in cancer patients treated with oncolytic adenovirus. Mol Ther 19, 1737–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Prestwich RJ et al. (2008) Oncolytic viruses: a novel form of immunotherapy. Expert Rev Anticancer Ther 8, 1581–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Saha D et al. (2020) Temozolomide antagonizes oncolytic immunovirotherapy in glioblastoma. J Immunother Cancer 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ghonime MG and Cassady KA (2018) Combination Therapy Using Ruxolitinib and Oncolytic HSV Renders Resistant MPNSTs Susceptible to Virotherapy. Cancer Immunol Res 6, 1499–1510. [DOI] [PubMed] [Google Scholar]
- 103.Ishikawa H et al. (2009) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lau L et al. (2015) DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350, 568–571 [DOI] [PubMed] [Google Scholar]
- 105.Barrueto L et al. (2020) Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl Oncol 13, 100738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Puzanov I et al. (2016) Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J Clin Oncol 34, 2619–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chesney J et al. (2018) Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J Clin Oncol 36, 1658–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tähtinen S et al. (2015) Adenovirus Improves the Efficacy of Adoptive T-cell Therapy by Recruiting Immune Cells to and Promoting Their Activity at the Tumor. Cancer Immunol Res 3, 915–925. [DOI] [PubMed] [Google Scholar]
- 109.Eckert EC et al. (2020) Generation of a tumor-specific chemokine gradient using oncolytic vesicular stomatitis virus encoding CXCL9. Molecular Therapy-Oncolytics 16, 63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li J et al. (2011) Chemokine Expression From Oncolytic Vaccinia Virus Enhances Vaccine Therapies of Cancer. Molecular Therapy 19, 650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bridle BW et al. (2013) Oncolytic vesicular stomatitis virus quantitatively and qualitatively improves primary CD8+ T-cell responses to anticancer vaccines. Oncoimmunology 2, e26013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pol JG et al. (2014) Maraba virus as a potent oncolytic vaccine vector. Molecular Therapy 22, 420–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Havunen R et al. (2017) Oncolytic Adenoviruses Armed with Tumor Necrosis Factor Alpha and Interleukin-2 Enable Successful Adoptive Cell Therapy. Mol Ther Oncolytics 4, 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cervera-Carrascon V et al. (2020) Comparison of Clinically Relevant Oncolytic Virus Platforms for Enhancing T Cell Therapy of Solid Tumors. Mol Ther Oncolytics 17, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Park AK et al. (2020) Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Science Translational Medicine 12, eaaz1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Evgin L et al. (2022) Oncolytic virus-mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice. Sci Transl Med 14, eabn2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu E et al. (2020) Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med 382, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ran GH et al. (2022) Natural killer cell homing and trafficking in tissues and tumors: from biology to application. Signal Transduct Target Ther 7, 205. 10.1038/s41392-022-01058-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hodgins JJ et al. (2019) Killers 2.0: NK cell therapies at the forefront of cancer control. The Journal of Clinical Investigation 129, 3499–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang Z et al. (2020) Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579, 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tang X et al. (2018) First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res 8, 1083–1089 [PMC free article] [PubMed] [Google Scholar]
- 122.Liu E et al. (2020) Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. New England Journal of Medicine 382, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chen X et al. (2016) A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 7, 27764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Twumasi-Boateng K et al. (2018) Oncolytic viruses as engineering platforms for combination immunotherapy. Nature Reviews Cancer 18, 419–432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.