

Summary
The entorhinal cortex (EC) is the brain region that often exhibits the earliest histological alterations in Alzheimer’s disease (AD), including the formation of neurofibrillary tangles and cell death. Recently, brain imaging studies from preclinical AD patients and electrophysiological recordings from AD animal models have shown that impaired neuronal activity in the EC precedes neurodegeneration. This implies that memory impairments and spatial navigation deficits at the initial stage of AD are likely caused by activity dysfunction, rather than cell death. This review will focus on recent findings on EC dysfunction in AD, and discuss the potential pathways for mitigating AD progression by protecting the EC.
Histological alterations in the EC emerge early in Alzheimer’s disease progression
Alzheimer’s disease (AD) is a brain disorder that slowly destroys memory and thinking skills, and eventually, the ability to carry out simple daily tasks. AD typically progresses over decades, through multiple stages (Fig. 1). First, at the preclinical (asymptomatic) stage, patients have no cognitive symptoms. However, at this stage, amyloid-β (Aβ) deposition, neurofibrillary tangles (NFT) of Tau proteins, as well as physiological alterations in neurons have already begun in focal regions of the brain, and worsen gradually over ~20 years [1–4]. Second, at the mild cognitive impairment (MCI) stage, patients display moderate memory-related symptoms, such as losing items frequently and missing appointments [5]. Finally, patients’ memory loss becomes more severe, and they start having difficulty managing their daily life activities. At this point, patients will be diagnosed with “dementia”. Now neurons in the brain are dying, and patients will eventually lose the ability to recognize beloved ones and their own identity. Sadly, no cure is currently available for AD. Because curing the disease would be far more difficult after neuronal death occurs, it is gradually becoming a consensus in the AD research field that strategies for preventing AD during the preclinical/MCI stages, rather than curing it after dementia, need to be developed [1]. What types of changes occur in the brain at the early preclinical stage? What brain regions can be targeted for preventing AD?
Figure 1. Putative time course of AD symptom progression.
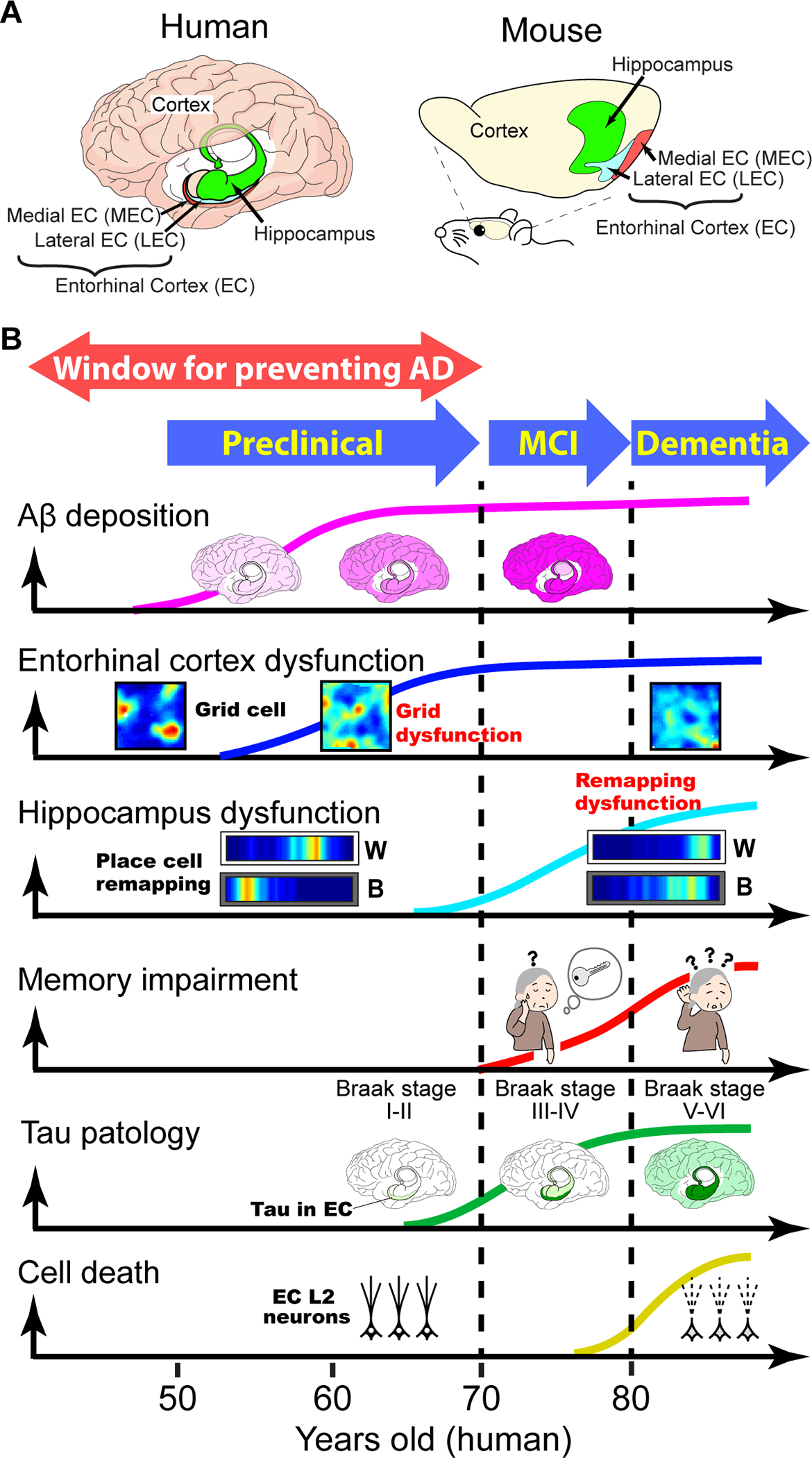
A, The entorhinal-hippocampal memory circuit in the human and mouse brain.
B, Hypothetical time course of AD symptom progression across three stages: preclinical, mild cognitive impairment (MCI), and dementia stages. Findings from human patients and AD animal models are putatively integrated. Aβ deposition: darker shades of pink representing greater Aβ accumulation [105]. Entorhinal cortex dysfunction: grid cell dysfunction at the preclinical stage (see Fig 3), based on findings in mouse models. Hippocampal dysfunction: Disruption of place cell remapping at the dementia stage), based on findings in mouse models (see also Fig. 4). Memory impairments: Memory impairments emerge in the MCI stage and worsen in the dementia stage. Tau pathology: Green depicts Tau accumulation starting in the EC (Braak stage I/II), spreading to the hippocampus (Braak stage III/IV) and to the cortex (Braak stage V/VI) [7]. Cell death: Neurodegeneration starts from entorhinal cortex (EC) layer 2 (L2) neurons [8, 10, 68].
Seminal past anatomical studies from postmortem AD patients showed that abnormal changes likely start from the entorhinal cortex (EC) [6–8]. Already at the preclinical stage, Tau tangle formation begins in the EC, especially in layer 2 neurons of the trans-entorhinal zone near the border to the perirhinal cortex (Braak stages I – II) [7, 8] (Fig. 1B). Tangles then propagate to medial temporal lobe structures including hippocampus at the MCI stage (Braak stages III – IV), and finally spread to various cortical areas at the dementia stage (Braak stages V – VI) [7, 8]. These Braak stages can now be assessed in living individuals using positron emission tomography (PET) imaging of Tau accumulation in the brain, or by detecting Tau in the cerebrospinal fluid (CSF) [9]. Correlated with tangle formation, the EC shows the severest cellular degeneration, eventually leading to structural atrophy (shrinkage of the brain region) [6]. During degeneration, EC layer 2 neurons undergo the severest cell death [10]. These findings helped formulate the idea that the EC is most plausibly the brain region where AD originate, at least in most cases. Findings that Aβ and Tau accumulated in the EC can be trans-synaptically transported to the hippocampus support this notion [11–14]. Functional imaging studies of human brains further showed that the EC exhibited the earliest brain activity impairment at the preclinical stage [15, 16]. Recently, by leveraging the development of mouse models of preclinical AD [17], we demonstrated early dysfunction of spike activity in the EC at the ‘preclinical stage’ (without overt memory impairments), followed by spike activity impairments in the hippocampus at the ‘dementia’ stage (that is, after the emergence of spatial memory impairments) [18]. Together, these results suggest that EC activity dysfunction emerges during the preclinical stage, then gradually affects the hippocampus and other cortical regions, leading to memory impairments. In this review, I will first summarize the functions of the EC in healthy brains. I will then discuss recent findings on EC dysfunction in AD, and current understanding of potential mechanisms of EC dysfunction. Last, I will discuss how these research venues may lead to future methods for AD prevention.
MEC grid cells and LEC object-reward cells in healthy brains
To identify brain dysfunctions in AD, one needs to first understand the precise functions of brain regions relevant to AD in heathy brains. Significant progress has been made in the past two decades in clarifying the anatomical architecture and function of the EC in healthy subjects (Fig. 2). The EC is anatomically positioned between the neocortex and the hippocampus, and its major role is to bridge information exchange between the two regions. The EC is anatomically divided into two distinct areas, the medial entorhinal cortex (MEC) and the lateral entorhinal cortex (LEC) [19]. The MEC receives major inputs from visual cortical areas via projections from the postrhinal cortex. By contrast, the LEC receives major input from the perirhinal cortex as well as from various olfactory regions, including the olfactory bulb [20–22]. This distinction between the MEC and LEC in their anatomical architecture and inputs is basically conserved across rodents, nonhuman primates and humans [19, 23, 24]. Neurons in the superficial layers (layers 2 and 3) of the EC receive input from multiple cortical regions and send axons to the hippocampus. Recently, it has become clear that EC layer 2 (L2) principal cells can be classified into two major cell types: stellate/fan cells and pyramidal cells [25, 26]. L2 stellate cells in the MEC (or fan cells in the LEC) express Reelin (RE), projecting their axons to the dentate gyrus via the perforant (indirect) pathway. The information in the dentate gyrus is further sent to CA3 and CA1, forming the tri-synaptic pathway. By contrast, L2 pyramidal cells express Calbindin (CB). L2 pyramidal cells in the MEC project to CA1 together with L3 pyramidal cells, via the temporoammonic (direct) pathway. L2 pyramidal cells in the LEC do not project to the hippocampus but rather make cortical projections to olfactory areas [27]. CA1 cells send information processed in the hippocampus through their axons projecting back to the deep layer (layer 5) of the EC, which in turn sends it back to cortical regions. This entorhinal-hippocampal circuit is critically involved in memory formation and retrieval, and damage to this circuit results in memory impairments [28, 29].
Figure 2. The entorhinal-hippocampal circuit, MEC grid cells, and LEC object + reward cells.
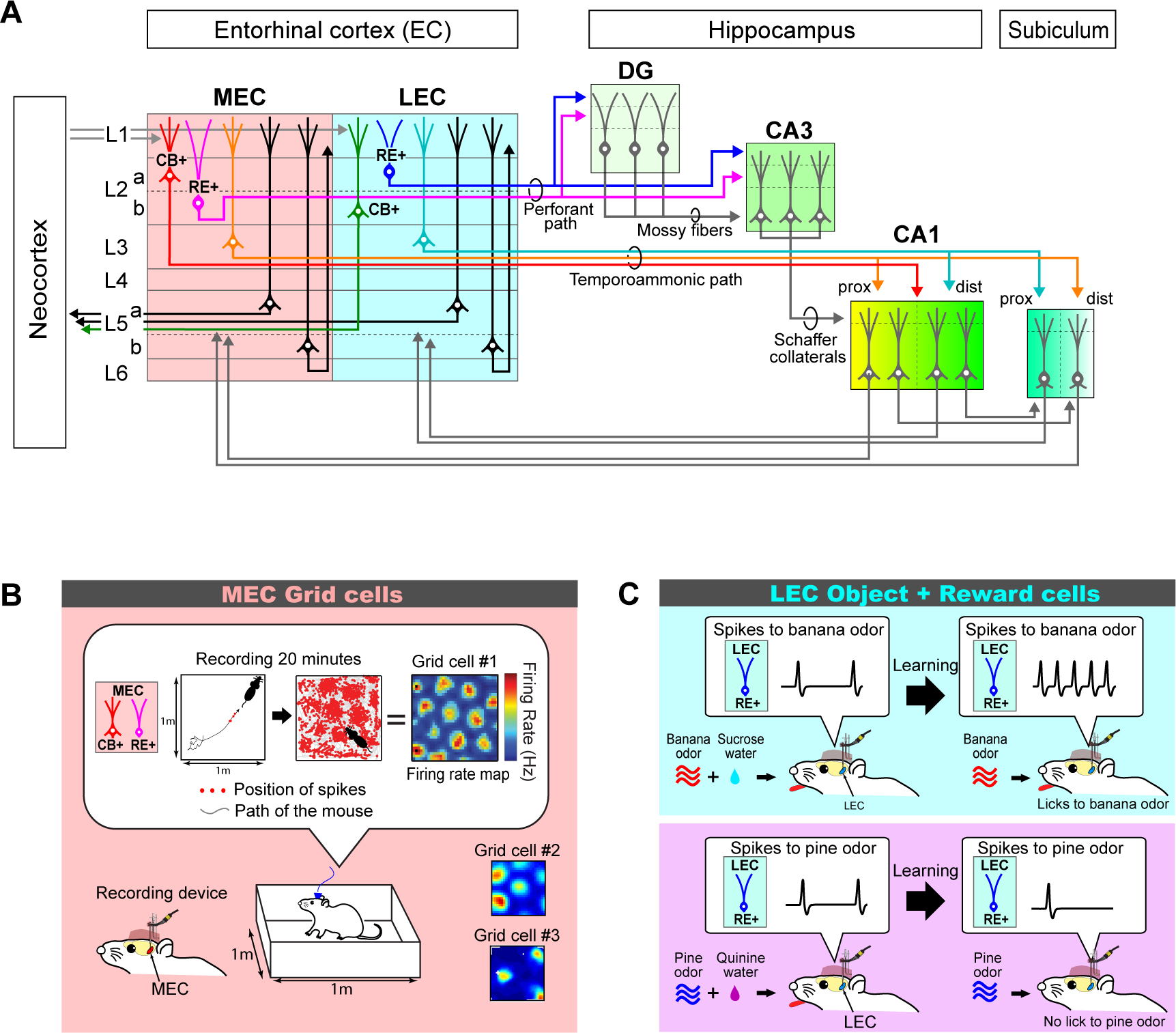
A, Diagram of the major connections of the mouse entorhinal-hippocampal circuit. The hippocampus receives information from the neocortex and sends information to it through the entorhinal cortex (EC). The medial entorhinal cortex (MEC) and the lateral entorhinal cortex (LEC) project to CA1 through the perforant and temporoammonic paths. In the perforant path, axons of layer 2 (L2) reelin-expressing (RE+) stellate cells (MEC) and fan cells (LEC) converge on the same population of cells in the dentate gyrus (DG) and CA3, but project to distal and proximal dendrites, respectively. This mixed information in DG and CA3 is conveyed to CA1 via mossy fibers and Schaffer collaterals. By contrast, EC layer 3 (L3) pyramidal cells and calbindin-expressing (CB+) MEC layer 2 (L2) pyramidal cells form the temporoammonic pathway. Pyramidal cells in MEC largely project to proximal CA1 (prox), whereas layer III cells in LEC project to distal CA1 (dist). Output from CA1 is conveyed directly to the EC layer 5 (L5) cells, or via the subiculum. CB+ L2 pyramidal cells in the LEC, as well as L5a cells in the MEC and LEC form output axonal projection to various cortical areas, whereas L5b neurons form recurrent projections in the EC.
B, In vivo electrophysiological recording of MEC grid cells in rodents. MEC Layer 2 single neurons were recorded using an implanted recording device having multiple tetrodes targeting the MEC. Spike activity was recorded for ~20 minutes while animals ran throughout an open field behavioral enclosure. Firing rate map was generated by calculating spike counts divided by time visited by the animal in each small spatial bin. Grid cells show equilateral triangular grid patterns with different spacings (grid cells #1, #2 and #3).. Modified from [18, 30, 31].
C, In vivo electrophysiological recording of LEC object + reward cells. For selectively recording LEC L2 RE+ fan cells, a cell-type-specific optogenetic-assisted electrophysiological recording technique was used [106]. Mice were trained in an odor object-reward associative memory task. When an odor (for example, banana odor) was paired with sucrose water reward, LEC L2 RE+ fan cells started to show enhanced firing, as depicted schematically in the single-neuron recording trace (top panel). By contrast (lower panel), this neuron came to exhibit decreased firing to an odor (for example, pine odor) paired with bitter quinine water. When fan cell activity was optogenetically inhibited, mice were no longer able to learn the associative memory task, indicating that LEC odor + reward cells support object memory. Modified from [49].
In the early 2000s, it was found that neurons in the MEC exhibit spike activity as grid cells, with triangular grid-like spatial patterns [30, 31]. These grid cells send spatial information to place cells, hippocampal pyramidal neurons that fire in a single position within the environment [32]. The MEC contains not only grid cells but also neurons carrying various types of information related to spatial navigation, including head-direction cells, border cells, speed cells and object-vector cells [33–36]. Manipulating grid cell activity induces impairment of place cell activity in the hippocampus and animal’s spatial memory impairment [37–39]. Thus, neuronal activity in the MEC provides spatial information that is critical for spatial memory and navigation [40, 41]. Importantly, place cell-like activity and grid cell-like activity are also found in the hippocampus and EC, respectively, of healthy human brains [42, 43].
By contrast, spike activity in the LEC does not appear to carry spatially-related information [44]. Rather, at least in rodents, LEC neurons increase spikes to odor cues [45] or fire when animals approach to objects placed in their environment [46, 47]. Similar object-related activities are also present in the LEC of healthy human brains [48]. We recently found that in mice, fan cells in the LEC represent conjunctive information of odors and reward, and blocking fan cell activity impairs odor-reward associative memory of the animals [49]. The LEC receives direct dopamine axon projections from the ventral tegmental area (VTA), which provide information whether a given olfactory object cue is associated with reward or not [49]. These findings suggest that the LEC provides information about memory of objects and their values as associative memory. It would be instrumental in future human imaging studies to examine whether the associative encoding between object sensory cues and reward value also exist in the human LEC.
Dysfunction of EC neuronal activity in preclinical AD
The finding of grid cells in healthy brains has recently triggered investigation of grid cell activity in human AD subjects and animal models of AD. First, using functional magnetic resonance imaging (fMRI), grid-cell-like activity was found to be reduced in the EC of young adult individuals of 18–30 years-of-age with the apolipoprotein (APOE)-ε4 allele, who have an increased risk for developing AD [16]. This finding suggests that EC activity dysfunction exists in human subjects even in the preclinical stage before the onset of dementia. Second, in a mouse model that expresses mutated human Tau in the EC (EC-Tau mice), MEC principal neurons exhibited decreased grid cell tuning at 30+ month-of-age (mo) [50]. In this mouse model, the mutated human Tau protein is expressed only in the EC using an EC-specific promoter. At 30+ mo, Tau propagates from the EC to the hippocampus, but not to neocortical regions, which presumably corresponds to Braak stage III-IV (MCI stage) of human AD patients. EC-Tau mice showed spatial memory deficits as well as significant degeneration of MEC L2 excitatory neurons, with ~75% reduction in cell number. In younger (14 mo) mice, grid cells and animals’ spatial memory were both intact. Degeneration of L2 excitatory neurons may have deteriorated excitatory-inhibitory (E-I) balance of the recurrent circuits in the MEC, which is critical for the formation of grid cells [51].
How does Aβ affect spike activity of EC neurons? Leveraging a newly developed mutated amyloid precursor protein (APP) knock-in mouse model, where slow Aβ accumulation enables tracking of longitudinal disease progression [17], we investigated the effect of Aβ on MEC grid cell activity [18]. The results showed that Aβ deposition and loss of grid cell tuning exist in the MEC as early as 3 mo, while mice showed normal spatial memory at this age, presumably corresponding to the preclinical stage. While ~20% of recorded principal neurons showed grid cell activity in control healthy mice, ~8% of principal cells had grid cell activity in APP knock-in mice. At 12 mo, the percentage of grid cells further decreased to ~2%, and mice showed spatial memory loss (thus presumably corresponding to the dementia stage). MEC neurons lost their confined spike firing at their grid fields, and instead displayed firing when animals were located all around the environment (Fig. 3). MEC neurons showed a tendency to exhibit increased spike firing rate, implying hyperexcitability, as previously reported in the hippocampus in AD [52]. The loss of grid cell activity was paralleled by diminished MEC gamma oscillation activity as well as impaired MEC-CA1 coordination of gamma oscillations (local field potential oscillations at 30–100 Hz frequency) and sharp-wave ripples (SWRs, 150–250 Hz) [18, 53, 54], both of which have critical roles in spatial memory in healthy animals [55, 56]. These results demonstrated that in mice, Aβ accumulation leads to grid cell impairment even at the preclinical stage. Subsequent recording studies from another Tau mouse model (rTg4510 mice) [57] and an APP transgenic mouse model (J20 mice) [58] observed grid cell impairment at the dementia stage. Together, these animal model studies suggest that both Tau pathology and amyloid pathology can induce grid cell dysfunction in the MEC. In human PET studies, however, cognitive dysfunction is shown to be related to Tau rather than amyloid pathology [9]. Future studies need to address whether APP mice accompany Tau pathology in their MEC.
Figure 3. Entorhinal cortex dysfunction in AD brains.
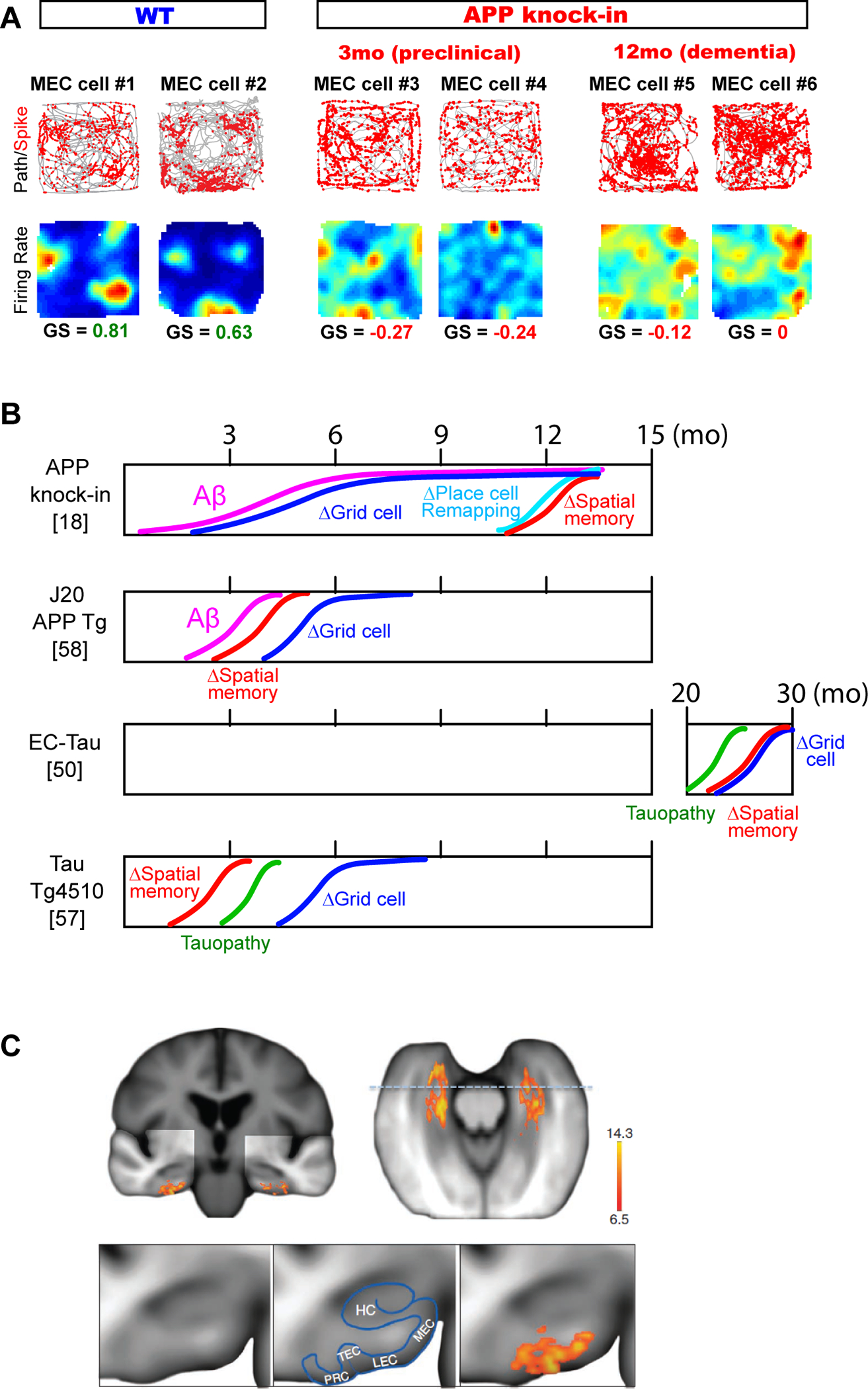
A, Dysfunction of MEC grid cells emerged at the preclinical period of APP knock-in mice [18]. Grid cell were defined as neurons with gridness score (GS) > ~0.4. At 3 mo, APP knock-in mice have intact spatial memory, but neurons in the MEC already lost their grid cell property. Adapted from [18]. Gridness score (GS) is a measure for repetitiveness of firing fields.
B, Schematic summary of time courses of grid cell dysfunction, spatial memory deficit and pathology found in AD mouse models. Grid cell impairment was found to emerge before spatial memory impairment in APP knock-in mice [18], whereas in other transgenic mouse models grid cell impairment was found only after memory impairment [50, 57, 58]. Δ denotes impairment.
C, Dysfunction of the lateral entorhinal cortex in the preclinical stage of AD patients [15]. A voxel-based analysis showed significantly lower cerebral blood volume (scale bar) in the LEC. Bottom, A higher magnification of the coronal view. TEC, transentorhinal cortex; HC, hippocampus proper; PRC, perirhinal cortex. Adapted from [15].
Although dysfunctions of the MEC are being revealed, it remains unclear if the object-related representation found in heathy LEC is impaired in AD. Imaging studies reported that the LEC showed more significant activity loss than the MEC in preclinical individuals (Fig. 3) [15]. Activity was lower in the LEC than MEC also in the EC-Tau mouse model, demonstrating the LEC as an origination point of activity dysfunction in AD. Because the LEC is adjacent to the trans-entorhinal zone that shows earlier Tau pathology in the EC (Fig. 3C), these findings suggest that LEC neurons exhibit earlier dysfunction as an effect from the trans-entorhinal zone. Future work is needed to clarify if the object-related spike representation of LEC becomes impaired earlier than the grid cell dysfunction.
Types of memory impairments caused by EC dysfunction
AD patients show impairments in various types of memory, including episodic memory (such as remembering daily events), semantic memory (such as memorizing facts and ideas), and spatial memory (such as recognizing your location) [59]. How are these memory impairment types related to dysfunction of the EC and hippocampus? Because much more is known about neuronal circuit mechanisms of spatial memory compared to episodic and semantic memory, the relationship between spatial memory impairments and MEC dysfunction will be discussed in the following section. Addressing the relationship between impairments of episodic/semantic memory and EC dysfunction would be an important goal for future work.
Spatial memory impairment is one of the major symptoms of AD, occurring in more than 60% of AD patients [60]. Does grid cell impairment in the MEC directly cause such spatial memory impairment in AD? In our study of APP knock-in mice, grid cell impairment was present both at 3 mo and 12 mo, whereas animals’ spatial memory, such as discriminating two distinct rooms, was intact at 3 mo and then impaired at 12 mo [18]. Thus, grid cell impairment may not be directly linked to spatial discrimination memory. Rather, it is likely that the function of the hippocampus is more directly linked to spatial memory. In APP knock-in mice, spatially-tuned firing properties of place cells were mostly intact, and these cells maintained their spatial-tuning property at both young (3 mo) and old (12 mo) ages. However, the remapping function of place cells, an ability of place cells to exhibit distinct firing patterns in different environments (see Box 1) [61, 62], was intact at 3 mo and then impaired at 12 mo. The remapping function is thought to be an underlying mechanism for spatial pattern separation between distinct environments [63]. Thus, impairment of place cell remapping may be linked to animals’ spatial memory impairment. We currently hypothesize that impaired grid cells, emerging (in mice) from 3 mo, increase in number as the disease progresses. This would lead to the emergence of remapping impairment of CA1 neurons at 12 mo, which ultimately causes impairment of spatial discrimination memory (i.e. grid cell impairment → hippocampal remapping impairment = spatial memory loss). This hypothesis, however, remains to be experimentally tested using circuit intervention techniques.
Box 1. Disrupted remapping of place cells and grid cells in AD.
In healthy brains, place cells in the hippocampal CA1 and CA3 show distinct firing patterns in different environments [61, 95–98]. Remapping experiments are normally performed by switching animals between two distinct behavioral enclosures, for example using black and white linear tracks (Black → White → White → Black, Fig. 4; see [99] for detailed protocol for remapping experiment). During switching, thousands of place cells simultaneously shift their firing field to new positions in the new environment (or sometimes they lose position-related firing), but the firing patterns are kept identical in the same environment (termed as stability of place cells). Most place cells showed distinct firing patterns in more than 10 different environments, suggesting that remapping provides subjects with combinatorial codes for distinguishing each environment [98]. Remapping of hippocampal activity has been also reported in healthy human subjects [100].
Like hippocampal place cells, grid cells in the MEC remap across environments as well [101]. Moreover, remapping of place cells and grid cells happens simultaneously [101], suggesting a shared underlying circuit mechanism, although the precise mechanisms of remapping remain unclear. Several reports suggested that MEC grid cells provide remapping signals to place cells [38, 102, 103], whereas other lines of evidence suggest an intrinsic remapping mechanism within the hippocampus [104].
The remapping function of place cells and grid cells is thought to provide subjects with spatial pattern separation, a cognitive ability to distinguish between distinct environments [63]. This remapping function was found to be disrupted in AD mice [18] (Fig. 4). At the preclinical period (3 mo) of APP knock-in mice, grid cells did not show remapping, as grid cells were disrupted and fired all across the environments. At the dementia stage (12 mo), place cells were still mostly present, but their remapping function was disrupted. These results suggest that place cell remapping, rather than grid cell remapping, provides a direct underlying circuit mechanism for spatial memory.
Are there any memory types that are directly linked to grid cell function? This question is relevant not only from a research perspective, but can have translational implications. Because grid cell impairment emerges even at the preclinical stage, such memory types could be impaired at the preclinical stage, and would be good candidates for a “behavioral biomarker” for identifying individuals who will likely develop dementia. Studies in healthy animals found that when grid cell activity is inhibited, animals lose path integration performance, an ability critical for spatial navigation to recognize spatial position by accumulating self-motion cues [64, 65]. The role of grid cells in path integration is also supported by theoretical work [40, 66]. Leveraging these basic findings, it was recently reported that path integration performance is impaired in preclinical APOE-ε4 carriers [67]. Together with the forementioned earlier fMRI study reporting impaired grid-cell-like activity in APOE-ε4 carriers [16], these findings point towards the exciting possibility that path integration testing could be used for diagnosing the earliest symptom of AD. If such a method becomes clinically available, the current definition of “preclinical” will likely have to be revised.
What causes entorhinal dysfunction: cell-type-specific vulnerabilities of EC neurons
Now that grid cell dysfunction clearly exists in the MEC at the preclinical stage, a logical next step toward preventing the dysfunction is to understand its underlying mechanisms. Are there any specific neuron types that cause grid cell dysfunction? Histological findings that EC L2 neurons show earlier cell death than neurons in other layers raises the idea that the EC contains neuron subtypes that are specifically vulnerable to degeneration in AD (that is, histological vulnerability) [7, 10, 68]. As an extension of this idea, there may be neuronal subtypes that show earliest spike dysfunction at the preclinical stage (that is, functional vulnerability). Studies toward understanding these two types of vulnerabilities are currently underway:
Cell-type-specific functional vulnerability.
Grid cells in healthy animals are shown to comprise both stellate-type and pyramidal-type principal cells in layers 2/3 [69, 70]. A study reported in vitro recording of MEC stellate cells in the 7–8 mo rTg4510 Tau mice [71], in which grid cell dysfunction was recently reported [57]. Afterhyperpolarization (AHP), an undershoot phase of action potentials that constitutes the spike refractory period, was found to be longer in MEC neurons, resulting in less high-frequency spike bursts important for forming grid fields [71]. Another study further found longer AHP in MEC stellate cells of APP transgenic rats even at the preclinical stage, but also reported hyperexcitability of stellate cells especially early in the spike trains [72]. Together, these studies suggest vulnerability of MEC stellate cells, but whether stellate cells show hypoactivity or hyperactivity remains unclear. Also, studies are needed to decipher if MEC pyramidal cells show functional vulnerability. In the LEC, in vitro recording studies from Tg2576 APP transgenic mice [73] and APOE-ε4 mice [74] showed hypoactivity of LEC neurons. In the hippocampus, extensive studies using 2-photon imaging in AD mice showed neuronal hyperexcitability of pyramidal cells near amyloid plaques [52, 75]. Detailed future studies using cell-type-specific recording methods are needed to elucidate if EC neurons show hyperactivity or hypoactivity.
Cell-type-specific histological vulnerability.
A pioneering study previously reported that Reelin-expressing EC L2 stellate/fan cells show neurodegeneration in AD patients, as well as in J20 APP transgenic mice [76]. Following up on this study, it was showed that LEC L2 fan cells accumulate intracellular Aβ in an APP transgenic rat at the preclinical stage, suggesting that this toxic species of Aβ may specifically harm fan cells [77]. By contrast, a histological study using postmortem brain sections of AD patients showed a decreased number of Calbindin-expressing layer 2 pyramidal cells in the EC [78]. Interestingly, surviving pyramidal cells showed high-level expression of Calbindin, suggesting a vulnerability of low-Calbindin-expressing neurons. Collectively, the results so far suggest histological vulnerability of both stellate/fan cells and pyramidal cells in the EC.
Links between functional vulnerability and histological vulnerability.
It is so far unclear whether a given EC cell type shows both functional vulnerability and histological vulnerability. Recently, several optogenetic and chemogenetic manipulation studies showed that the enhancement of hyperexcitability of EC neurons could facilitate Aβ deposition and tauopathy in synaptically connected neurons in the hippocampus [13, 79], whereas the attenuation of hyperexcitability reduced these pathologies [80, 81]. These results imply a potential causal link between functional vulnerability and histological vulnerability of EC neurons.
Possible molecular pathways causing vulnerability.
The studies discussed above suggest histological and functional vulnerability of EC neurons, primary in L2. A logical next step would be to find molecular mechanisms of neuronal vulnerability of EC L2 neurons, and this quest has become a significant focus of AD research. The first line of work proposed an idea that Reelin and Calbindin genes may be involved in causing vulnerability (or resilience) of L2 neurons.. As mentioned above, specific accumulation of intracellular Aβ in stellate cells has been shown [77]. Because intracellular Aβ has been shown to inhibit the Reelin pathway which is critical for long-term potentiation (LTP), it seems logical to propose that Reelin could cause functional vulnerability. By contrast, as Calbindin is shown to have neuroprotective functions against Aβ toxicity [82], it has been proposed that low-Calbindin-expressing neurons carry vulnerability because of low protection to Aβ [78]. These interesting hypotheses need to be further tested in future studies. Recently, advances in single-cell RNA sequencing techniques have enabled identification of genes specifically expressed in EC L2 neurons of postmortem AD patient brains. In a recent study, five groups of excitatory neurons were identified in MEC L2 using clustering of gene expression profiles, and three of them showed decrease in number in AD brains [83]. It was found that the RORB gene was highly expressed in two histologically vulnerable neuron groups, pointing to RORB as a specific marker for histologically vulnerable L2 neurons. Another study searched genes expressed in EC L2 neurons using RNA sequencing in healthy mice and postmortem human brains and identified Tau and the PTB gene, which encodes a regulator of Tau splicing [84]. Future work is needed to address whether neuron types expressing these genes overlap with reelin-expressing stellate cells and calbindin-expressing pyramidal cells, and whether these genes have causal roles in the vulnerability of EC neurons.
Neuromodulatory inputs to the EC as potential causes of EC dysfunction.
The EC receives massive neuromodulatory inputs, including noradrenergic fibers from the locus coeruleus, cholinergic fibers from the basal forebrain, and dopaminergic fibers from the ventral tegmental area (VTA) [19]. Accumulating evidence suggests that these neuromodulatory fibers contribute to EC dysfunction in AD. First, noradrenergic inputs from the locus coeruleus may contribute to the early tauopathy in the EC. Using an antibody for phosphorylated Tau (p-Tau) which detects earlier Tau pathological stage than conventional silver staining methods, it was recently shown that the locus coeruleus exhibited p-Tau accumulation earlier than the EC, even in human brains before the age of 30 [85, 86]. These results suggest that Tau pathology may originate in the locus coeruleus, which could be synaptically transported to the EC and cause dysfunction in this brain region at the preclinical stage. Second, cholinergic inputs may directly cause EC dysfunction. In healthy animal studies, cholinergic inputs were shown to be critical for persistent activity as well as grid cell formation of EC neurons [87, 88]. In AD human brains, the nucleus basalis of Meynert in the basal forebrain, which provides the heaviest cholinergic inputs to the limbic system, exhibits early Tau pathology at the preclinical stage, and neurodegeneration at later stages [89–91]. Given that cholinergic inputs play critical roles not only in the EC but also in the hippocampus [92], these findings have led to the “Cholinergic Hypothesis of AD” and the testing of cholinesterase inhibitors as symptomatic therapies for AD [93]. Although the Cholinergic Hypothesis is still under debate, the vulnerability of cholinergic inputs may contribute to EC dysfunction. Lastly, dopaminergic inputs from the VTA were recently shown to be critical for memory encoding in LEC neurons [49]. However, the role of dopamine in AD remains largely unknown, and future studies are needed to elucidate dopamine’s potential contribution to EC dysfunction.
Concluding Remarks and Future Perspectives
Recent imaging studies in human participants at increased risk of developing AD, as well as electrophysiological recording studies in AD animal models coherently demonstrate that activity dysfunction emerges in the EC at the preclinical stage. The idea of early neuronal dysfunction in AD is not new, and has been suggested over two decades ago [94]. From the treatment perspective, the early activity dysfunction stage can provide a window for preventing further progression of the disease, as it might be too late for treatments to be effective once neuronal degeneration occurs. Better understanding of the molecular and cellular mechanisms of EC dysfunctions, as well as the physiological and circuit mechanisms of EC dysfunctions (see Outstanding Questions) might pave the way toward a brighter future for those at risk of developing AD.
Outstanding questions raised from animal circuit research.
What types of functional spike activity in the LEC are lost in AD? An fMRI study showed earlier dysfunction in the LEC than in the MEC, and studies in healthy animals showed that LEC neurons represent object-reward information. Is object-reward spike activity in the LEC lost in AD patients and mouse models at the preclinical stage?
Is the remapping function of place cells and grid cells lost in AD patients? Disruption of place cell remapping and grid cell remapping was recently found in an AD mouse model. Methods for assessing hippocampal pattern separation of visual objects in humans are available, but this may be distinct from spatial remapping function. To investigate remapping of the hippocampus in AD patients, a method to assess remapping across distinct spatial environments needs to be developed.
Which cell types in the EC exhibit functional vulnerability and histological vulnerability? It is generally thought that EC L2 neurons exhibit both kinds of vulnerability, but it remains unclear whether pyramidal neurons and stellate/fan neurons both show such vulnerability in AD.
What are the molecular mechanisms that cause functional and histological vulnerabilities of EC neurons? Once candidate genes are identified, loss-of-function and gain-of-function experiments are needed to elucidate their exact roles in functional and histological vulnerabilities of EC neurons.
How do neuromodulatory systems contribute to entorhinal cortex dysfunction? Circuit-level investigations are required to precisely describe the contribution of neuromodulatory systems in AD.
Is it possible to develop clinical measurements detecting EC dysfunction of human subjects at the preclinical stage? Behavioral testing of path integration function, as well as recent work of subjective cognitive decline may lead to future detection methods for early EC dysfunction.
Figure 4. Remapping of place cells and grid cells, and their disruption in APP knock-in mice.
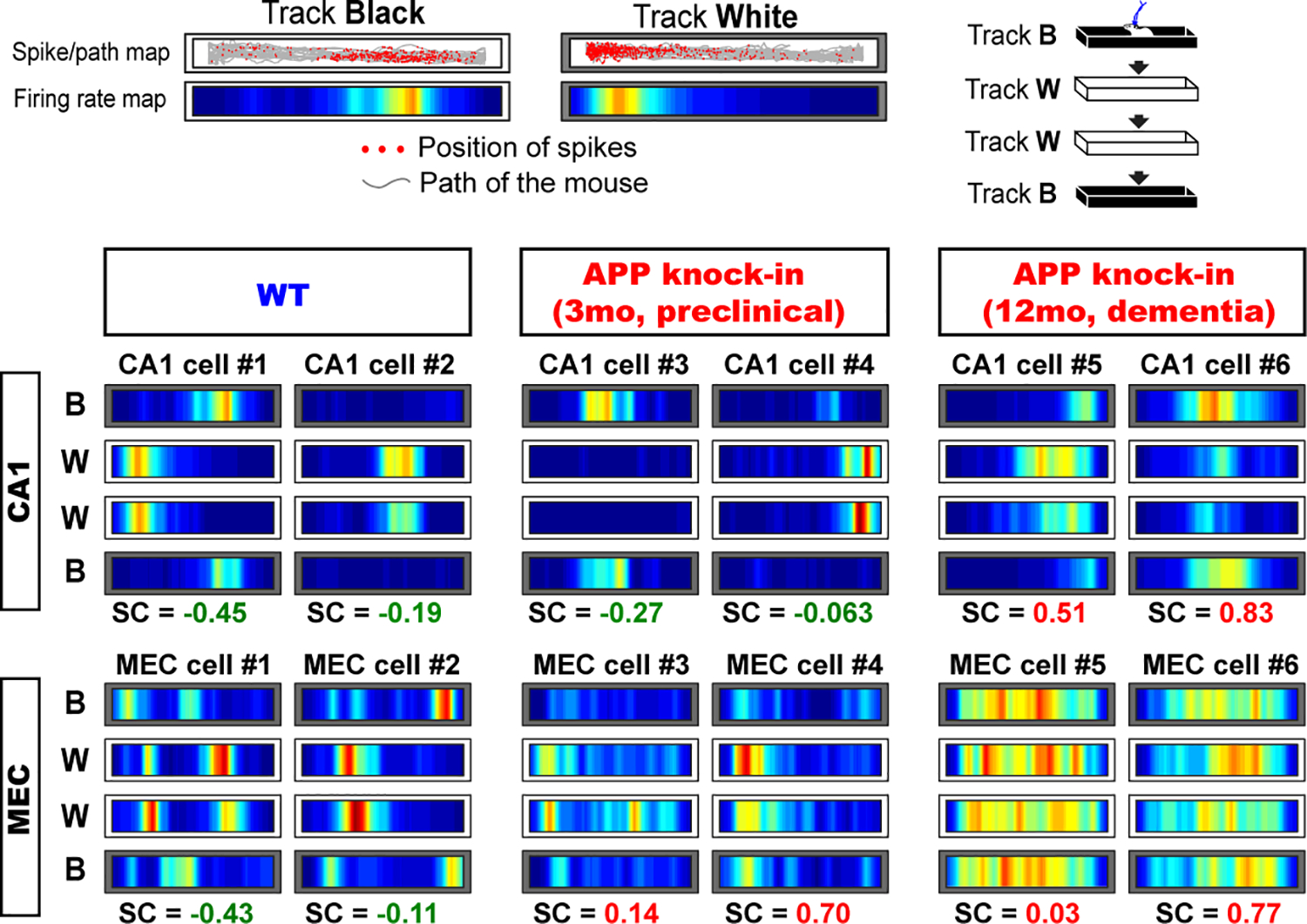
Remapping of place cells and grid cells were tested using black vs. white linear tracks, and assessed by calculating spatial correlation (SC) of firing maps between two tracks. Negative spatial correlation of neurons (that is, distinct firing patterns across two tracks) denotes strong remapping, whereas positive spatial correlation (that is, similar firing patterns across two tracks) means impaired remapping. Grid cells showed remapping impairment from the preclinical stage, whereas place cells showed remapping impairment only from the dementia stage. Adapted from [18].
Highlights.
It is becoming increasingly clear that dysfunction of the entorhinal cortex (EC) may play a role in early stages of Alzheimer’s disease (AD). Evidence for this notion comes from studies using in vivo electrophysiological recordings in mouse models of AD and brain imaging of AD patients.
Deficits of memory and spatial navigation related to EC dysfunction are also being elucidated. Such deficits, for instance in path integration, have a potential to be used as early behavioral biomarkers for the detection of AD, even at the early period currently defined as the “preclinical stage.”
Layer 2 neurons in the EC are one of the candidate neuron types causing EC dysfunction, and studies are underway to clarify their exact roles in the pathogenesis of EC dysfunction.
Studies to elucidate molecular pathways causing EC dysfunction are also currently underway. High-throughput sequencing techniques are starting to pinpoint candidate genes.
Better understanding of the network, cellular, and molecular mechanisms of EC dysfunction may lead to future prevention of disease progression at the early stage of AD.
Acknowledgments:
I thank Dr. Takaomi Saido at RIKEN Center for Brain Science, and Jason Lee, Heechul Jun, Jiayun L Xie, Sharon Lim and Tatsuki Nakagawa in the Igarashi lab for discussions and comments. I sincerely appreciate constructive comments by anonymous reviewers that significantly strengthened the manuscript. This work was supported by NIH R01 grants (R01MH121736, R01AG063864, R01AG066806), PRESTO grant from Japan Science and Technology Agency (JPMJPR1681), Brain Research Foundation Fay-Frank Seed Grant (BRFSG-2017-04), Whitehall Foundation Research Grant (2017-08-01), BrightFocus Foundation Research grant (A2019380S), Alzheimer’s Association Research Grant (AARG-17-532932) and Donors Cure Foundation New Vision Award (CCAD201902) to K.M.I.
Footnotes
Competing interests: The author declares no competing interests.
REFERENCES
- 1.Sasaguri H, et al. , APP mouse models for Alzheimer’s disease preclinical studies. EMBO J, 2017. 36(17): p. 2473–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bateman RJ, et al. , Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med, 2012. 367(9): p. 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Funato H, et al. , Quantitation of amyloid beta-protein (A beta) in the cortex during aging and in Alzheimer’s disease. Am J Pathol, 1998. 152(6): p. 1633–40. [PMC free article] [PubMed] [Google Scholar]
- 4.Dubois B, et al. , Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement, 2016. 12(3): p. 292–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelley BJ and Petersen RC, Alzheimer’s disease and mild cognitive impairment. Neurol Clin, 2007. 25(3): p. 577–609, v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Hoesen GW, Hyman BT, and Damasio AR, Entorhinal cortex pathology in Alzheimer’s disease. Hippocampus, 1991. 1(1): p. 1–8. [DOI] [PubMed] [Google Scholar]
- 7.Braak H and Braak E, Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol, 1991. 82(4): p. 239–59. [DOI] [PubMed] [Google Scholar]
- 8.Braak H and Braak E, The human entorhinal cortex: normal morphology and lamina-specific pathology in various diseases. Neurosci Res, 1992. 15(1–2): p. 6–31. [DOI] [PubMed] [Google Scholar]
- 9.Bjorkli C, Sandvig A, and Sandvig I, Bridging the Gap Between Fluid Biomarkers for Alzheimer’s Disease, Model Systems, and Patients. Front Aging Neurosci, 2020. 12: p. 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomez-Isla T, et al. , Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci, 1996. 16(14): p. 4491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris JA, et al. , Transsynaptic progression of amyloid-beta-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron, 2010. 68(3): p. 428–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Calignon A, et al. , Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron, 2012. 73(4): p. 685–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu JW, et al. , Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci, 2016. 19(8): p. 1085–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee WJ, et al. , Regional Abeta-tau interactions promote onset and acceleration of Alzheimer’s disease tau spreading. Neuron, 2022. 110(12): p. 1932–1943 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan UA, et al. , Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat Neurosci, 2014. 17(2): p. 304–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kunz L, et al. , Reduced grid-cell-like representations in adults at genetic risk for Alzheimer’s disease. Science, 2015. 350(6259): p. 430–3. [DOI] [PubMed] [Google Scholar]
- 17.Saito T, et al. , Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci, 2014. 17(5): p. 661–3. [DOI] [PubMed] [Google Scholar]
- 18.Jun H, et al. , Disrupted Place Cell Remapping and Impaired Grid Cells in a Knockin Model of Alzheimer’s Disease. Neuron, 2020. 107(6): p. 1095–1112 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Witter MP and Amaral DG, Hippocampal Formation, in The Rat Nervous System 3rd edn (ed. Paxinos G), Paxinos G, Editor. 2004, Elsevier: Amsterdam, The Netherlands. [Google Scholar]
- 20.Burwell RD, The parahippocampal region: corticocortical connectivity. Ann N Y Acad Sci, 2000. 911: p. 25–42. [DOI] [PubMed] [Google Scholar]
- 21.Igarashi KM, et al. , Parallel mitral and tufted cell pathways route distinct odor information to different targets in the olfactory cortex. J Neurosci, 2012. 32(23): p. 7970–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Insausti R, et al. , Comparative aspects of the olfactory portion of the entorhinal cortex and its projection to the hippocampus in rodents, nonhuman primates, and the human brain. Brain Res Bull, 2002. 57(3–4): p. 557–60. [DOI] [PubMed] [Google Scholar]
- 23.Garcia AD and Buffalo EA, Anatomy and Function of the Primate Entorhinal Cortex. Annu Rev Vis Sci, 2020. 6: p. 411–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Insausti R, Comparative anatomy of the entorhinal cortex and hippocampus in mammals. Hippocampus, 1993. 3 Spec No: p. 19–26. [PubMed] [Google Scholar]
- 25.Kitamura T, et al. , Island cells control temporal association memory. Science, 2014. 343(6173): p. 896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Surmeli G, et al. , Molecularly Defined Circuitry Reveals Input-Output Segregation in Deep Layers of the Medial Entorhinal Cortex. Neuron, 2015. 88(5): p. 1040–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leitner FC, et al. , Spatially segregated feedforward and feedback neurons support differential odor processing in the lateral entorhinal cortex. Nat Neurosci, 2016. 19(7): p. 935–44. [DOI] [PubMed] [Google Scholar]
- 28.Scoville WB and Milner B, Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry, 1957. 20(1): p. 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Squire LR, Stark CE, and Clark RE, The medial temporal lobe. Annu Rev Neurosci, 2004. 27: p. 279–306. [DOI] [PubMed] [Google Scholar]
- 30.Fyhn M, et al. , Spatial representation in the entorhinal cortex. Science, 2004. 305(5688): p. 1258–64. [DOI] [PubMed] [Google Scholar]
- 31.Hafting T, et al. , Microstructure of a spatial map in the entorhinal cortex. Nature, 2005. 436(7052): p. 801–6. [DOI] [PubMed] [Google Scholar]
- 32.O’Keefe J and Dostrovsky J, The hippocampus as a spatial map. Preliminary evidence from unit activity in the freely-moving rat. Brain Res, 1971. 34(1): p. 171–5. [DOI] [PubMed] [Google Scholar]
- 33.Sargolini F, et al. , Conjunctive representation of position, direction, and velocity in entorhinal cortex. Science, 2006. 312(5774): p. 758–62. [DOI] [PubMed] [Google Scholar]
- 34.Solstad T, et al. , Representation of geometric borders in the entorhinal cortex. Science, 2008. 322(5909): p. 1865–8. [DOI] [PubMed] [Google Scholar]
- 35.Kropff E, et al. , Speed cells in the medial entorhinal cortex. Nature, 2015. 523(7561): p. 419–24. [DOI] [PubMed] [Google Scholar]
- 36.Hoydal OA, et al. , Object-vector coding in the medial entorhinal cortex. Nature, 2019. 568(7752): p. 400–404. [DOI] [PubMed] [Google Scholar]
- 37.Steffenach HA, et al. , Spatial memory in the rat requires the dorsolateral band of the entorhinal cortex. Neuron, 2005. 45(2): p. 301–13. [DOI] [PubMed] [Google Scholar]
- 38.Kanter BR, et al. , A Novel Mechanism for the Grid-to-Place Cell Transformation Revealed by Transgenic Depolarization of Medial Entorhinal Cortex Layer II. Neuron, 2017. 93(6): p. 1480–1492 e6. [DOI] [PubMed] [Google Scholar]
- 39.Brun VH, et al. , Impaired spatial representation in CA1 after lesion of direct input from entorhinal cortex. Neuron, 2008. 57(2): p. 290–302. [DOI] [PubMed] [Google Scholar]
- 40.McNaughton BL, et al. , Path integration and the neural basis of the ‘cognitive map’. Nat Rev Neurosci, 2006. 7(8): p. 663–78. [DOI] [PubMed] [Google Scholar]
- 41.Buzsaki G and Moser EI, Memory, navigation and theta rhythm in the hippocampal-entorhinal system. Nat Neurosci, 2013. 16(2): p. 130–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ekstrom AD, et al. , Cellular networks underlying human spatial navigation. Nature, 2003. 425(6954): p. 184–8. [DOI] [PubMed] [Google Scholar]
- 43.Doeller CF, Barry C, and Burgess N, Evidence for grid cells in a human memory network. Nature, 2010. 463(7281): p. 657–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hargreaves EL, et al. , Major dissociation between medial and lateral entorhinal input to dorsal hippocampus. Science, 2005. 308(5729): p. 1792–4. [DOI] [PubMed] [Google Scholar]
- 45.Igarashi KM, et al. , Coordination of entorhinal-hippocampal ensemble activity during associative learning. Nature, 2014. 510(7503): p. 143–7. [DOI] [PubMed] [Google Scholar]
- 46.Deshmukh SS and Knierim JJ, Representation of non-spatial and spatial information in the lateral entorhinal cortex. Front Behav Neurosci, 2011. 5: p. 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsao A, Moser MB, and Moser EI, Traces of experience in the lateral entorhinal cortex. Curr Biol, 2013. 23(5): p. 399–405. [DOI] [PubMed] [Google Scholar]
- 48.Reagh ZM and Yassa MA, Object and spatial mnemonic interference differentially engage lateral and medial entorhinal cortex in humans. Proc Natl Acad Sci U S A, 2014. 111(40): p. E4264–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee JY, et al. , Dopamine facilitates associative memory encoding in the entorhinal cortex. Nature, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fu H, et al. , Tau Pathology Induces Excitatory Neuron Loss, Grid Cell Dysfunction, and Spatial Memory Deficits Reminiscent of Early Alzheimer’s Disease. Neuron, 2017. 93(3): p. 533–541 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Couey JJ, et al. , Recurrent inhibitory circuitry as a mechanism for grid formation. Nat Neurosci, 2013. 16(3): p. 318–24. [DOI] [PubMed] [Google Scholar]
- 52.Busche MA, et al. , Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science, 2008. 321(5896): p. 1686–9. [DOI] [PubMed] [Google Scholar]
- 53.Nakazono T, et al. , Impaired in vivo gamma oscillations in the medial entorhinal cortex of knock-in Alzheimer model. Front Syst Neurosci, 2017. 11: p. 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Funane H, et al. , Impaired sharp-wave ripple coordination between the medial entorhinal cortex and hippocampal CA1 of knock-in model of Alzheimer’s Disease. Front Syst Neurosci, 2022: p. DOI 10.3389/fnsys.2022.955178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fernandez-Ruiz A, et al. , Gamma rhythm communication between entorhinal cortex and dentate gyrus neuronal assemblies. Science, 2021. 372(6537). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamamoto J, et al. , Successful execution of working memory linked to synchronized high-frequency gamma oscillations. Cell, 2014. 157(4): p. 845–57. [DOI] [PubMed] [Google Scholar]
- 57.Ridler T, et al. , Impaired speed encoding and grid cell periodicity in a mouse model of tauopathy. Elife, 2020. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ying J, et al. , Disruption of the grid cell network in a mouse model of early Alzheimer’s disease. Nat Commun, 2022. 13(1): p. 886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bondi MW, et al. , Neuropsychological contributions to the early identification of Alzheimer’s disease. Neuropsychol Rev, 2008. 18(1): p. 73–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hope T, et al. , The Structure of Wandering in Dementia. International Journal of Geriatric Psychiatry, 1994. 9(2): p. 149–155. [Google Scholar]
- 61.Muller RU and Kubie JL, The effects of changes in the environment on the spatial firing of hippocampal complex-spike cells. J Neurosci, 1987. 7(7): p. 1951–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Colgin LL, Moser EI, and Moser MB, Understanding memory through hippocampal remapping. Trends Neurosci, 2008. 31(9): p. 469–77. [DOI] [PubMed] [Google Scholar]
- 63.Yassa MA and Stark CE, Pattern separation in the hippocampus. Trends Neurosci, 2011. 34(10): p. 515–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gil M, et al. , Impaired path integration in mice with disrupted grid cell firing. Nat Neurosci, 2018. 21(1): p. 81–91. [DOI] [PubMed] [Google Scholar]
- 65.Tennant SA, et al. , Stellate Cells in the Medial Entorhinal Cortex Are Required for Spatial Learning. Cell Rep, 2018. 22(5): p. 1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burak Y and Fiete IR, Accurate path integration in continuous attractor network models of grid cells. PLoS Comput Biol, 2009. 5(2): p. e1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bierbrauer A, et al. , Unmasking selective path integration deficits in Alzheimer’s disease risk carriers. Sci Adv, 2020. 6(35): p. eaba1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stranahan AM and Mattson MP, Selective vulnerability of neurons in layer II of the entorhinal cortex during aging and Alzheimer’s disease. Neural Plast, 2010. 2010: p. 108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tang Q, et al. , Pyramidal and stellate cell specificity of grid and border representations in layer 2 of medial entorhinal cortex. Neuron, 2014. 84(6): p. 1191–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rowland DC, et al. , Functional properties of stellate cells in medial entorhinal cortex layer II. Elife, 2018. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Booth CA, et al. , Electrical and Network Neuronal Properties Are Preferentially Disrupted in Dorsal, But Not Ventral, Medial Entorhinal Cortex in a Mouse Model of Tauopathy. J Neurosci, 2016. 36(2): p. 312–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heggland I, Kvello P, and Witter MP, Electrophysiological Characterization of Networks and Single Cells in the Hippocampal Region of a Transgenic Rat Model of Alzheimer’s Disease. eNeuro, 2019. 6(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marcantoni A, et al. , Firing properties of entorhinal cortex neurons and early alterations in an Alzheimer’s disease transgenic model. Pflugers Arch, 2014. 466(7): p. 1437–50. [DOI] [PubMed] [Google Scholar]
- 74.Nuriel T, et al. , Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat Commun, 2017. 8(1): p. 1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zott B, et al. , A vicious cycle of beta amyloid-dependent neuronal hyperactivation. Science, 2019. 365(6453): p. 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chin J, et al. , Reelin depletion in the entorhinal cortex of human amyloid precursor protein transgenic mice and humans with Alzheimer’s disease. J Neurosci, 2007. 27(11): p. 2727–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kobro-Flatmoen A, Nagelhus A, and Witter MP, Reelin-immunoreactive neurons in entorhinal cortex layer II selectively express intracellular amyloid in early Alzheimer’s disease. Neurobiol Dis, 2016. 93: p. 172–83. [DOI] [PubMed] [Google Scholar]
- 78.Thorns V, Licastro F, and Masliah E, Locally reduced levels of acidic FGF lead to decreased expression of 28-kda calbindin and contribute to the selective vulnerability of the neurons in the entorhinal cortex in Alzheimer’s disease. Neuropathology, 2001. 21(3): p. 203–11. [DOI] [PubMed] [Google Scholar]
- 79.Yamamoto K, et al. , Chronic optogenetic activation augments abeta pathology in a mouse model of Alzheimer disease. Cell Rep, 2015. 11(6): p. 859–865. [DOI] [PubMed] [Google Scholar]
- 80.Yuan P and Grutzendler J, Attenuation of beta-Amyloid Deposition and Neurotoxicity by Chemogenetic Modulation of Neural Activity. J Neurosci, 2016. 36(2): p. 632–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rodriguez GA, et al. , Chemogenetic attenuation of neuronal activity in the entorhinal cortex reduces Abeta and tau pathology in the hippocampus. PLoS Biol, 2020. 18(8): p. e3000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Phillips RG, et al. , Calbindin D28K gene transfer via herpes simplex virus amplicon vector decreases hippocampal damage in vivo following neurotoxic insults. J Neurochem, 1999. 73(3): p. 1200–5. [DOI] [PubMed] [Google Scholar]
- 83.Leng K, et al. , Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat Neurosci, 2021. 24(2): p. 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roussarie JP, et al. , Selective Neuronal Vulnerability in Alzheimer’s Disease: A Network-Based Analysis. Neuron, 2020. 107(5): p. 821–835 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Braak H, et al. , Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol, 2011. 70(11): p. 960–9. [DOI] [PubMed] [Google Scholar]
- 86.Braak H and Del Tredici K, The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol, 2011. 121(2): p. 171–81. [DOI] [PubMed] [Google Scholar]
- 87.Newman EL, Climer JR, and Hasselmo ME, Grid cell spatial tuning reduced following systemic muscarinic receptor blockade. Hippocampus, 2014. 24(6): p. 643–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Egorov AV, et al. , Graded persistent activity in entorhinal cortex neurons. Nature, 2002. 420(6912): p. 173–8. [DOI] [PubMed] [Google Scholar]
- 89.Mesulam MM, Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer’s disease. J Comp Neurol, 2013. 521(18): p. 4124–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Whitehouse PJ, et al. , Alzheimer disease: plaques, tangles, and the basal forebrain. Ann Neurol, 1982. 12(5): p. 494. [DOI] [PubMed] [Google Scholar]
- 91.Whitehouse PJ, et al. , Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science, 1982. 215(4537): p. 1237–9. [DOI] [PubMed] [Google Scholar]
- 92.Hasselmo ME, The role of acetylcholine in learning and memory. Curr Opin Neurobiol, 2006. 16(6): p. 710–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hampel H, et al. , Revisiting the Cholinergic Hypothesis in Alzheimer’s Disease: Emerging Evidence from Translational and Clinical Research. J Prev Alzheimers Dis, 2019. 6(1): p. 2–15. [DOI] [PubMed] [Google Scholar]
- 94.Selkoe DJ, Alzheimer’s disease is a synaptic failure. Science, 2002. 298(5594): p. 789–91. [DOI] [PubMed] [Google Scholar]
- 95.Leutgeb S, et al. , Distinct ensemble codes in hippocampal areas CA3 and CA1. Science, 2004. 305(5688): p. 1295–8. [DOI] [PubMed] [Google Scholar]
- 96.Leutgeb S, et al. , Independent codes for spatial and episodic memory in hippocampal neuronal ensembles. Science, 2005. 309(5734): p. 619–23. [DOI] [PubMed] [Google Scholar]
- 97.Leutgeb JK, et al. , Progressive transformation of hippocampal neuronal representations in “morphed” environments. Neuron, 2005. 48(2): p. 345–58. [DOI] [PubMed] [Google Scholar]
- 98.Alme CB, et al. , Place cells in the hippocampus: eleven maps for eleven rooms. Proc Natl Acad Sci U S A, 2014. 111(52): p. 18428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jun H, et al. , Protocol for remapping of place cells in disease mouse models. STAR Protoc, 2021. 2(3): p. 100759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kyle CT, et al. , Successful retrieval of competing spatial environments in humans involves hippocampal pattern separation mechanisms. Elife, 2015. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fyhn M, et al. , Hippocampal remapping and grid realignment in entorhinal cortex. Nature, 2007. 446(7132): p. 190–4. [DOI] [PubMed] [Google Scholar]
- 102.Miao C, et al. , Hippocampal Remapping after Partial Inactivation of the Medial Entorhinal Cortex. Neuron, 2015. 88(3): p. 590–603. [DOI] [PubMed] [Google Scholar]
- 103.Zutshi I, et al. , Extrinsic control and intrinsic computation in the hippocampal CA1 circuit. Neuron, 2022. 110(4): p. 658–673 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schlesiger MI, et al. , Hippocampal Global Remapping Can Occur without Input from the Medial Entorhinal Cortex. Cell Rep, 2018. 22(12): p. 3152–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Thal DR, et al. , Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology, 2002. 58(12): p. 1791–800. [DOI] [PubMed] [Google Scholar]
- 106.Cohen JY, et al. , Neuron-type-specific signals for reward and punishment in the ventral tegmental area. Nature, 2012. 482(7383): p. 85–8. [DOI] [PMC free article] [PubMed] [Google Scholar]