

Abstract
α-Synuclein is a neuronal protein that is enriched in presynaptic terminals. Under physiological conditions, it binds to synaptic vesicle membranes and functions in neurotransmitter release, although the molecular details remain unclear, and it is controversial whether α-synuclein inhibits or facilitates neurotransmitter release. Pathologically, in synucleinopathies including Parkinson’s disease, α-synuclein forms aggregates that recruit monomeric α-synuclein and spread throughout the brain, which triggers neuronal dysfunction at molecular, cellular and organ levels. Here, we present an overview of the effects of α-synuclein on SNARE-complex assembly, neurotransmitter release and synaptic vesicle pool homeostasis, and discuss how the observed divergent effects of α-synuclein on neurotransmitter release can be reconciled. We also discuss how gain-of-function versus loss-of-function of α-synuclein may contribute to pathogenesis in synucleinopathies.
Keywords: Synapse, Neurotransmission, SNARE, Synaptic vesicle, Synucleinopathies, Parkinson’s disease
The importance of α-synuclein
α-Synuclein belongs to a family of highly conserved proteins that includes β- and γ-synucleins [1]. α-Synuclein is highly expressed throughout the brain, and is particularly enriched in the synaptic terminals of neurons [2, 3]. It has gained notoriety due to its link to Parkinson’s disease (PD) and other neurodegenerative diseases termed synucleinopathies [4]. Therefore, research on the pathological role of α-synuclein has grown rapidly, while fewer studies have focused on the physiological function(s) of α-synuclein. Multiple therapeutic strategies aim at reducing α-synuclein levels but have failed so far in clinical trials, indicating a need for better understanding of α-synuclein’s biology and pathology. Interestingly, evolutionarily, α-synuclein has emerged only in vertebrates, demonstrating that it is not strictly required for neuron function and viability per se. However, modulation of α-synuclein expression during learning [5] and the phenotype(s) from removal of α-synuclein, as discussed below, demonstrate its importance in synaptic function and plasticity as well as in the long-term viability and functioning of the neuron.
Here, we provide an overview of the roles of α-synuclein pertaining to its synaptic function and dysfunction. We then discuss how loss-of-function and gain-of-toxic function of α-synuclein may contribute, either alone or together, to synucleinopathies.
α-Synuclein structure determines its localization and function
α-Synuclein is a small, 140 amino acid protein that is encoded by the SNCA gene. It is abundant in the brain and specifically enriched in presynaptic terminals of neurons [2, 3]. As an intrinsically disordered protein, it can adopt multiple conformations, ranging from a largely unstructured state in the cytosol, to an α-helical state upon membrane binding, to β-sheet rich oligomers and fibrils that deposit in Lewy bodies or Lewy neurites, the pathological hallmarks of PD [1, 4, 6]. In its physiologically relevant state, α-synuclein is a synaptic vesicle-bound multimer, mediated by its N-terminal domain which includes seven lysine-rich imperfect 11-residue repeats that form into an amphipathic α-helix (Figure 1) [7–9], with the 14 most N-terminal residues appearing to be particularly important for membrane localization [10]. The N-terminal region also contains the aggregation-prone region termed non-amyloid-β component (NAC) which was identified in Alzheimer’s disease pathology and forms the center of the fibril structure (Figure 1) [11, 12]. The C-terminal domain is negatively charged and mediates interactions with proteins including VAMP2 and calcium (Figure 1) (reviewed in [13]).
Figure 1. α-Synuclein domain structure.

(A, B) α-Synuclein is composed of an N-terminal membrane binding region that mediates its association with synaptic vesicles through formation of an amphipathic α-helix, and a C-terminal region that binds to VAMP2 (A). The N-terminal region can be further divided into eleven imperfect KTKEGV repeats (B), and contains the aggregation-prone NAC domain. Several mutations in α-synuclein have been identified that lead to disease (purple) or with yet unknown significance (pink). (C, D) Analysis of the position of mutations within the repetitive KTKEGV regions highlights that mutations cluster in repeat position 5, 10 and 11, while other positions are spared (C). Clusters at positions 10 and 11 face towards the membrane, while cluster 5 faces towards the cytosol (D).
The mechanisms regulating α-synuclein’s membrane/cytosol equilibrium are largely unknown but are important for physiological and pathological conditions. Membrane-binding has been mostly associated with α-synuclein’s physiological function and protection from aggregation, while aggregation is readily triggered from its cytosolic pool [13, 14]. Interestingly, disease-linked mutations in α-synuclein all cluster in the membrane binding domain (Figure 1), suggesting an intimate relationship between membrane binding, function, and pathology. There are also several variants of unknown significance located throughout the protein (Figure 1), but it remains unclear if these are linked to pathology.
α-Synuclein’s role in synaptic transmission
The list of potential interactors of α-synuclein is large and growing. Interactors include α-synuclein itself, lipids, synaptic vesicles, VAMP2, β- and γ-synucleins, synapsin-Ia, synapsin-III, proteins involved in calcium homeostasis, and many others reviewed elsewhere [13]. Interactions ascribed to modulation of dopamine-metabolism and -transport do not account for the wide-spread expression of α-synuclein but suggest specific functions in dopaminergic neurons in addition to a generic neuronal function [13]. Overall, most of the described interactors are found at the presynaptic terminal. Therefore, the majority of studies have focused on the effects of α-synuclein on synaptic vesicle cycling and neurotransmitter release.
As mentioned earlier, synucleins are restricted to vertebrates, and accordingly, they cannot be essential components for neurotransmission. In support of this argument, α-synuclein deficiency does not result in an overt phenotype that mimics phenotypes of proteins known to be essential for neurotransmitter release (reviewed in [15]). Relatedly, while α-synuclein is enriched in presynaptic terminals in adulthood, during development, α-synuclein is mostly somatic, and only concentrates in presynaptic boutons over time, being one of the last proteins to enrich in the presynapse [16]. Interestingly, α-synuclein expression in neurons is upregulated after injury [17]. Further, in songbird, α-synuclein expression correlates with song acquisition [5], suggesting a role in synaptic plasticity. Overall, this points to a modulatory function of α-synuclein.
α-Synuclein has been proposed to affect several steps in the synaptic vesicle cycle, including vesicle clustering [18–20], vesicle docking, SNARE-complex formation [21], and vesicle pool homeostasis [18, 22–24]. These roles can be divided into two distinct activities, which may or may not be linked: (i) organization of synaptic vesicle pools, and (ii) chaperoning of SNARE-complex assembly and modulation of fusion pore dynamics.
Synaptic vesicle pools are divided by functionality into readily releasable vesicles, recycling vesicles, and reserve pool vesicles (Figure 2) [25]. Changes in the dynamics of these pools play important roles in regulating synaptic strength and plasticity [26]. Mice lacking α-synuclein reveal a reduced docked pool and deficits in replenishing the docked pool from the reserve pool, resulting in impaired responses to long-lasting stimulation [27]. In mouse hippocampal neurons, acute knockdown of α-synuclein, in contrast, reduced the reserve pool without affecting the number of docked vesicles [28]. In lamprey reticulospinal synapses, antibody-mediated disruption of α-synuclein caused a dose-dependent loss of synaptic vesicles in the reserve pool, and depletion of the docked pool [20]. Deletion of all three synucleins in triple knockout mice resulted in an altered synaptic structure and transmission, a 30% decrease of the size of excitatory synapses [29], increased vesicle tethering to the active zone, and a reduction in the yet unidentified inter-linking connectors of synaptic vesicles, whose identity may be synapsins, synucleins, and/or other proteins (Figure 2) [30–32].
Figure 2. The physiological role of α-synuclein in the presynaptic nerve terminal.

Synaptic vesicles can be functionally divided into a reserve pool, recycling pool and readily releasable pool (RRP), which includes docked and fusing vesicles. Upon invasion of an action potential, RRP vesicles fuse with the plasma membrane to release neurotransmitters, and are retrieved via kiss-and-run, ultrafast, or clathrin-independent endocytosis (CIE) to reform the recycling pool of synaptic vesicles, or via clathrin-mediated endocytosis (CME), upon sorting through a recycling compartment, to reform the reserve pool of synaptic vesicles. The recycling pool is supplemented from the reserve pool under conditions of high synaptic activity, to fill the demand in synaptic vesicles. α-Synuclein maintains pool homeostasis via clustering synaptic vesicles, through interaction with the vesicle SNARE protein VAMP2/Synaptobrevin-2 and synapsins, which restricts synaptic vesicle mobility (Inset A). Separately, α-synuclein acts on the fusion pore formed between the synaptic vesicle membrane and the presynaptic plasma membrane where it stabilizes SNARE-complex assembly, which may result in an increased pore size and/or longer pore opening, and increased amount of neurotransmitter release (Inset B). Note that for simplicity, synaptic vesicle pools are depicted as spatially separated.
How can α-synuclein affect synaptic vesicle pools? Simultaneous binding of α-synuclein to the synaptic vesicle protein VAMP2/synaptobrevin-2 and synaptic vesicle phospholipids triggers vesicle clustering (Figure 2) [18, 19, 24]. This clustering involves also interactions with synapsins (Figure 2). In lamprey reticulospinal synapses, antibody-blockade of synapsin function depleted the reserve pool while leaving the readily releasable pool intact [33]. Recently, in vitro and in cultured neurons, a synapsin liquid phase was reported that can mediate synaptic vesicle clustering [34, 35], and which recruits α-synuclein [36], with a concentration-dependent effect on vesicle clustering and requiring a specific ratio of α-synuclein and synapsin-1 (Figure 2) [37]. Thus, synapsins may enhance vesicle clustering by recruiting or stabilizing α-synuclein at the synapse [38].
α-Synuclein is surprisingly mobile within the cytoplasm. Upon neuronal stimulation, both synapsins and α-synuclein dissociate from synaptic vesicles, disperse from presynaptic terminals, and re-cluster rapidly after signal cessation [39–41]. While synapsins detach from synaptic vesicles during stimulation, α-synuclein dispersion occurs only after exocytosis [40, 42, 43], possibly when the highly curved synaptic vesicle membrane collapses into the plasma membrane during fusion. This is supported by α-synuclein’s preference towards vesicles of high curvature [44, 45]. Loss of synaptic vesicle binding of synapsins and α-synuclein is expected to remove the brakes on release instituted by vesicle clustering. These findings are consistent with an inhibitory effect of α-synuclein on neurotransmitter release.
The net effect of α-synuclein on synaptic transmission is debated. Three possible scenarios have been proposed: that α-synuclein has no overall effect on neurotransmitter release, that it enhances synaptic transmission, or that it inhibits synaptic transmission (reviewed in [13]). The inconsistency in the interpretations may partially be due to compensatory mechanisms in germline deletion models, overexpression artefacts, or inherent differences in the interrogated systems. However, besides modulating synaptic vesicle pools upstream of exocytosis, α-synuclein has been shown to affect neurotransmitter release more directly. αβγ-Synuclein triple-knockout mice reveal age- and activity-dependent deficits in SNARE-complex assembly and neurological impairments [21, 46], presynaptic SNARE-complex levels are reduced in post-mortem brain tissue of individuals with PD [47], and α-synuclein rescued the SNARE-complex deficits and lethality that occurs in CSPα-knockout mice [48]. This activity is mediated by binding of α-synuclein’s C-terminal residues [21, 24] to N-terminal residues of VAMP2 (Figure 1 and 2) that do not participate in SNARE-complex formation [21]. Interactions of α-synuclein with both, VAMP2 and the synaptic vesicle membrane, are required for the SNARE-complex chaperoning activity (Figure 2) [21]. SNARE-complex assembly catalyzes the physical reaction of membrane fusion during vesicle release [49]. An increase in SNARE-complex assembly therefore indicates a function that promotes neurotransmitter release.
Could α-synuclein both inhibit and promote neurotransmitter release? Such dual activity is not unusual for molecules with regulatory functions. For example, Munc18–1 both inhibits and facilitates neurotransmitter release [49]. While α-synuclein may inhibit the release of clustered vesicles, once a vesicle is docked, α-synuclein may facilitate its fusion. α-Synuclein has been shown to stimulate vesicle docking in vitro [50]. And during individual exocytotic events, loss of synucleins slowed the rate of peptide secretion in adrenal chromaffin cells and neurons [51], suggesting that α-synuclein may act on the fusion pore to accelerate transmitter release. Interestingly, the PD-linked mutations A30P and A53T blocked this activity [51].
The synaptic fusion pore is a nanoscale connection between the synaptic vesicle and presynaptic plasma membrane, and a key intermediate during exocytosis of neurotransmitters [52]. Fusion pore formation requires zippering of the three synaptic SNARE proteins VAMP2, SNAP-25 and syntaxin-1 into a four-helix bundle, the SNARE-complex, along with the coordinated action of multiple other synaptic proteins (reviewed in [49]), to ensure tight regulation of this otherwise spontaneous SNARE-zippering driven process. The initial fusion pore is metastable, flickers between open and closed states, and can reseal without dilating beyond 1–2 nm [52], leading to a transient kiss-and-run exocytosis [53], an established mode of exocytosis for hormone secretion [54]. The pertinence of this mode of exocytosis for synaptic vesicle fusion is debated [53]. Alternatively, the pore may dilate and lead to a collapse of the synaptic vesicle membrane into the plasma membrane, termed full fusion [54]. While recent studies demonstrate that the fusion pore can enter into a relatively stable state [55], the underlying mechanisms that regulate and drive fusion pore dilation and stability remain poorly understood.
Irrespective of the mode of endocytosis, would an increase in SNARE-complex assembly trigger longer lasting neurotransmitter release? One could envision that chaperoning of SNARE-complex assembly by α-synuclein would increase the SNARE-driven force to drive fusion pore opening, by enabling a higher number of assembled SNARE-complexes for exocytosis, in particular with the observed multimerization of α-synuclein on the synaptic vesicle membrane [56] that can engage more VAMP2 molecules. In fact, while fusion pore nucleation required a minimum of two v-SNAREs and was unaffected by increasing v-SNARE copy number, the probability of fusion pore dilation increased with increasing v-SNARE copies and was far from saturation at 15 v-SNARE copies using nanolipoprotein particles and flipped t-SNARE expressing cells [57]. Moreover, a small increase in the number of SNARE-complexes enhanced the rate of release from single pores by three orders of magnitude in reconstituted fusion assays, via affecting both pore size and stability [58]. Accordingly, a reduced number of SNARE-complexes reduced the fusion rate [59]. Another in vitro study found that SNARE-complexes alone are inefficient at fusion pore dilation, and that α-synuclein enhanced both the probability as well as the duration of large pores in its membrane-bound state [60]. In contrast, a recent in vitro study found that membrane-bound α-synuclein accessed vSNAREs within the trans-SNARE complex to form an inhibitory complex leading to a reduction in pore open probability, which was overcome by calcium entry [61]. Another possibility would be that α-synuclein inhibits SNARE-complex disassembly, although this was found not to be the case [21]. Interestingly, although the three synucleins show an overall similar regional distribution in the brain with neuronal co-expression in many areas [28, 62–66], their relative levels differ. Evolutionarily recent regions express predominantly α-synuclein and β-synuclein, whereas more ancient structures have higher levels of γ-synuclein [11, 64, 67–71]. Single-cell RNA-sequencing analyses reveal expression ratios of α-synuclein/β-synuclein and α-synuclein/γ-synuclein varying between 0.5–2.3 in mouse cortex and hippocampus, and between 0.002–350 in the human cortex, with no detection of γ-synuclein in most cells [72, 73]. It remains to be tested if neurons with different ratios of α/β- or α/γ-synucleins vary in terms of SNARE-complex levels, fusion pore dilation, and neurotransmitter release, as β- and γ-synuclein heteromerization with α-synuclein reduces the vesicle-bound pool of α-synuclein [44]. In addition, the clustering activity of α-synuclein may provide a reserve pool of α-synuclein that becomes important during lasting synaptic activity, enabling sustained neurotransmitter release. This function is supported by the activity-dependance of SNARE-complex assembly in synuclein knockout mice [21].
Synaptic vesicles contain ~70 VAMP2 molecules [74], but it remains unknown how many of these copies are acting together in the formation of the fusion pore, and how many SNARE-complexes are necessary for synaptic vesicle fusion. In reconstituted systems, one SNARE-complex was sufficient to promote vesicle fusion, two SNARE-complexes were necessary for fast synaptic transmission in cultured hippocampal neurons, and at least three SNARE-complexes were required for fast membrane fusion kinetics, while a theoretical model based on measurements by atomic force microscopy proposed that at least four SNARE-complexes are required for fusion (reviewed in [75]). Eight SNARE-complexes were needed to trigger fusion on a millisecond time scale in a single vesicle fusion assay, and 15 SNARE-complexes were proposed for exocytosis using botulinum toxins [75]. It is possible that the required number of SNARE-complexes for vesicle fusion depends on the physiological conditions such as the vesicle and synapse type. Studying the role of α-synuclein as a function of synapse type will therefore be a logical next step to assess its need for fusion pore dilation. It is striking that the number of VAMP2 molecules per synaptic vesicle closely mimics that of α-synuclein on the vesicle surface [76], suggesting that α-synuclein levels may scale with the number of synaptic vesicles in each terminal.
α-Synuclein’s role in pathology
PD is conventionally characterized by a progressive loss of dopaminergic neurons in the substantia nigra pars compacta (reviewed in [77]). The surviving neurons accumulate α-synuclein in the form of Lewy bodies and Lewy neurites. When the loss of dopamine-producing neurons reaches 60–80%, the cardinal signs of bradykinesia, resting tremor, muscular rigidity, and postural and gait impairments begin to manifest, and worsen progressively [77]. However, the clinical spectrum of PD also includes non-motor features, such as olfactory dysfunction, dementia, depression, sleep disorder, autonomic dysfunction, pain and fatigue, many of which precede the classic motor features by years [77]. Therefore, brain regions other than the substantia nigra and neurotransmitters other than dopamine are affected as well. In addition, α-synuclein pathology occurs early in the peripheral nervous system, and aggregated α-synuclein has been detected in the GI tract, spinal cord, sympathetic ganglia, and sciatic and vagus nerves [77], which may account for the non-motor PD symptoms mentioned above. In fact, the Braak staging of PD postulates that Lewy pathology begins in the peripheral nervous system (PNS), with progressive involvement of the central nervous system (CNS), from caudal to rostral brain areas [78]. Concordantly, in a number of studies, vagotomy blocked α-synuclein spread from the gut to the brain and prevented neurodegeneration (reviewed in [79]). However, other studies have questioned the reduction of PD risk by vagotomy, and found that pathology does not always follow the gut-brain route; it has also been noted that pathological findings in Lewy body dementia patients support a CNS to PNS spread [79]. For addressing these lingering controversies, there is a need for specific, sensitive and reproducible methods for detecting α-synuclein pathology and its path of spread across the CNS and PNS, so that a more definitive timeline for the spread of pathology could be established.
Most PD cases are idiopathic, whereas about 10% cases are monogenic. Rare missense mutations (Figure 1), some of which are of unknown significance or have conflicting data regarding their propensity to aggregate and induce pathology, and SNCA gene triplication leads to early-onset disease (reviewed in [80, 81]). SNCA copy number gains occur also via mosaic patterns [82], and polymorphisms in the SNCA locus are associated with risk of developing PD, altering gene transcription, mRNA stability or processing, and modifying the age of onset [83, 84]. In addition, several posttranslational modifications of α-synuclein have been linked to PD, including phosphorylation at residues Y125, Y133, Y136, S87 and S129, of which S129 has been most consistently associated with pathology and affects α-synuclein aggregation and fibril structure [85–87]. α-Synuclein ubiquitination and sumoylation affect clearance and aggregation rates, nitration at residues Y39, Y125, Y133, and Y136 results in accelerated oligomerization, O-GlcNAcylation at multiple threonine residues inhibits aggregation and can alter the structure of α-synuclein aggregates, and C-terminal truncation, which is present in Lewy bodies, is proposed to accelerate α-synuclein aggregation (reviewed in [88, 89]). α-Synuclein self-assembles into β-sheet rich structures and insoluble α-synuclein aggregates (reviewed in [6]). Once aggregated, α-synuclein pathology is proposed to spread cell-to-cell throughout the brain, via templating of native α-synuclein into aggregates [90]. While this has been primarily established in animal models and via post-mortem imaging of transplanted cells or tissues in humans, definitive proof is still lacking in the normal disease course. Formation of intracellular α-synuclein aggregates upon seeding with exogenous fibrils is more pronounced in neurons that express high endogenous levels of α-synuclein [91]. This has raised the possibility that misfolded α-synuclein may originate in the gut or olfactory system, with environmental factors triggering the disease process.
Overall, elevated α-synuclein levels seem to accelerate aggregation and onset of pathology, which has prompted the development of α-synuclein lowering strategies as therapeutic targets [92]. However, these have yielded no clinical benefit so far [93, 94]. Noteworthy, it is unclear if α-synuclein levels are elevated in the brains of sporadic PD patients as evidence for both increased and decreased α-synuclein mRNA levels in surviving neurons was found [95]. These conflicting findings likely relate to artifacts of measuring end-stage α-synuclein expression, and were possibly also affected by the long course of PD treatment, which highlights the importance of measuring early changes in α-synuclein levels during disease progression.
How do α-synuclein aggregates trigger pathology? Many suspected mechanisms have been proposed, but a definitive picture is still lacking. Calcium dyshomeostasis predisposes to neuronal death [96], and formation of pore-like α-synuclein oligomers increases membrane permeability [97]. Such dysfunction would interfere significantly with synaptic function. Overexpression of WT, A53T and E46K, but not of A30P impairs synaptic vesicle exocytosis [22, 43, 98–100], possibly by interfering with a late step in exocytosis [43], or affecting vesicle reclustering following endocytosis [22]. With the reduced membrane binding by A30P, this highlights the impact of the N-terminal membrane binding region of α-synuclein also for its dysfunction. Other proposed mechanisms of α-synuclein toxicity include protein misfolding and aggregation, apoptosis, mitochondrial dysfunction, oxidative stress, impairments of the ubiquitin-proteasome system, lysosomal dysfunction, neuroinflammation, and activation of the innate and adaptive immune system, where α-synuclein may be a primary trigger for its activation (Figure 3) (reviewed in [101, 102]). It remains unclear, however, whether these are the first steps in the pathology, or alternatively follow other dysfunctional neuronal pathways (see Outstanding Questions).
Figure 3. Contributions of loss-of-function of versus gain-of-function of α-synuclein to disease.
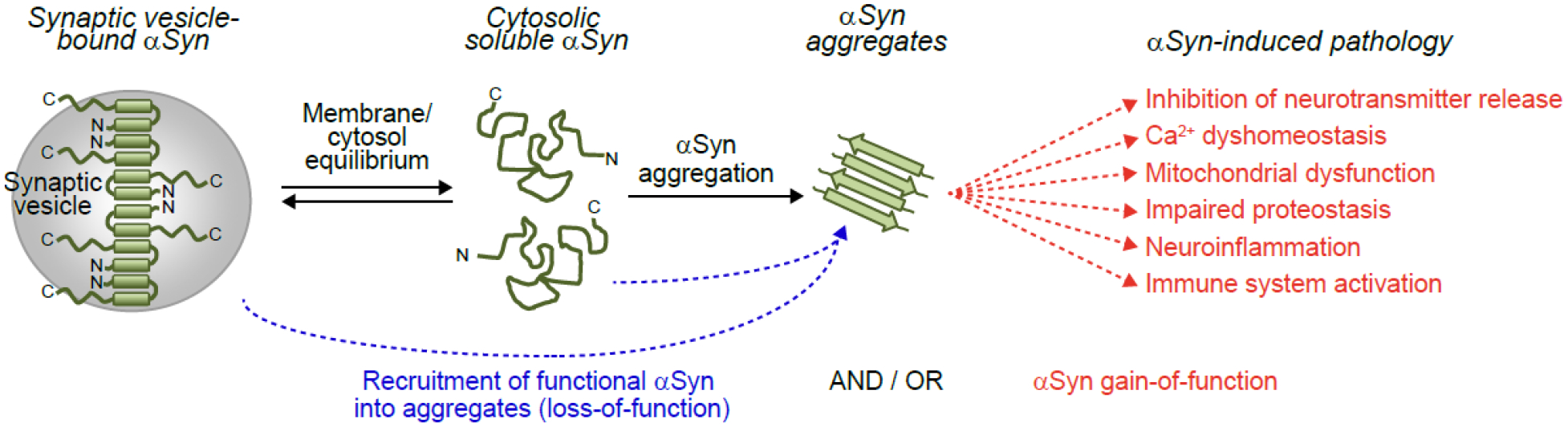
α-Synuclein exists in equilibrium between a membrane-bound α-helical pool that mediates its physiological activities (see also Figure 2), and a largely unstructured state in cytosol. At some point during disease pathogenesis, α-synuclein aggregates form from its cytosolic state. α-Synuclein aggregates sequester functional α-synuclein in the same neuron and connected neurons through spread, resulting in loss-of-function of α-synuclein and dysfunctional neurotransmitter release. In addition, or alternatively, α-synuclein aggregates directly lead to inhibition of neurotransmitter release, and result in calcium dyshomeostasis, mitochondrial dysfunction, impaired proteostasis and neuroinflammation, as well as immune system activation through a gain-of-toxic activity. The timeline and contribution of these events to disease remain unknown.
Outstanding Questions.
Are current animal models reflective of human α-synuclein pathology? Based on the topics discussed in this review, it is becoming clear that better animal models must be developed that (i) overcome compensation artefacts caused by germline knockout or constitutive overexpression, and that (ii) develop pathology slowly, to resolve causative changes from downstream effects.
Are high α-synuclein levels driving accelerated pathology or do they provide a buffer of functional α-synuclein? How much α-synuclein is required for its synaptic function? Studying the role of α-synuclein as a function of synapse type and its synaptic levels in an activity-dependent manner and in combination with age- and disease-specific changes will enable us to better address these questions. This includes improving current ways to identify the species of α-synuclein that contribute to total levels, likely comprising a heterogeneous population at varied ratios of modified and truncated, oligomeric and functional species.
What is the role of α-synuclein aggregation versus loss of its function at the synapse in disease? Likely, both, loss-of-function and gain-of-toxic function of α-synuclein contribute to synucleinopathies. Measuring the relative contribution of each in a time-, neuron- and synapse-resolved manner may help us to also better understand the selective vulnerability of certain neuron subtypes.
The impact of α-synuclein loss-of-function on disease pathogenesis
As discussed above, there is a strong link between α-synuclein dose and disease outcome, which has led to the gain-of-toxic-function hypothesis of α-synuclein. However, aggregation of α-synuclein can also result in loss-of-function of α-synuclein by recruitment of physiologically active α-synuclein into pathological aggregates, thereby diminishing its function(s) (Figure 3). In fact, little synaptic and soluble α-synuclein is found upon administration of α-synuclein fibrils in animals [90, 103, 104], suggesting that newly produced α-synuclein is sequestered by the aggregation process. Multiple lines of evidence support this scenario. First, studies using germline deletion of α-synuclein may be problematic due to compensatory mechanisms. Knockout of α-, β- or α/β-synucleins in mice had no effect on excitatory neurotransmission [105], but triggered compensatory increases in β- or γ-synuclein levels [105, 106]. In mice, removal of α-synuclein triggered an upregulation of synapsin-III [107], and loss of synapsin-III increases dopamine release [108]. Therefore, upregulation of synapsin-III may reduce dopamine levels, which was observed in α-synuclein knockout mice [109]. In addition, α/γ-synuclein knockout in mice increased dopamine release [110], and αβγ-synuclein triple-knockout mice showed more dopamine depletion compared to the α/β-double knockout mice [29]. An unbiased screen identified over 300 transcripts differentially regulated in α-synuclein germline knockouts versus WT mice [111]. However, the ability of synucleins to bind to synaptic vesicles differs significantly when directly compared [44], and γ-synuclein cannot interact with presynaptic SNARE-complexes [112], suggesting that β- or γ-synuclein cannot compensate, at least not fully, for lack of α-synuclein.
Can loss of α-synuclein cause neurodegeneration? While loss of α-synuclein in mice reduced midbrain dopaminergic neuron counts [113], studies in different α-synuclein knockout models [109, 114, 115], and αβγ-synuclein triple-knockout mice [29] did not find impaired survival. Furthermore, knockout of all synucleins in mice did not produce neurodegeneration or Parkinsonism, but resulted in hyperactivity and motor impairments, visual issues and changes in presynaptic morphology [21, 29, 46]. It remains unclear if knockout of α-synuclein over the short lifespan of mice can produce PD-like pathobiology, and if lack of α-synuclein in humans would produce PD symptoms. In contrast to constitutive α-synuclein deletion, acute reduction of α-synuclein levels in adult animals produced neurodegeneration in mice [116], rats [117], and non-human primates [116–120]. A caveat to consider is that most of these studies were conducted using RNA interference, which carries the possibility of toxicity or off-target effects. However, in some of these studies, neurodegeneration was rescued by re-introducing α-synuclein [116], which partly addresses the limitations of the RNA interference approach. Neuronal loss was not found in hippocampal mouse neurons [121], or in the midbrain of squirrel monkeys [122] where RNA levels remained higher. This suggests that once α-synuclein levels fall below a certain threshold (i.e., 40–50% in models lacking neurodegeneration versus 14–30% in models developing neurodegeneration), lack of synuclein function can produce neurodegeneration, at least in some neuronal populations which may depend more on the presence of functional α-synuclein. As a rough estimate, 50% of α-synuclein appear to be sufficient for maintaining normal physiological function, as there are no reports (to our knowledge) of PD or PD-like symptoms in humans heterozygous for α-synuclein [123] or in mice. It remains to be determined how much α-synuclein is needed to maintain its function at the synapse, and at what dose and age the impact of losing α-synuclein is most severe, which likely depends on the neuron and synapse type (see Outstanding Questions).
Initial studies of α-synuclein expression in sporadic PD patients have yielded conflicting results, because SNCA expression levels were normalized to housekeeping gene(s) that changed themselves. Utilizing a strategy that applies a normalization factor based on four expression-characterized housekeeping genes revealed a reduction of α-synuclein mRNA in the substantia nigra pars compacta of PD patients [95]. In further support to this notion, soluble α-synuclein protein decreased with disease progression in PD patients and became increasingly insoluble and S129-phosphorylated [124]. Yet, whether these changes are directly relevant to the progression of PD pathology remains unclear. It is possible, instead, that these changes reflect a compensatory reduction due to somatic α-synuclein accumulation, either due to an age-dependent redistribution of α-synuclein from the presynaptic terminal to the soma, observed in aging humans and monkeys [125], or due to perinuclear accumulation of aggregated α-synuclein. With aging being the largest risk factor for developing PD, decreased α-synuclein levels, in particular functional levels at the synapse, may participate in PD pathogenesis, or represent an independent process that contributes to disease. In addition, despite the doubled mRNA levels in brains of triplication patients, there is no increase in soluble α-synuclein, but only an increase in aggregated α-synuclein [126], likely due to sequestration of newly produced α-synuclein into aggregates. Further, α-synuclein accumulates in the brains of synucleinopathy patients, but the majority is aggregated [126]. Interestingly, all identified pathogenic missense mutations in α-synuclein cluster in the N-terminal membrane binding region (Figure 1), leading to either loss of membrane binding and thereby function, or gain of aggregation which in itself leads to loss of function via sequestration of functional α-synuclein (reviewed in [80, 81]). Lastly, patients with REP1 polymorphisms that result in lower α-synuclein expression, have worse motor and cognitive outcomes once they develop PD, while polymorphisms that result in higher α-synuclein levels, have better outcomes [127].
In contrast, mice overexpressing α-synuclein produce varied levels of neurodegeneration. Currently, based on diverse promoter usage for expression, α-synuclein levels vary between 0.3–30-fold and differ in the temporal onset of expression (reviewed in [128]). While some of these models recapitulate α-synuclein aggregation, loss of dopaminergic neurons, and motor impairments, many have not produced PD-like neuropathology with Lewy bodies, and the question arises whether such an overexpression probes for PD-relevant pathways or non-physiological responses to α-synuclein overexpression. This also highlights that compensation should be considered not only in germline knockout models, but also in constitutive overexpression models.
Toxic effects of α-synuclein include mitochondrial dysfunction, calcium dyshomeostasis and impairments in proteostasis (Figure 3) [101]. But are these effects independent of α-synuclein’s physiological function? Both aggregation and knockdown of α-synuclein have been shown to impair mitochondrial calcium buffering [101], and mitochondrial dysfunction would be expected to have dramatic effects on neurotransmitter release, due to the high energy-demand of this process as well as the relevance of cytosolic/synaptic calcium to its proper function. However, aggregated α-synuclein and thereby its toxicity are located mainly somatically and perisomatically [129], therefore likely having no direct effect on synaptic mitochondria. So far, it remains unclear if these pathological changes are causative for the pathology or alternatively reflecting downstream effects of losing α-synuclein’s function (see Outstanding Questions).
Inter-species comparisons provide additional insights into α-synuclein’s physiology and pathophysiology. Evolutionarily, α-Synuclein expression is limited to vertebrates. Yet, transgenic flies and worms expressing human α-synuclein developed α-synuclein aggregates and toxicity [130, 131]. Further, in α-synuclein knockout mice, overexpression of A53T α-synuclein caused toxicity [132]. In the forementioned three models, there is no endogenous α-synuclein, and therefore these models circumvent the possibility of loss-of-function as a potential mechanism when examining the effects of α-synuclein expression. Another line of evidence for α-synuclein toxicity is its ability to permeabilize membranes when in an oligomeric state [97, 133], with effects as described earlier. Furthermore, α-synuclein aggregates inhibit the autophagy-lysosome pathway [101], leading to accumulation of not only aggregated α-synuclein but also other potentially toxic undegraded molecules, causing proteostatic failure. And binding of α-synuclein aggregates to VAMP2 render VAMP2 unable to form SNARE-complexes [134], thereby inhibiting neurotransmitter release.
While animal models produce/display pathology at accelerated speed due to α-synuclein overexpression, this does not reflect PD pathology in humans. Aggregates and Lewy body pathology build up over a long time, and are present in pre-symptomatic PD patients, while neuron loss and widespread symptoms appear later [78, 135, 136]. It remains to be clarified whether this is due to the ability of cells to tolerate some α-synuclein toxicity, or whether loss of α-synuclein happens earlier than α-synuclein toxicity. Of note, ~10% of clinically normal individuals over the age of 60 have Lewy body pathology in the brain without notable neuron loss or PD symptoms [137], but it remains unclear whether these patients were, in fact, in a trajectory to develop PD later in life but were at a sufficiently early stage to remain symptom-free.
There is recent evidence for both, gain-of-toxic function and loss-of-normal function of α-synuclein in cortico-basolateral amygdala glutamatergic transmission [104]. In mice, α-synuclein aggregation selectively affected vGluT1-expressing neurons, where endogenous α-synuclein levels are high, while vGluT2 terminals that do not contain much α-synuclein were unaffected [104], and appeared more resilient to neurodegeneration [138, 139]. This suggests that terminals with higher native α-synuclein levels are more prone to pathology due to a higher requirement of functional α-synuclein which depletes with aggregation. With technological advances, determining the synaptic levels of α-synuclein across brain regions and neuron types will help addressing this hypothesis.
Concluding remarks and future perspectives
Current evidence suggests that pathology in PD and related synucleinopathies is likely due to a combination of losing α-synuclein’s physiological function at the synapse and of its gain of toxic activity which leads to dysfunction of multiple intracellular pathways within neurons and surrounding cells. This combined effect likely triggers dysfunction at molecular, cellular and organ levels, culminating in the multifaceted central and peripheral nervous system symptoms of PD.
The question of whether pathology in synucleinopathies is due to α-synuclein toxic gain-of-function, a loss-of-function, or a combination of both, has important translational and therapeutic implications. Currently, several therapeutic strategies being explored aim at reducing α-synuclein levels, either by reducing α-synuclein transcription or translation, or by increasing its degradation. Such approaches will reduce functional α-synuclein levels further, potentially with deleterious outcomes. Antibodies targeted at oligomeric α-synuclein are expected to overcome this limitation, since they do not target the physiological α-synuclein pool. However, two recent antibody trials have failed with respect to PD outcomes [93, 94]. The reasons for the lack of therapeutic benefit in these trials remain unknown. Target availability and disease stage may play a role.
Overall, more work is needed to understand why and when α-synuclein transitions from a physiologically functional conformation to a pathogenic state (see Outstanding Questions). Addressing this knowledge gap will help clarify why some individuals develop PD and others do not, and may provide better targets for therapeutic intervention.
Highlights.
α-Synuclein carries multiple functions in neurotransmitter release at neuronal synapses. Its net impact on synaptic release is likely a balance between an inhibitory effect mediated by synaptic vesicle clustering, and a release-promoting effect mediated by SNARE-complex chaperoning and fusion pore opening.
Pathologically, α-synuclein aggregates are a hallmark of synucleinopathies including Parkinson’s disease. α-Synuclein dosage and disease outcome are strongly linked, which has led to the gain-of-toxic-function hypothesis. However, loss-of-function of α-synuclein, through sequestering of physiological α-synuclein into aggregates, has recently gained attention as a potential pathogenic mechanism, supported by reduced functional α-synuclein in human PD tissue.
Current therapeutic strategies have mostly aimed at reducing α-synuclein levels, with little benefit so far. If α-synuclein loss-of-function contributes to disease pathogenesis, these strategies may be even expected to worsen disease outcome.
Acknowledgements
This work was supported by funding from NIH (1R01NS095988 and 1R01AG052505 to M.S.; R01NS121077, R01NS113960, R21NS127939, and R01NS102181 to J.B.; RF1NS126342 to M.S. and J.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1.George JM (2002) The synucleins. Genome Biol 3, REVIEWS3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maroteaux L, et al. (1988) Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 8, 2804–2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iwai A, et al. (1995) The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 14, 467–475 [DOI] [PubMed] [Google Scholar]
- 4.Spillantini MG, et al. (1997) Alpha-synuclein in Lewy bodies. Nature 388, 839–840 [DOI] [PubMed] [Google Scholar]
- 5.George JM, et al. (1995) Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 15, 361–372 [DOI] [PubMed] [Google Scholar]
- 6.Uversky VN (2007) Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J Neurochem 103, 17–37 [DOI] [PubMed] [Google Scholar]
- 7.Burre J, et al. (2013) Properties of native brain alpha-synuclein. Nature 498, E4–6; discussion E6–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chandra S, et al. (2003) A broken alpha -helix in folded alpha -Synuclein. J Biol Chem 278, 15313–15318 [DOI] [PubMed] [Google Scholar]
- 9.Ferreon AC, et al. (2009) Interplay of alpha-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc Natl Acad Sci U S A 106, 5645–5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cholak E, et al. (2020) Avidity within the N-terminal anchor drives alpha-synuclein membrane interaction and insertion. FASEB J 34, 7462–7482 [DOI] [PubMed] [Google Scholar]
- 11.Ueda K, et al. (1993) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 90, 11282–11286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hashimoto M, et al. (2000) The role of NAC in amyloidogenesis in Alzheimer’s disease. Am J Pathol 156, 734–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao V, et al. (2022) Functional and Pathological Effects of alpha-Synuclein on Synaptic SNARE Complexes. J Mol Biol, 167714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burre J, et al. (2015) Definition of a molecular pathway mediating alpha-synuclein neurotoxicity. J Neurosci 35, 5221–5232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melland H, et al. (2021) Disorders of synaptic vesicle fusion machinery. J Neurochem 157, 130–164 [DOI] [PubMed] [Google Scholar]
- 16.Withers GS, et al. (1997) Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res Dev Brain Res 99, 87–94 [DOI] [PubMed] [Google Scholar]
- 17.Busch DJ and Morgan JR (2012) Synuclein accumulation is associated with cell-specific neuronal death after spinal cord injury. J Comp Neurol 520, 1751–1771 [DOI] [PubMed] [Google Scholar]
- 18.Wang L, et al. (2014) alpha-synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr Biol 24, 2319–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diao J, et al. (2013) Native alpha-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. Elife 2, e00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fouke KE, et al. (2021) Synuclein Regulates Synaptic Vesicle Clustering and Docking at a Vertebrate Synapse. Front Cell Dev Biol 9, 774650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burre J, et al. (2010) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nemani VM, et al. (2010) Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65, 66–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott D and Roy S (2012) alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J Neurosci 32, 10129–10135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun J, et al. (2019) Functional cooperation of alpha-synuclein and VAMP2 in synaptic vesicle recycling. Proc Natl Acad Sci U S A 116, 11113–11115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rizzoli SO and Betz WJ (2005) Synaptic vesicle pools. Nat Rev Neurosci 6, 57–69 [DOI] [PubMed] [Google Scholar]
- 26.Alabi AA and Tsien RW (2012) Synaptic vesicle pools and dynamics. Cold Spring Harb Perspect Biol 4, a013680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cabin DE, et al. (2002) Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J Neurosci 22, 8797–8807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murphy DD, et al. (2000) Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci 20, 3214–3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anwar S, et al. (2011) Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J Neurosci 31, 7264–7274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vargas KJ, et al. (2017) Synucleins Have Multiple Effects on Presynaptic Architecture. Cell Rep 18, 161–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siksou L, et al. (2007) Three-dimensional architecture of presynaptic terminal cytomatrix. J Neurosci 27, 6868–6877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandez-Busnadiego R, et al. (2010) Quantitative analysis of the native presynaptic cytomatrix by cryoelectron tomography. J Cell Biol 188, 145–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pieribone VA, et al. (1995) Distinct pools of synaptic vesicles in neurotransmitter release. Nature 375, 493–497 [DOI] [PubMed] [Google Scholar]
- 34.Milovanovic D, et al. (2018) A liquid phase of synapsin and lipid vesicles. Science 361, 604–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pechstein A, et al. (2020) Vesicle Clustering in a Living Synapse Depends on a Synapsin Region that Mediates Phase Separation. Cell Rep 30, 2594–2602 e2593 [DOI] [PubMed] [Google Scholar]
- 36.Brodin L, et al. (2022) alpha-Synuclein in the Synaptic Vesicle Liquid Phase: Active Player or Passive Bystander? Front Mol Biosci 9, 891508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoffmann C, et al. (2021) Synapsin Condensates Recruit alpha-Synuclein. J Mol Biol 433, 166961. [DOI] [PubMed] [Google Scholar]
- 38.Atias M, et al. (2019) Synapsins regulate alpha-synuclein functions. Proc Natl Acad Sci U S A 116, 11116–11118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chi P, et al. (2001) Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci 4, 1187–1193 [DOI] [PubMed] [Google Scholar]
- 40.Fortin DL, et al. (2005) Neural activity controls the synaptic accumulation of alpha-synuclein. J Neurosci 25, 10913–10921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tao-Cheng JH (2006) Activity-related redistribution of presynaptic proteins at the active zone. Neuroscience 141, 1217–1224 [DOI] [PubMed] [Google Scholar]
- 42.Unni VK, et al. (2010) In vivo imaging of alpha-synuclein in mouse cortex demonstrates stable expression and differential subcellular compartment mobility. PLoS One 5, e10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Larsen KE, et al. (2006) Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci 26, 11915–11922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carnazza KE, et al. (2022) Synaptic vesicle binding of alpha-synuclein is modulated by beta- and gamma-synucleins. Cell Rep 39, 110675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Middleton ER and Rhoades E (2010) Effects of curvature and composition on alpha-synuclein binding to lipid vesicles. Biophys J 99, 2279–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Greten-Harrison B, et al. (2010) alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A 107, 19573–19578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharma M, et al. (2012) Proteasome inhibition alleviates SNARE-dependent neurodegeneration. Sci Transl Med 4, 147ra113 [DOI] [PubMed] [Google Scholar]
- 48.Chandra S, et al. (2005) Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396 [DOI] [PubMed] [Google Scholar]
- 49.Rizo J (2022) Molecular Mechanisms Underlying Neurotransmitter Release. Annu Rev Biophys 51, 377–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hawk BJD, et al. (2019) Alpha-Synuclein Continues to Enhance SNARE-Dependent Vesicle Docking at Exorbitant Concentrations. Front Neurosci 13, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Logan T, et al. (2017) alpha-Synuclein promotes dilation of the exocytotic fusion pore. Nat Neurosci 20, 681–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lindau M and Alvarez de Toledo G (2003) The fusion pore. Biochim Biophys Acta 1641, 167–173 [DOI] [PubMed] [Google Scholar]
- 53.He L and Wu LG (2007) The debate on the kiss-and-run fusion at synapses. Trends Neurosci 30, 447–455 [DOI] [PubMed] [Google Scholar]
- 54.Alabi AA and Tsien RW (2013) Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Annu Rev Physiol 75, 393–422 [DOI] [PubMed] [Google Scholar]
- 55.Shin W, et al. (2018) Visualization of Membrane Pore in Live Cells Reveals a Dynamic-Pore Theory Governing Fusion and Endocytosis. Cell 173, 934–945 e912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burre J, et al. (2014) alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A 111, E4274–4283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu Z, et al. (2017) Dilation of fusion pores by crowding of SNARE proteins. Elife 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bao H, et al. (2018) Dynamics and number of trans-SNARE complexes determine nascent fusion pore properties. Nature 554, 260–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karatekin E, et al. (2010) A fast, single-vesicle fusion assay mimics physiological SNARE requirements. Proc Natl Acad Sci U S A 107, 3517–3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khounlo R, et al. (2021) Membrane Binding of alpha-Synuclein Stimulates Expansion of SNARE-Dependent Fusion Pore. Front Cell Dev Biol 9, 663431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nellikka RK, et al. (2021) alpha-Synuclein kinetically regulates the nascent fusion pore dynamics. Proc Natl Acad Sci U S A 118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lavedan C, et al. (1998) Identification, localization and characterization of the human gamma-synuclein gene. Hum Genet 103, 106–112 [DOI] [PubMed] [Google Scholar]
- 63.Ninkina NN, et al. (1998) Organization, expression and polymorphism of the human persyn gene. Hum Mol Genet 7, 1417–1424 [DOI] [PubMed] [Google Scholar]
- 64.Lavedan C (1998) The synuclein family. Genome Res 8, 871–880 [DOI] [PubMed] [Google Scholar]
- 65.Ahmad M, et al. (2007) Gamma-synuclein and the progression of cancer. FASEB J 21, 3419–3430 [DOI] [PubMed] [Google Scholar]
- 66.Jeannotte AM, et al. (2009) Desipramine modulation of alpha-, gamma-synuclein, and the norepinephrine transporter in an animal model of depression. Neuropsychopharmacology 34, 987–998 [DOI] [PubMed] [Google Scholar]
- 67.Jakes R, et al. (1994) Identification of two distinct synucleins from human brain. FEBS Lett 345, 27–32 [DOI] [PubMed] [Google Scholar]
- 68.Buchman VL, et al. (1998) Persyn, a member of the synuclein family, has a distinct pattern of expression in the developing nervous system. J Neurosci 18, 9335–9341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jakowec MW, et al. (2001) Postnatal expression of alpha-synuclein protein in the rodent substantia nigra and striatum. Dev Neurosci 23, 91–99 [DOI] [PubMed] [Google Scholar]
- 70.Maroteaux L and Scheller RH (1991) The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res Mol Brain Res 11, 335–343 [DOI] [PubMed] [Google Scholar]
- 71.Ueda K, et al. (1994) Tissue-dependent alternative splicing of mRNA for NACP, the precursor of non-A beta component of Alzheimer’s disease amyloid. Biochem Biophys Res Commun 205, 1366–1372 [DOI] [PubMed] [Google Scholar]
- 72.Hawrylycz MJ, et al. (2012) An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lein ES, et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 [DOI] [PubMed] [Google Scholar]
- 74.Takamori S, et al. (2006) Molecular anatomy of a trafficking organelle. Cell 127, 831–846 [DOI] [PubMed] [Google Scholar]
- 75.van den Bogaart G and Jahn R (2011) Counting the SNAREs needed for membrane fusion. J Mol Cell Biol 3, 204–205 [DOI] [PubMed] [Google Scholar]
- 76.Fakhree MA, et al. (2016) The number of alpha-synuclein proteins per vesicle gives insights into its physiological function. Sci Rep 6, 30658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bloem BR, et al. (2021) Parkinson’s disease. Lancet 397, 2284–2303 [DOI] [PubMed] [Google Scholar]
- 78.Braak H, et al. (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24, 197–211 [DOI] [PubMed] [Google Scholar]
- 79.Schaeffer E, et al. (2020) Alpha Synuclein Connects the Gut-Brain Axis in Parkinson’s Disease Patients - A View on Clinical Aspects, Cellular Pathology and Analytical Methodology. Front Cell Dev Biol 8, 573696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guan Y, et al. (2020) Pathogenic Mutations Differentially Regulate Cell-to-Cell Transmission of alpha-Synuclein. Front Cell Neurosci 14, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fevga C, et al. (2021) A new alpha-synuclein missense variant (Thr72Met) in two Turkish families with Parkinson’s disease. Parkinsonism Relat Disord 89, 63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mokretar K, et al. (2018) Somatic copy number gains of alpha-synuclein (SNCA) in Parkinson’s disease and multiple system atrophy brains. Brain 141, 2419–2431 [DOI] [PubMed] [Google Scholar]
- 83.Nalls MA, et al. (2019) Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 18, 1091–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Blauwendraat C, et al. (2019) Parkinson’s disease age at onset genome-wide association study: Defining heritability, genetic loci, and alpha-synuclein mechanisms. Mov Disord 34, 866–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fujiwara H, et al. (2002) alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4, 160–164 [DOI] [PubMed] [Google Scholar]
- 86.Awa S, et al. (2022) Phosphorylation of endogenous alpha-synuclein induced by extracellular seeds initiates at the pre-synaptic region and spreads to the cell body. Sci Rep 12, 1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kawahata I, et al. (2022) Pathogenic Impact of alpha-Synuclein Phosphorylation and Its Kinases in alpha-Synucleinopathies. Int J Mol Sci 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang J, et al. (2019) The Roles of Post-translational Modifications on alpha-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front Neurosci 13, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Manzanza NO, et al. (2021) Alpha-Synuclein Post-translational Modifications: Implications for Pathogenesis of Lewy Body Disorders. Front Aging Neurosci 13, 690293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Luk KC, et al. (2009) Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 106, 20051–20056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Courte J, et al. (2020) The expression level of alpha-synuclein in different neuronal populations is the primary determinant of its prion-like seeding. Sci Rep 10, 4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Menon S, et al. (2022) Alpha-Synuclein Targeting Therapeutics for Parkinson’s Disease and Related Synucleinopathies. Front Neurol 13, 852003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pagano G, et al. (2022) Trial of Prasinezumab in Early-Stage Parkinson’s Disease. N Engl J Med 387, 421–432 [DOI] [PubMed] [Google Scholar]
- 94.Lang AE, et al. (2022) Trial of Cinpanemab in Early Parkinson’s Disease. N Engl J Med 387, 408–420 [DOI] [PubMed] [Google Scholar]
- 95.Dachsel JC, et al. (2007) The ups and downs of alpha-synuclein mRNA expression. Mov Disord 22, 293–295 [DOI] [PubMed] [Google Scholar]
- 96.Wojda U, et al. (2008) Calcium ions in neuronal degeneration. IUBMB Life 60, 575–590 [DOI] [PubMed] [Google Scholar]
- 97.Stockl MT, et al. (2013) alpha-Synuclein oligomers: an amyloid pore? Insights into mechanisms of alpha-synuclein oligomer-lipid interactions. Mol Neurobiol 47, 613–621 [DOI] [PubMed] [Google Scholar]
- 98.Yavich L, et al. (2005) Locomotor activity and evoked dopamine release are reduced in mice overexpressing A30P-mutated human alpha-synuclein. Neurobiol Dis 20, 303–313 [DOI] [PubMed] [Google Scholar]
- 99.Gaugler MN, et al. (2012) Nigrostriatal overabundance of alpha-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathol 123, 653–669 [DOI] [PubMed] [Google Scholar]
- 100.Lundblad M, et al. (2012) Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc Natl Acad Sci U S A 109, 3213–3219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wong YC and Krainc D (2017) alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med 23, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tansey MG, et al. (2022) Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol 22, 657–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Volpicelli-Daley LA, et al. (2011) Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen L, et al. (2022) Synaptic location is a determinant of the detrimental effects of alpha-synuclein pathology to glutamatergic transmission in the basolateral amygdala. Elife 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chandra S, et al. (2004) Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc Natl Acad Sci U S A 101, 14966–14971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Robertson DC, et al. (2004) Developmental loss and resistance to MPTP toxicity of dopaminergic neurones in substantia nigra pars compacta of gamma-synuclein, alpha-synuclein and double alpha/gamma-synuclein null mutant mice. J Neurochem 89, 1126–1136 [DOI] [PubMed] [Google Scholar]
- 107.Zaltieri M, et al. (2015) alpha-synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J Cell Sci 128, 2231–2243 [DOI] [PubMed] [Google Scholar]
- 108.Kile BM, et al. (2010) Synapsins differentially control dopamine and serotonin release. J Neurosci 30, 9762–9770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Abeliovich A, et al. (2000) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252 [DOI] [PubMed] [Google Scholar]
- 110.Senior SL, et al. (2008) Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both alpha-synuclein and gamma-synuclein. Eur J Neurosci 27, 947–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kuhn M, et al. (2007) Whole genome expression analyses of single- and double-knock-out mice implicate partially overlapping functions of alpha- and gamma-synuclein. Neurogenetics 8, 71–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lytkina OA, et al. (2014) Gamma-synuclein binds synaptic vesicles but does not interact with SNARE-complex proteins. Dokl Biochem Biophys 456, 108–110 [DOI] [PubMed] [Google Scholar]
- 113.Garcia-Reitboeck P, et al. (2013) Endogenous alpha-synuclein influences the number of dopaminergic neurons in mouse substantia nigra. Exp Neurol 248, 541–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dauer W, et al. (2002) Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci U S A 99, 14524–14529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schluter OM, et al. (2003) Role of alpha-synuclein in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in mice. Neuroscience 118, 985–1002 [DOI] [PubMed] [Google Scholar]
- 116.Gorbatyuk OS, et al. (2010) In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol Ther 18, 1450–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Benskey MJ, et al. (2018) Silencing Alpha Synuclein in Mature Nigral Neurons Results in Rapid Neuroinflammation and Subsequent Toxicity. Front Mol Neurosci 11, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Khodr CE, et al. (2011) An alpha-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson’s disease, but displays toxicity in dopamine neurons. Brain Res 1395, 94–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Collier TJ, et al. (2016) Is Alpha-Synuclein Loss-of-Function a Contributor to Parkinsonian Pathology? Evidence from Non-human Primates. Front Neurosci 10, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kanaan NM and Manfredsson FP (2012) Loss of functional alpha-synuclein: a toxic event in Parkinson’s disease? J Parkinsons Dis 2, 249–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lewis J, et al. (2008) In vivo silencing of alpha-synuclein using naked siRNA. Mol Neurodegener 3, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.McCormack AL, et al. (2010) Alpha-synuclein suppression by targeted small interfering RNA in the primate substantia nigra. PLoS One 5, e12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Blauwendraat C, et al. (2021) A population scale analysis of rare SNCA variation in the UK Biobank. Neurobiol Dis 148, 105182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhou J, et al. (2011) Changes in the solubility and phosphorylation of alpha-synuclein over the course of Parkinson’s disease. Acta Neuropathol 121, 695–704 [DOI] [PubMed] [Google Scholar]
- 125.Chu Y and Kordower JH (2007) Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease? Neurobiol Dis 25, 134–149 [DOI] [PubMed] [Google Scholar]
- 126.Miller DW, et al. (2004) Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62, 1835–1838 [DOI] [PubMed] [Google Scholar]
- 127.Markopoulou K, et al. (2014) Does alpha-synuclein have a dual and opposing effect in preclinical vs. clinical Parkinson’s disease? Parkinsonism Relat Disord 20, 584–589; discussion 584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cenci MA and Bjorklund A (2020) Animal models for preclinical Parkinson’s research: An update and critical appraisal. Prog Brain Res 252, 27–59 [DOI] [PubMed] [Google Scholar]
- 129.Osterberg VR, et al. (2015) Progressive aggregation of alpha-synuclein and selective degeneration of lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep 10, 1252–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Feany MB and Bender WW (2000) A Drosophila model of Parkinson’s disease. Nature 404, 394–398 [DOI] [PubMed] [Google Scholar]
- 131.Lakso M, et al. (2003) Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J Neurochem 86, 165–172 [DOI] [PubMed] [Google Scholar]
- 132.Cabin DE, et al. (2005) Exacerbated synucleinopathy in mice expressing A53T SNCA on a Snca null background. Neurobiol Aging 26, 25–35 [DOI] [PubMed] [Google Scholar]
- 133.Volles MJ and Lansbury PT Jr. (2002) Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 41, 4595–4602 [DOI] [PubMed] [Google Scholar]
- 134.Choi BK, et al. (2013) Large alpha-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc Natl Acad Sci U S A 110, 4087–4092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Halliday GM, et al. (2006) Critical appraisal of brain pathology staging related to presymptomatic and symptomatic cases of sporadic Parkinson’s disease. J Neural Transm Suppl, 99–103 [DOI] [PubMed] [Google Scholar]
- 136.Jellinger KA (2008) A critical reappraisal of current staging of Lewy-related pathology in human brain. Acta Neuropathol 116, 1–16 [DOI] [PubMed] [Google Scholar]
- 137.Dickson DW, et al. (2008) Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol 115, 437–444 [DOI] [PubMed] [Google Scholar]
- 138.Buck SA, et al. (2021) VGLUT2 Is a Determinant of Dopamine Neuron Resilience in a Rotenone Model of Dopamine Neurodegeneration. J Neurosci 41, 4937–4947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Steinkellner T, et al. (2022) Dopamine neurons exhibit emergent glutamatergic identity in Parkinson’s disease. Brain 145, 879–886 [DOI] [PMC free article] [PubMed] [Google Scholar]