

Abstract
Severe cases of COVID‐19 present hyperinflammatory condition that can be fatal. Little is known about the role of regulatory responses in SARS‐CoV‐2 infection. In this study, we evaluated the phenotype of regulatory T cells in the blood (peripheral blood mononuclear cell) and the lungs (broncho‐alveolar) of adult patients with severe COVID‐19 under invasive mechanical ventilation. Our results show important dynamic variation on Treg cells phenotype during COVID‐19 with changes in number and functional parameters from the day of intubation (Day 1 of intensive care unit admission) to Day 7. We observed that compared with surviving patients, non‐survivors presented lower numbers of Treg cells in the blood. In addition, lung Tregs of non‐survivors also displayed higher PD1 and lower FOXP3 expressions suggesting dysfunctional phenotype. Further signs of Treg dysregulation were observed in non‐survivors such as limited production of IL‐10 in the lungs and higher production of IL‐17A in the blood and in the lungs, which were associated with increased PD1 expression. These findings were also associated with lower pulmonary levels of Treg‐stimulating factors like TNF and IL‐2. Tregs in the blood and lungs are profoundly dysfunctional in non‐surviving COVID‐19 patients.
Keywords: COVID‐19, invasive mechanical ventilation, regulatory T cells
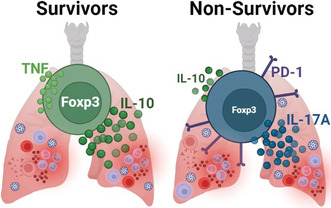
Deceased patients of COVID‐19 present profound Treg dysfunction in lung and blood when compared to severe COVID‐19 survivors. Non‐survivors' Tregs display high expression of PD1, high production of IL‐17A and low production of IL‐10. Treg dysfunction was associated with low levels of lung TNF.
Abbreviations
- ACE2
angiotensin‐converting enzyme 2
- APACHE II
acute physiological assessment and chronic health evaluation
- ARDS
acute respiratory distress syndrome
- BAL
mini broncho‐alveolar aspirate
- COVID‐19
coronavirus disease 2019
- FOXP3
factor Forkhead Box P3
- ICU
intensive care unit
- KLGR1
killer‐cell lectin‐like receptor G1
- MV
invasive mechanical ventilation
- SOFA
Sequential Organ Failure Assessment
- Tregs
regulatory T cells
INTRODUCTION
Coronavirus disease 2019 (COVID‐19) is a viral infection that emerged in Wuhan, China, at the end of 2019 and spread worldwide [1]. It is caused by the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) that is estimated to have caused over 520 million cases, with over 6 million deaths by May 2022 [2]. SARS‐CoV‐2 infection present as a mild disease in the majority of patients, with fever, chills, cough, dyspnoea, fatigue, headache, anosmia, ageusia, sore throat, nasal congestion and gastrointestinal disorders being the most common symptoms [3, 4]. Severe disease is characterized by intense dyspnoea, chest pain, confusion, reduced oxygen saturation levels, acute respiratory distress syndrome (ARDS), shock and acute kidney injury. Around 5% of patients require intensive care unit (ICU) admission [5], with mortality exceeding 70% among intubated patients. Many underlying conditions have been identified as risk factors for severity and higher mortality in COVID‐19, such as obesity, diabetes, high blood pressure and age over 60 years old. The pathogenesis of severe COVID‐19 is still elusive, but some mechanisms have been postulated, such as hyper‐inflammatory syndrome [6], cytokine storm [4, 7, 8, 9], defects in innate [10] or adaptive immunity [11, 12, 13, 14].
During infections, in general, inflammation is a reaction of the immune system to control pathogen proliferation and to restore damaged tissue back to homeostasis [15, 16]. The human innate and adaptive immune response against pathogens, like viruses, activate a variety of cellular and molecular mechanisms that lead to the secretion of pro‐inflammatory cytokines, activation of leukocytes and synthesis of anti‐pathogens molecules, like antibodies and complement factors [17, 18, 19]. Such coordinated response aims to impair viral replication and spread into tissues. However, excessive immune reaction may trigger immunopathogenesis, damaging tissues and impacting systemic homeostasis. For example, the cytokine storm, an overproduction of cytokines observed in some infections like dengue and COVID‐19, is thought to be one of the causes of severe presentation of both diseases [20, 21, 22].
To restrict the pro‐inflammatory reaction within physiological limits, the immune system employs a variety of regulatory mechanisms, of which, the most important is considered the regulatory T cells (Tregs) [23]. Tregs are CD4+ T cells that express high levels of CD25 and factor Forkhead Box P3 (FOXP3), which is a key nuclear transcription factor that determines Treg differentiation and function [24, 25, 26]. Moreover, Tregs suppress the activation, proliferation and effector mechanisms of CD4+ T cells and CD8+ T cells, as well of innate cells. To promote its regulatory functions, Tregs are able to express surface molecules, secrete cytokines with suppressive roles, including interleukin‐10 (IL‐10), or consume or modify lymphocyte survival factors, like IL‐2 and tryptophane [27, 28]. Although severe COVID‐19 is considered a hyperinflammatory complication of the SARS‐CoV‐2 infection, the information about how patients with COVID‐19 limit their inflammatory response is scarce.
Few studies have appreciated the role of Treg on SARS‐CoV‐2 infection [29, 30, 31, 32, 33, 34, 35]. It has been found that Tregs in severe COVID‐19 patients display increased expression of PD‐1 and several disturbances in transcriptional signatures [29]. In addition, therapy with Treg in severe COVID‐19 patients seems to be beneficial promoting recovery [36]. However, how Tregs are regulated during severe COVID‐19 infection and its implications on disease evolution is still a matter of debate. In this study, we evaluated the phenotype and functional differences of blood and lung Tregs in COVID‐19 patients admitted to the ICU. Our results suggest a major dysfunction of Tregs associated with fatal COVID‐19 cases.
MATERIALS AND METHODS
Patients and ethical aspects
This was a single‐centre prospective cohort, carried out from May 2020 to August 2021, at Hospital das Clínicas of the Universidade Federal de Minas Gerais (HC‐UFMG), in Belo Horizonte, MG, Brazil. The research received Institutional Review Board approval (CAAE: 30437020.9.3001.5124) and all the patients or their representatives signed an informed consent before the sample and data collection. This study selected a subset of 40 severe COVID‐19 patients admitted to the ICU, all of them undergoing invasive mechanical ventilation within the first 24 h of ICU stay. Participants were adults (18 years or older) with confirmed SARS‐COV‐2 infection by RT‐PCR in lower respiratory secretion or nasal swab. Patients with forecast of death within 48 h of admission were excluded from the study. Patients were enrolled at Day 1 (within 24 h of ICU admission and intubation) and followed until death or hospital discharge. Biological samples, that is, heparinized blood and mini broncho‐alveolar lavage (BAL), were collected at Days 1, 3 and/or 7 of intubation, which correspond to the days since admission to the ICU. We could not collect all time points from all participants because some patients were released from ICU or died before Day 7. Because of that, additional patients were enrolled to replace early withdraws at later time points (Figure S1).
Collection and processing of biological material
Peripheral blood samples (5 ml) were collected in heparinized tubes at Day 1 (ICU admission and intubation) and Days 3 and 7. Plasma and peripheral blood mononuclear cells (PBMCs) were separated by centrifugation using Histopaque‐1077 gradient (Sigma‐Aldrich). Plasma was collected and frozen at −80°C until use. PBMCs were washed and resuspended in RPMI 1640 (Sigma‐Aldrich) containing 10% fetal bovine serum (FBS) (Sigma‐Aldrich), 2 mM L‐glutamine (Sigma‐Aldrich), 100 IU/ml Penicillin (GIBCO) and 100 μg/ml Streptomycin (GIBCO) for cell culture. BAL samples were collected at the same time points as blood with the instillation of 10 ml of phosphate‐buffered saline (PBS) which was immediately aspirated and dispensed in a closed system secretion collector. BAL was processed within 2 h of collection as described previously [37] with minor modifications. Briefly, an aliquot of 500 μl of non‐processed BAL was separated and frozen at −80°C until use. The remaining BAL was diluted 2× in PBS containing 0.1% of Dithiothreitol (DTT) (LGC Biotecnologia), the sample was then vortexed for 15 s, rested for 10 min and vortexed again for 15 s. Samples were further diluted 4× in PBS and submitted to centrifugation to collect supernatants and cells. Cells were washed one more time with PBS and resuspended in RPMI 1640 (Sigma‐Aldrich) containing 10% FBS (Sigma‐Aldrich), 2 mM L‐glutamine (Sigma‐Aldrich), 100 IU/ml Penicillin (GIBCO) and 100 μg/ml Streptomycin (GIBCO).
Cells from blood and BAL were then cultured in a U‐bottom 96‐well plate (1 × 106 cells/well) in a 5% CO2 incubator at 37°C for 12 h in the presence of Brefeldin A (1.0 μg/ml) (BioLegend) for the accumulation of intracellular cytokines but without stimulation or in the presence of phorbol myristate acetate (PMA) (50 nM) and ionomycin (1 μM) as positive control (Tables S1 and S2). No specific antigen stimulation was used.
Immunophenotyping and intracellular cytokine assessment
After the incubation period, cells were washed in PBS, and labelled with surface antibodies against CD3 BV650 (clone OKT3, BioLegend), CD4 BV510 (RPA‐T4, BioLegend), CD8 eFluor450 (SK1, eBioscience), CD25 Alexa Fluor 700 (M‐A251, BioLegend), CD127 APC/Fire750 (A019D5, BioLegend), PD1 BV605 (EH12.2H7, BioLegend), GITR FITC (110416, R&D Systems), CD200 PE‐Cy7 (OX104, eBioscience) and LAP PerCP (27232, R&D Systems). Cells were washed, fixed and permeabilized using FOXP3 staining buffer Set (eBioscience) according to the manufacturer's instructions. Then proceed with the intracellular labelling of antibodies against FOXP3 PE (clone 236A/E7, BD), TNF PE/Dazzle (MAb11, BioLegend), IL‐10 APC (JES3‐19F1, BioLegend) and IL‐17A BV570 (BL168, BioLegend). After three washes, cells were resuspended in FACS buffer (PBS with 5% FBS) and acquired on a BD LSR‐FORTESSA using the BD FACSDiva software.
Flow cytometry
Analyses were performed using FlowJo Program version 10 (LLC) and the gate strategies are shown in Figure S2. The populations of lymphocytes (SSC‐A × FSC‐A), singlet (FSC‐H × FSC‐A) and time (SSC‐A × Time) were selected. The Boolean Gate tool ‘Make and Gate’ was used to obtain the intersection of these three initial gates, from which CD3+CD4+CD8− was selected (Figure S2). Then, the CD25+FOXP3+ and CD25+CD127low/neg populations were gated and intersected using the Boolean Gate: Make and Gate tool to define the Treg cell population (CD4+CD25+CD127lowFOXP3+). Finally, gates for PD1, GITR, CD200, LAP, IL‐10, TNF and IL‐17A were selected from the Treg cell population using FMO as a reference gate of negative populations. To evaluate the production of IL‐17A and IL‐10 cytokines by Tregs that express or not PD1, we used the Boolean gate tool ‘make and gate’ between Treg cells that produced IL‐17A and expressed or not PD1. In the same way, the Boolean gate ‘make and gate’ was also used between Treg cells that produced IL‐10 and expressed or not PD1. Samples with less than 100 events at the gate of Tregs (CD4+CD25+CD127lowFOXP3+) were excluded from the analysis. GraphPad PrismV5.0 (GraphPad‐Software) was used for data analysis and graphic presentation.
Quantification of cytokines and chemokines
Plasma and supernatant of BAL levels of IL‐6, IL‐2 and TNF were assessed by Bio‐Plex Pro™ Human Cytokine 17‐plex Assay, Cat #M5000031YV (Bio‐Rad) according to the manufacturer's instructions.
Statistical analysis
Statistical analysis was performed using GraphPad Prism program version 6.05 software. The results were analysed using appropriate statistical tests, as indicated in figure legends. The outliers were analysed using the ROUT (Q = 2%) method of GraphPad Prism V6.05 software.
RESULTS
Study population
Severe COVID‐19 patients (n = 40) admitted to the ICU of Hospital das Clínicas of the Universidade Federal de Minas Gerais, Belo Horizonte, Brazil, were recruited between 2020 and 2021. All patients were under invasive mechanical ventilation (MV) and were classified into two groups: survivor and non‐survivor groups. Blood samples and mini‐BAL were collected on Days 1, 3 and/or 7 of intubation which correspond to the day of admission to the ICU. Both groups were comparable for gender and severity scores, namely SOFA and APACHE II, and pulmonary viral load at admission (Table 1). Non‐survivors were older (median age of 48 years old in survivors' group versus 65 years old among non‐survivors, p = 0.01) and showed higher frequency of comorbidities or other risk factors for severe COVID‐19 when compared to survivors (68.4% vs. 95.2%, p = 0.04), mainly cardiovascular diseases, which included cardiac insufficiency, coronary disease, arrhythmias and others (p = 0.01). Non‐survivors also used higher frequencies of drugs that can cause ACE2 hyperexpression, such as ACE2 inhibitors, angiotensin 2 blockers and ibuprofen (26.3% vs. 61.9%, p = 0.03). The frequency of hypertension, diabetes and obesity was similar between survivors and non‐survivors. All patients received antibiotics at some point of ICU stay, often associated with secondary bacterial infection, and most of them received dexamethasone, vasopressor, or inotropic drugs (Table 1). Mean days of COVID‐19 symptoms at the time of admission to the ICU was 9 days (between 6 and 16 days) for both groups and median length of ICU stay was 19 days for survivors and 14 days for non‐survivors.
TABLE 1.
Demographics, clinical and laboratory characteristics of severe COVID‐19 patients with MV survivor or non‐survivor groups during the pandemic of 2020 and 2021
| Survivors (n = 19) | Non‐survivors (n = 21) | |
|---|---|---|
| Baseline ICU admission: | ||
| Age – median (IQR) | 48 (41–62) | 65 (55–75)* |
| Gender (F/M) | 6/13 | 8/13 |
| SOFA at admission – median (IQR) | 7 (3–9) | 7 (5–9) |
| APACHE II at admission – median (IQR) | 16 (10–20) | 13.5 (11–15) |
| Viral load – median (CT of target/CT of endogenous control) (IQR) | 1.02 (0.89–1.37) | 0.84 (0.66–1.12) |
| Patients with some comorbidities – no. (%) | 13 (68.4) | 20 (95.2)* |
| Systemic arterial hypertension – no. (%) | 8 (42.1) | 12 (57.1) |
| Other Cardiovascular diseases – no. (%) | 2 (11.0) | 10 (47.6)* |
| Diabetes mellitus – no. (%) | 4 (21.1) | 8 (38.1) |
| COPD – no. (%) | 3 (15.8) | 4 (19.0) |
| Asthma – no. (%) | 2 (11.0) | 1 (4.7) |
| CKD Dialytic – no. (%) | 0 | 2 (9.5) |
| Active neoplasm – no. (%) | 1 (5.2) | 3 (14.3) |
| Transplanted – no. (%) | 1 (5.2) | 0 |
| Obesity: BMI > 30 (weight (kg)/height (m2)) – no. (%) | 5 (26.3) | 3 (14.3) |
| Other comorbidities – no. (%) | 2 (11.0) | 6 (28.6) |
| Smoking – no. (%) | 3 (15.8) | 4 (19.0) |
| Use of immunosuppressants – no. (%) | 2 (11.0) | 1 (4.7) |
| Use of ACE2‐hyperexpressing drugs – no. (%) | 5 (26.3) | 13 (61.9)* |
| During the study follow‐up: | ||
| Use of dexamethasone – no. (%) | 16 (84.2) | 16 (76.2) |
| Vasopressor or inotropic – no. (%) | 18 (94.7) | 21 (100) |
| Antibiotic use – no. (%) | 19 (100) | 21 (100) |
| Number of days after onset of symptoms at study entry (Day 1 intubation/ICU) – median (IQR) | 10 (7–16) | 8 (6–11) |
| Number of days of ICU stay – median (IQR) | 19 (11–33) | 14 (9–27) |
Abbreviations: ACE2, angiotensin‐converting enzyme 2; BMI, body mass index; CKD, chronic kidney disease; COPD, chronic obstructive pulmonary disease; ICU, intensive care unit; IQR, interquartile range.
Indicates p < 0.05 by Mann–Whitney test.
Non‐survivor patients have fewer Tregs, with lower expression of FOXP3 when compared to survivors
To investigate Tregs (CD3+CD4+CD25hiCD127lowFOXP3+) in COVID‐19, we evaluated their levels and phenotype in the PBMCs and lungs cells (mini‐BAL) after a 12‐h culture without stimulation. Despite the culture, the absolute number of Tregs in the blood and BAL were in accordance with previously published data for COVID‐19 patients [29, 35, 38]. The absolute numbers of Tregs in the blood (PBMCs) of survivors were higher on Days 3 and 7 after ICU admission, but not in the lungs (Figure 1a,b). When the frequency of Treg in CD4+ T cell population was accessed, no difference between groups was observed either in the blood or lungs (Figure 1c,d). We also investigated the levels of expression of FOXP3 by Tregs of survivors and non‐survivors. While we did not find a difference in the GMFI levels of FOXP3 expression in the blood Tregs between survivors and non‐survivors (Figure 1e), there was a trend (p = 0.051) to reduced expression by lung Tregs of non‐survivor patients on Day 7 (Figure 1f).
FIGURE 1.
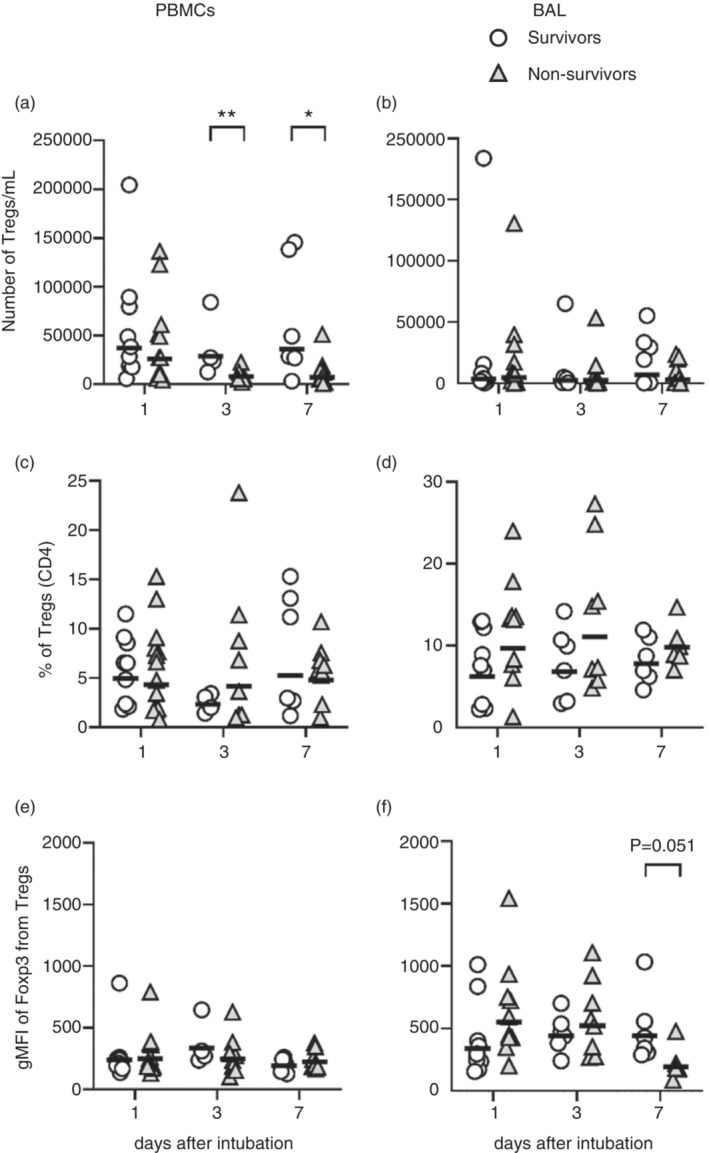
Number, frequency and FOXP3 MFI of blood and lung Tregs. PBMCs and BAL cells from patients with severe COVID‐19 were incubated for 12 h in the presence of BFA without stimulation. Number of Tregs per ml of blood (a) or BAL (b), frequencies of Tregs in CD4+ T cell population (c, d), and geometric means of FOXP3 expression by Tregs (e, f) were evaluated by flow cytometry. Survivors (open circles) n = 9 (D1), n = 4 or 6 (D3) and n = 6 (D7). Non‐survivors (grey triangles) n = 11 (D1), n = 8 (D3) and n = 8 (D7). The lines represent the geometric mean of each group. Differences between these two COVID‐19 groups were analysed by Mann–Whitney test and are indicated by asterisks (* for p < 0.05 and ** for p < 0.01) when statistically significant. BAL, broncho‐alveolar aspirate; PBMC, peripheral blood mononuclear cell.
Lung Tregs of non‐survivor COVID‐19 patients are phenotypically dysfunctional
To better characterize Treg cells, we analysed the expression of functional surface markers like GITR, CD200, LAP and PD1, known to be important for Treg function or dysfunction. Tregs of survivors and non‐survivors expressed similar levels of GITR, CD200 and LAP molecules, both in the blood and lungs, from the day of ICU admission through Day 7 of follow‐up (Figure S3a–f). On the other hand, lung Tregs of non‐survivors presented higher frequencies of PD1 expression when compared to survivors (Figure 2a–d) on Day 7.
FIGURE 2.
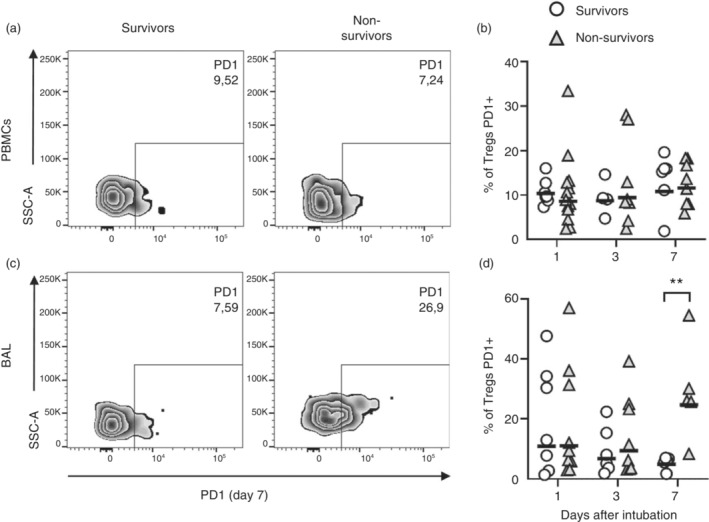
Tregs of fatal COVID‐19 cases express higher frequencies of PD1 in the lungs, but not in the blood, than survivor patients. PBMCs and BAL cells from patients with severe COVID‐19 were incubated for 12 h in the presence of BFA without stimulation. Frequency of PD1 expression by Tregs was evaluated by flow cytometry (a–d). Representative dot plots of Tregs expressing PD1 on Day 7 (a, c). Survivors (open circles) n = 9 (D1), n = 4 or 6 (D3) and n = 6 (D7). Non‐survivors (grey triangles) n = 10 (D1), n = 8 (D3) and n = 8 (D7). The lines represent the geometric mean of each group (b, d). Differences between severe COVID‐19 groups were analysed by Mann–Whitney test and are indicated by asterisks (**) when statistically significant (p < 0.01). BAL, broncho‐alveolar aspirate; PBMC, peripheral blood mononuclear cell.
We, then, analysed the expression of IL‐10, an important effector cytokine of Tregs. PBMCs and lung cells were cultured overnight without stimulation; therefore, we could capture spontaneous cytokine production. IL‐10 production by Tregs could be detected in the blood (Figure 3a,b) and in the lungs (Figure 3c,d). Although at admission, the blood and lung Tregs of both survivors and non‐survivors expressed similar frequencies of IL‐10 (Figure 3b,d), differences on IL‐10 production could be noticed at later time points. For example, starting on Day 3 of ICU admission, lung Tregs of survivors expressed higher frequencies of IL‐10 than non‐survivors (Figure 3d), which was even more evident at Day 7. Interestingly, while survivors increased or maintained the production of IL‐10 by lung Tregs relative to those found at Day 1, non‐survivors showed a significant decrease in IL‐10 production after ICU admission (Figure 3c,d), with most of Treg cells producing no IL‐10 by Day 7. In contrast, blood Treg of non‐survivors displayed higher IL‐10 production than Treg of survivors at Day 7 (Figure 3a,b).
FIGURE 3.
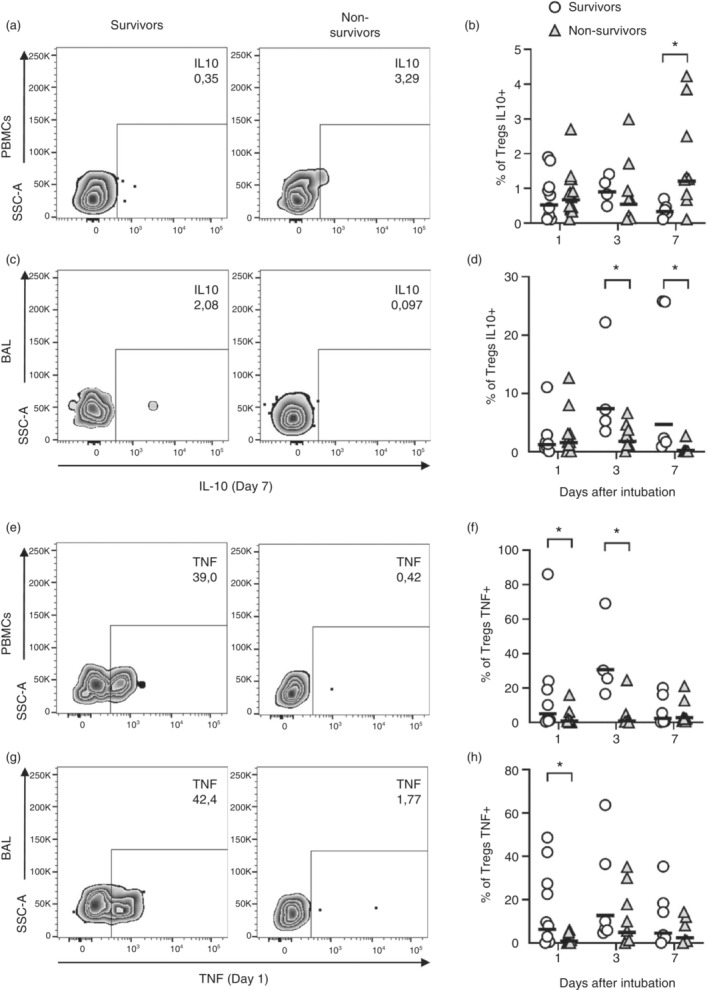
Tregs of non‐survivor COVID‐19 patients presented reduced frequencies of IL‐10 and TNF production. PBMCs and BAL cells from patients with severe COVID‐19 were incubated for 12 h in the presence of BFA without stimulation. Frequency of IL‐10 (a–d) and TNF (e–h) were evaluated by flow cytometry. Representative dot plots of Tregs producing IL‐10 on Day 7 (a, c) or TNF on Day 1 (e, f). Frequency of cytokine production by survivors (open circles) n = 9 (D1), n = 4 or 6 (D3) and n = 6 (D7) and non‐survivors (grey triangles) n = 10 (D1), n = 8 (D3) and n = 8 (D7) are represented by geometric means represented by horizontal lines (b, d, f and h). Differences between severe COVID‐19 groups were analysed by Mann–Whitney test and are indicated by asterisks (*) when statistically significant (p < 0.05). BAL, broncho‐alveolar aspirate; PBMC, peripheral blood mononuclear cell.
We also analysed the production of TNF, which is considered an autocrine survival and functional factor for active Tregs [39, 40, 41, 42]. We found higher frequencies of Tregs producing TNF in the blood (Days 1 and 3) and in the lungs (Day 1) of survivors when compared to non‐survivors (Figure 3e–h). These differences reduced and at Day 7, both groups expressed similar levels of TNF production by Tregs (Figure 4f,h). Since TNF and IL‐2 are important factors for Treg survival, we analysed their levels in plasma and supernatant of BAL obtained on Day 1 of intubation, and on Day 7. TNF levels were reduced at the first day of intubation in the blood and lungs of non‐survivors when compared to survivors (Figure S4a,b), while IL‐2 was reduced in lungs of non‐survivors at Day 7 after intubation (Figure S4d). These data suggest that Tregs of non‐survivors were not only dysfunctional, but also subject to impaired stimulatory factors.
FIGURE 4.
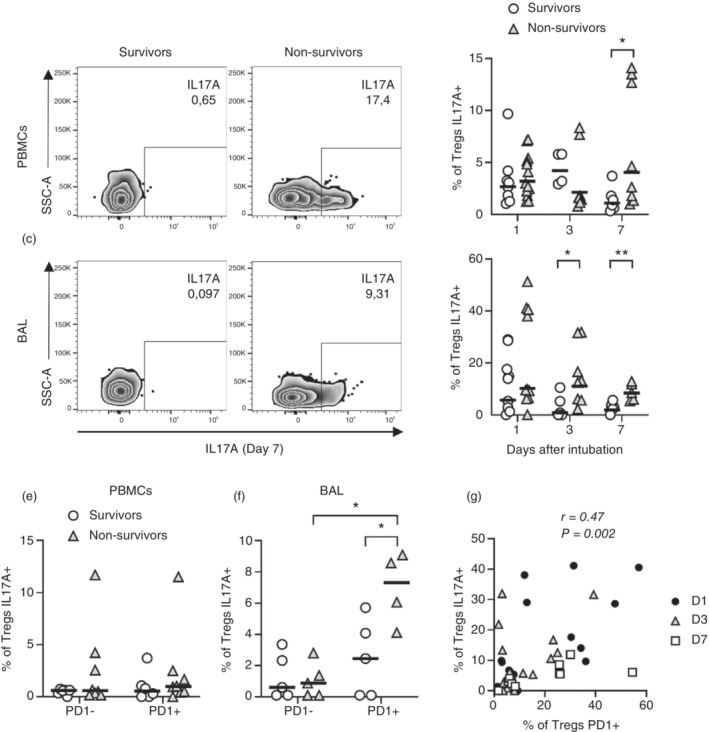
Tregs of non‐survivors display higher production of IL‐17A associated with PD1 expression. PBMCs and BAL cells from patients with severe COVID‐19 were incubated for 12 h in the presence of BFA without stimulation and subjected to flow cytometry. Frequency of IL‐17A+ Tregs (a–d), influence of PD1 expression on IL‐17A production at Day 7 (e, f) and correlation between the frequency of Tregs producing IL‐17A and expressing PD1 (g) were evaluated. Representative dot plots of Tregs producing IL‐17A on Day 7 (a, c) are shown. Frequencies of Tregs in survivors (open circles) n = 9 (D1), n = 4 or 6 (D3) and n = 6 (D7) and non‐survivors (grey triangles) n = 10 (D1), n = 8 (D3) and n = 8 (D7) (b, d, e and f) were evaluated by geometric means and represented by horizontal lines. Differences between severe COVID‐19 groups were analysed by Mann–Whitney test and are indicated by asterisks (* for p < 0.05 and ** for p < 0.01) when statistically significant. All patients were included on correlation analysis from Days 1 (black circles), 3 (white triangles) or 7 (white square) after intubation (g). BAL, broncho‐alveolar aspirate; PBMC, peripheral blood mononuclear cell.
Tregs are reported to produce proinflammatory cytokines, like IL‐17A, during infections or intense inflammatory processes [43] being IL‐6 one of the major drivers of Treg dysfunction and IL‐17A production in mice [44, 45, 46], while in humans, IL‐6 alone might not be sufficient [47]. On the other hand, there are reports showing that IL‐6 may increase the conversion of IL‐17A‐producing Tregs induced by IL‐1b [43, 48, 49]. We therefore evaluated the levels of IL‐6 and production of IL‐17A by Tregs of COVID‐19 survivors and non‐survivors. We found elevated levels of IL‐6 mainly in plasma from non‐survivors (Figure S4e). Indeed, Tregs of COVID‐19 patients, expressed reasonable levels of IL‐17A in the blood (Figure 4a,b) and lungs (Figure 4c,d) on the first day of intubation, that is, the day of ICU admission. However, survivor patients downregulated IL‐17A expression by Tregs from Days 1 to 7 (Figure 4b,d). On the other hand, Tregs of non‐survivors kept high levels of IL‐17A production by Tregs and presented significantly higher frequencies of IL‐17A+ Tregs in the blood (Day 7) and lungs (Days 3 and 7) when compared to survivors (Figure 4b,d). In addition, lung Tregs of non‐survivors also expressed higher per cell IL‐17A levels, as measured by GMFI, at Day 7 when compared to survivors (Figure S5a,b).
PD1 has been considered a marker of dysfunctional Tregs [50, 51]. Selective deletion of PD1 expression by Tregs enhanced Treg proliferation and suppressor activity, improving experimental autoimmune conditions in vivo [52]. In addition, increased activity of PD1‐expressing Tregs has also been implicated in failure with PD1 blockade as cancer therapy [53]. Therefore, we analysed if PD1 expression could be associated with Treg dysfunction. Indeed, IL‐17A was expressed by lung Tregs, but not blood Tregs, that also expressed surface PD1 on Day 7 after intubation (Figure 4e,f). In addition, aggregated data from survivors and non‐survivors showed that IL‐17A expression correlated with PD1 (Figure 4g) expression by Tregs. On the other hand, IL‐10 production by Tregs was negatively associated with the level of PD1 expression, that is, high PD1‐expressing Tregs expressed lower levels of IL‐10 on Day 7 (Figure S6a,b). The frequency of lung PD1+ Treg also correlated positively with SOFA score (Table S3) on Days 1 and 7. In contrast, on Day 1, lung Tregs producing TNF were negatively associated with SOFA. These data suggest that PD1 expression is associated with Treg dysfunction, that is, decreased IL‐10 production capability and increased production of IL‐17A, and with worse clinical outcomes.
DISCUSSION
Tregs are major regulators of peripheral tolerance, including tolerance to infectious diseases. During a severe infection, a balance between pro‐inflammatory and regulatory responses is very important for a favourable outcome. For example, while the lack of inflammation may cause impaired resistance to pathogens, exacerbated inflammatory response might be associated with the development of immune pathology. Infection by SARS‐CoV‐2 causes a mild flu‐like syndrome in most of the infected individuals, but it is estimated to cause severe respiratory manifestations in around 5% of cases, with a death rate of 1.2% in the United States and 2.2% in Brazil [2]. Fatal COVID‐19 evolution has been associated with defective activation of innate [10] and especially adaptive [11, 13, 14, 54] immunity, and cytokine storm [4, 8, 9]. Interestingly, when measuring cytokines associated with cytokine storms, such as IL‐6 and TNF, we found that non‐survivors displayed similar (IL‐6) or even lower (TNF) levels of these cytokines in the lungs. On the other hand, IL‐6 was increased in the blood, which suggests that although the lungs may be an important site for the infection, it may not be the only site responsible for the systemic cytokine storm. These observations suggest that severe COVID‐19 might be associated with the activation of a defective local and systemic inflammation associated with an important dysfunction of regulatory mechanisms.
Dysfunction of Tregs in COVID‐19 has been suggested by previous publications [29, 34, 35]. For example, some data suggest that the numbers of systemic Tregs and the level of FOXP3 expression are lower in severe COVID‐19 patients [32, 35, 38, 55]. Conversely, others have found that Tregs of severe COVID‐19 patients displayed increased regulatory signature, expressed higher FOXP3 levels and displayed increased expression of several regulatory and inflammatory markers like Tbet, KLGR1, PD1, ICOS, GITR, CTLA‐4 [29, 56] and also CXCR3, suggesting the ability to migrate to the lungs [56]. These differences may be associated with a dynamic regulation of Treg function along with the infection. Our data suggest that important changes of Treg phenotype and function may happen in the course of a few days. For example, on the first day of ICU admission, the numbers of Tregs in the blood and lungs were similar between survivors and non‐survivors, but 7 days later, Tregs of non‐survivors were found in lower numbers in the blood and expressed lower levels of FOXP3 in the lungs. Similar dynamics could be observed for IL‐10 and IL‐17A production, which were not different between groups at admission, and changed drastically on the course of 3 or 7 days resulting in significant differences between groups. Interestingly, differences on Treg phenotype were associated not only with time of infection, but also with tissue of evaluation, that is, differences were observed between blood or lungs. For example, on Day 7, Tregs of non‐survivors displayed higher IL‐10 production in the blood when compared to survivors, suggesting higher activity of these cells; however, in the lungs, Tregs were found to produce almost no IL‐10 in non‐survivors, suggesting dysfunction. Similarly, PD1 was highly expressed by lung Tregs of non‐survivors at Day 7, but not in the blood. These data suggest that Tregs were adapting to tissue‐specific environment and to time‐dependent stimulus in the course of infection.
Despite the variations observed in phenotype between blood and lung Tregs and time‐associated modulation, our data suggest that Treg in non‐survivors were undergoing a major dysfunctional phenotype as they stopped producing IL‐10 in the lungs and started to produce IL‐17A. IL‐17A production by FOXP3‐positive T cells may arise based on two mechanisms: (1) under pro‐inflammatory stimulation, Tregs lose FOXP3 expression and acquire IL‐17A production capacity, the exTregs [57], and (2) conventional T cells can transiently express FOXP3 while undergoing activation [58]. The phenotype characterization of peripheral and lung Tregs evaluated in this study suggests a conventional Tregs pattern, that is, CD4+CD25hiCD127lowFOXP3+. While IL‐17A expression by Tregs may not necessarily imply the loss of its regulatory capacity [59], it has often been associated with worsening of inflammatory conditions. We found that although IL‐17A production by Tregs was a common feature in severe COVID‐19 patients, especially in the lungs, Tregs of survivors presented a temporal decrease of its production, while Tregs of non‐survivors maintained high levels of IL‐17A production in the blood and the lungs throughout the observation period. We speculate that such sustained production of a pro‐inflammatory cytokine by a cell which should regulate the inflammatory response may contribute to aggravated pathology, either local or systemic, in severe COVID‐19 patients.
The role of PD1 expression by Tregs is controversial. There is data to show the importance of PD1 for Treg development [60, 61], expression and maintenance of FOXP3 [60] and general regulatory function [62]. On the other hand, PD1 can be associated with dysfunctional Tregs in the context of an infection [63] or cancer [50]. Indeed, Tregs lacking PD1 show better regulatory capacity and control of inflammatory diseases than PD1‐expressing Tregs [52] and blockage of PD1 during cancer treatment can activate PD1+ Tregs, which were dormant before, and offset the activation of PD1‐expressing effector T lymphocytes [53]. Our findings suggest that PD1 expression by Tregs is associated with dysfunction of these cells. PD1‐expressing Tregs not only showed increased IL‐17A levels, but also lower IL‐10 expression in the lungs of non‐survivors. In addition, PD1 expression by lung Tregs correlated with IL17A production and with clinical score SOFA, that is, the higher the expression of PD1, the worse the clinical score of the patients. Interestingly, non‐survivors also lacked factors to sustain Treg function in the lungs. For example, Tregs are highly dependent on TNF [39, 40, 41, 42, 64] and IL‐2 [27] as survival factors. Accordingly, we found that the level of both factors was decreased in the lungs of non‐survivors on Day 1 (TNF) and Day 7 (IL‐2).
Our findings, that Tregs dysfunction is associated with poor COVID‐19 evolution, support previous reports of Tregs in ARDS. For instance, increased levels of Tregs in BAL, but not in the blood or tracheal aspirate, are associated with faster resolution ARDS in intubated patients [65]. Another group found that Tregs were increased in the blood and reduced in the BAL of individuals with ARDS, when compared to non‐ARDS patients [66]. In addition, higher levels of BAL Tregs expressing activation markers were associated with increased survival outcome of ARDS [66] confirming the beneficial role of Tregs in ARDS. In contrast, Treg levels have also been associated with increased mortality by ARDS [67]. However, no characterization of the Tregs has been performed in this study [67] and it is not known if the Tregs associated with poor ARDS outcome were properly functional. Taken together, data from ARDS also suggests that the proper function of Tregs is important for a favourable outcome, as we found in COVID‐19.
Severe COVID‐19 is considered a hyperinflammatory complication of the SARS‐COV‐2 infection suggesting an imbalance between inflammatory and regulatory mechanisms. Therefore, establishing the phenotype of Tregs is important to understand the physiopathology of severe COVID‐19 evolution. In this work, we showed that Tregs of fatal COVID‐19 cases display important dysfunctional markers, especially high expression of PD1 and high levels of IL‐17A production associated with defective production of IL‐10, especially in lungs and to a lesser degree in the periphery. Our results suggest that the loss of proper capacity to control inflammation of dysfunctional Tregs may be one of the mechanisms of immunopathology associated with severe and fatal evolution of COVID‐19.
AUTHOR CONTRIBUTIONS
Marcela Helena Gonçalves‐Pereira and Luciana Santiago contributed equally. Marcela Helena Gonçalves‐Pereira, Luciana Santiago, Cecilia Gómez Ravetti, Paula Frizera Vassallo, Vandack Nobre and Helton da Costa Santiago designed experiments. Marcela Helena Gonçalves‐Pereira, Luciana Santiago, Cecilia Gómez Ravetti, Paula Frizera Vassallo, Marcus Vinicius Melo de Andrade, Mariana Sousa Vieira, Fernanda de Fátima Souza de Oliveira and Adriano de Paula Sabino performed experiments. Marcela Helena Gonçalves‐Pereira, Luciana Santiago, Vandack Nobre and Helton da Costa Santiago analysed the data. Marcela Helena Gonçalves‐Pereira and Helton da Costa Santiago wrote the manuscript. All authors reviewed the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
FIGURE S1. Distribution of biological samples of each participant in the different time points
FIGURE S2. Flow Cytometry Analysis Strategy
FIGURE S3. Frequency of GITR, CD200 and LAP from Tregs
FIGURE S4. Measurement of cytokines TNF, IL‐2 and IL‐6 in plasma and BAL.
FIGURE S5. GMFI of IL‐17A from Tregs
FIGURE S6. Frequency of Tregs IL‐10+ PD1− or IL‐10+ PD1+
TABLE S1. Mean ± SD of frequency of blood Tregs expressing CD200, GITR, LAP, PD1, IL‐10, IL‐17A and TNF with or without stimulation with PMA/IONOMYCIN
TABLE S2. Mean ± SD of frequency of lung Tregs expressing CD200, GITR, LAP, PD1, IL‐10, IL‐17A and TNF with or without stimulation with PMA/IONOMYCIN
TABLE S3. Correlation of SOFA with frequencies of Tregs PD1+ or Tregs TNF+
ACKNOWLEDGEMENTS
The authors are grateful to study participants and healthy volunteers for their participation. The authors are thankful to the Institutional Laboratory of Biomarkers Research (LINBIO) for support with the flow cytometry analyses. The authors also thank the Hospital das Clínicas and Departamento de Pós‐Graduação em Bioquímica e Imunologia of the Universidade Federal de Minas Gerais for the structure to carry out the collection of samples and assays. This work was funded by Ministério da Educação (SESU‐MEC—Public notice ‘Fighting COVID‐19’) and Instituto Nacional de Ciência e Tecnologia em Vacinas (INCT‐Vacinas).
Gonçalves‐Pereira MH, Santiago L, Ravetti CG, Vassallo PF, de Andrade MVM, Vieira MS, et al. Dysfunctional phenotype of systemic and pulmonary regulatory T cells associate with lethal COVID‐19 cases. Immunology. 2022. 10.1111/imm.13603
Marcela Helena Gonçalves‐Pereira and Luciana Santiago contributed equally to this study.
Funding information Instituto Nacional de Ciencia e Tecnologia em Vacinas; Ministério da Educação
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Wang C, Horby PW, Hayden FG, Gao GF. A novel coronavirus outbreak of global health concern. Lancet. 2020;395:470–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coronavirus Resource Center, 2022. https://coronavirus.jhu.edu/data/mortality
- 3. Dixon BE, Wools‐Kaloustian KK, Fadel WF, Duszynski TJ, Yiannoutsos C, Halverson PK, et al. Symptoms and symptom clusters associated with SARS‐CoV‐2 infection in community‐based populations: results from a statewide epidemiological study. PLoS One. 2021;16:e0241875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Phua J, Weng L, Ling L, Egi M, Lim CM, Divatia JV, et al. Intensive care management of coronavirus disease 2019 (COVID‐19): challenges and recommendations. Lancet Respir Med. 2020;8:506–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical course and outcomes of critically ill patients with SARS‐CoV‐2 pneumonia in Wuhan, China: a single‐centered, retrospective, observational study. Lancet Respir Med. 2020;8:475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39:529–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen R, Lan Z, Ye J, Pang L, Liu Y, Wu W, et al. Cytokine storm: the primary determinant for the pathophysiological evolution of COVID‐19 deterioration. Front Immunol. 2021;12:589095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Qi Y, et al. Pathogenic T‐cells and inflammatory monocytes incite inflammatory storms in severe COVID‐19 patients. Natl Sci Rev. 2020;7:998–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Galani IE, Andreakos E. Impaired innate antiviral defenses in COVID‐19: causes, consequences and therapeutic opportunities. Semin Immunol. 2021;55:101522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang W, Berube J, McNamara M, Saksena S, Hartman M, Arshad T, et al. Lymphocyte subset counts in COVID‐19 patients: a meta‐analysis. Cytometry A. 2020;97:772–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu Z, Long W, Tu M, Chen S, Huang Y, Wang S, et al. Lymphocyte subset (CD4+, CD8+) counts reflect the severity of infection and predict the clinical outcomes in patients with COVID‐19. J Infect. 2020;81:318–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mahmoudi S, Rezaei M, Mansouri N, Marjani M, Mansouri D. Immunologic features in coronavirus disease 2019: functional exhaustion of T cells and cytokine storm. J Clin Immunol. 2020;40:974–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sette A, Crotty S. Adaptive immunity to SARS‐CoV‐2 and COVID‐19. Cell. 2021;184:861–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cronkite DA, Strutt TM. The regulation of inflammation by innate and adaptive lymphocytes. J Immunol Res. 2018;2018:1467538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of inflammation: what controls its onset? Front Immunol. 2016;7:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aoshi T, Koyama S, Kobiyama K, Akira S, Ishii KJ. Innate and adaptive immune responses to viral infection and vaccination. Curr Opin Virol. 2011;1:226–32. [DOI] [PubMed] [Google Scholar]
- 18. Bordallo B, Bellas M, Cortez AF, Vieira M, Pinheiro M. Severe COVID‐19: what have we learned with the immunopathogenesis? Adv Rheumatol. 2020;60:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hosseini A, Hashemi V, Shomali N, Asghari F, Gharibi T, Akbari M, et al. Innate and adaptive immune responses against coronavirus. Biomed Pharmacother. 2020;132:110859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cloutier M, Nandi M, Ihsan AU, Chamard HA, Ilangumaran S, Ramanathan S. ADE and hyperinflammation in SARS‐CoV2 infection – comparison with dengue hemorrhagic fever and feline infectious peritonitis. Cytokine. 2020;136:155256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nanaware N, Banerjee A, Mullick Bagchi S, Bagchi P, Mukherjee A. Dengue virus infection: a tale of viral exploitations and host responses. Viruses. 2021;13:1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP. The trinity of COVID‐19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20:363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Campbell C, Rudensky A. Roles of regulatory T cells in tissue pathophysiology and metabolism. Cell Metab. 2020;31:18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roncador G, Brown PJ, Maestre L, Hue S, Martinez‐Torrecuadrada JL, Ling KL, et al. Analysis of FOXP3 protein expression in human CD4+CD25+ regulatory T cells at the single‐cell level. Eur J Immunol. 2005;35:1681–91. [DOI] [PubMed] [Google Scholar]
- 25. Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol. 2008;9:239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yagi H, Nomura T, Nakamura K, Yamazaki S, Kitawaki T, Hori S, et al. Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:1643–56. [DOI] [PubMed] [Google Scholar]
- 27. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T cells and human disease. Annu Rev Immunol. 2020;38:541–66. [DOI] [PubMed] [Google Scholar]
- 28. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg‐mediated T cell suppression. Front Immunol. 2012;3:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Galvan‐Pena S, Leon J, Chowdhary K, Michelson DA, Vijaykumar B, Yang L, et al. Profound Treg perturbations correlate with COVID‐19 severity. Proc Natl Acad Sci U S A. 2021;118(37):e2111315118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alahyari S, Rajaeinejad M, Jalaeikhoo H, Amani D. Regulatory T cells in immunopathogenesis and severity of COVID‐19: a systematic review. Arch Iran Med. 2022;25:127–32. [DOI] [PubMed] [Google Scholar]
- 31. Liu Y, Qi G, Bellanti JA, Moser R, Ryffel B, Zheng SG. Regulatory T cells: a potential weapon to combat COVID‐19? MedComm. 2020;1(2):157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meckiff BJ, Ramirez‐Suastegui C, Fajardo V, Chee SJ, Kusnadi A, Simon H, et al. Imbalance of regulatory and cytotoxic SARS‐CoV‐2‐reactive CD4(+) T cells in COVID‐19. Cell. 2020;183:1340–1353.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saghafi N, Rezaee SA, Momtazi‐Borojeni AA, Tavasolian F, Sathyapalan T, Abdollahi E, et al. The therapeutic potential of regulatory T cells in reducing cardiovascular complications in patients with severe COVID‐19. Life Sci. 2022;294:120392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang H, Wang Z, Cao W, Wu Q, Yuan Y, Zhang X. Regulatory T cells in COVID‐19. Aging Dis. 2021;12:1545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Y, Zheng J, Islam MS, Yang Y, Hu Y, Chen X. The role of CD4(+)FoxP3(+) regulatory T cells in the immunopathogenesis of COVID‐19: implications for treatment. Int J Biol Sci. 2021;17:1507–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stephen‐Victor E, Das M, Karnam A, Pitard B, Gautier JF, Bayry J. Potential of regulatory T‐cell‐based therapies in the management of severe COVID‐19. Eur Respir J. 2020;56:2002182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pizzichini E, Pizzichini MM, Efthimiadis A, Evans S, Morris MM, Squillace D, et al. Indices of airway inflammation in induced sputum: reproducibility and validity of cell and fluid‐phase measurements. Am J Respir Crit Care Med. 1996;154:308–17. [DOI] [PubMed] [Google Scholar]
- 38. Jimenez‐Cortegana C, Liro J, Palazon‐Carrion N, Salamanca E, Sojo‐Dorado J, de la Cruz‐Merino L, et al. Increased blood monocytic myeloid derived suppressor cells but low regulatory T lymphocytes in patients with mild COVID‐19. Viral Immunol. 2021;34:639–45. [DOI] [PubMed] [Google Scholar]
- 39. He X, Landman S, Bauland SC, van den Dolder J, Koenen HJ, Joosten I. A TNFR2‐agonist facilitates high purity expansion of human low purity Treg cells. PLoS One. 2016;11:e0156311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Urbano PCM, He X, van Heeswijk B, Filho OPS, Tijssen H, Smeets RL, et al. TNFalpha‐signaling modulates the kinase activity of human effector Treg and regulates IL‐17A expression. Front Immunol. 2019;10:3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Urbano PCM, Koenen H, Joosten I, He X. An autocrine TNFalpha‐tumor necrosis factor receptor 2 loop promotes epigenetic effects inducing human Treg stability In vitro. Front Immunol. 2018;9:573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang S, Wang J, Brand DD, Zheng SG. Role of TNF‐TNF receptor 2 signal in regulatory T cells and its therapeutic implications. Front Immunol. 2018;9:784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jung MK, Kwak JE, Shin EC. IL‐17A‐producing Foxp3(+) regulatory T cells and human diseases. Immune Netw. 2017;17:276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8. [DOI] [PubMed] [Google Scholar]
- 45. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL‐17‐producing T cells. Immunity. 2006;24:179–89. [DOI] [PubMed] [Google Scholar]
- 46. Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25‐Foxp3‐T cells or are self‐induced to become Th17 cells in the absence of exogenous TGF‐beta. J Immunol. 2007;178:6725–9. [DOI] [PubMed] [Google Scholar]
- 47. Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL‐17‐producing cells. Blood. 2008;112:2340–52. [DOI] [PubMed] [Google Scholar]
- 48. Afzali B, Mitchell P, Lechler RI, John S, Lombardi G. Translational mini‐review series on Th17 cells: induction of interleukin‐17 production by regulatory T cells. Clin Exp Immunol. 2010;159:120–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bhaskaran N, Faddoul F, Paes da Silva A, Jayaraman S, Schneider E, Mamileti P, et al. IL‐1beta‐MyD88‐mTOR Axis promotes immune‐protective IL‐17A(+)Foxp3(+) cells during mucosal infection and is dysregulated with aging. Front Immunol. 2020;11:595936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lowther DE, Goods BA, Lucca LE, Lerner BA, Raddassi K, van Dijk D, et al. PD‐1 marks dysfunctional regulatory T cells in malignant gliomas. JCI Insight. 2016;1:e85935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perry JA, Shallberg L, Clark JT, Gullicksrud JA, DeLong JH, Douglas BB, et al. PD‐L1‐PD‐1 interactions limit effector regulatory T cell populations at homeostasis and during infection. Nat Immunol. 2022;23:743–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tan CL, Kuchroo JR, Sage PT, Liang D, Francisco LM, Buck J, et al. PD‐1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J Exp Med. 2021;218:e20182232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, et al. PD‐1(+) regulatory T cells amplified by PD‐1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U S A. 2019;116:9999–10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen Z, John WE. T cell responses in patients with COVID‐19. Nat Rev Immunol. 2020;20:529–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hoffmann AD, Weinberg SE, Swaminathan S, Chaudhuri S, Mubarak HF, Schipma MJ, et al. Unique molecular signatures sustained in circulating monocytes and regulatory T cells in convalescent COVID‐19 patients. bioRxiv. 2022:485922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vick SC, Frutoso M, Mair F, Konecny AJ, Greene E, Wolf CR, et al. A regulatory T cell signature distinguishes the immune landscape of COVID‐19 patients from those with other respiratory infections. Sci Adv. 2021;7:eabj0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh‐hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. 2014;20:62–8. [DOI] [PubMed] [Google Scholar]
- 58. Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, et al. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity. 2012;36:262–75. [DOI] [PubMed] [Google Scholar]
- 59. Voo KS, Wang YH, Santori FR, Boggiano C, Wang YH, Arima K, et al. Identification of IL‐17‐producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci U S A. 2009;106:4793–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Amarnath S, Costanzo CM, Mariotti J, Ullman JL, Telford WG, Kapoor V, et al. Regulatory T cells and human myeloid dendritic cells promote tolerance via programmed death ligand‐1. PLoS Biol. 2010;8:e1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD‐L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, et al. The programmed death‐1 (PD‐1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med. 2003;198:63–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Franceschini D, Paroli M, Francavilla V, Videtta M, Morrone S, Labbadia G, et al. PD‐L1 negatively regulates CD4+CD25+Foxp3+ Tregs by limiting STAT‐5 phosphorylation in patients chronically infected with HCV. J Clin Invest. 2009;119:551–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kleijwegt FS, Laban S, Duinkerken G, Joosten AM, Zaldumbide A, Nikolic T, et al. Critical role for TNF in the induction of human antigen‐specific regulatory T cells by tolerogenic dendritic cells. J Immunol. 2010;185:1412–8. [DOI] [PubMed] [Google Scholar]
- 65. Norton DL, Ceppe A, Tune MK, McCravy M, Devlin T, Drummond MB, et al. Bronchoalveolar Tregs are associated with duration of mechanical ventilation in acute respiratory distress syndrome. J Transl Med. 2020;18:427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Halter S, Aimade L, Barbie M, Brisson H, Rouby JJ, Langeron O, et al. T regulatory cells activation and distribution are modified in critically ill patients with acute respiratory distress syndrome: a prospective single‐Centre observational study. Anaesth Crit Care Pain Med. 2020;39:35–44. [DOI] [PubMed] [Google Scholar]
- 67. Adamzik M, Broll J, Steinmann J, Westendorf AM, Rehfeld I, Kreissig C, et al. An increased alveolar CD4+ CD25+ Foxp3+ T‐regulatory cell ratio in acute respiratory distress syndrome is associated with increased 30‐day mortality. Intensive Care Med. 2013;39:1743–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1. Distribution of biological samples of each participant in the different time points
FIGURE S2. Flow Cytometry Analysis Strategy
FIGURE S3. Frequency of GITR, CD200 and LAP from Tregs
FIGURE S4. Measurement of cytokines TNF, IL‐2 and IL‐6 in plasma and BAL.
FIGURE S5. GMFI of IL‐17A from Tregs
FIGURE S6. Frequency of Tregs IL‐10+ PD1− or IL‐10+ PD1+
TABLE S1. Mean ± SD of frequency of blood Tregs expressing CD200, GITR, LAP, PD1, IL‐10, IL‐17A and TNF with or without stimulation with PMA/IONOMYCIN
TABLE S2. Mean ± SD of frequency of lung Tregs expressing CD200, GITR, LAP, PD1, IL‐10, IL‐17A and TNF with or without stimulation with PMA/IONOMYCIN
TABLE S3. Correlation of SOFA with frequencies of Tregs PD1+ or Tregs TNF+
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.