

Abstract
Severe acute respiratorysyndrome coronavirus‐2 (SARS‐CoV‐2) pandemic spread rapidly and this scenario is concerning worldwide, presenting more than 590 million coronavirus disease 2019 cases and 6.4 million deaths. The emergence of novel lineages carrying several mutations in the spike protein has raised additional public health concerns worldwide during the pandemic. The present study review and summarizes the temporal spreading and molecular evolution of SARS‐CoV‐2 clades and variants worldwide. The evaluation of these data is important for understanding the evolutionary histories of SARSCoV‐2 lineages, allowing us to identify the origins of each lineage of this virus responsible for one of the biggest pandemics in history. A total of 2897 SARS‐CoV‐2 whole‐genome sequences with available information from the country and sampling date (December 2019 to August 2022), were obtained and were evaluated by Bayesian approach. The results demonstrated that the SARS‐CoV‐2 the time to the most recent common ancestor (tMRCA) in Asia was 2019‐12‐26 (highest posterior density 95% [HPD95%]: 2019‐12‐18; 2019‐12‐29), in Oceania 2020‐01‐24 (HPD95%: 2020‐01‐15; 2020‐01‐30), in Africa 2020‐02‐27 (HPD95%: 2020‐02‐21; 2020‐03‐04), in Europe 2020‐02‐27 (HPD95%: 2020‐02‐20; 2020‐03‐06), in North America 2020‐03‐12 (HPD95%: 2020‐03‐05; 2020‐03‐18), and in South America 2020‐03‐15 (HPD95%: 2020‐03‐09; 2020‐03‐28). Between December 2019 and June 2020, 11 clades were detected (20I [Alpha] and 19A, 19B, 20B, 20C, 20A, 20D, 20E [EU1], 20F, 20H [Beta]). From July to December 2020, 4 clades were identified (20J [Gamma, V3], 21 C [Epsilon], 21D [Eta], and 21G [Lambda]). Between January and June 2021, 3 clades of the Delta variant were detected (21A, 21I, and 21J). Between July and December 2021, two variants were detected, Delta (21A, 21I, and 21J) and Omicron (21K, 21L, 22B, and 22C). Between January and June 2022, the Delta (21I and 21J) and Omicron (21K, 21L, and 22A) variants were detected. Finally, between July and August 2022, 3 clades of Omicron were detected (22B, 22C, and 22D). Clade 19A was first detected in the SARS‐CoV‐2 pandemic (Wuhan strain) with origin in 2019‐12‐16 (HPD95%: 2019‐12‐15; 2019‐12‐25); 20I (Alpha) in 2020‐11‐24 (HPD95%: 2020‐11‐15; 2021‐12‐02); 20H (Beta) in 2020‐11‐25 (HPD95%: 2020‐11‐13; 2020‐11‐29); 20J (Gamma) was 2020‐12‐21 (HPD95%: 2020‐11‐05; 2021‐01‐15); 21A (Delta) in 2020‐09‐20 (HPD95%: 2020‐05‐17; 2021‐02‐03); 21J (Delta) in 2021‐02‐26 (2020‐11‐02; 2021‐04‐24); 21M (Omicron) in 2021‐01‐25 (HPD95%: 2020‐09‐16; 2021‐08‐08); 21K (Omicron) in 2021‐07‐30 (HPD95%: 2021‐05‐30; 2021‐10‐19); 21L (Omicron) in 2021‐10‐03 (HPD95%: 2021‐04‐16; 2021‐12‐23); 22B (Omicron) in 2022‐01‐25 (HPD95%: 2022‐01‐10; 2022‐02‐05); 21L in 2021‐12‐20 (HPD95%: 2021‐05‐16; 2021‐12‐31). Currently, the Omicron variant predominates worldwide, with the 21L clade branching into 3 (22A, 22B, and 22C). Phylogeographic data showed that Alpha variant originated in the United Kingdom, Beta in South Africa, Gamma in Brazil, Delta in India, Omicron in South Africa, Mu in Colombia, Epsilon in the United States of America, and Lambda in Peru. The COVID‐19 pandemic has had a significant impact on global health worldwide and the present study provides an overview of the molecular evolution of SARS‐CoV‐2 lineage clades (from the Wuhan strain to the currently circulating lineages of the Omicron).
Keywords: Bayesian, COVID‐19, molecular evolution, SARS‐CoV‐2
1. INTRODUCTION
The pandemic caused by coronavirus disease 19 (COVID‐19) has had an overwhelming impact, with more than 614 825 354 cases and 6 536 284 deaths worldwide as of September 24, 2022. 1 Considering the situation by region, Europe has a total of 252 176 618 cases, the Americas have 177 751 530 cases, the Western Pacific 88 872 987 cases, South‐East Asia 60 235 604 cases, the Eastern Mediterranean 23 061 879 cases, and Africa 9 322 404 cases. 2 At the country level, the highest numbers of new cases were reported in Japan (605 919 new cases), the United States of America (395 117 new cases), the Republic of Korea (389 579 new cases), the Russian Federation (372 485 new cases), and China (297 693 new cases). 3
COVID‐19 is caused by the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), a virus of the Coronaviridae family, which infects animals and humans. 4 Tissue tropism and symptoms of infection caused by this virus may vary among different host species. 5 The SARS‐CoV‐2 virus is from the Coronaviridae family of the betacoronavirus B lineage. 6 , 7 Coronaviruses are viruses with a single‐stranded positive‐sense RNA (ssRNA)—ready for translation and subsequent synthesis of its proteins—and a nucleocapsid with helical symmetry. 8 The genome is considered large, with 29 903 base pairs. 9 There are at least 50 different sites where translation can begin. 10 The genomic sequence mainly encodes proteins necessary for virus synthesis and structure. Its genome is covered by a nucleocapsid protein, which makes up the viral capsid, consisting of three structural proteins: membrane protein, an envelope protein, and a spike protein. 11 In addition to these structural proteins, the virus contains about 16 nonstructural proteins, which participate in different functions within the viral replication process. 12
Interindividual variability regarding the incidence, severity, and mortality rate of the COVID‐19 outbreak, was recently recorded. Human genetics, where different genetic polymorphisms of specific genes, might account for higher susceptibility and unexpected outcomes of COVID‐19 infections in different populations. It was found that both angiotensin converting enzyme 2 (ACE2) and transmembrane protease serine‐type 2 (TMPRSS2) are playing a crucial role for virus entry into host cells. 13
The role and effect of variants alleles of human leukocyte antigen (HLA) on the severity and incidence of infection with COVID‐19 and its management were investigated. HLA‐DQA1 may induce the production of anti‐drug antibodies against anti‐TNF drugs like infliximab, and adalimumab; which are used as options in the management of COVID‐19, and therefore might result in management failure. Also, HLA‐B*46:01 carrier individuals are more vulnerable to COVID‐19. 14
Several genetic mutations were discovered in several gene locations; c.222G>C (p.Leu74Leu) in the E structural protein and c.213C>T(p.Tyr71Tyr) in the M structural protein, D614 G, and other additional variants have been found which might decrease the main host antibody response. The probability of fast generation of treatment‐resistant strains, including antiviral resistance, is highly possible and consequently is an important raised issue concerning vaccine effectiveness and its benefits in immunization against SARS‐COV‐2. 15
The established nomenclature systems for naming and tracking SARS‐CoV‐2 genetic lineages are GISAID (which has allowed the comparison of viral genome variation in various regions of the world), Nextstrain, and Pango. These are currently and will remain in use by scientists and scientific research. 16 The main classifications to assess the clinical and virological relevance between the different variants, according to the WHO are five variants of concern (VOCs), namely Alpha (B.1.1.7 lineage); Beta (B.1.351 lineage); Gamma (P.1 lineage); Delta (B.1.617.2 lineage) and Delta (B.1.617.2 lineage); and, eight variants of interest (VOIs), namely Epsilon (B.1.427 and B.1.429); Zeta (P.2); Eta (B.1.525); Theta (P.3); Iota (B.1,526); Kappa (B.1.617.1); Lambda (C.37); and Mu (B.1.621). In terms of VOCs, the B.1.1.7 strain (Alpha variant) was initially a UK strain, associated with worrisome variants such as N501Y, P681H, and several other mutations. 17 Lineage B.1.351 (Beta variant) was first identified in South Africa. The P.1 lineage (Gamma variant) was identified for the first time in Brazil with the biologically significant variants E484K, N501Y, and K417T. Lineage B.1.617.2 (Delta variant) was first detected in India. The B.1.1.529 strain (Omicron variant) was detected in several countries with a high number of spike protein mutations. 18
According to the public database of the GISAID, three major SARS‐CoV‐2 lineages/clades could be initially identified and they were named G (variant of the spike protein S‐D614G), V (variant of the ORF3 according protein NS3‐G251), and S (variant ORF8‐L84S). 19 All viruses, including SARS‐CoV‐2, vary over time. Most of these changes have little, if any, impact on the properties of the virus. However, some changes can affect the properties of the virus, such as the ease with which it spreads, the severity of the associated disease, or the performance of vaccines, drugs, diagnostic tools, or even other social prevention and prevention measures of public health. 16 Therefore, it is extremely important to know the characteristics of the pathogen and its origins, as well as its classification into clades and lineages. This aspect is important, as it allows a better understanding of the speed of dissemination, geographic routes, as well as possible clinical aggressiveness of the strains.
The present study aims to review and summarizes the temporal spreading and evolution of SARS‐CoV‐2 clades and variants worldwide by Bayesian method evaluating 2897 whole genome sequences (WGS) of SARS‐CoV‐2 obtained between December 2019 and August 2022. The evaluation of these data is important for understanding the evolutionary histories of SARS‐CoV‐2 lineages, allowing us to identify the origins of each lineage, speed of dissemination, and geographic routes of this virus responsible for one of the biggest pandemics in history.
2. METHODS
2.1. Data collection and phylogenetic tree
SARS‐CoV‐2 WGS with available country and year of sampling were downloaded from GenBank and nextstrain 20 , 21 (Supporting Information material). All SARS‐CoV‐2 WGS were aligned by the MAFFT v7 and visually inspected with AliView v1.26. The best‐fitting nucleotide substitution (GTR) model was selected using a hierarchical likelihood ratio, Akaike information criterion, and Bayesian information criterion tests with Model Finder in IQ‐TRE web server (http://iqtree.cibiv.univie.ac.at/). Maximum likelihood phylogenetic tree was inferred according to the best‐fitting model using IQ‐TRE web server (http://iqtree.cibiv.univie.ac.at/). We used this tree to obtain root‐to‐tip regressions in TempEst v1.5.
2.2. Bayesian coalescent inference
Time‐scaled phylogenetic trees, evolutionary rates, and demographic histories of SARS‐CoV‐2 WGS were evaluated using the Bayesian coalescent framework implemented in BEAST v2.6.2, which uses a Markov Chain Monte Carlo (MCMC) sampling method to obtain posterior distributions of tree topologies and parameter estimates. Bifurcating nodes with posterior probability greater than 0.95 were considered statistically well supported. For each run of 500 million of MCMC, the marginal likelihood was estimated via path sampling (PS) and stepping stone (SS) methods and the resulting Bayes factors (BF) (ratio of marginal likelihoods) was used to select the best‐fitting clock/demographic model. The models can be compared to evaluate the strength of evidence against the null hypothesis (H0) defined in the following way: 2lnBF <2 indicates no evidence against H0; 2−6, weak evidence; 6−10, strong evidence; and >10 very strong evidence. Both SS and PS estimators indicated the uncorrelated lognormal relaxed molecular clock (BF = 25.1) as the best‐fitted model to the data set under analysis. Besides, we have used the GTR substitution model.
2.3. Viral dynamics
MCMC were run for 500 million generations to ensure stationary and adequate effective sample size for all statistical parameters. Tracer v.1.6 software was used to diagnose MCMC, adjust initial burn‐in. Bayesian coalescent analyses were performed to estimate the viral dynamics and the time to the most recent common ancestor (tMRCA). The time‐scale calibration was based on the isolation date of samples. Uncertainty in parameter estimates was evaluated in the 95% highest posterior density (HPD95%) interval. TreeAnnotator v1.8.2 was used to summarize the maximum clade credibility tree from the posterior distribution of trees.
2.4. Phylogeographic approach
Phylogeographic analysis, incorporating both spatial and temporal information, was performed with BEAST v2.6.2 using a discrete trait, symmetric substitution model with Bayesian stochastic search variable selection. The reversible discrete Bayesian phylogeographic model with a continuous‐time Markov chain rate reference prior was performed. The number of viral migrations between locations was estimated using “Markov Jump” counts of location‐state transitions along with the posterior tree distribution. Migratory events across time were summarized using the SPREAD v.1.0.7. BFs >10 were considered as well‐supported diffusion rates constituting the migration graph.
3. RESULTS
In the present study, 2897 WGS of SARS‐CoV‐2 collected between December 2019 and August 2022 were evaluated. Figure 1 shows the phylogenetic tree of SARS‐CoV‐2 WGS in the world, and additionally, Figure 2 demonstrates the global distribution of SARS‐CoV‐2. The SARS‐CoV‐2 tMRCA in Asia was 2019‐12‐26 (HPD95%: 2019‐12‐18; 2019‐12‐29), in Oceania 2020‐01‐24 (HPD95%: 2020‐01‐15; 2020‐01‐30), in Africa 2020‐02‐27 (HPD95%: 2020‐02‐21; 2020‐03‐04), in Europe 2020‐02‐27 (HPD95%: 2020‐02‐20; 2020‐03‐06), in North America 2020‐03‐12 (HPD95%: 2020‐03‐05; 2020‐03‐18), and in South America 2020‐03‐15 (HPD95%: 2020‐03‐09; 2020‐03‐28).
Figure 1.
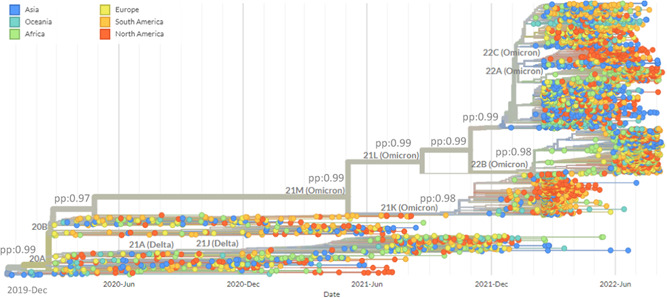
Time‐scaled maximum clade credibility tree from the evolutionary reconstruction by Bayesian analysis of SARS‐CoV‐2 complete genome sequences by countries obtained from GenBank and GISAID between December 2019 and August 2022. pp, posterior probability; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2.
Figure 2.
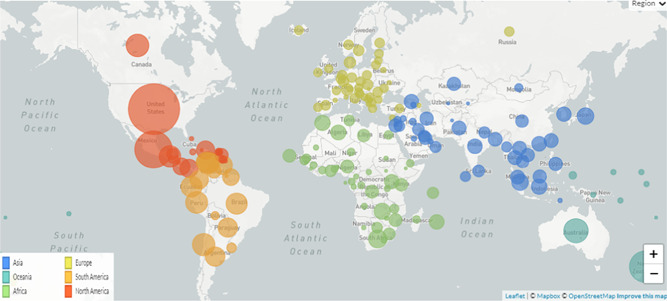
SARS‐CoV‐2 dissemination in worldwide detected between December 2019 and August 2022. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2.
The correlation between the nucleotide divergence and the years of sequence collection was positive (R 2 = 0.88; p < 0.01) (Figure 3). Figure 4 demonstrates the phylogenetic tree of SARS‐CoV‐2 clades and variants detected up to August 2022. While Figure 5 shows the global distribution of these clades and variants. Between December 2019 and June 2020, another 11 clades were detected (20I [Alpha] and 19A, 19B, 20B, 20C, 20A, 20D, 20E [EU1], 20F, 20H [Beta]). From July to December 2020, 4 clades were identified (20J [Gamma, V3], 21C [Epsilon], 21D [Eta], and 21G [Lambda]). Between January and June 2021, 3 clades of the Delta variant were detected (21A, 21I, and 21J). Between July and December 2021 two variants were detected, Delta (21A, 21I, and 21J) and Omicron (21K, 21L, 22B, and 22C). Between January and June 2022, the Delta (21I and 21J) and Omicron (21K, 21L, and 22A) variants were detected. Finally, between July and August 2022, 3 clades of Omicron were detected (22B, 22C, and 22D).
Figure 3.
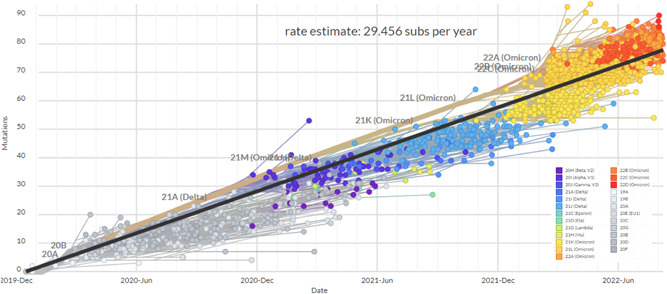
Root‐to‐tip regression of 2897 complete genomes of SARS‐CoV‐2 by countries between December 2019 and August 2022. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2.
Figure 4.
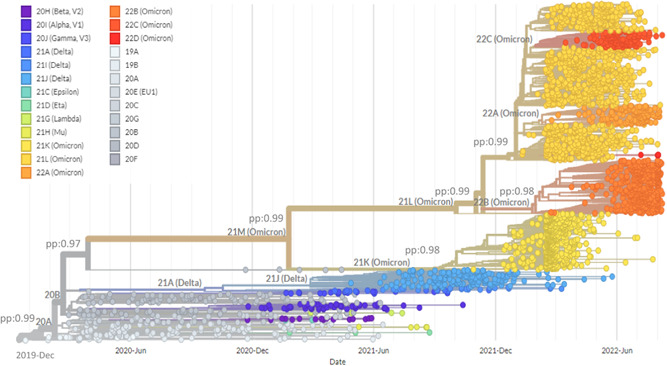
Time‐scaled maximum clade credibility tree from the evolutionary reconstruction by Bayesian analysis of SARS‐CoV‐2 complete genome sequences by clades obtained from GenBank and GISAID between December 2019 and August 2022. pp, posterior probability; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2.
Figure 5.
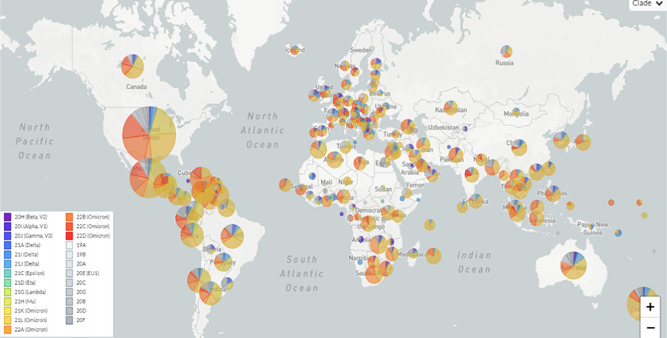
Frequency of SARS‐CoV‐2 clades in worldwide detected between December 2019 and August 2022. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2.
Clade 19A was first detected in the SARS‐CoV‐2 pandemic (Wuhan strain) and had a tMRCA of 2019‐12‐16 (HPD95%: 2019‐12‐15; 2019‐12‐25). The tMRCA of 20I (Alpha) 2020‐11‐24 (HPD95%: 2020‐11‐15; 2021‐12‐02); 20H (Beta) was 2020‐11‐25 (HPD95%: 2020‐11‐13; 2020‐11‐29); 20J (Gamma) was 2020‐12‐21 (HPD95%: 2020‐11‐05; 2021‐01‐15), 21A (Delta) tMRCA was 2020‐09‐20 (HPD95%: 2020‐05‐17; 2021‐02‐03); 21J (Delta) was 2021‐02‐26 (2020‐11‐02; 2021‐04‐24); 21M (Omicron) was 2021‐01‐25 (HPD95%: 2020‐09‐16; 2021‐08‐08); 21K (Omicron) was 2021‐07‐30 (HPD95%: 2021‐05‐30; 2021‐10‐19); 21L (Omicron) was 2021‐10‐03 (HPD95%: 2021‐04‐16; 2021‐12‐23); 22B (Omicron) was 2022‐01‐25 (HPD95%: 2022‐01‐10; 2022‐02‐05); 21L was 2021‐12‐20 (HPD95%: 2021‐05‐16; 2021‐12‐31) (Figure 4). Currently, the omicron variant predominates worldwide, with the 21L clade branching into three (22A, 22B, and 22C) (Figure 4).
Phylogeographic data demonstrate the origin of the main variants that circulated in the world (Figure 5). Alpha variant originated in the United Kingdom (BF = 125), Beta in South Africa (BF = 102), Gamma in Brazil (BF = 117), Delta in India (BF = 99), Omicron in South Africa (BF = 101), Mu in Colombia (BF = 31), Epsilon in the United States of America (BF = 28), and Lambda in Peru (BF = 39) (Figure 5). Table 1 shows the characteristics of the main SARS‐CoV‐2 strains (Alpha, Beta, Gamma, Delta, and Omicron). The transmissibility of the strains increased progressively over time, while the Gamma and Delta variants were the most clinically aggressive.
Table 1.
Characteristics of the main variants detected during the COVID‐19 pandemic
| Variants | Origin | tMRCA | Transmissibility | Clinical severity | Clinical response to vaccines |
|---|---|---|---|---|---|
| Alpha | UK | 2020‐11‐25 | Higher than previous variants | Possibly similar incidence of severe COVID‐19 and death | Vaccines prevent infection and adverse events |
| Beta | South Africa | 2020‐11‐24 | Higher than previous variants | Possibly similar incidence of severe COVID‐19 and death | Vaccines prevent infection and adverse events (possibly less effective in preventing infection) |
| Gamma | Brazil | 2020‐12‐21 | Higher than previous variants | Possibly similar incidence of severe COVID‐19 and death | Vaccines prevent infection and adverse events (possibly less effective in preventing infection) |
| Delta | India | 2020‐09‐20 | Much higher than previous variants | Increased incidence of severe COVID‐19 and death | Vaccines prevent infection and adverse events (slightly lower efficacy in preventing infection) |
| Omicron | South Africa | 2021‐01‐25 | Extremely higher than previous variants | Lower incidence of severe COVID‐19 and death | Preliminary data suggest that vaccines prevent infection and adverse events (possible lower efficacy vs. other VOCs) |
Abbreviations: tMRCA, time to the most recent common ancestorp; UK, United Kingdom; VOCs, variants of concern.
4. DISCUSSION
The phylogenetic characterization of an emerging viral infection can help in understanding and monitoring the pandemic progression. Currently, there is a very large amount of WGS data to study the recent SARS‐CoV‐2 spreading. More evolutionary and dissemination studies are necessary to understand the SARS‐CoV‐2 genetic diversity and to identify the main epidemic findings. These data are essential to define public health measures to control the current pandemic.
The present study reviewed the phylodynamic data of SARS‐CoV‐2 lineages in the world between December 2019 and August 2022. According to our findings, SARS‐CoV‐2 presented different dates of origin on the continents: Asia (2019‐12‐26), Oceania (2020‐01‐24), Africa (2020‐02‐27), Europe (2020‐02‐27), North America (2020‐03‐12), and South America (2020‐03‐15). The rapid spread of SARS‐CoV‐2 worldwide, from China, was due to the intense air traffic present in the 21st century, which spread the virus widely in the world. This scenario was particular since we do not experience a pandemic in modern times, the last one being caused by the Spanish flu at the beginning of the 20th century, a historical moment when the world population was smaller and the means of transport were completely different from those present today.
The first lineage of SARS‐CoV‐2 (clade 19A) was detected in Wuhan China dated 2019‐12‐16. The Alpha, Beta, Gamma, Delta, and Omicron strains were initially spread during the period of the pandemic. Alpha on 2020‐11‐24, Beta on 2020‐11‐25, Gamma on 2020‐12‐21, Delta on 2020‐09‐20 and 2021‐02‐26, Omicron on 2021‐01‐25 and 2022‐01‐25. The Alpha lineage originated in the United Kingdom, Beta in South Africa, Gamma in Brazil, Delta in India, Omicron in South Africa, Mu in Colombia, Epsilon in the United States of America, and Lambda in Peru.
In November 2020, Alpha lineage was identified in the United Kingdom based on a sample obtained in September 2020. This lineage stood out from those previously observed due to the more significant increase in cases and the high number of observed mutations of potential epidemiological impact. 22 On 2020‐12‐18, Alpha lineage was designated as a VOC. 16 This lineage has at least 22 mutations, including 13 non‐synonymous, 6 synonymous, and 3 deletions. 23 These mutations include the N501Y of the spike protein, which increases the affinity of RBD with the host cell's receptor (ACE2), the P681H, which would facilitate the entry of the virus into the cell, 22 and the D614G in the spike protein which increases infectivity. 24 The D614G mutation was initially detected in early 2020 and has also been identified in all the VOCs that have appeared after lineage Alpha. 25 Concerning transmissibility, this lineage has a higher transmission capacity than previous lineages of SARS‐COV‐2, between 50% and 100% higher. 26 , 27 Regarding the severity of the disease, the evidence is not conclusive. Although initial studies have associated this lineage with a higher rate of severe COVID‐19 and death, 28 , 29 other studies have shown no differences in adverse outcomes. 30 Regarding the efficacy of vaccines, the available evidence indicates that there would be no decrease in efficacy to prevent infection and adverse outcomes. 31 , 32
In December 2020, Beta lineage was detected in South Africa, identified for the first time in September 2020 in the same country, which presented a rapid expansion in the region as compared to previous lineages, 33 , 34 receiving the VOC designation on 2020‐12‐18. 16 This lineage has at least 18 important mutations, including some previously described in the Alpha, such as N501Y in protein S. 35 Among the mutations of this lineage, K417N and E484K stand out, present in the RBM region of the RBD domain of the spike protein, which is associated with increased infectivity. 36 This variant presents higher transmissibility compared to other previous variants. 34 , 36 Regarding the severity of the disease, as with variant Alpha, the evidence is not conclusive. Previous studies have shown that it would be associated with a more significant number of hospitalizations. However, an increase in mortality has not been observed. 37 , 38 Additionally, regarding the efficacy of vaccines, the available data indicate that they prevent infection and adverse outcomes, 31 , 32 but some reports indicate less efficacy to prevent infection as compared to previous lineages. 39
At the beginning of January 2021, Gamma lineage was detected for the first time in Tokyo, Japan, in four people from the Brazilian Amazon in December 2020. Its extension was subsequently demonstrated in Brazil. 40 , 41 On 2021‐01‐11, it was designated as VOC by the WHO. 16 This lineage has at least 23 important mutations, including some reported in other variants such as N501Y and E484K in protein S, which increase the infective capacity of the virus on the host cell. 36 , 42 Regarding transmissibility, data reported from Brazil indicate higher transmissibility as compared to other previous variants. 40 , 43 However, it has not been demonstrated increase in the severity of the disease. Although an increase in deaths was initially reported in Brazil, 44 epidemiological data from Europe do not show an increase in deaths but an increase in hospitalizations. 37 Therefore, it has been proposed that the higher mortality in Brazil was more associated with the region's socioeconomic and public health limitations. 45 , 46 Finally, on the efficacy of the vaccines, there is less information as compared to the Alpha and Beta lineages. However, the available data indicate that the vaccines prevent infection and adverse outcomes, 31 , 32 similar to variant Beta. 33
On 2021‐03‐24, the Indian Ministry of Health reported Delta lineage, with mutations associated with potential immune escape. 47 On 2021‐04‐04, the WHO assigned the VOI classification, and on 2021‐05‐11, its classification was changed to VOC. 16 This variant presents at least 21 important mutations, such as D614G, L452R (which gives higher stability to the RBD‐ACE2 complex, increasing infectivity), and P681R (which optimizes spike protein cleavage, with the potential to increase transmissibility). 48 , 49 All these mutations provide a higher infective capacity of the virus than the previously reported variants. The transmissibility of Delta is higher than reported in previous variants, 33 , 50 with high transmissibility indoors. 51 Therefore, as of 2021‐12‐31, it is the dominant lineage of SARS‐CoV‐2 worldwide. 52 Furthermore, regarding severity, it has been observed that patients infected with this lineage have a higher rate of adverse outcomes, including hospitalizations, ICU requirements, and deaths, compared to those infected with variant Alpha. 53 , 54 Finally, vaccines have a slight decrease in their efficacy to prevent infection but similar efficacy to prevent severe disease when compared with other variants, including previous VOCs. 32 , 33
On 2021‐11‐11, Omicron lineage was reported in South Africa which was detected on a traveler from Botswana. Later, this variant began to be detected in patients from South Africa. 55 The appearance of this lineage was associated with an abrupt increase in COVID‐19 cases in the country, from an average of 280 daily cases to 800 daily cases in the following weeks. Especially relevant was the situation in the Gauteng province of South Africa, where the doubling time of cases decreased as compared to previous COVID‐19 waves in the area. 55 , 56 On 26‐11‐2021, the WHO classified this lineage as VOC. 16 This lineage presents a higher number of mutations as compared to other VOCs, having identified at least 48 important mutations by 2021‐12‐31, including several mutations previously not described in other variants. 57 , 58 Among the most relevant mutations are RBD mutations, which would confer increased binding to ACE2 and infectivity of the host cell. 59 , 60 Previous studies suggest that lineage Omicron could infect between three and six times more people than lineage Delta, 56 , 60 making it the lineage of SARS‐CoV‐2 with the highest transmissibility identified up to date. Given this, Omicron become the dominant variant worldwide in short term, surpassing lineage Delta. Regarding severity, preliminary data suggest that it would be lower than previous lineages. 60 , 61 Data from the UK suggest that hospitalization rates would be 50%–70% lower than for variant Delta. 61 This lineage showed higher transmissibility and the number of hospitalizations was relatively high. Also, preliminary data suggest that vaccines would continue to be effective in preventing infection and adverse outcomes. 62 Reports from Pfizer‐BioNTech 63 and AstraZeneca 64 indicate that a vaccine booster produces neutralizing antibodies to the Omicron. However, recent data show that the reinfection rate of lineage Omicron in COVID‐19 survivors would be higher than lineage Beta or Delta. 65 This suggests that the immune response after infection or vaccination could be lower than in other lineages. It should be noticed that for 2021‐12‐3, the two most prevalent SARS‐CoV‐2 lineages are Delta and Omicron, while variants Alpha, Beta, and Gamma have significantly reduced, being practically absent in current COVID‐19 patients evaluated. 66 In 2022, the Omicron variant prevailed in the majority of SARS‐CoV‐2 infections. 67 , 68
Competition among viral strains of changing virulence is being evidenced during the current SARS‐CoV‐2 dissemination and COVID‐19 pandemic. The continuous monitoring of the most frequent SARSCoV‐2 clades, lineages, and strains as well as their specific dynamic evolution processes are now imperative for epidemiologists to define public health measures, such as limiting or relaxing the social movement. Furthermore, this information will be necessary to develop more appropriate diagnostic tests (as the molecular biology methods) and vaccines for the circulating SARS‐CoV‐2 strains. Deeply surveillance of viral transmission at local and global scales and the evaluation of the effect of the different control measures on COVID‐19 transmission will offer assistance to decide an ideal mitigation procedure to minimize infections and decrease public healthcare demand. Therefore, continued monitoring of the SARS‐CoV‐2 genetic and antigenic diversity is already essential for public human health in the World.
Finally, SARS‐CoV‐2 lineages with different virulence and pathogenicity features are continuously emerging in the COVID‐19 pandemic. The continuous monitoring of the most frequent viral clades, lineages, VOIs, and VOCs as well as the study of the specific dynamic evolution processes are really necessary to define public health measures. All this information will be also necessary to develop more appropriate diagnostic tests (as the molecular biology methods) and vaccines for the circulating SARS‐CoV‐2 lineages. Deeply surveillance of viral transmission at local and global scales and the evaluation of the effect of the different control measures on COVID‐19 transmission will offer assistance to decide an ideal mitigation procedure to minimize infections and decrease public healthcare demand.
This review presents the different genetic variants of COVID‐19 from the start to date which is an important collective review for a pandemic worldwide. These data are useful to understand the transmission routes, origins, and evolutionary rates of this virus. The review is very beneficial for the literature to get the whole history of mutation of viral infection which gives an overview of the whole story and readers could expect what will happen in the future concerning this pandemic.
AUTHOR CONTRIBUTIONS
Jonas M. Wolf and Lucas M. Wolf designed the study. Jonas M. Wolf performed the bioinformatic analyses. Jonas M. Wolf, Lucas M. Wolf, Graziele L. Bello, Juçara G. Maccari, and Luiz A. Nasi wrote the first draft of the manuscript and contributed to the literature review and discussion of the results. All authors contributed to and have approved the final manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supplementary information.
Wolf JM, Wolf LM, Bello GL, Maccari JG, Nasi LA. Molecular evolution of SARS‐CoV‐2 from December 2019 to August 2022. J Med Virol. 2022;95:e28366. 10.1002/jmv.28366
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.John Hopkins University of Medicine. Coronavirus Resource Center. 2022. Accessed September 23, 2022. https://coronavirus.jhu.edu/map.html
- 2. WHO (World Health Organization) . Coronavirus (COVID‐19) dashboard. 2022. Accessed September 24, 2022.
- 3. WHO (World Health Organization) . Weekly epidemiological update on COVID. 2022. Accessed September 23, 2022. https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---21-september-2022
- 4. Sharma A, Ahmad Farouk I, Lal SK. COVID‐19: a review on the novel coronavirus disease evolution, transmission, detection, control and prevention. Viruses. 2021;13(2):202. 10.3390/v13020202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. 2015;1282:1‐23. 10.1007/978-1-4939-2438-7_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang MY, Zhao R, Gao LJ, Gao XF, Wang DP, Cao JM. SARS‐CoV‐2: structure, biology, and structure‐based therapeutics development. Front Cell Infect Microbiol. 2020;10:587269. 10.3389/fcimb.2020.587269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fox D. What you need to know about the novel coronavirus. Nature. Published online January 24, 2020. 10.1038/d41586-020-00209-y [DOI] [PubMed] [Google Scholar]
- 8. Parra‐Lucares A, Segura P, Rojas V, Pumarino C, Saint‐Pierre G, Toro L. Emergence of SARS‐CoV‐2 variants in the world: how could this happen? Life. 2022;12(2):194. 10.3390/life12020194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raskin S. Genetics of COVID‐19. J Pediatr. 2021;97(4):378‐386. 10.1016/j.jped.2020.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ye Q, West AMV, Silletti S, Corbett KD. Architecture and self‐assembly of the SARS‐CoV‐2 nucleocapsid protein. Protein Sci. 2020;29(9):1890‐1901. 10.1101/2020.05.17.100685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mandala VS, McKay MJ, Shcherbakov AA, Dregni AJ, Kolocouris A, Hong M. Structure and drug binding of the SARS‐CoV‐2 envelope protein transmembrane domain in lipid bilayers. Nat Struct Mol Biol. 2020;27(12):1202‐1208. 10.21203/rs.3.rs-77124/v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zheng Y, Zhuang MW, Han L, et al. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) membrane (M) protein inhibits type I and III interferon production by targeting RIG‐I/MDA‐5 signaling. Signal Transduct Target Ther. 2020;5(1):299. 10.1038/s41392-020-00438-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alshahawey M, Raslan M, Sabri N. Sex‐mediated effects of ACE2 and TMPRSS2 on the incidence and severity of COVID‐19. The need for genetic implementation. Curr Res Translational Med. 2020;68(4):149‐150. 10.1016/j.retram.2020.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raslan MA, Alshahawey M, Shehata EM, Sabri NA. Does human leukocyte antigen gene polymorphism affect management of COVID‐19 patients? A review article. Scientific J Genet Gene Ther. 2020;6(1):001‐003. 10.17352/sjggt.000018 [DOI] [Google Scholar]
- 15. Nagwa Ali S, Mohamed Ahmed R, Eslam Mansour S, Sara Ahmed R. Genetic variants of COVID‐19 and vaccination. Is there a correlation? Open J Proteomics Genomics. 2022;7(1):001‐005. 10.17352/ojpg [DOI] [Google Scholar]
- 16. WHO (World Health Organization) . Tracking SARS‐CoV‐2 variants. 2022. Accessed September 29, 2022. https://www.who.int/activities/tracking-SARS-CoV-2-variants
- 17. Aleem A, Akbar SAB, Slenker AK. Emerging variants of SARS‐CoV‐2 and novel therapeutics against coronavirus (COVID‐19). StatPearls [Internet]. StatPearls Publishing; 2022.
- 18. O'Toole Á, Scher E, Underwood A, et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021;7(2):veab064. 10.1093/ve/veab064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Forster P, Forster L, Renfrew C, Forster M. Phylogenetic network analysis of SARS‐CoV‐2 genomes. Proc Natl Acad Sci. 2020;117(17):9241‐9243. 10.1073/pnas.2004999117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hadfield J, Megill C, Bell SM, et al. Nextstrain: real‐time tracking of pathogen evolution. Bioinformatics. 2018;34(23):4121‐4123. 10.1093/bioinformatics/bty407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sagulenko P, Puller V, Neher RA. TreeTime: maximum‐likelihood phylodynamic analysis. Virus Evol. 2018;4(1):vex042. 10.1093/ve/vex042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Walker AS, Vihta KD, Gethings O, et al. Tracking the emergence of SARS‐CoV‐2 Alpha variant in the United Kingdom. N Engl J Med. 2021;385(27):2582‐2585. 10.1056/NEJMc2103227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Latif AA, Mullen JL, Alkuzweny M, et al. Hughes, and the center for viral systems biology. B.1.1.7 lineage report. Outbreak.Info. 2022. Accessed September 28, 2022. https://outbreak.info/situationreports?>pango=B.1.1.7
- 24. Korber B, Fischer WM, Gnanakaran S, et al. Tracking changes in SARS‐CoV‐2 spike: evidence that D614G increases infectivity of the COVID‐19 virus. Cell. 2020;182(4):812‐827. 10.1016/j.cell.2020.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chakraborty C, Saha A, Sharma AR, Bhattacharya M, Lee SS, Agoramoorthy G. D614G mutation eventuates in all VOI and VOC in SARS‐CoV‐2: is it part of the positive selection pioneered by Darwin? Mol Therapy—Nuc Acids. 2021;26:237‐241. 10.1016/j.omtn.2021.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davies NG, Abbott S, Barnard RC, et al. Estimated transmissibility and impact of SARS‐CoV‐2 lineage B.1.1.7 in England. Science. 2021;372(6538):eabg3055. 10.1126/science.abg3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Volz E, Mishra S, Chand M, et al. Assessing transmissibility of SARS‐CoV‐2 lineage B.1.1.7 in England. Nature. 2021;593(7858):266‐269. 10.1038/s41586-021-03470-x [DOI] [PubMed] [Google Scholar]
- 28. Davies NG, Jarvis CI, CMMID COVID‐19 Working Group , et al. Increased mortality in community‐tested cases of SARS‐CoV‐2 lineage B.1.1.7. Nature. 2021;593(7858):270‐274. 10.1038/s41586-021-03426-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Challen R, Brooks‐Pollock E, Read JM, Dyson L, Tsaneva‐Atanasova K, Danon L. Risk of mortality in patients infected with SARS‐CoV‐2 variant of concern 202012/1: matched cohort study. BMJ. 2021;372:n579. 10.1136/bmj.n579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Graham MS, Sudre CH, May A, et al. Changes in symptomatology, reinfection, and transmissibility associated with the SARS‐CoV‐2 variant B.1.1.7: an ecological study. Lancet Pub Health. 2021;6(5):e335‐e345. 10.1016/S2468-2667(21)00055-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Charmet T, Schaeffer L, Grant R, et al. Impact of original, B.1.1.7, and B.1.351/P.1 SARS‐CoV‐2 lineages on vaccine effectiveness of two doses of COVID‐19 mRNA vaccines: results from a nationwide case‐control study in France. Lancet Reg Health—Europe. 2021;8:100171. 10.1016/j.lanepe.2021.100171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cevik M, Grubaugh ND, Iwasaki A, Openshaw P. COVID‐19 vaccines: keeping pace with SARS‐CoV‐2 variants. Cell. 2021;184(20):5077‐5081. 10.1016/j.cell.2021.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Choi JY, Smith DM. SARS‐CoV‐2 variants of concern. Yonsei Med J. 2021;62(11):961‐968. 10.3349/ymj.2021.62.11.961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tegally H, Wilkinson E, Giovanetti M, et al. Detection of a SARS‐CoV‐2 variant of concern in South Africa. Nature. 2021;592(7854):438‐443. 10.1038/s41586-021-03402-9 [DOI] [PubMed] [Google Scholar]
- 35. Latif AA, Mullen JL, Alkuzweny M, et al. Hughes, and the center for viral systems biology. B.1.351 lineage report. Outbreak.Info . 2022. Accessed on 28 September, 2022. https://outbreak.info/situationreports
- 36. Khan A, Zia T, Suleman M, et al. Higher infectivity of the SARS‐CoV‐2 new variants is associated with K417N/T, E484K, and N501Y mutants: an insight from structural data. J Cell Physiol. 2021;236(10):7045‐7057. 10.1002/jcp.30367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Funk T, Pharris A, Spiteri G, et al. Characteristics of SARS‐CoV‐2 variants of concern B.1.1.7, B.1.351 or P.1: data from seven EU/EEA countries, weeks 38/2020 to 10/2021. Euro Surveill. 2021;26(16):2100348. 10.2807/1560-7917.ES.2021.26.16.2100348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nyberg T, Twohig KA, Harris RJ, et al. Risk of hospital admission for patients with SARS‐CoV‐2 variant B.1.1.7: cohort analysis. BMJ. 2021;373:n1412. 10.1136/bmj.n1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mahase E. Covid‐19: novavax vaccine efficacy is 86% against UK variant and 60% against South African variant. BMJ. 2021;372:n296. 10.1136/bmj.n296 [DOI] [PubMed] [Google Scholar]
- 40. Naveca FG, Nascimento V, de Souza VC, et al. COVID‐19 in Amazonas, Brazil, was driven by the persistence of endemic lineages and P.1 emergence. Nature Med. 2021;27(7):1230‐1238. 10.1038/s41591-021-01378-7 [DOI] [PubMed] [Google Scholar]
- 41. Wolf JM, Kipper D, Borges GR, Streck AF, Lunge VR. Temporal spread and evolution of SARS‐CoV‐2 in the second pandemic wave in Brazil. J Med Virol. 2022;94(3):926‐936. 10.1002/jmv.27371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Latif AA, Mullen JL, Alkuzweny M, et al. Hughes, and the center for viral systems biology. P.1 lineage report Outbreak.Info . 2022. Accessed 28, September, 2022. https://outbreak.info/situation-reports
- 43. Sabino EC, Buss LF, Carvalho MPS, et al. Resurgence of COVID‐19 in Manaus, Brazil, despite high seroprevalence. Lancet. 2021;397(10273):452‐455. 10.1016/S0140-6736(21)00183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Freitas ARR, Beckedorff OA, Cavalcanti LPG, et al. The emergence of novel SARS‐CoV‐2 variant P.1 in Amazonas (Brazil) was temporally associated with a change in the age and sex profile of COVID‐19 mortality: a population based ecological study. Lancet Regional Health—Americas. 2021;1:100021. 10.1016/j.lana.2021.100021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taylor L. Covid‐19: Brazil's spiralling crisis is increasingly affecting young people. BMJ. 2021;373:n879. 10.1136/bmj.n879 [DOI] [PubMed] [Google Scholar]
- 46.The Catastrophic Brazilian response to COVID‐19 may amount to a crime against humanity. 2022. Accessed September 28, 2022. https://blogs.bmj.com/bmj/2021/04/05/the-catastrophic-brazilian-response-to-COVID-19-may-amount-to-a-crime-against-humanity/
- 47.Genome sequencing by INSACOG shows variants of concern and a novel variant in India. 2022. Accessed September 28, 2022. https://pib.gov.in/PressReleaseIframePage.aspx?PRID=1707177
- 48. Alaa Abdel Latif JL, Mullen M, Alkuzweny G, et al. Hughes, and the center for viral systems biology. B.1.617.2 lineage report. Outbreak.Info. 2022. Accessed September 28, 2022. https://outbreak.info/situationreports
- 49. Teng S, Sobitan A, Rhoades R, Liu D, Tang Q. Systemic effects of missense mutations on SARS‐CoV‐2 spike glycoprotein stability and receptor‐binding affinity. Brief. Bioinform. 2021;22(2):1239‐1253. 10.1093/bib/bbaa233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen J, Wang R, Wang M, Wei GW. Mutations strengthened SARS‐CoV‐2 infectivity. J Mol Biol. 2020;432(19):5212‐5226. 10.1016/j.jmb.2020.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dougherty K, Mannell M, Naqvi O, Matson D, Stone J. SARS‐CoV‐2 B.1.617.2 (Delta) variant COVID‐19 outbreak associated with a gymnastics facility—Oklahoma, April‐May 2021. MMWR Morb Mortal Wkly Rep. 2021;70(28):1004‐1007. 10.15585/mmwr.mm7028e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Australian Government Department of Health . 2022. Accessed September 28, 2022. https://www.health.gov.au/health-alerts/covid-19/symptomsand-variants/delta
- 53. Ong SWX. Clinical and virological features of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) variants of concern: a retrospective cohort study comparing B.1.1.7 (Alpha), B.1.351 (Beta), and B.1.617.2 (Delta). Clin Infect Dis. 2022;75(1):e1128‐e1136. 10.1093/cid/ciab721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Twohig KA, Nyberg T, Zaidi A, et al. Hospital admission and emergency care attendance risk for SARS‐CoV‐2 delta (B.1.617.2) compared with alpha (B.1.1.7) variants of concern: a cohort study. Lancet Infect Dis. 2022;22(1):35‐42. 10.1016/S1473-3099(21)00475-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fisman DN, Tuite AR. Evaluation of the relative virulence of novel SARS‐CoV‐2 variants: a retrospective cohort study in Ontario, Canada. Can Med Assoc J. 2021;193(42):E1619‐E1625. 10.1503/cmaj.211248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Department of Health . Government of South Africa. COVID‐19. 2022. Accessed September 28, 2022. https://sacoronavirus.co.za/
- 57. Alaa Abdel Latif JL, Mullen M, Alkuzweny G, et al. Hughes, and the center for viral systems biology. BA.1 lineage report. Outbreak.Info . 2022. Accessed September 28, 2022. https://outbreak.info/situation-reports?pango=BA.1
- 58. Torjesen I. Covid‐19: Omicron may be more transmissible than other variants and partly resistant to existing vaccines, scientists fear. BMJ. 2021;375:n2943. 10.1136/bmj.n2943 [DOI] [PubMed] [Google Scholar]
- 59. Meng B, Abdullahi A, Ferreira IATM, et al. Altered TMPRSS2 usage by SARS‐CoV‐2 Omicron impacts infectivity and fusogenicity. Nature. 2022;603(7902):706‐714. 10.1038/s41586-022-04474-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Callaway E, Ledford H. How bad is omicron? What scientists know so far. Nature. 2021;600(7888):197‐199. 10.1038/d41586-021-03614-z [DOI] [PubMed] [Google Scholar]
- 61. CDC . Omicron variant: what you need to know. 2020. Accessed September 28, 2022. https://www.cdc.gov/coronavirus/2019-ncov/variants/ omicron‐variant.html
- 62. Burki TK. Omicron variant and booster COVID‐19 vaccines. Lancet Resp Med. 2022;10(2):e17. 10.1016/S2213-2600(21)00559-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Maas A. Pfizer and BioNTech provide update on omicron variant . 2022. Accessed September 28, 2022. https://www.pfizer.com/news/press
- 64. Gruvris C, Sparks P, Sanford M. Vaxzevria significantly boosted antibody levels against Omicron . 2022. Accessed September 28, 2022. https://www.astrazeneca.com/content
- 65. Pulliam JRC, van Schalkwyk C, Govender N, et al. Increased risk of SARS‐CoV‐2 reinfection associated with emergence of Omicron in South Africa. Science. 2022;376(6593):eabn4947. 10.1126/science.abn4947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pan American Health Organization . COVID‐19 genomic surveillance regional network. 2022. Accessed September 28, 2022. https://www.paho.org/en/topics/influenza-and-other-respiratory-viruses/covid-19-genomic-surveillance-regional-network
- 67. K A, Sharma A, Kumar D, et al. Molecular aspects of Omicron, vaccine development, and recombinant strain XE: a review. J Med Virol. 2022;94(10):4628‐4643. 10.1002/jmv.27936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Le TTB, Vasanthakumaran T, Thi Hien HN, et al. SARS‐CoV‐2 Omicron and its current known unknowns: A narrative review. Rev Med Virol. 2022;23:e2398. 10.1002/rmv.2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.