

Key Points
Question
Are lipid-lowering drugs associated with reduced risk of psoriasis?
Findings
In this mendelian randomization study of 12 116 patients with psoriasis and 278 303 controls across 2 European populations, genetically proxied inhibition of proprotein convertase subtilisin/kexin type 9 (PCSK9) was consistently associated with lower risk of psoriasis.
Meaning
The study results suggest that PCSK9 is implicated in psoriasis pathogenesis, and existing PCSK9 inhibitors that are licensed to treat hypercholesterolemia may be used for preventing psoriasis.
Abstract
Importance
Lipid pathways have been implicated in the pathogenesis of psoriasis, and some lipid-lowering drugs, such as statins, are hypothesized to have disease-modifying properties. However, large population-level studies are scarce, and causal interpretation of results from traditional observational designs is limited by confounding.
Objective
To investigate the causal association between genetically proxied lipid-lowering drugs and psoriasis risk.
Design, Setting, and Participants
This 2-sample mendelian randomization study was performed from August to October 2022 and included population-level genome-wide association studies of psoriasis in the UK Biobank and FinnGen studies and low-density lipoprotein (LDL) by the Global Lipids Genetics Consortium. The inverse variance–weighted method was used with pleiotropy robust methods and colocalization as sensitivity analyses.
Exposures
Genetically proxied inhibition of 3-hydroxy-3-methylglutaryl CoA reductase (HMGCR, targeted by statins), Niemann-Pick C1–like 1 (NPC1L1, targeted by ezetimibe), and proprotein convertase subtilisin/kexin type 9 (PCSK9, targeted by, eg, alirocumab), using LDL as the biomarker.
Main Outcomes and Measures
Risk of psoriasis.
Results
Data from 12 116 patients with psoriasis and approximately 1.3 million individuals with LDL measurement were analyzed. Genetically proxied PCSK9 inhibition was associated with reduced risk of psoriasis (odds ratio, 0.69 per standard deviation reduction in LDL; 95% CI, 0.55-0.88; P = .003), which was replicated in FinnGen (odds ratio, 0.71; 95% CI, 0.57-0.88; P = .002). Sensitivity analyses did not provide statistical evidence of bias from pleiotropy or genetic confounding. No robust association was found for HMGCR or NPC1L1 inhibition.
Conclusions and Relevance
This mendelian randomization study suggests that PCSK9 is implicated in psoriasis pathogenesis, and its inhibition is associated with reduced psoriasis risk. These findings potentially pave the way for future studies that may allow personalized selection of lipid-lowering drugs for those at risk of psoriasis.
This mendelian randomization examines the association between genetically proxied lipid-lowering drugs and psoriasis risk.
Introduction
Disordered lipid metabolism is common among people with psoriasis and contributes to the increased risk of cardiovascular morbidity and mortality.1 Screening for dyslipidemia is recommended for those with moderate to severe psoriasis,2,3 and lipid-lowering drugs play an important role in cardiovascular risk management. Some lipid-lowering drugs, such as statins4 and proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors,5,6 have also been hypothesized to have disease-modifying properties for psoriasis.
Investigating the disease-modifying potential of manipulating lipid pathways in psoriasis is appealing for several reasons. First, the pathogenesis of psoriasis is incompletely understood; therefore, discovery of causal pathways that implicate lipids may improve our understanding of mechanisms that lead to psoriasis development. Second, selecting lipid-lowering drugs with dual action on lipids and psoriasis promotes personalized treatment (eg, for individuals with dyslipidemia and severe or refractory psoriasis who require adjunctive therapy). Such agents may also be preferred for preventing psoriasis in high-risk individuals (eg, those with strong family history) with dyslipidemia. Third, evidence for disease-modifying properties of lipid-lowering drugs may also encourage repurposing for psoriasis treatment (eg, development of topical PCSK9 inhibitors5). Targeting lipid pathways may offer lower theoretical risk of adverse events associated with direct immunosuppression. However, generating supportive evidence can be challenging. Traditional pharmacoepidemiologic designs are susceptible to confounding (ie, unmeasured characteristics that affect prescription of lipid-lowering drugs and psoriasis risk) and reverse causation (psoriasis causing dyslipidemia). Studying relatively uncommon outcomes (psoriasis incidence) may be practically challenging, particularly with rare exposures (eg, prescription of PCSK9 inhibitors).
Natural variation in the gene that encodes a protein drug target can offer insight into potential clinical effects.7 Because genetic variants are randomly allocated at conception, genetic instrumental variable analyses, or mendelian randomization (MR), can be conceptualized as a quasirandomized natural experiment that is more robust against biases in traditional epidemiologic designs.8 Our aim was to use 2-sample MR to investigate the association between the risk of developing plaque psoriasis and 3 genetically proxied lipid-lowering drugs: 3-hydroxy-3-methylglutaryl–CoA reductase (HMGCR) inhibitors (ie, statins), Niemann-Pick C1–like 1 (NPC1L1) inhibitors (ie, ezetimibe), and PCSK9 inhibitors (eg, alirocumab).
Methods
This study used publicly available, deidentified summary data from prior genome-wide association studies (GWAS); ethical approval was obtained in all original studies with relevant citations detailed. The UK Biobank study received ethical approval from the North West Multicenter Research Ethics Committee, and participants provided informed consent.
Genetic Association for Psoriasis
We obtained genetic associations for psoriasis by performing a GWAS of 6495 patients with psoriasis and 25 980 randomly selected controls without psoriasis from the UK Biobank study (1:4 ratio to reduce case-control imbalance that can increase type I error9,10). Details of genotyping and quality control methods for the UK Biobank study were described previously.11 Full details of the current GWAS are provided in the eMethods in Supplement 1. We restricted the data to unrelated (kinship coefficient <0.0884) participants of self-reported White British ancestry, with concordant submitted and genetically inferred sex. Psoriasis was defined using any of the following: International Statistical Classification of Diseases and Related Health Problems, Tenth Revision (ICD-10) code L40 (from fields 41270, 40001, or 40002); self-report (code 1453 in field 20002); and Read code in linked primary care data (code M160, M161, or M16..) that were available for a subset of approximately 225 000 participants covering a period of 29 years. To minimize misclassification, we removed cases that had ICD-10 codes for atopic eczema (L20). The genetic correlation coefficient with a previous psoriasis GWAS12 that used physician-diagnosed cases was high (rg = 0.95; P = 9.4 × 10−11) using linkage disequilibrium score regression; the correlation coefficient would be low if a significant proportion of the UK Biobank psoriasis cases were misclassified (eMethods in Supplement 1).
To replicate the analyses, we obtained genetic associations for psoriasis from the FinnGen study (release 6), which comprised 5621 patients with psoriasis and 252 323 controls. Cases were defined using ICD-10 code L40. The genetic correlation with the GWAS using physician-diagnosed cases was rg = 0.89 (P = 1.7 × 10−25). We used genetic association data from the UK Biobank for the primary analysis due to greater confidence regarding case definition (compared with FinnGen, which used an automated pipeline) and the greater number of available variants (compared with the 2012 GWAS by Tsoi and colleagues12). Consistency in estimates using each outcome data sets was tested using heterogeneity indices as in meta-analysis, ie, I2 based on the Q statistic and P value for the χ2 test.13
Genetic Proxies for Lipid-Lowering Drugs
We chose low-density lipoprotein (LDL) as the biomarker because each of the 3 lipid-lowering drugs were shown to reduce levels of LDL cholesterol. Genetic associations with LDL were taken from what is to our knowledge the largest GWAS meta-analysis to date by the Global Lipids Genetics Consortium, comprising approximately 1.3 million individuals of European ancestry.14 Data excluding UK Biobank (n = 842 660) or Finnish (n = 1 177 987) participants were used for analyses using outcomes from UK Biobank and FinnGen to reduce potential bias from sample overlap.
We selected variants associated with LDL at genome-wide significance (P < 5 × 10−8) that were minimally correlated (linkage disequilibrium threshold of r2 < 0.1 using PLINK and phase 3 version 5 of the 1000 genomes project as a reference panel) within ±100 kb of the HMGCR gene (build GRCh37/hg19: chromosome 5: 74632154-74657929) to instrument statins, NPC1L1 gene (chromosome 7: 44552134-44580914) to instrument ezetimibe, and PCSK9 gene (chromosome 1: 55505221-55530525) for PCSK9 inhibitors, such as alirocumab or evolocumab.
Given the observational association between lipids and psoriasis, we investigated whether genetically predicted LDL overall (rather than specifically through lipid-lowering drug targets) was associated with psoriasis risk. We instrumented LDL using genome-wide significant variants throughout the genome that had pairwise correlations of r2 < 0.001, excluding variants within the 3 drug target gene regions described previously.
Statistical Analysis and MR Assumptions
We applied the inverse variance–weighted method using multiplicative random effects to obtain a weighted average of individual variant estimates.15 The MR estimates were scaled to 1 SD, ie, approximately 6.7 mmol/mol, reduction in LDL. Multiple testing was accounted for using a P value threshold of .05/3 = .017 for 3 drug targets.
Valid instrumental variables are defined by 3 assumptions.16 First, variants must be associated with the exposure of interest. We estimated the F statistics of drug target instruments (using the square of the β divided by square of the standard error17) with an F greater than 10 being suggestive of adequate instrument strength.18
Second, the variants should share no common cause with the outcome. Confounding arising from underlying population structure was reduced through use of European ancestry populations. Confounding is also possible through linkage disequilibrium, ie, a variant that causes the outcome via a nonlipid pathway being closely correlated with the true causal variant. Correlation is more likely when using instruments from a single gene region, while traditional sensitivity analyses for violation of MR assumptions can be limited in this setting.19 We performed a bayesian colocalization analysis to examine the potential for such genetic confounding. The primary output of interest is the probability of colocalization (ie, exposure and outcomes traits being affected by the same causal variant) conditional on the presence of a causal variant for the outcome. Default prior probabilities were used,20 with detailed methods provided in the eMethods in Supplement 1.
Third, variants should not affect the outcome except through the risk factor. We applied MR Egger,21 weighted median,22 and weighed mode23 methods to test the robustness of the main inverse variance–weighted estimates to horizontal pleiotropy (ie, when instruments affect the outcome through traits other than the exposure). We used a curated genotype-phenotype database (PhenoScanner)24,25 to search for associations between variants used to instrument each drug target and other traits that may represent pleiotropic pathways; specific examples were traits associated with smoking and body fat that are recognized risk factors for psoriatic disease.26 Variants associated with these and other traits (using the software default threshold of P < 1 × 10−5) that are plausible sources of pleiotropy were excluded in sensitivity analyses.
Supplementary Analysis
We performed a series of supplementary analyses to examine the robustness of, and provide context to, the primary analyses. First, we examined the validity of the instruments by using coronary artery disease as a positive control outcome, given the recognized benefits of lipid-lowering drugs in this context. Genetic associations were taken from a GWAS of 60 801 clinically confirmed cases (eg, myocardial infarction, acute coronary syndrome, chronic stable angina, or coronary stenosis >50%) and 123 504 controls.27
Second, we attempted to replicate the previously described analyses (that used LDL as biomarker) with genetic instruments for circulating protein levels of each drug target (ie, protein quantitative trait loci [pQTL]). In preliminary analyses, only PCSK9 cis-pQTLs (ie, variants near the PCSK9 gene) yielded adequate instruments (no cis instruments for HMGCR; no pQTL for NPC1L1). Circulating PCSK9 was previously studied in a plasma proteome study of 35 559 European individuals.28 We selected instruments from within ±100 kb of the PCSK9 gene ( r2 < 0.1; P < 5 × 10−8). Analyses were performed in R using the TwoSampleMR and coloc packages (R Foundation).20,29 This study was reported according to Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) MR guidelines.
Results
For the primary analysis that used psoriasis data from the UK Biobank, 19 variants were selected to proxy LDL lowering through inhibition of HMGCR (mean F statistic of 187), 9 for NPC1L1 (F statistic of 105), and 34 for PCSK9 (F statistic of 246). Genetically proxied PCSK9 inhibition was associated with reduced risk of psoriasis (odds ratio [OR], 0.69 per SD reduction in LDL; 95% CI, 0.55-0.88; P = .003), which was replicated using data from the FinnGen study (OR, 0.77; 95% CI, 0.66-0.91; P = .001); there was no statistical heterogeneity between the 2 estimates (P = .46) (Figure 1). Sensitivity analyses showed consistent estimates, with no statistical evidence for bias from horizontal pleiotropy (Figure 2; eTables 2 and 3 Supplement 1). The posterior probability of colocalization between LDL and psoriasis in the PCSK9 gene region using UK Biobank data was 79% conditional on the presence of a causal variant for the outcome (eTable 4 in Supplement 1).
Figure 1. Associations Between Genetically Proxied Lipid-Lowering Drugs and Psoriasis Risk.

HMGCR indicates 3-hydroxy-3-methylglutaryl CoA reductase; LDL, low-density lipoprotein; NPC1L1, Niemann-Pick C1–like 1; OR, odds ratio; PCSK9, proprotein convertase subtilisin/kexin type 9; UKB, UK Biobank.
Figure 2. Summary of Psoriasis Risk Results From Pleiotropy Robust Sensitivity Analyses.
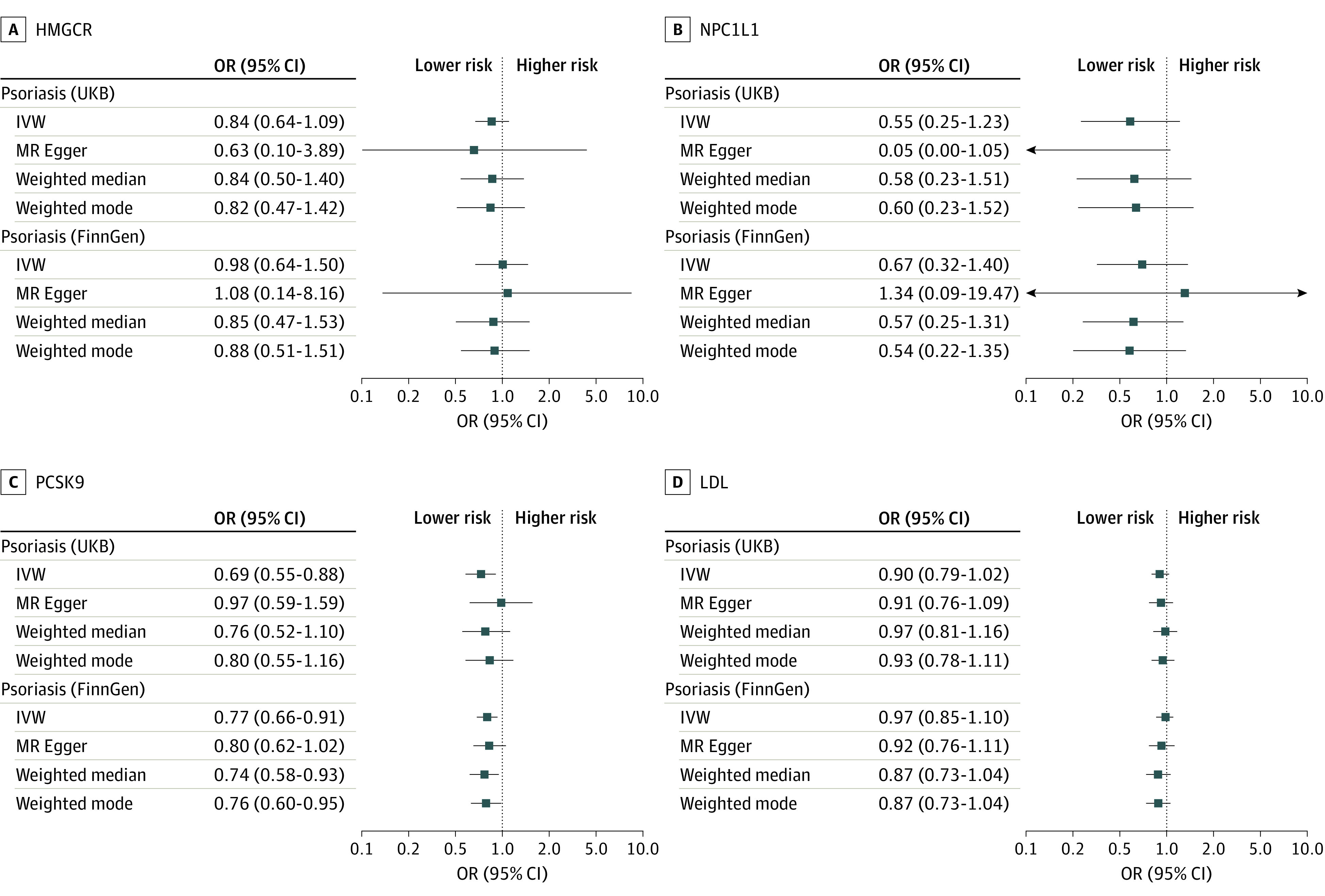
HMGCR indicates 3-hydroxy-3-methylglutaryl CoA reductase; IVW, inverse variance–weighted method; LDL, low-density lipoprotein; MR, mendelian randomization; NPC1L1, Niemann-Pick C1–like 1; PCSK9, proprotein convertase subtilisin/kexin type 9.
There was little statistical evidence of an association between HMGCR and psoriasis risk in either data set (Figure 1 and Figure 2). Some variants selected to instrument HMGCR were associated with body composition traits (eTable 1 in Supplement 1); excluding these variants did not change the conclusion (eTable 2 in Supplement 1). No associations with such potentially pleiotropic traits were found for other exposures. Point estimates for NPC1L1 suggested reduced psoriasis risk, but confidence intervals included the null. Genetically proxied reduction in LDL (instrumented using 320 genetic variants with mean F statistic of 172) was not associated with psoriasis in UK Biobank or FinnGen.
Supplementary Analyses
Genetically proxied inhibition of all 3 drug targets and LDL were all associated with reduced risk of the positive control outcome of coronary artery disease (Figure 3; eTable 2 in Supplement 1). Genetically proxied reduction in circulating PCSK9 levels was associated with reduced psoriasis risk with concordant estimates in UK Biobank (OR, 0.86; 95% CI, 0.49-1.52; P = .61) and FinnGen (OR, 0.83; 95% CI, 0.74-0.93; P = .001), although the former lacked precision due to only 2 of the 3 instruments being available in the UK Biobank data (eTable 5 in Supplement 1).
Figure 3. Associations Between Genetically Proxied Lipid-Lowering Drugs and Risk of Coronary Artery Disease as the Positive Control.

HMGCR indicates 3-hydroxy-3-methylglutaryl CoA reductase; LDL, low-density lipoprotein; NPC1L1, Niemann-Pick C1–like 1; OR, odds ratio; PCSK9, proprotein convertase subtilisin/kexin type 9.
Discussion
This MR study suggests that PCSK9 inhibition is causally associated with reduced risk of psoriasis. This association appeared to be independent of circulating LDL levels, since we did not observe an overall association of LDL with psoriasis risk. Genetically proxied HMGCR (targeted by statins) and NPC1L1 inhibition (targeted by ezetimibe) were not associated with psoriasis risk.
Proprotein convertase subtilisin/kexin type 9, a member of proprotein convertase family, promotes LDL receptor degradation; higher PCSK9 level is associated with greater incidence of cardiovascular events.30 Two PCSK9 inhibitors (alirocumab and evolocumab) are licensed for treating primary hypercholesterolemia or mixed dyslipidemia in those who have not responded adequately to treatment with other lipid-lowering therapies. Circulating PCSK9 levels are higher in patients with psoriasis than healthy controls and may be associated with atherosclerosis and cardiovascular risk in psoriasis.31 There is evidence to suggest that PCSK9 is also implicated in inflammatory pathways, such as tumor necrosis factor and interleukin (IL) 17; in hyperlipidemic mouse models, PCSK9 knockout reduced circulating IL-17 levels and differentiation of IL-17–producing cells.32 Human recombinant PCSK9 induced macrophage activation in vitro with increased expression of tumor necrosis factor and IL-6,33 while suppressing PCSK9 reduced inflammation in stimulated macrophages by inhibiting nuclear factor κB.34 A recent study by Luan and colleagues5 showed that PCSK9 expression was higher in psoriatic plaques than normal skin in keratinocytes and epithelial cells of dermis blood vessels. The authors provided further supportive results using an imiquimod-induced mouse model of psoriasis to show that PCSK9 knockout mice had suppressed imiquimod reaction and small interfering RNA that reduced PCSK9 expression suppressed the imiquimod reaction. Taken together, these results provide compelling evidence that PCSK9 is implicated in psoriasis pathophysiology. Although the results of the current analysis only provide inference for psoriasis prevention, the aforementioned animal models support PCSK9 as a potential target for treatment. Clinical studies of PCSK9 inhibitors are needed to test efficacy and safety in humans.
The largest body of literature on lipid-lowering drugs and psoriasis risk relates to statins. Several suggestive observational studies motivated a few small randomized clinical trials to compare disease-modifying effects of statins on psoriasis severity.4 Interpretations of these trials are limited by power and potential biases. The current MR analysis did not find evidence in support of a causal association between HMCGR inhibition and psoriasis risk, with the caveat that risk factors for disease onset may not coincide with prognosis.
To our knowledge, prior studies of NPC1L1 and psoriasis are limited. There was a consistent association between NPC1L1 inhibition and psoriasis risk, but estimates had wide confidence intervals that included the null. Meta-analysis of UK Biobank and FinnGen estimates would produce a statistically significant result, but we caution against overinterpretation because this was not part of our analysis plan. Mechanistic and better powered epidemiologic studies are needed to examine this association further.
Strengths and Limitations
The main strengths of this study are the robustness of results to replication (using UK Biobank, FinnGen, and PCSK9 pQTL data) and several sensitivity analyses. Investigating the association of lipid-lowering drugs with psoriasis risk and prognosis using cohort designs would likely be limited by indication bias and reverse causation; in the case of PCSK9 inhibition, the small number of patients prescribed these drugs would provide further challenges. When instrumental variable assumptions are met, MR estimates the causal exposure-outcome association with less bias from unmeasured confounding.8
This MR study had several limitations. First, the risk factors for disease onset are not always the same as those for disease severity or prognosis; that is, the direct inference of our results relates to prevention rather than treatment of psoriasis. However, the experiments of Luan and colleagues5 provide evidence supporting the therapeutic potential for treating (rather than preventing) psoriasis. Second, MR estimates cannot be directly compared with pharmacological inhibition. Subtle differences in PCSK9 levels through genetic variation cannot be compared with effects of pharmacological inhibition. Lifelong exposure in MR differs from shorter duration of pharmacological intervention. Genetic variation in PCSK9 throughout the body may differ from tissues targetable by intervention.7 Third, as with all MR studies, instrumental variable assumptions are not empirically verifiable. It is possible that pleiotropy or confounding may bias the current estimates, although sensitivity analyses that examine these sources of bias were reassuring. Fourth, the study population only included participants of European ancestry; future studies among other ethnic populations are needed to examine the generalizability of the current findings. Fifth, the posterior probability of the shared causal variant was low (less than the conventional 80% threshold) in colocalization analysis, which was underpowered. Data from larger GWAS of psoriasis are needed to verify these results. Lastly, the L40 ICD-10 code used in both data sets may include a heterogenous group of psoriasis phenotypes. However, nonvulgaris phenotypes are rare (eg, pustular, palmoplantar and guttate types together form approximately 1% of patients with psoriasis in the UK Biobank), and significant outcome misclassification is likely to bias estimates toward the null.
Conclusions
The results of this MR study suggest that PCSK9 inhibition is causally associated with reduced risk of psoriasis. Existing PCSK9 inhibitors hold potential as therapeutic targets for prevention, and possibly treatment, of psoriasis, although further clinical studies are needed.
eMethods.
eFigure 1. Illustration of instrumental variable assumptions
eFigure 2. Plots from the psoriasis GWAS using UK Biobank data
eTable 1. Variants in the HMGCR gene associated with body composition traits excluded in sensitivity analyses
eTable 2. Associations between genetically proxied exposures (drug targets and LDL) and outcomes (psoriasis using UK Biobank and FinnGen and control outcomes)
eTable 3. MR Egger intercepts
eTable 4. Colocalization results
eTable 5. Associations between circulating PCSK9 level and psoriasis risk
eTable 6. Genetic variants used to instrument LDL and each lipid lowering drug target for the primary analysis
eTable 7. STROBE-MR checklist of recommended items to address in reports of Mendelian randomization studies
Data sharing statement
References
- 1.Miller IM, Ellervik C, Yazdanyar S, Jemec GBE. Meta-analysis of psoriasis, cardiovascular disease, and associated risk factors. J Am Acad Dermatol. 2013;69(6):1014-1024. doi: 10.1016/j.jaad.2013.06.053 [DOI] [PubMed] [Google Scholar]
- 2.Friedewald VE, Cather JC, Gelfand JM, et al. AJC editor’s consensus: psoriasis and coronary artery disease. Am J Cardiol. 2008;102(12):1631-1643. doi: 10.1016/j.amjcard.2008.10.004 [DOI] [PubMed] [Google Scholar]
- 3.Elmets CA, Leonardi CL, Davis DMR, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with awareness and attention to comorbidities. J Am Acad Dermatol. 2019;80(4):1073-1113. doi: 10.1016/j.jaad.2018.11.058 [DOI] [PubMed] [Google Scholar]
- 4.Socha M, Pietrzak A, Grywalska E, et al. The effect of statins on psoriasis severity: a meta-analysis of randomized clinical trials. Arch Med Sci. 2019;16(1):1-7. doi: 10.5114/aoms.2019.90343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luan C, Chen X, Zhu Y, et al. Potentiation of psoriasis-like inflammation by PCSK9. J Invest Dermatol. 2019;139(4):859-867. doi: 10.1016/j.jid.2018.07.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gudjonsson JE, Ding J, Li X, et al. Global gene expression analysis reveals evidence for decreased lipid biosynthesis and increased innate immunity in uninvolved psoriatic skin. J Invest Dermatol. 2009;129(12):2795-2804. doi: 10.1038/jid.2009.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gill D, Georgakis MK, Walker VM, et al. Mendelian randomization for studying the effects of perturbing drug targets. Wellcome Open Res. 2021;6:16. doi: 10.12688/wellcomeopenres.16544.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89-R98. doi: 10.1093/hmg/ddu328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zengini E, Hatzikotoulas K, Tachmazidou I, et al. Genome-wide analyses using UK Biobank data provide insights into the genetic architecture of osteoarthritis. Nat Genet. 2018;50(4):549-558. doi: 10.1038/s41588-018-0079-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou W, Nielsen JB, Fritsche LG, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet. 2018;50(9):1335-1341. doi: 10.1038/s41588-018-0184-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203-209. doi: 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsoi LC, Spain SL, Knight J, et al. ; Collaborative Association Study of Psoriasis; Genetic Analysis of Psoriasis Consortium; Psoriasis Association Genetics Extension; Wellcome Trust Case Control Consortium 2 . Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet. 2012;44(12):1341-1348. doi: 10.1038/ng.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The Cochrane Collaboration . Cochrane handbook for systematic reviews of interventions. Accessed May 1, 2022. https://training.cochrane.org/handbook/current
- 14.Graham SE, Clarke SL, Wu KHH, et al. ; VA Million Veteran Program; Global Lipids Genetics Consortium . The power of genetic diversity in genome-wide association studies of lipids. Nature. 2021;600(7890):675-679. doi: 10.1038/s41586-021-04064-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658-665. doi: 10.1002/gepi.21758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. doi: 10.1136/bmj.k601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li B, Martin EB. An approximation to the F distribution using the chi-square distribution. Comput Stat Data Anal. 2002;40:21-26. doi: 10.1016/S0167-9473(01)00097-4 [DOI] [Google Scholar]
- 18.Burgess S, Thompson SG; CRP CHD Genetics Collaboration . Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40(3):755-764. doi: 10.1093/ije/dyr036 [DOI] [PubMed] [Google Scholar]
- 19.Zuber V, Grinberg NF, Gill D, et al. Combining evidence from Mendelian randomization and colocalization: review and comparison of approaches. Am J Hum Genet. 2022;109(5):767-782. doi: 10.1016/j.ajhg.2022.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383. doi: 10.1371/journal.pgen.1004383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512-525. doi: 10.1093/ije/dyv080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304-314. doi: 10.1002/gepi.21965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46(6):1985-1998. doi: 10.1093/ije/dyx102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Staley JR, Blackshaw J, Kamat MA, et al. PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics. 2016;32(20):3207-3209. doi: 10.1093/bioinformatics/btw373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamat MA, Blackshaw JA, Young R, et al. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. 2019;35(22):4851-4853. doi: 10.1093/bioinformatics/btz469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao SS, Bellou E, Verstappen SMM, et al. Association between psoriatic disease and lifestyle factors and comorbidities: cross-sectional analysis and Mendelian randomisation. Rheumatology (Oxford). Published online July 21, 2022. doi: 10.1093/rheumatology/keac403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nikpay M, Goel A, Won HH, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47(10):1121-1130. doi: 10.1038/ng.3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferkingstad E, Sulem P, Atlason BA, et al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet. 2021;53(12):1712-1721. doi: 10.1038/s41588-021-00978-w [DOI] [PubMed] [Google Scholar]
- 29.Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408. doi: 10.7554/eLife.34408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leander K, Mälarstig A, Van’t Hooft FM, et al. Circulating proprotein convertase subtilisin/kexin type 9 (PCSK9) predicts future risk of cardiovascular events independently of established risk factors. Circulation. 2016;133(13):1230-1239. doi: 10.1161/CIRCULATIONAHA.115.018531 [DOI] [PubMed] [Google Scholar]
- 31.Garshick MS, Baumer Y, Dey AK, et al. Characterization of PCSK9 in the blood and skin of psoriasis. J Invest Dermatol. 2021;141(2):308-315. doi: 10.1016/j.jid.2020.05.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim YU, Kee P, Danila D, Teng BB. A critical role of PCSK9 in mediating IL-17–producing T cell responses in hyperlipidemia. Immune Netw. 2019;19(6):e41. doi: 10.4110/in.2019.19.e41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ricci C, Ruscica M, Camera M, et al. PCSK9 induces a pro-inflammatory response in macrophages. Sci Rep. 2018;8(1):2267. doi: 10.1038/s41598-018-20425-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruscica M, Ricci C, Macchi C, et al. Suppressor of cytokine signaling-3 (SOCS-3) induces proprotein convertase subtilisin kexin type 9 (PCSK9) expression in hepatic hepG2 cell line. J Biol Chem. 2016;291(7):3508-3519. doi: 10.1074/jbc.M115.664706 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods.
eFigure 1. Illustration of instrumental variable assumptions
eFigure 2. Plots from the psoriasis GWAS using UK Biobank data
eTable 1. Variants in the HMGCR gene associated with body composition traits excluded in sensitivity analyses
eTable 2. Associations between genetically proxied exposures (drug targets and LDL) and outcomes (psoriasis using UK Biobank and FinnGen and control outcomes)
eTable 3. MR Egger intercepts
eTable 4. Colocalization results
eTable 5. Associations between circulating PCSK9 level and psoriasis risk
eTable 6. Genetic variants used to instrument LDL and each lipid lowering drug target for the primary analysis
eTable 7. STROBE-MR checklist of recommended items to address in reports of Mendelian randomization studies
Data sharing statement