

Abstract
The development of efficient computational methods for drug target protein identification can compensate for the high cost of experiments and is therefore of great significance for drug development. However, existing structure‐based drug target protein‐identification algorithms are limited by the insufficient number of proteins with experimentally resolved structures. Moreover, sequence‐based algorithms cannot effectively extract information from protein sequences and thus display insufficient accuracy. Here, we combined the sequence‐based self‐supervised pretraining protein language model ESM1b with a graph convolutional neural network classifier to develop an improved, sequence‐based drug target protein identification method. This complete model, named QuoteTarget, efficiently encodes proteins based on sequence information alone and achieves an accuracy of 95% with the nonredundant drug target and nondrug target datasets constructed for this study. When applied to all proteins from Homo sapiens, QuoteTarget identified 1213 potential undeveloped drug target proteins. We further inferred residue‐binding weights from the well‐trained network using the gradient‐weighted class activation mapping (Grad–Cam) algorithm. Notably, we found that without any binding site information input, significant residues inferred by the model closely match the experimentally confirmed drug molecule‐binding sites. Thus, our work provides a highly effective sequence‐based identifier for drug target proteins, as well to yield new insights into recognizing drug molecule‐binding sites. The entire model is available at https://github.com/Chenjxjx/drug-target-prediction.
Keywords: binding site inference, deep learning, druggable protein, graph convolutional network, sequence‐based, transformer
1. INTRODUCTION
With the development of high‐throughput sequencing technology, the number of known proteins has increased exponentially (Nucleic Acids Res, 2021). However, despite this increase, only about 3000 proteins have been identified as targets for drug molecules in clinical use (Wishart et al., 2018) far fewer than the total number of proteins that have been discovered. Indeed, due to the significant time and economic cost of drug discovery (DiMasi et al., 2016; Scannell et al., 2012; Paul et al., 2010) process of target protein identification is much slower and costlier than that of protein discovery (Swaminathan et al., 2018; Alfaro et al., 2021; Wouters et al., 2020). This has highlighted the need for developing efficient computational tools to accurately identify new drug target proteins (Gashaw et al., 2011) which can compensate for the deficiency of experimental screening and accelerate drug development.
Several databases, such as DrugBank (Wishart et al., 2018) and the Therapeutic Target Database (TTD) (Wang et al., 2020) had been constructed, which contain critical information on clinically available drugs, including additional information such as mechanisms of action, known target proteins, and metabolism. Based on these databases, computational tools to identify putative drug targets were subsequently developed (Tian et al., 2018; Xu et al., 2018; Le Guilloux et al., 2009; Hussein et al., 2015; Zhang et al., 2022a). Among them, some methods rely on the conformational structure of proteins (Volkamer et al., 2010) while others depict proteins using their sequence characteristics and physicochemical properties (Thangudu et al., 2012). These methods have been reported to identify drug target proteins with accuracies of nearly 90% (Yu et al., 2022). However, as the number of currently known drug target proteins is still small, and the dataset of putative nondrug target proteins may consist of potentially undeveloped drug target proteins (Thangudu et al., 2012) these drug targets prediction methods may need further evaluation on larger external datasets, even though they demonstrated high accuracies on individual datasets (Jamali et al., 2016; Sun et al., 2018; Li & Lai, 2007).
Notably, the release of AlphaFold2 (Jumper et al., 2021) a tool for predicting protein structures based on artificial intelligence, has inspired the development of data‐driven approaches that integrate information on protein coevolution, phylogenetic relationships, and conserved sites from multiple sequence alignment (MSA) to uncover meaningful embeddings for amino acid and secondary structures (Rao et al., 2019; Rives et al., 2021; Zhang et al., 2022b; Rao et al., 2021; Jing et al., 2021). These methods perform better than traditional natural language processing algorithms in several different downstream tasks (Kulmanov & Hoehndorf, 2021; Gligorijević et al., 2021). For instance, ESM1b (Rives et al., 2021) a sequence‐based, self‐supervised pretraining transformer protein language model based on positional context, has been used to predict both protein function and folding categories, demonstrating a strong generalization ability (Zhang et al., 2022c; Guo et al., 2022). This highlights the tremendous potential for performing drug target protein identification and addressing the urgent need for new druggable targets by combining effective pretraining models with deep learning‐based frameworks.
Here, we constructed a complete algorithm flow called QuoteTarget (Sequence‐based transformer protein language model to identify potential druggable protein targets), which includes both a protein representation method and a classifier for identifying potential drug target proteins. By combining the sequence‐based pretraining model ESM1b with a graph convolutional neural network (GCN)‐based classifier, our algorithm achieves 95% classification accuracy on the nonredundant dataset. QuoteTarget outperformed existing methods and demonstrated a strong generalization ability on multiple datasets. In addition, QuoteTarget was used to identify 1213 undeveloped putative drug target proteins in Homo sapiens, thereby providing a valuable reference for future experimental studies. Using the gradient‐weighted class activation mapping (Grad–Cam) algorithm, we calculated residue‐binding weights from the well‐trained model, which are consistent with experimentally confirmed drug molecule‐binding sites. Our study, therefore, provides an efficient model for extracting features from amino acid sequences alone to perform drug target protein identification, with potential implications for recognizing drug‐binding sites.
2. RESULTS
2.1. Composition and basic features of the dataset
To compile data for developing a comprehensive drug target prediction algorithm, we first collected and integrated datasets containing known drug target proteins and nondrug target proteins. Drug target proteins were obtained from the DrugBank and TTD databases. DrugBank includes comprehensive molecular information about drugs, including their mechanisms, interactions, and targets (Wishart et al., 2018) and TTD is a database consisting of therapeutic targets (Wang et al., 2020). After removing duplicated proteins and proteins with sequence identities larger than 95%, we obtained 6582 drug target proteins, including 4056 entries from H. sapiens. This group contains proteins bound by molecules in DrugBank from the following categories: approved, small molecule, biotech, experimental, nutraceutical, illicit, withdrawn, and investigational. A total of 2837 target proteins (2183 from H. sapiens) for drug molecules approved by the US Food and Drug Administration (FDA) were included in the drug target group (Figure 1a).
FIGURE 1.
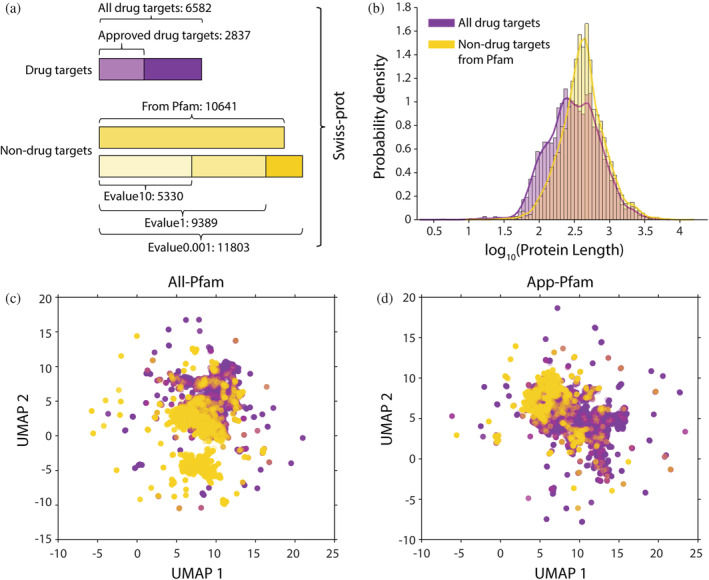
The composition and basic features of the datasets constructed in this study. (a) we utilized two drug‐target datasets: (1) all targets and (2) Food and Drug Administration (FDA)‐approved drug targets, as well as four nondrug‐target datasets, constructed using different extraction methods. (b) Length distribution of all drug‐target proteins and nondrug‐target proteins from Pfam. (c) Dimensionality reduction of protein sequences based on uniform manifold approximation and projection (UMAP). Purple dots represent all drug‐target proteins and yellow dots represent nondrug‐target proteins from Pfam. (d) Data analyzed as in (c), including only FDA‐approved drug targets
To compile the nondrug target protein datasets, we first removed the known drug target proteins from the Swiss‐Prot database (Boutet et al., 2007). We then removed similar sequences using several different methods. Based on the Pfam (Mistry et al., 2021) database, we constructed a dataset of nondrug target proteins with protein family redundancy removed, which contains 10,641 proteins. In addition, we constructed three other datasets of nondrug target proteins with redundant proteins removed based on varying levels of sequence similarity. By using three different E‐value (same meaning as BLAST E‐value) cutoffs of 0.001 (Evalue0.001), 1 (Evalue1), and 10 (Evalue10), we obtained datasets containing 11,803, 9389, and 5330 proteins, respectively (Figure 1a); of these, 7900, 5941, and 3078 proteins, respectively, are from H. sapiens. In conclusion, we obtained two distinct datasets of drug target proteins and four distinct datasets of nondrug target proteins. Combining positive and negative samples, we got a total of eight datasets: All‐Pfam, All‐Evalue0.001, All‐Evalue1, All‐Evalue10, App‐Pfam, App‐Evalue0.001, App‐Evalue1, and App‐Evalue10.
To determine whether we could detect a clear distinction between features of the drug target proteins compared to the nondrug target proteins, we measured the length of amino acid sequences for proteins in both groups (Figure 1b). We found that using protein length alone cannot distinguish the two clusters of proteins. We then encoded the proteins based on their sequences and reduced dimensionality to a two‐dimensional (2D) plane using Uniform Manifold Approximation and Projection (UMAP). It showed that neither all drug target proteins nor FDA‐approved drug target proteins could be clearly distinguished from nondrug target proteins (Figure 1c,d). These results indicated that a powerful classifier is needed to distinguish drug target proteins from nondrug target proteins.
2.2. Flow of the drug target protein‐identification algorithm
To computationally identify drug target proteins, we constructed a complete algorithm flow from protein representation to classification. To this end, a sequence‐based protein representation method was first built using a pretraining model by encoding each protein sequence into a protein representation matrix with size LM and a contact map matrix with size L × L (Figure 2a, top panel). We adopted the pretraining model from ESM1b in this step, which is a large‐scale, self‐supervised, and transformer‐based protein language framework from Rives et al. (Rives et al., 2021) (Figure 2a, bottom panel).
FIGURE 2.
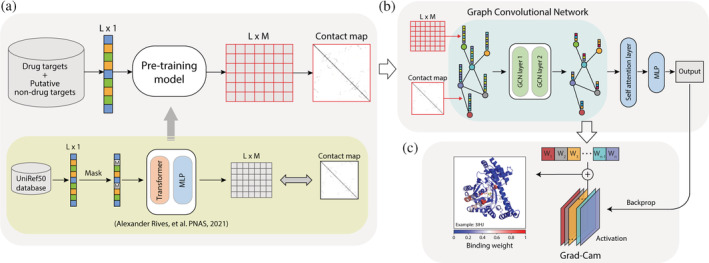
Flowchart for the drug target protein‐prediction algorithm QuoteTarget developed in this study. (a) Protein pretraining model and coding steps. All protein sequences were encoded into a matrix and a contact map as inputs for subsequent classifiers (top panel). This pretraining model was adapted from Rives et al. (bottom panel). (b) Training graph convolutional neural network (GCN) for drug target protein classification. The matrices generated in the coding step were used as the nodes of the graph, and the contact maps were used as the edges of the graph. (c) The drug molecular‐binding weight of each residue was calculated by grad–cam algorithm. Left panel shows an example of protein 3IHJ. The color represents the drug's molecular‐binding weight
To better extract features of proteins for subsequent classification, we then constructed a classifier based on a GCN (Figure 2b). Each amino acid in a protein was represented as a node of the graph, with the contact map matrix serving as the adjacency matrix (see Methods). The inputs then passed through two identically structured stacked graph convolutional layers and one self‐attention layer. Finally, the classification results were output through a full‐connection layer.
To determine the essential residue sites for drug–protein interactions, we also calculated the binding weight of each residue using the Grad–Cam algorithm. In this step, the output of the classifier was backpropagated to the last layer of the GCN, and the obtained gradients were then multiplied by the activation matrix elements for that layer to get the binding weight for each residue (Figure 2c, see Methods for details). The complete algorithm was named QuoteTarget and is available at https://github.com/Chenjxjx/drug-target-prediction.
2.3. Results on 5‐fold cross‐validation and an external test dataset
Using the aforementioned protein representation method and GCN, we then trained models on randomly extracted datasets and tested them on external test datasets. To verify the robustness of QuoteTarget, we showed the average index with a standard deviation of all models rather than the optimal index from the 5‐fold cross‐validation. The results revealed that QuoteTarget demonstrated excellent performance on both validation datasets and external test dataset, with an accuracy reaching 0.95 (95%). In addition, we found that QuoteTarget also performed well on other metrics, including precision, F1 score, Mcc (Matthews correlation coefficient), sensitivity, and specificity (Table 1). To eliminate possible effects of our de‐redundancy approaches, we also trained models separately on datasets of All‐Pfam, All‐Evalue0.001, All‐Evalue1, and All‐Evalue10. The results showed that QuoteTarget performed equally well on all these datasets (Table 1). What's more, QuoteTarget performed well in receiver operating characteristic (ROC) curve analysis on both validation datasets and external test dataset, and areas under the curve (AUC) were all above 0.98 (Figure 3a). These results indicated that our algorithm identified drug target proteins accurately and reliably with a strong generalization ability.
TABLE 1.
Classification results of QuoteTarget on the all drug target dataset
| Dataset | Acc | Precision | F1 | Mcc | Sensitivity | Specificity | |
|---|---|---|---|---|---|---|---|
|
5‐fold cross‐validation |
All‐Pfam | 0.95 ± 0.00 | 0.96 ± 0.01 | 0.94 ± 0.00 | 0.90 ± 0.01 | 0.92 ± 0.01 | 0.97 ± 0.01 |
| All‐Evalue0.001 | 0.96 ± 0.01 | 0.97 ± 0.01 | 0.94 ± 0.00 | 0.90 ± 0.00 | 0.90 ± 0.01 | 0.98 ± 0.00 | |
| All‐Evalue1 | 0.95 ± 0.00 | 0.97 ± 0.01 | 0.94 ± 0.01 | 0.90 ± 0.01 | 0.91 ± 0.02 | 0.98 ± 0.01 | |
| All‐Evalue10 | 0.94 ± 0.00 | 0.97 ± 0.01 | 0.95 ± 0.00 | 0.89 ± 0.01 | 0.93 ± 0.01 | 0.96 ± 0.02 | |
| External test | All‐Pfam | 0.95 ± 0.00 | 0.96 ± 0.01 | 0.94 ± 0.00 | 0.90 ± 0.01 | 0.91 ± 0.01 | 0.98 ± 0.00 |
| All‐Evalue0.001 | 0.96 ± 0.00 | 0.97 ± 0.01 | 0.93 ± 0.01 | 0.90 ± 0.01 | 0.90 ± 0.01 | 0.98 ± 0.00 | |
| All‐Evalue1 | 0.95 ± 0.00 | 0.96 ± 0.01 | 0.93 ± 0.00 | 0.89 ± 0.00 | 0.91 ± 0.01 | 0.97 ± 0.01 | |
| All‐Evalue10 | 0.94 ± 0.00 | 0.97 ± 0.01 | 0.95 ± 0.00 | 0.89 ± 0.01 | 0.93 ± 0.01 | 0.96 ± 0.01 |
Note: The indexes in the table are the mean values and standard deviations are from the 5‐fold cross‐validation.
Abbreviations: Acc, accuracy; F1, F1‐score; Mcc, Matthews correlation coefficient.
FIGURE 3.
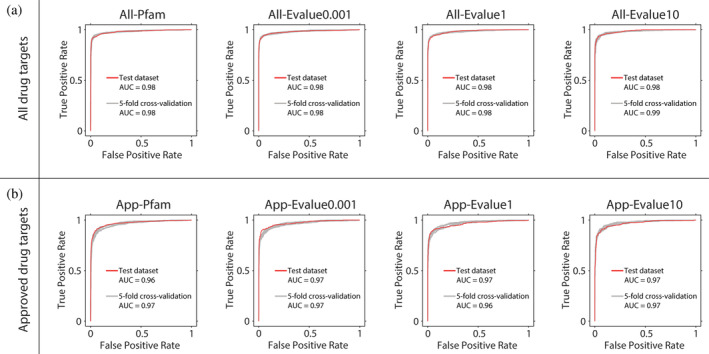
Receiver operating characteristic (ROC) curves for classification results with the QuoteTarget algorithm on different datasets. (a) Classification results on datasets of all‐Pfam, all‐Evalue0.001, all‐Evalue1, and all‐Evalue10. Red line represents the ROC curve of the external test dataset, and gray line represents the ROC curve of 5‐fold cross‐validation. (b) Classification results for data analyzed as (a) but using datasets of app‐Pfam, app‐Evalue0.001, app‐Evalue1, and app‐Evalue10
To further verify the robustness of the algorithm, we trained and tested QuoteTarget on four other datasets of App‐Pfam, App‐Evalue0.001, App‐Evalue1, and App‐Evalue10. As above, we found the accuracy was close to 95%, and the algorithm also performed well on other metrics (Table 2). Moreover, ROC curves showed good performance for both validation datasets and external test dataset, with AUC values above 0.96 (Figure 3b).
TABLE 2.
Classification results of QuoteTarget on the FDA‐approved drug target dataset
| Dataset | Acc | Precision | F1 | Mcc | Sensitivity | Specificity | |
|---|---|---|---|---|---|---|---|
|
5‐fold cross‐validation |
App‐Pfam | 0.94 ± 0.00 | 0.91 ± 0.01 | 0.86 ± 0.01 | 0.82 ± 0.01 | 0.81 ± 0.02 | 0.98 ± 0.00 |
| App‐Evalue0.001 | 0.95 ± 0.00 | 0.91 ± 0.01 | 0.86 ± 0.01 | 0.83 ± 0.01 | 0.82 ± 0.02 | 0.98 ± 0.00 | |
| App‐Evalue1 | 0.94 ± 0.00 | 0.91 ± 0.03 | 0.87 ± 0.01 | 0.83 ± 0.01 | 0.84 ± 0.02 | 0.97 ± 0.01 | |
| App‐Evalue10 | 0.93 ± 0.00 | 0.92 ± 0.02 | 0.89 ± 0.01 | 0.84 ± 0.01 | 0.87 ± 0.01 | 0.96 ± 0.01 | |
| External Test | App‐Pfam | 0.95 ± 0.00 | 0.93 ± 0.01 | 0.87 ± 0.01 | 0.84 ± 0.01 | 0.81 ± 0.02 | 0.98 ± 0.00 |
| App‐Evalue0.001 | 0.96 ± 0.00 | 0.89 ± 0.02 | 0.88 ± 0.01 | 0.86 ± 0.01 | 0.87 ± 0.01 | 0.97 ± 0.00 | |
| App‐Evalue1 | 0.95 ± 0.00 | 0.94 ± 0.02 | 0.89 ± 0.01 | 0.86 ± 0.01 | 0.84 ± 0.03 | 0.98 ± 0.01 | |
| App‐Evalue10 | 0.93 ± 0.00 | 0.93 ± 0.01 | 0.90 ± 0.01 | 0.85 ± 0.01 | 0.87 ± 0.02 | 0.97 ± 0.01 |
Note: The indexes in the table are the mean values and standard deviations are from the 5‐fold cross‐validation.
Given that there are more nontarget proteins than drug‐target proteins in our datasets, we performed additional analysis to eliminate possible effects from the different dataset sizes. To this end, we retrained QuoteTarget with randomly extracted nondrug target samples so that the numbers of target and nontarget proteins were comparable. We found that regardless of the dataset, QuoteTarget displayed high accuracy and stability, other than a slight decrease in accuracy observed with the decreased training data (Tables S1 and S2). These results demonstrated that our algorithm was robust for various data extraction methods and can identify drug target proteins accurately.
2.4. Performance comparison with different protein representation methods and different classifiers
QuoteTarget algorithm can be divided into two parts: protein representation and classifier. We first tested and compared the performance of QuoteTarget with word2vec for protein representation method. word2vec is a typical encoding algorithm for language processing, which has also been utilized for encoding protein sequences in previous studies (Li & Lai, 2007; Chu et al., 2022; Saar et al., 2021). For this analysis, we used the hyperparameter of word2vec, which was shown to be optimal for protein classification (see Methods). As the output of word2vec cannot be adapted by GCN, we chose traditional machine learning methods as classifiers, including decision tree, K‐nearest neighbor, GaussionNB, SVM, logistic regression, and random forest. Taking the dataset of All‐Pfam as an example, we found that the performance of word2vec was significantly inferior to that of ESM1b in both 5‐fold cross‐validation and the external test (Figure 4). Detailed analyses showed that word2vec combined with traditional machine learning classifiers cannot extract features effectively, and sometimes all samples were classified as negative. We also performed an ablation of ESM1b pretraining model, that was, replacing the complete ESM1b with randomly initialized ESM1b, and then re‐training QuoteTarget on our dataset. The performance of the algorithm after ESM1b ablation was significantly inferior to that using the complete ESM1b (Figure 4, Figure S1a,b, Table S3). In summary, as a powerful protein encoding method, ESM1b significantly improved the performance of QuoteTarget.
FIGURE 4.
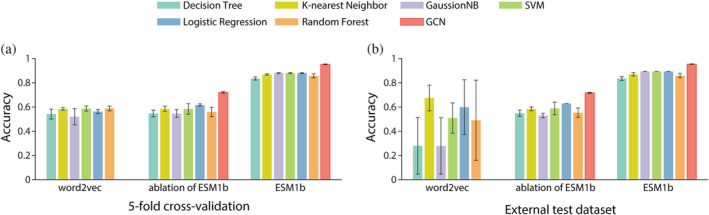
Comparison of different protein encoding methods combined with different classification algorithms. (a) Classification results on 5‐fold cross‐validation with word2vec, randomly initialized ESM1b, and complete ESM1b protein‐encoding, respectively. All‐Pfam dataset was used for this analysis. (b) Data analyzed as in (a) but showing the results from the external test dataset
AlphaFold2 has made a great breakthrough in structural prediction (Jumper et al., 2021). Next, we wanted to explore whether the use of contact maps predicted by AlphaFold2 could be better than ESM1b. We retrained QuoteTarget using the contact maps predicted by AlphaFold2 (see Methods for details). On both All‐Pfam and App‐Pfam datasets, the results with AlphaFold2 were very close to those with ESM1b (Table 3). In most statistical indexes, there was only a 0.01 fluctuation between AlphaFold2 and ESM1b. However, predicting contact maps using AlphaFold2 consumed 36 times more time than ESM1b with the same computational resources (see Methods). These results highlighted the high efficiency of ESM1b in protein encoding.
TABLE 3.
Classification results of quoteTarget using contact maps predicted by AlphaFold2 instead of ESM1b
| Dataset | Acc | Precision | F1 | Mcc | Sensitivity | Specificity | |
|---|---|---|---|---|---|---|---|
|
5‐fold cross‐validation |
All‐Pfam | 0.96 ± 0.00 | 0.96 ± 0.00 | 0.94 ± 0.01 | 0.90 ± 0.01 | 0.92 ± 0.01 | 0.97 ± 0.00 |
| App‐Pfam | 0.95 ± 0.00 | 0.93 ± 0.01 | 0.87 ± 0.01 | 0.85 ± 0.01 | 0.82 ± 0.02 | 0.99 ± 0.00 | |
| External Test | All‐Pfam | 0.95 ± 0.00 | 0.94 ± 0.00 | 0.93 ± 0.00 | 0.90 ± 0.00 | 0.93 ± 0.01 | 0.96 ± 0.00 |
| App‐Pfam | 0.94 ± 0.00 | 0.94 ± 0.00 | 0.86 ± 0.01 | 0.83 ± 0.01 | 0.79 ± 0.01 | 0.98 ± 0.00 |
Note: The indexes in the table are the mean values and standard deviations are from the 5‐fold cross‐validation.
As for the classifier, we then compared the performance of GCN to the traditional machine learning methods combined with the ESM1b protein representation. The results on multiple datasets showed that the traditional machine learning methods performed well with accuracies higher than 0.8 (80%), but not as good as GCN (Figure S1c–f, Table S4).
2.5. Comparison of QuoteTarget with other methods
Next, we compared QuoteTarget with two best‐performing drug target protein prediction algorithms that have been reported recently (Yu et al., 2022). One was developed by Lin et al. based on protein physicochemical properties and bagging‐SVM classifier, which achieved the highest accuracy compared to previous studies (Lin et al., 2019). The other was constructed by Yu et al. based on hybrid deep learning model, which performed well in multiple statistical indexes (Yu et al., 2022). In their studies, several different combinations of protein features and hyperparameters have been used. We selected the best‐performing feature combinations and hyperparameters for both methods. We downloaded the source codes and retrained the two models using their original datasets. Indeed, we achieved exactly the same accuracy as originally reported in their articles, respectively, indicating that we were able to reproduce their models exactly. We then retrained and tested the two algorithms using their source codes on our datasets, so that we could make a fair comparison. The results showed that the accuracy of QuoteTarget (95%) was much better than the other two retrained models (67%–84%) on both All‐Pram and App‐Pfam datasets. And our algorithm was more robust in multiple statistical indexes (Table 4). In summary, QuoteTarget performed pretty well on a variety of datasets and outperformed the existing algorithms.
TABLE 4.
Comparison of QuoteTarget with other algorithms
| Algorithm | Dataset | Acc | Precision | F1 | Mcc | Sensitivity | Specificity |
|---|---|---|---|---|---|---|---|
| BLAST | All‐Pfam | 0.28 | 0.84 | 0.42 | −0.35 | 0.00 | 0.84 |
| App‐Pfam | 0.16 | 0.82 | 0.27 | −0.39 | 0.00 | 0.82 | |
| Lin et al. (Lin et al., 2019) | All‐Pfam | 0.73 | 0.67 | 0.61 | 0.41 | 0.56 | 0.72 |
| App‐Pfam | 0.67 | 0.92 | 0.21 | 0.25 | 0.12 | 0.93 | |
| Yu et al. (Yu et al., 2022) | All‐Pfam | 0.76 | 0.67 | 0.77 | 0.50 | 0.83 | 0.67 |
| App‐Pfam | 0.84 | 0.93 | 0.83 | 0.65 | 0.81 | 0.93 | |
| QuoteTarget | All‐Pfam | 0.95 | 0.96 | 0.94 | 0.90 | 0.91 | 0.98 |
| App‐Pfam | 0.95 | 0.93 | 0.87 | 0.84 | 0.81 | 0.98 |
We also used protein sequence similarity (through BLAST) as a baseline method. The classification results with different BLAST parameters on multiple datasets showed that drug target proteins could not be effectively distinguished from sequence similarity alone (Table 4, Table S5).
2.6. Identification of undeveloped drug target proteins in H. sapiens
The above results demonstrated that QuoteTarget efficiently extracted features from sequences for identifying drug target proteins, with high‐classification accuracy and strong generalization ability. Next, we attempted to identify undeveloped drug target proteins in H. sapiens to provide references for future experimental studies (Figure 5a). Based on the nonredundant datasets compiled at the start of the study, we extracted 4056 drug target proteins from H. sapiens as positive samples. A total of 6861 nondrug target proteins from H. sapiens were then equally divided into three portions, and three iterations were performed. In each iteration, two portions were selected as negative samples (approximately the same as the number of positive samples in the drug target protein dataset) for 5‐fold cross‐validation. The remaining portion was used as the test samples. This way, all potential undeveloped targets among the nondrug target proteins of H. sapiens could be identified.
FIGURE 5.
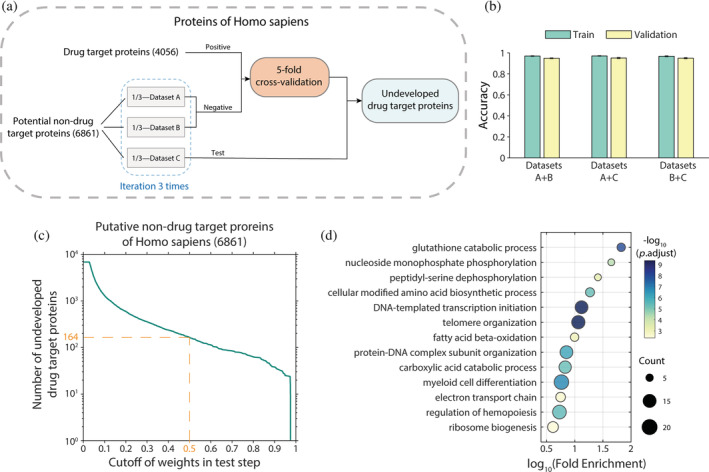
Identification of undeveloped drug targets in Homo sapiens. (a) Schematic overview of the experimental strategy for identifying undeveloped drug target proteins. (b) Results of 5‐fold cross‐validation analysis. The different combinations of datasets correspond to the three randomly divided groups of nondrug target (negative) proteins in (a). (c) Graph showing the number of proteins obtained using different weight cutoffs in the test step. (d) Functional enrichment gene ontology (GO) analysis of putative drug target proteins obtained with a p‐value cutoff of 0.5. In total, we identified 164 proteins. Color of the dots represents the −log10 adjusted p‐value, and the size represents the number of proteins
The results showed that the accuracies of all three iterations reached 0.95, with strong robustness (Figure 5b, Table S6). In addition, although the vast majority of proteins were identified as nondrug target proteins, a small number of them were identified as drug target proteins with high QuoteTarget scores. Among the 6861 nondrug target proteins tested, 164 proteins were judged to be drug targets with a cutoff of 0.5 (Figure 5c, Table S7). Notably, these proteins were not classified as drug target proteins in any known database. Gene ontology (GO) enrichment analysis revealed that they are involved in basic metabolic processes, genome assembly, and ribosomal synthesis (Figure 5d).
Next, we tested our model on larger datasets to uncover more drug target candidates. To this end, we extracted all H. sapiens proteins from the unreviewed UniProt (TrEMBL) database (The UniProt, 2021). After removing known drug target proteins and corresponding protein families, a large dataset containing 62,037 nondrug target proteins was compiled. Using the well‐trained model for prediction, 60,824 (98%) proteins from this group were identified as nondrug target proteins by QuoteTarget, with a scoring cutoff of 0.5, supporting the robustness of our model. In addition, we identified 1213 proteins in total with a score above 0.5, which may represent undeveloped drug target proteins (Figure S2). Detailed gene lists and scoring values are available in the Supplementary File.
2.7. Identification of key residues in drug target proteins by grad–cam
Lastly, we attempted to determine whether QuoteTarget can successfully learn information about key residue‐binding sites and extract this information from complex network parameters. For this analysis, we used Grad–Cam, a feature weight visualization method for image recognition (Selvaraju et al., 2020) which was also applied in protein function identification to calculate residue weights (Gligorijević et al., 2021) and applied it to our well‐trained drug target protein‐identification model (Figure 1c).
Key residue sites for drug target protein identification form the binding sites for drug molecules and thus have clear biological relevance. Therefore, residue‐binding weights calculated by Grad–Cam were compared with experimentally confirmed drug molecule‐binding sites from the BioLiP database (Yang et al., 2013a). For example, 3UCD is an asymmetric complex of human neuron‐specific enolase‐2‐PGA/PEP (Qin et al., 2012). It can bind to the drug molecule 2PG (Knox et al., 2011) and the 3D structure has been resolved through structural biology experiments (Figure 6a). Here, we found that residue‐binding weights from Grad–Cam are in good agreement with experimentally confirmed drug molecule‐binding sites (Figure 6b). To further confirm the credibility of Grad–Cam scoring, we randomly scored the residue‐binding weights of the protein 10 times and compared these values with BioLiP‐derived drug molecule‐binding sites. Based on ROC curve analysis, we found that random scoring was far inferior to scoring based on the trained model (Figure 6c). Moreover, in similar analyses with the proteins 1A4I, 1R3T, and 1Z5V, Grad–Cam scoring peaks were found to be consistent with experimentally confirmed drug molecule‐binding sites (Figure 6d–l). We then collected 1571 proteins from the intersection of the BioLiP database and our drug target protein dataset. By analyzing the Grad‐Cam scores of all these proteins, we found that >80% proteins have experimentally confirmed drug molecule‐binding sites near the peaks of Grad–Cam scoring (Figure S3a). In total, these data suggested that QuoteTarget can successfully learn information about drug molecule‐binding sites, and the residual binding weights can be inferred from complex network parameters using Grad–Cam.
FIGURE 6.
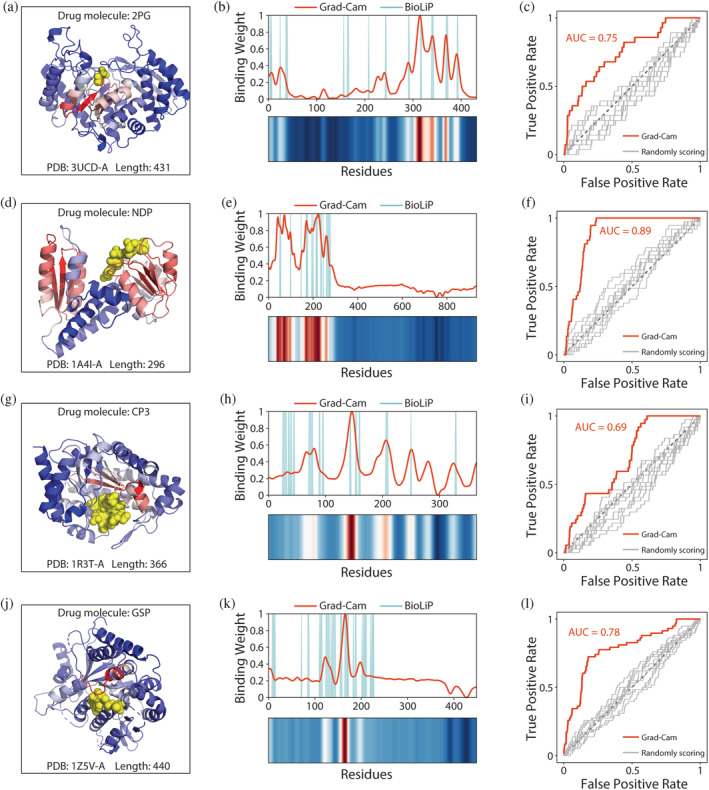
Residue‐binding weights calculated by grad–cam match experimentally confirmed binding sites. (a) 3D conformation of 3UCD binding to the drug molecule 2PG. Yellow spheres represent drug molecules. Color of the cartoon indicates the residue‐binding weights calculated by grad–cam. Higher scored regions are shown in red, and lower scored regions are shown in blue. (b) Comparison between residue‐binding weights calculated by grad–cam and experimentally confirmed binding sites from BioLiP. Gaussian smoothing was performed for the residue‐binding weight curve. (c) ROC curves comparing predicted residue‐binding weights and true binding sites. Gray curves represent the residue‐binding weights obtained by random scoring. Results for the protein (d–f) 1A4I, (g–i) 1R3T, and (j–l) 1Z5V, analyzed as described in (a–c)
3. DISCUSSION
QuoteTarget performed better than previous sequence‐based deep learning methods (Jamali et al., 2016; Li & Lai, 2007). This may derive from the use of the large‐scale protein pretraining model ESM1b (Rives et al., 2021). For drug target protein identification, the contact map predicted by ESM1b with much fewer computing resources can achieve the same effect as AlphaFold2. To some extent, this compensates for the lack of experimentally determined protein structure. Probably due to the efficient protein representation and the strong generalization ability of ESM1b, we obtained consistently good results on different datasets without tailoring hyperparameters. On the other hand, GCN has strong data understanding and high‐cognitive ability (Wu et al., 2021) and the structure of edges and nodes makes it easier to parse connections between data. With the combination of ESM1b and GCN, we have developed a well‐performed drug target protein prediction model with interpretability.
There are numerous advantages to sequence‐based algorithms. In particular, the application of structure‐based algorithms is limited by the fact that there are far fewer protein structures than sequences. Moreover, our sequence‐based feature extraction may have the potential to be applied to proteins containing disordered fragments (Chu et al., 2022) which can deepen our understanding of the fundamental features of proteins embedded in sequences.
The druggability of a protein comes from that the protein can bind to an approved drug molecule with a therapeutic benefit (Liu & Altman, 2014). In this sense, protein druggability is associated with the binding pocket and the binding affinity of drug molecules (Owens, 2007). In our results, the majority of proteins had actual binding sites close to the Grad‐Cam score peaks (Figure S3). The position with high Grad‐Cam score is the key position for classification, indicating that the ability to bind to drug molecules is an important criterion for identifying druggable proteins. Given that we did not use any structural information in QuoteTarget, nor did we train it with any binding site information, it was remarkable that the residue‐binding weights were consistent with experimentally confirmed drug molecule‐binding sites (Figure 6). This may be because the evolutionary information contained in a large number of sequences is related to protein function, which may be essential for drug target identification.
It is worth noting that for a considerable number of proteins, there are more peaks based on Grad–Cam scoring than known binding sites in BioLiP (Figure S4). In these cases, some peaks of Grad‐Cam scoring still corresponded to the experimentally confirmed drug molecule‐binding sites. Considering that there may be binding sites for more than one drug molecule, we speculate that these additional peaks in Grad–Cam scoring may be potentially unexplored drug molecule‐binding sites. Thus, our algorithm not only provided a tool for identifying drug target proteins precisely but also had the non‐negligible potential to reveal previously unidentified drug molecule‐binding sites from amino acid sequences alone.
4. METHODS
4.1. Dataset integration and preprocessing
Drug target proteins were obtained from the DrugBank and TTD databases (Wishart et al., 2018; Wang et al., 2020). DrugBank database contains 5696 drug target proteins, including 3061 FDA‐approved drug target proteins. The TTD database contains 3473 drug target proteins, including 594 FDA‐approved drug target proteins. Redundant sequences within each database were removed using seqkit (Shen et al., 2016) and sequences with more than 95% identity were removed using CD‐HIT (Fu et al., 2012). After filtering, the DrugBank database retained 5260 drug target proteins, including 2682 FDA‐approved drug target proteins, and the TTD database retained 3265 drug target proteins, including 588 FDA‐approved drug target proteins. Integration of the two databases yielded a total of 6582 drug target proteins, including 2837 FDA‐approved drug target proteins. These proteins represent 3494 different protein families based on Pfam.
Nondrug target proteins were obtained from Swiss‐Prot databases. After removing repeated sequences from the 27,278 total sequences, 26,714 sequences remained. We then constructed datasets of nondrug target proteins in two ways. In the first way, we removed all drug target protein families from the Pfam database according to the type of “Family” under the family layer, leaving 10,641 proteins as nondrug target proteins. In the second way, we removed known drug target proteins from DrugBank and TTD and then removed proteins with similar sequences based on different E‐values from BLAST. After removing similar sequences with E‐values less than 0.001, 1, and 10, we obtained 11,803, 9389, and 5330 nondrug target proteins, respectively. Potential nondrug target proteins in H. sapiens were obtained from the Swiss‐Prot and TreEMBL databases. After removing known drug target proteins and corresponding protein families from each group, these contained 6861 and 62,037 nontarget proteins, respectively.
4.2. Protein encoding with ESM1b
Proteins were encoded by calling the pretraining model esm1b_t33_650M_UR50S from the ESM1b framework, which contains 27.1 million UniRef50 sequences. The maximum protein sequence length that can be used by the ESM1b model is 1024 residues. For sequences with a length greater than 1024 residues, tokens exceeding 1024 were removed. For sequences with a length less than 1024 residues, zero‐values (missing) were added at the end to make the matrix reach the length of 1024 for subsequent calculation. We took the values of “representation” in the 33rd layer tuple of positional embedding as the protein representation matrix, and then used this representation to calculate the contact map (threshold at 8 Å). In QuoteTarget, the L in Figure 1 is 1024 and the M is 1280.
When AlphaFold2 was used to calculate the contact maps, the single protein sequence was used as input. The size of the contact map matrix calculated by AlphaFold2 was . Then we performed argmax and normalized the matrix to obtain the contact map matrix with size . It took 24 hours to calculate the contact map of 18,000 proteins using AlphaFold2 on 8 x Nvidia 3090 GPUs. The same task took 40 minutes with ESM1b.
4.3. Protein encoding with word2vec
word2vec, a typical natural language processing method, contains two models of continuous bag‐of‐words (CBOW) and continuous skip‐gram (Mikolov et al., 2013; Rong, 2014). We used the skip‐gram model, with a window size of three. After a 3‐gram overlapped model, each residue was encoded in 200 dimensions.
4.4. Machine learning classifiers
For the GCN‐based classifier, we used contact maps predicted by ESM1b as the adjacency matrix and the representation vectors of each amino acid as the nodes of the network. In detail, a protein with L residues was represented as for nodes, contact map for edge. The graph neural network we used was as following:
where was a diagonal degree matrix with , and normalized to sum up to 1 in each row. was the adjacency matrix. added the predicted contact map in graph network with the identity matrix for self‐loops. was the activation hidden matrix for the layer. The initial state defined as . was a weight matrix of layer‐specific trainable parameters which map the features from size of M to a lower dimension space with a size of . denoted a nonlinear activation function . Normalization layer was added after each GCN layer. the final output of the GCN layers was: . where was a p dimensional vector token embedding for the node. integrated all token embeddings with .
Then we used the self‐attention mechanism to compute the weight coefficients . was the number of attention groups:
where was the transposition of . and were two learned attention matrices with the hyper‐parameters and . The SoftMax function normalized the weight sum of each row to 1. The final output was represented by the product of T and H:
Hyperparameters during training were as follows: Learning rate = 1 e‐4, Batch size = 64, Weight_decay = 1 e‐5, GCN_feature_dim = 1280, GCN_hidden_dim = 256, and GCN_output_dim = 64. Hyperparameters of the self‐attention layer were as follows: Dense_dim = 16 and Attention_heads = 4. Loss function: mean square error loss. The model was trained for a total of 10 epochs. Hyperparameters of the models for traditional machine learning classifiers are shown in Table S8.
4.5. 5‐fold cross‐validation and external test dataset
We randomly extracted one‐fifth of the total dataset containing drug and nondrug target samples as the external test dataset. The remaining data were then used for 5‐fold cross‐validation. For each fold cross‐validation, we trained 10 models with batch sizes from 1 to 10, and the optimal model was then selected based on the one with a minimum loss of validation. Accuracy and other reported indices are the average values of 5‐fold cross‐validation with the standard deviation. The results of the external test were derived by testing the 5‐fold models on the same external test dataset.
To ensure that the numbers of positive and negative samples were comparable, we randomly extracted 65%, 55%, 70%, and 100% of proteins from the four nondrug target datasets, Pfam, Evalue0.001, Evalue1, Evalue10, respectively, and then analyzed these subsets with the all drug target dataset. We also randomly extracted 25%, 25%, 30%, and 50% of proteins from the four nondrug target datasets, Pfam, Evalue0.001, Evalue1, Evalue10, respectively, and then analyzed these subsets with the FDA‐approved drug target dataset. Results from 5‐fold cross‐validation and tests on external datasets were consistent with those described above.
4.6. GO analysis
Functional enrichment analysis was performed using the function enrichGO in the R package clusterProfiler, with the items of biological process (Yu et al., 2012). In addition, genome‐wide annotation org.Hs.eg.db for H. sapiens was used. All enrichment results were filtered with an adjusted p‐value <0.05.
4.7. Extraction of residue‐binding weights based on Grad–Cam
The output t of the classifier was back propagated to the last layer of the GCN. The obtained gradients were then used to calculate the importance of each filter in the layer, and the weight α was obtained. Weighted summations were performed for filter data of each feature layer G by α. Finally, a matrix representing important sites with the same length as the protein was obtained by the ReLU activation function:
where G represents the feature of the convolutional network layer output of the last graph, t represents the category of the target (1 for drug target, and 0 for nontarget), k represents the kth filter, and represents the weight on . We then calculated as follows:
where is the score of the drug target identified by the classifier, and represents the parameter of the ith residue in the kth filter.
4.8. Experimentally confirmed drug molecule‐binding sites
Experimentally confirmed drug molecule‐binding sites were collected from the BioLiP database (Yang et al., 2013). We integrated all sites that bind to known drug molecules. The BioLiP database, updated as of January 5, 2022, contains 1571 proteins and 6089 drug molecules that overlap with our drug target protein dataset. For polymeric proteins with multiple chains, we showed the 3D structure of binding between the A‐chain and drug molecules.
AUTHOR CONTRIBUTIONS
Jiaxiao Chen: Data curation (lead); formal analysis (lead); investigation (lead); methodology (lead); visualization (lead); writing – original draft (lead). Zhonghui Gu: Resources (supporting); software (supporting); validation (supporting). Youjun Xu: Methodology (supporting); resources (supporting). Minghua Deng: Supervision (equal). Luhua Lai: Project administration (equal); supervision (equal); writing – review and editing (equal). Jianfeng Pei: Conceptualization (lead); methodology (supporting); project administration (lead); supervision (equal); writing – review and editing (equal).
CONFLICT OF INTEREST
The authors declare no competing interests.
Supporting information
Data S1 Supporting information
Data S2 Supporting information
ACKNOWLEDGMENT
This work has been supported in part by the National Natural Science Foundation of China (22033001) and the Chinese Academy of Medical Sciences (2021‐I2M‐5‐014).
Chen J, Gu Z, Xu Y, Deng M, Lai L, Pei J. QuoteTarget: A sequence‐based transformer protein language model to identify potentially druggable protein targets. Protein Science. 2023;32(2):e4555. 10.1002/pro.4555
Review Editor: Nir Ben‐Tal
Funding information Chinese Academy of Medical Sciences, Grant/Award Number: 2021‐I2M‐5‐014; National Natural Science Foundation of China, Grant/Award Number: 22033001
DATA AVAILABILITY STATEMENT
The entire model is available at https://github.com/Chenjxjx/drug-target-prediction.
REFERENCES
- Alfaro JA, Bohländer P, Dai M, et al. The emerging landscape of single‐molecule protein sequencing technologies. Nat Methods. 2021;18:604–17. 10.1038/s41592-021-01143-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutet E, Lieberherr D, Tognolli M, Schneider M, Bairoch A. UniProtKB/swiss‐Prot. Methods Mol Biol. 2007;406:89–112. 10.1007/978-1-59745-535-0_4 [DOI] [PubMed] [Google Scholar]
- Chu X, Sun T, Li Q, et al. Prediction of liquid‐liquid phase separating proteins using machine learning. BMC Bioinform. 2022;23:72. 10.1186/s12859-022-04599-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ. 2016;47:20–33. 10.1016/j.jhealeco.2016.01.012 [DOI] [PubMed] [Google Scholar]
- Fu L, Niu B, Zhu Z, Wu S, Li W. CD‐HIT: accelerated for clustering the next‐generation sequencing data. Bioinformatics. 2012;28:3150–2. 10.1093/bioinformatics/bts565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gashaw I, Ellinghaus P, Sommer A, Asadullah K. What makes a good drug target? Drug Discov Today. 2011;16:1037–43. 10.1016/j.drudis.2011.09.007 [DOI] [PubMed] [Google Scholar]
- Gligorijević V, Renfrew PD, Kosciolek T, et al. Structure‐based protein function prediction using graph convolutional networks. Nat Commun. 2021;12:3168. 10.1038/s41467-021-23303-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Wu J, Ma H, Huang J. Self‐supervised pre‐training for protein embeddings using tertiary structures. Proc AAAI Conf Artif Intelligence. 2022;36:6801–9. 10.1609/aaai.v36i6.20636 [DOI] [Google Scholar]
- Hussein HA, Borrel A, Geneix C, Petitjean M, Regad L, Camproux AC. PockDrug‐server: a new web server for predicting pocket druggability on holo and apo proteins. Nucleic Acids Res. 2015;43:W436–42. 10.1093/nar/gkv462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamali AA, Ferdousi R, Razzaghi S, Li J, Safdari R, Ebrahimie E. DrugMiner: comparative analysis of machine learning algorithms for prediction of potential druggable proteins. Drug Discov Today. 2016;21:718–24. 10.1016/j.drudis.2016.01.007 [DOI] [PubMed] [Google Scholar]
- Jing B, Eismann S, Suriana P, Townshend RJL, Dror RO. Learning from protein structure with geometric vector Perceptrons. ArXiv. 2021;2019.01411. [Google Scholar]
- Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9. 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox C, Law V, Jewison T, et al. DrugBank 3.0: a comprehensive resource for 'omics' research on drugs. Nucleic Acids Res. 2011;39:D1035–41. 10.1093/nar/gkq1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulmanov M, Hoehndorf R. DeepGOPlus: improved protein function prediction from sequence. Bioinformatics. 2021;37:1187. 10.1093/bioinformatics/btaa763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guilloux V, Schmidtke P, Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics. 2009;10:168. 10.1186/1471-2105-10-168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Lai L. Prediction of potential drug targets based on simple sequence properties. BMC Bioinform. 2007;8:353. 10.1186/1471-2105-8-353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Chen H, Li S, Liu Y, Li X, Yu B. Accurate prediction of potential druggable proteins based on genetic algorithm and bagging‐SVM ensemble classifier. Artif Intell Med. 2019;98:35–47. 10.1016/j.artmed.2019.07.005 [DOI] [PubMed] [Google Scholar]
- Liu T, Altman RB. Identifying druggable targets by protein microenvironments matching: application to transcription factors. CPT Pharmacometrics Syst Pharmacol. 2014;3:e93. 10.1038/psp.2013.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikolov T, Chen K, Corrado G, Dean J. Efficient estimation of word representations in vector space. arXiv. 2013;1301.3781. [Google Scholar]
- Mistry J, Chuguransky S, Williams L, et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021;49:D412–d419. 10.1093/nar/gkaa913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480–d489. 10.1093/nar/gkaa1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens J. Determining druggability. Nat Rev Drug Discov. 2007;6:187–7. 10.1038/nrd2275 [DOI] [Google Scholar]
- Paul SM, Mytelka DS, Dunwiddie CT, et al. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat Rev Drug Discov. 2010;9:203–14. 10.1038/nrd3078 [DOI] [PubMed] [Google Scholar]
- Qin J, Chai G, Brewer JM, Lovelace LL, Lebioda L. Structures of asymmetric complexes of human neuron specific enolase with resolved substrate and product and an analogous complex with two inhibitors indicate subunit interaction and inhibitor cooperativity. J Inorg Biochem. 2012;111:187–94. 10.1016/j.jinorgbio.2012.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R, Bhattacharya N, Thomas N, et al. Evaluating protein transfer learning with TAPE. Adv Neural Inf Process Syst. 2019;32:9689–701. [PMC free article] [PubMed] [Google Scholar]
- Rao R, Liu J, Verkuil R, et al. MSA transformer. PMLR. 2021;139:8844–56. [Google Scholar]
- Rives A, Meier J, Sercu T, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc Natl Acad Sci U S A. 2021;118(15):e2016239118. 10.1073/pnas.2016239118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong X. word2vec parameter learning explained. arXiv. 2014;1411.2738. [Google Scholar]
- Saar KL, Morgunov AS, Qi R, et al. Learning the molecular grammar of protein condensates from sequence determinants and embeddings. Proc Natl Acad Sci U S A. 2021;118(15)e2019053118. 10.1073/pnas.2019053118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannell JW, Blanckley A, Boldon H, Warrington B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov. 2012;11:191–200. 10.1038/nrd3681 [DOI] [PubMed] [Google Scholar]
- Selvaraju RR, Cogswell M, das A, Vedantam R, Parikh D, Batra D. Grad‐CAM: visual explanations from deep networks via gradient‐based localization. Int J Comput Vision. 2020;128:336–59. 10.1007/s11263-019-01228-7 [DOI] [Google Scholar]
- Shen W, Le S, Li Y, Hu F. SeqKit: a cross‐platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One. 2016;11:e0163962. 10.1371/journal.pone.0163962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Lai L, Pei J. Analysis of protein features and machine learning algorithms for prediction of druggable proteins. Quant Biol. 2018;6:334–43. 10.1007/s40484-018-0157-2 [DOI] [Google Scholar]
- Swaminathan J, Boulgakov AA, Hernandez ET, et al. Highly parallel single‐molecule identification of proteins in zeptomole‐scale mixtures. Nat Biotechnol. 2018;36:1076–82. 10.1038/nbt.4278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangudu RR, Bryant SH, Panchenko AR, Madej T. Modulating protein‐protein interactions with small molecules: the importance of binding hotspots. J Mol Biol. 2012;415:443–53. 10.1016/j.jmb.2011.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W, Chen C, Lei X, Zhao J, Liang J. CASTp 3.0: computed atlas of surface topography of proteins . Nucleic Acids Res. 2018;46:W363–w367. 10.1093/nar/gky473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkamer A, Griewel A, Grombacher T, Rarey M. Analyzing the topology of active sites: on the prediction of pockets and subpockets. J Chem Inf Model. 2010;50:2041–52. 10.1021/ci100241y [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang S, Li F, et al. Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 2020;48:D1031–d1041. 10.1093/nar/gkz981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–d1082. 10.1093/nar/gkx1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters OJ, McKee M, Luyten J. Estimated Research and Development investment needed to bring a new medicine to market, 2009‐2018. JAMA. 2020;323:844–53. 10.1001/jama.2020.1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Pan S, Chen F, Long G, Zhang C, Yu PS. A comprehensive survey on graph neural networks. IEEE Trans Neural Netw Learn Syst. 2021;32:4–24. 10.1109/tnnls.2020.2978386 [DOI] [PubMed] [Google Scholar]
- Xu Y, Wang S, Hu Q, et al. CavityPlus: a web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018;46:W374–w379. 10.1093/nar/gky380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Roy A, Zhang Y. BioLiP: a semi‐manually curated database for biologically relevant ligand‐protein interactions. Nucleic Acids Res. 2013a;41:D1096–103. 10.1093/nar/gks966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L‐G, Han Y, He Q‐Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. J Integr Biol. 2012;16:284–7. 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Xue L, Liu F, Li Y, Jing R, Luo J. The applications of deep learning algorithms on in silico druggable proteins identification. J Adv Res. 2022;41:219–31. 10.1016/j.jare.2022.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Chen L, Zhong F, et al. Graph neural network approaches for drug‐target interactions. Curr Opin Struct Biol. 2022a;73:102327. 10.1016/j.sbi.2021.102327 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Xu M, Jamasb A, et al. Protein representation learning by geometric structure pretraining. ArXiv. 2022b;2203.06125. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Supporting information
Data S2 Supporting information
Data Availability Statement
The entire model is available at https://github.com/Chenjxjx/drug-target-prediction.