

Abstract
At the cellular level cardiac alternans is observed as beat-to-beat alternations in contraction strength, action potential (AP) morphology and Ca2+ transient (CaT) amplitude and represents a risk factor for cardiac arrhythmia. The (patho)physiological roles of small conductance Ca2+-activated K+ (SK) channels in ventricle are poorly understood. We tested the hypothesis that in single rabbit ventricular myocytes pharmacological modulation of SK channels plays a causative role for the development of pacing-induced CaT and AP duration (APD) alternans. SK channel blockers (apamin, UCL1684) had only a minor effect on AP repolarization. However, SK channel activation by NS309 resulted in significant APD shortening, demonstrating that functional SK channels are well expressed in ventricular myocytes. Effects of NS309 were prevented or reversed by apamin and UCL1684, indicating that NS309 acted on SK channels. SK channel activation abolished or reduced the degree of pacing induced CaT and APD alternans. Inhibition of KV7.1 (with HMR1556) and KV11.1 (with E4031) channels was used to mimic conditions of long QT syndromes type-1 and type-2, respectively. Both HMR1556 and E4031 enhanced CaT alternans that was prevented by SK channel activation. In AP voltage-clamped cells the SK channel activator had no effect on CaT alternans, confirming that suppression of CaT alternans was caused by APD shortening. APD shortening contributes to protection from alternans by lowering sarcoplasmic reticulum Ca2+ content and curtailing Ca2+ release. The data suggest SK activation as a potential intervention to avert development of alternans with important ramifications for arrhythmia prevention and therapy for patients with LQT syndrome.
Graphical Abstract
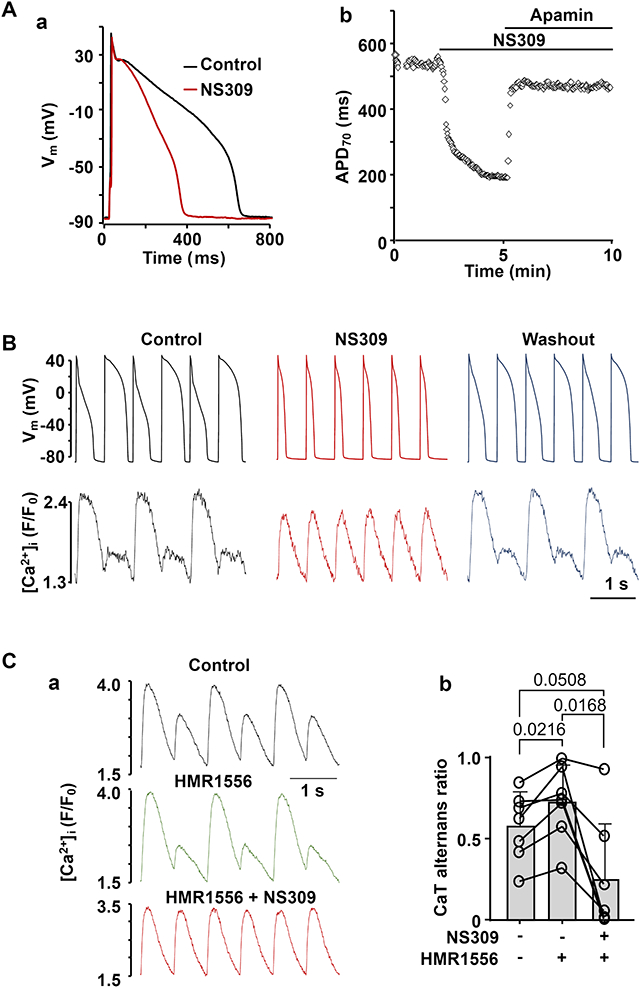
SK channel activation shortens ventricular action potentials (APs) and abolishes CaT and AP duration (APD) alternans. A: (a) activation of SK channels by NS309 (2 μm) leads to APD shortening. (b) APD70 recorded from an individual ventricular myocyte during the course of an experiment where APD shortening caused by NS309 was eliminated by application of apamin (100 nm), demonstrating that effect of NS309 is elicited due to activation of SK channels. B: AP and CaT traces simultaneously recorded from the same current-clamped ventricular myocyte in control, in presence of SK channel activator NS309 (2 μm) and after washout of NS309. Activation of SK channels abolishes CaT and APD alternans. C: Activation of SK channels reduced risk of alternans in ventricular cells with drug-induced LQTS. (a) CaT alternans observed in control and in the presence of KV7.1 channel blocker HMR1556 (1 μm), subsequently were abolished by NS309 application in the presence of HMR1556. (b) Mean and individual cell CaT alternans ratios recorded from field stimulated ventricular myocytes in control, in the presence of HMR1556, followed by simultaneous application of HMR1556 and NS309.
Introduction
Cardiac alternans is observed as beat-to-beat alternation in contraction strength (mechanical alternans), cytosolic Ca2+ transient (CaT) amplitude (CaT alternans) and action potential duration (APD or electrical alternans) at constant beating frequency. Clinically, alternans has been identified as a significant risk factor for atrial as well as ventricular arrhythmia with clinical manifestations such as pulsus alternans and repolarization or T-wave alternans in the ECG (Walker & Rosenbaum, 2003). AP repolarization is governed by the time-dependent activity of various membrane K+ channels. Disturbances in K+ conductances have been linked to electro-mechanical alternans (Fossa et al., 2004; Ni et al., 2019). In a previous study we have shown that AP shortening induced by pharmacological activators of two K+ channels (KV7.1 and KV11.1) abolished both APD and CaT alternans in field-stimulated and current-clamped myocytes and activation of KV11.1 channels attenuated atrial T-wave alternans in isolated Langendorff perfused hearts (Kanaporis et al., 2019). The goal of the current study was to test whether activation of small conductance Ca2+-activated K+ (SK) channels could reduce the risk for pro-arrhythmic cardiac alternans. SK channels are expressed in both atria (Xu et al., 2003; Tuteja et al., 2005; Skibsbye et al., 2014) and ventricles (Haugaard et al., 2015; Weisbrod, 2020), including in humans (Xu et al., 2003; Skibsbye et al., 2014). However, physiological and pathophysiological roles of SK channels are poorly understood. In atria SK channels were shown to contribute to AP repolarization (Skibsbye et al., 2014; Diness et al., 2020) and have been linked to atrial fibrillation (Skibsbye et al., 2014; Haugaard et al., 2015; Diness et al., 2020). In contrast to atria, several studies have shown no or little effect of SK channel inhibition on ventricular repolarization (Xu et al., 2003; Nagy et al., 2009; Chen et al., 2018). However, it was suggested that in ventricle expression of SK channels increases in pathologies associated with elevated cytosolic [Ca2+]i ([Ca2+]i), such as heart failure (Chang et al., 2013; Weisbrod, 2020), and that SK channels may play a protective role by increasing repolarization reserve (Hsueh et al., 2013). Activation of SK channels was shown to prevent ventricular arrhythmias in hypertrophy (Kim et al., 2017), while block of SK channels is arrhythmogenic in ventricle (Chang et al., 2013; Chan et al., 2015) and atrium (Hsueh et al., 2013), but has also been shown to reduce the propensity for arrhythmias during muscarinic and β-adrenergic stimulation in rat atria (Skibsbye et al., 2018). In addition, block of SK channels was suggested to contribute to acquired long QT syndrome (LQTS) in murine hearts pretreated with the late Na+ current enhancer ATX-II (Ko et al., 2018). Cardiac repolarization alternans are typically observed in patients with LQTS (Takasugi et al., 2016; Gadage, 2018) and in experimental LQTS studies (Nemec et al., 2010; Liu et al., 2018). APD prolongation is one of the most frequent electrical cardiac disorders. Mutations in 17 genes have been linked to congenital LQTS, while acquired LQTS results from adverse effects of a wide range of medications and cardiac pathologies (Goldenberg et al., 2008; Perez-Riera et al., 2018). LQTS carries a high arrhythmia risk. In LQTS sarcoplasmic reticulum (SR) Ca2+ overload (Lindegger et al., 2009; Qi et al., 2009; Kim et al., 2015), increased activity of the ryanodine receptor (RyR) SR Ca2+ release channel (Terentyev et al., 2014; Frommeyer et al., 2018), higher propensity of spontaneous Ca2+ release events and waves (Terentyev et al., 2014; Kim et al., 2015; Wilson et al., 2017) were suggested to trigger early and delayed afterdepolarizations (Jost et al., 2013; Zhong et al., 2018). Current therapies, involving administration of beta blockers, cardiac sympathetic denervation surgery and cardioverter-defibrillator implantation, significantly reduce the risk of lethal arrhythmias. However, treatments that directly restore normal AP morphology currently are unavailable.
Our previous studies have demonstrated that AP duration plays an important role in modulating the onset and degree of APD and CaT alternans (Kanaporis & Blatter, 2017b; Kanaporis et al., 2019). Here we investigate how modulation of SK channel activity affects the risk for CaT and APD alternans in ventricular myocytes. While SK channel blockers have just a minor effect on APD in rabbit ventricular myocytes, we demonstrate that pharmacological activation of SK channels shortened the AP and suppressed or completely eliminated CaT and APD alternans in single ventricular myocytes. In addition, activation of SK channels prevented CaT alternans in cells with pharmacologically induced APD prolongation mimicking LQTS. Therefore, we suggest that SK channel activation could serve as a potential intervention to avert development of alternans with important ramifications for arrhythmia prevention and therapy for patients with LQTS.
Methods
Ethical approval
All procedures and protocols were approved by the Institutional Animal Care and Use Committee of Rush University Chicago and comply with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All the experiments of the present study also comply with the policy and regulations on animal experimentation of The Journal of Physiology (Grundy, 2015). The animals were housed in dedicated animal housing facility with a maintained light/dark cycle of 12/12h. Animals were provided with a constant access to drinking water and were fed ad libitum.
Myocyte isolation
Ventricular myocytes were isolated from male New Zealand White rabbits (2.5-2.9 kg; 51 rabbits; Envigo, Indianapolis, IN). Rabbits were anaesthetized with an intravenous injection of sodium pentobarbital (100 mg/kg), together with heparin (1000 UI/kg). The depth of the anesthesia was evaluated by foot pinch or checking corneal reflexes. Hearts were excised, mounted on a Langendorff apparatus and retrogradely perfused via the aorta. After an initial 5-10 min perfusion with oxygenated Ca2+-free Tyrode solution (in mM: 140 NaCl, 4 KCl, 10 D-Glucose, 5 Hepes, 1 MgCl2, 1000 UI/l Heparin; pH 7.4 with NaOH), the heart was perfused with minimal essential medium Eagle (MEM) solution containing 20 μM Ca2+ and 22.5 μg/ml Liberase TH (Roche Diagnostic Corporation, Indianapolis, IN, USA) for ~20 min at 37° C. The left ventricle was dissected from the heart and minced, filtered and washed in MEM solution containing 50 μM Ca2+ and 10 mg/ml bovine serum albumin. Isolated cells were washed and kept in MEM solution with 50 μM Ca2+ at room temperature (20-24°C) and were used within 1-8 h after isolation.
Patch clamp experiments
The external Tyrode solution was composed of (in mM): 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 Hepes, 10 D-glucose; pH 7.4 with NaOH. All chemicals and reagents were from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise stated. Patch clamp pipettes (1.5-3 MΩ filled with internal solution) were pulled from borosilicate glass capillaries (WPI, Sarasota, FL, USA) with a horizontal puller (model P-97; Sutter Instruments, Novato, CA, USA). For all voltage- and current-clamp experiments pipettes were filled with an internal solution containing (in mM): 130 K+ glutamate, 10 NaCl, 10 KCl, 0.33 MgCl2, 4 MgATP, and 10 Hepes with pH adjusted to 7.2 with KOH. For simultaneous cytosolic [Ca2+]i measurements 100 μM Fluo-4 pentapotassium salt (Thermo Fisher Scientific, Waltham MA, USA) was added to the internal solution. The internal solution was filtered through 0.22-μm pore filters. Electrophysiological signals were recorded from single ventricular myocytes in the whole-cell ruptured patch clamp configuration using an Axopatch 200A patch clamp amplifier, the Axon Digidata 1440A interface and pCLAMP 10.7 software (Molecular Devices, Sunnyvale, CA, USA). AP recordings were low-pass filtered at 5 kHz and digitized at 10 kHz. All patch clamp experiments were performed at room temperature (20–24°C).
For AP measurements the whole-cell 'fast' current clamp mode of the Axopatch 200A was used and APs were evoked by 4 ms stimulation pulses with a magnitude ~1.5 times higher than AP activation threshold. Membrane potential (Vm) measurements were corrected for a junction potential error of −10 mV. APD alternans were induced by increasing the stimulation frequency until stable alternans was observed. To quantify the degree of APD alternans we calculated ratios of the integral (area under the curve) of AP recordings, and ratios of APD50, APD70 and APD90 of pairs of alternating APs. For AP-clamp experiments a voltage command in form of a ventricular AP was generated from previously recorded ventricular APs at a pacing frequency of 1.3 Hz (Kanaporis & Blatter, 2015). In AP-clamp experiments the stimulation frequency was varied by changing the diastolic interval between AP voltage commands.
[Ca2+] measurements
For [Ca2+]i measurements in field stimulation experiments ventricular myocytes were loaded with 5 μM Cal520/AM (AAT Bioquest, Sunnyvale, CA, USA) or 5 μM Fluo-4/AM in the presence of 0.05% Pluronic F-127 (Thermo Fisher Scientific, Waltham MA, USA) for 20-30 min at room temperature, and then twice washed for 10 min in Tyrode solution to allow for de-esterification of the dye. For field stimulation experiments, glass coverslips were coated with 1 mg/ml laminin to increase adhesion of the cells. CaTs were triggered by electrical field stimulation with a pair of platinum electrodes. The electrical stimulus was set at a voltage ~50% greater than the threshold to induce myocyte SR Ca2+ release. During the course of experiments cells were continuously superfused with Tyrode solution. Fluo-4 or Cal520 fluorescence was excited at 485 nm with a Xe arc lamp and signals were collected at 515 nm using a photomultiplier tube. Background-subtracted fluorescence emission signals (F) were normalized to resting fluorescence (F0) recorded under steady-state conditions at the beginning of an experiment, and changes of [Ca2+]i are presented as changes of F/F0. Data recording and digitization were achieved using the Axon Digidata 1440A interface and pCLAMP 10.7 software. Fluorescence signals were low-pass filtered at 30 Hz.
For SR [Ca2+] ([Ca2+]SR) measurements ventricular myocytes were loaded with 5 μM Cal520N/AM (AAT Bioquest, Sunnyvale, CA, USA) for 30 min at room temperature in the presence of 0.05% Pluronic F-127. Cal520N fluorescence was excited at 485 nm with a Xe arc lamp and signals were collected at 515 nm using a photomultiplier tube. Background-subtracted fluorescence emission signals (F) were normalized to diastolic fluorescence (F0) recorded in field stimulated cells at 0.5 Hz. Changes of [Ca2+]SR are presented as changes of F/F0. Fluorescence signals were low-pass filtered at 30 Hz.
As an alternative method to assess SR Ca2+ content, SR Ca2+ release was induced with rapid application of caffeine (10 mM) and the amplitude of the caffeine-induced cytosolic CaT was used to quantify [Ca2+]SR. Cells were paced at 0.5 Hz for at least 2 min before adding caffeine to ensure consistent SR Ca2+ loading.
CaT alternans
CaT alternans was induced by incrementally increasing the pacing frequency until stable alternans was observed (typical range where stable CaT were observed was 1.6 - 2.5 Hz). The degree of CaT alternans was quantified as the alternans ratio (AR). AR = 1 - [Ca2+]i,Small/[Ca2+]i,Large, where [Ca2+]i,Large and [Ca2+]i,Small are the amplitudes of the large and small CaTs of a pair of alternating CaTs. By this definition AR values fall between 0 and 1, where AR=0 indicates no CaT alternans and AR=1 indicates a situation where SR Ca2+ release is completely abolished on every other beat. CaTs were considered alternating when the beat-to-beat difference in CaT amplitude exceeded 10% (AR >0.1) (Kanaporis & Blatter, 2015). The amplitude of a CaT was measured as the difference in F/F0 (ΔF/F0) measured immediately before the stimulation pulse and at the peak of the CaT.
Drugs
10 mM NS309 (Cayman Chemical, Ann Harbor, MI, USA), 5 mM UCL1684 (Tocris/Bio-Techne, Minneapolis, MN, USA), and 5 mM HMR1556 (Sigma-Aldrich, St. Louis, MO, USA) stock solutions were prepared in DMSO. 1 mM stock solution of apamin (Alomone Labs, Jerusalem, Israel)) and 1 mM stock of E4031 hydrochloride (Cayman Chemical, Ann Harbor, MI, USA) was prepared in deionized water. Stock solutions were diluted to final concentration in external solutions. Corresponding amount of DMSO was added to control solution (final DMSO concentration ≤0.06%).
Data analysis and presentation
Results are presented as individual observations or as mean ± SD, n represents the number of individual cells and N is the number of animals. Data was collected in paired manner. Statistical significance was evaluated using one-way ANOVA with Tukey’s test for comparison of multiple groups or paired Student’s t test when two groups were compared. Wilcoxon nonparametric test was used to compare AP integral, APD50, APD70 and APD90 ratios (Fig. 4B). Dunn’s multiple group nonparametric comparison test was used for analysis of diastolic [Ca2+]SR data. Grubb’s (α=0.05) and ROUT statistical tests were applied to APD data sets to identify possible outliers. As a result, one cell with abnormally long APD was identified by both tests as an outlier and was removed from the further analysis. Statistical analysis was performed with GraphPad Prism 9.0 (San Diego, CA, USA). Differences were considered significant at P<0.05.
Fig. 4. Activation of SK channels abolishes APD alternans.

A: AP and CaT traces simultaneously recorded from the same current-clamped ventricular myocyte in control, in presence of SK channel activator NS309 (2 μM) and after washout of NS309.
B: Mean and individual ratios of AP integrals (a), and ratios of APD50 (b), APD70 (c) and APD90 (d) of pairs of alternating APs in control and in the presence of NS309 (n/N=9/5). Dashed line indicates ratio of 1 when there is no alternans. Statistical analysis was performed paired Wilcoxon nonparametric test.
Results
Effect of SK activation and inhibition on ventricular AP repolarization
The physiological function of SK channels in the healthy ventricle is poorly understood, and several studies have reported that block of SK channels has little or no effect on ventricular repolarization (Xu et al., 2003; Nagy et al., 2009; Chen et al., 2018). Therefore, to determine the effect of SK channel activation on ventricular repolarization at the cellular level, APs were measured from current-clamped rabbit ventricular myocytes paced at 0.5 Hz in control and in the presence of SK channel activator NS309 (2 μM). Our data show that application of NS309 led to a pronounced APD shortening (Fig. 1A). APD was quantified at 50, 70 and 90% repolarization levels (Fig. 1B). Activation of SK channels shortened APD50 from 447 ± 78 ms in control to 240 ± 88 ms in the presence of NS309 (n/N=14/5, P<0.0001; paired t-test). Similarly, APD70 shortened from 562 ± 73 to 298 ± 96 ms after SK activation (P<0.0001) and APD90 decreased from 595 ± 76 to 328 ± 95 ms (P<0.0001). NS309 had no significant effect on resting Vm (−86.9 ± 4.9 mV in control; −87.3 ± 5.4 mV in NS309; n/N=14/5, P=0.3208; paired t-test) or AP amplitude (136.8 ± 4.3 mV in control; 136.5 ± 4.2 mV in NS309; n/N=14/5, P=0.6402).
Fig. 1. Effect of SK channel activation on APD.

A: Overlay of APs recorded from the same ventricular myocyte in control and after application of SK channel activator NS309 (2 μM).
B: Individual cell measurements and average APD50, APD70 and APD90 values observed in control and after application of NS309 (n/N=14/5; paired t test).
In addition to SK channel activation, we also investigated the effect of SK channel blockers on APD of ventricular myocytes (Fig. 2). To block SK channels two different inhibitors were used: apamin (100 nM) and UCL1684 (1 μM). Application of SK channel blockers produced only a minor increase in APD. The most pronounced APD prolongation was observed for APD50 however the effect reached statistical significance only for UCL1684 (Fig. 2C; n/N=6/3; P=0.0454). Changes in APD70 and APD90 were not statistically significant. Block of SK channels had no significant effect on resting Vm (−88.1 ± 1.8 mV in control; −88.3 ± 2.2 mV in apamin; n/N=8/2, P=0.4015; and resting Vm was −86.2 ± 5.2 mV before and −86.4 ± 6.0 mV after application of UCL1684; n/N=6/3, P=0.7394; paired t-test). Inhibition of SK channels also had no significant effect on AP amplitude (138.5 ± 1.5 mV before and 138.4 ± 1.1 mV after application of apamin; n/N=8/2, P=0.8750; 136.8 ± 4.1 mV before and 137.2 ± 6.3 mV after application of UCL1684; n/N=6/3, P=0.8001; paired t-test). Overall, these observations in rabbit ventricular myocytes are consistent with previous studies suggesting that under normal physiological conditions SK channels play no or only a minor role in ventricular repolarization (Xu et al., 2003; Nagy et al., 2009; Chen et al., 2018). In contrast, pharmacological activation of SK channels elicited a pronounced APD shortening (Fig. 1). These results indicate the presence of functional SK channels in ventricular myocytes, even though the majority of them appear not to be activated during normal ventricular excitation.
Fig. 2. Effect of SK channel blockers on APD.

A: (a) Overlay of APs recorded in control and after application of SK channel blocker apamin (100 nM). (b) Time course of changes of APD50 during application of apamin recorded from the same myocyte as in panel Aa.
B: APD50, APD70 and APD90 in control and in the presence of apamin (n/N=9/3).
C: APD50, APD70 and APD90 in control and in the presence of 1 μM UCL1684 (n/N=6/3).
Statistical analysis was performed using paired t test.
Activation of SK channels suppresses CaT and APD alternans
We tested the hypothesis that SK channel activation decreases CaT alternans susceptibility through shortening of the AP. Fig. 3A shows an example of CaT traces recorded from the same field stimulated ventricular cell in control, in the presence of the SK channel activator NS309 (2 μM) and after washout of NS309. The changes of CaT AR over the course of this experiment is shown in Fig. 3B. Application of NS309 consistently abolished or greatly reduced the degree of CaT alternans. On average SK channel activation reduced mean CaT AR from 0.58 ± 0.20 to 0.15 ± 0.19 (Fig. 3C; n/N=18/6; P<0.0001; ANOVA with Tukey’s multigroup comparison statistical test). Washout of NS309 was attempted in 9 cells which restored CaT alternans and increased AR on average to 0.56 ± 0.20 (P=0.0004).
Fig. 3. Activation of SK channels abolishes CaT alternans.

A: CaT traces recorded from the same ventricular myocyte in control, in presence of SK channel activator NS309 (2 μM) and after washout of NS309. CaT alternans was induced by increasing pacing frequency in field stimulated ventricular myocytes.
B: CaT AR monitored over time in control and during SK activation calculated from CaT data in panel A.
C: Mean and individual cell CaT AR values in control (Ctrl), during NS309 exposure (2 μM; n/N=18/6) and after washout in a subset of cells (n/N=9/3). Statistical analysis was performed using Tukey’s multiple group comparison test.
D: CaT ARs recorded in field stimulated ventricular myocytes in control and after application of SK channel blockers (a) apamin (n/N=6/2; paired t test) and (b) UCL1684 (n/N=11/3; paired t test).
Although SK channels blockers had only a minor effect on APD, we tested whether inhibition of SK channels affected the development of CaT alternans (Fig. 3D). Ventricular myocytes were field stimulated and pacing frequency was increased until stable CaT alternans was observed. Cells were then subjected to apamin (100 nM) or UCL1684 (1 μM). Both SK channel blockers had no statistically significant effect on the degree of CaT alternans. Mean CaT ARs observed in control was 0.58 ± 0.19 and 0.74 ± 0.10 after application of 100 nM apamin (Fig. 3Da, n/N=6/2, P=0.1387, paired t-test). Similarly, CaT ARs were 0.47 ± 0.15 before and 0.49 ± 0.25 after application of 1 μM UCL1684 (Fig. 3Db; n/N=11/3, P=0.7534, paired t-test). The results show that SK channel inhibition has no significant effect on the degree of CaT alternans and are consistent with the findings that inhibition of SK channels has only a minor effect on APD (Fig. 2).
Activation of SK channels by NS309 also abolished or reduced the degree of pacing induced APD alternans. Fig. 4A shows simultaneously recorded APs and CaTs in current-clamped myocyte in control, after application of NS309 and after NS309 wash out. To quantify AP alternans we calculated ratios of the AP integral, and ratios of APD50, APD70 and APD90 of pairs of alternating APs (Fig. 4B). NS309 reduced AP integral ratio from 1.41 ± 0.34 to 1.06 ± 0.09 (n/N=9/5; P=0.0391; paired Wilcoxon nonparametric test). APD50 ratio in NS309 decreased from 1.58 ± 0.64 to 1.15 ± 0.16 (P=0.0977); APD70 ratio decreased from 1.34 ± 0.21 to 1.06 ± 0.09 (P=0.0273); APD90 ratio: from 1.31 ± 0.20 to 1.05 ± 0.06 (P=0.0195). In six cells [Ca2+]i was monitored simultaneously with APs (Fig. 4A). In these cells the mean CaT AR in control was 0.92 ± 0.06 which in the presence of NS309 was reduced to 0.28 ± 0.25 (n=6/4; P=0.0009; paired t test).
Our data clearly demonstrates that activation of SK channels abolished pacing induced CaT and APD alternans in ventricular myocytes.
SK channel inhibition prevents APD shortening by SK activators and abolishes the effect of NS309 on CaT alternans
To determine whether the effects of NS309 were elicited selectively by the activation of SK channels, we performed a series of experiments where SK channel blockers were applied together with NS309. Application of apamin (100 nM) reverted NS309 induced APD shortening (Fig. 5A, n/N=5/3). Similarly, the effect of NS309 on APD was also strongly antagonized by UCL1684 (1 μM) (Fig. 5B; n/N=4/3). Our results show that inhibition of SK channels by two different SK channels blockers efficiently prevents NS309 induced APD shortening, demonstrating that NS309 indeed induces APD shortening by SK channel activation.
Fig. 5. Inhibition of SK channels prevents APD shortening in the presence of NS309.

A: (a) APD70 recorded from an individual ventricular myocyte during the course of an experiment where APD shortening caused by NS309 was eliminated by application of apamin (100 nM). (b) APD70 recorded in individual cells and mean APD70 in control, in the presence of NS309 and during simultaneous application of NS309 and apamin (n/N=5/3). Statistical analysis was performed by Tukey’s multiple group comparison test.
B: (a) APD70 recorded during the course of an experiment in a ventricular myocyte subjected to the SK channel blocker UCL1684 alone, UCL1684 together with NS309 and NS309 alone. Presence of UCL1684 eliminated the effect of NS309 on APD. (b) APD70 recorded in individual cells and mean APD70 in control, in the presence of UCL1684, during simultaneous application of NS309 and UCL1684 and NS309 alone (n/N=4/3; Tukey’s multiple group comparison test).
We also tested if SK channel inhibition could prevent the abolishment of CaT alternans by NS309. In the first series of experiments we induced CaT alternans by incrementally increasing pacing frequency. Once stable alternans was established, we recorded CaT alternans for at least 1 min under control conditions. Subsequently, cells were exposed to NS309 for 3-4 min, followed by simultaneous superfusion with NS309 and apamin (Fig. 6A). Mean CaT AR of 0.54 ± 0.16 in control decreased to 0.11 ± 0.15 in NS309 (n/N=9/4, P=0.0001; Tukey’s multiple groups comparison test), while subsequent application of apamin in the presence of NS309 restored CaT AR to 0.36 ± 0.34 which was not significantly different from control AR (P=0.1950). In the next series of experiments, after stable pacing-induced alternans was established, cells were first superfused with apamin and after 3-4 minutes were subjected simultaneously to apamin and NS309 (Fig. 6B). With this protocol mean CaT AR was 0.62 ± 0.20 in control, 0.73 ± 0.10 in apamin and 0.76 ± 0.15 in the presence of apamin + NS309 (n/N=5/2, no statistically significant differences between CaT ARs; Tukey’s multiple groups comparison test). These results show that in the presence of apamin, NS309 had no effect on CaT AR. This was distinctly different from the situation when NS309 was applied alone where it was very efficient in reducing the degree of CaT alternans (Figs.3 and 6A). We also performed experiments (Fig. 6C) using the alternative SK channel blocker UCL1684. Similar to apamin, in cells exhibiting pacing-induced alternans in control (AR = 0.58 ± 0.13, n/N=6/3), UCL1684 (AR = 0.68 ± 0.19), followed by simultaneous exposure to UCL1684 + NS309 (AR = 0.58 ± 0.24) NS309 had no statistically significant effect on AR. Subsequent removal of UCL1684 after 3-4 min, but in the maintained presence of NS309 led to a drastic decrease of CaT AR (AR = 0.14 ± 0.16; P=0.0049; Tukey’s multiple groups comparison test). Finally, washout of NS309 (5-6 min) restored CaT alternans to a similar degree as under control conditions (AR = 0.76 ± 0.20).
Fig. 6. Inhibition of SK channels abolishes effect of NS309 on CaT alternans.

A: CaT ARs (mean and individual cell data) recorded from the same field stimulated ventricular myocytes in control, and in the presence of NS309 followed by simultaneous application of NS309 and SK channel blocker apamin (n/N=9/4).
B: CaT ARs recorded from field stimulated ventricular myocytes in control, and in the presence of apamin, followed by simultaneous application of apamin and NS309 (n/N=5/2).
C: (a) CaTs recorded from the same field stimulated cell in control, in the presence of UCL1684, during simultaneous application of NS309 and UCL1684, in NS309 alone and after NS309 washout. (b) Mean and individual cell CaT ARs recorded using the same experimental protocol as in panel Ca (n/N=6/3).
All statistics using Tukey’s multiple group comparison test.
Increased risk for CaT alternans through APD prolongation is abolished by activation of SK channels
T-wave alternans is commonly observed in the patients with LQT syndrome (Takasugi et al., 2016; Gadage, 2018). Here we tested whether during simulated LQTS type-1 and type-2 the risk of CaT alternans is increased and whether activation of SK channels can rescue CaT alternans under these conditions. The KV7.1 blocker HMR1556 (1 μM) was used to simulate the most common manifestation of congenital LQTS type-1. HMR1556 (1 μM) caused a significant APD prolongation at 50, 70 and 90% repolarization (Figs. 7Aa and 7Ab; n/N=8/4). Fig. 7Ba shows an example of the time course of changes of APD70 in control, in the presence of HMR1556 and during SK channel activation with NS309 in the presence of HMR1556. In this series of experiments application of HMR1556 prolonged APD70 on average from 631 ± 112 to 713 ± 83 ms (Fig. 7Bb; n/N=6/3; P=0.0093), while subsequent application of NS309 in the presence of HMR1556 led to APD70 shortening to 428 ± 150 ms (P=0.0088). In addition, KV7.1 channel inhibition enhanced CaT alternans (Fig. 7C). In these experiments the field stimulation frequency was gradually increased until ventricular myocytes exhibited stable CaT alternans. After monitoring stable CaT alternans in control (mean CaT AR 0.42 ± 0.25) for at least 1 minute, cells were exposed to HMR1556 (1 μM) which increased CaT AR to 0.62 ± 0.26 (n/N =17/7; P =0.0029; paired t test). Furthermore, we tested if activation of SK channels during KV7.1 channel inhibition can reduce the degree of CaT alternans (Fig. 7Da). In field stimulated myocytes application of HMR1556 increased CaT AR from 0.58 ± 0.21 to 0.73 ± 0.23 (Fig. 7Db; n/N=7/3; P=0.0216). Application of NS309 during KV7.1 inhibition suppressed CaT alternans and reduced AR to 0.25 ± 0.35 (P=0.0168, Tukey’s multiple comparison test).
Fig. 7. Activation of SK channels reduces risk for CaT alternans in myocytes with simulated LQTS type-1 and type-2.

A: (a) Overlay of APs recorded from the same ventricular myocyte in control and after application of Kv7.1 blocker HMR1556 (1 μM). (b) Mean and individual cell APD50, APD70 and APD90 data in control and in the presence of HMR1556 (n/N=8/4; paired t test).
B: (a) Time course of APD70 changes during application of HMR1556 alone and simultaneously with NS309. (b) Mean and individual cell APD70 data recorded in control, in the presence of HMR1556, followed by simultaneous application of NS309 and HMR1556 (n/N=6/3).
C: Mean and individual cell CaT ARs observed in field stimulated ventricular myocytes in control and after application of HMR1556 (n/N = 17/7; paired t test).
D: (a) CaT alternans observed in control and in the presence of 1 μM HMR1556, subsequently abolished by NS309 application in the presence of HMR1556.
(b) Mean and individual cell CaT ARs recorded from field stimulated ventricular myocytes in control, in the presence of HMR1556, followed by simultaneous application of HMR1556 and NS309 (n/N=7/3, Tukey’s multiple group comparison test).
E: Mean and individual cell CaT ARs observed in control, in the presence of 1 μM E4031, and during simultaneous application of E4031 and NS309 (n/N=8/3).
In addition, we also investigated the effect of NS309 on CaT alternans in myocytes where LQTS type-2 was simulated by the application of KV11.1 channel blocker E4031 (1 μM) (Fig. 7E). E4031 increased CaT AR from 0.41 ± 0.30 to 0.64 ± 0.33 (n/N=8/3; P=0.0348). subsequent application of NS309 in the presence of E4031 reduced mean CaT AR to 0.20 ± 0.22 (P=0.0144).
Reduction of alternans risk by SK channel activation involves a decrease in SR Ca2+ load
Finally, we attempted to determine the mechanisms by which activation of SK channels reduces CaT alternans risk in ventricular myocytes. First, we tested if the observed changes in AP morphology after SK channel activation (Fig. 1) were responsible for the reduced degree of CaT alternans in the presence of NS309 (Fig. 3). For this purpose the beat-to-beat AP waveform was kept constant by applying an AP voltage-clamp protocol (Fig. 8Aa) which precludes any potential pharmacological manipulation of the AP. Under constant AP-clamp conditions application of NS309 failed to rescue CaT alternans (Fig. 8Ab; n/N=7/3). These results support the conclusion that NS309 prevents alternans through the changes in AP shape.
Fig. 8. Effect of SK channel activation on CaT alternans in AP voltage-clamped myocytes, CaT amplitude and SR Ca2+ load.

A: (a) CaT traces recorded in control and in the presence of NS309 in AP voltage-clamped myocytes (bottom trace: AP voltage commands). (b) Individual cell and mean CaT ARs recorded in AP-clamped ventricular myocytes in control and in the presence of NS309 (2 μM). Under AP-clamp conditions the SK channel activator had no effect on CaT AR (n/N=7/3; paired t test).
B: (a) Overlay of CaTs recorded from the same field stimulated (0.5 Hz; no alternans) ventricular myocyte in control (black) and in NS309 (red). (b) Individual cell and mean CaT amplitudes (ΔF/F0) before and during application of NS309 (n/N=10/3, paired t test).
C: (a) [Ca2+]SR traces recorded in a ventricular myocyte loaded with the low affinity Ca2+ dye Cal520N/AM and field stimulated at 0.5 Hz. Top trace shows changes of [Ca2+]SR over the course of the experiment in control, during application of 2 μM NS309 and during washout of the drug. Bottom: depletions of [Ca2+]SR recorded in control, in NS309 and during washout. (b) Individual and mean amplitudes of [Ca2+]SR depletions recorded in control, in NS309 and during washout (n/N=12/2, Tukey’s multiple group comparison test). (c) Individual and mean changes in diastolic [Ca2+]SR (n/N=12/2, Dunn’s multiple group nonparametric comparison test).
D: Amplitudes of caffeine (10 mM) induced CaTs before and during application of NS309 demonstrating reduced SR Ca2+ load in the presence of NS309 (n/N=10/3; paired t test).
Furthermore, we tested the effect of NS309 on CaT properties. Effects of NS309 (2 μM) on AP-induced CaT amplitude was tested in rabbit ventricular myocytes paced at 0.5 Hz (i.e. in the absence of CaT alternans). In control the CaT amplitude (ΔF/F0) was 2.45 ± 0.67 and decreased to 1.70 ± 0.53 (n/N=10/3; P=0.0002, paired t-test) after 3 min exposure to NS309 (Fig. 8B). We further explored whether the decrease in CaT amplitude was the result of reduced SR Ca2+ load. For [Ca2+]SR measurements Cal520N/AM loaded myocytes were field stimulated at 0.5 Hz (Fig. 8Ca). Application of NS309 resulted in lower diastolic [Ca2+]SR (Fig. 8Cc) and significantly reduced the amplitude of pacing induced [Ca2+]SR depletions (Fig. 8Cb). SR Ca2+ load was restored after washout of NS309. Alternatively, to assess SR Ca2+ load caffeine (10 mM) induced CaTs were monitored in a paired manner in control and after application of NS309. Summarized data is presented in Fig. 8D, demonstrating that SK activation results in lower SR Ca2+ content (caffeine induced CaT amplitude (ΔF/F) in control was 4.43 ± 0.97 and 3.30 ± 0.72 in NS309; n/N=10/3; P=0.0003, paired t test).
In summary we demonstrate that SK channel activation suppresses CaT alternans via changes in AP shape. Also, activation of SK channels leads to reduced SR Ca2+ load which acts as a contributing factor to the reduced alternans risk.
Discussion
In this study we investigated the effect of SK channel modulators on AP morphology and development of CaT and APD alternans in rabbit ventricular myocytes. The main findings are: (i) While inhibition of SK channels induced just a minor prolongation of ventricular APs, activation of SK channels led to substantial APD shortening; (ii) activation of SK channels suppressed pacing-induces CaT and APD alternans; (iii) the degree of CaT alternans was not affected by SK channel inhibition; (iv) block of KV7.1 and KV11.1 K+ channels, mimicking conditions observed in LQTS type-1 and LQTS type-2, increased the risk of CaT alternans which could be eliminated by activation of SK channels; (v) activation of SK channels suppressed CaT alternans due to changes in AP shape, with contribution from reduced SR Ca2+ load.
Activity of SK channels in ventricular myocytes
There are three subtypes of SK channels expressed in the heart (genes KCNN1, KCNN2 and KCNN3) (Tuteja et al., 2005; Zhang et al., 2021). SK channels have been mostly studied in the nervous system where their function has been attributed to control of neuronal excitability and AP firing rate through membrane hyperpolarization, with ramifications for synaptic plasticity, learning and memory and possible involvement in the pathogenesis of Parkinson's disease (Lam et al., 2013; Sun et al., 2020). In the heart SK channels are expressed in both atria (Xu et al., 2003; Tuteja et al., 2005; Skibsbye et al., 2014) and ventricles (Haugaard et al., 2015; Weisbrod, 2020), including in humans (Xu et al., 2003; Skibsbye et al., 2014). In the atria SK channels were shown to contribute to AP repolarization (Skibsbye et al., 2014; Diness et al., 2020) and have been linked to atrial fibrillation (Skibsbye et al., 2014; Haugaard et al., 2015; Diness et al., 2020). The functional role of SK channels in ventricle, however remains poorly understood. Several studies have failed to demonstrate changes in AP morphology during SK inhibition and, consequently, it was proposed that SK channels do not play a role in repolarization of healthy ventricles. However, expression of SK channels increases in pathologies associated with elevated [Ca2+]i, such as heart failure (Chang et al., 2013; Weisbrod, 2020), and SK channels may play a protective role by increasing repolarization reserve of the myocardium (Hsueh et al., 2013). Our data in rabbit ventricular myocytes fall in line with aforementioned observations as block of SK channels by apamin or UCL1684 induced only a minor prolongation of the AP (Fig. 2). The modest APD prolongations were most pronounced at the APD50 level, while we did not observe statistically significant changes at APD70 or APD90 levels.
Interestingly, in contrast to SK channel inhibition, pharmacological activation of SK channels by NS309 led to significant APD shortening (by ~46%, Fig. 1). These results clearly indicate that fully functional SK channels are well expressed in ventricular myocytes, even though just a small fraction of these channels appears to be activated during the physiological excitation-contraction coupling cycle. We further confirmed experimentally that NS309 effects are indeed elicited due to SK activation. For this purpose, we conducted a series of experiments where NS309 was applied to the cells together with selective SK blockers apamin or UCL1684. Both SK channel blockers almost completely eliminated the effect of NS309 on APD (Fig. 5) and on CaT alternans (Fig. 6) demonstrating that NS309 acted indeed through SK channel activation. We did not attempt to distinguish between SK channel subtypes because available pharmacological modulators of SK channels have limited selectivity towards different subtypes.
Cardiac alternans
Cardiac alternans is characterized by repolarization and Ca2+ signaling irregularities throughout the myocardium and is closely linked to the development of cardiac arrhythmias. It is well established that APD and CaT alternans typically coincide in time and space (Pruvot et al., 2004; Kanaporis & Blatter, 2015) and it is generally agreed that the bi-directional coupling between [Ca2+]i and Vm regulation plays a key role in the development of cardiac alternans. Vm directly affects voltage-dependent Ca2+ handling mechanisms such as Ca2+ entry through L-type Ca2+ channels and Ca2+ removal from the cells by Na+/Ca2+ exchange. On the other hand, [Ca2+]i dynamics modulate Vm through Ca2+-dependent ion currents and transporters. Computational (Nolasco & Dahlen, 1968; Tolkacheva et al., 2004; Jordan & Christini, 2007) and several experimental studies (Koller et al., 2005; Tolkacheva et al., 2006) have suggested that self-sustaining beat-to-beat oscillations of APD can occur if diastolic APD restitution has a steep slope (Jordan & Christini, 2007; Bayer et al., 2010). However other studies found only a weak relationship between experimentally determined APD restitution kinetics and inducibility of alternans (Saitoh et al., 1988; Kalb et al., 2004; Pruvot et al., 2004). Alternative to the Vm hypothesis, the Ca2+ alternans hypothesis proposes that alternans originates from a primary disturbance of [Ca2+]i regulation and causal mechanisms for CaT alternans involve beat-to-beat fluctuations in SR Ca2+ load (Diaz et al., 2004; Eisner et al., 2006; Nivala & Qu, 2012) and refractoriness of SR Ca2+ release (Shkryl et al., 2012; Lugo et al., 2014; Wang et al., 2014). Major support for Ca2+ driven alternans is provided by the observations that CaT alternans can still be induced in voltage-clamped cardiac myocytes (Chudin et al., 1999; Kanaporis & Blatter, 2015, 2017a) when Vm is kept constant on every beat, i.e. in the absence of APD alternans. In addition, beat-to-beat alternation in AP morphology was abolished when intracellular Ca2+ release was blocked (Kanaporis & Blatter, 2015), suggesting that electrical or APD alternans is a consequence rather than the cause of cardiac alternans.
K+ channels as potential targets for alternans and arrhythmia prevention
Irrespective of whether Vm or [Ca2+]i dysregulation plays the leading role in the initiation of alternans, we have demonstrated previously, with AP voltage-clamp experiments employing different combinations of AP waveforms, that AP morphology and beat-to-beat APD alternation are pivotal for development and sustainability of atrial alternans (Kanaporis & Blatter, 2015, 2017a; Kanaporis et al., 2019). We also demonstrated the crucial importance of K+ channels for the development of alternans, as pharmacological activation of KV7.1 and KV11.1 K+ channels in atrial myocytes reduced alternans susceptibility (Kanaporis et al., 2019). Here we demonstrate that activation of SK channels with NS309 effectively reduced the degree of pacing induced alternans in ventricular myocytes (Figs. 3, 4, 6 and 7). Simultaneous application of SK channel blockers (apamin, UCL1684) abolished the effect of NS309 confirming that prevention of alternans is achieved by SK channel activation (Fig. 6). Inhibition of SK channels alone had no significant effect on CaT AR (Fig. 3D) which is consistent with our observations that SK channel blockers resulted in just a minor change in APD morphology (Fig. 2).
Furthermore, we provide evidence that change in AP morphology is essential for abolishing CaT alternans. When ventricular myocytes were voltage-clamped and CaTs were triggered with AP-shaped voltage commands (in the absence of APD alternans), NS309 had no effect on the degree of pacing-induced CaT alternans (Fig. 8A). We propose that altered AP morphology due to SK channels activation leads to reduced magnitude of SR Ca2+ release (Fig. 8B) which results from lower SR Ca2+. The latter was confirmed by direct [Ca2+]SR measurements (Fig. 8C) and by the amplitudes of caffeine induced CaTs (Fig. 8D). As mentioned above, many factors are known to contribute to alternans development. We propose that a decrease in [Ca2+]SR constitutes one of the mechanisms resulting in the reduced risk for alternans during SK activation. Previously, we have also demonstrated that the shape of the AP affects kinetics and peak of L-type Ca2+ current (ICa,L). AP waveforms during SK activation have faster repolarization and lower Vm at AP plateau leading to larger ICa,L and thus increases efficiency of CaT triggering which may in turn also contribute to lower alternans risk (Kanaporis & Blatter, 2017b). Increase in peak ICa,L also might contribute to SR Ca2+ depletion.
Acquired LQT syndrome and SK channel activation
APD shortening, achieved by SK channel activation as demonstrated in this study in ventricular myocytes, may have complex consequences for arrhythmia risk. APD shortening itself could be arrhythmogenic by increasing the diastolic interval and potentially increasing the risk of premature beats. On the other hand AP shortening has the potential to normalize AP morphology under pathological conditions where APD is abnormally long. APD prolongation is one of the most common electrical cardiac disorders. Mutations in 17 genes have been linked to congenital LQTS, while acquired LQTS results from adverse effects of a wide range of medications and cardiac pathologies (Goldenberg et al., 2008; Antoniou et al., 2017; Perez-Riera et al., 2018). LQTS carries a high arrhythmia risk that was attributed to SR Ca2+ overload (Lindegger et al., 2009; Qi et al., 2009; Kim et al., 2015), increased activity of ryanodine receptors (Terentyev et al., 2014; Frommeyer et al., 2018), and higher propensity of spontaneous Ca2+ release events (Terentyev et al., 2014; Kim et al., 2015; Wilson et al., 2017) leading to early and delayed afterdepolarizations (Jost et al., 2013; Terentyev et al., 2014; Zhong et al., 2018). In addition, cardiac alternans is regularly observed in clinical (Takasugi et al., 2016; Gadage, 2018) and experimental LQTS studies (Nemec et al., 2010; Liu et al., 2018) and has been directly linked to the initiation of arrhythmias (Ziv et al., 2009; Liu et al., 2018). To mimic LQTS conditions we conducted experiments where we prolonged APD using the KV7.1 channel blocker HMR1556 and the KV11.1 channel blocker E4031. Loss of function mutations of KV7.1 channels result in LQTS type-1, the most common form of congenital LQTS, accounting for 40-50% of all congenital LQTS cases (Shimizu & Horie, 2011). As expected, the HMR1556 induced APD prolongation (Fig. 7A-B) was accompanied by an increased risk for CaT alternans (Fig. 7C-D). Under these conditions, activation of SK channels reduced both AP duration and the degree of CaT alternans. Block of KV11.1 channels results in LQTS type-2, which is the most common type of drug-induced LQTS. Similarly to HMR1556, application of E4031 increased the degree of CaT alternans that could be prevented by SK channel activation (Fig. 7E).
Limitations and future perspectives
Here we report that ventricular myocytes express functional SK channels. Activation of SK channels results in APD shortening and we show that SK channels may serve as a potential anti-arrhythmic target. However, our study has some limitations and raises new questions that would need be addressed in future studies. It was proposed that recruitment and activity of SK channels is increased by sympathetic simulation (Hamilton et al., 2020) which was not investigated here. Also, it was suggested that expression levels of SK channels in the heart is sex-dependent with females exhibiting higher SK channel expression (Chen et al., 2018). In this study only male rabbits were used. Therefore, it remains to be established how effects of SK channel activation on AP morphology and cardiac alternans depends on sex and sympathetic stimulation.
We demonstrate that activation of SK channels can reduce the risk of alternans in normal myocytes (Figs 3, 4 and 6) and in myocytes with drug-induced LQTS type-1 and type-2 (Fig. 7). Activation of SK channels by 2 μM NS309 induced substantial shortening of APD (Fig. 1). However, the extreme shortening of APD is potentially arrhythmogenic, therefore it would be important to establish the dosage of SK channel agonists at which they normalize APD during LQTS but do not lead to excessive APD shortening.
In this study all experiments were performed at room temperature (20–24°C). The pacing frequency threshold for the occurrence of alternans decreases with lower temperatures (Huser et al., 2000). By conducting experiments at room temperature, alternans could be studied in isolated myocytes with pacing protocols that were significantly less stressful, allowing for prolonged stimulation protocols and extended cell viability over time (Figs 2, 3, 5 and 7). Furthermore, kinetic limitations of fluorescent Ca2+ indicator dyes make reliable recording of high-frequency CaTs difficult at higher temperatures, and in the case of CaT alternans the small-amplitude CaT typically starts to fuse with the declining phase of the preceding large-amplitude transient and thus becomes difficult to detect and to be quantified. However, Ca2+ handling mechanisms and pathways are temperature-dependent, particularly ion channels showing faster kinetics at physiological temperatures. Thus, AP morphology during alternans might be affected differently at higher temperature. Nonetheless, previous studies found that electrophysiological properties of the myocardium that are potentially relevant for the development of alternans are not significantly different whether experiments were performed at 30° or 37°C (Myles et al., 2011), consistent with a study that found no mechanistic differences between alternans at 27° and 37°C (Pastore et al., 1999).
Conclusions
In this study we demonstrated that pharmacological manipulation of cardiac K+ channels provides a tool for correction of pathologically altered AP morphology that allows to minimize the risk of electrical and CaT alternans, and thus to reduce a potential arrhythmogenic substrate. This might be especially true for LQTS patients where activation of ventricular SK channels could serve as a potential strategy to reduce arrhythmia risk.
Supplementary Material
Key points.
At the cellular level cardiac alternans is observed as beat-to-beat alternations in contraction strength, action potential (AP) morphology and intracellular Ca2+ release amplitude and represents a risk factor for cardiac arrhythmia.
The (patho)physiological roles of small conductance Ca2+-activated K+ (SK) channels in ventricle are poorly understood.
We investigated if pharmacological modulation of SK channels affects the development of cardiac alternans in normal ventricular cells and in cells with drug-induced long QT syndrome (LQTS).
While SK channel blockers have only a minor effect on AP morphology, their activation leads to AP shortening and abolishes or reduces the degree of pacing induced Ca2+ and AP alternans. AP shortening contributed to protection against alternans by lowering sarcoplasmic reticulum Ca2+ content and curtailing Ca2+ release.
The data suggest SK activation as a potential intervention to avert development of alternans with important ramifications for arrhythmia prevention for patients with LQTS.
Funding:
This work was supported by National Institutes of Health grants HL057832, HL132871, HL134781, HL155762 and HL164453.
Abbreviations and Acronyms
- AP
action potential
- APD
action potential duration
- APD50, APD70, APD90
action potential duration at 50, 70 and 90% AP repolarization
- AR
alternans ratio
- [Ca2+]i
cytosolic Ca2+ concentration
- [Ca2+]SR
SR Ca2+ concentration
- CaT
Ca2+ transient
- Kv7.1
slow delayed rectifying K+ channel
- KV11.1
rapid delayed rectifying K+ channel
- LQTS
long QT syndrome
- RyR
ryanodine receptor
- SK channels
small conductance Ca2+-activated K+ channels
- SR
sarcoplasmic reticulum
- Vm
membrane potential
Biography

Giedrius Kanaporis received his PhD in Biophysics from Kaunas University of Medicine, Lithuania. He worked as postdoctoral research fellow at Stony Brook University in the USA and Kaunas University of Medicine in Lithuania. Currently he is an Assistant Professor at Rush University in Chicago, USA. His research interests are cell-to-cell communication, gap junctions, calcium signaling, electrophysiology of the heart, pathophysiological cardiac remodeling and mechanisms of cardiac arrhythmias.
Footnotes
Conflict of Interest: none declared.
Data availability statement:
The datasets analyzed in this study are available from the corresponding author on reasonable requests.
References
- Antoniou CK, Dilaveris P, Manolakou P, Galanakos S, Magkas N, Gatzoulis K & Tousoulis D. (2017). QT Prolongation and Malignant Arrhythmia: How Serious a Problem? Eur Cardiol 12, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer JD, Narayan SM, Lalani GG & Trayanova NA. (2010). Rate-dependent action potential alternans in human heart failure implicates abnormal intracellular calcium handling. Heart Rhythm 7, 1093–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YH, Tsai WC, Ko JS, Yin D, Chang PC, Rubart M, Weiss JN, Everett THt, Lin SF & Chen PS. (2015). Small-Conductance Calcium-Activated Potassium Current Is Activated During Hypokalemia and Masks Short-Term Cardiac Memory Induced by Ventricular Pacing. Circulation 132, 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PC, Hsieh YC, Hsueh CH, Weiss JN, Lin SF & Chen PS. (2013). Apamin induces early afterdepolarizations and torsades de pointes ventricular arrhythmia from failing rabbit ventricles exhibiting secondary rises in intracellular calcium. Heart Rhythm 10, 1516–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Yin D, Guo S, Xu DZ, Wang Z, Chen Z, Rubart-von der Lohe M, Lin SF, Everett Iv TH, Weiss JN & Chen PS. (2018). Sex-specific activation of SK current by isoproterenol facilitates action potential triangulation and arrhythmogenesis in rabbit ventricles. J Physiol 596, 4299–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudin E, Goldhaber J, Garfinkel A, Weiss J & Kogan B. (1999). Intracellular Ca2+ dynamics and the stability of ventricular tachycardia. Biophys J 77, 2930–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz ME, O'Neill SC & Eisner DA. (2004). Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ Res 94, 650–656. [DOI] [PubMed] [Google Scholar]
- Diness JG, Kirchhoff JE, Speerschneider T, Abildgaard L, Edvardsson N, Sorensen US, Grunnet M & Bentzen BH. (2020). The KCa2 Channel Inhibitor AP30663 Selectively Increases Atrial Refractoriness, Converts Vernakalant-Resistant Atrial Fibrillation and Prevents Its Reinduction in Conscious Pigs. Front Pharmacol 11, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner DA, Li Y & O'Neill SC. (2006). Alternans of intracellular calcium: mechanism and significance. Heart Rhythm 3, 743–745. [DOI] [PubMed] [Google Scholar]
- Fossa AA, Wisialowski T, Wolfgang E, Wang E, Avery M, Raunig DL & Fermini B. (2004). Differential effect of HERG blocking agents on cardiac electrical alternans in the guinea pig. Eur J Pharmacol 486, 209–221. [DOI] [PubMed] [Google Scholar]
- Frommeyer G, Krawczyk J, Ellermann C, Bogeholz N, Kochhauser S, Dechering DG, Fehr M & Eckardt L. (2018). Ryanodine-receptor inhibition by dantrolene effectively suppresses ventricular arrhythmias in an ex vivo model of long-QT syndrome. J Cardiovasc Electrophysiol 29, 471–476. [DOI] [PubMed] [Google Scholar]
- Gadage SN. (2018). T-wave alternans in long QT syndrome. Ann Pediatr Cardiol 11, 219–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg I, Zareba W & Moss AJ. (2008). Long QT Syndrome. Curr Probl Cardiol 33, 629–694. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton S, Polina I, Terentyeva R, Bronk P, Kim TY, Roder K, Clements RT, Koren G, Choi BR & Terentyev D. (2020). PKA phosphorylation underlies functional recruitment of sarcolemmal SK2 channels in ventricular myocytes from hypertrophic hearts. J Physiol 598, 2847–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugaard MM, Hesselkilde EZ, Pehrson S, Carstensen H, Flethoj M, Praestegaard KF, Sorensen US, Diness JG, Grunnet M, Buhl R & Jespersen T. (2015). Pharmacologic inhibition of small-conductance calcium-activated potassium (SK) channels by NS8593 reveals atrial antiarrhythmic potential in horses. Heart Rhythm 12, 825–835. [DOI] [PubMed] [Google Scholar]
- Hsueh CH, Chang PC, Hsieh YC, Reher T, Chen PS & Lin SF. (2013). Proarrhythmic effect of blocking the small conductance calcium activated potassium channel in isolated canine left atrium. Heart Rhythm 10, 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huser J, Wang YG, Sheehan KA, Cifuentes F, Lipsius SL & Blatter LA. (2000). Functional coupling between glycolysis and excitation-contraction coupling underlies alternans in cat heart cells. J Physiol 524 Pt 3, 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan PN & Christini DJ. (2007). Characterizing the contribution of voltage- and calcium-dependent coupling to action potential stability: implications for repolarization alternans. Am J Physiol Heart Circ Physiol 293, H2109–2118. [DOI] [PubMed] [Google Scholar]
- Jost N, Nagy N, Corici C, Kohajda Z, Horvath A, Acsai K, Biliczki P, Levijoki J, Pollesello P, Koskelainen T, Otsomaa L, Toth A, Papp JG, Varro A & Virag L. (2013). ORM-10103, a novel specific inhibitor of the Na+/Ca2+ exchanger, decreases early and delayed afterdepolarizations in the canine heart. Br J Pharmacol 170, 768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalb SS, Dobrovolny HM, Tolkacheva EG, Idriss SF, Krassowska W & Gauthier DJ. (2004). The restitution portrait: a new method for investigating rate-dependent restitution. J Cardiovasc Electrophysiol 15, 698–709. [DOI] [PubMed] [Google Scholar]
- Kanaporis G & Blatter LA. (2015). The mechanisms of calcium cycling and action potential dynamics in cardiac alternans. Circ Res 116, 846–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaporis G & Blatter LA. (2017a). Membrane potential determines calcium alternans through modulation of SR Ca2+ load and L-type Ca2+ current. J Mol Cell Cardiol 105, 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaporis G & Blatter LA. (2017b). Membrane potential determines calcium alternans through modulation of SR Ca2+ load and L-type Ca2+ current. J Mol Cell Cardiol 105, 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaporis G, Kalik ZM & Blatter LA. (2019). Action potential shortening rescues atrial calcium alternans. J Physiol 597, 723–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Nemec J, Li Q & Salama G. (2015). Synchronous systolic subcellular Ca2+-elevations underlie ventricular arrhythmia in drug-induced long QT type 2. Circ Arrhythm Electrophysiol 8, 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TY, Terentyeva R, Roder KH, Li W, Liu M, Greener I, Hamilton S, Polina I, Murphy KR, Clements RT, Dudley SC Jr., Koren G, Choi BR & Terentyev D. (2017). SK channel enhancers attenuate Ca2+-dependent arrhythmia in hypertrophic hearts by regulating mito-ROS-dependent oxidation and activity of RyR. Cardiovascular research 113, 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JS, Guo S, Hassel J, Celestino-Soper P, Lynnes TC, Tisdale JE, Zheng JJ, Taylor SE, Foroud T, Murray MD, Kovacs RJ, Li X, Lin SF, Chen Z, Vatta M, Chen PS & Rubart M. (2018). Ondansetron blocks wild-type and p.F503L variant small-conductance Ca2+-activated K+ channels. Am J Physiol Heart Circ Physiol 315, H375–H388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller ML, Maier SK, Gelzer AR, Bauer WR, Meesmann M & Gilmour RF Jr. (2005). Altered dynamics of action potential restitution and alternans in humans with structural heart disease. Circulation 112, 1542–1548. [DOI] [PubMed] [Google Scholar]
- Lam J, Coleman N, Garing AL & Wulff H. (2013). The therapeutic potential of small-conductance KCa2 channels in neurodegenerative and psychiatric diseases. Expert Opin Ther Targets 17, 1203–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindegger N, Hagen BM, Marks AR, Lederer WJ & Kass RS. (2009). Diastolic transient inward current in long QT syndrome type 3 is caused by Ca2+ overload and inhibited by ranolazine. J Mol Cell Cardiol 47, 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Kim TY, Huang X, Liu MB, Koren G, Choi BR & Qu Z. (2018). Mechanisms linking T-wave alternans to spontaneous initiation of ventricular arrhythmias in rabbit models of long QT syndrome. J Physiol 596, 1341–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugo CA, Cantalapiedra IR, Penaranda A, Hove-Madsen L & Echebarria B. (2014). Are SR Ca content fluctuations or SR refractoriness the key to atrial cardiac alternans?: insights from a human atrial model. Am J Physiol Heart Circ Physiol 306, H1540–1552. [DOI] [PubMed] [Google Scholar]
- Myles RC, Burton FL, Cobbe SM & Smith GL. (2011). Alternans of action potential duration and amplitude in rabbits with left ventricular dysfunction following myocardial infarction. J Mol Cell Cardiol 50, 510–521. [DOI] [PubMed] [Google Scholar]
- Nagy N, Szuts V, Horvath Z, Seprenyi G, Farkas AS, Acsai K, Prorok J, Bitay M, Kun A, Pataricza J, Papp JG, Nanasi PP, Varro A & Toth A. (2009). Does small-conductance calcium-activated potassium channel contribute to cardiac repolarization? J Mol Cell Cardiol 47, 656–663. [DOI] [PubMed] [Google Scholar]
- Nemec J, Kim JJ, Gabris B & Salama G. (2010). Calcium oscillations and T-wave lability precede ventricular arrhythmias in acquired long QT type 2. Heart Rhythm 7, 1686–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni H, Zhang H, Grandi E, Narayan SM & Giles WR. (2019). Transient outward K+ current can strongly modulate action potential duration and initiate alternans in the human atrium. Am J Physiol Heart Circ Physiol 316, H527–H542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivala M & Qu Z. (2012). Calcium alternans in a couplon network model of ventricular myocytes: role of sarcoplasmic reticulum load. Am J Physiol Heart Circ Physiol 303, H341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolasco JB & Dahlen RW. (1968). A graphic method for the study of alternation in cardiac action potentials. J Appl Physiol 25, 191–196. [DOI] [PubMed] [Google Scholar]
- Pastore JM, Girouard SD, Laurita KR, Akar FG & Rosenbaum DS. (1999). Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation 99, 1385–1394. [DOI] [PubMed] [Google Scholar]
- Perez-Riera AR, Barbosa-Barros R, Daminello Raimundo R, da Costa de Rezende Barbosa MP, Esposito Sorpreso IC & de Abreu LC. (2018). The congenital long QT syndrome Type 3: An update. Indian Pacing Electrophysiol J 18, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruvot EJ, Katra RP, Rosenbaum DS & Laurita KR. (2004). Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circulation research 94, 1083–1090. [DOI] [PubMed] [Google Scholar]
- Qi X, Yeh YH, Chartier D, Xiao L, Tsuji Y, Brundel BJ, Kodama I & Nattel S. (2009). The calcium/calmodulin/kinase system and arrhythmogenic afterdepolarizations in bradycardia-related acquired long-QT syndrome. Circ Arrhythm Electrophysiol 2, 295–304. [DOI] [PubMed] [Google Scholar]
- Saitoh H, Bailey JC & Surawicz B. (1988). Alternans of action potential duration after abrupt shortening of cycle length: differences between dog Purkinje and ventricular muscle fibers. Circ Res 62, 1027–1040. [DOI] [PubMed] [Google Scholar]
- Shimizu W & Horie M. (2011). Phenotypic manifestations of mutations in genes encoding subunits of cardiac potassium channels. Circ Res 109, 97–109. [DOI] [PubMed] [Google Scholar]
- Shkryl VM, Maxwell JT, Domeier TL & Blatter LA. (2012). Refractoriness of sarcoplasmic reticulum Ca2+ release determines Ca2+ alternans in atrial myocytes. Am J Physiol Heart Circ Physiol 302, H2310–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibsbye L, Bengaard AK, Uldum-Nielsen AM, Boddum K, Christ T & Jespersen T. (2018). Inhibition of Small Conductance Calcium-Activated Potassium (SK) Channels Prevents Arrhythmias in Rat Atria During beta-Adrenergic and Muscarinic Receptor Activation. Front Physiol 9, 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibsbye L, Poulet C, Diness JG, Bentzen BH, Yuan L, Kappert U, Matschke K, Wettwer E, Ravens U, Grunnet M, Christ T & Jespersen T. (2014). Small-conductance calcium-activated potassium (SK) channels contribute to action potential repolarization in human atria. Cardiovascular research 103, 156–167. [DOI] [PubMed] [Google Scholar]
- Sun J, Liu Y, Baudry M & Bi X. (2020). SK2 channel regulation of neuronal excitability, synaptic transmission, and brain rhythmic activity in health and diseases. Biochim Biophys Acta Mol Cell Res 1867, 118834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasugi N, Goto H, Takasugi M, Verrier RL, Kuwahara T, Kubota T, Toyoshi H, Nakashima T, Kawasaki M, Nishigaki K & Minatoguchi S. (2016). Prevalence of Microvolt T-Wave Alternans in Patients With Long QT Syndrome and Its Association With Torsade de Pointes. Circ Arrhythm Electrophysiol 9, e003206. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Rees CM, Li W, Cooper LL, Jindal HK, Peng X, Lu Y, Terentyeva R, Odening KE, Daley J, Bist K, Choi BR, Karma A & Koren G. (2014). Hyperphosphorylation of RyRs underlies triggered activity in transgenic rabbit model of LQT2 syndrome. Circ Res 115, 919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolkacheva EG, Anumonwo JM & Jalife J. (2006). Action potential duration restitution portraits of mammalian ventricular myocytes: role of calcium current. Biophys J 91, 2735–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolkacheva EG, Romeo MM, Guerraty M & Gauthier DJ. (2004). Condition for alternans and its control in a two-dimensional mapping model of paced cardiac dynamics. Phys Rev E Stat Nonlin Soft Matter Phys 69, 031904. [DOI] [PubMed] [Google Scholar]
- Tuteja D, Xu D, Timofeyev V, Lu L, Sharma D, Zhang Z, Xu Y, Nie L, Vazquez AE, Young JN, Glatter KA & Chiamvimonvat N. (2005). Differential expression of small-conductance Ca2+-activated K+ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am J Physiol Heart Circ Physiol 289, H2714–2723. [DOI] [PubMed] [Google Scholar]
- Walker ML & Rosenbaum DS. (2003). Repolarization alternans: implications for the mechanism and prevention of sudden cardiac death. Cardiovasc Res 57, 599–614. [DOI] [PubMed] [Google Scholar]
- Wang L, Myles RC, De Jesus NM, Ohlendorf AK, Bers DM & Ripplinger CM. (2014). Optical mapping of sarcoplasmic reticulum Ca2+ in the intact heart: ryanodine receptor refractoriness during alternans and fibrillation. Circ Res 114, 1410–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisbrod D (2020). Small and Intermediate Calcium Activated Potassium Channels in the Heart: Role and Strategies in the Treatment of Cardiovascular Diseases. Frontiers in Physiology 11, 590534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson D, Ermentrout B, Nemec J & Salama G. (2017). A model of cardiac ryanodine receptor gating predicts experimental Ca2+-dynamics and Ca2+-triggered arrhythmia in the long QT syndrome. Chaos 27, 093940. [DOI] [PubMed] [Google Scholar]
- Xu Y, Tuteja D, Zhang Z, Xu D, Zhang Y, Rodriguez J, Nie L, Tuxson HR, Young JN, Glatter KA, Vazquez AE, Yamoah EN & Chiamvimonvat N. (2003). Molecular identification and functional roles of a Ca2+-activated K+ channel in human and mouse hearts. J Biol Chem 278, 49085–49094. [DOI] [PubMed] [Google Scholar]
- Zhang XD, Thai PN, Lieu DK & Chiamvimonvat N. (2021). Cardiac small-conductance calcium-activated potassium channels in health and disease. Pflugers Arch 473, 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M, Rees CM, Terentyev D, Choi BR, Koren G & Karma A. (2018). NCX-Mediated Subcellular Ca2+ Dynamics Underlying Early Afterdepolarizations in LQT2 Cardiomyocytes. Biophys J 115, 1019–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv O, Morales E, Song YK, Peng X, Odening KE, Buxton AE, Karma A, Koren G & Choi BR. (2009). Origin of complex behaviour of spatially discordant alternans in a transgenic rabbit model of type 2 long QT syndrome. J Physiol 587, 4661–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analyzed in this study are available from the corresponding author on reasonable requests.