

Abstract
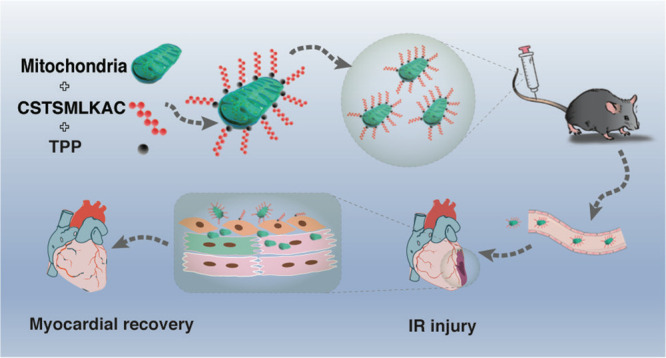
It is known that mitochondrial dysfunction is a critical factor involved in myocardial ischemia–reperfusion injury. Mitochondrial transplantation has been suggested as an effective therapeutic strategy to protect against myocardial ischemia–reperfusion injury. However, its clinical translation remains limited because it requires the local injection of mitochondria into the myocardium. Here, a polypeptide, CSTSMLKAC (PEP), bound to triphenylphosphonium cations (TPP+) effectively binds mitochondria to form a PEP–TPP–mitochondrial compound. Further investigation of this compound has revealed that the ischemia-sensing properties of PEP promote its translocation into the ischemic myocardium. Additionally, the targeting peptide, PEP, readily dissociates from the PEP–TPP–mitochondrial compound, allowing for the transplanted mitochondria to be efficiently internalized by cardiomyocytes or transferred to cardiomyocytes by endothelial cells. Mitochondrial transplantation promotes cardiomyocyte energetics and mechanical contraction, subsequently reducing cellular apoptosis, macrophage infiltration, and the pro-inflammatory response, all of which lead to attenuation of ischemia–reperfusion injury. Thus, this study provides promising evidence that the PEP–TPP–mitochondrial compound effectively promotes intravenous mitochondrial transplantation into the ischemic myocardium and subsequently ameliorates myocardial ischemia–reperfusion injury.
Keywords: targeting peptide, myocardial ischemia−reperfusion injury, mitochondrial transplantation, PEP−TPP−mitochondrial compound, cardiomyocytes
Introduction
Myocardial infarction, as the consequence of coronary artery occlusion, usually leads to ischemic mitochondrial injury due to a severe imbalance in energy supply and demand. This ultimately leads to irreversible cell death, adverse myocardial remodeling, heart failure, electrical instability, and malignant arrythmia.1−3 Timely vascular reperfusion by cardiac catheterization and coronary stenting as well as coronary artery bypass surgery are effective strategies to attenuate the ischemic insult after myocardial infarction. However, the restoration of oxygen and substrates often leads to myocardial ischemia–reperfusion (IR) injury, which is hallmarked by mitochondrial dysfunction,4,5 excessive reactive oxygen species (ROS), mitochondrial calcium dysregulation, and cardiomyocyte death.4,6 Recent efforts have been made to evaluate the efficacy of supplementation with functional mitochondria during reperfusion to attenuate myocardial IR injury. Studies by McCully et al.7−10 verified that mitochondrial transplantation is beneficial in both animals and humans. Our previous study using a mouse model of IR injury has also demonstrated the cardioprotective effects of mitochondrial transplantation after activation of aldehyde dehydrogenase 2.11 However, mitochondrial transplantation is usually achieved by infusion of mitochondria into the coronary arteries or direct injection of mitochondria into the ischemic myocardium after sternotomy. These procedures significantly limit their wide clinical application for patients who may benefit from mitochondrial transplantation. It is therefore essential to explore clinically feasible delivery routes for mitochondria to ameliorate IR injury.
The selective homing of intravenously injected mitochondria to the ischemic region is challenging, but it is the most effective strategy for mitochondrial transplantation. To achieve targeted delivery of therapeutic mitochondria to the ischemic area of the heart, tissue-specific vehicles must be recognized. CSTSMLKAC (PEP), a polypeptide with the sequence Cys-Ser-Thr-Ser-Met-Leu-Lys-Ala-Cys, has been reported to have significant selectivity for ischemic myocardium by using phage display. This preferential binding of the polypeptide to the ischemic myocardium may mimic natural ligands that bind to titin receptors exposed by the ischemic injury.12 Intravenous injection of proteins or exosomes fused with PEP have exhibited preferential binding to ischemic heart tissues compared with normal heart tissues as well as other control organs in a mouse ischemic model.13 Instead of invasive catheter or sternotomy-based procedures, targeting peptides represent a molecular tool that may be useful to deliver mitochondria to the injured myocardium by systemic intravenous administration. Additionally, triphenylphosphonium cations (TPP+), which have been used to deliver probes, antioxidants, and pharmacophores of interest to mitochondria, possess a specific mitochondria-targeting feature.14−16 Given the ischemic sensitivity of PEP and the mitochondria-targeting characteristics of TPP+, we speculate that binding the PEP with TPP+ and mitochondria to create a PEP–TPP–mitochondrial compound may be a potential strategy to efficiently transport mitochondria into the ischemic myocardium. Furthermore, a study by Thierry et al.17 has verified that blood contains circulating cell-free, respiratory-competent mitochondria, which may favor intravenous transplantation. We have thus tested the feasibility and efficacy of intravenous transplantation of an ischemic tissue-specific PEP–TPP–mitochondrial compound in a myocardial IR injury mouse model.
This study has resulted in the successful creation of the PEP–TPP–mitochondrial compound and has verified the effects of this compound in a mouse model of myocardial IR injury. Furthermore, our findings demonstrate that PEP effectively transports respiratory-competent mitochondria into the ischemic myocardium via intravenous injection. The transplanted mitochondria readily dissociate from the PEP–TPP–mitochondrial compound and are subsequently internalized by cardiomyocytes with the help of the endothelial cells. The internalization of exogenous mitochondria by cardiomyocytes effectively attenuates myocardial IR injury by reducing apoptosis, macrophage infiltration, and pro-inflammatory factors.
Results and Discussion
Intravenous Injection of Mitochondria without Modification Fails to Improve Myocardial IR Injury
Because the translational application of mitochondrial transplantation is limited due to the challenges of local injection, we proposed the therapeutic strategy of mitochondrial transplantation via intravenous injection to treat myocardial IR injury. A mouse model of myocardial IR injury was used to determine the cardioprotective effects of this procedure. In brief, active mitochondria were isolated from donor mice heart and injected intravenously to receptor mice with myocardial IR injury by ligation the left anterior descending branch for 45 min ischemia and following 24 h reperfusion (Figure 1A,B). Echocardiography was then used to evaluate the cardiac function of the mice as an indicator of the therapeutic potential of the intravenous mitochondrial transplantation. However, no significant differences in the overall left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) parameters were observed between the mice of IR injury that received intravenous mitochondrial transplantation and those that did not (Figure 1C–E). Then the cardiac infarction size of IR injury was assessed by using Evan’s blue and triphenyltetrazolium choride (TTC) staining (Figure 1F). Consistently, no therapeutic difference was found between these mice in area at risk/left ventricle (AAR/LV) (Figure 1G). Furthermore, evaluation of pro-apoptotic protein cleaved caspase 3 as well as the BAX/BCL2 ratio indicated increased apoptosis in response to IR injury, which was not ameliorated by intravenous mitochondrial transplantation. Specifically, compared with the control sham mice, a 2.5- and 3.2-fold increase in cleaved caspase 3 expression and an 8.8- and 7.8-fold increase in the BAX/BCL2 ratio were observed in the mice that received intravenous mitochondrial transplantation after IR injury and those that did not, respectively (Figure 1H–J). Additionally, the expression of inflammatory factors, including NLRP3, IL6, and cleaved IL 1β, was significantly upregulated in mice after IR injury in comparison with sham (change ratios: 4.9, 3.9, and 1.1). Moreover, intravenous mitochondrial transplantation exacerbated the expression of these pro-inflammatory factors (change ratios: 12.5, 6.8, and 1.6) (Figure 1K–N). Altogether, these results indicated that the intravenous injection of mitochondria without modification elicited no therapeutic effects on myocardial IR injury. Nonetheless, given the important role of mitochondria in sustaining myocardial function, these findings led us to explore resources that may be used to specifically target the ischemic myocardium and thus promote mitochondrial transplantation.
Figure 1.

Intravenous injection of mitochondria shows no therapeutic effect on mice myocardial IR injury. (A) Schematic representation of the mitochondrial transplantation strategy. (B) Representative transmission electron microscopy (TEM) images of isolated mitochondria from donor mice heart. Scale bar = 1 μm. (C) Representative left ventricular (LV) M-mode echocardiographic tracings. Images are representative of independent mice. (D,E) Statistical analysis of echocardiographic left ventricular ejection fraction (LVEF) and shortening fraction (LVFS). (F) Representative images and (G) statistical analysis of Evan’s blue and TTC staining in IR heart treated with or without mitochondria. (H–J) Western blots and statistical analyses for expression changes of apoptosis proteins cleaved caspase 3, Bax, and Bcl2. (K–N) Western blots and statistical analyses for expression changes of inflammatory factors, including NLRP3, IL6, and cleaved IL 1β. Mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001. Statistical analysis was carried out by a one-way ANOVA analysis followed by Tukey’s test for post hoc analysis.
Generation and Characteristics of the PEP–TPP–Mitochondrial Compound
To spatiotemporally transport mitochondria, we adopted PEP as a delivery tool to guide mitochondria into the ischemic myocardium.12 Additionally, TPP+ exhibit low chemical reactivity toward cellular components of biological systems, both lipophilic and hydrophilic properties, and are relatively simple to synthesize and purify.16,18,19 Therefore, TPP+ were used as the moiety to connect the PEP to mitochondria. The PEP–TPP–mitochondrial compound was generated by proportionally mixing the components at 4 °C. However, due to the mitochondrial binding property of TPP+, the PEP–TPP compound was synthesized first (Figure S1) before being mixed with mitochondria to form the PEP–TPP–mitochondrial compound (Figure 1A). To observe mitochondrial localization, the fluorescent compound, Cy5, was added onto the lysine (K) of the PEP sequence to generate PEP(Cy5). MitoTracker Green-labeled mitochondria were then incubated with PEP(Cy5). Confocal microscopy revealed changes in the fluorescence of mitochondria and the PEP(Cy5) upon conjugation (Figure 2B). The PEP(Cy5)–TPP–mitochondrial compound was next incubated with both AC16 and adult cardiomyocytes to assess its cellular internalization characteristics in vitro. Specially, AC16 and adult cardiomyocyte resident mitochondria were labeled with MitoTracker Red, and transplanted mitochondria were labeled with MitoTracker Green or PEP(Cy5) when bound to the compound. The internalization characteristics of the PEP(Cy5)–TPP–mitochondrial compound were imaged after incubation with AC16 or adult cardiomyocytes for 3 h. The results indicated that the targeting peptide was excluded from the cells, and only the mitochondria were internalized by both the AC16 and adult cardiomyocytes (Figure 2C). Real-time detection of the internalization of the mitochondria with or without PEP(Cy5)–TPP conjugation was used to preliminarily determine the influence of the compound on mitochondrial transplantation efficiency in vitro. Compared with nonmodified mitochondria, the accelerated transfer into cardiomyocytes was observed in the PEP–TPP–mitochondria compound (Figure 2D,E). The significant statistical difference was determined after mitochondrial transplantation for 1 h (Figure 2F).
Figure 2.

Conjugation process and characteristic analyses of targeting peptide and mitochondria. (A) Schematic diagram of peptide and mitochondrial conjugation. (B) Representative confocal images before and after the combination of isolated mitochondria with PEP(Cy5). Scale bar = 20 μm. Mitochondria were labeled with MitoTracker Green. (C) In vitro analyses of the internalization process of mitochondria and peptide compound into AC16 and cardiomyocytes. Transplanted mitochondria were labeled with MitoTracker Green. AC16 and adult cardiomyocytes were stained with MitoTracker Red. Scale bar = 20 μm. (D–F) Dynamic internalization of mitochondria into with or without peptide conjugation into cardiomyocytes. Mitochondria was labeled with MitoTracker Green. Scale bar = 100 μm. Mean ± SEM, *P < 0.05. Statistical analysis was carried out by a one-way ANOVA analysis followed by Tukey’s test for post hoc analysis.
TPP+ have been initially used as probes to study the coupling mechanism between mitochondrial membrane potential and oxidative phosphorylation (OXPHOS), and they have been subsequently used to determine mitochondrial membrane potential.20−26 This indicates that TPP+ more readily bind to mitochondria with higher membrane potentials. Based on this, we have created an automatic screening process for high-quality mitochondria because the mitochondria in the PEP(Cy5)–TPP–mitochondrial compound are suggested to have higher membrane potentials and OXPHOS capacities. Therefore, the PEP(Cy5)–TPP–mitochondrial compound has a dual role, in which it can effectively target the ischemic myocardium and also screen for high-quality mitochondria.
The proliferating human cardiomyocyte cell line, AC16, retains the nuclear and mitochondrial DNA of the primary cardiomyocytes, which is potentially useful for in vitro studies.27 Examination of the differences between AC16 and adult cardiomyocytes has revealed that the internalization of exogenous mitochondria is associated with cellular and mitochondrial volume. Compared with AC16 cells, adult cardiomyocytes exhibit a larger volume, which may have provided greater contact area for the internalization of the exogenous mitochondria (Figure 2C). Additionally, mitochondria account for approximately 30% of the volume of adult cardiomyocytes,28 which is much more than that of AC16 cells. This may be because AC16 cells are generated by fusing human ventricular cardiomyocytes with an SV40-transformed fibroblast cell line that is devoid of mitochondrial DNA,27 resulting in a smaller mitochondrial volume. The difference in mitochondrial volume between the AC16 and primary cardiomyocytes may affect the fusion efficiency of endogenous and exogenous mitochondria.
Transplantation with the PEP–TPP–Mitochondrial Compound Improves the Respiratory and Mechanical Contraction Capacities of Cardiomyocytes
The internalization of exogenous mitochondria was expected to increase OXPHOS in cardiomyocytes. The expression and activity of mitochondrial OXPHOS complexes were therefore assessed in cardiomyocytes after mitochondrial transplantation with or without PEP–TPP conjugation. The OXPHOS complexes contain five representative subunits: CI subunit NDUFB8, CII subunit SDHB, CIII subunit UQCRC2, CIV subunit MTCO1, and CV subunit ATP5A. Compared with the control, mitochondrial transplantation increased the expression of cellular OXPHOS complexes subunit NDUFB8, UQCRC2, MTCO1, and ATP5A by 1.23-, 1.2-, 1.05-, and 1.13-fold, respectively, whereas this expression was increased by 1.92-, 1.33-, 1.27-, and 1.28-fold, respectively, after PEP–TPP conjugation (Figure 3A). Oxygen consumption rate (OCR) was used as an indicator of cardiomyocyte respiratory capacity. As expected, mitochondrial transplantation increased the overall OCR of cardiomyocytes, but it was significantly increased after PEP–TPP conjugation (Figure 3B,C). Furthermore, resting cell length, maximal velocity of shortening (−dl/dt), time to peak shortening (TPS-s), peak shortening (% cell lengthening), maximal velocity of relengthening (+dl/dt), and time to 90% relengthening (TR90-s) were used to analyze cardiomyocyte mechanical contraction. Single-cell analysis revealed that the maximal velocity of shortening and relengthening were significantly increased in cardiomyocytes after transplantation with the PEP–TPP–mitochondrial compound, whereas the TPS-s and TR90-s were significantly decreased (Figure 3D–I). Altogether, these findings indicated that conjugation of mitochondria with PEP–TPP prior to transplantation effectively enhanced the respiratory capacities and mechanical contractility of cardiomyocytes.
Figure 3.

Transplantation with the PEP–TPP–mitochondria compound enhances respiration-associated mechanical function of cardiomyocytes. (A) Expression of OXPHOS complexes of cardiomyocytes after transplantation of mitochondria with and without PEP–TPP conjugation for 6 h. (B) OCR changes of cardiomyocytes transplanted with different mitochondria. (C) Statistical analysis of basal respiration in cardiomyocytes. (D) Resting cell length. (E) Maximal velocity of shortening (−dl/dt). (F) Time to peak shortening (TPS-s). (G) Peak shortening (% cell lengthening). (H) Maximal velocity of relengthening (+dl/dt). (I) Time to 90% relengthening (TR90-s). Mean ± SEM, *P < 0.05, **P < 0.01, ****P < 0.0001. Statistical analysis was carried out by a one-way ANOVA analysis followed by Tukey’s test for post hoc analysis.
Intravenous Transplantation with the PEP–TPP–Mitochondrial Compound Ameliorates Myocardial IR Injury
The mouse model of myocardial IR injury was next used to investigate the efficacy of the PEP–TPP–mitochondrial compound in vivo (Figure 4A). Specially, the PEP–TPP–mitochondrial compound was prepared by incubating the PEP–TPP solution with mitochondria in a shaker at 4 °C for 20 min before being incubated quiescently at 4 °C for 20 min. The PEP–TPP–mitochondrial compound was then quantified to a final concentration of 7.5–10 × 104/mL, and 200 μL of the PEP–TPP–mitochondrial compound was injected into the mice via the tail vein before myocardial ischemia and 24 h reperfusion. Cardiac function was evaluated after 24 h reperfusion and indicated that transplantation using the PEP–TPP–mitochondrial compound effectively protected against IR injury. This was evidenced by a significantly increased overall LVEF and LVFS compared with the mice that underwent IR injury without PEP–TPP–mitochondrial transplantation (60.18% and 31.62% vs 43.01% and 20.75%) (Figure 4B–D). Additionally, Evan’s blue and TTC staining revealed that the PEP–TPP–mitochondrial compound significantly decreased myocardial infarction size in the mice after IR injury (Figure 4E–H). Next, we investigated the long-term cardioprotective effects of the PEP–TPP–mitochondrial compound in the mice 2 and 4 weeks after intravenous mitochondrial transplantation. The mice persistently exhibited an increase in their overall LVEF and LVFS, thus indicating that the PEP–TPP–mitochondrial compound elicited long-term protection against myocardial IR injury (Figure 4I,J).
Figure 4.

PEP–TPP–mitochondria compound intravenously transplantation ameliorated myocardial IR injury in mice. (A) Schematic representation of the PEP–TPP–mitochondria compound intravenously transplantation strategy. (B) Echocardiography analyses of mice: sham (sham group), IR (IR injury group), IR + PEP-MITO (IR injury + PEP–TPP–mitochondrial transplantation group). (C) Ejection fraction and (D) fractional shortening mechanism. (E) Representative images of Evan’s blue and TTC staining. (F–H) Changes in infarction size induced by PEP–TPP–mitochondria compound transplantation. (I) Long-term analyses of ejection fraction and (J) fractional shortening in IR mice with or without PEP–TPP–mitochondria compound transplantation. Mean ± SEM, *P < 0.05, ***P < 0.001, ****P < 0.0001. Statistical analysis was carried out by a one-way ANOVA analysis followed by Tukey’s test for post hoc analysis.
Targeting Peptide PEP Promotes the Homing of Mitochondria to the Ischemic Myocardium
Next, we used isolated mitochondria from mice with green fluorescent protein-labeled cytochrome c oxidase subunit 4 (Cox IV-GFP mice) and PEP(Cy5) to investigate the localization of the compound components in vivo. Therefore, the PEP(Cy5)–TPP–mitochondrial compound was double-labeled with both Cy5 and GFP. After transplantation, mitochondrial localization was monitored at different time points using a fluorescence-tracking imaging system. Both the Cy5 and GFP fluorescence were detected in the ischemic myocardium after intravenous transplantation with the PEP(Cy5)–TPP–mitochondrial compound. However, significantly elevated Cy5 fluorescence was observed 3 and 6 h after transplantation, which diminished after 24 h (Figure 5A,C). In contrast, GFP fluorescence was significantly increased 24 h after transplantation, which was demonstrated by both the fluorescence-tracking system (Figure 5B,D) and frozen-section imaging (Figure 5E). Furthermore, cardiac troponin T (cTnT) was used to visualize cardiomyocytes and observe the specific location of the GFP-labeled mitochondria. Frozen-section imaging at a higher magnification revealed that the GFP-labeled mitochondria colocalized with cTnT around cardiac microvessels (Figure 5F). These results verified our previous findings (Figure 2c) that PEP effectively targeted the ischemic myocardium without being internalized by the cardiomyocytes.
Figure 5.

Cardiac location of PEP(Cy5)–TPP–mitochondria after transplantation in IR mice heart. (A) Representative hearts in each group for the Cy5 location of transplanted mitochondria in the myocardium at 3 and 6 h (red fluorescence). (B) Representative hearts in each group for the GFP fluorescent location of transplanted mitochondria in the myocardium at 24 h (green fluorescence). (C) Statistical analysis of average radiant efficiency of Cy5 after PEP–TPP–mitochondrial transfer for 3 and 6 h. (D) Statistical analysis of average radiant efficiency of GFP after PEP–TPP–mitochondrial transfer for 24 h. (E) GFP fluorescence imaging of frozen section from IR heart tissue after PEP–TPP–mitochondrial transfer for 0, 3, 6, and 24 h. Scale bar = 2000 μm. (F) Fluorescent colocalization imaging of GFP–mitochondria and cTnT–cardiomyocytes after PEP(Cy5)–TPP–mitochondria transplantation for 24 h. Mean ± SEM, *P < 0.05, **P < 0.01. Statistical analysis was carried out by a one-way ANOVA analysis followed by Tukey’s test for post hoc analysis.
The combination of PEP and TPP+ represents an effective strategy for the dual recognition of both mitochondria and ischemic myocardial tissues. Under the guidance of PEP, the conjugated mitochondria preferentially home to the ischemic myocardium. The precise mechanism of PEP that allows for its ischemic sensitivity may be associated with its role in binding natural ligands and receptors exposed by ischemic injury.12 Short peptide sequences can be incorporated into fusion proteins,29,30 which implies that this approach may be used with mitochondria to target the injured myocardium. However, this fusion between the targeting peptide and mitochondrial membrane proteins may cause other issues, even if the ischemic myocardium is effectively targeted. Further studies are warranted to identify the effects of using mitochondria that have been modified in such a way to target the ischemic myocardium.
Transplantation with the PEP–TPP–Mitochondrial Compound Reduces Cellular Apoptosis, Immune Cell Infiltration, and the Inflammatory Response
The expression of proteins associated with cellular apoptosis were next evaluated to further determine the cardioprotective role of the PEP–TPP–mitochondrial compound. Cleaved caspase 3, pro-apoptotic BAX, and anti-apoptotic BCL2 were detected in the ischemic myocardial tissues of mice with or without transplantation with the PEP–TPP–mitochondrial compound. Intravenous transplantation with the PEP–TPP–mitochondrial compound significantly reduced the expression of cleaved caspase 3 as well as the BAX/BCL2 ratio after myocardial IR injury, thus indicating reduced apoptosis (Figure 6A–C). Specifically, PEP–TPP–mitochondrial transplantation resulted in a 2.3-fold decrease in the expression of cleaved caspase 3 and a 2.2-fold decrease in the BAX/BCL2 ratio compared with that of mice that did not receive the transplantation. Consistently, TUNEL assay using frozen sections of the myocardial tissues revealed a 1.54-fold decrease in apoptosis after intravenous transplantation with the PEP–TPP–mitochondrial compound (Figure 6D,E). Next, we evaluated the effect of intravenous PEP–TPP–mitochondrial transplantation on immune cell infiltration and the inflammatory response because these factors play an important role in myocardial IR injury. Flow cytometry revealed that the infiltration of CD11b+F4/80+ macrophages was significantly enhanced in response to IR injury, but this was inhibited by intravenous transplantation with the PEP–TPP–mitochondrial compound (66.1% vs 34.6%) (Figure 6F,G). Furthermore, the expression of pro-inflammatory cytokines, including NLRP3, IL6, and cleaved IL1β, was significantly upregulated after IR injury. However, PEP–TPP–mitochondrial transplantation resulted in a 1.6-, 2.8-, and 2-fold decrease in the expression of these cytokines, respectively, after IR injury (Figure 6H–K), Altogether, these results indicated that PEP–TPP–mitochondrial transplantation significantly ameliorated cardiomyocyte apoptosis and the inflammatory response after myocardial IR injury.
Figure 6.

Protective role of PEP–TPP–mitochondria in myocardial IR injury by inhibiting apoptosis, immune cell infiltration, and proinflammatory reaction. (A) Cleaved caspase 3 levels and the ratios of pro-apoptotic Bax and anti-apoptotic Bcl-2 were analyzed by Western blotting in mice heart. Sham (left), IR (middle), and IR and PEP–TPP–mitochondria transplantation (IR+PEP-MITO, right). (B) Statistical assay of relative expression levels of cleaved caspase 3. (C) Statistical assay of ratios of pro-apoptotic Bax and anti-apoptotic Bcl2. (D,E) Apoptotic cardiomyocytes (red) were quantified by TUNEL assay after PEP–TPP–mitochondrial compound transplantation and myocardial IR injury. Cell nuclei were stained by DAPI (blue). Scale bars, 50 μm (n ≥ 3 per group). (F) Flow cytometry analysis of single-cell suspension isolated from fresh heart tissues after staining for macrophage markers CD45, F4/80, and CD11b. (G) Statistical significance of CD45+CD11b+F4/80+ macrophages. (H–K) Western blots and statistical analyses for expression changes of proinflammatory cytokines, including NLRP3, IL6, and cleaved IL 1β. Mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001. Statistical analysis was carried out by a one-way ANOVA analysis followed by Tukey’s test for post hoc analysis.
Transplanted Mitochondria Are Transferred to Cardiomyocytes by Endothelial Cells
Our results so far have demonstrated that the PEP–TPP–mitochondrial compound helps home mitochondria to the ischemic myocardium with the guidance of targeting peptide PEP. However, because the transplanted mitochondria localized around cardiac microvessels (Figure 5F), we further investigated the mechanism associated with this mitochondrial translocation. To accomplish this, we established two in vitro coculture systems using endothelial cells (bEnd.3) and cardiomyocytes. First, mitochondria were isolated from cardiomyocytes and labeled with MitoTracker Green. Then, these mitochondria were cocultured with endothelial cells for 6 h so that the cells could internalize as many exogenous mitochondria as possible. This also allowed the endothelial cells to be labeled with green fluorescence. The endothelial cells were then cocultured with cardiomyocytes isolated from adult mice (Figure 7A). Real-time imaging of the coculture system was conducted using a live-cell fluorescence imager. The continuous images showed that the green fluorescence was gradually transferred to the cardiomyocytes (Figure 7B). These results demonstrated that the mitochondria that were internalized by the endothelial cells were ultimately transferred to the cardiomyocytes. Consistent results were observed using mitochondria conjugated with PEP–TPP (Figure 7C). To further demonstrate this selective transfer of mitochondria, endothelial cells were cocultured with cardiomyocytes, and then isolated MitoTracker Green-labeled mitochondria were either directly added to the culture medium or conjugated to the PEP–TPP compound and then added to the culture medium (Figure 7D). Real-time imaging of the coculture system showed that the MitoTracker Green-labeled mitochondria accumulated in cardiomyocytes (Figure 7E). These results collectively demonstrated that transplanted mitochondria were selectively transferred to and also preferentially internalized by cardiomyocytes. Furthermore, the endothelial cells acted as a carrier and transported the transplanted mitochondria to cardiomyocytes, the main workstations requiring them.
Figure 7.

Translocation mechanism of mitochondria into cardiomyocytes. (A) Schematic representation of the coculture strategy of murine endothelial cell line (bEnd.3) with cardiomyocytes. After coincubated with mitochondria, the bEnd.3 were cocultured with cardiomyocytes. (B,C) Living cell imaging detection of dynamic changes of transplanted mitochondria or PEP–TPP–mitochondria in both bEnd.3 and cardiomyocytes. Scale bars, 200 μm. (D) Schematic representation of the coculture strategy of bEnd.3 with cardiomyocytes. (E) Green fluorescence was imaged after mitochondria or PEP–TPP–mitochondria adding into the bEnd.3 and cardiomyocytes coculture system. Scale bars, 200 μm.
The reasons behind this selective transfer and uptake of transplanted mitochondria may be related to mitochondrial distribution and cellular functional characteristics that differ based on cell type. Specifically, only 2–6% of the endothelial cell volume consists of mitochondria compared with 32% of that of cardiomyocytes.31 The mitochondrial content of endothelial cells indicates that these cells predominantly rely on anaerobic glycolysis rather than mitochondrial OXPHOS. However, the mechanical contraction of cardiomyocytes requires large amounts of energy and therefore heavily relies on mitochondrial energetics. Taking this into account, it makes sense that the transplanted mitochondria are preferentially taken up by cardiomyocytes.
To accurately mimic the interactions between endothelial cells and cardiomyocytes, the cardiac microvascular endothelial cell was the optimal candidate, whereas it reported that the cardiac microvascular endothelial cells are highly calcium-sensitive, and calcium-overload evokes the mitochondrial dysfunction.32 Calcium released from cardiomyocytes may challenge the survival of endothelial cells. Additionally, the culture medium for cardiomyocyte containing 1.8 mM Ca2+ was not applicable to the culture of cardiac microvascular endothelial cells. Thus, it is difficult to overcome the difficulties associated with coculturing cardiac microvascular endothelial cells and cardiomyocytes. We therefore chose the murine endothelial cell line (bEnd.3) to coincubated with adult cardiomyocytes in our study. The coculture system efficiently elicited the priority of mitochondria into cardiomyocytes. Furthermore, the colocalization results of fluorescent cTnT and GFP in our in vivo study also showed the directly evidence that the transplanted mitochondria were localization cardiomyocyte around microvessels (Figure 5F). Therefore, these findings suggest that the endothelium has a carrier effect during mitochondrial transplantation.
Mitochondrial quality control the machineries responding to a broad array of stress stimuli to regulate fission, fusion, mitophagy, and biogenesis in mitochondria.33 Except for cardiomyocytes, damages of mitochondrial quality control elements also contribute to endothelial death in cardiac IR injury.34 It is known that endothelial dysfunction could impair the blood supply to the heart and exacerbate myocardial reperfusion injury. Maintaining mitochondrial function could attenuate endothelial cell damage and thereby improve cardiac function.35 Given the important role of endothelial cells in maintaining the structural integrity and microcirculatory function of the coronary microvasculature, mitochondrial transplantation specifically directed toward endothelial cells may also hold therapeutic potential and warrants further investigation. Fibroblasts are abundant in the heart, and accumulated evidence has indicated that metabolic remodeling of fibroblasts promotes fibroblast phenotype transformation.36 However, the extent to which mitochondria transplantation is involved in fibroblast phenotype transformation remains unknown. Future studies are needed to explore the impact of exogenous mitochondrial transplantation on the interaction between fibroblasts, cardiomyocytes, macrophages, and other cell types within the injured myocardium.
PEP–TPP–Mitochondrial Compound Elicits a Cardioprotective Effect on Cardiomyocytes In Vitro
Additional in vitro studies were performed to gain more insights into the cardioprotection elicited by the PEP–TPP–mitochondrial compound. Cardiomyocytes underwent 45 min of hypoxia and 3 h of reoxygenation (H/R injury) to mimic in vivo myocardial IR injury. Real-time imaging was then conducted after the internalization of exogenous mitochondria with or without conjugation to PEP–TPP (Figure 8A). H/R injury significantly induced cardiomyocyte death, which was evidenced by a change in morphology from a rod to round shape. However, H/R injury was partially ameliorated by mitochondrial transplantation, and it was significantly ameliorated by PEP–TPP–mitochondrial transplantation (Figure 8B). Additionally, PEP–TPP–mitochondrial transplantation resulted in an accelerated cardiomyocyte internalization rate of the transplanted mitochondria (Figure 8C), which may explain its enhanced cardioprotective effect. There was a significant increase in the density of MitoTracker Green-labeled mitochondria 3 h after PEP–TPP–mitochondrial transplantation compared with mitochondrial transplantation alone. Furthermore, a previous study demonstrated that the majority of transplanted exogenous mitochondria had fused with the endogenous mitochondrial network, which was associated with an increase in mitochondrial metabolism.11,37 Therefore, we used MitoTracker Green and Red to visualize such events within cardiomyocytes after internalization of exogenous mitochondria, thus illustrating the recovery mechanism of the cells. Specifically, resident mitochondria of AC16 and adult cardiomyocytes were labeled with MitoTracker Red, and the cells were then exposed to H/R conditions in vitro. Then, isolated mitochondria were labeled with MitoTracker Green and were subsequently used for transplantation with the cardiomyocytes. PEP–TPP conjugation promoted the adherence of the mitochondria to the cells and also enhanced mitochondrial internalization. Confocal imaging also revealed enhanced mitochondrial fusion (Figure 8D,E). Because increased fusion should result in increased mitochondrial respiration, OCR was analyzed in the adult cardiomyocytes after H/R injury and with or without mitochondrial transplantation. As expected, H/R injury significantly decreased the overall OCR of the cardiomyocytes, indicating significantly impaired mitochondrial respiration. However, this decrease was partially inhibited by mitochondrial transplantation (Figure 8F). These results collectively suggested that the cardioprotection elicited by mitochondrial transplantation was highly dependent on the recovery of mitochondrial fusion-regulated cardiomyocyte respiration. Additionally, PEP conjugation significantly enhanced mitochondrial fusion between the exogenous and endogenous mitochondria, leading to enhanced respiration and the subsequent inhibition of cardiomyocyte death after H/R injury.
Figure 8.

PEP–TPP–mitochondria transplantation alleviates cardiomyocytes H/R injury by increasing cellular internalization and mitochondrial respiration. (A) Representative images of cardiomyocytes viability induced by hypoxia–reoxygenation (H/R) treatment with PEP–TPP–mitochondria transplantation or mitochondrial transplantation alone. Green represents the transplanted mitochondria. Red represents peptide. Scale bar = 200 μm. (B) Statistical analysis of cellular death rate (circle cardiomyocyte represents death one). (C) Statistical analysis of mitochondrial internalization rate/green fluorescence density. (D,E) Representative images of mitochondrial internalization into H/R cardiomyocyte with or without peptide conjugation. (F) OCR changes of adult cardiomyocytes after H/R exposure and mitochondrial transplantation with or without PEP conjunction. Mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001. Statistical analysis was carried out by a one-way ANOVA analysis followed by Tukey’s test for post hoc analysis.
Conclusion
Our study was designed to explore the therapeutic application of intravenous mitochondrial transplantation for treating myocardial IR injury. We used the ischemic-targeting peptide, PEP, and mitochondria-targeting cations, TPP+, to establish the PEP–TPP–mitochondrial compound to effectively transport mitochondria to the ischemic myocardium after IR injury. Our results indicated that intravenous administration of the PEP–TPP–mitochondrial compound not only effectively transported the mitochondria to the ischemic myocardium, but the targeting peptide also readily dissociated from the mitochondria, thus promoting cardiomyocyte internalization. Additionally, we observed that the transplanted mitochondria were also selectively transferred to cardiomyocytes via the endothelium. Nonetheless, transplantation with the PEP–TPP–mitochondrial compound effectively reduced immune cell infiltration, the inflammatory response, and ultimately cardiomyocyte apoptosis. The application and targeted delivery of viable mitochondria via the PEP–TPP–mitochondrial compound allows for the spatiotemporal transfer of active mitochondria to damaged cardiomyocytes. This study suggests the potential translational value of the intravenous delivery of mitochondria to the ischemic heart to stimulate cardiac repair after IR injury. This intravenous mitochondrial transplantation strategy may enhance the clinical applicability of mitochondrial transplantation for patients with myocardial mitochondria loss and dysfunction.
Materials and Methods
Animals
Eight-week-old male C57BL/6J mice (weighing 20–22 g) were provided by Cavens Biogle Model Animal Research Co., Ltd. (Suzhou, China). All animal studies were performed according to the guidelines on the Use and Care of Laboratory Animals for Biomedical Research published by National Institutes of Health (No. 85-23, revised 1996). The experimental protocol was evaluated and approved by the Animal Ethics Committee of Fudan University.
Isolation of Adult Primary Cardiomyocytes
For isolation of adult primary cardiomyocytes, 8-week-old male mice were anesthetized with 1% pentobarbital (70 mg/kg), and then the mouse heart was perfused accordingly to the Langendorff-free method.38 In brief, the heart was injected with EDTA buffer into the right ventricle after cutting the descending aorta. Then the ascending aorta was clamped and the heart was transferred into a culture dish. EDTA buffer, perfusion buffer, and collagenase buffer were sequentially injected into the left ventricle. The collected single cardiomyocyte solution was passed through a 100 μm filter and restored calcium concentration to physiological levels by using 3 intermediate calcium reintroduction buffers. Finally, the viable rod-shaped cells were resuspended in prewarmed medium and plated into a laminin-precoated culture dish (5 μg/mL).
Cell Culture
AC16 cells and the murine endothelial cell line (bEnd.3) were purchased from the Chinese Academy of Science Cell Bank (Shanghai, China) and cultured in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum (Gibco, Grand Island, NY, USA). Primary cardiomyocytes were isolated from adult mice and incubated in culture medium. All cells were maintained for use in a humidified 37 °C, 5% CO2 culture incubator (Thermo Fisher Scientific, Waltham, MA, USA).
Myocardial Ischemia–Reperfusion (IR) Injury Model
Myocardial IR injury model was performed as previously reported. Briefly, 8-week-old male mice were anesthetized with 2% isoflurane gas inhalation without intubation. Mice myocardial ischemia was established by ligation of the left anterior descending artery with a 6–0 silk suture slipknot after temporarily exteriorizing the heart. Then the heart was carefully returned to the chest cavity. After ligation for 45 min, the reperfusion was performed by releasing the slipknot.
Mitochondrial Isolation
Mitochondria were isolated from C57BL/6J and Cox4i1-GFP mice heart by using a tissue mitochondrial isolation kit (Beyotime Biotechnology, China, #C3606) according to the manufacturer’s instruction. The isolated mitochondria for transplantation were suspended and then quantized by a Beckman counter.
PEP(Cy5)–TPP–Mitochondria Compound Preparation and Transplantation
To track the PEP and realize the combination with mitochondria, the Cy5 fluorophore and triphenylphosphine were added. The composition flowchart is provided in Supporting Informations. To establish the PEP(Cy5)–TPP–mitochondria compound, the PEP(Cy5)–TPP suspension was prepared by dissolving PEP(Cy5)–TPP dry powder with mitochondrial isolation solution at 10 mg/mL, which was used to dissolve the purified mitochondria. To obtain the compound, PEP(Cy5)–TPP and mitochondria solution was incubated in a shaker at 4 °C for 20 min and then incubated quiescently at 4 °C for 20 min. In the end, the PEP(Cy5)–TPP–mitochondria compound was quantified to the final concentration of 7.5–10 × 104/mL. 200 μL of PEP(Cy5)–TPP–mitochondria compound in 7.5–10 × 104/mL was transplanted by tail vein before myocardial ischemia. For placebo-treated ischemic reperfusion mice, 200 μL of PBS was transplanted by tail vein.
Echocardiography
To measure the cardiac function, mice were anesthetized with 2% isoflurane for induction and 1.5% isoflurane for maintenance. Then the mice heart function was evaluated with a Vevo 2100 high-frequency ultrasound system (Visual Sonics, Toronto, ON, Canada). M-mode images from the parasternal short-axis view at papillary muscle level were acquired for heart parameter analysis. The echocardiographer was blinded to this study.
Mitochondrial Fluorescence Tracking
For in vitro imaging, MitoTracker Green and Red (Cell Signaling Technology, Danvers, MA, USA) were used to incubate with isolated mitochondria and receptor cells, respectively. The combination of Cy5 marked PEP–TPP and MitoTracker Green-labeled mitochondria was imaged by a confocal microscope (Thermo Fisher Scientific). To observe the dynamic internalization of transplanted mitochondria, PEP(Cy5)–TPP–mitochondria was added into the culture medium. Then the fluorescence intensity of MitoTracker Green and Cy5 was detected by the Lionheart FX living cell imaging analysis system (BioTek, Winooski, VT, USA).
For in vivo imaging, to detect the retention of transplanted PEP(Cy5)–TPP–mitochondria (GFP), mouse hearts were imaged by a fluorescence detection system (IVIS Lumina XRMS, USA) after reperfusion for 3, 6, and 24 h. Besides, the Cox4i1-GFP marked mitochondria of formalin-fixed heart tissue sections were imaged with a fluorescence microscope (Thermo Fisher Scientific).
Cell Shortening/Relengthening Assay
To determine the mechanical properties of cardiomyocytes, a SOFTEDGE MYOCAM system (IonOptix Corporation, Milton, MA, USA) was used in our study. Specifically, the adult primary cardiomyocytes were prepared by seeding on Costar 6-well plates after plating the 24 × 24 mm microscope cover glass. Then the mitochondria conjugated with or without peptide were coincubated with cardiomyocytes. After mitochondrial transplantation, the cardiomyocytes were translocated into a Warner chamber of an inverted microscope (Olympus, IX-70). The shortening and relengthening properties were recorded by IonOptix MyoCam camera after stimulating with 0.5 Hz frequency of FHC stimulator (Brunswick, NE, USA). The mechanical parameters of cardiomyocytes were analyzed by IONOPTIX SOFTEDGE software: bl-resting cell length, dep v-maximal velocity of shortening (−dl/dt), dep vt–time to peak shortening (TPS-s), bl % peak h-peaking shortening (% cell lengthening), ret v-maximal velocity of relengthening (+dl/dt), t to bl 90.0%–time to 90% relengthening (TR90-s).
Hypoxia–Reoxygenation Model of Cardiomyocytes In Vitro
To mimic ischemic reaction, adult primary cardiomyocytes were plated in laminin-precoated culture dish and cultured with ischemic buffer (118 mmol/L NaCl, 24 mmol/L NaHCO3, 1 mmol/L NaH2PO4, 2.5 mmol/L CaCl2–2H2O, 1.2 mmol/L MgCl2, 20 mmol/L sodium lactate, 16 mmol/L KCl, and 10 mmol/L 2-deoxyglucose, pH adjusted to 6.2). Then the culture dish was immediately transferred in a 0.5% hypoxic incubator and incubated for 45 min. To mimic the process of reperfusion of mice, the ischemic buffer in the culture dish was replaced with culture medium. At the same time, transplanted mitochondria were added to the culture medium.
Mitochondrial Respiratory Capacity
Mitochondrial respiratory capacity of cardiomyocytes was measured by the oxygen consumption rates. Briefly, the adult primary cardiomyocytes were seed in Seahorse plate. After hypoxia–reoxygenation treatment or mitochondrial transplantation, the cells were analyzed by XFe96 extracellular flux analyzer (Seahorse Bioscience, Billerica, MA, USA) by adding oligomycin A (1 μM), 1 μM FCCP, antimycin A (1 μM), and rotenone (1 μM).
Infarct Size Assessment
Twenty-four hours after myocardial IR injury, the mice were anesthetized with 1% pentobarbital (70 mg/kg). The left anterior descending coronary artery of mice was retied and subsequently injected with 1 mL of 1% Evan’s blue dye (Sigma-Aldrich, St. Louis, MO, USA) into the right ventricle. Then, the heart was quickly acquired and frozen at −80 °C for 30 min. Afterward, the frozen heart was immediately sliced into 4–5 short-axis sections and incubated in 1% triphenyltetrazolium chloride (TTC, Sigma-Aldrich) solution at 37 °C for 30 min. The infarct size was calculated as previously described.11 Briefly, the white area that was not stained by Evan’s blue dye or TTC represented the myocardial infarction area. The red area that was stained by TTC instead of Evan’s blue dye represented the myocardial ischemic area. The blue area that was stained by both Evan’s blue dye and TTC represented the nonischemic area. The area at risk (AAR) included both the ischemic and myocardial infarction areas, whereas the areas not at risk were indicated by phthalocyanine blue staining. Image quantification was performed by segmenting the stained areas of each section using ImageJ software. The infarction area was expressed as the percentage of the AAR.
Myocardial Apoptosis Assay
For the myocardial apoptosis assay, the TUNEL staining was performed. After anesthetizing with 1% pentobarbital (70 mg/kg), the mouse heart was excised and fixed in 4% paraformaldehyde and cut into 6 μm thick sections. Then, a One Step TUNEL apoptosis assay kit (Beyotime Biotechnology, China) was used to detect apoptotic cells. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, Beyotime Biotechnology, China), Five fields or more in >3 different sections/animals were examined by an Olympus microscope.
Flow Cytometric Analysis
Single-cell suspensions were prepared by using the multitissue dissociation kit 2 (Miltenyi Biotec). Briefly, after deeply anesthetizing, the mouse heart was harvested and transferred into a 10 cm dish containing PBS. The hearts were cut into small pieces (1–2 mm3) and the tissue transferred into the gentle MACS C Tube. Then the preheated enzyme mix (prepared according to the manufacturer’s instruction) was added into the C Tube. The C Tube was attached onto the sleeve of the gentle MACS dissociator. Following the trituration, the tissue samples were passed through a 70 μm cell strainer (BD Falcon, NJ, USA). The debris was removed and erythrocyte was lytically from obtained cells. For cytometric analyses, the cells were incubated with a mixture of antibodies at 4 °C for 20 min. The antibodies used in the present study are listed: PE-F4/80 monoclonal antibody (eBioscience) (Catalog No. 12-4801-82), FITC rat anti-mouse CD45 antibody (BD Pharmingen) (Catalog No. 553079), PerCP-Cy5.5 rat anti-CD11b (BD Pharmingen) (Catalog No. 561114). The obtained results were expressed as the percent or cell number per microgram of tissue. Flow cytometric analysis and cell sorting were performed on a LSRFORTESSA and FACS Aria instruments (BD Biosciences, San Jose, CA, USA) and analyzed using FlowJo software (Tree Star).
Western Blot Analysis
For total protein extraction, heart tissue samples were homogenized in lysis buffer using a homogenizer. Then the protein concentration was determined by a BCA protein assay kit (Bio-Rad, 5000006JA). Total protein was separated by 10 and 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Merck, Darmstadt, Germany). After blocking the membranes with 5% nonfat milk in Tris-buffered saline with 0.1% Tween 20 (TBST) for 1 h at room temperature, the membranes were incubated with primary antibodies at 4 °C overnight and then horseradish peroxidase-conjugated secondary antibodies (1:4000) for 1 h at room temperature. Afterward, specific bands were imaged using a Bio-Rad detection system (Bio-Rad Laboratories, Hercules, CA, USA), and the obtained images were analyzed using ImageJ. The primary antibodies Bax (#2772), Bcl2 (#3498), NLRP3 (#15101), and IL6 (#12912) were obtained from Cell Signaling, Danvers, MA. Cleaved caspase 3 (#19677-1-AP) was obtained from Proteintech, and IL1β (#A1112) was obtained from ABclonal.
Statistics Analysis and Software
All experiments were repeated at least three times. All values are presented as mean ± standard error of mean (SEM) or median with interquartile ranges as appropriate. Data are presented as the mean ± SEM. One-way ANOVA analysis followed by Tukey’s post hoc test was applied for comparisons among multiple groups. Unpaired Student’s t tests were used for comparisons between two groups; values of P < 0.05 were considered statistically significant. Statistical analyses were performed using GraphPad Prism 8.01 (GraphPad Prism Software Inc., San Diego, CA, USA) software.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81900353, 82270264, 81700347, T2288101), the National Science Fund for Distinguished Young Scholars (817200010), and Funding for Top Hospital and Specialty Excellence of Fujian Province.
Data Availability Statement
The main data supporting the results in this study are available within the paper and its Supporting Information. Source data for the figures are provided within this paper. Other raw data generated during the study are available from the corresponding authors on reasonable request.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.2c05286.
Schematic diagrams of the synthetic routes of CSTSMLKAC (PEP), cy5, and TPP (PDF)
Author Contributions
X.S., Z.C., and A.S. conceived and designed the experiments. X.S., H.C., R.G., Y.Q., Y.H., N.Z., S.H., and F.F. performed the experiments. X.S., H.C., R.G., Y.Q., Y.H., N.Z., S.H., F.F., Y.Z., K.H., and J.G. analyzed the data and discussed the results. X.S., Z.C., and A.S. wrote the manuscript. All of the authors read, revised, and approved the manuscript. X.S., H.C., R.G., and Y.Q. contributed equally to this work.
The authors declare no competing financial interest.
Notes
All animal studies were performed according to the guidelines on the Use and Care of Laboratory Animals for Biomedical Research published by National Institutes of Health (No. 85–23, revised 1996). The experimental protocol was evaluated and approved by the Animal Ethics Committee of Fudan University (AECFU).
Supplementary Material
References
- Yellon D. M.; Hausenloy D. J. Myocardial Reperfusion Injury. N Engl J. Med. 2007, 357 (11), 1121–1135. 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- Lesnefsky E. J.; Chen Q.; Tandler B.; Hoppel C. L. Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies. Annu. Rev. Pharmacol Toxicol 2017, 57, 535–565. 10.1146/annurev-pharmtox-010715-103335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt D. L.; Lopes R. D.; Harrington R. A. Diagnosis and Treatment of Acute Coronary Syndromes: A Review. JAMA 2022, 327 (7), 662–675. 10.1001/jama.2022.0358. [DOI] [PubMed] [Google Scholar]
- Murphy E.; Steenbergen C. Mechanisms Underlying Acute Protection from Cardiac Ischemia-Reperfusion Injury. Physiol Rev. 2008, 88 (2), 581–609. 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani E. T.; Pell V. R.; Gaude E.; Aksentijevic D.; Sundier S. Y.; Robb E. L.; Logan A.; Nadtochiy S. M.; Ord E. N. J.; Smith A. C.; et al. Ischaemic Accumulation of Succinate Controls Reperfusion Injury through Mitochondrial ROS. Nature 2014, 515 (7527), 431–435. 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q.; Camara A. K.; Stowe D. F.; Hoppel C. L.; Lesnefsky E. J. Modulation of Electron Transport Protects Cardiac Mitochondria and Decreases Myocardial Injury during Ischemia and Reperfusion. Am. J. Physiol Cell Physiol 2007, 292 (1), C137–147. 10.1152/ajpcell.00270.2006. [DOI] [PubMed] [Google Scholar]
- Emani S. M.; Piekarski B. L.; Harrild D.; Del Nido P. J.; McCully J. D. Autologous Mitochondrial Transplantation for Dysfunction after Ischemia-Reperfusion Injury. J. Thorac Cardiovasc Surg 2017, 154 (1), 286–289. 10.1016/j.jtcvs.2017.02.018. [DOI] [PubMed] [Google Scholar]
- Guariento A.; Piekarski B. L.; Doulamis I. P.; Blitzer D.; Ferraro A. M.; Harrild D. M.; Zurakowski D.; Del Nido P. J.; McCully J. D.; Emani S. M. Autologous Mitochondrial Transplantation for Cardiogenic Shock in Pediatric Patients Following Ischemia-Reperfusion Injury. J. Thorac Cardiovasc Surg 2021, 162 (3), 992–1001. 10.1016/j.jtcvs.2020.10.151. [DOI] [PubMed] [Google Scholar]
- McCully J. D.; Levitsky S.; Nido P. J.; Cowan D. B. Mitochondrial Transplantation for Therapeutic Use. Clin Transl Med. 2016, 5 (1), 16. 10.1186/s40169-016-0095-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollihue J. L.; Rabchevsky A. G. Prospects for Therapeutic Mitochondrial Transplantation. Mitochondrion 2017, 35, 70–79. 10.1016/j.mito.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Gao R.; Li W.; Zhao Y.; Yang H.; Chen H.; Jiang H.; Dong Z.; Hu J.; Liu J.; et al. Alda-1 Treatment Promotes the Therapeutic Effect of Mitochondrial Transplantation for Myocardial Ischemia-Reperfusion Injury. Bioact Mater. 2021, 6 (7), 2058–2069. 10.1016/j.bioactmat.2020.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki S.; Jaalouk D. E.; Lee S.; Yu A. Y.; Gannon J.; Lee R. T. Identification of Targeting Peptides for Ischemic Myocardium by in vivo Phage Display. Journal of molecular and cellular cardiology 2011, 50 (5), 841–848. 10.1016/j.yjmcc.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Chen Y.; Zhao Z.; Meng Q.; Yu Y.; Sun J.; Yang Z.; Chen Y.; Li J.; Ma T.; et al. Engineered Exosomes With Ischemic Myocardium-Targeting Peptide for Targeted Therapy in Myocardial Infarction. J. Am. Heart Assoc 2018, 7 (15), e008737 10.1161/JAHA.118.008737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G.; Zielonka J.; Ouari O.; Lopez M.; McAllister D.; Boyle K.; Barrios C. S.; Weber J. J.; Johnson B. D.; Hardy M.; et al. Mitochondria-Targeted Analogues of Metformin Exhibit Enhanced Antiproliferative and Radiosensitizing Effects in Pancreatic Cancer Cells. Cancer Res. 2016, 76 (13), 3904–3915. 10.1158/0008-5472.CAN-15-2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy M.; Poulhes F.; Rizzato E.; Rockenbauer A.; Banaszak K.; Karoui H.; Lopez M.; Zielonka J.; Vasquez-Vivar J.; Sethumadhavan S.; et al. Mitochondria-Targeted Spin Traps: Synthesis, Superoxide Spin Trapping, and Mitochondrial Uptake. Chem. Res. Toxicol. 2014, 27 (7), 1155–1165. 10.1021/tx500032e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielonka J.; Joseph J.; Sikora A.; Hardy M.; Ouari O.; Vasquez-Vivar J.; Cheng G.; Lopez M.; Kalyanaraman B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117 (15), 10043–10120. 10.1021/acs.chemrev.7b00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Amir Dache Z.; Otandault A.; Tanos R.; Pastor B.; Meddeb R.; Sanchez C.; Arena G.; Lasorsa L.; Bennett A.; Grange T.; et al. Blood Contains Circulating Cell-Free Respiratory Competent Mitochondria. FASEB J. 2020, 34 (3), 3616–3630. 10.1096/fj.201901917RR. [DOI] [PubMed] [Google Scholar]
- Murphy M. P.; Smith R. A. Targeting Antioxidants to Mitochondria by Conjugation to Lipophilic Cations. Annu. Rev. Pharmacol Toxicol 2007, 47, 629–656. 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- Ross M. F.; Kelso G. F.; Blaikie F. H.; James A. M.; Cocheme H. M.; Filipovska A.; Da Ros T.; Hurd T. R.; Smith R. A.; Murphy M. P. Lipophilic Triphenylphosphonium Cations As Tools in Mitochondrial Bioenergetics and Free Radical Biology. Biochemistry (Mosc) 2005, 70 (2), 222–230. 10.1007/s10541-005-0104-5. [DOI] [PubMed] [Google Scholar]
- Liberman E. A.; Skulachev V. P. Conversion of Biomembrane-Produced Energy into Electric Form. IV. General discussion. Biochimica et biophysica acta 1970, 216 (1), 30–42. 10.1016/0005-2728(70)90156-8. [DOI] [PubMed] [Google Scholar]
- Walsh Kinnally K.; Tedeschi H.; Maloff B. L. Use of Dyes to Estimate the Electrical Potential of the Mitochondrial Membrane. Biochemistry 1978, 17 (16), 3419–3428. 10.1021/bi00609a036. [DOI] [PubMed] [Google Scholar]
- Lichtshtein D.; Kaback H. R.; Blume A. J. Use of a Lipophilic Cation for Determination of Membrane Potential in Neuroblastoma-Glioma Hybrid Cell Suspensions. Proc. Natl. Acad. Sci. U. S. A. 1979, 76 (2), 650–654. 10.1073/pnas.76.2.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek J. B.; Nicholls D. G.; Williamson J. R. Determination of the Mitochondrial Protonmotive Force in Isolated Hepatocytes. J. Biol. Chem. 1980, 255 (4), 1458–1464. 10.1016/S0021-9258(19)86052-0. [DOI] [PubMed] [Google Scholar]
- Hiller R.; Schaefer A.; Zibirre R.; Kaback H. R.; Koch G. Factors Influencing the Accumulation of Tetraphenylphosphonium Cation in HeLa Cells. Mol. Cell. Biol. 1984, 4 (1), 199–202. 10.1128/mcb.4.1.199-202.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G. C.; Brand M. D. Thermodynamic Control of Electron Flux through Mitochondrial Cytochrome bc1 Complex. Biochem. J. 1985, 225 (2), 399–405. 10.1042/bj2250399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holian A.; Wilson D. F. Relationship of Transmembrane pH and Electrical Gradients with Respiration and Adenosine 5′-Triphosphate Synthesis in Mitochondria. Biochemistry 1980, 19 (18), 4213–4221. 10.1021/bi00559a012. [DOI] [PubMed] [Google Scholar]
- Davidson M. M.; Nesti C.; Palenzuela L.; Walker W. F.; Hernandez E.; Protas L.; Hirano M.; Isaac N. D. Novel Cell Lines Derived from Adult Human Ventricular Cardiomyocytes. Journal of molecular and cellular cardiology 2005, 39 (1), 133–147. 10.1016/j.yjmcc.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Schaper J.; Meiser E.; Stammler G. Ultrastructural Morphometric Analysis of Myocardium from Dogs, Rats, Hamsters, Mice, and from Human Hearts. Circ. Res. 1985, 56 (3), 377–391. 10.1161/01.RES.56.3.377. [DOI] [PubMed] [Google Scholar]
- Patel D. K.; Menon D. V.; Patel D. H.; Dave G. Linkers: A Synergistic Way for the Synthesis of Chimeric Proteins. Protein Expr Purif 2022, 191, 106012. 10.1016/j.pep.2021.106012. [DOI] [PubMed] [Google Scholar]
- Lin C. Y.; Liu J. C. Incorporation of Short, Charged Peptide Tags Affects the Temperature Responsiveness of Positively-Charged Elastin-Like Polypeptides. J. Mater. Chem. B 2019, 7 (34), 5245–5256. 10.1039/C9TB00821G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth E.; Stammler G.; Speiser B.; Schaper J. Ultrastructural Quantitation of Mitochondria and Myofilaments in Cardiac Muscle from 10 Different Animal Species Including Man. Journal of molecular and cellular cardiology 1992, 24 (7), 669–681. 10.1016/0022-2828(92)93381-S. [DOI] [PubMed] [Google Scholar]
- Li S.; Chen J.; Liu M.; Chen Y.; Wu Y.; Li Q.; Ma T.; Gao J.; Xia Y.; Fan M.; et al. Protective Effect of HINT2 on Mitochondrial Function via Repressing MCU Complex Activation Attenuates Cardiac Microvascular Ischemia-Reperfusion Injury. Basic Res. Cardiol 2021, 116 (1), 65. 10.1007/s00395-021-00905-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang X.; Lochner A.; Wang H. H.; Wang S.; Zhu H.; Ren J.; Zhou H. Coronary Microvascular Injury in Myocardial Infarction: Perception and Knowledge for Mitochondrial Quality Control. Theranostics 2021, 11 (14), 6766–6785. 10.7150/thno.60143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Toan S.; Zhou H. New Insights into the Role of Mitochondria in Cardiac Microvascular Ischemia/Reperfusion injury. Angiogenesis 2020, 23 (3), 299–314. 10.1007/s10456-020-09720-2. [DOI] [PubMed] [Google Scholar]
- Wang J.; Zhou H. Mitochondrial Quality Control Mechanisms As Molecular Targets in Cardiac Ischemia-Reperfusion Injury. Acta Pharm. Sin B 2020, 10 (10), 1866–1879. 10.1016/j.apsb.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb A. A.; Lazaropoulos M. P.; Elrod J. W. Myofibroblasts and Fibrosis: Mitochondrial and Metabolic Control of Cellular Differentiation. Circ. Res. 2020, 127 (3), 427–447. 10.1161/CIRCRESAHA.120.316958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacak C. A.; Preble J. M.; Kondo H.; Seibel P.; Levitsky S.; Del Nido P. J.; Cowan D. B.; McCully J. D. Actin-Dependent Mitochondrial Internalization in Cardiomyocytes: Evidence for Rescue of Mitochondrial Function. Biol. Open 2015, 4 (5), 622–626. 10.1242/bio.201511478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackers-Johnson M.; Li P. Y.; Holmes A. P.; O’Brien S. M.; Pavlovic D.; Foo R. S. A Simplified, Langendorff-Free Method for Concomitant Isolation of Viable Cardiac Myocytes and Nonmyocytes From the Adult Mouse Heart. Circ. Res. 2016, 119 (8), 909–920. 10.1161/CIRCRESAHA.116.309202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the results in this study are available within the paper and its Supporting Information. Source data for the figures are provided within this paper. Other raw data generated during the study are available from the corresponding authors on reasonable request.