

Abstract
Lysosomes are subjected to physiological and patho-physiological insults over the course of their life cycle and are accordingly repaired or recycled. Lysophagy, the selective degradation of lysosomes via autophagy, occurs upon unrepairable lysosomal membrane rupture; galectins bind to glycosylated macromolecules in the lysosome lumen, orchestrating a series of cellular responses to promote autophagic recycling of damaged lysosomes and transcriptional upregulation of lysosomal genes. Damaged lysosomes are ubiquitylated, resulting in the recruitment of ubiquitin-binding autophagy receptors, which promote assembly of an autophagosome around damaged lysosomes for delivery to healthy lysosomes for degradation. Here, we review the current state of our understanding of mechanisms used to mark and eliminate damaged lysosomes, and discuss the complexities of galectin function and ubiquitin-chain linkage types. Finally, we discuss the limitations of available data and challenges with the goal of understanding the mechanistic basis of key steps in lysophagic flux.
Introduction:
Lysosomes are the degradative endpoints within eukaryotic cells, but also function as complex signaling organelles linking the recycling of cellular building blocks to a myriad of metabolic pathways. Acidic hydrolases within the lysosome degrade diverse macromolecular substrates derived from cellular and extracellular compartments. These substrates are delivered to the lysosome primarily through the endocytic pathway wherein endosomes containing plasma membrane-derived proteins and other substrates fuse with lysosomes. In addition, damaged or surplus organelles and proteins are recycled via autophagy. These components are surrounded by a double membrane structure called an autophagosome, which subsequently fuses with a lysosome, allowing the contents to be degraded within the lysosomal lumen. Thus, the lysosome constitutes a central cellular hub maintaining protein and organelle homeostasis.
Reactive oxygen species or lipid metabolites can permeabilize the lysosomal limiting membrane that results in the leakage of hydrolases into the cytosol triggering cell death. As such, the maintenance of lysosome integrity is indispensable for cellular health. Upon damage, a sequence of endo-lysosomal damage response pathways are activated, resulting in either the repair of partially permeabilized membranes via re-sealing or the degradation of unrepairable lysosomes [1]. ESCRT III-mediated membrane resealing systems provide an acute response [2,3], but if membrane damage persists, such lysosomes are sequestered within autophagosomes, which subsequently fuse with healthy lysosomes in a selective autophagy pathway termed “lysophagy” [4,5]. Understanding the degree of damage required and subsequent signals for driving mechanisms that result in lysophagy as opposed to repair is an active area of research.
Lysosomal membrane integrity is lost in a variety of disease states and stress conditions. Damaged lysosomes are observed in tissues from patients with hyperuricemic nephropathy [6,7] and in tissues of patients with inclusion body myopathy associate with frontotemporal dementia[8]. Oxidative stress, proteases, specific types of lipids, and urate can all result in lysosome membrane permeabilization[9,10]. Damaged lysosomes display: 1) reduced acidity, 2) altered lipid composition, 3) reduced proteolytic capacity, and/or 4) increased propensity for rupture of the limiting lysosomal membrane [11]. Growing evidence indicates that endocytosed neurotoxic aggregates including α-synuclein, Huntington, Aβ, or tau fibrils can promote rupture of endolysosomal membranes, potentially allowing release of toxic aggregation-prone proteins into the cytosol if the damaged organelle isn’t rapidly eliminated [8,12–15]. Despite the many different physiological ways that lysosomes can be ruptured, the majority of studies employ small molecule lysosomotropic agents such as LLOMe (L-leucyl-L-leucine methyl ester) or GPN (glycyl-l-phenylalanine 2-naphthylamide) that promote largely synchronous rupture of lysosomal membranes, allowing kinetic and mechanistic dissection of downstream events [11]. LLOMe enters the lysosomal system via endocytosis and forms conjugates that can specifically rupture lysosomal membranes on a subset of lysosomes to initiate lysophagy, while GPN promotes lysosomal osmotic swelling and rupture. Membrane rupture can initiate a series of steps that facilitate lysophagy: 1) damage sensing by galectins, 2) amplification of the damage signal via ubiquitin conjugation onto lysosome-associated proteins, and 3) ubiquitin-dependent recruitment of autophagy machinery for subsequent capture and elimination of irreversibly damaged lysosomes [16, 17] (Figure 1). Elements within the lysophagy pathway have parallels with, and were in some cases initially discovered in the context of xenophagy, where bacteria-containing damaged vacuoles or phagosomes are targeted and degraded via autophagy [18,19]. Here we describe our mechanistic understanding of the aforementioned steps of lysophagy and elaborate on several areas where gaps in our understanding continue to exist.
Figure 1.
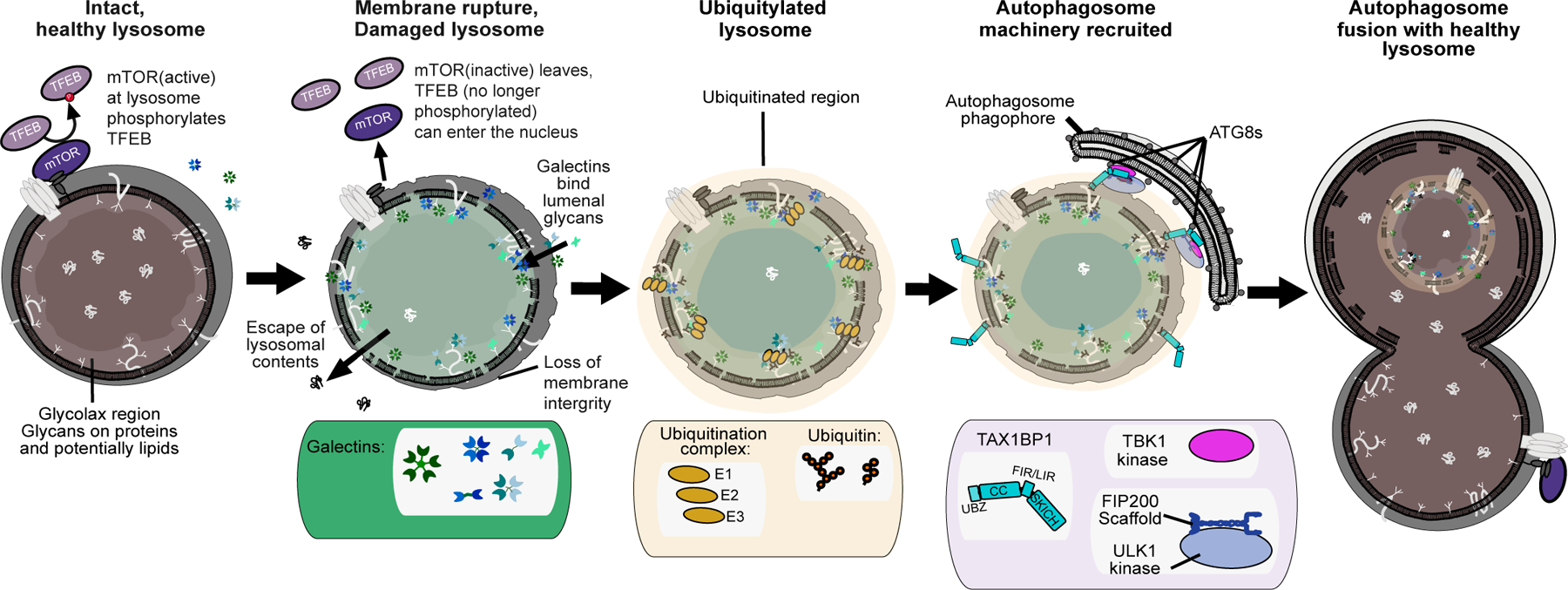
Overview of lysophagy. Healthy lysosomes have an intact membrane with an acidified lumen. Upon rupture of the limiting membrane, cytosolic galectins get access into the lumen where they associate with glycans. Rupture also results in the recruitment of ubiquitin ligase machinery which promotes the ultimate recruitment of autophagy receptors, thereby nucleating assembly of an autophagosome around the damaged lysosome. The autophagosome ultimately fuses with a healthy lysosome, thereby allowing recycling of the materials present in the damaged lysosome.
Galectin recruitment signals lysosomal membrane rupture
Macromolecules within the lysosomal lumen are modified with glycans, which become exposed upon rupture of the limiting membrane. Specific cytosolic galectins serve a surveillance function and rapidly access the lumen of ruptured lysosomes, where they bind glycans (preferentially beta-galactosides) using a conserved carbohydrate recognition domain (CRD). The galectin family of proteins, with 12 members in humans, conform to three general prototypes with distinct glycan specificities: homodimeric CDRs, tandem repeat CDRs typically binding unique glycans, and a chimera containing a CDR fused with a domain that interacts with other proteins (Figure 2). Interestingly, unique galectin types are selective to which glycan modifications they will bind [20], and galectins also display distinct patterns of expression across cell types and tissues [20–22].
Figure 2.
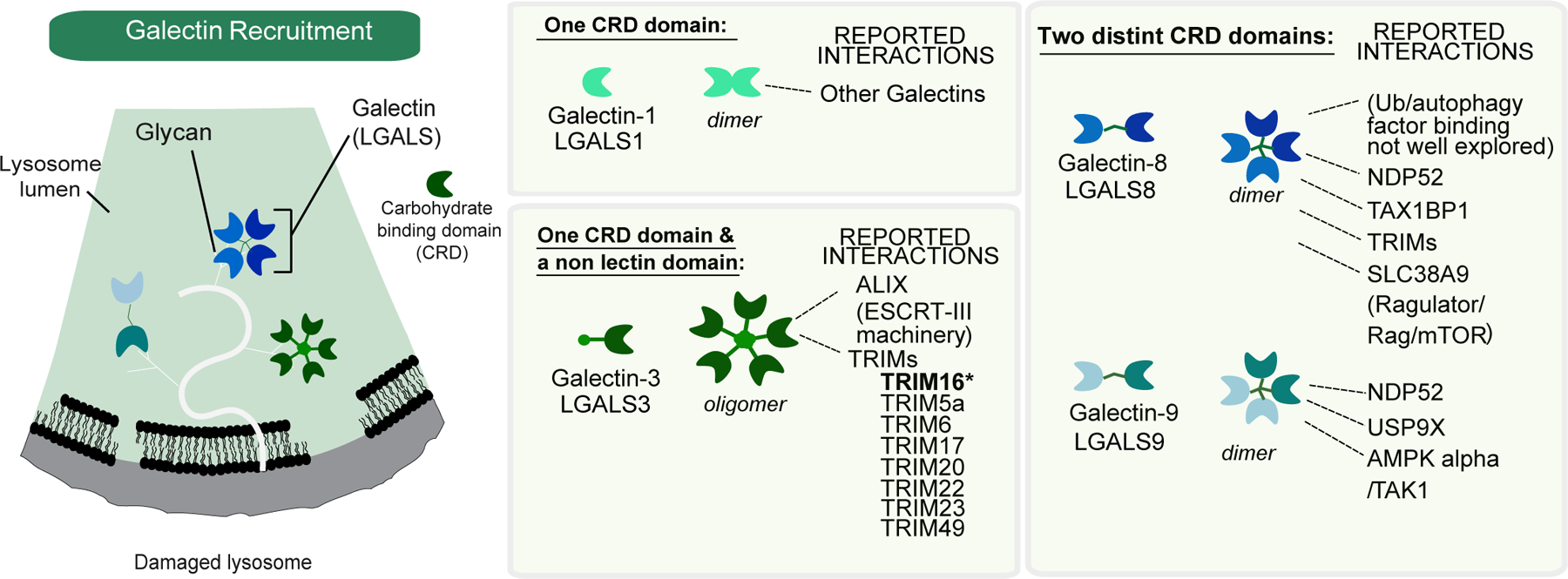
Overview of galectins. Galectins are rapidly recruited to the lumen of lysosomes upon membrane rupture, where they associate with glycans. Multiple classes of galectins have been shown to be recruited to damaged lysosomes, including LGALS3, LGALS8, and LGALS9. Galectins associate with glycans through their carbohydrate recognition domains (CRD). While some galectins have single CRDs, others have two CRD domains in a single polypeptide while others oligomerize to create a cluster of CRDs. Various galectins have been reported to associate with multiple classes of proteins, ranging from autophagy receptors and components of the membrane repair apparatus to members of the TRIM family of ubiquitin ligases.
Four Galectins (LGALS1, LGALS3, LGALS8, and LGALS9) are rapidly recruited to ruptured lysosomes and initiate the process of lysophagy [23–28] (Figure 1,2). However, to date, removal of no single galectin or combination of galectins, has been shown to completely block lysophagic flux, although defects in lysosomal ubiquitylation and recruitment of autophagy machinery (ATG13, ATG16L1, LC3B) to membrane-compromised lysosomes have been reported [24–27]. As such, the precise contributions of specific galectins to downstream processes and the extent of redundancy is incompletely understood.
LGALS3 and LGALS8 (Figure 2), while not required for lysosomal ubiquitylation [29], nevertheless contribute to repair and/or recycling of damaged lysosomes but appear to play roles that are independent of lysosomal ubiquitylation [17,25,26,29]. ALIX – a regulator of the ESCRT III membrane resealing complex – is rapidly recruited to damaged lysosomes and this recruitment is largely abolished in cells lacking LGALS3, indicating a role for LGALS3 in membrane resealing [25]. Indeed, cells lacking LGALS3 fail to effectively repair lysosomal membranes, have reduced capability to recycle damaged lysosomes (as indicated by reduced recruitment of LC3B) but instead display increased nuclear TFEB, indicating a prolonged transcriptional upregulation of lysosomal biogenesis genes [25]. LGALS8 recruitment to damaged lysosomes leads to inhibition of mTOR via interactions with the Ragulator-SLC38A9 system [26]. Ragulator is a lysosomal membrane associated complex that associates with a heterodimeric Rag GTPase complex whose GTPase activity is required for mTOR activation in response to amino acids. In the absence of LGALS8, mTOR is not effectively released from damaged lysosomes, as assessed by immunofluorescence [26]. Consistent with this, proteomic analysis of purified lysosomes in response to lysosomal damage revealed rapid loss of mTORC1 complex subunits (mTOR, Raptor, MLST8) and Rag GTPase subunits, reflecting dynamic release from damaged lysosomes [27]. Negative regulation of mTOR via LGALS8 may act synergistically with AMPK activation to promote clearance of damaged lysosomes via autophagy, as indicated by reduced LC3B lipidation upon lysosomal damage in cells lacking LGALS8 [26].
While cells lacking LGALS3 or LGALS8 display wild-type levels of lysosomal ubiquitylation (ubiquitin puncta as detected by immunofluorescence), deletion of LGALS9 results in a 50–60% reduction in lysosomal ubiquitylation, which is rescued by expression of wild-type LGALS9 but not a glycan binding mutant [29]. Whether this partial reduction in lysosomal ubiquitylation reflects compensatory functions for other galectins is unknown. Interestingly, LGALS9 can associate with the USP9X deubiquitylating enzyme (DUB) (Figure 2), an interaction that is lost upon lysosomal damage, and depletion of USP9X in cells lacking LGALS9 results in rescue of lysosomal ubiquitylation [29] (Figure 3). This finding suggests that LGALS9 itself is not essential for recruitment of ubiquitylation machinery to damaged lysosomes, but may indirectly control access of USP9X to ubiquitylated lysosomal proteins. LGALS9 also appears to regulate additional signaling systems on lysosomal membranes by displacing USP9X from its associated TAK1 subunit, which in turn activates adenosine monophosphate (AMP)-activated protein kinase (AMPK) and ULK1 phosphorylation to activate autophagy [29] (Figure 3).
Figure 3.
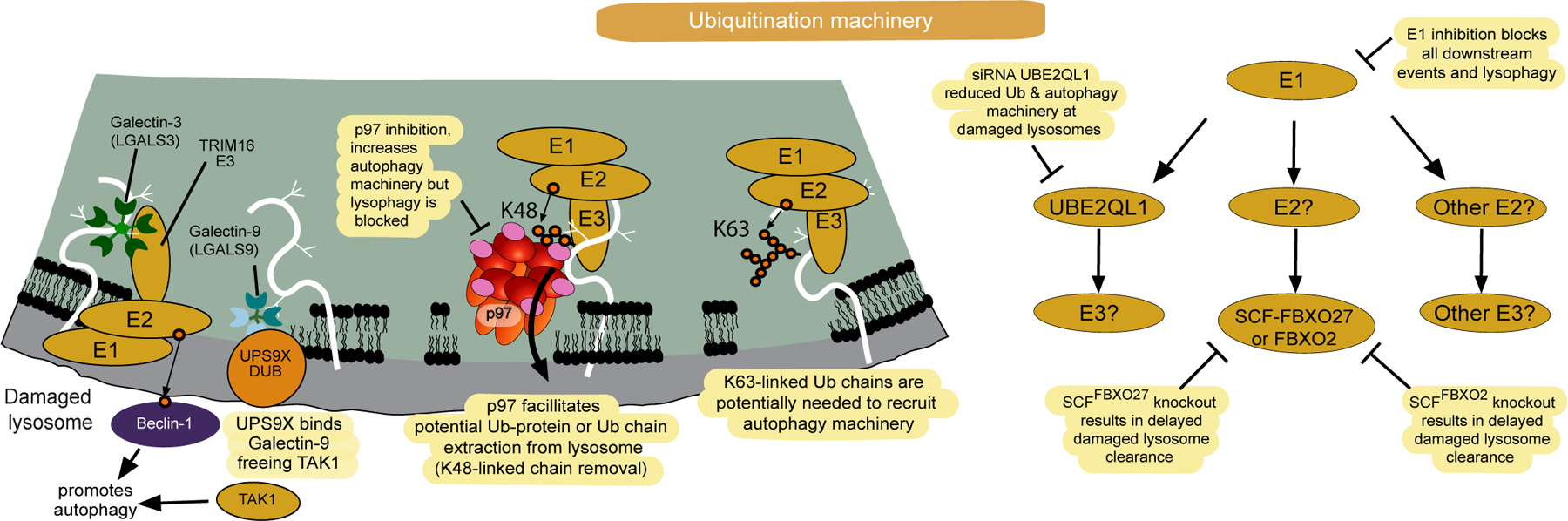
Lysosome ubiquitylation in response to membrane rupture. In response to membrane rupture, lysosomal proteins are ubiquitylated thereby promoting recruitment of autophagy receptors (left panel). Multiple E2 and E3 enzymes have been reported to function in lysosomal ubiquitylation or ubiquitylation of autophagy machinery, including the E2 enzyme UBE2QL1, and the E3 ligases SCFFBXO27 and TRIM16 (left and right panels). TRIM16, which is recruited via LGALS3, has been reported to ubiquitylate the Beclin subunit of the VPS34 PI3 kinase complex. SCFFBXO27 is reported to ubiquitylate LAMP1 and LAMP2 in response to membrane rupture but cells lacking FBXO27 have only a modest defect in clearance. p97 promotes extraction of proteins labeled with K48-chains likely downstream of the UBE2QL1 E2 enzyme.
Lysosomal ubiquitylation in response to membrane rupture
One of the earliest responses to lysosomal membrane damage is the accumulation of ubiquitin on lysosomes [8,16] (Figure 1). Indeed, pharmacological inhibition of the ubiquitin-activating enzyme (UBA1) completely blocks lysophagic flux in response to damage [27]. In HeLa cells, lysosomal membrane damage is associated with rapid accumulation of K63-linked Ub chains and delayed assembly of K48-linked chains on a subset of damaged lysosomes [8] (Figure 3). Whether these ubiquitylation patterns based on immunostaining reflect distinct states, for example reflecting the extent of initial damage, or simply the maturation state of the process, is currently unclear. Lysosomal ubiquitylation promotes the recruitment of ubiquitin-binding autophagy receptors as described below. During lysosomal ubiquitylation, the p97/VCP AAA+-ATPase – a ubiquitin-dependent segregase – is recruited to a subset of ubiquitylated lysosomes and pharmacological inhibition or genetic depletion of p97 function blocks lysophagy [8] (Figure 3). p97 employs a number of accessory factors to regulate its targets and in the case of lysophagy, the PLAA adaptor protein and YOD1 deubiquitylating enzyme are thought to be associated with p97 on damaged lysosomes. Interestingly, a Ub-binding site on YOD1 is necessary for recruitment to K48-linked conjugates, leading to the idea that YOD1 serves as a sensor for this form of damaged lysosomes [8]. Indeed, current models posit that p97 functions to extract K48-linked ubiquitylated target proteins from the damaged lysosomal membrane which precedes it’s clearance [16] (Figure 3). This hypothesis is supported by the findings that in the presence of catalytically inactive p97, K48-linked conjugates and LC3B accumulate on damaged lysosomes that are not effectively cleared [8]. Interestingly, subsequent proteasomal degradation of the K48-Ub modified proteins is not required, as proteasome inhibition does not block lysophagy [8]. Nevertheless, the underlying biochemical mechanisms are yet to be established and precisely how this step could be required for “licensing” autophagy remains unclear, especially given the apparent role of K63-Ub chains in recruiting autophagy adaptors as described below.
While ubiquitylation is essential for lysophagic flux, our understanding of this process is fragmentary. Active areas of research include 1) identifying lysosomal proteins or possibly non-proteinaceous components that are ubiquitylated in response to membrane rupture 2) identifying relevant ubiquitylation machinery, and 3) understanding how relevant machinery is either directed to damaged lysosomes or activated upon loss of lysosomal membrane integrity. As mentioned above, LGALS9 deletion reduces lysosomal ubiquitylation upon damage but this effect is eliminated upon co-depletion of USP9X, indicating that LGALS9 is not an essential gate-keeper for lysosomal ubiquitylation [29]. LGALS3 has been linked with recruitment of the TRIM16 ubiquitin ligase to damaged lysosomes, but in this context, TRIM16 appears to associate with ULK1, Beclin and ATG16 and to ubiquitylate Beclin1, but has not been shown to directly ubiquitylate components of the lysosome itself [24] (Figure 3). Indeed, cells genetically engineered to lack LGALS3 have no apparent defect in lysosomal ubiquitylation as assessed using immunofluorescence [25]. Thus, the role for TRIM16 as Ub ligase directly regulating lysophagy through ubiquitylation of lysosomally localized proteins remains unclear.
The primary candidate E2 and E3 enzymes for ubiquitylation of damaged lysosomes are the E2 conjugating enzyme UBE2QL1, and two E3 ligases - SCFFBXO27, and SCFFBXO2[30–32] (Figure 3). However, whether each of these components function independently or in concert is unclear, and there is no evidence that UBE2QL1 can function in the context of an SCF ubiquitin ligase. UBE2QL1 accumulates on damaged lysosomes with kinetics that correlate with assembly of K48-linked chains (~1–2 hours), and depletion of UBE2QL1 by siRNA partially reduces both the recruitment of p97 and the clearance of LGALS3-positive lysosomes but does not eliminate K63 ubiquitylation [30] (Figure 3). Therefore, while UBE2QL1 appears to play an important modulatory role, the E3 ligase(s) that function together with this E2 in this context are unknown.
SCFFBXO27 and SCFFBXO2 are composed of the scaffold protein CUL1, the RING domain protein RBX1, and the SKP1-FBXO27 (or FBXO2) substrate adaptor module, and the activity of SCF complexes requires that CUL1 be specifically neddylated by the NAE1 NEDD8 activation and transfer machinery [33]. Interestingly, FBXO27 and FBXO2 contain an “F-box associated domain” capable of binding to carbohydrate moieties on proteins [34] and are recruited to damaged lysosomes, within minutes of LGALS3 recruitment in the case of FBXO27 [31,32]. Overexpression of SCFFBXO27 promotes K48-linked ubiquitylation of LAMP2 and to a lesser extent LAMP1 [31], but whether this E3 ligase is responsible for assembly of K48-linked chains that are removed by p97-YOD1 is unknown (Figure 3). Deletion of FBXO27 in PANC-1 cells leads to a modest reduction in ubiquitin-binding receptor SQSTM1 and LC3B localization at damaged lysosomes and clearance of LGALS3-positive lysosomes [31]. However, significant lysosome clearance was still present, indicating that SCFFBXO27 is not absolutely essential for this process [31]. SCFFBXO2 has been studied in the context of a Nieman Pick type C disease model [32]. In NPC1 mutant fibroblasts, unesterified cholesterol accumulates in late endosomes and lysosomes, rendering these organelles more susceptible to membrane damage and recruitment of LGALS3. Overexpressed FBXO2 is recruited to damaged lysosomes in fibroblasts and Fbxo2−/− cortical neurons display a slight delay in turnover of LGALS3 upon lysosomal damage [30]. NPC1 mutant mice additionally deficient in FBXO2 exhibited significantly worse motor function and decreased survival [32], but whether this is a reflection of defects in in lysophagy is unknown. However, the general and absolute requirement for these E3 ligases in lysophagic flux is brought into question by the finding that this process is not blocked by the cullin neddylation inhibitor MLN4924 under conditions that eliminate CUL1 neddylation in HeLa cells (M.J.H., Julia Paoli, J.W.H., unpublished results), but is blocked by inhibition of the ubiquitin E1 enzyme with an analogous small molecule inhibitor MLN7243 both HeLa and fibroblasts [27]. Given that cullin neddylation is required for SCF activation and indeed all cullin-RING E3 ligases [33], it would appear that this broad class of E3 ligases is not required for lysophagic flux in HeLa cells.
Taken together, the available data suggest that further studies are needed to identify additional ubiquitylation machinery responsible for marking damaged lysosomes, and for understanding any cell-type dependent control of the process, for example via distinct E2 and E3 enzymes. In particular, the identification of machinery capable of assembly of K63-linked chains on damaged lysosomes would represent a step forward, given the apparent role of K63-linked Ub chains in recruitment of autophagy receptors, as discussed below.
Role of Ub-binding autophagy receptors in lysophagy
Previous studies have identified a small group of related proteins as key receptors linking cargo ubiquitylation with assembly of autophagosomes in situ on the ubiquitylated organelle [35]. These proteins – typified by OTPN (also called optineurin), TAX1BP1, NBR1, CALCOCO2 (also called NDP52), and SQSTM1 (also called p62) contain C-terminal Ub-interacting domains and extensive coiled-coil domains (except for SQSTM1) that typically serve as dimerization domains (Figure 4A). Multiple types of Ub-binding domains are found within these proteins, including UBZ1, UBA, Znf, and UBAN domains [36], and while systematic data is lacking, these domains appear to generally prefer association with linear or K63-linked ubiquitin chains, as measured in vitro [37–39], although in some studies CALCOCO2 was found to bind M1, K48, and K63 chains equally well [39]. These Ub-binding domains bind to ubiquitylated cargo to promote autophagosome formation [35]. In addition, these receptors contain short motifs that interact with either ATG8 proteins on the surface of growing phagophores and/or the C-terminal “claw” domain of FIP200 (also called RB1CC1) (Figure 4A–D). Historically, the LC3 interacting region (LIR) was demonstrated to bind directly to ATG8 proteins, but more recently, LIR-like motifs have been shown to also bind to the FIP200 claw (referred to as FIR motifs), thereby recruiting the FIP200-ULK1 kinase to ubiquitylated cargo [40–43] (Figure 4C). In addition, CALCOCO2 and TAX1BP1 contain N-terminal SKICH domains that can independently bind the coiled-coil region of FIP200 (Figure 4B,C) [41,44,45]. It is thought that ULK1 kinase activity in the proximity of the autophagic cargo leads to activation of PI3P kinase activity and WIPI protein recruitment, thereby initiating autophagosome assembly in situ (Figure 4B,D). This mechanism explains how autophagosome formation is limited to the target organelle for elimination [46,47].
Figure 4.
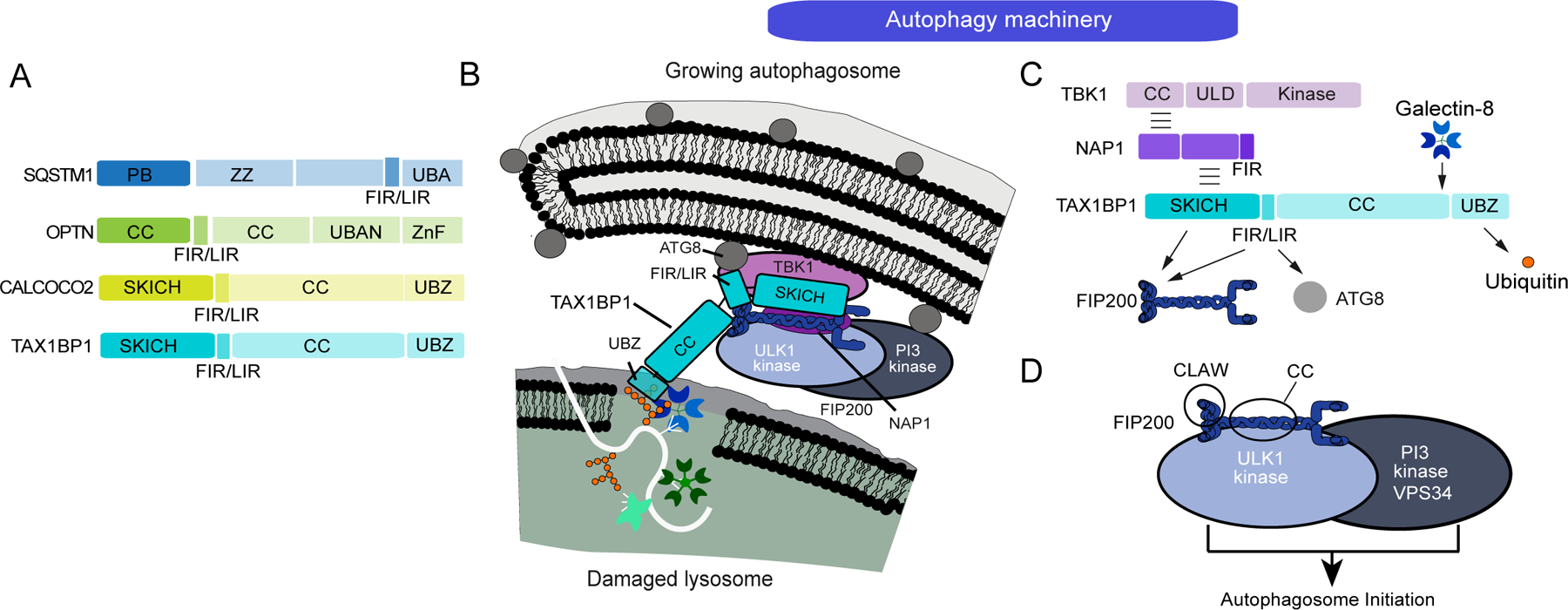
Recognition of ubiquitylated lysosomes by the autophagy receptor TAX1BP1. (A) Domain organization for ubiquitin-binding autophagy receptors. (B) Model depicting molecular interactions between damaged lysosomes and the autophagosome initiation machinery. TAX1BP1 interacts with the FIP200-ULK1 complex, which in turn interacts with the VPS34 PI3 kinase complex. (C) Schematic showing interactions between TAX1BP1 and the autophagy machinery. TAX1BP1 contains an N-terminal SKICH domain and FIR motifs that both associate with the FIP200 claw domain and sequences within the coiled-coil. The SKICH domain also associates with the TBK1 kinase complex via the adaptor proteins NAP1 and SINTBAD. The C-terminal UBZ domain binds ubiquitin chains. (D) Schematic of the FIP200-ULK1 complex and its association with the VPS34 PI3 kinase complex, whose recruitment to autophagic cargo can initiate autophagosome formation via formation of PI3P on target membranes.
Early studies using RNAi indicated that SQSTM1 promotes clearance of LGALS3-positive lysosomes [1,8]. However, subsequent studies in HeLa cells indicate that lysosomal damage leads to rapid (<30 min) recruitment of multiple receptors – including OTPN, TAX1BP1, CALCOCO2, and SQSTM1. Using GFP-RFP-LGALS3 or mKeima-LGALS3 reporters to measure lysophagic flux, it was found that deletion of SQSTM1, OPTN, or CALCOCO2 did not block lysophagic flux [27]. In contrast, cells engineered to lack TAX1BP1 (Figure 4B) were profoundly defective in lysophagy, as determined using both flux reporters and galectin puncta clearance assays in HeLa cells or induced neurons [27]. A common feature of these receptors is association with the TBK1 protein kinase, either directly in the case of OPTN or through one of two adaptor proteins (NAP1 and SINTBAD) in the case of SKICH domain receptors (Figure 4B,C), and TBK1 is required for other types of autophagic flux for cargo ranging from intracellular bacteria to mitochondria [39,41,48,49]. Indeed, HeLa cells or induced neurons either lacking the TBK1 gene or TBK1 activity via pharmacological inhibition also display defects in lysophagic flux [27].
Taken together, the current model for cargo receptor function in lysophagy is that upon membrane rupture and lysosome ubiquitylation, TAX1BP1 in association with TBK1 is recruited to lysosomes (Figure 4B). In this context, TAX1BP1 can recruit the ULK1-FIP200 complex to initiate in situ phagophore initiation. Evidence for this model includes the finding that deletion of the SKICH domain, or mutations that render SKICH unable to associate with FIP200, inactivates TAX1BP1-dependent lysophagic flux [27]. As with CALCOCO2 in bacterial autophagy [44], TAX1BP1 can also interact with LGALS8 [50] and could therefore be brought to damaged lysosomes independently of a ubiquitin signal. However, mutation of the C-terminal UBZ1 in TAX1BP1 impairs lysophagic flux [27], indicating that lysosomal ubiquitylation plays a role in amplifying autophagic signaling. In contrast, mutation of sequences in TAX1BP1 that bind LGALS8 do not impact lysophagic flux [27]. Interestingly, in HeLa cells lacking OPTN, CALCOCO2, and TAX1BP1, ectopic expression of OPTN – but not CALCOCO2 – can rescue lysophagic flux and this activity is absolutely dependent on the ability of OPTN to bind ubiquitin [27].
Open questions in lysophagy
Data accumulated thus far indicates a complex interplay between galectins, membrane repair machinery, the ubiquitin system, and autophagy machinery in the cellular decisions as to the fate of damaged lysosomes, but several aspects remain mechanistically unresolved. Central to this is the decision of whether to repair damaged lysosomal membranes via ESCRT-related pathways. Although ESCRT proteins are known to be rapidly recruited to damaged lysosomes[2,3,25,27,51], what determines the damage threshold for repair versus degradation is unclear, as are any mechanisms that control “switching” between the two pathways [17]. Analysis of the earliest stages of lysosomal damage has primarily relied on clearance of LGALS3-positive puncta using immunofluorescence [28], which provided an indirect readout of the process. The development of tandem GFP-RFP or mKeima lysosome flux assays allows for quantitative analysis of the delivery of damaged lysosome to healthy lysosomes for degradation via a change in fluorescent property upon autophagosome-lysosome fusion but not complete loss of fluorescence [7,11,27], Such assays should now be used to facilitate a more rigorous analysis of the pathway. Among the most pressing questions concerns the identity of the enzymatic machinery necessary for K63-linkage ubiquitylation of damaged lysosomes. As described above, the E3 ligases responsible for tagging lysosomes, as well as the relevant targets within the organelle, are not clearly defined (Figure 3). The finding that cells depleted of UBE2QL1 maintain significant K63-linked chains [30] indicates that additional E2s are likely to be involved, and the expectation is that such E2s would function together with relevant E3 ligases to produce K63-linked chains. We note that lysosomes harbor several resident RING-domain containing E3 ligases including RNF13, RNF167, and RNF152, although none of these has been linked with lysophagy. In the case of bacterial autophagy, two Ub ligases – RNF213 and LUBAC (linear Ub assembly complex) – have been demonstrated to work in sequence to ubiquitylate both the LPS molecule on the bacterial surface as well as proteins on the bacteria or vacuole membrane [52–54]. Both of these enzymes are capable of linking Ub to non-proteinaceous moieties via oxyester linkages [52,55] and it is conceivable that glycans present within the lysosomal membrane could also be modified by relevant lysophagy E3 ligases rather than canonical lysine residues in proteins. The ability to elucidate the extent to which mechanisms of lysophagy are cell type specific, and the machinery responsible for pathway activation, will require identification of relevant E3 ligases across a range of cell types and damaging agents. Finally, the identification of the relevant enzymes is critical for providing mechanistic clarity to the underlying process, as well as eventual goals of reconstituting key steps in the pathway [46]. This includes a determination of the lysosome-associated molecules that are directly conjugated with ubiquitin as a first step in understanding whether target specificity plays a role in the ultimate recruitment of the ubiquitylation machinery.
Also unclear is the extent to which galectins themselves support recruitment of the ubiquitylation machinery. Although deletion of LGALS9 reduces lysosomal ubiquitylation, this activity is completely rescued upon co-depletion of USP9X [29]. As such, no single galectin has been demonstrated to be required for ubiquitylation of damaged lysosomes. Systematic genetic analysis of galectin mutants may be required to understand relevant dependences once relevant ubiquitylation machinery is identified. The development of a more comprehensive understanding of the molecular mechanisms involved will likely facilitate future studies aimed at understanding the identity of endogenous triggers of lysosomal damage, and how pathogenic states may contribute to lysosomal dysfunction.
ACKNOWLEDGMENTS
J.W.H. acknowledges research support from the NIH (AG011085, NS083524, NS110395, U24 HG006673), Aligning Science Across Parkinson’s (ASAP), Chan-Zuckerberg Neurodegeneration Challenge Network Collaborative Grant, and the Bluefield Project. The Michael J. Fox Foundation administers the grant ASAP-000282 on behalf of ASAP and itself. For the purpose of open access, the author has applied a CC-BY public copyright license to the Author Accepted Manuscript (AAM) version arising from this submission. M.J.H. was supported by a postdoctoral fellowship from Jane Coffin Childs Foundation.
Footnotes
CONFLICT OF INTERESTS
J.W.H. is a consultant and co-founder of Caraway Therapeutics and is a founding board member of Interline Therapeutics.
REFERENCES
- 1.Papadopoulos C, Meyer H: Detection and Clearance of Damaged Lysosomes by the Endo-Lysosomal Damage Response and Lysophagy. Curr Biol 2017, 27:R1330–R1341. [DOI] [PubMed] [Google Scholar]
- 2.Radulovic M, Schink KO, Wenzel EM, Nahse V, Bongiovanni A, Lafont F, Stenmark H: ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J 2018, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skowyra ML, Schlesinger PH, Naismith TV, Hanson PI: Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 2018, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hung YH, Chen LM, Yang JY, Yang WY: Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat Commun 2013, 4:2111. [DOI] [PubMed] [Google Scholar]
- 5.Hasegawa J, Maejima I, Iwamoto R, Yoshimori T: Selective autophagy: lysophagy. Methods 2015, 75:128–132. [DOI] [PubMed] [Google Scholar]
- 6.Emmerson BT, Cross M, Osborne JM, Axelsen RA: Reaction of MDCK cells to crystals of monosodium urate monohydrate and uric acid. Kidney Int 1990, 37:36–43. [DOI] [PubMed] [Google Scholar]
- 7.Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, et al. : Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 2013, 32:2336–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, Poehler R, Dressler A, Fengler S, Arhzaouy K, et al. : VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J 2017, 36:135–150. p97 translocates to damaged lysosomes, p97 mutations disrupt lysosome clearance, and damaged lysosomes accumulate in patient mutation tissues. In their model, p97 acts downstream of K63-linked ubiquitination and p62 recruitment to selectively remove K48-linked ubiquitin conjugates using cofactors UBXD1, PLAA, and the deubiquitinating enzyme YOD1. This K48-linked ubiquitin removal is thought to promote autophagosome formation and drive damaged lysosome clearance.
- 9.Gomez-Sintes R, Ledesma MD, Boya P: Lysosomal cell death mechanisms in aging. Ageing Res Rev 2016, 32:150–168. [DOI] [PubMed] [Google Scholar]
- 10.Nixon RA: The aging lysosome: An essential catalyst for late-onset neurodegenerative diseases. Biochim Biophys Acta Proteins Proteom 2020, 1868:140443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Otomo T, Yoshimori T: Lysophagy: A Method for Monitoring Lysosomal Rupture Followed by Autophagy-Dependent Recovery. Methods Mol Biol 2017, 1594:141–149. [DOI] [PubMed] [Google Scholar]
- 12.Falcon B, Noad J, McMahon H, Randow F, Goedert M: Galectin-8-mediated selective autophagy protects against seeded tau aggregation. J Biol Chem 2018, 293:2438–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bussi C, Peralta Ramos JM, Arroyo DS, Gallea JI, Ronchi P, Kolovou A, Wang JM, Florey O, Celej MS, Schwab Y, et al. : Alpha-synuclein fibrils recruit TBK1 and OPTN to lysosomal damage sites and induce autophagy in microglial cells. J Cell Sci 2018, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siew JJ, Chen HM, Chen HY, Chen HL, Chen CM, Soong BW, Wu YR, Chang CP, Chan YC, Lin CH, et al. : Galectin-3 is required for the microglia-mediated brain inflammation in a model of Huntington’s disease. Nat Commun 2019, 10:3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ditaranto K, Tekirian TL, Yang AJ: Lysosomal membrane damage in soluble Abeta-mediated cell death in Alzheimer’s disease. Neurobiol Dis 2001, 8:19–31. [DOI] [PubMed] [Google Scholar]
- 16.Papadopoulos C, Kravic B, Meyer H: Repair or Lysophagy: Dealing with Damaged Lysosomes. J Mol Biol 2020, 432:231–239. [DOI] [PubMed] [Google Scholar]
- 17.Jia J, Claude-Taupin A, Gu Y, Choi SW, Peters R, Bissa B, Mudd MH, Allers L, Pallikkuth S, Lidke KA, et al. : MERIT, a cellular system coordinating lysosomal repair, removal and replacement. Autophagy 2020, 16:1539–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tripathi-Giesgen I, Behrends C, Alpi AF: The ubiquitin ligation machinery in the defense against bacterial pathogens. EMBO Rep 2021, 22:e52864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyle KB, Randow F: The role of ‘eat-me’ signals and autophagy cargo receptors in innate immunity. Curr Opin Microbiol 2013, 16:339–348. [DOI] [PubMed] [Google Scholar]
- 20.Johannes L, Jacob R, Leffler H: Galectins at a glance. J Cell Sci 2018, 131. [DOI] [PubMed] [Google Scholar]
- 21.Punt S, Thijssen VL, Vrolijk J, de Kroon CD, Gorter A, Jordanova ES: Galectin-1, −3 and −9 Expression and Clinical Significance in Squamous Cervical Cancer. PLoS One 2015, 10:e0129119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thijssen VL, Heusschen R, Caers J, Griffioen AW: Galectin expression in cancer diagnosis and prognosis: A systematic review. Biochim Biophys Acta 2015, 1855:235–247. [DOI] [PubMed] [Google Scholar]
- 23.Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F: Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012, 482:414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, Choi SW, Peters R, Mandell M, Bruun JA, et al. : TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-direct Autophagy in Endomembrane Damage Homeostasis. Dev Cell 2016, 39:13–27. A previous screen to uncover the broad role of TRIM proteins in autophagy pointed to interactions between TRIMs and Galectins. In basal conditions TRIM5α, TRIM6, TRIM17, TRIM20, TRIM22, TRIM23, and TRIM49 immunoprecipitated with both LGALS3 and LGALS8. Upon lysosomal damage, TRIM16 interacts with LGALS3. In TRIM16 knock out HeLa cells, ubiquitin puncta still form when lysosomal damage reagent is introduced, but there is a reduction in LC3B immunofluorescence at these puncta. Through LGALS3 interaction, TRIM16 appears to associate with ULK1 and ATG16 and to ubiquitylate Beclin1. Subsequently, TRIM16 affects mTOR and TFEB to upregulate autophagy and lysosome biogenesis, respectively.
- 25. Jia J, Claude-Taupin A, Gu Y, Choi SW, Peters R, Bissa B, Mudd MH, Allers L, Pallikkuth S, Lidke KA, et al. : Galectin-3 Coordinates a Cellular System for Lysosomal Repair and Removal. Dev Cell 2020, 52:69–87 e68. The ESCRT membrane resealing complex does not get properly recruited to damaged lysosomes when cells lack LGALS3. Additionally, LGALS3 knockout cells fail to effectively repair lysosomal membranes and display persistent nuclear TFEB, indicating a prolonged transcriptional response for lysosomal biogenesis. Autophagy component puncta formation was diminished in LGALS3 knockout cells. At later lysosome damage time points LGALS3 interacts with TRIM16, a scaffold protein that potentially promotes autophagic clearance of lysosomes.
- 26. Jia J, Abudu YP, Claude-Taupin A, Gu Y, Kumar S, Choi SW, Peters R, Mudd MH, Allers L, Salemi M, et al. : Galectins Control mTOR in Response to Endomembrane Damage. Mol Cell 2018, 70:120–135 e128. Generated LGALS3, LGALS8, LGALS9 knockout cells. co-IPs of LGALS3, LGALS8, and LGALS9 reveal that only LGALS8 interacts with mTOR and the Ragulator Rag SLC38A9 system. There is increased binding of LGALS8 to SLC38A9 and Ragulator components upon lysosomal damage which affects Rag GTPases, inhibits mTOR activity, and promotes autophagy. co-IPs of LGALS3, LGALS8, and LGALS9 reveal that only LGALS9 interacts with AMPKα. There is increased binding of LGALS9 to AMPKα upon lysosomal damage. LGALS9 engagement with TAK1 and AMPKα upregulates AMPKα to promote autophagy.
- 27. Eapen VV, Swarup S, Hoyer MJ, Paulo JA, Harper JW: Quantitative proteomics reveals the selectivity of ubiquitin-binding autophagy receptors in the turnover of damaged lysosomes by lysophagy. Elife 2021, 10. Proteomic and immunofluorescence analysis detected the recruitment of multiple ubiquitin binding receptors – including OTPN, TAX1BP1, CALCOCO2, and SQSTM1. GFP-RFP-LGALS and Keima-LGALS reporter systems were generated to measure damaged lysosome flux to healthy lysosomes through autophagy (lysophagic flux). Pharmacological inhibition or knock out of TBK1 resulted in defective lysophagic flux (TBK1 is a protein kinase previously shown to interact with these receptors and link these receptors to autophagy components). OTPN or TAX1BP1 overexpression in a triple receptor (CALCOCO2, OPTN, TAX1BP1) knockout cell line were both capable of regaining lysophagic flux. Single knock out of TAX1BP1 (but not SQSTM1, OPTN, or CALCOCO2) blocked lysophagic flux in HeLa cells. TAX1BP1-driven lysophagic flux required a N-terminal SKICH domain, which binds both TBK1 and the autophagy regulatory factor RB1CC1, and requires upstream ubiquitylation events. TAX1BP1, with its associated kinase TBK1, are both necessary and sufficient to promote lysophagic flux in both HeLa cells and induced neurons.
- 28.Aits S, Kricker J, Liu B, Ellegaard AM, Hamalisto S, Tvingsholm S, Corcelle-Termeau E, Hogh S, Farkas T, Holm Jonassen A, et al. : Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 2015, 11:1408–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jia J, Bissa B, Brecht L, Allers L, Choi SW, Gu Y, Zbinden M, Burge MR, Timmins G, Hallows K, et al. : AMPK, a Regulator of Metabolism and Autophagy, Is Activated by Lysosomal Damage via a Novel Galectin-Directed Ubiquitin Signal Transduction System. Mol Cell 2020, 77:951–969 e959. LGALS9 knock out results in a 50–60% reduction in ubiquitylation of damaged lysosomes (LGALS3 and LGALS8 depletion did not reduce ubiquitylation). Regaining wildtype ubiquitylation levels is dependent on LGAS9’s ability to recognize lysosome lumenal glycans. In untreated cells LGALS9 associates with USP9X deubiquitylating enzyme (DUB), but this interaction is lost upon lysosomal damage. LGALS9 deletion reduces damaged lysosome ubiquitylation but this effect is eliminated upon co-depletion of USP9X. These findings suggest LGALS9 itself is not essential for access or recruitment of ubiquitylation machinery to damaged lysosomes, but may indirectly control access of this DUB to ubiquitylated lysosomal proteins. LGALS9 controls additional signaling systems on the lysosomal membrane by displacing USP9X from its associated TAK1 subunit, which in turn activates adenosine monophosphate (AMP)-activated protein kinase (AMPK) and ULK1 phosphorylation to activate autophagy
- 30. Koerver L, Papadopoulos C, Liu B, Kravic B, Rota G, Brecht L, Veenendaal T, Polajnar M, Bluemke A, Ehrmann M, et al. : The ubiquitin-conjugating enzyme UBE2QL1 coordinates lysophagy in response to endolysosomal damage. EMBO Rep 2019, 20:e48014. A microscopy based siRNA screen for E2 ubiquitin protein ligases detected altered levels of ubiquitin at damaged lysosomes when UBE2QL1 was depleted. UBE2QL1 translocates to damaged lysosomes. UBE2QL1 knockdown reduces ubiquitination, autophagy receptor p62, autophagosome component LC3B and p97 levels at damaged lysosomes. Recruitment of LGALS3 or LGALS8 to damaged lysosomes does not require the Ub pathway downstream of UBE2QL1. In untreated cells, depletion of UBE2QL1 results in increased lysosomal damage, mTOR dissociation from lysosomes (signals inactivation of mTOR and subsequent upregulation of autophagy), and TFEB activation (TFEB translocates to the nucleus to upregulate lysosomal biogenies).
- 31. Yoshida Y, Yasuda S, Fujita T, Hamasaki M, Murakami A, Kawawaki J, Iwai K, Saeki Y, Yoshimori T, Matsuda N, et al. : Ubiquitination of exposed glycoproteins by SCF(FBXO27) directs damaged lysosomes for autophagy. Proc Natl Acad Sci U S A 2017, 114:8574–8579. FBXO27 is recruited to damaged lysosomes and this recruitment is dependent on a glycoprotein binding region within FBXO27. Overexpression of SCFFBXO27 promotes K48-linked ubiquitylation of LAMP2 and to a lesser extent LAMP1. Deletion of FBXO27 in PANC-1 cells results in reduced ubiquitylation of LAMP2, a decrease in autophagy machinery at damaged lysosomes and reduced clearance of LGALS3-positive lysosomes. However, a small amount of ubiquitylation of LAMP2, autophagosome machinery recruitment, and damaged lysosome clearance still occurred in the FBXO27 knock out cells.
- 32.Liu EA, Schultz ML, Mochida C, Chung C, Paulson HL, Lieberman AP: Fbxo2 mediates clearance of damaged lysosomes and modifies neurodegeneration in the Niemann-Pick C brain. JCI Insight 2020, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harper JW, Schulman BA: Cullin-RING Ubiquitin Ligase Regulatory Circuits: A Quarter Century Beyond the F-Box Hypothesis. Annu Rev Biochem 2021, 90:403–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW: Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev 2004, 18:2573–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stolz A, Ernst A, Dikic I: Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 2014, 16:495–501. [DOI] [PubMed] [Google Scholar]
- 36.Kirkin V, McEwan DG, Novak I, Dikic I: A role for ubiquitin in selective autophagy. Mol Cell 2009, 34:259–269. [DOI] [PubMed] [Google Scholar]
- 37.Xie X, Li F, Wang Y, Wang Y, Lin Z, Cheng X, Liu J, Chen C, Pan L: Molecular basis of ubiquitin recognition by the autophagy receptor CALCOCO2. Autophagy 2015, 11:1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ordureau A, Heo JM, Duda DM, Paulo JA, Olszewski JL, Yanishevski D, Rinehart J, Schulman BA, Harper JW: Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc Natl Acad Sci U S A 2015, 112:6637–6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thurston TL, Boyle KB, Allen M, Ravenhill BJ, Karpiyevich M, Bloor S, Kaul A, Noad J, Foeglein A, Matthews SA, et al. : Recruitment of TBK1 to cytosol-invading Salmonella induces WIPI2-dependent antibacterial autophagy. EMBO J 2016, 35:1779–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ravenhill BJ, Boyle KB, von Muhlinen N, Ellison CJ, Masson GR, Otten EG, Foeglein A, Williams R, Randow F: The Cargo Receptor NDP52 Initiates Selective Autophagy by Recruiting the ULK Complex to Cytosol-Invading Bacteria. Mol Cell 2019, 74:320–329 e326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, Randow F, Youle RJ: Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol Cell 2019, 74:347–362 e346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu T, Liu J, Wang Y, Xie X, Hu S, Pan L: Mechanistic insights into the interactions of NAP1 with the SKICH domains of NDP52 and TAX1BP1. Proc Natl Acad Sci U S A 2018, 115:E11651–E11660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou Z, Liu J, Fu T, Wu P, Peng C, Gong X, Wang Y, Zhang M, Li Y, Wang Y, et al. : Phosphorylation regulates the binding of autophagy receptors to FIP200 Claw domain for selective autophagy initiation. Nat Commun 2021, 12:1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F: The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol 2009, 10:1215–1221. [DOI] [PubMed] [Google Scholar]
- 45.Shi X, Chang C, Yokom AL, Jensen LE, Hurley JH: The autophagy adaptor NDP52 and the FIP200 coiled-coil allosterically activate ULK1 complex membrane recruitment. Elife 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang C, Jensen LE, Hurley JH: Autophagosome biogenesis comes out of the black box. Nat Cell Biol 2021, 23:450–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goodall EA, Kraus F, Harper JW: Mechanisms Underlying Ubiquitin-driven Selective Mitochondrial and Bacterial Autophagy. Mol Cell 2022, in press. [DOI] [PMC free article] [PubMed]
- 48.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ: The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524:309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW: The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell 2015, 60:7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huttlin EL, Bruckner RJ, Navarrete-Perea J, Cannon JR, Baltier K, Gebreab F, Gygi MP, Thornock A, Zarraga G, Tam S, et al. : Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 2021, 184:3022–3040 e3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bohannon KP, Hanson PI: ESCRT puts its thumb on the nanoscale: Fixing tiny holes in endolysosomes. Curr Opin Cell Biol 2020, 65:122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Otten EG, Werner E, Crespillo-Casado A, Boyle KB, Dharamdasani V, Pathe C, Santhanam B, Randow F: Ubiquitylation of lipopolysaccharide by RNF213 during bacterial infection. Nature 2021, 594:111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Wijk SJL, Fricke F, Herhaus L, Gupta J, Hotte K, Pampaloni F, Grumati P, Kaulich M, Sou YS, Komatsu M, et al. : Linear ubiquitination of cytosolic Salmonella Typhimurium activates NF-kappaB and restricts bacterial proliferation. Nat Microbiol 2017, 2:17066. [DOI] [PubMed] [Google Scholar]
- 54.Noad J, von der Malsburg A, Pathe C, Michel MA, Komander D, Randow F: LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-kappaB. Nat Microbiol 2017, 2:17063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kelsall IR, McCrory EH, Xu Y, Scudamore CL, Nanda SK, Mancebo-Gamella P, Wood NT, Knebel A, Matthews SJ, Cohen P: HOIL-1 ubiquitin ligase activity targets unbranched glucosaccharides and is required to prevent polyglucosan accumulation. EMBO J 2022, 41:e109700. [DOI] [PMC free article] [PubMed] [Google Scholar]