

Abstract
Background: Individuals with a phenotype of early-onset severe obesity associated with intellectual disability can have molecular diagnoses ranging from monogenic to complex genetic traits. Severe overweight is the major sign of a syndromic physical appearance and predicting the influence of a single gene and/or polygenic risk profile is extremely complicated among the majority of the cases. At present, considering rare monogenic bases as the principal etiology for the majority of obesity cases associated with intellectual disability is scientifically poor. The diversity of the molecular bases responsible for the two entities makes the appliance of the current routinely powerful genomics diagnostic tools essential.
Objective: Clinical investigation of these difficult-to-diagnose patients requires pediatricians and neurologists to use optimized descriptions of signs and symptoms to improve genotype correlations.
Methods: The use of modern integrated bioinformatics strategies which are conducted by experienced multidisciplinary clinical teams. Evaluation of the phenotype of the patient’s family is also of importance.
Results: The next step involves discarding the monogenic canonical obesity syndromes and considering infrequent unique molecular cases, and/or then polygenic bases. Adequate management of the application of the new technique and its diagnostic phases is essential for achieving good cost/efficiency balances.
Conclusion: With the current clinical management, it is necessary to consider the potential coincidence of risk mutations for obesity in patients with genetic alterations that induce intellectual disability. In this review, we describe an updated algorithm for the molecular characterization and diagnosis of patients with a syndromic obesity phenotype.
Keywords: Syndromic obesity, classical obesity syndrome, non-canonical obesity syndrome, whole-genome array, nonsyndromic monogenic obesity, exome sequencing
1. INTRODUCTION
Syndromic obesity is the manifestation of severe and early onset overweight, with a specific set of clinical signs and symptoms, including the presence of at least one of these traits: intellectual disability, congenital malformations and/or abnormal facies [1]. It is more frequently observed in patients with mild developmental delay; characterized by a decrease in language skills and cognitive function, as well as motor impairment [2]. It is quite rare for genetic obesity to be a result of a mutation with a Mendelian pattern of inheritance [3]. In most cases of genetic obesity, the patients are affected by complex features as a result of different patterns of genetic alterations, linked to other individual non-genetic conditions [4, 5]. These non-genetic factors involve interactions with multiple behavioral and/or environmental factors [6]. Consequently, even with the increasing impact of genomic medicine in healthcare practice, the clinical management of most cases of obesity does not involve identifying the molecular bases or genetic patterns of inheritance [7]. However, although complex traits do not readily follow predictable patterns of inheritance, it is indisputable that severe, early and maintained obesity is a sign that heritability is evidently superior to that of entities of comparable molecular complexity, such as susceptibility to cancer [8]. Even traits that appear to be monogenic could also be influenced by variation in other modifier genes, altering the expected phenotype [9].
This review outlines the current understanding of the genetic architecture of two complex genetic entities related to extreme and maintained obesity, which is intimately associated with mental disability [10]. The factors accepted as contributors to the syndromic obesity phenotype and its complex development (from a monogenic Mendelian inheritance to those generated by non-Mendelian patterns) are discussed with respect to evaluating an individual and/or group of patients. Genomic medicine is particularly focused on the identification of pathogenic genetic variants in the maximum number of patients. However, reaching a molecular diagnosis in this type of individuals is difficult, as the percentage of genetic tests without any pathogenic finding in this group is very high. A high level of diagnostic inefficiency is evident even among adults, whose obesity phenotypes may be fully developed.
In general terms, it seems clear that obese patients with mental delay could be an adequate model to study the development of early-onset and maintained obesity, mainly due to an altered hypothalamic satiety control. The altered neurodevelopment compromises the regulation of hormonal responses and the satiety axis. Although hyperphagia is an extremely difficult symptom to demonstrate and quantify (both in quantity and type of preferred food), it is clinically evident in most patients and obviously constitutes the basis for the unstoppable gain of weight and adipose tissue.
Alteration in hypothalamic satiety is considered a concrete trait in a remarkable percentage of patients with syndromic obesity, and is correlated with specific genotypes within the high heterogeneity of causes of genetic obesity and mental disability. The molecular diagnosis of these patients initiates the understanding of the main functional pathways that causes their obesity [11]. Conversely, although canonical obesity syndromes only represent a tiny percentage of 0.05% to 0.1% of all types of obesity, the etiopathogenesis and inheritance patterns (dominant, recessive, epigenetic and printed) have already been widely established%. Despite the wide range of obesity syndromes, only 30 types have been well studied, whilst the rest have unknown origins [1, 3].
The classification and study of non-canonical obesity syndromes is the main challenge of researchers and clinical experts. Non-canonical obesity syndromes have a number of different genetic causes, including specific genes with a putative Mendelian pattern of inheritance, or chromosome rearrangements, or duplications and/or deletions with a huge variability in size and localization in the human genome [3]. It has been calculated that these new entities would represent around 1% of characterized syndromic obese cases. Although these non-canonical obesity syndromes are less well studied than classical obesity syndromes, the majority of the clinical manifestation of obesity corresponds to this very heterogeneous group [5, 12-17]. The diversity of the underlying genetic causes results in a high variety of distinct clinical manifestations, which makes it crucial that expert clinicians conduct an exhaustive and methodical phenotypic evaluation for each patient [18]. In recent years, the discovery of new genes implicated in non-canonical obesity syndromes, together with the development of powerful technologies in biosciences (which are widely used in both research and the diagnosis of patients) have changed the perspective for the treatment of these patients [19-22].
The SIRT1 genes (* 604479) have been suggested as epigenetic regulator in human complex diseases. A significant decrease in the SIRT1 expression levels has been correlated with enhancement in the fatty acid oxidation in individuals affected with severe overweight [23]. Sirtuin 1 (SIRT1) has been suggested as anti-aging gene, and its inactivation has been linked to obesity with mental disability. Diet, lifestyle and/or environmental conditions evidence the capability to modify Sirtuin 1 expression, inducing then susceptibility to obesity, diabetes and neurodegenerative diseases [24, 25].
Due to the absence of a globally accepted “pipeline” for the molecular diagnosis of syndromic obesity patients, this review describes a detailed strategy to follow, as a structured algorithm, to determine the diagnosis of these individuals.
In order to achieve an efficient and complete genetic diagnosis when the molecular basis is the primary etiopathogenesis, it is essential to ensure a more proximate scientific management and precise genetic counseling of patients. Finally, we highlight the importance of detailed phenotypic evaluation by expert neurologists, neuropediatricians, endocrinologists and expert geneticists. The latter are directly involved in the application of efficient new techniques for the discovery of new candidate loci involved in the pathogenesis of non-canonical obesity syndromes [26-29].
1.1. First Step: Familial Background and Pregnancy History of Mental Disability and/or Obesity Antecedent
The starting point for evaluating the complex causes of mental disability associated with severe obesity is a medical history, including pregnancy history, mental disability and/or familial obesity antecedents. Offspring of obese mothers have elevated risks of obesity and central nervous system developmental problems, among others [30]. There are various environmental factors in utero that are linked to offspring obesity and mental disability, including maternal alcoholism, smoking and nutritional status (which can cause fetal undernutrition and overnutrition), infections and inflammation, environmental chemicals, maternal stress, and gut microbiota, and paternal factors; such as smoking and obesity, which result in epigenetic changes in sperm cells [31-34]. Affective deprivation, lack of resources and no adequate methods during the first months of life could also influence the development of intellectual disability in newborns, manifested with or without obesity [35, 36].
After exhaustively evaluating pregnancy history, newborn history and the availability of resources in the family, it is time to determine if the patient carries an inherited genetic susceptibility for syndromic obesity. Cases of syndromic obesity can appear as isolated or familial incidences. In a large percentage of cases, a specific genetic test confirms the suspected diagnosis, familial genetic alterations can result in obesity inheritance patterns which affect multiple generations; in these instances, it is important to identify the most appropriate family member to be considered as the index case. However, it is widely accepted that individuals with similar obesity phenotypes resulting from familial aggregation of genetic alterations might have different molecular bases for their obesity.
When an individual belonging to a family affected by syndromic obesity cases is referred, the most difficult aspect of the evaluation is describing precisely the phenotype of the individuals considered as patients. It is essential to know the severity of the phenotype in relation to familial relationships, and to try and compare them according to the syndromes each family member has manifested. In other words, it is important to collate a rigorous list of developed signs and symptoms per affected individual, emphasizing features of intellectual disability, congenital malformations, abnormal facies, and characteristics of overweight.
2. IDENTIFICATION AND DIAGNOSIS OF CLASSICAL OBESITY SYNDROMES
After reviewing the aforementioned personal and family antecedents, the next step is to rule out a classical obesity syndrome. These monogenic types of obesity are widely studied and characterized using efficient diagnosis “pipelines” involving methodical phenotypic descriptions by systems and organs. There are clinical signs and symptoms which require special evaluation, as they are often present across multiple distinct syndromes. In general, although obesity and intellectual disability would be the central axis in the diagnosis of classical obesity syndromes, other differential features also form part of the phenotype, and it will be crucial to include these as we develop the first diagnostic approach.
The Human Phenotype Ontology (HPO) website and Face2Gene software (Fig. 1) are useful tools for specialists in aiding the diagnosis of these complex conditions. The main aim of HPO is to provide standardized vocabulary to describe patients with phenotypic abnormalities and the associated traits of human diseases [37]. This provides clinicians with a standardized method to describe the main traits that a patient has, from the first consultation, and aids understanding across all professional services. Each HPO term describes a feature or phenotypic abnormality. HPO currently contains 11,000 terms and 115,000 annotations for hereditary diseases, as well as annotations for 4,000 common diseases (https://hpo.jax.org/app/). The HPO website is currently being developed using the medical literature, Orphanet (https://www.orpha.net/consor/cgi-bin/index.php), DatabasE of genomiC varIation and Phenotype in Humans using Ensembl Resources (DECIPHER) (https://decipher.sanger.ac.uk/) and Online Mendelian Inheritance in Man (OMIM) (https://www.omim.org/). The website differentiates sets of signs and symptoms according to their genetic origin. The description and characterization of each condition is remarkably stringent and exhaustive. Face2Gene software (FDNA Inc, MA, USA) is a free online tool for phenotyping that facilitates the detection of medical syndromes, predominately based on front and side photographs of the patient’s face, as well as physical traits. The software detects phenotypes and significant facial and non-facial features and matches them to known syndromes (https://www.face2 gene.com) (Fig. 1). As such, the Face2Gene enables a comprehensive and precise basis for genetic evaluation and diagnosis.
Fig. (1).
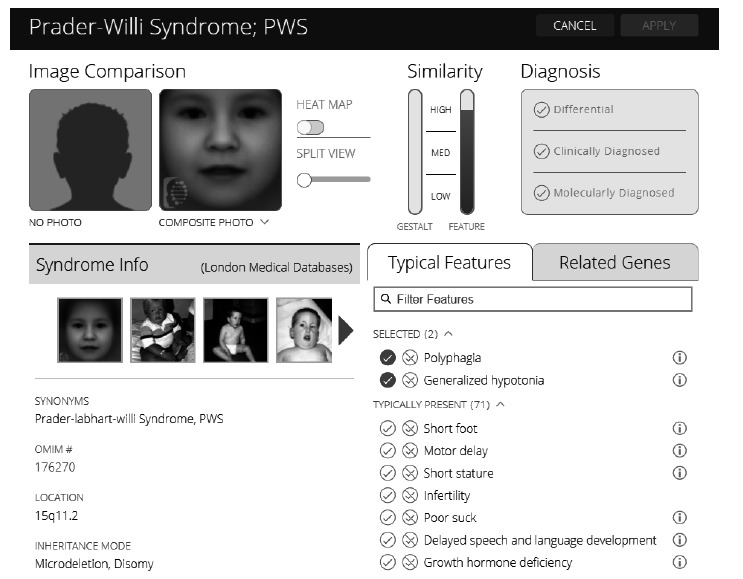
An example of the Face2Gene software consultation related to Prader-willy Syndrome. An example of the Face2Gene software consultation related to Prader-Willi Syndrome. The main use of this computer-assisted facial recognition is the prediction of the etiology of patients with global developmental delay and dysmorphic features and to determine a diagnosis. This is achieved through the comparison of photographs of the patients with photographs of the syndrome model (left upper panel). The clinician is able to indicate the type of diagnosis made; a differential diagnosis, clinical diagnosis or molecular diagnosis (right upper panel). The software provides information on the chromosomal location involved, the inheritance mode and the OMIM reference (left lower panel). The Face2Gene search can be refined by selecting the phenotypic traits that the patient has, enabling a perfected diagnostic approach (right lower panel).
The diagnostic strategy for suspected classical syndromes of obesity is quite different from diagnoses of other types of syndromic obesity but is still focused on a syndrome approach. Approximately 20 to 30 early-onset obesity syndromes have been reported in the literature. The most common forms with an established genetic cause are Down’s syndrome and Fragile X syndrome, followed by Prader-Willi, Bardet-Bield, Alström and WAGR syndromes, among others [9, 38].
Down’s syndrome (DS, OMIM #190685) is generally caused by a full chromosome 21 trisomy (94% of DS cases). It can also be caused by mosaic chromosome 21 trisomy (2.5% of cases) or a translocation of chromosome 21 (in 3.5% of cases). DS usually manifests phenotypically with overweight [39] and is associated with very specific dimorphic facies, varying severities of intellectual disability, learning retardation, memory defects, short stature and congenital heart defects. The link to the HPO website for DS is https://hpo.jax.org/app/browse/disease/OMIM:190685.
Fragile X syndrome (FXS, OMIM #300624) is an X-linked genetic disorder caused by the unstable expansion of the CGG repeat in the FMR1 gene or an abnormal methylation pattern in this gene. This alteration results in a decrease in FMR1 expression in the brain, which manifests in a phenotype with recognizable abnormal facies, moderate to severe intellectual disability, macroorchidism and frequently obesity [40]. The link to the HPO website for FXS is https://hpo.jax.org/app/browse/disease/OMIM:300624.
The most well studied classical obesity-related syndrome is Prader-Willi syndrome (PWS OMIM#176270). PWS involves the 15q11-13 region and the failure of the SNRPN, NDN, MAGEL2 genes in the father’s copy [41]. The main causes of this are 1. Paternal deletion, which occurs in about 70% of cases, 2. Maternal uniparental disomy, which occurs in about 25% of cases, and 3. An imprinting defect, which occurs in less than 5% of cases. The absence of function of these paternal genes is manifested in a peculiar phenotype with principal features of central obesity and severe hyperphagia, as well as hypothalamic hypogonadism, neonatal hypotonia, mild intellectual disability, short stature and abnormal facies [9, 42-44]. The link to the HPO website for PWS is https://hpo.jax.org/app/browse/disease/OMIM: 176270.
Bardet-Bieldt syndrome (BBS, OMIM#209900) is considered a classical autosomal recessive obesity syndrome, which shows genetic heterogeneity characterized by mutations in approximately 14 BBS genes; the principal genes involved are BBS1-16, ARL6, MKKS, MKS1, CEP290 [38]. Early-onset obesity is a constant feature, with associated decreased learning skills, dyslexia, abnormal finger morphology, progressive cone-rod dystrophy and renal disease [44, 45]. The link to the HPO website for BBS is https://hpo.jax.org/app/browse/disease/OMIM:209900.
Alström syndrome (ALMS, OMIM#203800) is a monogenic autosomal recessive obesity-related syndrome caused by dysfunctional mutations affecting the ALMS1 gene [41, 46]. This genetic alteration is clinically manifested by mild truncal obesity, intellectual disability, deafness, blindness, type 2 diabetes, hyperinsulinemia and other hormonal alterations [44, 47]. The link to the HPO website for ALMS is https://hpo.jax.org/app/browse/disease/OMIM:203800.
WAGR syndrome (OMIM#194072) is a genetic disorder caused by deletions in the 11p13 region of the WT1 and PAX6 genes. These genetic mutations result in the classical form of WAGR Syndrome, when the adjacent BDNF gene is also altered, obesity appears as the predominant sign, and is classified as WAGRO Syndrome [44, 48, 49]. The link to the HPO website for WAGR syndrome is https://hpo.jax.org/app/browse/disease/ORPHA:893.
Albright’s Hereditary Osteodystrophy (OMIM#103580) is caused by a mutation in GNAS1, leading to a defect in the alpha subunit of G proteins (Gαs) coupled to transmembrane receptors. Gαs deficiency in imprinted regions of the hypothalamus results in early onset excess weight gain, round facies, brachydactyly, metacarpia of the hands and/or feet, and heterotopic ossifications, as well as t hormonal alterations [50]. The link to the HPO website for Albright’s Hereditary Osteodystrophy is https://hpo.jax.org/app/browse/disease/OMIM:103580.
Other minor monogenic obesity syndromes recognizable by their associated phenotype, OMIM# code and their susceptibility gene are:
- Those with an autosomal dominant hereditary pattern, including: Rubinstein-Taybi syndrome (#180849, CREBBP gene; #613684, EP300 gene); Ulnar-mammary syndrome (#181450, TBX3 gene).
- Those with an autosomal recessive hereditary pattern, including: Carpenter syndrome (#201000, RAB23 gene; #614976, CRPT2 gene); Cohen syndrome (#216550, VPS13B gene); Majewski osteodysplastic primordial dwarfism type II (#210720, PCNT gene); Mental retardation, truncal Obesity, Retinal dystrophy, and Micropenis (MORM) syndrome (#610156, INPP5E gene); Macrosomia, Obesity, Macrocephaly, and Ocular abnormalities (MOMO) syndrome (#157980, unknown gene/s).
- Those with an X-linked hereditary pattern, including: Börjeson-Forssman-Lehman syndrome (#301900, PHF6 gene); Coffin–Lowry syndrome (#303600, RPS6KA3 gene).
If a specific genetic test identifies a molecular cause of classic obesity syndrome, the diagnosis “pipeline” would finish here, and family members would also be tested for the identified mutation. If these syndromes are phenotypically or genetically discarded, the next step in the diagnosis algorithm should be taken.
3. IDENTIFICATION AND DIAGNOSIS OF NON-CANONICAL OBESITY SYNDROMES
The other manifestation of syndromic obesity occurs when early-onset, severe and maintained overweight is associated with a non-affiliated complex phenotype characterized by intellectual disability, congenital malformations and/or abnormal facies. There is wide variability in the phenotypic features presented with this type of patient, and the absence of a correlation with a typical trait of a classical syndrome, makes a diagnosis much more difficult. Again, it is essential to compile an exhaustive review of familial antecedents of obesity along with the patient´s characteristics, in order to help determine the most efficient technique to identify the potential molecular etiopathogenesis.
3.1. Whole-genomic DNA Array Hybridization
Whole-genomic DNA array hybridization is the first line technique in the diagnosis of syndromic patients with non-canonical obesity syndrome. It allows the study of variability in the human genome using DNA probes and can identify and characterize duplications and deletions of a wide range of sizes, from kilobases to megabases, as well as mosaic lines and regions with a loss of heterozygosis (LOH) [51]. This technology is growing in both its specificity and sensibility and is currently considered to be the most suitable methodology for studying patients with syndromic obesity, especially those with a phenotype characterized by intellectual disability, neuromotor dysfunctions, severe alterations of behavior and/or congenital anomalies [52, 53]. A significant percentage of this patient group develop a phenotype of severe overweight, which is the predominant sign of syndromic obesity. The use of whole-genome hybridization arrays is increasing in parallel with the development of different databases that detail variants related to pathology and healthy populations. The Database of Genomic Variants (DGV, http://dgv.tcag.ca/) describes CNVs and SNPS variants which have been identified in the general population, and is, therefore a powerful database which can be used as a control to correlate the variants with the phenotype.
OMIM is the largest database that correlates human genes with genetic diseases; it even associates the OMIM inputs of the precisely described CNVs, with the phenotype of the patient. The International Standards for Cytogenomic Arrays (ISCA, http://dbsearch.clinicalgenome.org/search) includes variants detected in patients with rare chromosomal disorders, including microdeletions and microduplications. DECIPHER, contains information about submicroscopic chromosomal imbalances related to a particular phenotype. Finally, the UCSC Genome Browser (http://genome.ucsu.edu/) contains reference sequences and draft sequences of different genomes, and is a very useful tool because it contains information on gene expression and homology [52].
The high specificity and sensibility of whole-genome hybridization microarrays and the increasing amount of variant information in the databases enable the molecular characterization of syndromic patients with previous non-concluding genetic studies. Now, even single-exon deletion and duplication events can be detected with high sensitivity, providing an impressive whole-genome coverage and offering results capable of confirming CNV findings. As such, it provides a strong complement to mutation analysis performed by the increasingly popular Next-Generation Sequencing (NGS). NGS is able to powerfully detect LOH regions; LOH are chromosomal segments without the presence of biallelic variants, due to similarity of the information of both parental alleles. LOH regions also enable the calculation of the consanguinity of the patient. Populations with high endogamy and in geographic regions with a reduced genetic variability often develop incidences of recessive syndromes such as Alström syndrome [46]. It is essential to highlight the importance of considering the homozygous regions of the genome because mutations in these regions could manifest a recessive inheritance pattern responsible for a syndrome. Furthermore, study of homozygous regions of the genome could determine new candidate genes which might be key elements or modifiers in syndromic obesity phenotypes. Another application of LOH is to detect a uniparental disomy, which is an important mutation in syndromes such as PWS or BBS.
All reported CNVs have to be analyzed by a highly experienced expert who classifies them into the five currently agreed types of variation: benign, likely benign, uncertain significance (VUS), likely pathogenic, or pathogenic variations [54, 55]. When a deleterious CNV is found, such as a duplication or deletion, the diagnosis is clear and the patient is informed. However, when a variant of uncertain significance is carried by a syndromic patient related or non-related to obesity, it has to be compared with the reported CNVs in the various databases detailed above in order to determine if it is deleterious or not. If a variant of uncertain significance that could be deleterious is found, it is recommended (and should be mandatory) to investigate its familial segregation of it. This segregation is an efficient method in determining if the variant is inherited or if it is a de novo alteration, and moreover, if it is associated with the phenotype of the syndromic patient and the presented obesity (Fig. 2).
Fig. (2).
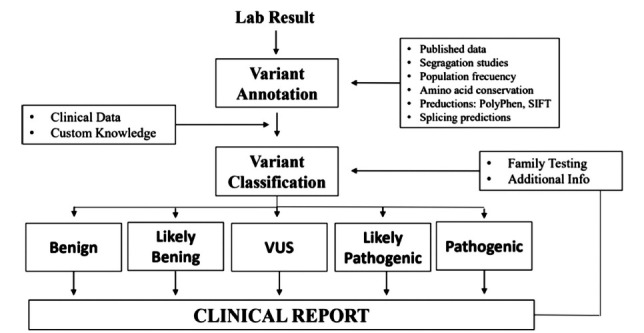
Variant assessment workflow. Variant assessment workflow. Genetic variants identified by laboratory testing are annotated with information from various sources including publications, computational prediction algorithms, and public, collaborative and internal databases. After evaluation of all pertinent information, and in conjunction with patient-specific clinical and familial information, a professionally trained individual will classify the variant into one of the five clinical categories and combine all the findings into a clinical report [55]. “Adapted from Duzkale 2013”.
An example of a non-canonical obesity syndrome detected by whole-genome microarray hybridization is the 16p11.2 distal microdeletion syndrome [56, 57]. Other chromosome regions and alterations detected by whole-genome array hybridization have also been clinically related to causing or predisposing an individual to syndromic obesity. Examples include microdeletions in the 1p21.3 region, 2p25.3 deletions, 6q14.1q15 and 11p14.1 interstitial deletions, deletions that involve the 6q22, 12q subtelomeric deletions, microdeletions affecting the 17q24.2 region, and duplications of 19q [21]. Furthermore, deletions of 6q16, 1p36, 2q37 and 9q34 have also been linked to obesity [58]. Gimeno-Ferrer et al. detailed a previously non-described 10-Mb duplication in the 16p13.2p12.3 region which is considered to cause intellectual disability and severe overweight. They also describe eleven 16p11.2 CNVs (nine proximal and two distal) which have a low prevalence but a frequent recurrence in syndromic patients with a severe alteration in Body Mass Index (BMI) [27].
If the whole-genome microarray hybridization is analyzed and all the CNVs are considered to be benign, the result of the array is considered negative. The next step is to perform (NGS) of the monogenic genes which have attributed susceptibility to syndromic and/or non-syndromic severe overweight, as familial antecedents of obesity may be present. NGS analyzes the possible existence of a molecular cause of the non-affiliated obesity syndrome.
3.2. Molecular Analysis of Classified Non-syndromic and Syndromic Monogenic Causes of Obesity
Until now, there remains a global insistence on the classification of patients with severe obesity as syndromic or non-syndromic individuals, whether they have a recognizable phenotype. For inclusion in the second category, the patients must not show any signs or symptoms of any neurological disorder or congenital malformation. As has been previously detailed, incidences of syndromic obesity are an important part of the molecular causes of severe overweight but only represent a low percentage of approximately (1%) of total incidences of obesity. In this review, we aim to highlight the set of molecular bases that are capable of causing severe overweight in individuals who do not meet the criteria that has been developed and is currently accepted as a syndromic obesity phenotype. A patient with an obese phenotype and intellectual disability could manifest both traits, but these could be caused by different origins. Effectively, non-canonical syndromic obese patients show an intellectual coefficient very slightly below that that can be considered or recognized as normal. Other individuals appear to have a slight deficit in neurodevelopment, without presenting dysmorphic features or congenital malformations. In addition, other patients may be diagnosed with mild brain immaturity but not be considered patients with intellectual disability. Therefore, sometimes it is difficult to distinguish the existence of an intellectual disability, attitudes or borderline personality disorders, or behavioral traits that a patient might have. Clinical evaluation becomes even more complex as the patient ages due to the confusion of comorbidities that are acquired and appear over the years, as well as the lack of training neurologists, have in identifying genetically-based intellectual disability in adults.
Most articles with the highest impact factors are focused on the search for unique causative mutations of severe obesity and analyze so-called “non-syndromic obesity” patients according to the principal inclusion criteria of: non-syndromic features, age of onset of obesity <5 years (or up to prepubertal onset in adult subgroups), high risk familial antecedents of severe obesity among first and second degree relatives, maintained extreme obesity (BMI >40 kg/m2) and inefficient response to endocrine intervention and/or bariatric surgery determined as weight regain or insufficient weight loss. However, mutations in these genes are likely to be found in patients with evident intellectual disability, with a similar frequency to that of the rest of the population. The effect on weight should be comparable or even more powerful, given the factor of greater inhibition of hypothalamic control of satiety in the context of intellectual disability.
The literature reflects the fact that the identified molecular causes of monogenic non-syndromic (rare alleles with high or moderated attributed risk) and polygenic obesity (more common alleles with low or moderated attributed risk) cover an interesting percentage of genetic obesity (7% and 20%, respectively). Monogenic syndromic or non-syndromic obesity, and the different inheritance patterns involved (dominant, recessive and co-dominant), could be an alternative origin of the obesity in syndromic patients with or without familial antecedents of obesity [41].
Monogenic non-syndromic obesity is an inherited genetic obesity that shows a clear genotype-phenotype correlation. The molecular mechanism underlying this is the alteration in hypothalamic satiety control mediated by the Leptin/Melanocortin pathway, which has a significant role in the correct functioning of appetite regulation [38, 59]. This control begins after an individual has eaten, with the release of the leptin hormone (encoded by the LEP gene) from the adipose tissue, which initiates negative feedback in the brain with an important response, to stop eating [60]. Therefore, leptin is the first regulator of this system. The target of leptin is the leptin receptor (encoded by the LEPR gene), which is located in the Cocaine- and Amphetamine-Related Transcript (CART) and Pro-opiomelanocortin (POMC) neurons of the Arcuate Nucleus (ARC) in the hypothalamus [61]. LEPR, with the help of Tubby neuropeptide (encoded by the TUB gene) [62-64], is responsible for signal transduction through the Janus Kinase/Signal Transducers and Activators of Transcription (JAK/STATs) pathway, resulting in the activation of the Proopiomelanocortin Precursor Polypeptide (encoded by the POMC gene), which is also expressed in the ARC [65]. The activation of JAK/STAT is regulated by the SH2B adaptor protein 1 (encoded by the SH2B1 gene) [66]. This precursor is enzymatically cleaved by the Proprotein Convertase Subtilisin/Kexin Type 1 (encoded by the PCSK1 gene) [67] and regulated by the activity of Carboxypeptidase E (encoded by the CPE gene) [68], which is involved in the production of α- and β-Melanocyte-stimulating Hormones (MSH). Melanocortin 3 Receptor and Melanocortin 4 Receptor (encoded by the MC3R and MC4R genes respectively) have an important role in the satiety control pathway. MC3R is located in POMC neurons and acts as an auto-receptor for POMC-derived peptides, generating negative feedback [69]. While, MC4R is located in the Paraventricular Nucleus (PVN) and acts as a receptor for the α-Melanocyte-stimulating hormone (α-MSH) ligand [70]. The binding of the α-MSH ligand to the receptor is regulated by the Melanocortin Receptor Accessory Protein 2 (encoded by MRAP2) [71-74]. The activation of MC4R receptors induces the expression of the Brain-derived Neurotrophic Factor (encoded by the BDNF gene) [75] through the cyclic AMP-protein kinase A signaling pathway, in which Adenylate Cyclase 3 (encoded by the ADCY3 gene) has a crucial role [75-77]. This MC4R activity is facilitated by Single-minded 1 (encoded by the SIM1 gene) [78]. Finally, BDNF activates the Neurotrophic receptor Tyrosine kinase 2 (encoded by NTRK2) [79] which results in the liberation of oxytocin in the PVN of the hypothalamus, causing activation of fatty acids β-oxidation and a clear response in the organism; an increase in energy expenditure and a decrease in food intake [59].
Another important factor involved in the POMC/CART neurons of the ARC nucleus and the pathogenesis of obesity is CART (encoded by the CARTPT gene). This gene could have a major role in feeding, stress and reward [79]. CART neurons are activated after the action of leptin in the hypothalamus, highlighting the involvement of CARTPT in satiety control [80]. Mutations in this gene have been reported to cause the aggregation of familial obesity through generations [81, 82].
A parallel pathway in ARC nucleus of the hypothalamus involves the NPY-AgRP neurons [83]. Neuropeptide Y (encoded by the NPY gene) is expressed in these neurons and induces food intake by binding to the Neuropeptide Y Receptor Y2 (encoded by NPY2R). After nutrient ingestion, leptin blocks the NPY-AgRP neurons, inducing satiety [84]. Mutations in both NPY and NPY2R are involved in the development of an obese phenotype [85]. Further, the analogous appetite-suppressing hormone Peptide YY (encoded by PYY) is released from the gut after nutrient ingestion [83-86]. PPY secretion stimulates satiety by exerting its effect directly in the ARC nucleus through a Y2 receptor, inhibiting food intake [87]. Mutations in the PYY gene have also been reported to cause an obese phenotype. The PYY gene is associated with satiety control regulation via the NPY-AgRP neurons of the ARC nucleus, but not the POMC cells [88].
In monogenic non-syndromic obesity, the genes that encode most of the regulators involved in the hypothalamic control of satiety are mutated and the signaling pathway results in inefficient control of the appetite feedback loop. The genes responsible for early-onset monogenic non-syndromic obesity include LEP, LEPR, POMC, PCSK1, MC3R, MC4R, CARTPT, PYY, NPY and ADCY3. Mutations in these genes are reported to be responsible for less than 10% of incidences of obesity and have different inheritance patterns; dominant, co-dominant or recessive, depending on which gene is involved [38, 44].
There are also a set of alterations or mutations in these genes which are involved in the manifestation of obesity and associated with mental delay. The genes with a known role in monogenic syndromic obesity with mild intellectual disability and without abnormal facies are TUB, CPE, MRAP2, SH2B1, SIM1, NTRK2 and BDNF (which are included among the susceptibility genes for WAGRO syndrome).
Effectors and regulators of classic and new signaling pathways involved in satiety control have been discovered in the same way that technology and research studies elucidated the molecular mechanisms. These newly discovered genes and their coding products are also involved in this intricate system, including POU domain class 3 transcription factor 2 (POU3F2), Uncoupling Protein 3 (UCP3), Centrosomal Protein 19 (CEP19), and Dual Specificity Tyrosine Phosphorylation Regulated Kinase 1B (DYRK1B) genes.
POU3F2 is a gene that encodes a pro-neuronal transcription factor which is important for hypothalamic development and function. The functional characterization of a knockout POU3F2 zebrafish mutant model demonstrates that the POU3F2 regulator is downstream to SIM1 in the Leptin/Melanocortin pathway, as the same phenotype was developed. POU3F2 controls the expression of oxytocin in the hypothalamus, particularly in the neuroendocrine preoptic area and PVN in the model [87]. As such, POU3F2 influences the β-oxidation of fatty acids, as mediated by leptin, which introduces a new player to the energy balance, satiety and body mass signaling pathways. The POU3F2 gene is adjacent to the SIM1 gene on chromosome 6. Deletions encompassing one or both genes result in the same Prader-Willi-like phenotype [89].
UCP3 is a mitochondrial inner membrane anion carrier protein involved in the uncoupling of oxidative phosphorylation. It is not involved in the satiety pathway, but it is involved in cellular fatty acid metabolism in the mitochondria. Heterozygous mutations in this gene can cause a phenotype characterized by obesity and type 2 diabetes [90, 91].
CEP19 is a gene that encodes a centrosomal protein. Genetic alteration in CEP19 results in a phenotype characterized by ciliopathy, obesity, hyperphagia, glucose intolerance, insulin resistance and mental delay, with a functional and molecular characterization that is observed in both mice models and patients [92, 93]. The function of the gene in the Leptin/Melanocortin pathway is not known. However, MC4R and ADCY3 are co-expressed in neuron cilia and this could explain the connection [77]. Like the CEP19 gene, DYRK1B is a newly discovered gene involved in obesity which is involved in a signaling pathway for adipocyte differentiation. A homozygous nonsense mutation in the CEP19 gene was identified and strongly associated with obesity in a large consanguineous family who had morbid obesity and spermatogenic failure [92]. Additionally, members of 3 families who had a common ancestor were affected with autosomal dominant metabolic syndrome and early-onset coronary artery disease, related to a heterozygous missense mutation in the DYRK1B gene. Other infrequent missense mutations in the DYRK1B gene have also been detected in morbidly obese Caucasian individuals with coronary artery disease and multiple metabolic phenotypes. The predominant phenotype is characterized by a mild intellectual delay, early onset coronary artery disease, central obesity, hypertension and diabetes [94].
The genes discussed above participate in satiety control and other important signaling pathways. Reported mutations in these genes result in the development of a phenotype of monogenic syndromic obesity. However, the position of these genes in the cascade regulation is still unknown.
Therefore, the strategy for patients with non-canonical syndromic obesity with negative whole-genome hybridization array results is to analyze this group of familial monogenic non-syndromic genes, as well as those known to cause monogenic syndromic obesity coexisting with intellectual disability. The most efficient strategy to do this is through the analysis of a panel including the aforementioned genes using NGS technologies (Table 1). In the case that a monogenic non-syndromic mutation is detected that could explain the development of the obese phenotype due to familial aggregation, then the intellectual disability would have an alternative etiology, as we have previously discussed.
Table 1.
Updated list of principal monogenic genes responsible for syndromic obesity phenotypes.
| Gene Symbol | Omim Number | Name | Location | Phenotype |
|---|---|---|---|---|
| ADCY3 | 600291 | Adenylate Cyclase 3 | 2p23.3 | Non-Syndromic |
| BDNF | 113505 | Brain-derived Neurotrophic Factor | 11p14.1 | Syndromic |
| CARTPT | 602606 | CART Prepropeptide | 5q13.2 | Non-Syndromic |
| CEP19 | 615586 | Centrosomal Protein, 19-kd | 3q29 | Both |
| CPE | 114855 | Carboxypeptidase E | 4q32.3 | Syndromic |
| DYRK1B | 604556 | Dual-specificity Tyrosine Phosphorylation-regulated Kinase 1B | 19q13.2 | Syndromic |
| LEP | 164160 | Leptin | 7q32.1 | Non-Syndromic |
| LEPR | 614963 | Leptin Receptor | 1p31.3 | Non-Syndromic |
| MC3R | 155540 | Melanocortin 3 Receptor | 20q13.2 | Non-Syndromic |
| MC4R | 155541 | Melanocortin 4 Receptor | 18q21.32 | Non-Syndromic |
| MRAP2 | 615410 | Melanocortin Receptor Accessory Protein 2 | 6q14.2 | Syndromic |
| NPY | 162640 | Neuropetide Y | 7p15.3 | Non-Syndromic |
| NTRK2 | 600456 | Neurotrophic Receptor Tyrosine Kinase 2 | 9q21.33 | Syndromic |
| PCSK1- PC1 | 162150 | Proprotein Convertase 1 | 5q15 | Non-Syndromic |
| POMC | 176830 | Proopiomelanocortin | 2p23.3 | Non-Syndromic |
| POU3F2 | 600494 | Pou Domain, Class 3, Transcription Factor 2 | 6q16.1 | Syndromic |
| PYY | 600781 | Peptide YY | 17q21.31 | Non-Syndromic |
| SH2B1 | 608937 | SH2B Adaptor Protein 1 | 16p11.2 | Syndromic |
| SIM1 | 603128 | Single-minded 1 | 6q16.3 | Syndromic |
| TUB | 601197 | Tubby | 11p15.4 | Syndromic |
| UCP3 | 602044 | Uncoupling Protein 3 | 11q13.4 | Non-Syndromic |
Note: An updated list of principal monogenic genes that have been accepted as responsible for the majority of non-recognizable mental delay with obesity and non-syndromic obesity phenotypes, from the OMIM catalogue.
These panels of genes have been designed to identify mutations in individuals with severe obesity with greater efficiency. A manuscript has recently emerged which avoided classifying obese patients [n=1230] as having non-syndromic or syndromic phenotypes [95]. For inclusion in the study, patients had an obesity phenotype and at least one of the following criteria: an age of obesity onset <5 years (or up to prepubertal onset in the adult subgroup), a high risk familial history of obesity, hyperphagia, intellectual disability or developmental delay, congenital malformations, visual impairment and/or deafness, abnormal growth parameters (determined by head circumference and height), maintained extreme obesity (BMI >50 kg/m2), and being unresponsive to treatment which was defined by weight regain or insufficient weight loss. The candidate genes that were analyzed included most of the genes previously mentioned in this manuscript. These genes were selected due to their association with an obese phenotype per the OMIM catalogue, or because they had been associated with obesity in Genome-Wide Association Studies (GWAS), or involved in obesity or diabetes pathways as described by the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database, as well as those identified from known obesity CNVs including: ALMS1, ARL6, BBS1, BBS2, BBS4, BBS5, BBS7, BBS9, BBS10, BBS12, BDNF, CCDC28B, CEP290, CRHR2, FLOT1, G6PC, GNAS, IRS1, IRS2, IRS4, KIDINS220, LEP, LEPR, LZTFL1, MAGEL2, MC3R, MC4R, MCHR1, MKKS, MKRN3, MKS1, MRAP2, NDN, NTRK2, PAX6, PCK1, PCSK1, PHF6, POMC, PRKAR1A, PTEN, SIM1, SNRPD2, SNRPN, SPG11, TBX3, THRB, TMEM67, TRIM32, TTC8, TUB and WDPCP.
Kleinendorst et al. demonstrated that analysis of mixed syndromic and non-syndromic obese (n=1230 affected individuals) using a gene panel that includes genes simply associated with severe overweight, within the framework of any phenotype, resulted in the genetic diagnosis of the expected percentage of syndromic individuals (3.9% globally and 7.3% in the pediatric subgroup). A definitive molecular diagnosis was established in 2.7% of the patients in the adult subgroup (12.5% of adult patients had a definitive molecular diagnosis of syndromic obesity and the rest were linked to non-syndromic obesity). The mutations identified as responsible for syndromic obesity in these adult patients were located in 11 genes (previously marked in bold). In addition, 5.4% of individuals carried VUS in 44 genes, which had been identified through familial segregation and functional studies.
There were two very interesting conclusions to this study. Firstly, MC4R mutations were found to be the most frequent genetic cause of obesity in the cohort from the 11 different genes in which mutations were found to lead to a definitive diagnosis. Secondly, 5% of the total cohort were identified as carriers of a known heterozygous pathogenic mutation that only leads to an obesity phenotype in autosomal recessive modes of inheritance (ALMS1, PCK1, SPG11, TUB, BBS genes and modifiers). An additional 6.2% of the total cohort were carriers of a VUS in one of those genes, with the Bardet-Biedl susceptibility genes the most common. This suggests a stronger predisposition for common obesity in individuals who carry the heterozygote BBS gene mutation compared to the general population [95].
Everything discussed in this section leads us to determine that using the panel of genes with a known relationship to obesity is a good strategy for the analysis of all patients affected by severe early overweight, that is maintained and related to any phenotype. All of the genes currently classified as susceptibility genes for syndromic obesity and non-syndromic obesity should be considered (Table 1). It must also be proven that these genes are inherited with a Mendelian pattern of inheritance. However, we still consider that this strategy must be carried out, once the previous steps of the diagnostic algorithm that we describe in this manuscript have been carried out.
If, at this point a genetic diagnosis has been determined, the diagnosis “pipeline” would finish here. However, if the origin of the non-canonical syndromic obesity has still not been elucidated, the next step would be exome sequencing.
3.3. The Contribution of Clinical and Complete Exome Sequencing in Patients with Obesity within the Framework of Intellectual Disability
If all the previously discussed techniques have obtained negative results and the patient remains without a clear cause for syndromic obesity, the next step in the diagnosis “pipeline” is to sequence the clinical exome (OMIM genes) or the complete exome.
Clinical exome sequencing is used for patients characterized by an extreme phenotype, with or without a Mendelian pattern of genetic inheritance, after all previously associated single or groups of candidate genes have been ruled out and an aberrant cytogenetic etiology is suspected. Clinical exome sequencing involves analyzing all the encoding regions and intron boundaries of all OMIM genes by NGS [96]. In other words, all genes with a possible clinical relevance are sequenced. The aim is to determine a monogenic cause of the syndromic obesity phenotype and, therefore, a conclusion to the pathophysiology presented in the patient [97]. This technique generates a huge amount of data and the analysis of the variants is highly complex. In some cases, a single variant is detected and is the unique cause of the presented phenotype. However, in other cases, several genetic variants are identified and their impact on the clinical manifestation of the phenotype must be investigated. Finally, in other cases, any putative causal variants are detected.
When a variant is detected through sequencing of the clinical exome, it is mandatory to perform familial segregation of the variant and an analytic and clinical validation. Previously, the first validation involved corroborating the identification of NGS variants using another technique, though this is not currently necessary. Validation involves determining that the gene with the detected variant is involved in the disease, and that the specific variant has a pathogenic role in the development of the phenotype [98, 99]. Therefore, this technology allows the analysis of a variety y of candidate genes and the identification and addition of new locus/loci as possible causes of syndromic obesity. The global data strongly supports the value of clinical exome sequencing as an effective diagnostic tool, particularly for patients with obesity related to pediatric neurological diseases, and much more so than for individuals with non-syndromic obesity.
If clinical exome sequencing identifies a clear variant which could cause the phenotype, the diagnosis “pipeline” would finish here. However, if clinical exome sequencing does not yield results, the next step is whole exome sequencing. Whole exome sequencing, particularly within proband-parent trios, will help to advance clinical and research progress into extreme syndromic and non-affiliated phenotypes [100].
Using whole exome sequencing, as a genetic diagnostic test involves the extension of the clinical exome, so that the targeted sequence includes both OMIM and non-OMIM genes. The strategy of detecting and validating variants using whole exome sequencing has the same aim as clinical exome sequencing; familial segregation of the alleged variant, corroboration of the variant using another technology, and proving that the phenotype is a consequence of the detected genetic variant [101].
If whole exome sequencing identifies a clear variant which is responsible for the syndromic obesity phenotype, the diagnosis “pipeline” would finish here. However, whole exome sequencing is sometimes unable to determine the genetic cause of the syndromic phenotype and returns a negative result. In this instance, the algorithm “pipeline” is re-started and the medical history is re-investigated and re-evaluated for a potential complication during pregnancy or birth, including maternal alcohol or drug use or a lack of oxygen during pregnancy or birth.
In recent years, a considerable number of mutations have been detected in the general population which has complicated the validation of the causality of mutations identified in the patient group. Some mutations have been found which, despite their molecular characteristics, should be the cause of diseases with a Mendelian inheritance pattern but the carriers of these gene mutations do not exhibit the disease trait to any extent [102].
The international effort to develop genetic databases has been impressive and proved an invaluable tool in genetic counseling. The information contained in the databases facilitates rigorous genetic diagnostics and counseling, at the extremely high standard required. The table below details the online tools that are most frequently consulted by the professionals who work in the discipline of translational genomics (Table 2).
Table 2.
Accesses to the genetic databases most frequently consulted for genetic diagnosis and counseling in hereditary diseases.
Note: The most frequently accessed genetic databases by professionals working in the field of genetic diagnostics and couseling for hereditary diseases, with their name and their main utility. These online tools have become essential in the scientific research and exhaustive clinical management of obesity, and provide the basis for the study of syndromic obesity [55]. “Adapted from Duzkale 2013”.
3.4. Differential Gene Expression Profiles in Obese Subjects
Preliminary results have already shown sufficient differences in the gene expression between the obese and the non-obese subjects; the main objective is to identify the correlations between gene profiles and clinical obesity indices [103]. SIRT1 gene is considered an obesity biomarker, as its compensatory increase brings some additional metabolic dysfunctions in the body due to altered peptides in the endocrine system.
SIRT1 upregulates the level of orexin receptor specifically in the lateral hypothalamic area and the ventromedial nucleus of the hypothalamus, and also regulates the expression of BDNF (*113505) in the brain. It was found that increased SIRT1 level diminished BDNF signal which resulted in severe hyperphagia and obesity both in humans and animals. That coincidence between neurodevelopment and metabolic pathways throught SIRT1, in the same manner that was widely corroborated for the BDNF gene, presume that SIRT expression analysis could be an obesity biomarker to take into account among clinical parameters to evaluate in the management of obesity. Recent studies have shown that age-related diseases or endocrine system dysfunctions are associated with an increase in SIRT1 expression levels, but with a decrease in their activity [23].
CONCLUSION
The application of a normalized algorithm for the genetic diagnosis of syndromic obesity is essential due to the high heterogeneity of causes. We have described a revised and updated homogeneous algorithm for diagnosing future patients affected by syndromic obesity. The starting point for diagnosis in this patient group is to fully characterize familial antecedents, the history of pregnancy and newborn, and the phenotype of the individual. When a complex non-affiliated disorder is encountered, consideration should be given to non-genetic or acquired causes. This is particularly important for alterations observed in the field of neurodevelopment and obesity, and the approach should be the same whether alterations are expressed in isolation or in combination. Alcohol, drugs, tobacco and fever during pregnancy can have a teratogenic effect that could be responsible for the manifestation of the observed phenotype.
The genetic etiology of syndromic obesity is strikingly different for the two main subgroups; classical obesity syndromes and non-canonical obesity syndromes. The first group has well-characterized phenotypes of obesity alongside intellectual disability, among other traits. Examples of classical obesity syndromes include Prader-willi, Alström, Bardet-Bield, WAGR, Down and Fragile X syndromes. The causes vary from trinucleotide repeat expansion for Fragile X syndrome, uniparental disomies for UPD14 and methylation abnormalities for Prader-willi syndrome. Each of the classical obesity syndromes has a determined inheritance pattern and a specific gene responsible for the signs and symptoms. On the other hand, the non-canonical obesity syndromes have a wide heterogeneity of phenotypes with different genetic origins, including chromosome rearrangements, deletions and duplications; monogenic, and sometimes familial, variants with different inheritance patterns in specific obesity genes (with syndromic and non-syndromic causes); and genetic variants in non-candidate genes which are detected through exhaustive genome sequencing. Due to this huge variability, determination of the precise cause of non-canonical obesity is difficult. The proposed diagnosis “pipeline” described in this manuscript is the most suitable algorithm to diagnose syndromic obesity with non-canonical phenotypes and provides a comprehensive methodology which is able to solve the majority of cases.
The first step is to collate all of the clinical data, including any tests that the patient may have had to date. Laboratory tests are of particular importance, especially those performed through cytogenetic methods or molecular diagnostic techniques. Previous tests must be carefully assessed to determine their relevance and may need to be repeated with current technology. Of course, it is important not to repeat previous genetic analysis if they meet current standards, and the results from previous Sanger directed sequencing, methylation studies, genomic DNA hybridization analysis with arrays and massive sequencing should be exhaustively assessed to determine their significance. For this, it is essential to have access to the original report issued by the laboratory that performed the test.
Additionally, much importance should be given to any information that is available from other affected individuals in the family, particularly if they have undergone genetic testing.
In most cases, cytogenetic alterations are the main cause of these non-canonical phenotypes and whole-genome microarray hybridization is used as the principal technology to confirm or discard this cause. The most frequently diagnosed form of non-canonical obesity syndrome is the 16p11.2 microdeletion syndrome. The second most common cause of non-canonical obesity is the presence of mutations or alterations in the monogenic genes related to any kind of severe obesity. This type of obesity would be early-onset with a familial phenotype non-related to intellectual disability (mainly caused by the genes LEP, LEPR, POMC, PCSK1, MC4R, ADCY3 and CEP19) or manifested with intellectual deficits (mainly caused by the genes CEP19, TUB, CPE, POU3F2, MRAP2, SH2B1, SIM1, NTRK2, BDNF and DYRK1B). When the origin of these monogenic causes is determined, familial segregation is required to predict phenotype development, penetrance and variability in order to prevent incidences of the disease in future generations. Finally, if these investigations have not been able to determine the genetic cause and make a diagnosis. Clinical exome or whole exome sequencing could enable the detection of alleged variants that might be responsible for the syndromic obesity phenotype. If a monogenic cause of obesity is genetically determined by exome sequencing, it is mandatory to perform segregation studies of the mutation in all members of the family in order to confirm or discard a de novo origin, and to identify family members who carry the mutation. This facilitates better genetic counseling for future generations.
Research into obesity and mental delay genetics is rapidly progressing. NGS gene panel testing is continually identifying obesity-associated genes altered by mutations, as well as functional CNV and cumulative genetic risk profiles could provide a sufficient molecular basis to cause a specific phenotype in a single individual.
Newly identified genes will be added to future versions of the gene panels to aid the diagnosis of syndromic obesity. Nowadays, most of the mutations identified are related to non-syndromic monogenic forms of obesity, due to the inclusion bias of patients. It is a very common occurrence that patients who present a syndromic form of obesity are genetically analyzed for their mental disability and/or congenital anomalies only, rather than for their characteristics of obesity, even when obesity is extreme.
Available data suggests that modification of these diagnostic strategies is important, given that an established diagnosis of genetic obesity could influence the choice of treatment, e.g. bariatric surgery [94]. An effective and globally accepted NGS-based gene panel analysis in patients with obesity might be a useful tool for diagnosing genetic obesity and could have serious impact on the treatment of patients.
Although in our experience this genetic algorithm is successful in determining an accurate diagnosis, sometimes there will be cases where a final diagnosis is not achieved and the next step in the algorithm is to return to the beginning. Therefore, we emphasize the importance of neuropediatrics, neurologists and genetic consultations in performing an exhaustive medical history including pregnancy and family history.
ACKNOWLEDGEMENTS
To CeGen and especially Ines Quintela for her inestimable contribution since 2006, for the realization of full genome arrays in all diagnostic cases of syndromic obesity in our laboratories. To Cristina Torreira for providing us with the data from her syndromic obesity series to increase our experience in the existence of CNVs associated with the risk of severe overweight.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This study has been funded by the Instituto de Salud Carlos III through the project “PI17/01551 ISCIII”; Title: “Integration of omic techniques for the identification of polygenic profiles of susceptibility to obesity”; principal research RRL (Co-funded by the European Regional Development Fund/European Social Fund “A way to make Europe”/”Investing in your future”).
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Campbell Am L.V. Genetics of obesity. Aust. Fam. Physician. 2017;46(7):456–459. [PubMed] [Google Scholar]
- 2.Pratt H.D., Greydanus D.E. Intellectual disability (mental retardation) in children and adolescents. Prim. Care. 2007;34(2):375–386. doi: 10.1016/j.pop.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 3.Kaur Y., de Souza R.J., Gibson W.T., Meyre D. A systematic review of genetic syndromes with obesity. Obes. Rev. an Off J. Int. Assoc. Study. Obes. 2017;18(6):603–634. doi: 10.1111/obr.12531. [DOI] [PubMed] [Google Scholar]
- 4.Singh R.K., Kumar P., Mahalingam K. Molecular genetics of human obesity: A comprehensive review. C. R. Biol. 2017;340(2):87–108. doi: 10.1016/j.crvi.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Dasouki M.J., Youngs E.L., Hovanes K. Structural chromosome abnormalities associated with obesity: Report of four new subjects and review of literature. Curr. Genomics. 2011;12(3):190–203. doi: 10.2174/138920211795677930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weihrauch-Blüher S., Wiegand S. Risk factors and implications of childhood obesity. Curr. Obes. Rep. 2018;7(4):254–259. doi: 10.1007/s13679-018-0320-0. [DOI] [PubMed] [Google Scholar]
- 7.Albuquerque D., Nóbrega C., Manco L., Padez C. The contribution of genetics and environment to obesity. Br. Med. Bull. 2017;123(1):159–173. doi: 10.1093/bmb/ldx022. [DOI] [PubMed] [Google Scholar]
- 8.Vasileva L.V., Marchev A.S., Georgiev M.I. Causes and solutions to “globesity”: The new fa(s)t alarming global epidemic. J. Publ. Br. Ind. Biol. Res. Assoc. 2018;121:173–193. doi: 10.1016/j.fct.2018.08.071. [DOI] [PubMed] [Google Scholar]
- 9.Albuquerque D., Stice E., Rodríguez-López R., Manco L., Nóbrega C. Current review of genetics of human obesity: From molecular mechanisms to an evolutionary perspective. Mol. Genet. Genomics. 2015;290(4):1191–1221. doi: 10.1007/s00438-015-1015-9. [DOI] [PubMed] [Google Scholar]
- 10.Maïano C. Prevalence and risk factors of overweight and obesity among children and adolescents with intellectual disabilities. Obes. Rev. 2011;12(3):189–197. doi: 10.1111/j.1467-789X.2010.00744.x. [DOI] [PubMed] [Google Scholar]
- 11.Delrue M.A., Michaud J.L. Fat chance: Genetic syndromes with obesity. Clin. Genet. 2004;66(2):83–93. doi: 10.1111/j.0009-9163.2004.00300.x. [DOI] [PubMed] [Google Scholar]
- 12.D’Angelo C.S., Kohl I., Varela M.C., de Castro C.I., Kim C.A., Bertola D.R., Lourenço C.M., Perez A.B., Koiffmann C.P. Obesity with associated developmental delay and/or learning disability in patients exhibiting additional features: Report of novel pathogenic copy number variants. Am. J. Med. Genet. A. 2013;161A(3):479–486. doi: 10.1002/ajmg.a.35761. [DOI] [PubMed] [Google Scholar]
- 13.Lespinasse J., Bugge M., Réthoré M.O., North M.O., Lundsteen C., Kirchhoff M. De novo Complex Chromosomal Rearrangements (CCR) involving chromosome 1, 5, and 6 resulting in microdeletion for 6q14 in a female carrier with psychotic disorder. Am. J. Med. Genet. A. 2004;128A(2):199–203. doi: 10.1002/ajmg.a.30064. [DOI] [PubMed] [Google Scholar]
- 14.Menten B., Buysse K., Vandesompele J., De Smet E., De Paepe A., Speleman F., Mortier G. Identification of an unbalanced X-autosome translocation by array CGH in a boy with a syndromic form of chondrodysplasia punctata brachytelephalangic type. Eur. J. Med. Genet. 2005;48(3):301–309. doi: 10.1016/j.ejmg.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 15.Probst F.J., Roeder E.R., Enciso V.B., Ou Z., Cooper M.L., Eng P., Li J., Gu Y., Stratton R.F., Chinault A.C., Shaw C.A., Sutton V.R., Cheung S.W., Nelson D.L. Chromosomal Microarray Analysis (CMA) detects a large X chromosome deletion including FMR1, FMR2, and IDS in a female patient with mental retardation. Am. J. Med. Genet. A. 2007;143A(12):1358–1365. doi: 10.1002/ajmg.a.31781. [DOI] [PubMed] [Google Scholar]
- 16.Vuillaume M-L., Naudion S., Banneau G., Diene G., Cartault A., Cailley D., Bouron J., Toutain J., Bourrouillou G., Vigouroux A., Bouneau L., Nacka F., Kieffer I., Arveiler B., Knoll-Gellida A., Babin P.J., Bieth E., Jouret B., Julia S., Sarda P., Geneviève D., Faivre L., Lacombe D., Barat P., Tauber M., Delrue M.A., Rooryck C. New candidate loci identified by array-CGH in a cohort of 100 children presenting with syndromic obesity. Am. J. Med. Genet. A. 2014;164A(8):1965–1975. doi: 10.1002/ajmg.a.36587. [DOI] [PubMed] [Google Scholar]
- 17.Zung A., Rienstein S., Rosensaft J., Aviram-Goldring A., Zadik Z. Proximal 19q trisomy: A new syndrome of morbid obesity and mental retardation. Horm. Res. 2007;67(3):105–110. doi: 10.1159/000096419. [DOI] [PubMed] [Google Scholar]
- 18.D’Angelo C.S., Koiffmann C.P. Copy number variants in obesity-related syndromes: Review and perspectives on novel molecular approaches. J. Obes. 2012;2012:845480. doi: 10.1155/2012/845480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marenne G., Hendricks A.E., Perdikari A., Bounds R., Payne F., Keogh J.M., Lelliott C.J., Henning E., Pathan S., Ashford S., Bochukova E.G., Mistry V., Daly A., Hayward C., Wareham N.J., O’Rahilly S., Langenberg C., Wheeler E., Zeggini E., Farooqi I.S., Barroso I. Exome sequencing identifies genes and gene sets contributing to severe childhood obesity, linking PHIP variants to repressed POMC transcription. Cell Metab. 2020;31(6):1107–1119.e12. doi: 10.1016/j.cmet.2020.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Angelo C.S., Varela M.C., de Castro C.I., Otto P.A., Perez A.B.A., Lourenço C.M., Kim C.A., Bertola D.R., Kok F., Garcia-Alonso L., Koiffmann C.P. Chromosomal microarray analysis in the genetic evaluation of 279 patients with syndromic obesity. Mol. Cytogenet. 2018;11(1):14. doi: 10.1186/s13039-018-0363-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D’Angelo C.S., Varela M.C., de Castro C.Ie., Kim C.A., Bertola D.R., Lourenço C.M., Perez A.B., Koiffmann C.P. Investigation of selected genomic deletions and duplications in a cohort of 338 patients presenting with syndromic obesity by multiplex ligation-dependent probe amplification using synthetic probes. Mol. Cytogenet. 2014;7(1):75. doi: 10.1186/s13039-014-0075-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiao H., Arner P., Gerdhem P., Strawbridge R.J., Näslund E., Thorell A., Hamsten A., Kere J., Dahlman I. Exome sequencing followed by genotyping suggests SYPL2 as a susceptibility gene for morbid obesity. Eur. J. Hum. Genet. 2015;23(9):1216–1222. doi: 10.1038/ejhg.2014.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elibol B., Kilic U. High levels of SIRT1 expression as a protective mechanism against disease-related conditions. Front. Endocrinol. (Lausanne) 2018;9:614. doi: 10.3389/fendo.2018.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martins I.J. Anti-aging genes improve appetite regulation and reverse cell senescence and apoptosis in global populations. Adv. Aging Res. 2016;5(1):9–26. doi: 10.4236/aar.2016.51002. [DOI] [Google Scholar]
- 25.Martins I.J. Single gene inactivation with implications to diabetes and multiple organ dysfunction syndrome. J. Clin. Epigenetics. 2017;3(3):24. [Google Scholar]
- 26.Bachmann-Gagescu R., Mefford H.C., Cowan C., Glew G.M., Hing A.V., Wallace S., Bader P.I., Hamati A., Reitnauer P.J., Smith R., Stockton D.W., Muhle H., Helbig I., Eichler E.E., Ballif B.C., Rosenfeld J., Tsuchiya K.D. Recurrent 200-kb deletions of 16p11.2 that include the SH2B1 gene are associated with developmental delay and obesity. Genet. Med. 2010;12(10):641–647. doi: 10.1097/GIM.0b013e3181ef4286. [DOI] [PubMed] [Google Scholar]
- 27.Gimeno-Ferrer F., Albuquerque D., Guzmán Luján C., Marcaida Benito G., Torreira Banzas C., Repáraz-Andrade A., Ballesteros Cogollos V., Aleu Pérez-Gramunt M., Galán Gómez E., Quintela I., Rodríguez-López R. The effect of copy number variations in chromosome 16p on body weight in patients with intellectual disability. J. Hum. Genet. 2019;64(3):221–231. doi: 10.1038/s10038-018-0545-5. [DOI] [PubMed] [Google Scholar]
- 28.Pettersson M., Viljakainen H., Loid P., Mustila T., Pekkinen M., Armenio M., Andersson-Assarsson J.C., Mäkitie O., Lindstrand A. Copy number variants are enriched in individuals with early-onset obesity and highlight novel pathogenic pathways. J. Clin. Endocrinol. Metab. 2017;102(8):3029–3039. doi: 10.1210/jc.2017-00565. [DOI] [PubMed] [Google Scholar]
- 29.Redaelli S., Maitz S., Crosti F., Sala E., Villa N., Spaccini L., Selicorni A., Rigoldi M., Conconi D., Dalprà L., Roversi G., Bentivegna A. Refining the phenotype of recurrent rearrangements of chromosome 16. Int. J. Mol. Sci. 2019;20(5):E1095. doi: 10.3390/ijms20051095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casas M., Chatzi L., Carsin A.E., Amiano P., Guxens M., Kogevinas M., Koutra K., Lertxundi N., Murcia M., Rebagliato M., Riaño I., Rodríguez-Bernal C.L., Roumeliotaki T., Sunyer J., Mendez M., Vrijheid M. Maternal pre-pregnancy overweight and obesity, and child neuropsychological development: Two Southern European birth cohort studies. Int. J. Epidemiol. 2013;42(2):506–517. doi: 10.1093/ije/dyt002. [DOI] [PubMed] [Google Scholar]
- 31.Memo L., Gnoato E., Caminiti S., Pichini S., Tarani L. Fetal alcohol spectrum disorders and fetal alcohol syndrome: The state of the art and new diagnostic tools. Early Hum. Dev. 2013;89(Suppl. 1):S40–S43. doi: 10.1016/S0378-3782(13)70013-6. [DOI] [PubMed] [Google Scholar]
- 32.Scott-Goodwin A.C., Puerto M., Moreno I. Toxic effects of prenatal exposure to alcohol, tobacco and other drugs. Reprod. Toxicol. 2016;61:120–130. doi: 10.1016/j.reprotox.2016.03.043. [DOI] [PubMed] [Google Scholar]
- 33.Denny L., Coles S., Blitz R. Fetal alcohol syndrome and fetal alcohol spectrum disorders. Am. Fam. Physician. 2017;96(8):515–522. [PubMed] [Google Scholar]
- 34.Fernandez-Twinn D.S., Hjort L., Novakovic B., Ozanne S.E., Saffery R. Intrauterine programming of obesity and type 2 diabetes. Diabetologia. 2019;62(10):1789–1801. doi: 10.1007/s00125-019-4951-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Charlin A., Vexliard D., Guerre D., Peuteuil P. Severe retardation of affective origin, curable by environmental psychotherapy. Ethiological approach. Ann. Med. Psychol. (Paris) 1975;1(3):356–362. [PubMed] [Google Scholar]
- 36.Hofstatter L., Hofstatter L. Emotional problems of the child with mental retardation and his family. South. Med. J. 1969;62(5):583–587. doi: 10.1097/00007611-196905000-00019. [DOI] [PubMed] [Google Scholar]
- 37.Köhler S., Vasilevsky N.A., Engelstad M., Foster E., McMurry J., Aymé S., Baynam G., Bello S.M., Boerkoel C.F., Boycott K.M., Brudno M., Buske O.J., Chinnery P.F., Cipriani V., Connell L.E., Dawkins H.J.S., DeMare L.E., Devereau A.D., de Vries B.B.A., Firth H.V., Freson K., Greene D., Hamosh A., Helbig I., Hum C., Jähn J.A., James R., Krause R., F Laulederkind. S.J.; Lochmüller, H.; Lyon, G.J.; Ogishima, S.; Olry, A.; Ouwehand, W.H.; Pontikos, N.; Rath, A.; Schaefer, F.; Scott, R.H.; Segal, M.; Sergouniotis, P.I.; Sever, R.; Smith, C.L.; Straub, V.; Thompson, R.; Turner, C.; Turro, E.; Veltman, M.W.; Vulliamy, T.; Yu, J.; von Ziegenweidt, J.; Zankl, A.; Züchner, S.; Zemojtel, T.; Jacobsen, J.O.; Groza, T.; Smedley, D.; Mungall, C.J.; Haendel, M.; Robinson, P.N. The human phenotype ontology in 2017. Nucleic Acids Res. 2017;45(D1):D865–D876. doi: 10.1093/nar/gkw1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chung W.K. An overview of mongenic and syndromic obesities in humans. Pediatr. Blood Cancer. 2012;58(1):122–128. doi: 10.1002/pbc.23372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asim A., Kumar A., Muthuswamy S., Jain S., Agarwal S. Down syndrome: An insight of the disease. J. Biomed. Sci. 2015;22(1):41. doi: 10.1186/s12929-015-0138-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Vries B.B., Mohkamsing S., van den Ouweland A.M., Mol E., Gelsema K., van Rijn M., Tibben A., Halley D.J., Duivenvoorden H.J., Oostra B.A., Niermeijer M.F. Screening for the fragile X syndrome among the mentally retarded: A clinical study. J. Med. Genet. 1999;36(6):467–470. [PMC free article] [PubMed] [Google Scholar]
- 41.González-Jiménez E., Aguilar Cordero M.J., Padilla López C.A., García García I. Monogenic human obesity: Role of the leptin-melanocortin system in the regulation of food intake and body weight in humans. An. Sist. Sanit. Navar. 2012;35(2):285–293. doi: 10.4321/s1137-66272012000200010. [DOI] [PubMed] [Google Scholar]
- 42.Butler M.G. Prader-Willi syndrome: Obesity due to genomic imprinting. Curr. Genomics. 2011;12(3):204–215. doi: 10.2174/138920211795677877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cassidy S.B., Dykens E., Williams C.A. Prader-Willi and Angelman syndromes: Sister imprinted disorders. Am. J. Med. Genet. 2000;97(2):136–146. doi: 10.1002/1096-8628(200022)97:2<136:AID-AJMG5>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 44.Farooqi I.S., O’Rahilly S. Monogenic obesity in humans. Annu. Rev. Med. 2005;56(1):443–458. doi: 10.1146/annurev.med.56.062904.144924. [DOI] [PubMed] [Google Scholar]
- 45.Suspitsin E.N., Imyanitov E.N. Bardet-Biedl syndrome. Mol. Syndromol. 2016;7(2):62–71. doi: 10.1159/000445491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monzó C., Gimeno-Ferrer F., Ferrer J.C., Amadoz A., Albuquerque D., Barros F., Marcaida G., Rodríguez-López R. Alström syndrome caused by deletion in ALMS1 gene fixed in a Northern Pakistan recurrent haplotype. Indian J. Case Reports. 2017;3(4):171–174. [Google Scholar]
- 47.Álvarez-Satta M., Castro-Sánchez S., Valverde D. Alström syndrome: Current perspectives. Appl. Clin. Genet. 2015;8:171–179. doi: 10.2147/TACG.S56612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rodríguez-López R., Pérez J.M., Balsera A.M., Rodríguez G.G., Moreno T.H., García de Cáceres M., Serrano M.G., Freijo F.C., Ruiz J.R., Angueira F.B., Pérez P.M., Estévez M.N., Gómez E.G. The modifier effect of the BDNF gene in the phenotype of the WAGRO syndrome. Gene. 2013;516(2):285–290. doi: 10.1016/j.gene.2012.11.073. [DOI] [PubMed] [Google Scholar]
- 49.Fischbach B.V., Trout K.L., Lewis J., Luis C.A., Sika M. WAGR syndrome: A clinical review of 54 cases. Pediatrics. 2005;116(4):984–988. doi: 10.1542/peds.2004-0467. [DOI] [PubMed] [Google Scholar]
- 50.Leclercq V., Benoit V., Lederer D., Delaunoy M., Ruiz M., de Halleux C., Robaux O., Wanty C., Maystadt I. Case report: An infantile lethal form of Albright hereditary osteodystrophy due to a GNAS mutation. Clin. Case Rep. 2018;6(10):1933–1940. doi: 10.1002/ccr3.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Le Scouarnec S., Gribble S.M. Characterising chromosome rearrangements: Recent technical advances in molecular cytogenetics. Heredity. 2012;108(1):75–85. doi: 10.1038/hdy.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaminsky E.B., Kaul V., Paschall J., Church D.M., Bunke B., Kunig D., Moreno-De-Luca D., Moreno-De-Luca A., Mulle J.G., Warren S.T., Richard G., Compton J.G., Fuller A.E., Gliem T.J., Huang S., Collinson M.N., Beal S.J., Ackley T., Pickering D.L., Golden D.M., Aston E., Whitby H., Shetty S., Rossi M.R., Rudd M.K., South S.T., Brothman A.R., Sanger W.G., Iyer R.K., Crolla J.A., Thorland E.C., Aradhya S., Ledbetter D.H., Martin C.L. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet. Med. 2011;13(9):777–784. doi: 10.1097/GIM.0b013e31822c79f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weise A., Mrasek K., Klein E., Mulatinho M., Llerena J.C., Jr, Hardekopf D., Pekova S., Bhatt S., Kosyakova N., Liehr T. Microdeletion and microduplication syndromes. J. Histochem. Cytochem. Off J. Histochem. Soc. 2012;60(5):346–358. doi: 10.1369/0022155412440001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nowakowska B. Clinical interpretation of copy number variants in the human genome. J. Appl. Genet. 2017;58(4):449–457. doi: 10.1007/s13353-017-0407-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duzkale H., Shen J., McLaughlin H., Alfares A., Kelly M.A., Pugh T.J., Funke B.H., Rehm H.L., Lebo M.S. A systematic approach to assessing the clinical significance of genetic variants. Clin. Genet. 2013;84(5):453–463. doi: 10.1111/cge.12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Egger G., Dixon J. Beyond obesity and lifestyle: A review of 21st century chronic disease determinants. BioMed Res. Int. 2014;2014:731685. doi: 10.1155/2014/731685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perrone L., Marzuillo P., Grandone A., del Giudice E.M. Chromosome 16p11.2 deletions: Another piece in the genetic puzzle of childhood obesity. Ital. J. Pediatr. 2010;36(1):43. doi: 10.1186/1824-7288-36-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goldenberg P. An update on common chromosome microdeletion and microduplication syndromes. Pediatr. Ann. 2018;47(5):e198–e203. doi: 10.3928/19382359-20180419-01. [DOI] [PubMed] [Google Scholar]
- 59.Yazdi F.T., Clee S.M., Meyre D. Obesity genetics in mouse and human: Back and forth, and back again. PeerJ. 2015;3:e856. doi: 10.7717/peerj.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y., Chua S., Jr Leptin function and regulation. Compr. Physiol. 2017;8(1):351–369. doi: 10.1002/cphy.c160041. [DOI] [PubMed] [Google Scholar]
- 61.Pan W.W., Myers M.G.J., Jr Leptin and the maintenance of elevated body weight. Nat. Rev. Neurosci. 2018;19(2):95–105. doi: 10.1038/nrn.2017.168. [DOI] [PubMed] [Google Scholar]
- 62.Nies V.J.M., Struik D., Wolfs M.G.M., Rensen S.S., Szalowska E., Unmehopa U.A., Fluiter K., van der Meer T.P., Hajmousa G., Buurman W.A., Greve J.W., Rezaee F., Shiri-Sverdlov R., Vonk R.J., Swaab D.F., Wolffenbuttel B.H.R., Jonker J.W., van Vliet-Ostaptchouk J.V. TUB gene expression in hypothalamus and adipose tissue and its association with obesity in humans. Int. J. Obes. 2018;42(3):376–383. doi: 10.1038/ijo.2017.214. [DOI] [PubMed] [Google Scholar]
- 63.Wang Y., Wang A., Donovan S.M., Terán-García M. Individual genetic variations related to satiety and appetite control increase risk of obesity in preschool-age children in the STRONG kids program. Hum. Hered. 2013;75(2-4):152–159. doi: 10.1159/000353880. [DOI] [PubMed] [Google Scholar]
- 64.Borman A.D., Pearce L.R., Mackay D.S., Nagel-Wolfrum K., Davidson A.E., Henderson R., Garg S., Waseem N.H., Webster A.R., Plagnol V., Wolfrum U., Farooqi I.S., Moore A.T. A homozygous mutation in the TUB gene associated with retinal dystrophy and obesity. Hum. Mutat. 2014;35(3):289–293. doi: 10.1002/humu.22482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rubinstein M., Low M.J. Molecular and functional genetics of the proopiomelanocortin gene, food intake regulation and obesity. FEBS Lett. 2017;591(17):2593–2606. doi: 10.1002/1873-3468.12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ren D., Zhou Y., Morris D., Li M., Li Z., Rui L. Neuronal SH2B1 is essential for controlling energy and glucose homeostasis. J. Clin. Invest. 2007;117(2):397–406. doi: 10.1172/JCI29417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramos-Molina B., Martin M.G., Lindberg I. PCSK1 variants and human obesity. Prog. Mol. Biol. Transl. Sci. 2016;140:47–74. doi: 10.1016/bs.pmbts.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alsters S.I.M., Goldstone A.P., Buxton J.L., Zekavati A., Sosinsky A., Yiorkas A.M., Holder S., Klaber R.E., Bridges N., van Haelst M.M., le Roux C.W., Walley A.J., Walters R.G., Mueller M., Blakemore A.I.F. Truncating homozygous mutation of carboxypeptidase E (CPE) in a morbidly obese female with type 2 diabetes mellitus, intellectual disability and hypogonadotrophic hypogonadism. PLoS One. 2015;10(6):e0131417. doi: 10.1371/journal.pone.0131417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Millington G.W. The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr. Metab. (Lond.) 2007;4(1):18. doi: 10.1186/1743-7075-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ju S.H., Cho G.B., Sohn J.W. Understanding melanocortin-4 receptor control of neuronal circuits: Toward novel therapeutics for obesity syndrome. Pharmacol. Res. 2018;129:10–19. doi: 10.1016/j.phrs.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 71.Geets E., Zegers D., Beckers S., Verrijken A., Massa G., Van Hoorenbeeck K., Verhulst S., Van Gaal L., Van Hul W. Copy Number Variation (CNV) analysis and mutation analysis of the 6q14.1-6q16.3 genes SIM1 and MRAP2 in Prader Willi like patients. Mol. Genet. Metab. 2016;117(3):383–388. doi: 10.1016/j.ymgme.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 72.Chaly A.L., Srisai D., Gardner E.E., Sebag J.A. The melanocortin receptor accessory protein 2 promotes food intake through inhibition of the prokineticin receptor-1. eLife. 2016;5:5. doi: 10.7554/eLife.12397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jackson D.S., Ramachandrappa S., Clark A.J., Chan L.F. Melanocortin receptor accessory proteins in adrenal disease and obesity. Front. Neurosci. 2015;9:213. doi: 10.3389/fnins.2015.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu T., Elmquist J.K., Williams K.W. Mrap2: An accessory protein linked to obesity. Cell Metab. 2013;18(3):309–311. doi: 10.1016/j.cmet.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gray J., Yeo G.S.H., Cox J.J., Morton J., Adlam A.L.R., Keogh J.M., Yanovski J.A., El Gharbawy A., Han J.C., Tung Y.C.L., Hodges J.R., Raymond F.L., O’rahilly S., Farooqi I.S. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes. 2006;55(12):3366–3371. doi: 10.2337/db06-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grarup N., Moltke I., Andersen M.K., Dalby M., Vitting-Seerup K., Kern T., Mahendran Y., Jørsboe E., Larsen C.V.L., Dahl-Petersen I.K., Gilly A., Suveges D., Dedoussis G., Zeggini E., Pedersen O., Andersson R., Bjerregaard P., Jørgensen M.E., Albrechtsen A., Hansen T. Loss-of-function variants in ADCY3 increase risk of obesity and type 2 diabetes. Nat. Genet. 2018;50(2):172–174. doi: 10.1038/s41588-017-0022-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Siljee J.E., Wang Y., Bernard A.A., Ersoy B.A., Zhang S., Marley A., Von Zastrow M., Reiter J.F., Vaisse C. Subcellular localization of MC4R with ADCY3 at neuronal primary cilia underlies a common pathway for genetic predisposition to obesity. Nat. Genet. 2018;50(2):180–185. doi: 10.1038/s41588-017-0020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stanikova D., Buzga M., Krumpolec P., Skopkova M., Surova M., Ukropcova B., Ticha L., Petrasova M., Gabcova D., Huckova M., Piskorova L., Bozensky J., Mokan M., Ukropec J., Zavacka I., Klimes I., Stanik J., Gasperikova D. Genetic analysis of single-minded 1 gene in early-onset severely obese children and adolescents. PLoS One. 2017;12(5):e0177222. doi: 10.1371/journal.pone.0177222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gray J., Yeo G., Hung C., Keogh J., Clayton P., Banerjee K., McAulay A., O’Rahilly S., Farooqi I.S. Functional characterization of human NTRK2 mutations identified in patients with severe early-onset obesity. Int. J. Obes. 2007;31(2):359–364. doi: 10.1038/sj.ijo.0803390. [DOI] [PubMed] [Google Scholar]
- 80.Elias C.F., Lee C., Kelly J., Aschkenasi C., Ahima R.S., Couceyro P.R., Kuhar M.J., Saper C.B., Elmquist J.K. Leptin activates hypothalamic CART neurons projecting to the spinal cord. Neuron. 1998;21(6):1375–1385. doi: 10.1016/S0896-6273(00)80656-X. [DOI] [PubMed] [Google Scholar]
- 81.del Giudice E.M., Santoro N., Cirillo G., D’Urso L., Di Toro R., Perrone L. Mutational screening of the CART gene in obese children: Identifying a mutation (Leu34Phe) associated with reduced resting energy expenditure and cosegregating with obesity phenotype in a large family. Diabetes. 2001;50(9):2157–2160. doi: 10.2337/diabetes.50.9.2157. [DOI] [PubMed] [Google Scholar]
- 82.Echwald S.M., Sørensen T.I., Andersen T., Hansen C., Tommerup N., Pedersen O. Sequence variants in the human Cocaine and Amphetamine-Regulated Transcript (CART) gene in subjects with early onset obesity. Obes. Res. 1999;7(6):532–536. doi: 10.1002/j.1550-8528.1999.tb00710.x. [DOI] [PubMed] [Google Scholar]
- 83.Suzuki K., Jayasena C.N., Bloom S.R. Obesity and appetite control. Exp. Diabetes Res. 2012;2012:824305. doi: 10.1155/2012/824305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wen S., Wang C., Gong M., Zhou L. An overview of energy and metabolic regulation. Sci. China Life Sci. 2019;62(6):771–790. doi: 10.1007/s11427-018-9371-4. [DOI] [PubMed] [Google Scholar]
- 85.Aerts E., Geets E., Sorber L., Beckers S., Verrijken A., Massa G., Van Hoorenbeeck K., Verhulst S.L., Van Gaal L.F., Van Hul W. Evaluation of a role for NPY and NPY2R in the pathogenesis of obesity by mutation and copy number variation analysis in obese children and adolescents. Ann. Hum. Genet. 2018;82(1):1–10. doi: 10.1111/ahg.12211. [DOI] [PubMed] [Google Scholar]
- 86.Tatemoto K., Mutt V. Isolation of two novel candidate hormones using a chemical method for finding naturally occurring polypeptides. Nature. 1980;285(5764):417–418. doi: 10.1038/285417a0. [DOI] [PubMed] [Google Scholar]
- 87.Ashby D., Bloom S.R. Recent progress in PYY research--an update report for 8th NPY meeting. Peptides. 2007;28(2):198–202. doi: 10.1016/j.peptides.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 88.Kasher P.R., Schertz K.E., Thomas M., Jackson A., Annunziata S., Ballesta-Martínez M.J., Campeau P.M., Clayton P.E., Eaton J.L., Granata T., Guillén-Navarro E., Hernando C., Laverriere C.E., Liedén A., Villa-Marcos O., McEntagart M., Nordgren A., Pantaleoni C., Pebrel-Richard C., Sarret C., Sciacca F.L., Wright R., Kerr B., Glasgow E., Banka S. Small 6q16.1 deletions encompassing pou3f2 cause susceptibility to obesity and variable developmental delay with intellectual disability. Am. J. Hum. Genet. 2016;98(2):363–372. doi: 10.1016/j.ajhg.2015.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Spikol E.D., Laverriere C.E., Robnett M., Carter G., Wolfe E.M., Glasgow E. Zebrafish models of prader-willi syndrome: Fast track to pharmacotherapeutics. Diseases. 2016;4(1):13. doi: 10.3390/diseases4010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ricquier D., Bouillaud F. Mitochondrial uncoupling proteins: From mitochondria to the regulation of energy balance. J. Physiol. 2000;529 Pt 1(Pt 1):3–10. doi: 10.1111/j.1469-7793.2000.00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jia J.J., Zhang X., Ge C.R., Jois M. The polymorphisms of UCP2 and UCP3 genes associated with fat metabolism, obesity and diabetes. Obes. Rev. 2009;10(5):519–526. doi: 10.1111/j.1467-789X.2009.00569.x. [DOI] [PubMed] [Google Scholar]
- 92.Shalata A., Ramirez M.C., Desnick R.J., Priedigkeit N., Buettner C., Lindtner C., Mahroum M., Abdul-Ghani M., Dong F., Arar N., Camacho-Vanegas O., Zhang R., Camacho S.C., Chen Y., Ibdah M., DeFronzo R., Gillespie V., Kelley K., Dynlacht B.D., Kim S., Glucksman M.J., Borochowitz Z.U., Martignetti J.A. Morbid obesity resulting from inactivation of the ciliary protein CEP19 in humans and mice. Am. J. Hum. Genet. 2013;93(6):1061–1071. doi: 10.1016/j.ajhg.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yıldız Bölükbaşı, E.; Mumtaz, S.; Afzal, M.; Woehlbier, U.; Malik, S.; Tolun, A. Homozygous mutation in CEP19, a gene mutated in morbid obesity, in Bardet-Biedl syndrome with predominant postaxial polydactyly. J. Med. Genet. 2018;55(3):189–197. doi: 10.1136/jmedgenet-2017-104758. [DOI] [PubMed] [Google Scholar]
- 94.Keramati A.R., Fathzadeh M., Go G.W., Singh R., Choi M., Faramarzi S., Mane S., Kasaei M., Sarajzadeh-Fard K., Hwa J., Kidd K.K., Babaee Bigi M.A., Malekzadeh R., Hosseinian A., Babaei M., Lifton R.P., Mani A. A form of the metabolic syndrome associated with mutations in DYRK1B. N. Engl. J. Med. 2014;370(20):1909–1919. doi: 10.1056/NEJMoa1301824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kleinendorst L., Massink M.P.G., Cooiman M.I., Savas M., van der Baan-Slootweg O.H., Roelants R.J., Janssen I.C.M., Meijers-Heijboer H.J., Knoers N.V.A.M., Ploos van Amstel H.K., van Rossum E.F.C., van den Akker E.L.T., van Haaften G., van der Zwaag B., van Haelst M.M. Genetic obesity: Next-generation sequencing results of 1230 patients with obesity. J. Med. Genet. 2018;55(9):578–586. doi: 10.1136/jmedgenet-2018-105315. [DOI] [PubMed] [Google Scholar]
- 96.Lee H., Deignan J.L., Dorrani N., Strom S.P., Kantarci S., Quintero-Rivera F., Das K., Toy T., Harry B., Yourshaw M., Fox M., Fogel B.L., Martinez-Agosto J.A., Wong D.A., Chang V.Y., Shieh P.B.S., Palmer C.G.S., Dipple K.M., Grody W.W., Vilain E., Nelson S.F. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312(18):1880–1887. doi: 10.1001/jama.2014.14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Biesecker L.G., Green R.C. Diagnostic clinical genome and exome sequencing. N. Engl. J. Med. 2014;370(25):2418–2425. doi: 10.1056/NEJMra1312543. [DOI] [PubMed] [Google Scholar]
- 98.MacArthur D.G., Manolio T.A., Dimmock D.P., Rehm H.L., Shendure J., Abecasis G.R., Adams D.R., Altman R.B., Antonarakis S.E., Ashley E.A., Barrett J.C., Biesecker L.G., Conrad D.F., Cooper G.M., Cox N.J., Daly M.J., Gerstein M.B., Goldstein D.B., Hirschhorn J.N., Leal S.M., Pennacchio L.A., Stamatoyannopoulos J.A., Sunyaev S.R., Valle D., Voight B.F., Winckler W., Gunter C. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508(7497):469–476. doi: 10.1038/nature13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Swaminathan R., Huang Y., Astbury C., Fitzgerald-Butt S., Miller K., Cole J., Bartlett C., Lin S. Clinical exome sequencing reports: Current informatics practice and future opportunities. J. Am. Med. Inform. Assoc. 2017;24(6):1184–1191. doi: 10.1093/jamia/ocx048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Córdoba M., Rodriguez-Quiroga S.A., Vega P.A., Salinas V., Perez-Maturo J., Amartino H., Vásquez-Dusefante C., Medina N., González-Morón D., Kauffman M.A. Whole exome sequencing in neurogenetic odysseys: An effective, cost- and time-saving diagnostic approach. PLoS One. 2018;13(2):e0191228. doi: 10.1371/journal.pone.0191228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bainbridge M.N., Wiszniewski W., Murdock D.R., Friedman J., Gonzaga-Jauregui C., Newsham I., Reid J.G., Fink J.K., Morgan M.B., Gingras M.C., Muzny D.M., Hoang L.D., Yousaf S., Lupski J.R., Gibbs R.A. Whole-genome sequencing for optimized patient management. Sci. Transl. Med. 2011;3(87):87re3. doi: 10.1126/scitranslmed.3002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Narasimhan V.M., Hunt K.A., Mason D., Baker C.L., Karczewski K.J., Barnes M.R., Barnett A.H., Bates C., Bellary S., Bockett N.A., Giorda K., Griffiths C.J., Hemingway H., Jia Z., Kelly M.A., Khawaja H.A., Lek M., McCarthy S., McEachan R., O’Donnell-Luria A., Paigen K., Parisinos C.A., Sheridan E., Southgate L., Tee L., Thomas M., Xue Y., Schnall-Levin M., Petkov P.M., Tyler-Smith C., Maher E.R., Trembath R.C., MacArthur D.G., Wright J., Durbin R., van Heel D.A. Health and population effects of rare gene knockouts in adult humans with related parents. Science. 2016;352(6284):474–477. doi: 10.1126/science.aac8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jang, K.; Tong, T.; Lee, J.; Park, T.; Lee, H. Altered gene expression profiles in peripheral blood mononuclear cells in obese subjects. Obes. Facts. 2020;13(3)(375):385. doi: 10.1159/000507817. [DOI] [PMC free article] [PubMed] [Google Scholar]