

Abstract
Genome sequences indicate a wide variety of characteristics, which include species and sub-species type, genotype, diseases, growth indicators, yield quality, etc. To analyze and study the characteristics of the genome sequences across different species, various deep learning models have been proposed by researchers, such as Convolutional Neural Networks (CNNs), Deep Belief Networks (DBNs), Multilayer Perceptrons (MLPs), etc., which vary in terms of evaluation performance, area of application and species that are processed. Due to a wide differentiation between the algorithmic implementations, it becomes difficult for research programmers to select the best possible genome processing model for their application. In order to facilitate this selection, the paper reviews a wide variety of such models and compares their performance in terms of accuracy, area of application, computational complexity, processing delay, precision and recall. Thus, in the present review, various deep learning and machine learning models have been presented that possess different accuracies for different applications. For multiple genomic data, Repeated Incremental Pruning to Produce Error Reduction with Support Vector Machine (Ripper SVM) outputs 99.7% of accuracy, and for cancer genomic data, it exhibits 99.27% of accuracy using the CNN Bayesian method. Whereas for Covid genome analysis, Bidirectional Long Short-Term Memory with CNN (BiLSTM CNN) exhibits the highest accuracy of 99.95%. A similar analysis of precision and recall of different models has been reviewed. Finally, this paper concludes with some interesting observations related to the genomic processing models and recommends applications for their efficient use.
Keywords: Machine learning, genome processing, classification, computational complexity, deep learning, precision and recall
1. INTRODUCTION
Extraction of species-specific information from genomic data is a multidomain task, which involves various signal processing, deep learning, post-processing, feedback-based learning, and performance tuning operations. In order to perform this task, a large amount of data is required to be collected for the given species, and this data must be tagged with species-specific information [1]. This tagged information is decided by the application scenario, and thus requires expert intervention for accurate analysis. A sample architecture that performs this task can be observed in Fig. (1), wherein macro steps for genomic sequence processing are showcased.
Fig. (1).
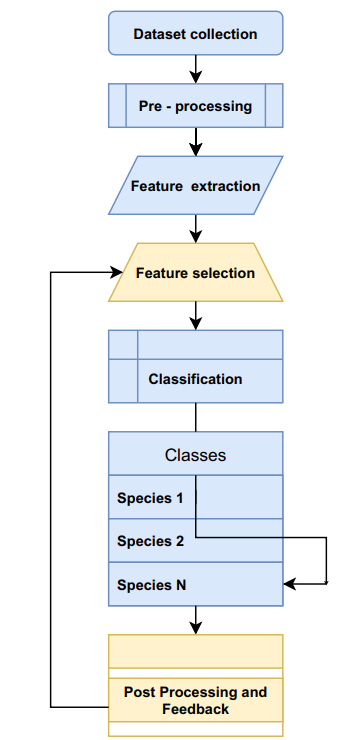
General purpose architecture for genomic data processing.
Based on this architecture, it can be observed that a wide variety of data must be collected in order to train the processing model. The collected data is input into a pre-processing model, wherein operations like denoising, missing value estimation, filtering, etc., are performed. The pre-processed genome sequences are given to a feature extraction block, wherein unigram, bigram, trigram, and other pattern features are extracted. These features must follow two main rules, such as (i) feature variance for sequences belonging to the same category must be as low as possible, and (ii) feature variance for sequences belonging to different categories must be as high as possible.
To facilitate application of these rules, the feature selection engine is used. This engine utilizes algorithms, like principal component analysis (PCA) and latent dirichlet analysis (LDA), on extracted features in order to widen the feature variance gap between different class sequences [2]. These algorithms aim to solve this problem by projecting the data from higher to lower dimensions. Results of the selection engine are given to a classifier, wherein machine learning algorithms, like convolutional neural network (CNN), recurrent neural network (RNN), artificial neural network (AiNN), etc., are used [3]. Machine learning methods are most effective when they optimize an appropriate performance measure. They also focus on algorithmically constructed models with optimal prediction as their goal rather than parametric data modeling [4]. These algorithms assist in the stratification of genome sequences into 1 of N genes [5]. Results of this engine are given to a post-processing block, wherein error estimation is performed, and based on this error performance of classifier, the feature selection engine is tuned. This engine also assists in the temporal analysis of genomic data for the prediction of future diseases (or events) based on historical classification [6]. The selection of an optimal subset of features improves the learning efficiency and increases the predictive performance [7]. In order to perform these tasks, a wide variety of system models have been proposed by researchers in the last decade. A survey of these algorithms, along with their nuances, advantages, limitations and characteristics, can be observed in the next section. This is followed by a performance evaluation of the reviewed models, which assists in the identification of best models for a given genome processing application. Finally, this review concludes with a comparative analysis of various machine learning and deep learning models and recommends various ways to improve their performance.
2. GENOME SEQUENCE PROCESSING MODELS
With the massive generation of data, the era known as ‘big’ data, deep learning (DL) approaches have appeared as a discipline of machine learning (ML) that are considered to be more efficient and effective when we deal with a big amount of data. The DL methodologies have helped provide high computation power to resolve complex research hypotheses in genomics [8]. Genomic data sequences are capable of representing a wide variety of information about the underlying species. For instance, in human beings, the genomic sequences assist in the presence of cancer, diabetes, heart and stroke issues, and other diseases. While, in plants, these sequences can also be used to analyze the types of species, the disease severity, yield quality, etc. [9].
2.1. Processing Models for Crops’ Genome Sequences
Deep learning techniques are useful in genome sequence analysis and classification. Deep Neural Network (DNN) has a vanishing gradient problem; as the number of layers increases, the number of connections increases persistently. The novel Spinal net model is the advancement of the DNN model for high-accuracy prediction of 6mA sites in rice genomes [5]. Abbas et al. proposed a novel model that uses the SpineNet-6mA network, which is capable of predicting DNA N6-methyladenine sites in genomes. The model is tested on rice genomes and is able to achieve an accuracy of 94.31%, precision of 92.92%, and recall of 95.71% on multiple datasets [5]. It uses a combination of batch normalization, along with max pooling and one-hot encoding in order to classify between 6m and non-6m genomes, as observed in Fig. (2), wherein entire internal architecture for this model is described. From the SpineNet-6mA network model, it is observed that each Spinal Net neuron comprises normal neurons, the outputs of which are combined in order to obtain the final activation. Due to this, the total number of processing elements is doubled, thereby improving the computational power of the network. This increase in computational power also increases accuracy, precision and recall of classification [10]. This novel architecture of the SpineNet-6mA model is able to receive large data that thereby achieve better efficiency. Hence, SpineNet-6mA model can have the best performance on rice genome processing due to its modified internal architecture when compared to the models iDNA6m, SNN Rice6m, MM 6mA, i6m, and DNA6m MINT, which showcase an accuracy of 91.7%, 92.04%, 83.6%, 90.9%, and 90.11%, a precision of 90.5%, 89.75%, 83.63%, 86.64%, and 93.24%, and recall of 93%, 94.33%, 89.32%, 86.7%, and 92.16%, respectively [5].
Fig (2).
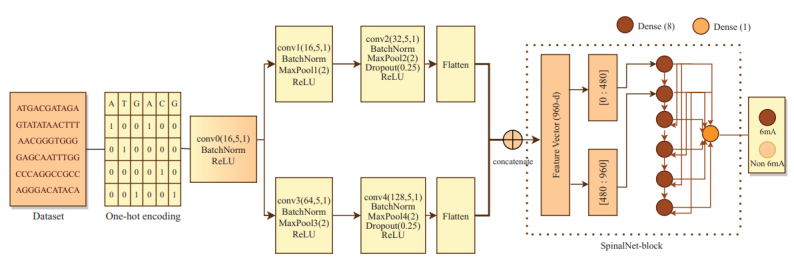
Design of the spinal net model for genome classification [5].
Various classification models have been proposed by researchers, which assist in deploying context-sensitive genome stratification engines [11]. Besides, emerging high-performance bioinformatics tools specific for plant research are explained by Martinez et al. [2016], where the authors have studied the genomic sequence of numerous plant species, including the main crop species. Here, the comparative analysis is focused on the common and specific computational tools developed to achieve the particular objectives of each database [10]. For instance, Yu et al. proposed the design of a maize micro phenotype classification model, wherein forward and reverse genomic prediction for shoot apical meristem (SAM) data has been described. The proposed model is capable of achieving an accuracy of 62% for seven different corn classes, including sweet corn, landraces, tropical, popcorn, stiff stalk, non-stiff stack, and unknown [9]. When evaluated on seven different classes, the model has been observed to have lower accuracy; thus, incorporation of deep learning and transfer learning must be done here for improving it. An example of a high-efficiency model for maternal haploid induction rate prediction in maize crops has been described by Almeida et al.; the genomic estimated breeding value (GEBV) was used for training and validation. This model used haploid induction rate, days to anthesis, the height of plant, the height of ear, the size of tassel, and self-induction rate in order to achieve an accuracy of 83% across different datasets, which can be improved via the use of multiple deep learning models [12]. Deep learning models have been widely used for crop classification, where the study of summer crops has been done using two types of models, i.e., Long Short-term Memory (LSTM), and the other based on one-dimensional convolutional (Conv1D) layers [13]. An increase in the delay in models’ selective phenotyping has been discussed by Michel et al., wherein pedigree and genomic (PG) information was combined with pre-existing phenotypic information to remove non-variant training samples. This information is represented using equation 1, where different phenotypes are combined to form the final quality measure.
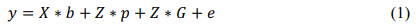
Where, b,p,e,x,y,G and Z represent various phenotypes and their respective genomic effects [14]. A higher value of output phenotype (y) indicates better selection quality of combination, thereby indicating better algorithmic performance. Due to the use of such strength evaluation attributes, the proposed PG model is capable of obtaining an accuracy of 59% on multiple datasets. An improved model for application-specific genomic data classification has been explained by Dai et al., wherein the non-homology analysis of gene functions for maize was conducted. The authors have discussed six different methods, and compared their accuracy, precision, recall and area under the curve (AUC) performance. Random Forest (RF), Gradient Boosting Machine (GBM), Partial Least Squares (PLS) and Lasso and Elastic-Net Regularized Generalized Linear Models (GLMNET) exhibited an accuracy of 97%, 96%, 95%, and 91%, respectively [15]. RF had the highest accuracy, but precision, recall and AUC performance were higher for GLMNET, which indicated that RF must be fused with GLMNET to improve overall genome classification performance.
Deep learning models have better classification performance when compared to linear models. This can also be observed from work done by Grinberg et al., where elastic net (EN), lasso regression (LaR), ridge regression (RiR), GBM, RF, support vector machines (SVM), two-step sequential method based on linear regression (TSSLR), and Genomic best linear unbiased prediction (GBLUP) have been used for yeast, wheat and rice genome classification [16]. Onda et al. observed that when cross-validation frameworks are added to these models, their accuracy is exponentially improved, because these models assist in variant feature selection, thereby improving their classification performance [17]. An accuracy of 78% was observed for EN, 77.9% for LaR, 56.3% for RiR, 60.9% for GBM, 63.6% for RF, 71.2% for SVM, 78.6% for TSSLR, and 71.1% for GBLUP, which is higher than Bloom filter and other linear models. Thus, these models must be used for clinical applications that classify the genomic data accurately. Another interesting approach that uses GBLUP modelling techniques for analysis of sugarcane clonal performance via analysis of non-additive genetic effects has been discussed by the authors Yadav et al. and Virnodkar et al.; this model uses different genomic traits for analysis, which allows them to measure cane per hectare, fibre content and commercial cane sugar properties with 65.9% accuracy, thereby suggesting the use of deep learning models like CNN, or LSTM based RNN for better performance [13, 18, 19]. This performance can be further tuned via the use of calibration and validation steps as suggested by Auinger et al., where they have analysed advanced cycle maize plants and predicted their genomic breeding values. The model uses the GBLUP method and employs population-specific calibration using the analysis of molecular variance (AMOVA) method, due to which, the proposed model is capable of achieving an accuracy of 71%, thereby making the model applicable for coarse-grained analysis [20].
Various species-specific models have also been proposed, where Lubanga et al. have analysed quality traits in the tea genome via genomic and pedigree-based prediction. They have compared various prediction models and concluded that Bayesian ridge regression (BRR) is superior to BayesA, BayesB, BayesC, and GBLUP, reproducing kernel Hilbert spaces (RKHS) models that use pedigree relationships, namely RKHS-pedigree (RKHSP), RKHS markers (RKHSM), and RKHS markers and pedigree (RKHSMP). Tea traits, including theogallin, theobromine and epicatechin gallate, were predicted with 73% accuracy via BRR, 72% accuracy via BayesA, 70% accuracy via GBLUP, and 68% accuracy via RKHSMP models [21]. Also, a deep learning model has been discussed by Knoch et al. (2021) for canola, where they have proposed the use of multi-omics-based predictive model (MOBPM). The MOBPM method replaces genetic markers with transcriptomic information, and uses reproducing Hilbert regression, which is based on Gaussian kernels [22]. This combination is capable of achieving better hybrid prediction accuracies for complex genomic canola traits. Researchers have been able to find different canola stages, including seed emergence, seed yield, oil yield, protein content, days to onset for flowering, oil content and seed glycosylates with 75% accuracy, thereby making it useful for coarse-grained analysis. This accuracy can be improved via the use of deep learning models designed by Montesinos et al., wherein models like RNNs, CNNs, multilayer perceptron (MLP), deep belief networks (DBNs), and their combinations for better classification performance have been compared. MLP has been observed to have an accuracy of 91%, CNNs an accuracy of 93%, RNNs an accuracy of 92%, while DBNs have been observed to have an accuracy of 94% on different genomic datasets, thereby making them useful for a wide variety of clinical applications [23]. An application of similar models for groundnut trait identification was carried out, where Bayesian Generalized Linear Regression (BGLR) with cross-validation schemes was used. The proposed model by Pandey et al. was capable of achieving an accuracy of 65% on different genomic sequences, thereby making it useful for analysis of flowering duration, maturity duration, seed weight analysis, oleic acid estimation, late leaf spot estimation, etc. [24].
The deep learning models are useful for a wide variety of applications, including cross-genomic prediction; one such work was done by Mellers et al., wherein oat breeding costs reduced by 15% due to genomic analysis. The model proposed uses BLUP and differentially penalized regression (DiPR) for analysis, which results in an accuracy of 75% across different data inputs [25]. The work by Basnet et al. explained such a model for hybrid wheat prediction using BLUP, general combining ability (GCA), specific combining ability (SCA), along with the gender of the species. Due to such a combination, the model has been observed to achieve an accuracy of 91.1% for different wheat yield classes [26]. This makes it useful for on-field analysis of days to flowering, days to heading, and days to maturity.
For all crop genome processing, a novel heuristic feature selection-based model can be designed, where context-specific feature selection model can be combined as a dual model using an SVM classifier and extra trees. The genome sequences will be initially transformed in the form of N-gram features. These feature sets will be chosen using a dual model that maximizes variance levels using a genetic algorithm. The parameters considered would be the optimization iterations, optimization solutions, features extracted by the N-gram process, and the learning rate of the model. With this, we could get highly accurate training and testing datasets. Furthermore, the ensembled deep learning models can be used for classification.
2.2. Processing Models for Disease Classification and Identification in Human Beings
In order to increase the scalability of the learning models, testing can be done on different genomic datasets as described by Sun et al., wherein The Cancer Genome Atlas (TCGA) cancer survival datasets and age-related eye disease studies (AREDS and AREDS2) datasets are described. It is observed that these datasets cover a wide variety of genomic data, and thus can be used for better evaluation of genome classification models [6]. The use of various deep learning models has been explained by Ramasamy et al., wherein an Adaptive Skipping Training model named Half of Threshold (HoT) was described. The model has been tested on various genomic datasets, including Hepatitis, Heart, SPeCT, Liver Disorders, Drug Consumption, Breast Cancer Wisconsin (Diagnostic), Cardiotocography, Thyroid Disease and Splice-junction Gene Sequences, thereby indicating its vast scalability [27].
The HoT model is used for parametric feedback and iterative training, which improves the overall accuracy of genomic classification. The model is showcased in Fig. (3), wherein results from the neural network are compared with target results, and based on this comparison, training weights are manipulated and non-variant inputs are skipped for improved classification efficiency. Due to the use of iterative learning, the proposed adaptive skipping HoT (SHoT) model showcases an accuracy of 92.6%, which is higher than the normal HoT model that achieves an accuracy of 85.3%, and back-propagation neural network (BPNN), which achieves an accuracy of 78.6%, on the same dataset [27].
Fig (3).
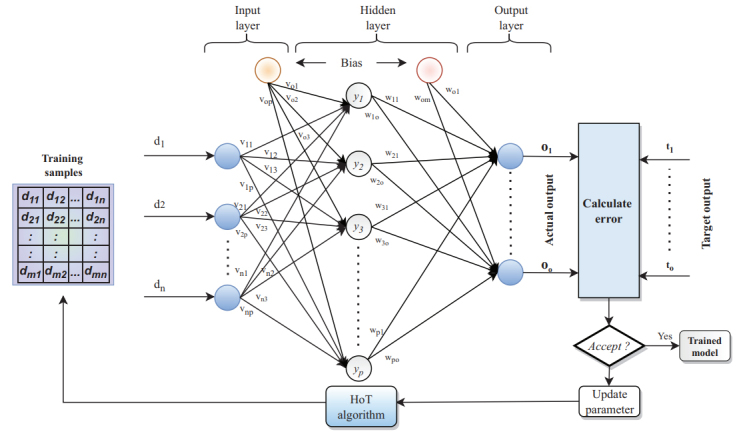
The Half of Threshold (HoT) algorithm for gene classification [27].
Due to adaptive skipping, the algorithm is showcased to have a faster response when compared to HoT and BPNN models, thereby improving its scalability and real-time clinical usage. The one-hot encoding is a sample strategy that uses n-bit state registers to encode n states. Each state has its own register bit, and only one register is valid at any time [28]. Zhang et al. worked on a CNN model named DeepDRBP-2L, which uses LSTM for the identification of DNA and RNA binding proteins. In this model of DeepDRBP-2L, initially, a pool of convolutional layers is used for effective feature extraction, followed by multiple pooling layers and convolutional pooling layers for effective feature selection. The selected features are given to a bidirectional LSTM model for feature activation, and the activated features are classified using a flatten neural network layer for improved classification accuracy [29]. The proposed DeepDRBP model has been observed to be capable of achieving an accuracy of 91%, a precision of 80.68%, and a recall of 81.14%, which is better than DNA binder having an accuracy of 89.5%, a precision of 62.45%, and recall of 89.1%, and Stack DP prediction model having an accuracy of 86.5%, a precision of 55.63%, and recall of 89.1% on the same datasets [29].
The convolutional layers can be used for the identification of disease-specific patterns in genomic data, which can be used for future analysis and disease prediction. Yu et al. proposed a model wherein essential methylation patterns and their related genes were identified using maximum relevance feature selection (MRFS), increment feature selection (IFS) and SVM-based classification. Internal architecture for MRFS and IFS utilizes feature convolutions in order to identify genes with the largest relevance, genes with robust and consensus ranks, and genes with optimal combination; this work was able to achieve an accuracy of 89.5% for stroke-related genome prediction [30]. A similar work has been done by Singh et al. and Xu et al., wherein DNNs and AiNN were used for the prediction of enhancer-promoter interaction and essential genes in prokaryotes with high efficiency. The DNN model is capable of achieving an accuracy of 97%, a precision of 91%, and recall of 90% [31], while AiNN showcases an accuracy of 83%, a precision of 80%, and recall of 79% for a wide variety of datasets [32]. The performance of these models is high enough for clinical usage, but they require large computational delays, which limits their deployment capabilities. Improved models have beenproposed by Liu et al. and Davi et al., wherein machine learning models have been used for the prediction of sigma-54 promoters and severe dengue promoters in human DNA and RNA sequences. The proposed models are capable of identifying the mentioned sequences via intelligent feature selection, which allows them to achieve high accuracy with minimum error rates. It is observed that RF [33] has an accuracy of 91.6%, while SVM [34] has an accuracy of 86%, which makes it useful for a wide variety of applications.
Many machine learning algorithms have been used for classification of Alzheimer's disease; these algorithms have their own application and challenges [35]. The proposed model for Alzheimer’s disease prediction used Sparse Regression Model (SRM) with Joint Projection Learning (JPL) on the standard Alzheimer’s Disease Neuroimaging Initiative (ADNI) dataset with over 91% accuracy [36]. Similarly, the model suggested by Sergeev et al. was capable of identifying tuberculosis using two-step cross-validation via a combination of LR, RF, GBM, Single Marker Test, and Elastic Net classifier. An accuracy of 84% was achieved by using the proposed model with an aggregated precision of 92.9% and recall of 75.2% [37]. The deep learning models have beenmentioned by Khorshed et al., wherein multiple tissue cancer prediction has been performed. These models used a specialized CNN architecture, namely GeneXNet, which is capable of achieving an accuracy of 98.9% on different cancer tumour types. The GeneXNet model uses a combination of CNN and transfer learning for analysis of data from multiple domains [38]. A series of GeneX blocks that consist of a deep learning block followed by a residual learning block were combined in order to form the GeneXNetwork, which is capable of highly accurate cancer classification for adrenal gland, bile duct, bladder, bone marrow, brain, cervix, colorectal, eyes, kidney, liver, lung, lymph nodes, and other body sites [38]. The model outperforms ResNet, which has an accuracy of 96.5%, DenseNet with an accuracy of 95.3%, NasNet with an accuracy of 93.5%, and MobileNet having an accuracy of 94.2% on the same datasets [38]. This model is currently capable of cancer detection but can be further used for detecting multiple types of genomic classes via the application of gene consensus modelling, wherein genes are selected depending on their applicability to the given context. An example of such a consensus model was provided by Wu et al., where PLS-based gene microarray analysis was performed for the Large B Cell Lymphoma dataset [39]. The model uses a combination of singular value decomposition (SVD) and thresholding for linear consensus classification. Genes selected after consensus were used for multiple disease type classification via a combination of ridge PLS, which resulted in an accuracy of 93.4%, being higher than SVM having an accuracy of 91.5% and RF-SVM, which has an accuracy of 91.9%, on the same cancer dataset [39].
This accuracy can be improved via the use of multivariate gene interaction analysis described by Knight et al., wherein they applied optimal Bayesian classification [40]. This model uses a combination of Poisson and Bayesian (P and B) analysis in order to achieve an accuracy of 91.5% on different gene types [40]. Similar models have been discussed where inverse projection representation (IP), comprehensive pathway activity analysis (CPAA) and hybrid heuristic dimensionality reduction (HHDR) have been used [41]. The IP model is capable of effective tumour classification and uses two-stage hybrid gene selections to achieve an accuracy of 93%, which is higher than SVM having an accuracy of 85%, and sparse representation-based classification (SRC) having an accuracy of 89% on the same datasets [41]. While the CPAA model is used for cancer classification via inferring gene interactions with an accuracy of 83% [42], the HHDR model is used for the classification of malaria via genetic algorithm (GA) and a combination of PCA, independent component analysis (ICA) and SVM to achieve an accuracy of 91.7%, which is higher than GA with PCA that has an accuracy of 85% and GA with ICA that has an accuracy of 90.3% [43].
The reviewed models utilize deep learning and perform feature selection via supervised learning. This requires a large amount of training data, thereby limiting their work to big data applications. The neural network is trained with gene expression profiles of genes that are predictive of recurrence in liver cancer; the ANNs have become capable of correctly classifying all samples and distinguishing the genes most suitable for the organization [44]. In order to reduce data requirement, Ye et al. proposed the use of an adaptive unsupervised feature learning (AUFL) model that is capable of gene signature identification for lung cancer. TCGA was used for its evaluation, and an accuracy of 92.2% was achieved when AUFL was combined with kNN, 92.1% with DT, 91.3% with SVM, and 91% with LDA, which makes it useful for a wide variety of applications [45]. Similar models have been discussed, wherein copy number variation (CNV) detection, tumour classification using AI, and cell subtype classification using denoising autoencoder (DAE) have been performed on multiple gene datasets [46]. The CNV approach uses Bayesian inference models for obtaining an accuracy of 99.27% [47], while AI classifies kidney renal clear cell carcinoma (KIRC), lung adenocarcinoma (LUAD) [48], lung squamous cell carcinoma (LUSC), and uterine corpus endometrial carcinoma (UCEC) with 96.9% accuracy, which makes it useful for clinical applications [49]. The artificial intelligent model uses a combination of binary particle swarm optimization and decision tree (PSODT) with CNN, which assists in optimum feature selection, thereby reducing computational delay [47]. The DAE model uses a combination of DNN with autoencoder that works by reducing the reconstruction error.
Due to the complex structure, the model is able to extract the most relevant features from the input set, thereby resulting in an accuracy of 98.59%, which is higher than SVM, RF and Active NN, which has an accuracy of 98.45%, 92.06% and 97.66%, respectively, on the same dataset. Algorithms, like robust trace norm multitask learning (TNML) [50], use logistic regression for efficient feature selection and are able to achieve an accuracy of 79.3% for cancer detection. PLS with novel TTZ feature vector [51] is applied to lung disease classification and is able to achieve an accuracy of 92.65%, while the LASSO SVM model is applied to cancer datasets and is able to achieve an accuracy of 91.3%, thereby making them useful for various applications. Similar models have been explained, where the authors have discussed the use of repeated incremental pruning to produce error reduction (RIPPER) [52], CNNs, RNNs [53], and RIPPER with SVM for multiple applications [54]. The RIPPER model is capable of achieving an accuracy of 80.8%, CNNs an accuracy of 96%, RNNs an accuracy of 96.2%, and SVM with RIPPER to achieve an accuracy of 99.7%, thereby improving their utility for real-time clinical and on-field deployments. Based on this review, deep learning and iterative learning models have been found to be most efficient for human gene sequence classification; also, these methods are able to take advantage of high dimensional input, which is an important asset for population genetics inference and often more robust than other statistical approaches [4].
2.3. Processing Models for Viral Genome Classification
Accurate genomic sequence classification and typing could help enhance the phylogenetics and functional studies of viruses [7]. The work done by Dasari et al. compared various CNN architectures, and concluded that LSTM models along with EdeepVPP models outperform other models in terms of accuracy of genomic classification. EdeepVPP model has been observed to be capable of achieving an accuracy of 99.2% on various CoVID-19 datasets, which makes it highly useful for viral genome prediction [55]. The EdeepVPP model utilizes one-hot encoding along with motif analysis for pattern evaluation, thereby assisting in formation of feature maps, as indicated in Fig. (4), wherein viral sequences along with their positions are shown. The CNN model utilizes five different-sized layer sets, each consisting of dropout, max pooling, 1D convolution, and activation layers, which assist in high-density feature extraction.
Fig. (4).

Deep learning model for genomic feature map generation [55].
The proposed model was compared on 2011 G5, 2011 N19, 2015 F and other genomic datasets; it was observed to outperform other models, including Vira Sorter having an accuracy of 74.2%, Vira Pipe having an accuracy of 79%, Vira Finder having an accuracy of 89.3%, Vira Miner having an accuracy of 92.3%, RNN Vira Seeker having an accuracy of 91.8%, and Deep Vira Finder with an accuracy of 93% on the same datasets [55]. Similar fused models have been proposed by Liu et al. and Ibba et al., wherein Virus Finding and Mining (VFM), Bayesian multi-trait multi-environment (BMTME), and multi-trait ridge regression (MTR) have been described. These models utilize a combination of different clustering and classification techniques in order to estimate genomic activity, thereby assisting in improved classification performance [56]. The VFM model has an accuracy of 97%, while the BMTME model has an accuracy of 90%, and the MTR model has an accuracy of 91% on different genomic datasets [57].
The Artificial Intelligence system learns to execute the interpretation task on new health data of the same type, which in clinical diagnostics is often the identification or forecasting of a disease state [58]. Various deep learning and artificial intelligence techniques have been discussed, wherein sequential pattern mining (SPM), along with co-occurrence matrix (CMSPAM), and All-K-Order-Markov (AKOM), is used for highly efficient pattern modelling. The results indicate that AKOM outperforms CMSPAM and other models in terms of accuracy and delay needed for evaluation. This is due to the fact that AKOM works on effective feature selection via inter-class variance maximization, which improves its real-time performance. The accuracy of AKOM was observed to be 91.2%, which is higher than CMSPAM having an accuracy of 89.5%, Compact Prediction Tree (CPT) having an accuracy of 86.5%, CPT+ having an accuracy of 90.2%, Dependency Graph (DG) having an accuracy of 80.5%, Transition Directed Acyclic Graph (TDAG) having an accuracy of 86.5%, and LZ78 having an accuracy of 85.4% on different datasets [59]. These models showcase moderate precision and recall performance due to inconsistency in output rule generation. This precision and recall performance can be further improved as Poran et al. suggested the models that use effective feature extraction and multiple feature selection, wherein mass spectrometry-based profiling of users is performed. These models are capable of performing this task with 94% accuracy, which is higher than the random selection accuracy of 89%, due to which the model is capable of real-time clinical analysis [60]. Similar to this work, the model that was studied by Xie et al. analysed the effects of Middle East respiratory syndrome-coronavirus (MERS CoV) via the artificial neural network-based linear B-Cell prediction (ABCPred) classification method. The ABCPred model is capable of achieving an accuracy of 84.5% on different epitopes [61], thereby making it useful for coarse-grained analysis of MERS-CoV. It was found that the machine learning classification method can be implemented to diagnose COVID-19 as an assistant system [62]. The BiLSTM CNN [63] was able to achieve an accuracy of 99.95% for COVID genome sequence classification.
2.4. Models for Classification of Various Gene Expressions and RNA Sequences of Multiple Genome Data
Ribonucleic acid (RNA) modifications are post-transcriptional chemical composition changes that have a fundamental role in regulating the main aspect of RNA function [64]. Barbeira et al. discussed various deep learning models, including EN, cross‐tissue gene expression imputation (CTGI), deterministic approximation of posteriors (DAP), and multivariate adaptive shrinkage (MASH) for genomic data classification [1]. These models are capable of classification of human genomes, plant genomes, and animal genomes with high accuracy. The EN model showcased a precision and recall of 80% and 85% with moderate computation complexity, while the CTGI model showcased a precision and recall of 83% and 86% with low complexity. Similarly, the DAP model showcased a precision and recall of 86% and 89% with high complexity, while the MASH model showcased a precision and recall of 75% and 79% with a moderate level of complexity [1]. All these models were applied to different kinds of genomic data and showcased good performance. Although these models exhibited good accuracy, they involved large delays for training and evaluation. These delays can be reduced using feature selection, as proposed by Seo et al., wherein GA was used to identify most variant features. The modelled GA utilizes a ratio of statistically identical k-words (SIWRs) in order to evaluate the fitness function of each genomic sequence, where SIWR is evaluated as follows in equation (2).

Where, S1 and S2 represent sequence occurrence counts for each of the feature vectors. The proposed SIWR GA model is able to identify plant and human genomes with 96% accuracy, 95% precision, and 94.5% recall rates, which is higher than the frequent pattern (FP) tree that has an accuracy of 93%, precision of 91%, and recall of 90%. Further, it showcases better efficiency when compared to FP SVM and spaced SVM, which exhibit an accuracy of 90% and 85%, a precision of 89% and 83%, and recall of 80% and 79%, respectively. All this performance is achieved while having low delay, thereby improving the deployment capability of the network for high-speed clinical applications. Animal-specific models are also suggested for highly efficient genomic prediction, wherein Australian sheep whole genome sequence data were processed using GBLUP and Bayesian classification [65]. The model is able to categorize between crossbred Border Leicester x Merino and purebred Merino sheep with an accuracy of 61.1%, which can be improved via reinforcement and deep learning models, as discussed by Zrimec et al., wherein an accuracy of 83% was achieved across 20k RNA datasets of seven different organisms. This accuracy is very high considering the fact that the used CNN model is evaluated for multiple organism types [66]. The development of classification models is highly recommended for the diagnosis and classification of diseases and disease monitoring at the molecular level [67]. Zhou and Ji discussed another example, wherein chromatin accessibility was evaluated using genomic data via the big data improved reliability (BIRD) method. The suggested model used genomic data along with chromatin accessibility and temporal genomic information in order to predict chromatic accessibility with an accuracy of 85%, thereby improving its deployment capabilities [68].
Machine learning models are also trained using 2D and 3D data hyperspectral imaging, where early prediction of biomass is performed in hybrid rye using GBLUP [69]. The GBLUP model has been observed to have moderate accuracy, but it is highly precise and has high recall values with low delay for multiple datasets. Patra et al. explained a novel Regulatory Enrichment Pathway Analysis (REPA) approach, which assists in the application of gene set analysis to genome-wide transcription factor binding data. The model is highly scalable and can be used for the analysis of ribosome, alcoholism, cancer pathways, bacterial invasion of epithelial cells, etc., with 83% accuracy, thereby making it useful for a wide variety of applications [70]. An interesting piece of research was done by Waldvogel et al., where evolutionary computational models were used for the estimation of species responses to global climate change. The identification of keystone species has been observed to be of utmost importance while performing this analysis [71]. These species can be identified using genomic data, phenotypic data, and ecological data, which assists in the classification of allele frequency changes, reaction norms, range shifts, etc., with high accuracy. Interactions between different genotypes and environmental elements also assist in the estimation of different properties in biological species [71].
The algorithms, like Least Absolute Shrinkage and Selection Operator (LASSO) with SVM [72] and Positive Matrix Factorization Method (PMFM) [73], are given for efficient gene sequence classification. The PMFM [73] model and nonoverlapping sequence pattern mining (NOSEP) model [74] work on effective feature selection and can be used on multiple types of datasets. The PMFM model is able to achieve an accuracy of 90.03%, while NOSEP model achieves an accuracy of 85.6% on different genomic datasets. The delay of these models is high, which can be reduced via the use of parallel processing as suggested by Khan et al., wherein large-scale RNAs are classified into piRNAs and non-piRNAs with an accuracy of 81.7%, which makes them useful for theoretical analysis [11, 75]. As explained by Wang et al., gene-gene (GG) interactions are used for clustering, classification and construction of inference networks, which can be used for single-cell RNA sequences [76]. The model was evaluated using RF, k-nearest neighbour (kNN), ANN, SVM with linear kernel, SVM with radial basis kernel function (RBF), and deep neural networks (DNNs). GG with RF has been observed to have an accuracy of 79.54%, kNN an accuracy of 74.53%, ANN an accuracy of 78.14%, LIN SVM an accuracy of 78.06%, and SVM RBF with an accuracy of 77.18%, while DNNs were reported to have an accuracy of 78.95% on BioCarta and Kyoto Encyclopaedia of Genes and Genomes (KEGG) datasets [76]. A statistical survey of these models, along with their suggested applications, is discussed in the next section, which will assist researchers in selecting the most optimum models for their cases.
3. EMPIRICAL MODEL ANALYSIS
From the previous section, it can be observed that deep learning models, like CNN, RNN, BiLSTM, DAE, etc., are capable of performing high-accuracy gene sequence classification. However, these models are applied to specific applications and specific datasets, which encapsulate their capabilities with respect to other fields of genomic pattern analysis. Thus, it is difficult to identify the best performing models for multiple genomic applications. By referring to this section, researchers would be able to identify most optimum models suited for their application, and use them to improve accuracy, precision, recall, and computational complexity of their deployments.
3.1. Accuracy for Specific Applications
In order to perform this, parametric values for accuracy (A) and recommended application (RA) are tabulated in Table 1. Due to such a wide variety of available applications, this accuracy comparison is divided into two different parts, such as for human diseases and crops. While the values of accuracy were evaluated for different disease types in their respective researches, for the purpose of comparison, we have evaluated the average values, which will assist in quantifying this comparison even for different disease and species types. For the purpose of this review, we did not augment any value, but used it directly from the reference texts, which assisted in maintaining its credibility for comparison purposes.
Table 1.
Accuracy comparison of genomic classification models along with their area of application.
| Sr. No | Recommended Application | Method | Accuracy (%) | References |
|---|---|---|---|---|
| 1. | Multiple genomes | EN | 86 | [1, 23, 31, 40, 53, 54, 57, 59, 60, 68] |
| DAP | 90 | |||
| BMTME | 90 | |||
| MTR | 91 | |||
| DNN | 97 | |||
| AiNN | 83 | |||
| AKOM | 91.2 | |||
| BIRD | 85 | |||
| P&B | 91.5 | |||
| CNN | 96 | |||
| RNN | 96.2 | |||
| RIPPER SVM | 99.7 | |||
| CMSPAM | 89.5 | |||
| CPT | 86.5 | |||
| CPT+ | 90.2 | |||
| DG | 80.5 | |||
| MLP | 91 | |||
| TDAG | 86.5 | |||
| Spectrometry | 94 | |||
| Random Selection | 89 | |||
| 2. | Plant and human genomes | GA SIWR | 96 | [2] |
| FP Tree | 93 | |||
| FP SVM | 90 | |||
| 3. | Rice | SpineNet-6m | 94.3 | [5] |
| iDNA6m | 91.7 | |||
| SNN Rice6m | 92.04 | |||
| i6m | 90.9 | |||
| DNA6m MINT | 90.11 | |||
| 4. | Cardiac | SHoT | 92.6 | [27] |
| HoT | 85.3 | |||
| BPNN | 78.6 | |||
| 5. | Covid | EdeepVPP | 99.2 | [55, 63] |
| Vira Sorter | 74.2 | |||
| Vira Pipe | 79 | |||
| Vira Finder | 89.3 | |||
| Vira Miner | 92.3 | |||
| Vira Seeker | 91.8 | |||
| Deep Vira Finder | 93 | |||
| BiLSTM CNN | 99.95 | |||
| 6. | Sheep species | GBLP | 61.1 | [65] |
| 7. | Maize | RF | 97 | [12, 15, 20] |
| GBM | 96 | |||
| PLS | 95 | |||
| GLMNET | 91 | |||
| LDA, PMLR, SVM | 89 | |||
| AMOVA | 71 | |||
| GEBV | 83 | |||
| 8. | Human disease | VFM | 97 | [16, 29, 33, 34, 56] |
| DeepDRBP-2L | 91 | |||
| RF | 91.6 | |||
| DNA Binder | 89.5 | |||
| Stack DP | 86.5 | |||
| EN | 78 | |||
| LaR | 77.9 | |||
| SVM | 86 | |||
| TSSLR | 78.6 | |||
| 9. | Biomass | GBLUP | 75 | [69] |
| 10. | Sugarcane | GBLUP | 65.9 | [18] |
| 11. | Tea | BRR | 73 | [21] |
| BayesA | 72 | |||
| GBLUP | 70 | |||
| 12. | Canola | MOBPM | 75 | [22] |
| 13. | Groundnut | BGLR | 65 | [24] |
| 14. | MERS-CoV | ABCPred | 84.5 | [61] |
| 15. | Stroke prediction | MRFS, IFS with SVM | 89.5 | [30] |
| 16. | Alzheimer’s disease | SRM with JPL | 91 | [36] |
| 17. | Tuberculosis | LR, RF & GB | 84 | [37] |
| 18. | Wheat | GCA & SCA | 91.1 | [26] |
| 19. | Cancer | GeneXNet | 98.9 | [38, 39, 41, 43, 45, 47, 48] |
| ResNet | 96.5 | |||
| DenseNet | 95.3 | |||
| NasNet | 93.5 | |||
| MobileNet | 94.2 | |||
| RPLS | 93.4 | |||
| FLD | 81.5 | |||
| TSP | 91.2 | |||
| RF-SVM | 91.9 | |||
| - | - | IP | 93 | - |
| SVM | 98.45 | |||
| SRC | 89 | |||
| RF | 92.06 | |||
| GA ICA | 90.3 | |||
| AUFL kNN | 92.2 | |||
| AUFL DT | 92.1 | |||
| PSODT | 96.9 | |||
| Active NN | 97.66 | |||
| DAE | 98.59 | |||
| CNV Bayesian | 99.27 | |||
| 20. | Lung disease | PLS TTZ | 92.65 | [51] |
| 21. | Homo Sapiens | GG with RF | 79.54 | [76] |
| - | GG with ANN | 78.14 | - | |
| - | GG with LIN SVM | 78.06 | - | |
| - | GG with SVM RBF | 77.18 | - | |
| - | GG with DNN | 78.95 | - |
3.1.1. Accuracy of Genomic Processing Models for Human Diseases
The models used for human diseases were analyzed, and each of them was bifurcated according to the genome data processed. To support the classification, Fig. (5) is presented, wherein the accuracy of different genome models for heart and brain sequence data analysis is visualized. It is observed that RIPPER SVM [54], SHoT [27], RNN [53], and CNN [53] outperform other models in terms of accuracy, and thus must be used for heart and brain dataset genome sequence analysis.
Fig. (5).
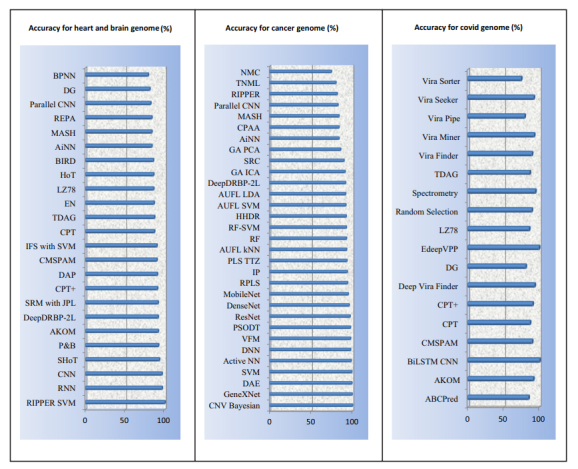
Accuracy of models for classification of heart and brain, cancer and Covid genome analysis (%).
A similar representation for Covid and cancer genome analysis can be observed (Fig. 5), wherein BiLSTM CNN [63] and EdeepVPP [55] have exhibited good accuracy for Covid genomic dataset, whereas VFM [56], CNV Bayesian [46], GeneXNet [38], DAE [48], SVM [48], and Active NN [48] have been shown to outperform others for cancer genome data analysis.
These models have been observed to have good accuracy because of their feature extraction and selection capabilities, which assist in improving genome representation efficiency levels. This, when combined with deep learning-based classification, assists in improving classification performance for different genome types. A weighted sum classifier can be built to combine these models, which will assist in further enhancing their performance for different disease types.
3.1.2. Accuracy of Genomic Processing Models for Crops
The genome sequence analysis of crops is classified as grains and non-grains, where rice, wheat, oats, etc., come under grains. It can be observed from Fig. (6) that RNN [53], CNN [53], GA SIWR [2], and SpineNet-6m [5] outperform others for grain genome dataset. A similar representation for genome analysis of non-grains can be observed (Fig. 6), wherein sugarcane, maize, tea, groundnut, etc., come under non-grain. It can be observed that RF [15], GBM [15], PLS [15], GLMNET [15], LDA, PMLR, SVM [15], GEBV [12], and AMOVA [20] outperform others.
Fig. (6).

Accuracy of models for classification of grain and non-grain genome analysis (%).
Crop genomes have simpler structures than human genomes, thus models that use simplified techniques are observed to have better classification performance levels. The identified models use simple feature extraction, and combine it with high-performance feature selection and classification, which assists in improving accuracy, precision, and recall performance for different genome types. A bioinspired model (like genetic algorithm or firefly optimization) with a context-specific classifier can be built to identify optimum features from these models, which will assist in further enhancing their performance for different disease types.
3.2. Precision, Recall and Computational Complexity of Genomic Models
A comparison was made for precision (P), recall (R) and computational complexity (CC) of the algorithms that have good accuracy, and results are tabulated in Table 2, where the computational complexity is divided into ranges of low (L), medium (M), high (H), and very high (VH) depending on internal architectures for the genomic model.
Table 2.
Comparison of precision, recall and computational complexity of the models.
| Sr. No. | Method |
Precision
(%) |
Recall (%) |
Computational
Complexity |
References |
|---|---|---|---|---|---|
| 1. | DAP | 85.5 | 83.57 | H | [1] |
| 2. | GA SIWR | 91.2 | 89.14 | M | [2] |
| 3. | FP Tree | 88.35 | 86.36 | M | |
| 4. | FP SVM | 85.5 | 83.57 | H | |
| 5. | i6m | 86.36 | 84.41 | H | [5] |
| 6. | EdeepVPP | 94.24 | 92.11 | VH | [55] |
| 7. | Vira Miner | 87.69 | 85.71 | H | - |
| 8. | Vira Seeker | 87.21 | 85.24 | H | |
| 9. | Deep Vira Finder | 88.35 | 86.36 | H | |
| 10. | RF | 92.15 | 90.07 | H | [15] |
| 11. | GBM | 91.2 | 89.14 | VH | |
| 12. | PLS | 90.25 | 88.21 | H | |
| 13. | VFM | 92.15 | 90.07 | H | [56] |
| 14. | BMTME | 85.5 | 83.57 | H | [57] |
| 15. | MTR | 86.45 | 84.5 | H | |
| 16. | AKOM | 86.64 | 84.69 | H | [59] |
| 17. | CMSPAM | 85.03 | 83.11 | H | |
| 18. | Spectrometry | 89.3 | 87.29 | H | [60] |
| 19. | Random Selection | 84.55 | 82.64 | M | |
| 20. | MLP | 86.45 | 84.5 | H | [23] |
| 21. | DNN | 92.15 | 90.07 | VH | [31] |
| 22. | RF | 87.02 | 85.06 | H | [33] |
| 23. | GCA & SCA | 86.55 | 84.59 | H | [26] |
| 24. | GeneXNet | 93.96 | 91.84 | VH | [38] |
| 25. | ResNet | 91.68 | 89.61 | VH | |
| 26. | DenseNet | 90.54 | 88.49 | VH | |
| 27. | RPLS | 84.06 | 84.5 | H | [39] |
| 28. | RF-SVM | 82.71 | 83.15 | H | |
| 29. | IP | 83.7 | 84.14 | H | [41] |
| 30. | GA ICA | 81.27 | 81.7 | H | [43] |
| 31. | AUFL DT | 82.89 | 83.33 | H | [45] |
| 32. | AUFL kNN | 82.98 | 83.42 | H | |
| 33. | CNV Bayesian | 89.34 | 89.82 | VH | [46] |
| 34. | PSODT | 87.21 | 87.67 | H | [47] |
| 35. | SVM | 88.61 | 89.07 | H | [48] |
| 36. | DAE | 88.73 | 89.2 | VH | |
| 37. | RF | 82.85 | 83.29 | H | |
| 39. | BiLSTM CNN | 89.96 | 90.43 | VH | [63] |
| 40. | PLS TTZ | 83.39 | 83.83 | H | [51] |
| 41. | RIPPER SVM | 89.73 | 90.2 | VH | [54] |
In Table 2, it can be observed that EdeepVPP [55], GeneXNet [38], RF [15], VFM [56], DNN [31], ResNet [38], GA SIWR [2], GBM [15], DenseNet [38], PLS [15], and BiLSTM CNN [63] outperform others in terms of precision performance, while EdeepVPP [55], GeneXNet [38], BiLSTM CNN [63], RIPPER SVM [54], RF [15], VFM [56], DNN [31], CNV Bayesian [46], ResNet [38], DAE [48], GA SIWR [2], GBM [15], and SVM [48] have better recall performance than others. While, in terms of computational complexity, GA SIWR [2], FP Tree [2], Random Selection [60], RF [15], VFM [56], SVM [48], PLS [15], PSODT [47], and Spectrometry [60] outperform others; thus, they are categorized as high-speed genome processing algorithms. Fig. (7) represents top 20 genomic models that outperform others in terms of precision and recall.
Fig. (7).
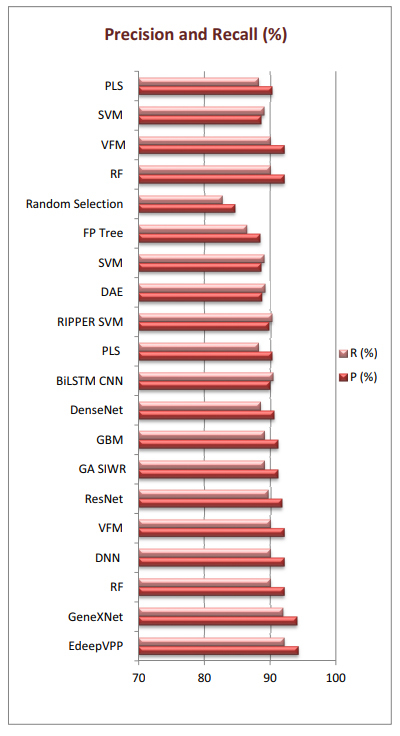
Top 20 genomic processing models that outperform in terms of precision and recall.
3.3. Algorithmic Rank of Genomic Models
As per the genomic application requirement, these models must be used by a programmer or end user for improving their system’s performance. Based on these metrics, a novel algorithmic rank is evaluated, which will further assist in model selection. The rank is evaluated using equation 3 as follows:

Where, (A) is Accuracy, (P) Precision, (R) Recall, and (CC) Computational complexity. The rank will assist readers in identifying algorithms with maximum accuracy, good precision, high recall and low computational complexity. Table 3 shows algorithmic rank (AR) for the top 30 genomic models. From this rank, it is observed that GA SIWR [2], FP Tree [2], and Random Selection [2] are the best-performing models for genomic data classification, and must be used for real-time clinical purposes.
Table 3.
Algorithmic rank (AR) and model rank for top 30 models.
| Sr. No. | Method | AR | Model Rank | References |
|---|---|---|---|---|
| 1. | DAP | 2.11 | 23 | [1] |
| 2. | GA SIWR | 2.59 | 1 | [2] |
| 3. | FP SVM | 2.11 | 24 | |
| 4. | FP Tree | 2.56 | 2 | |
| 5. | i6m | 2.12 | 20 | [5] |
| 6. | Vira Miner | 2.14 | 11 | [55] |
| 7. | Vira Seeker | 2.13 | 12 | |
| 8. | Deep Vira Finder | 2.14 | 10 | |
| 9. | RF | 2.18 | 4 | [15] |
| 10. | PLS | 2.16 | 7 | |
| 11. | VFM | 2.18 | 5 | [56] |
| 12. | BMTME | 2.11 | 25 | [57] |
| 13. | MTR | 2.12 | 17 | |
| 14. | AKOM | 2.13 | 14 | [59] |
| 15. | CMSPAM | 2.11 | 30 | |
| 16. | Spectrometry | 2.15 | 9 | [60] |
| 17. | Random Selection | 2.52 | 3 | |
| 18. | MLP | 2.12 | 18 | [23] |
| 19. | RF | 2.13 | 13 | [33] |
| 20. | SRM with JPL | 2.12 | 19 | [36] |
| 21. | GCA & SCA | 2.12 | 15 | [26] |
| 22. | RPLS | 2.12 | 16 | [39] |
| 23. | RF-SVM | 2.11 | 29 | |
| 24. | IP | 2.12 | 21 | [41] |
| 25. | AUFL kNN | 2.11 | 26 | [45] |
| 26. | AUFL DT | 2.11 | 27 | |
| 27. | PSODT | 2.16 | 8 | [47] |
| 28. | SVM | 2.17 | 6 | [48] |
| 29. | RF | 2.11 | 28 | |
| 30. | PLS TTZ | 2.12 | 22 | [51] |
CONCLUSION AND FUTURE PROSPECTS
From the in-depth comparative analysis, researchers will be able to identify the best-performing algorithms suited for a given category of genome sequences. In terms of accuracy, it is observed that RIPPER SVM, DNN, VFM, RNN, CNN, GA SIWR, DBN, Spectrometry, CNN, FP Tree, BiLSTM CNN, CNV Bayesian, EdeepVPP, GeneXNet, DAE, SVM, Active NN, PSODT, ResNet, SpineNet-6m, RF, GBM, and PLS have better performance when compared to other models. Thus, these models must be used for high-accuracy classification and genome processing applications. In terms of precision, EdeepVPP, GeneXNet, RF, VFM, DNN, ResNet, GA SIWR, GBM, DenseNet, PLS, BiLSTM CNN, RIPPER SVM, SpineNet-6m, CNV Bayesian, Spectrometry, and DBN outperform other models; thus, their use for highly precise applications is suggested. While, in terms of recall, EdeepVPP, GeneXNet, BiLSTM CNN, RIPPER SVM, RF, VFM, DNN, CNV Bayesian, ResNet, DAE, GA SIWR, GBM, SVM, DenseNet, Active NN, PLS, PSODT, SpineNet-6m, Spectrometry, DBN, and RNN showcase better performance across multiple types of datasets. Thus, these models must be used when genomic data has to be classified with a low error rate and high consistency. Furthermore, GA SIWR, FP Tree, Random Selection, RF, VFM, SVM, PLS, PSODT, and Spectrometry have the highest speed; therefore, their use is recommended for high-speed and moderate to high accuracy applications.
In the future, researchers can identify the best-performing algorithms for application-specific cases and create an ensemble model. This model must initially identify the type of genome and then process it using the highest performing classification model. Moreover, augmentation of genomic data must be done in order to improve the accuracy and precision performance via oversampling, which might incur greater computational delays, but would guarantee better performance than their original counterparts. The future goal is to build systems biology models of biological systems that faithfully reflect the area of biology, and which can be used for mechanistic predictions. Furthermore, they can also be used as recommender systems for gene sequence processing. This will assist in the identification of optimum sequence pairs for high-accuracy and low-delay computational system design, thereby assisting in improving overall system efficiency.
ACKNOWLEDGEMENTS
The authors are thankful to the Director, Department of Computer Science and Engineering, Visvesvaraya National Institute of Technology (VNIT), Nagpur, for providing the necessary facilities for this work.
LIST OF ABBREVIATIONS
- AiNN
Artificial Neural Network
- CNNs
Convolutional Neural Networks
- DBNs
Deep Belief Networks
- LDA
Latent Dirichlet Analysis
- MLPs
Multilayer Perceptrons
- PCA
Principal Component Analysis
- RNN
Recurrent Neural Network
AUTHORS’ CONTRIBUTION
Aditi Durge compiled the data and wrote the original draft; Deepti Shrimankar contributed to conceptualization, visualization, supervision, review and editing; and Ankush Sawarkar performed compiling of data, conceptualization, review and editing.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Barbeira A.N., Melia O.J., Liang Y., Bonazzola R., Wang G., Wheeler H.E., Aguet F., Ardlie K.G., Wen X. Im, H.K. Fine‐mapping and QTL tissue‐sharing information improves the reliability of causal gene identification. Genet. Epidemiol. 2020;44(8):854–867. doi: 10.1002/gepi.22346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seo H., Song Y.J., Cho K., Cho D.H. Specificity analysis of genome based on statistically identical K-words with same base combination. IEEE Open J. Eng. Med. Biol. 2020;1:214–219. doi: 10.1109/OJEMB.2020.3009055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Libbrecht M.W., Noble W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015;16(6):321–332. doi: 10.1038/nrg3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schrider D.R., Kern A.D. Supervised machine learning for population genetics: A new paradigm. Trends Genet. 2018;34(4):301–312. doi: 10.1016/j.tig.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abbas Z., Tayara H., Chong K. Spinenet-6MA: A novel deep learning tool for predicting DNA N6-methyladenine sites in genomes. IEEE Access. 2020;8:201450–201457. doi: 10.1109/ACCESS.2020.3036090. [DOI] [Google Scholar]
- 6.Sun T., Wei Y., Chen W., Ding Y. Genome‐wide association study‐based deep learning for survival prediction. Stat. Med. 2020;39(30):4605–4620. doi: 10.1002/sim.8743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Remita M.A., Halioui A., Malick Diouara A.A., Daigle B., Kiani G., Diallo A.B. A machine learning approach for viral genome classification. BMC Bioinformatics. 2017;18(1):208. doi: 10.1186/s12859-017-1602-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abass Y.A., Adeshina S.A. Deep learning methodologies for genomic data prediction:Review . Journal of Artificial Intelligence for Medical Sciences. 2021;2(1-2):1. doi: 10.2991/jaims.d.210512.001. [DOI] [Google Scholar]
- 9.Yu X., Leiboff S., Li X., Guo T., Ronning N., Zhang X., Muehlbauer G.J., Timmermans M.C.P., Schnable P.S., Scanlon M.J., Yu J. Genomic prediction of maize microphenotypes provides insights for optimizing selection and mining diversity. Plant Biotechnol. J. 2020;18(12):2456–2465. doi: 10.1111/pbi.13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez M. Computational tools for genomic studies in plants. Curr. Genomics. 2016;17(6):509–514. doi: 10.2174/1389202917666160520103447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo Q., Liu Q., Smith N.A., Liang G., Wang M.B. RNA silencing in plants: Mechanisms, technologies and applications in horticultural crops. Curr. Genomics. 2016;17(6):476–489. doi: 10.2174/1389202917666160520103117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Almeida V.C., Trentin H.U., Frei U.K., Lübberstedt T. Genomic prediction of maternal haploid induction rate in maize. Plant Genome. 2020;13(1):e20014. doi: 10.1002/tpg2.20014. [DOI] [PubMed] [Google Scholar]
- 13.Zhong L., Hu L., Zhou H. Deep learning based multi-temporal crop classification. Remote Sens. Environ. 2019;221:430–443. doi: 10.1016/j.rse.2018.11.032. [DOI] [Google Scholar]
- 14.Michel S., Löschenberger F., Sparry E., Ametz C., Bürstmayr H. Mitigating the impact of selective phenotyping in training populations on the prediction ability by multi‐trait pedigree and genomic selection models. Plant Breed. 2020;139(6):1067–1075. doi: 10.1111/pbr.12862. [DOI] [Google Scholar]
- 15.Dai X., Xu Z., Liang Z., Tu X., Zhong S., Schnable J.C., Li P. Non‐homology‐based prediction of gene functions in maize (Zea mays ssp. mays). Plant Genome. 2020;13(2):e20015. doi: 10.1002/tpg2.20015. [DOI] [PubMed] [Google Scholar]
- 16.Grinberg N.F., Orhobor O.I., King R.D. An evaluation of machine-learning for predicting phenotype: Studies in yeast, rice, and wheat. Mach. Learn. 2020;109(2):251–277. doi: 10.1007/s10994-019-05848-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Onda Y., Mochida K. Exploring genetic diversity in plants using high-throughput sequencing techniques. Curr. Genomics. 2016;17(4):358–367. doi: 10.2174/1389202917666160331202742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yadav S., Wei X., Joyce P., Atkin F., Deomano E., Sun Y., Nguyen L.T., Ross E.M., Cavallaro T., Aitken K.S., Hayes B.J., Voss-Fels K.P. Improved genomic prediction of clonal performance in sugarcane by exploiting non-additive genetic effects. Theor. Appl. Genet. 2021;134(7):2235–2252. doi: 10.1007/s00122-021-03822-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Virnodkar S.S., Pachghare V.K., Patil V.C. Application of machine learning on remote sensing data for sugarcane crop classification: A review BT-ICT analysis and applications. Singapore: Springer Singapore; 2020. pp. 539–555. [Google Scholar]
- 20.Auinger H.J., Lehermeier C., Gianola D., Mayer M., Melchinger A.E., da Silva S., Knaak C., Ouzunova M., Schön C.C. Calibration and validation of predicted genomic breeding values in an advanced cycle maize population. Theor. Appl. Genet. 2021;134(9):3069–3081. doi: 10.1007/s00122-021-03880-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lubanga N., Massawe F., Mayes S. Genomic and pedigree‐based predictive ability for quality traits in tea (Camellia sinensis (L.) O. Kuntze). Euphytica. 2021;217(3):32. doi: 10.1007/s10681-021-02774-3. [DOI] [Google Scholar]
- 22.Knoch D., Werner C.R., Meyer R.C., Riewe D., Abbadi A., Lücke S., Snowdon R.J., Altmann T. Multi-omics-based prediction of hybrid performance in canola. Theor. Appl. Genet. 2021;134(4):1147–1165. doi: 10.1007/s00122-020-03759-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montesinos-López O.A., Montesinos-López A., Pérez-Rodríguez P., Barrón-López J.A., Martini J.W.R., Fajardo-Flores S.B., Gaytan-Lugo L.S., Santana-Mancilla P.C., Crossa J. A review of deep learning applications for genomic selection. BMC Genomics. 2021;22(1):19. doi: 10.1186/s12864-020-07319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pandey M.K., Chaudhari S., Jarquin D., Janila P., Crossa J., Patil S.C., Sundravadana S., Khare D., Bhat R.S., Radhakrishnan T., Hickey J.M., Varshney R.K. Genome-based trait prediction in multi- environment breeding trials in groundnut. Theor. Appl. Genet. 2020;133(11):3101–3117. doi: 10.1007/s00122-020-03658-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mellers G., Mackay I., Cowan S., Griffiths I., Martinez-Martin P., Poland J.A., Bekele W., Tinker N.A., Bentley A.R., Howarth C.J. Implementing within‐cross genomic prediction to reduce oat breeding costs. Plant Genome. 2020;13(1):e20004. doi: 10.1002/tpg2.20004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basnet B.R., Crossa J., Dreisigacker S., Pérez-Rodríguez P., Manes Y., Singh R.P., Rosyara U.R., Camarillo-Castillo F., Murua M. Hybrid wheat prediction using genomic, pedigree, and environmental covariables interaction models. Plant Genome. 2019;12(1):180051. doi: 10.3835/plantgenome2018.07.0051. [DOI] [PubMed] [Google Scholar]
- 27.Ramasamy M.D., Periasamy K., Krishnasamy L., Dhanaraj R.K., Kadry S., Nam Y. Multi-disease classification model using Strassen’s Half of Threshold (SHoT) training algorithm in healthcare sector. IEEE Access. 2021;9:112624–112636. doi: 10.1109/ACCESS.2021.3103746. [DOI] [Google Scholar]
- 28.Li J., Huang Y., Zhou Y. A mini-review of the computational methods used in identifying RNA 5- methylcytosine sites. Curr. Genomics. 2020;21(1):3–10. doi: 10.2174/2213346107666200219124951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J., Chen Q., Liu B. DeepDRBP-2L: A new genome annotation predictor for identifying DNA-binding proteins and RNA-binding proteins using convolutional neural network and long short-term memory. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021;18(4):1451–1463. doi: 10.1109/TCBB.2019.2952338. [DOI] [PubMed] [Google Scholar]
- 30.Yu X., Gan Z., Xu Y., Wan S., Li M., Ding S., Zeng T. Identifying essential methylation patterns and genes associated with stroke. IEEE Access. 2020;8:96669–96676. doi: 10.1109/ACCESS.2020.2994646. [DOI] [Google Scholar]
- 31.Singh S., Yang Y., Póczos B., Ma J. Predicting enhancer-promoter interaction from genomic sequence with deep neural networks. Quant. Biol. 2019;7(2):122–137. doi: 10.1007/s40484-019-0154-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu L., Guo Z., Liu X. Prediction of essential genes in prokaryote based on artificial neural network. Genes Genomics. 2020;42(1):97–106. doi: 10.1007/s13258-019-00884-w. [DOI] [PubMed] [Google Scholar]
- 33.Liu B., Han L., Liu X., Wu J., Ma Q. Computational prediction of sigma-54 promoters in bacterial genomes by integrating motif finding and machine learning strategies. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019;16(4):1211–1218. doi: 10.1109/TCBB.2018.2816032. [DOI] [PubMed] [Google Scholar]
- 34.Davi C., Pastor A., Oliveira T., Neto F.B.L., Braga-Neto U., Bigham A.W., Bamshad M., Marques E.T.A., Acioli-Santos B. Severe dengue prognosis using human genome data and machine learning. IEEE Trans. Biomed. Eng. 2019;66(10):2861–2868. doi: 10.1109/TBME.2019.2897285. [DOI] [PubMed] [Google Scholar]
- 35.Li X., Qiu Y., Zhou J., Xie Z. Applications and challenges of machine learning methods in alzheimer’s disease multi-source data analysis. Curr. Genomics. 2021;22(8):564–582. doi: 10.2174/1389202923666211216163049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou T., Thung K.H., Liu M., Shen D. Brain-wide genome-wide association study for alzheimer’s disease via joint projection learning and sparse regression model. IEEE Trans. Biomed. Eng. 2019;66(1):165–175. doi: 10.1109/TBME.2018.2824725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sergeev R.S., Kavaliou I.S., Sataneuski U.V., Gabrielian A., Rosenthal A., Tartakovsky M., Tuzikov A.V. Genome-wide analysis of MDR and XDR tuberculosis from belarus: Machine-learning approach. IEEE/ACM Trans. Comput. Biol. Bioinformatics. 2019;16(4):1398–1408. doi: 10.1109/TCBB.2017.2720669. [DOI] [PubMed] [Google Scholar]
- 38.Khorshed T., Moustafa M.N., Rafea A. Deep learning for multi-tissue cancer classification of gene expressions (GeneXNet). IEEE Access. 2020;8:90615–90629. doi: 10.1109/ACCESS.2020.2992907. [DOI] [Google Scholar]
- 39.Wu H.C., Wei X.G., Chan S.C. Novel consensus gene selection criteria for distributed GPU partial least squares-based gene microarray analysis in Diffused Large B Cell Lymphoma (DLBCL) and related findings. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018;15(6):2039–2052. doi: 10.1109/TCBB.2017.2760827. [DOI] [PubMed] [Google Scholar]
- 40.Knight J.M., Ivanov I., Triff K., Chapkin R.S., Dougherty E.R. Detecting multivariate gene interactions in RNA-Seq data using optimal bayesian classification. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018;15(2):484–493. doi: 10.1109/TCBB.2015.2485223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang X., Tian L., Chen Y., Yang L., Xu S., Wu W. Inverse projection representation and category contribution rate for robust tumor recognition. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020;17(4):1262–1275. doi: 10.1109/TCBB.2018.2886334. [DOI] [PubMed] [Google Scholar]
- 42.Xu P., Zhao G., Kou Z., Fang G., Liu W. Classification of cancers based on a comprehensive pathway activity inferred by genes and their interactions. IEEE Access. 2020;8:30515–30521. doi: 10.1109/ACCESS.2020.2973220. [DOI] [Google Scholar]
- 43.Arowolo M.O., Adebiyi M.O., Adebiyi A.A., Okesola O.J. A hybrid heuristic dimensionality reduction methods for classifying malaria vector gene expression data. IEEE Access. 2020;8:182422–182430. doi: 10.1109/ACCESS.2020.3029234. [DOI] [Google Scholar]
- 44.Jujjavarapu S.E., Deshmukh S. Artificial neural network as a classifier for the identification of hepatocellular carcinoma through prognosticgene signatures. Curr. Genomics. 2018;19(6):483–490. doi: 10.2174/1389202919666180215155234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ye X., Zhang W., Sakurai T. Adaptive unsupervised feature learning for gene signature identification in non-small-cell lung cancer. IEEE Access. 2020;8:154354–154362. doi: 10.1109/ACCESS.2020.3018480. [DOI] [Google Scholar]
- 46.Yuan X., Bai J., Zhang J., Yang L., Duan J., Li Y., Gao M. CONDEL: Detecting copy number variation and genotyping deletion zygosity from single tumor samples using sequence data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020;17(4):1141–1153. doi: 10.1109/TCBB.2018.2883333. [DOI] [PubMed] [Google Scholar]
- 47.Khalifa N.E.M., Taha M.H.N., Ezzat Ali D., Slowik A., Hassanien A.E. Artificial intelligence technique for gene expression by tumor RNA-Seq Data: A novel optimized deep learning approach. IEEE Access. 2020;8:22874–22883. doi: 10.1109/ACCESS.2020.2970210. [DOI] [Google Scholar]
- 48.Choi J., Rhee J.K., Chae H. Cell subtype classification via representation learning based on a denoising autoencoder for single-cell RNA sequencing. IEEE Access. 2021;9:14540–14548. doi: 10.1109/ACCESS.2021.3052923. [DOI] [Google Scholar]
- 49.Sonea L., Buse M., Gulei D., Onaciu A., Simon I., Braicu C., Berindan-Neagoe I. Decoding the emerging patterns exhibited in non-coding rnas characteristic of lung cancer with regard to their clinical significance. Curr. Genomics. 2018;19(4):258–278. doi: 10.2174/1389202918666171005100124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang X., Zhu L., Huang D.S. Optimization of gene set annotations using robust trace-norm multitask learning. IEEE/ACM Trans. Comput. Biol. Bioinformatics. 2018;15(3):1016–1021. doi: 10.1109/TCBB.2017.2690427. [DOI] [PubMed] [Google Scholar]
- 51.He Q., Qiu Z., Tong Y., Song K. A new TTZ feature extracting algorithm to decipher tobacco related mutation signature genes for the personalized lung adenocarcinoma treatment. IEEE Access. 2020;8:89031–89040. doi: 10.1109/ACCESS.2020.2993118. [DOI] [Google Scholar]
- 52.Bian J., Modave F. The rapid growth of intelligent systems in health and health care. Health Informatics J. 2020;26(1):5–7. doi: 10.1177/1460458219896899. [DOI] [PubMed] [Google Scholar]
- 53.Ho T.K.K., Gwak J. Toward deep learning approaches for learning structure motifs and classifying biological sequences from RNA A-to-I editing events. IEEE Access. 2019;7:127464–127474. doi: 10.1109/ACCESS.2019.2939281. [DOI] [Google Scholar]
- 54.Chen L., Pan X., Zeng T., Zhang Y-H., Huang T., Cai Y-D. Identifying essential signature genes and expression rules associated with distinctive development stages of early embryonic cells. IEEE Access. 2019;7:128570–128578. doi: 10.1109/ACCESS.2019.2939556. [DOI] [Google Scholar]
- 55.Dasari C.M., Bhukya R. Explainable deep neural networks for novel viral genome prediction. Appl. Intell. 2021 doi: 10.1007/s10489-021-02572-3. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Q., Liu F., He J., Zhou M., Hou T., Liu Y. VFM: Identification of bacteriophages from metagenomic bins and contigs based on features related to gene and genome composition. IEEE Access. 2019;7:177529–177538. doi: 10.1109/ACCESS.2019.2957833. [DOI] [Google Scholar]
- 57.Ibba M.I., Crossa J., Montesinos-López O.A., Montesinos-López A., Juliana P., Guzman C., Delorean E., Dreisigacker S., Poland J. Genome‐based prediction of multiple wheat quality traits in multiple years. Plant Genome. 2020;13(3):e20034. doi: 10.1002/tpg2.20034. [DOI] [PubMed] [Google Scholar]
- 58.Dias R., Torkamani A. Artificial intelligence in clinical and genomic diagnostics. Genome Med. 2019;11(1):70. doi: 10.1186/s13073-019-0689-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nawaz M.S., Fournier-Viger P., Shojaee A., Fujita H. Using artificial intelligence techniques for COVID-19 genome analysis. Appl. Intell. 2021;51(5):3086–3103. doi: 10.1007/s10489-021-02193-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Poran A., Harjanto D., Malloy M., Arieta C.M., Rothenberg D.A., Lenkala D., van Buuren M.M., Addona T.A., Rooney M.S., Srinivasan L., Gaynor R.B. Sequence-based prediction of SARS-CoV-2 vaccine targets using a mass spectrometry-based bioinformatics predictor identifies immunogenic T cell epitopes. Genome Med. 2020;12(1):70. doi: 10.1186/s13073-020-00767-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xie Q., He X., Yang F., Liu X., Li Y., Liu Y., Yang Z., Yu J., Zhang B., Zhao W. Analysis of the genome sequence and prediction of B-Cell epitopes of the envelope protein of middle east respiratory syndrome-coronavirus. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018;15(4):1344–1350. doi: 10.1109/TCBB.2017.2702588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kushwaha S., Bahl S., Bagha A.K., Parmar K.S., Javaid M., Haleem A., Singh R.P. Significant applications of machine learning for COVID-19 pandemic. J. Indus. Integr. Manage. 2020;5(4):453–479. doi: 10.1142/S2424862220500268. [DOI] [Google Scholar]
- 63.Whata A., Chimedza C. Deep learning for SARS COV-2 genome sequences. IEEE Access. 2021;9:59597–59611. doi: 10.1109/ACCESS.2021.3073728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.El Allali A., Elhamraoui Z., Daoud R. Machine learning applications in RNA modification sites prediction. Comput. Struct. Biotechnol. J. 2021;19:5510–5524. doi: 10.1016/j.csbj.2021.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moghaddar N., Khansefid M., van der Werf J.H.J., Bolormaa S., Duijvesteijn N., Clark S.A., Swan A.A., Daetwyler H.D., MacLeod I.M. Genomic prediction based on selected variants from imputed whole-genome sequence data in Australian sheep populations. Genet. Sel. Evol. 2019;51(1):72. doi: 10.1186/s12711-019-0514-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zrimec J., Börlin C.S., Buric F., Muhammad A.S., Chen R., Siewers V., Verendel V., Nielsen J., Töpel M., Zelezniak A. Deep learning suggests that gene expression is encoded in all parts of a co-evolving interacting gene regulatory structure. Nat. Commun. 2020;11(1):6141. doi: 10.1038/s41467-020-19921-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kotsiantis S.B., Zaharakis I.D., Pintelas P.E. Machine learning: A review of classification and combining techniques. Artif. Intell. Rev. 2006;26(3):159–190. doi: 10.1007/s10462-007-9052-3. [DOI] [Google Scholar]
- 68.Zhou W., Ji H. Genome‐wide prediction of chromatin accessibility based on gene expression. Wiley Interdiscip. Rev. Comput. Stat. 2021;13(5):1–13. doi: 10.1002/wics.1544. [DOI] [Google Scholar]
- 69.Galán R.J., Bernal-Vasquez A.M., Jebsen C., Piepho H.P., Thorwarth P., Steffan P., Gordillo A., Miedaner T. Early prediction of biomass in hybrid rye based on hyperspectral data surpasses genomic predictability in less-related breeding material. Theor. Appl. Genet. 2021;134(5):1409–1422. doi: 10.1007/s00122-021-03779-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patra P., Izawa T., Pena-Castillo L. REPA: Applying pathway analysis to genome-wide transcription factor binding data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018;15(4):1270–1283. doi: 10.1109/TCBB.2015.2453948. [DOI] [PubMed] [Google Scholar]
- 71.Waldvogel A.M., Feldmeyer B., Rolshausen G., Exposito-Alonso M., Rellstab C., Kofler R., Mock T., Schmid K., Schmitt I., Bataillon T., Savolainen O., Bergland A., Flatt T., Guillaume F., Pfenninger M. Evolutionary genomics can improve prediction of species’ responses to climate change. Evol. Lett. 2020;4(1):4–18. doi: 10.1002/evl3.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sedaghat N., Fathy M., Modarressi M.H., Shojaie A. Combining supervised and unsupervised learning for improved mirna target prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018;15(5):1. doi: 10.1109/TCBB.2017.2727042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jung I., Choi J., Chae H. A non-negative matrix factorization-based framework for the analysis of multi-class time-series single-cell RNA-Seq data. IEEE Access. 2020;8:42342–42348. doi: 10.1109/ACCESS.2020.2977106. [DOI] [Google Scholar]
- 74.Wu Y., Tong Y., Zhu X., Wu X. NOSEP: Nonoverlapping sequence pattern mining with gap constraints. IEEE Trans. Cybern. 2018;48(10):2809–2822. doi: 10.1109/TCYB.2017.2750691. [DOI] [PubMed] [Google Scholar]
- 75.Khan S., Khan M., Iqbal N., Li M., Khan D.M. Spark-based parallel deep neural network model for classification of large scale RNAs into piRNAs and non-piRNAs. IEEE Access. 2020;8:136978–136991. doi: 10.1109/ACCESS.2020.3011508. [DOI] [Google Scholar]
- 76.Wang G., Pu P., Shen T. An efficient gene bigdata analysis using machine learning algorithms. Multimedia Tools Appl. 2020;79(15-16):9847–9870. doi: 10.1007/s11042-019-08358-7. [DOI] [Google Scholar]