

The gene mutation that causes Huntington’s Disease (HD) is associated with neurodevelopmental changes.1, 2 Our group has shown that children who are decades from their predicted age of onset of HD have striatal hypertrophy early in life followed by a faster rate of neurodegeneration later in life relative to children without the mutation that causes HD.2
Little is known about the neurodevelopment of patients with long CAG repeats who will likely manifest symptoms of HD in early adulthood or even those who will develop Juvenile-Onset HD (JOHD). Because these patients are so young, they generally present after symptoms have manifested. Consequently, their striatal volumes are significantly decreased at clinical presentation.3 Therefore, it is unknown if long CAG repeats in the huntingtin protein prevented the normal development of the striatum (i.e., the striatum never reached an age-appropriate volume) or if the striatum was enlarged or volumetrically comparable to similarly aged children. Characterization of striatal development in early-onset HD (EOHD) and JOHD participants could have significant implications for future clinical trials.
We used the Kids-HD2 and Kids-JOHD3 databases and identified 7 children with a CAG > 50 who did not have HD motor manifestations and were far from their predicted onset of symptoms based on their standardized CAG-Age-Product (CAP).4 Detailed methods of these studies have been previously published.2, 3 These children (referred to as pre-EOHD) were compared to 14 Gene Non-Expanded children (GNE group) who were similar in age and sex. The mean age of the pre-EOHD group was 8.64 (Standard Deviation, 1.73) and included 5 females and 2 males. Median CAG repeat length was 56 (range: 51–73). The GNE group’s mean age was 8.87 (S.D. 1.60) and included 10 females and 4 males. Analyses of Covariance that controlled for age and sex were used to compare baseline striatal volumes (corrected for intracranial volume [ICV]) between the pre-EOHD and GNE groups. The mean strial volume of the pre-EOHD group (1.35 % ICV, S.E.=0.03) was significantly higher than the GNE group (1.26 % ICV, S.E.=0.02; p=0.013, Figure 1A). We suspect the pre-EOHD participants will have a faster rate of degeneration. We explored the rate of degeneration in one individual with a CAG of 73 who participated in our at-risk study.2 The striatal volume of this individual was similar to the GNE group at the age of six (Figure 1B), but had decreased by nearly 20% approximately two years later. This rapid degeneration continued and the participant received a diagnosis of JOHD before the age of 12. Importantly, the linear trajectory of this participant suggests striatal hypertrophy prior to the age of six (Figure 1B).
Figure 1: Assessment of Striatal Volume Amongst HD Children with Long CAG Repeats.
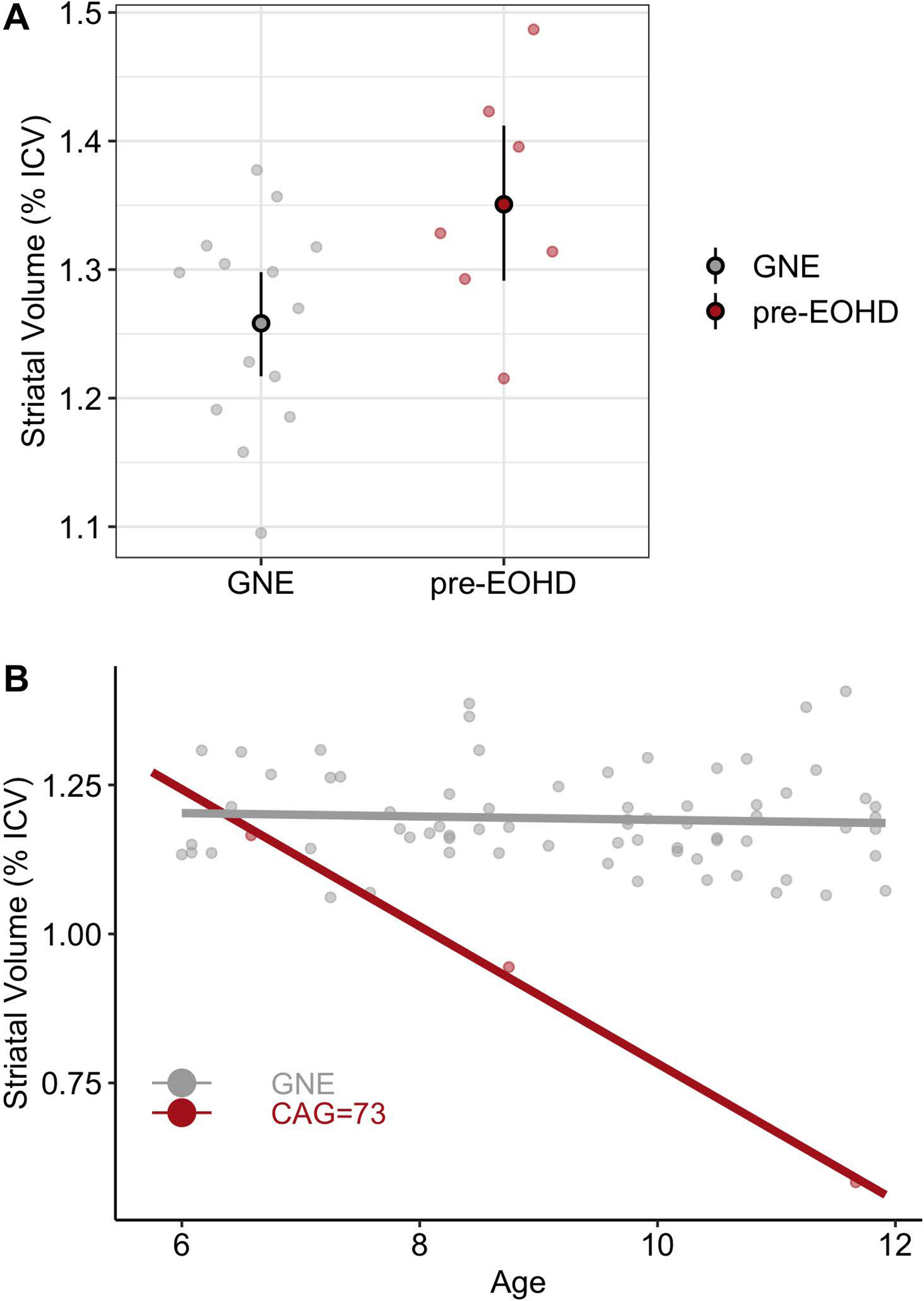
A) The mean striatal volume of the pre-EOHD children was significantly elevated compared to matched GNE participants. The large circles represent the adjusted group means and the 95% confidence interval.
B) A participant with a CAG of 73 who participated in the at-risk study from age 6–12 and their striatal volume is compared to a group of GNE participants comparable in age and matched for sex. At the age of 6, the participant was far from their predicted motor onset and had a striatal volume that was comparable to the GNE group.This participant’s striatal volume decreased quickly and they received a diagnosis of JOHD before the age of 12. Their trajectory suggests that this participant’s striatal volume was elevated prior to the age of 6 years.
EOHD, Early-Onset HD; GNE, Gene-non-expanded; HD, Huntington Disease; ICV, Intracranial Volume; JOHD, Juvenile-Onset Huntington Disease
This work implies that children with long CAG repeats in the huntingtin gene have early striatal hypertrophy. This expands on a growing body of literature suggesting that CAG repeat expansions in the huntingtin gene significantly impact neurodevelopment.1, 2, 5–7 Understanding of the pathophysiological mechanisms of HD in the developing brain will bring us closer to finding ways to mitigate or even prevent neurodegeneration.
ACKNOWLEDGEMENTS
We would like to thank all the participants and families who took part, providing their time, data, and samples to make this work possible.
FINANCIAL DISCLOSURES
This work was supported by the National Institute of Health (NIH) (R01-NS094387 and RO1-NS055903 to PN and K23-NS117736 to JLS) and the CHDI Foundation (to PCN [no award/grant numbers]). Images collected in this study were collected on a scanner funded by NIH Grant S10-OD025025. VAM receives salary support from the NIH (P50-HD103556). The funding organizations did not play a role in the analysis of data, nor did they play a role in the decision to publish these results.
The authors report no potential conflicts of interest related to this work.
REFERENCES
- 1.Barnat M, Capizzi M, Aparicio E, et al. Huntington’s disease alters human neurodevelopment. Science 2020;369(6505):787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Plas E, Langbehn DR, Conrad AL, et al. Abnormal brain development in child and adolescent carriers of mutant huntingtin. Neurology 2019;93(10):e1021–e1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tereshchenko A, Magnotta V, Epping E, et al. Brain structure in juvenile-onset Huntington disease. Neurology 2019;92(17):e1939–e1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Long JD, Mills JA, Warner JH, Lu W, Paulsen JS. Indexing disease progression at study entry with individuals at-risk for Huntington disease. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics 2011;156B(7):751–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schultz JL, van der Plas E, Langbehn DR, Conrad AL, Nopoulos PC. Age-Related Cognitive Changes as a Function of CAG Repeat in Child and Adolescent Carriers of Mutant Huntingtin. Ann Neurol 2021;89(5):1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Plas E, Schultz JL, Nopoulos PC. The Neurodevelopmental Hypothesis of Huntington’s Disease. J Huntingtons Dis 2020;9(3):217–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tereshchenko AV, Schultz JL, Bruss JE, Magnotta VA, Epping EA, Nopoulos PC. Abnormal development of cerebellar-striatal circuitry in Huntington disease. Neurology 2020;94(18):e1908–e1915. [DOI] [PMC free article] [PubMed] [Google Scholar]