

ABSTRACT
The Gram-negative outer membrane (OM) is an asymmetric bilayer with phospholipids in its inner leaflet and mainly lipopolysaccharide (LPS) in its outer leaflet and is largely impermeable to many antibiotics. In Enterobacterales (e.g., Escherichia, Salmonella, Klebsiella, and Yersinia), the outer leaflet of the OM also contains phosphoglyceride-linked enterobacterial common antigen (ECAPG). This molecule consists of the conserved ECA carbohydrate linked to diacylglycerol-phosphate (DAG-P) through a phosphodiester bond. ECAPG contributes to the OM permeability barrier and modeling suggests that it may alter the packing of LPS molecules in the OM. Here, we investigate, in Escherichia coli K-12, the reaction synthesizing ECAPG from ECA precursor linked to an isoprenoid carrier to identify the lipid donor that provides the DAG-P moiety to ECAPG. Through overexpression of phospholipid biosynthesis genes, we observed alterations expected to increase levels of phosphatidylglycerol (PG) increased the synthesis of ECAPG, whereas alterations expected to decrease levels of PG decreased the synthesis of ECAPG. We discovered depletion of PG levels in strains that could synthesize ECAPG, but not other forms of ECA, causes additional growth defects, likely due to the buildup of ECA precursor on the isoprenoid carrier inhibiting peptidoglycan biosynthesis. Our results demonstrate ECAPG can be synthesized in the absence of the other major phospholipids (phosphatidylethanolamine and cardiolipin). Overall, these results conclusively demonstrate PG is the lipid donor for the synthesis of ECAPG and provide a key insight into the reaction producing ECAPG. In addition, these results provide an interesting parallel to lipoprotein acylation, which also uses PG as its DAG donor.
IMPORTANCE The Gram-negative outer membrane is a permeability barrier preventing cellular entry of antibiotics. However, outer membrane biogenesis pathways are targets for small molecule development. Here, we investigate the synthesis of a form of enterobacterial common antigen (ECA), ECAPG, found in the outer membrane of Enterobacterales (e.g., Escherichia, Salmonella, and Klebsiella). ECAPG consists of the conserved ECA carbohydrate unit linked to diacylglycerol-phosphate—ECA is a phospholipid headgroup. The details of the reaction forming this molecule from polymerized ECA precursor are unknown. We determined the lipid donor providing the phospholipid moiety is phosphatidylglycerol. Understanding the synthesis of outer membrane constituents such as ECAPG provides the opportunity for development of molecules to increase outer membrane permeability, expanding the antibiotics available to treat Gram-negative infections.
KEYWORDS: cell envelope biogenesis, enterobacterial common antigen, glycolipids, outer membrane, phospholipids
INTRODUCTION
Gram-negative bacteria have a cell envelope that consists of an inner membrane (IM), aqueous periplasm containing the peptidoglycan cell wall, and an outer membrane (OM). In contrast to the IM and most biological membranes, the OM is an asymmetrical membrane with phospholipids in the inner leaflet and mainly lipopolysaccharide (LPS) in the outer leaflet (1). Beyond its lipid components, the OM is heavily populated with outer membrane proteins and OM lipoproteins (1–3). In addition, the OM contains lower abundance constituents, such as enterobacterial common antigen (ECA) (4), which is found throughout Enterobacterales.
The OM presents a strong permeability barrier capable of excluding both large molecules and hydrophobic molecules, including many antibiotics (1, 5, 6). Thus, the biogenesis pathways for OM components are potential targets for the development of small molecules to increase outer membrane permeability and the cell’s susceptibility to antibiotics (7, 8). In fact, several potential antimicrobials targeting biosynthesis of LPS and OM protein biosynthesis have recently been developed (8–15). Although these pathways are found throughout Gram-negative bacteria, the biosynthesis of ECA is a pathway that could allow development of small molecules to increase permeability in an order-specific manner, limiting off-target effects on bystander species during treatment.
ECA is an invariant carbohydrate moiety that is found in all Enterobacterales (e.g., Escherichia, Salmonella, Klebsiella, Shigella, Enterobacter, and Yersinia), with the exception of endosymbionts with greatly reduced genomes (reviewed in reference 4). The carbohydrate moiety consists of GlcNAc (N-acetylglucosamine), ManNAcA (N-acetyl-d-mannosaminuronic acid), and Fuc4NAc (4-acetamido-4,6-dideoxy-d-galactose) which form repeating units of →3)-α-Fuc4NAc-(1→4)-β-ManNAcA-(1→4)-α-GlcNAc-(1→ (Fig. 1A) (16, 17). ECA can be found in three forms: ECACYC, a cyclic form found soluble in the periplasm; ECAPG (ECA phosphoglyceride), a surface-exposed phospholipid form of ECA (see below); and ECALPS, LPS with ECA attached in place of O antigen (18–25). We have observed that ECACYC and ECAPG play roles in maintaining the OM permeability barrier in Escherichia coli K-12 (26). Modeling through molecular dynamics has suggested that the presence of ECAPG in the outer leaflet of the OM can lead to changes in packing of LPS with more space allotted per molecule (27, 28). In addition, work in Salmonella enterica serovar Typhimurium shows that ECA-deficient strains are nonvirulent and establish low-level persistent infections (29). Beyond its direct role in pathogenesis, exposure to ECA can lead to the development of antibodies that recognize species throughout Enterobacterales (30–32), likely playing an important protective or pathogenic role in Enterobacterales infection. These antibodies are most easily generated by exposure to rough Enterobacterales strains, which have high levels of ECALPS.
FIG 1.
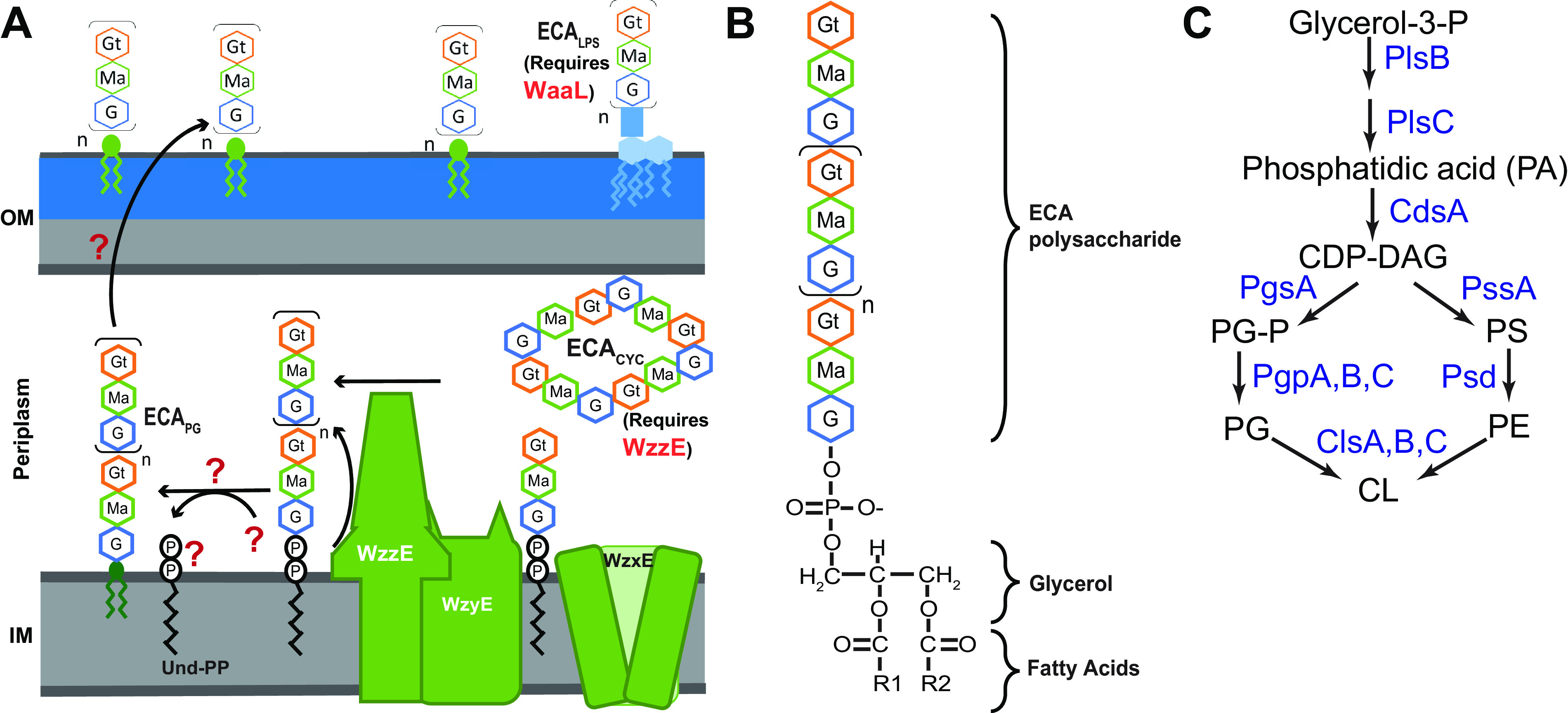
Structure and synthesis of ECAPG. (A) Subunits of ECA are assembled on the cytoplasmic face of the inner membrane, attached to an isoprenoid carrier, undecaprenyl-pyrophosphate (Und-PP). The subunits are then flipped across the membrane by WzxE and polymerized by WzyE. WzzE controls the chain length of the final ECA molecules. Polymerized ECA can be made into three forms: cyclic ECA (ECACYC), LPS-linked ECA (ECALPS), and ECAPG (see below). ECALPS synthesis requires the O-antigen ligase, WaaL, for synthesis, while ECACYC synthesis requires WzzE. The steps and genes required for the formation of ECAPG from polymerized ECA on Und-PP and for its subsequent surface exposure are unknown. (B) Structure of ECAPG glycolipid. The ECA carbohydrate chain is linked to diacylglycerol-phosphate (DAG-P) through a phosphodiester bond. Thus, ECA is linked to the phospholipid backbone in place of a headgroup. (C) E. coli phospholipid synthesis begins with the addition of acyl chains to glycerol-3-phosphate by PlsB and PlsC, forming phosphatidic acid (PA). Then, CdsA activates PA with CDP for form CDP-DAG, the precursor for all phospholipid synthesis. From this point, PgsA and PgpA, PgpB, or PgpC synthesize phosphatidylglycerol (PG), while PssA and Psd synthesize phosphatidylethanolamine (PE). Cardiolipin (CL) is synthesized by ClsA or ClsB from two PG molecules, while ClsC synthesizes CL from one PG and one PE molecule. PS, phosphatidylserine; PG-P, phosphatidylglycerol phosphate.
Given their differences in structure, localization, and phenotypes, it is likely that each form of ECA plays distinct roles in the cells. However, lack of understanding of the steps that differentiate the biosynthesis of the three forms of ECA impedes experimental approaches to investigate their function. ECA is synthesized in a Wzy pathway analogous to that of many O antigens (Fig. 1A) (33). ECA trisaccharide units are first assembled on undecaprenyl-pyrophosphate (Und-PP), an isoprenoid carrier, and then flipped to the outer leaflet of the inner membrane by WzxE (4, 34–36). ECA units are polymerized by WzyE with the chain length of the final ECA unit controlled by WzzE (37, 38). Once a polymerized ECA precursor is formed on Und-PP, it can be made into ECAPG, ECALPS, or ECACYC. Formation of ECACYC requires the chain length regulator, WzzE (24), while formation of ECALPS requires the O-antigen ligase WaaL (18). However, the details of the reaction forming ECAPG and leading to ECAPG surface exposure are completely unknown.
Here, we set out to identify, in E. coli K-12, the lipid donor(s) that provides the “phospholipid base” to ECAPG. ECAPG consists of the ECA carbohydrate moiety attached to diacylglycerol-phosphate (DAG-P; phosphatidic acid) through a phosphodiester bond (Fig. 1B) (20, 25). In essence, ECA is a large phospholipid headgroup. It seems biochemically likely that ECA is removed from the isoprenoid carrier freeing Und-PP, as occurs when O antigen and other forms of ECA are synthesized, and transferred to a specific subset of phospholipids or phospholipid precursors releasing the headgroup in favor of ECA (Fig. 1A).
In actively growing E. coli, the cell’s phospholipid composition is 75% phosphatidylethanolamine (PE), 20% phosphatidylglycerol (PG), and 5% cardiolipin (CL) with the amount of CL increasing in stationary phase at the expense of PG (reviewed in reference 39). The distribution of phospholipids in the IM is asymmetric with higher PE levels in the inner leaflet than the outer leaflet (40). Phospholipid synthesis begins with the sequential addition of fatty acids to glycerol-3-P by PlsB and PlsC to form phosphatidic acid (PA) (Fig. 1C) (41). Subsequently, CdsA activates PA through addition of CMP to form CDP-DAG, the precursor for phospholipid biosynthesis (42). From this point, the pathway splits between synthesis of PG and PE. PgsA exchanges CMP for glycerol-3-phosphate to form phosphatidylglycerol phosphate (PG-P) (43). Then, PG-P is dephosphorylated by PgpA, PgpB, or PgpC to form PG (44–46). While PgpA and PgpC are specific to PG-P dephosphorylation (45, 46), PgpB can also dephosphorylate DAG-PP, PA, lyso-PA, and Und-PP (44, 47). For PE synthesis, PssA (Pss) first synthesizes phosphatidylserine (PS) from CDP-DAG (48). Then, Psd decarboxylates PS to form PE (49). E. coli has three CL synthase ClsA, ClsB, and ClsC (50–53). ClsA and ClsB form CL from two molecules of PG (50, 54), while ClsC synthesizes CL from one PG molecule and one PE molecule (50). ClsA is mainly responsible for CL synthesis in exponential phase, while all three synthases contribute to CL synthesis in stationary phase.
We examined the effects of alterations in expression of phospholipid biosynthesis on levels of ECAPG. Our overexpression data suggest that increasing PG synthesis increases production of ECAPG. We determined that the donor for ECAPG synthesis is PG since ECAPG is still synthesized in the absence of CL or PE and depletion of PG leads to inhibited cell growth even in a strain where PG is normally not essential, likely due to the accumulation of ECA precursor on Und-PP inhibiting peptidoglycan biosynthesis. These data are the first to characterize the reaction(s) forming ECAPG and suggest a common approach to use of PG as a lipid donor.
RESULTS
Genetic alterations in phospholipid synthesis do not affect ECALPS levels.
The kinetics of biochemical reactions, and so the amount of product produced, often depend on the amounts of available substrates for the reaction. For ECAPG to be formed, at least two substrates are necessary: the polymerized ECA precursor on Und-PP and the donor phospholipid or phospholipid precursor. Thus, we hypothesized that changing the availability of the donor lipid would alter the kinetics of the reaction(s) producing ECAPG and so the amount of ECAPG produced. However, changes in phospholipid composition can alter the folding and topology of membrane proteins (reviewed in reference 55). Since the ECA biosynthesis pathway contains many membrane proteins (4), we sought to determine whether alteration of phospholipid composition would affect ECA synthesis in a manner unrelated to the ECAPG lipid donor. Therefore, we tested the effect of overexpressing genes in phospholipid biosynthesis on levels of ECALPS to identify any off-target effects of phospholipid level alteration on this form of ECA. Using our previously described WGA-staining protocol (56), we found that overexpression of genes in phospholipid biosynthesis caused only minor decreases in the levels of ECALPS (Fig. 2). This result is consistent with our previous observation that ECALPS levels are largely dependent on the availability of WaaL (56) and confirms that ECA can be successfully synthesized with our tested alterations in phospholipid biosynthesis.
FIG 2.

Alterations in phospholipid synthesis cause only minor changes in ECALPS levels. The levels of ECALPS were assayed by WGA staining in strains overexpressing the indicated genes in phospholipid biosynthesis. The data are displayed as fold values relative to the vector control. Only minor decreases and no significant increases in ECALPS levels were observed. The data are means of three biological replicates ± the standard errors of the mean (SEM). *, P < 0.05 (by paired t test); **, P < 0.005 (by paired t test).
The ECAPG lipid donor is not PA.
We investigated the effect of phospholipid gene overexpression on the accumulation of ECAPG and on activity of the Pwec promoter region responsible for expression of the wec operon that contains the genes necessary for synthesis of the polymerized ECA precursor. We reasoned overexpression of genes encoding enzymes functioning upstream of synthesis of the lipid donor in the phospholipid biosynthesis pathway would increase the amount of the lipid donor and so the amount of ECAPG. Conversely, overexpression of genes whose products lead to consumption of the lipid donor or which are on a different branch of the pathway would decrease the amount of the donor and so the amount of ECAPG. Changes in Pwec activity suggest changes to ECA precursor levels that may be occurring, in addition to any changes in lipid donor levels.
The lipid to which ECA is attached in ECAPG is DAG-P and so is most similar to PA. Therefore, we assayed the effect on ECAPG levels of overexpression under an IPTG inducible promoter of a His-tagged cdsA construct, whose product converts PA into CDP-DAG. Accumulation of CdsA was confirmed by immunoblot (see Fig. S1 in the supplemental material, lane 1 versus lanes 2 to 5). To allow specific detection of ECAPG, we tested ECAPG levels in a ΔwzzE ΔwaaL strain that only produces ECAPG and not the other two forms of ECA. At all tested IPTG (isopropyl-β-d-thiogalactopyranoside) concentrations, we observed a large increase in the level of ECAPG (Fig. 3A, lanes 2 versus lanes 3 to 6). In contrast, there were no significant increases in Pwec activity when cdsA was overexpressed (Fig. 3B). These data indicate that the lipid donor for ECAPG synthesis is CDP-DAG and/or a phospholipid but is not PA or an earlier biosynthetic intermediate.
FIG 3.
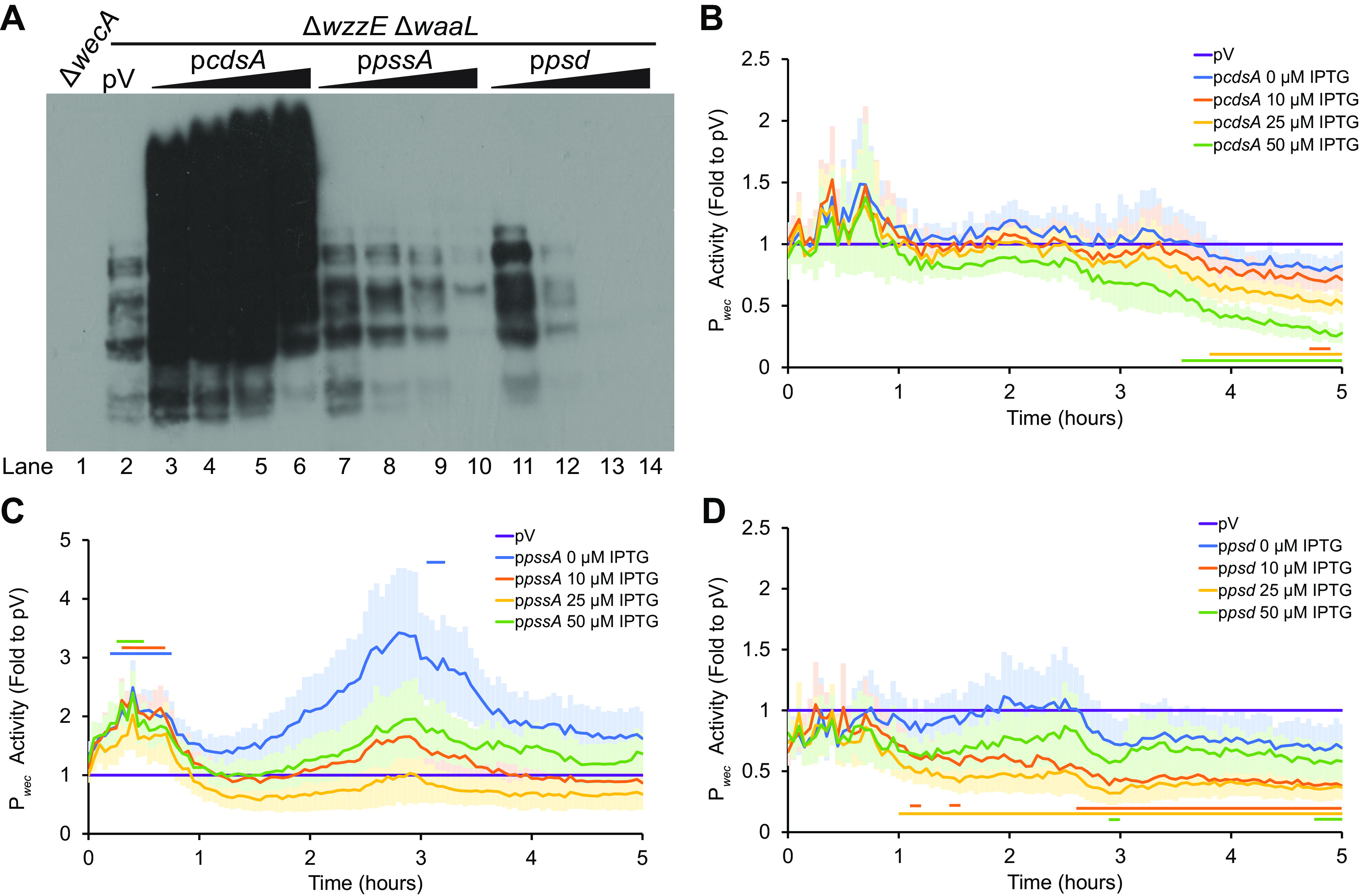
A phospholipid is the substrate for ECAPG synthesis. (A) The levels of ECAPG were assayed by immunoblotting in a strain that produces only ECAPG and not the other two forms of ECA (ΔwzzE ΔwaaL). The vector control (pV) treated with 50 μM IPTG is compared to strains overexpressing the indicated genes in phospholipid biosynthesis with increasing concentrations of IPTG (0, 10, 25, and 50 μM). A strain with a deletion of wecA, the first gene in the ECA biosynthesis pathway, serves as a negative control. Overexpression of cdsA causes large increases in the amounts of ECAPG, while overexpression of pssA and psd decrease ECAPG levels. Image is representative of three independent experiments. (B to D) Strains overexpressing genes in phospholipid biosynthesis and carrying a luciferase reporter for wec operon promoter (Pwec) activity were assayed for luminescence and OD600. The data are shown as fold value of the relative luminescence to the vector control and are means from six biological replicates ± the SEM. The empty vector (pV) sample contained the empty vector for phospholipid gene overexpression and the Pwec reporter and was treated with 50 μM IPTG. Horizontal bars: P < 0.05 by t test consistently for three or more time points. (B) Strains overexpressing cdsA have very similar Pwec activity to that of the empty vector control, with a decrease later in growth. (C) Strains overexpressing pssA have some increase in Pwec activity. (D) Overexpression of psd decreases Pwec activity.
PE synthesis is off the pathway of the lipid donor.
To determine whether PE could serve as the donor for ECAPG synthesis, we then overexpressed the genes in PE synthesis pathway, pssA and psd. We confirmed accumulation of PssA and Psd by immunoblot (see Fig. S1, lane 1 versus lanes 6 to 9 and 10 to 13, respectively). Overexpression of these genes caused a decrease in the accumulation of ECAPG in a manner that correlates with the concentration of IPTG (Fig. 3A, lane 2 versus lanes 7 to 10 and 11 to 14). Overexpression of pssA caused some significant increases in Pwec activity at early time points after induction but no decreases in activity (Fig. 3C). However, psd overexpression caused decreased Pwec activity at higher IPTG concentrations (Fig. 3D), confounding interpretations of decreased ECAPG accumulation observed with psd overexpression. Nevertheless, the decreased ECAPG levels without changes in Pwec activity with pssA overexpression demonstrate the pathway for synthesis of PE is not involved in synthesis of the ECAPG lipid donor.
Overexpression suggests the ECAPG lipid donor is part of the PG/CL synthesis pathway.
Since PE synthesis appeared off pathway for the synthesis of the ECAPG lipid donor, we then examined the effects of gene overexpression in the PG synthesis pathway. As PG is necessary for synthesis of CL, overexpression of these genes would also be expected to increase the synthesis of both PG and CL (50, 54). We confirmed the accumulation of proteins from the overexpression constructs by immunoblot (see Fig. S2). Overexpression of pgsA, the first dedicated gene in PG synthesis, caused an increase in ECAPG levels at the highest IPTG concentration we tested (Fig. 4A, lane 2 versus lanes 3 to 6). However, pgsA overexpression caused a consistent increase in Pwec activity (Fig. 4B), making these data difficult to interpret. Overexpression of pgpA led to higher levels of ECAPG at all IPTG concentrations assayed (Fig. 4A, lane 2 versus lanes 7 to 10), despite very little significant change in Pwec activity (Fig. 4C). This result suggests that the lipid donor for ECAPG synthesis may be downstream of PgpA in the biosynthesis pathway.
FIG 4.
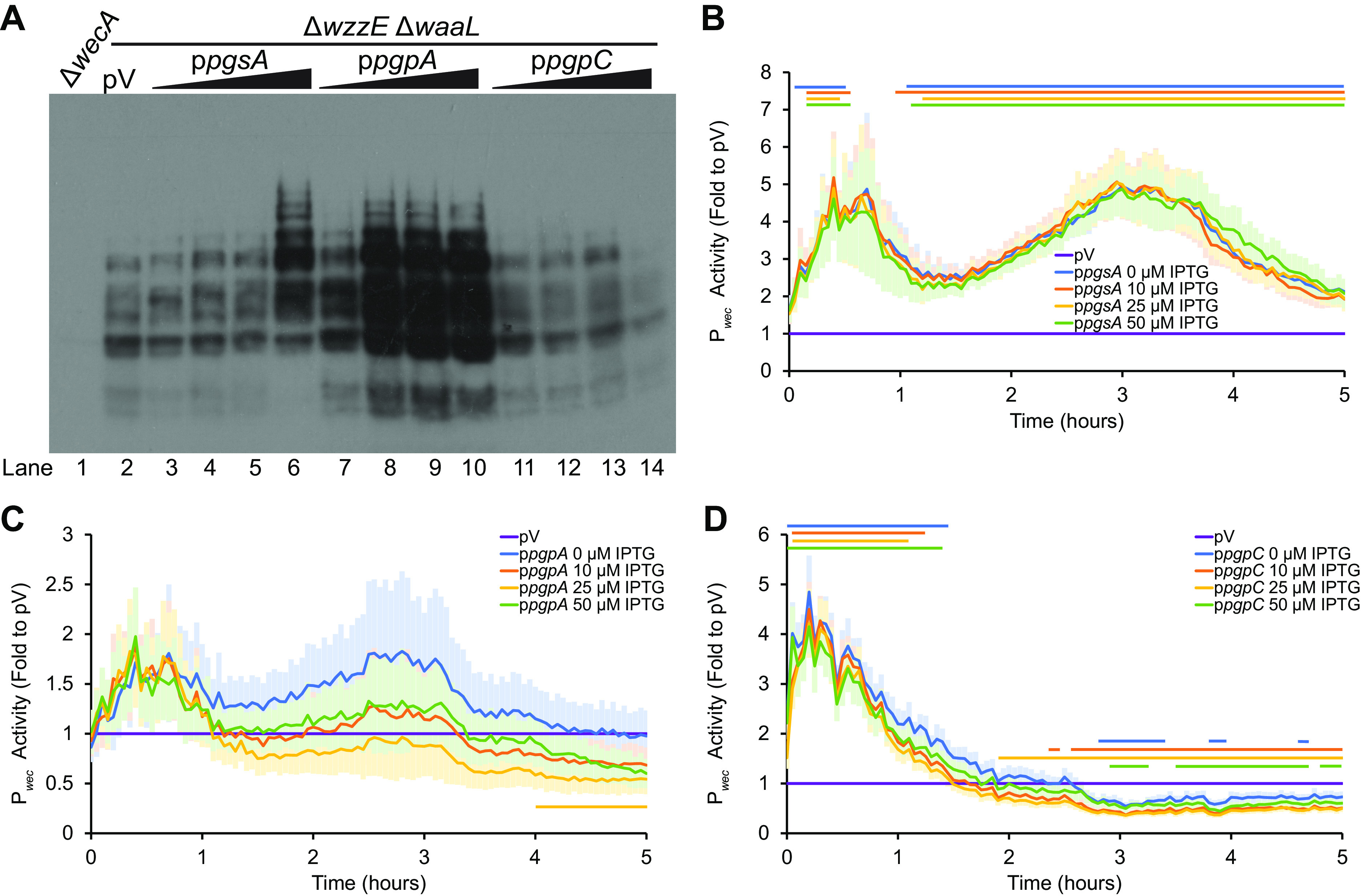
The substrate for ECAPG biosynthesis is in the PG/CL biosynthesis pathway. (A) ECAPG levels were assayed by immunoblot analysis in strains overexpressing the indicated genes in phospholipid biosynthesis. The ΔwzzE ΔwaaL strain background allows the production of ECAPG without the other forms of ECA. For phospholipid synthesis gene overexpression, cultures were treated with 0, 10, 25, and 50 μM IPTG. The vector control (pV) was treated with 50 μM IPTG. A ΔwecA strain serves as a negative control. The image is representative of three independent experiments. Overexpression of both pgsA and pgpA increase accumulation of ECAPG. Overexpression of pgpC causes a small decrease in ECAPG levels. (B to D) Pwec activity was assayed in strains overexpressing genes in phospholipid biosynthesis as in Fig. 3. The means ± the SEM for the relative luminescence as a fold value to the vector control are shown for six biological replicates. pV, empty vector sample carrying the Pwec reporter and the empty vector for the phospholipid synthesis gene overexpression and treated with 50 μM IPTG. Horizontal bars: P < 0.05 by t test consistently for three or more time points. (B) Overexpressing pgsA consistently increased Pwec activity. (C) Overexpression of pgpA did not change Pwec activity at most time points and IPTG concentrations. (D) Overexpressing pgpC causes early increases and later decreases in Pwec activity.
Interestingly, when we overexpressed pgpC, we observed the opposite phenotype, a decrease in ECAPG levels at the highest IPTG concentration (Fig. 4A, lane 2 versus lanes 11 to 14). There are two likely explanations for the contradictory results between pgpA and pgpC overexpression. The first is that the decrease in ECAPG levels with pgpC is due to the lower Pwec activity observed several hours after induction of pgpC overexpression (Fig. 4D). The second is due to the relative activities of PgpA, PgpB, and PgpC. Of the three enzymes, PgpA has the highest activity and the most effect on the levels of PG-P and PG (46). Thus, it is possible that overexpression of pgpC actually decreases flux through the PG/CL synthesis pathway. We did not overexpress pgpB, as PgpB is active on several different substrates including Und-PP (47). Overall, these results suggest that PG or CL may be the donor for ECAPG biosynthesis.
CL synthesis is downstream from the ECAPG lipid donor.
There are three CL synthases: ClsA, ClsB, and ClsC (50–53). When we overexpressed the gene for the CL synthase active in exponentially growing cells, clsA, we observed very little change in either ECAPG levels (Fig. 5A, lane 2 versus lanes 3 to 6) or in Pwec activity (Fig. 5B). This may be due to feedback inhibition of CL on ClsA activity (57), due to the low levels of ClsA from overexpression we observed compared to ClsB and ClsC (see Fig. S3), or due to inhibition of activity from His-tagging. However, when we overexpressed either clsB or clsC, which are normally active in stationary phase when CL levels increase, we observed a decrease in ECAPG levels that was more severe at higher IPTG concentrations (Fig. 5A, lane 2 versus lanes 7 to 10 and 11 to 14). Overexpression of clsB did not cause significant changes in Pwec activity (Fig. 5C), while overexpression of clsC caused some decreases in Pwec activity at higher IPTG concentrations (Fig. 5D). These data suggest that CL is downstream of the lipid donor for ECAPG synthesis.
FIG 5.
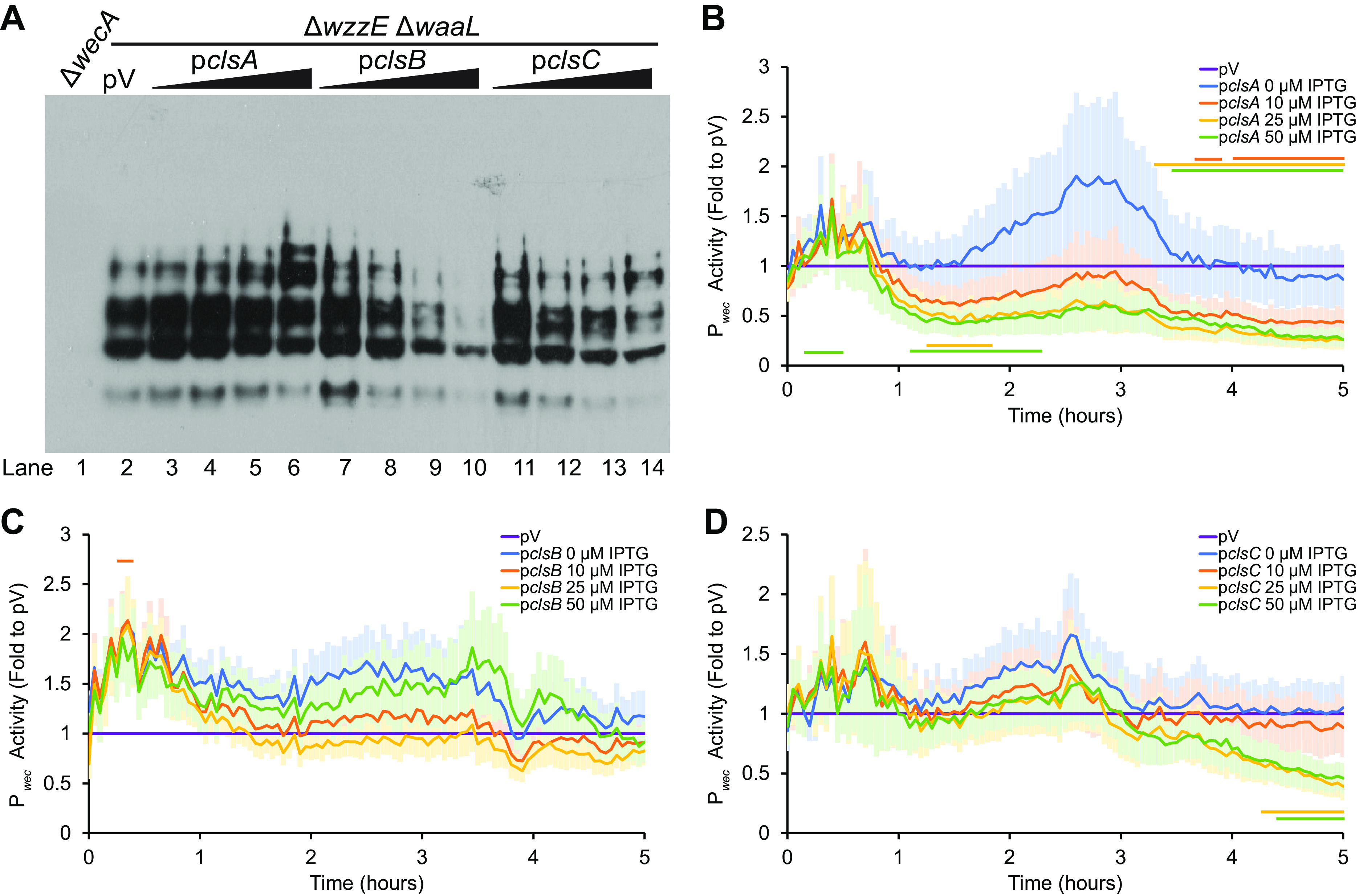
CL synthase overexpression decreases ECAPG levels. (A) ECAPG levels were assayed by immunoblot in strains overexpressing genes in CL biosynthesis. The ΔwzzE ΔwaaL strain produces only ECAPG and not the other two forms of ECA. The vector control (pV) was treated with 50 μM IPTG, while overexpression strains were treated with 0, 10, 25, and 50 μM IPTG. The ΔwecA strain serves as a negative control. The image is representative of three independent experiments. Overexpression of both clsB and clsC decreases ECAPG levels, while the levels of ECAPG are constant with clsA overexpression. (B to D) The activity of Pwec was assayed as in Fig. 3. The data are shown as the fold of the relative luminescence to the vector control (pV) and are means of 5 to 6 biological replicates ± the SEM. pV control carried the Pwec reporter and an empty vector for overexpression and was treated with 50 μM IPTG. Horizontal bars: P < 0.05 by t test consistently for three or more time points. (B) Overexpression of clsA causes some significant increases and decreases in Pwec activity at higher IPTG concentrations. (C) Overexpression of clsB does not change Pwec activity. (D) Overexpression of clsC causes some decrease in Pwec activity at higher IPTG concentrations and later time points.
PG is the substrate for ECAPG biosynthesis.
Taken as a whole, our overexpression data point to PG as the lipid donor for ECAPG synthesis: Overexpression of genes expected to increase levels of PE and CL at the expense of PG decreases levels of ECAPG, while overexpression of genes expected to increase levels of PG increases levels of ECAPG. Therefore, we decided to examine the growth of strains where genes in the PG/CL biosynthesis pathway could be depleted. ECA and peptidoglycan are both synthesized on Und-P (58, 59), and so interruption of ECA biosynthesis can lead to the buildup of Und-PP-linked intermediates, disrupting peptidoglycan biosynthesis (24, 56, 60–64). The severity of the peptidoglycan defect depends on the stage in biosynthesis interrupted. Defects in ECA biosynthesis leading to the accumulation of Und-PP-GlcNAc-ManNAcA (Lipid IIECA) produce cell shape defects, envelope permeability, and envelope stress response activation (60, 61, 63). Disruption of the flippases capable of flipping Und-PP-GlcNAc-ManNAcA-Fuc4NAc (Lipid IIIECA) ECA across the IM or of the ECA polymerase, wzyE, is lethal in E. coli K-12 (24). We have speculated that, in a strain that makes only ECAPG, disruption of the next step in ECAPG biosynthesis would be lethal as well (56). In fact, we have shown that dysregulation of ECAPG biosynthesis in a strain making only ECAPG is lethal (56). Therefore, we hypothesized that loss of the lipid donor for ECAPG biosynthesis would cause peptidoglycan synthesis defects, and so growth phenotypes, specifically when ECAPG is made without the other forms of ECA.
Our depletion strategy involved cloning phospholipid biosynthesis genes under the control of the PBAD promoter, which is arabinose inducible and subject to catabolite repression (65). We then deleted the chromosomal copies of the affected genes while maintaining expression from the plasmid-borne copy in either the wild-type background, the ΔwzzE ΔwaaL strain that produces ECAPG without the other two forms of ECA, or an isogenic background without ECA (ΔwecA-wzzE ΔwaaL) to control of effects of loss of ECA envelope strength. Treating cells with arabinose or glucose in the presence of the empty plasmid produced similar growth curves in the wild-type and ΔwzzE ΔwaaL strains (see Fig. S4A).
pgsA is essential due to the necessity for PG for lipoprotein processing (66–69). The essentiality of pgsA can be circumvented by deletion of lpp (66). Lpp (Braun’s lipoprotein) is highly abundant and IM localization is lethal due to cross-linking of the peptidoglycan to the IM (70). Δlpp ΔpgsA strains are still temperature sensitive, however (66). This temperature sensitivity can be relieved by inactivating the Rcs stress response (67), which is overactivated by localization of RcsF in the inner membrane (71). Therefore, we built PgsA depletion strains in ΔrcsF Δlpp and ΔrcsF Δlpp ΔwzzE ΔwaaL backgrounds to test the effect of PgsA depletion in conditions where PG is normally not essential. In the ΔrcsF Δlpp background, the PgsA depletion strain grew equally with arabinose or glucose, confirming PG is not essential in this strain (Fig. 6A). However, the ΔrcsF Δlpp ΔwecA-wzzE ΔwaaL background strain ceased growth when depleted, suggesting that the presence of ECA is important for the integrity of the cell envelope in the absence of PG. In contrast, the ΔrcsF Δlpp ΔwzzE ΔwaaL background strain grew to near stationary phase and then abruptly lysed, demonstrating a different phenotype than either the wild type and no ECA strains.
FIG 6.
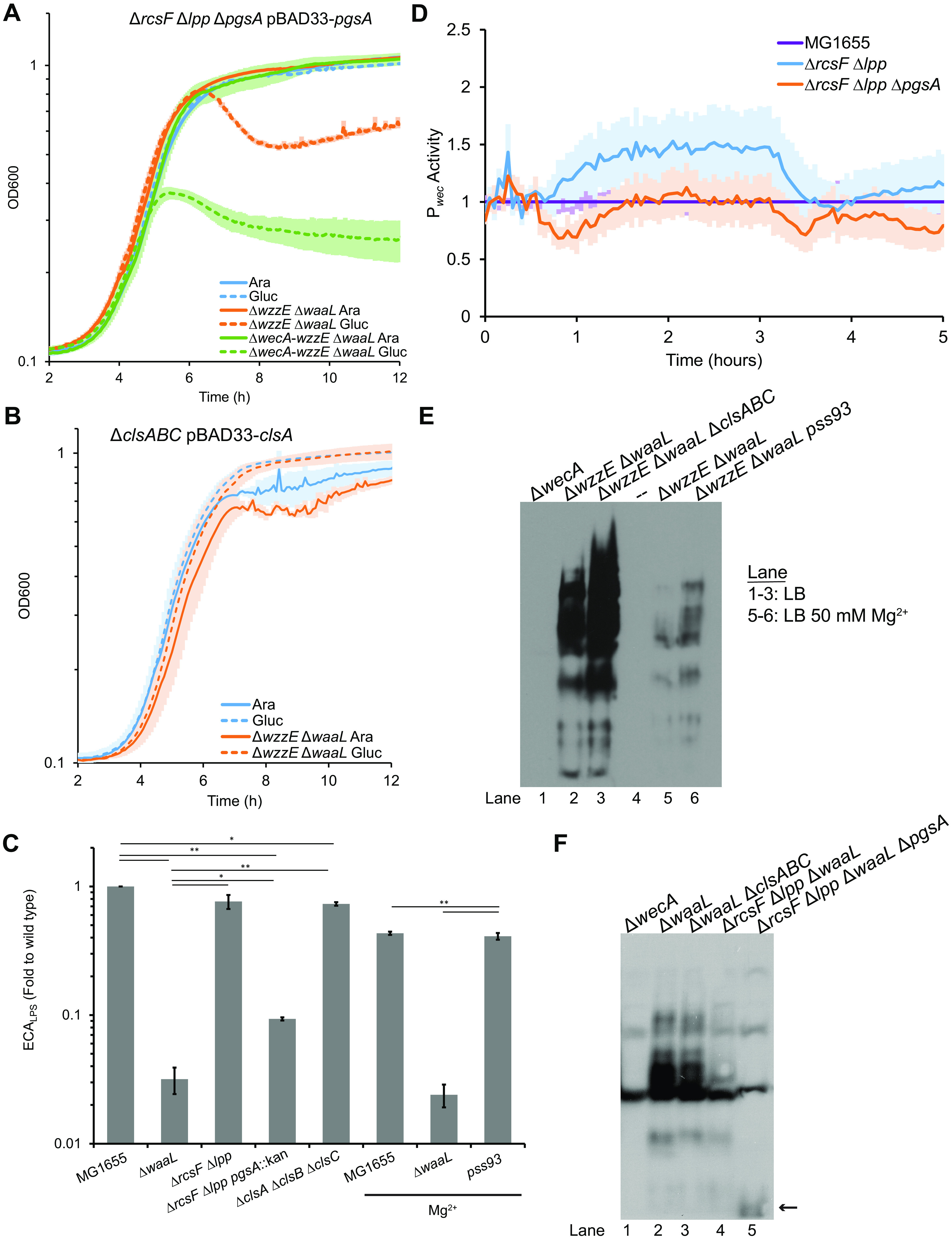
Phosphatidylglycerol is the substrate for the synthesis of ECAPG. (A) PG is not essential in strains where pressure on lipoprotein maturation is relieved (ΔrcsF Δlpp). Growth curves were performed with PgsA depletion strains in this background alone (blue), with wzzE and waaL deletions (orange), or an isogenic strain with a wecA deletion (green) to investigate growth phenotypes caused by loss of ECA and disruptions to peptidoglycan biosynthesis caused by accumulation of ECA precursor. Cultures were diluted 1:10,000 and treated with arabinose to induce expression from the PBAD promoter (solid lines) or glucose to repress expression (hashed lines). Depletion of PgsA in the ΔrcsF Δlpp background allowed full growth. Cells without ECA (ΔwecA-wzzE ΔwaaL) stopped growing when PG was depleted and then showed a slow decrease in OD600. However, cells making only ECAPG (ΔwzzE ΔwaaL) grew to stationary phase and then quickly lysed. The data are means of four biological replicates ± the SEM. (B) CL is not essential. Cultures for growth curves were grown as in panel A. Depletion of ClsA in strains with deletions of clsA, clsB, and clsC did not cause lysis either alone or when combined with ΔwzzE ΔwaaL. The data are means of three biological replicates ± the SEM. (C) ECALPS levels were assayed by WGA staining of strains with deletions in the indicated genes. Where indicated, cultures were grown with 50 mM Mg2+ to allow survival of strains lacking PE (pss93). Strains lacking CL and PE had small but significant decreases in ECALPS levels. The strain lacking PG had a large decrease in ECALPS; however, ECALPS levels remained detectable. The data are the mean of three biological replicates ± the SEM. *, P < 0.05 (by paired t test); **, P < 0.005 (by paired t test). (D) The activity of Pwec was assayed as in Fig. 3. The data are shown as the fold of the relative luminescence to the wild type and are means of six biological replicates ± the SEM. No significant changes determined by t test were observed for contiguous data points. (E) ECAPG levels in strains without CL (ΔclsABC) or without PE (pss93) were assayed in the ΔwzzE ΔwaaL (ECAPG only) strain background. Cultures for lanes 5 and 6 were grown with 50 mM Mg2+. The ΔwecA strain serves as a negative control. Both strains lacking CL and strains lacking PE retained production of ECAPG. The images are representative of two independent experiments. (F) Since depletion of PG causes lysis in an ECAPG only background, the effect of the ΔpgsA mutation was assayed in a ΔwaaL background where only ECAPG can be observed by immunoblotting. Immunoblot analysis was performed to assay ECAPG levels in strains without CL (ΔclsABC) or without PG (ΔrcsF Δlpp ΔpgsA). The ΔwecA strain serves as a negative control, indicating nonspecific bands. Deletion of waaL in the presence of ECACYC causes inhibition of ECAPG synthesis (56), leading to lower ECA levels overall. ECAPG is observed in the strain without CL. However, only a low-abundance, low-molecular-weight band (arrow) is observed in the strain without PG. The images are representative of three independent experiments.
Depletion of the phosphatidylglycerophosphatases (PgpA, PgpB, and PgpC) (see Fig. S4B) or of PgsA with Lpp and RcsF intact (see Fig. S4C) also lead to increased lysis in the ECAPG only (ΔwzzE ΔwaaL) background. These data confirm a second growth defect in this strain beyond that observed in the wild-type background, likely due to the buildup ECA intermediates on Und-PP. Depletion of either PgpABC or PgsA will reduce the level of both PG and CL. Thus, to confirm that the results we observed with depletion were the result of loss of PG and not loss of CL, we assayed the growth of a ClsA depletion strain in a ΔclsA ΔclsB ΔclsC background. CL is not essential and this strain grew fully when depleted in both the wild type and the ΔwzzE ΔwaaL strain (Fig. 6B). These data confirm that it is loss of PG not CL that causes increased lysis with PgsA and PgpABC depletion.
Depletion of PG in a background where it is normally nonessential leads to lysis in an ECAPG only background but not a background wild type for ECA, suggesting accumulation of ECA precursors on an isoprenoid carrier. As accumulation of Lipid IIIECA due to ΔwzyE or deletion of all the flippases capable of flipping ECA across the IM is lethal in E. coli K-12 (24), we can exclude complete inhibition of WzyE and/or flippase activity as the cause of the lysis since cells wild type for ECA synthesis survive depletion. However, it is possible that partial inhibition, due to improper membrane protein folding, plays a role in the lysis observed in the ECAPG only strain, in addition to specific inhibition of ECAPG synthesis.
To investigate whether the shared steps in ECA synthesis are intact in strains without specific phospholipids, we assayed ECALPS levels in strains lacking either PG, CL, or PE. Preventing synthesis of CL (ΔclsA ΔclsB ΔclsC) or PE (pss93) caused only minimal decreases in ECALPS levels (Fig. 6C); however, preventing PG synthesis (ΔrcsF Δlpp ΔpgsA) caused a large decrease in ECALPS levels. ECALPS levels were, nevertheless, detectable. Pwec activity was not changed in this strain, suggesting that the decrease is posttranscriptional (Fig. 6D) and may be due to regulation of synthesis or membrane protein misfolding in the absence of PG leading to decreased efficiency in ECA or LPS synthesis.
To finalize our conclusion that PG was the lipid donor for the synthesis of ECAPG, we attempted to build deletion strains for the PG, PE, and CL synthesis genes in the ΔwzzE ΔwaaL background. Although PE makes up 75% of the membrane in wild-type cells (39), strains with disruption of pssA are viable as long as they are maintained with 20 to 50 mM Ca2+ or Mg2+ (72). We were able to build strains with a pssA disruption allele (pss93) and with a ΔclsA ΔclsB ΔclsC background. When we assayed these strains, we found that both strains had increased ECAPG levels compared to the ΔwzzE ΔwaaL strain (Fig. 6E, lane 2 versus lane 3 and lane 5 versus lane 6). We also observed a large decrease in ECAPG levels with Mg2+ treatment. Consistent with the growth defects we observed in depletion strains (Fig. 6A), we were not successful in building a ΔrcsF Δlpp ΔwzzE ΔwaaL ΔpgsA background and reviving it from frozen stocks. Therefore, we investigated the effect of loss of PG and CL in a ΔwaaL background. ECACYC cannot be detected through immunoblot and so ECAPG levels can be assayed in this strain. However, posttranscriptional regulation of ECAPG synthesis occurs in with ΔwaaL that decreases ECAPG synthesis (56). ECAPG was observed in the strain lacking CL and the ΔrcsF Δlpp background strain (Fig. 6F, lanes 2 to 4). However, in the strain without PG, only one very low molecular weight band was observed other than those present in the ΔwecA strain (Fig. 6F, lane 1 versus lane 5). This band may represent an Und-PP-linked ECA precursor or a degradation product thereof. Together, our data demonstrate that PG is the lipid donor for the synthesis of ECAPG.
DISCUSSION
In this work, we investigated the identity of the lipid donor for synthesis of ECAPG. Our data demonstrate through several lines of evidence that this donor is PG and that other phospholipids cannot efficiently substitute when PG is lost. (i) Changes in flux through phospholipid biosynthesis expected to increase PG production increase ECAPG levels, while changes expected to decrease PG production decrease ECAPG levels. (ii) Depletion of PG levels when ECA precursors cannot be made into other forms of ECA causes cell lysis, even where PG is otherwise nonessential. (iii) ECAPG is produced in the absence of PE or CL and is not produced in the absence of PG. These results also suggest preventing transfer of ECA from Und-PP to the lipid donor may cause sufficient peptidoglycan stress to inhibit growth despite the Und-PP released when ECA subunits are polymerized.
In our previous investigations (56), we used this peptidoglycan stress phenotype to identify a system that regulates the production of ECAPG. ElyC and ECACYC work together to posttranscriptionally inhibit the production of ECA during normal growth conditions (56). We envision this system as a feedback pathway that regulates ECAPG production based on levels of ECACYC. We can now add the identity of the lipid donor for ECAPG synthesis to our model of ECAPG synthesis. In our model, polymerized ECA is removed from Und-PP and transferred to the DAG-P portion of a PG molecule, releasing glycerol into the periplasm and Und-PP into the outer leaflet of the IM. The protein(s) responsible for this reaction would be regulated by ElyC in a manner controlled by ECACYC. This model provides a framework for further investigations of the synthesis of ECAPG including the following: identification of the gene(s) involved in synthesizing ECAPG from PG and the ECA precursor, determining whether the synthesizing enzyme(s) have specific preferences for PG fatty acid composition, the details of the reaction forming ECAPG and of the mechanism of regulation by ElyC and ECACYC, and the pathway leading to ECAPG surface exposure.
In addition to regulation of ECAPG production by ElyC and ECACYC (56), our results have revealed other mechanisms through which ECAPG production can be regulated, specifically in strains lacking ECACYC and so full activity of the ElyC-ECACYC pathway. We relied in our overexpression experiments on the ability of ECAPG production to be altered by lipid donor availability and found the changes in ECAPG production to be robust, with both quite large increases and decreases in ECAPG levels (for example see Fig. 3A, lane 6 versus lane 10). These data are especially interesting given that the ratio of PG to CL varies based on growth phase with the amount of CL increasing in stationary phase at the expense of PG levels (39, 73). Our data suggest that this change may have a direct effect on the synthesis of ECAPG.
Beyond regulation by substrate availability, we observed several changes in phospholipid-synthesis gene expression that led to changes in the activity of the promoter region of the wec operon that encodes the genes necessary to synthesize the ECA precursor. The largest of these changes was the 4- to 5-fold increase in Pwec activity we observed when we overexpressed pgsA (Fig. 4B). These data demonstrate an interplay between phospholipid synthesis and ECA synthesis and suggest that there may be feedback from membrane conditions that affects ECA synthesis. We also observed a large decrease in ECAPG levels when cells were grown with a high concentration of Mg2+ (Fig. 6E). This decrease was present in both the strain with normal phospholipid composition and the strain lacking PE. It is possible that this decrease could be due to changes in Mg2+ sensing transcriptional regulation pathways, due to changes in OM order due to increase bridging between LPS molecules, or to changes in the activity of enzymes in the ECA synthesis pathway. Experiments investigating these phenotypes are ongoing in our lab.
As mentioned above, our results demonstrate, while cells producing only ECAPG and not the other forms of ECA lyse in the absence of PG, cells without ECA deplete earlier and do not continue to grow even when PG is nonessential (Fig. 6A). The lack of growth in the strain without ECA and the longer growth before lysis in the ECAPG-only strain are intriguing. Both of these strains lack ECACYC, which is important for maintaining the OM permeability barrier (26); however, it may be that ECAPG plays an important role in maintaining envelope integrity and that loss of ECAPG before depletion is the reason that the cells without ECA cease growth earlier. Alternatively, it may be that accumulation of Und-PP-linked ECA precursors lead to further growth before lysis either through envelope stress response activation (60, 61, 63) or through a direct role of ECA precursors in envelope integrity (74). These questions warrant future investigation.
It is interesting to compare the synthesis of ECAPG to another glycolipid with a very similar structure, membrane protein intergrase (MPIase) (reviewed in reference 75). MPIase consists of the same carbohydrate unit as ECAPG attached to DAG through a pyrophosphate diester bond (76). This molecule is essential and has been implicated in insertion of proteins into the IM in a Sec-independent manner and in Sec-dependent protein translocation (75–81). Unlike ECAPG, the MPIase sugar subunit is built on DAG-PP with the first step being attachment of GlcNAc-P to DAG-P, a reaction catalyzed by CdsA and YnbB (77). The lipid donor for this reaction is CDP-DAG. The synthesis of MPIase has previously been shown to be independent of ECA synthesis (82), and our results confirm that these molecules use distinct lipid donors. The ECA genes necessary for biosynthesis of the ECA sugars and the formation of the ECA chain has also been shown to be dispensable for MPIase formation (82), although later results suggest they may contribute to some extent (74). Exploring the differences and similarities between these biosynthetic pathways will provide interesting insights into both pathways.
Our identification of PG as the lipid donor for ECAPG biosynthesis provides an interesting parallel with lipoprotein synthesis: Lgt uses PG as the donor to transfer DAG to nascent lipoproteins (69). Despite this, PE is the most abundant phospholipid in the E. coli membrane. It is possible that these pathways use PG as the lipid donor due to the asymmetry of the IM, which contains more PE in the inner leaflet than the outer leaflet (40). It is also possible that it is advantageous to the cell to use a less abundant membrane constituent for lipid modification as it can be more easily regulated, such as by decreasing PG levels in favor of CL during stationary phase (39, 73). Finally, the use of PG as a donor may be due to relative ease of recycling the glycerol headgroup compared to recycling of ethanolamine. These are intriguing questions for future study.
MATERIALS AND METHODS
Strains and growth conditions.
The strains used in these experiments are listed in Table 1. Cultures were grown in LB Lennox at 37°C unless otherwise noted. Where necessary for plasmid maintenance, medium was supplemented with 20 mg/L chloramphenicol and/or 25 mg/L kanamycin. Where indicated, cultures were supplemented with 0.2% glucose, 0.2% arabinose, or 50 mM MgSO4. Unless otherwise noted, deletion alleles are from the Keio collection and were moved into our strains using P1vir transduction (83, 84). A deletion allele for pgsA was constructed using λ-Red recombineering and the primers listed in Table S1 (85). Kanamycin resistance cassettes were flipped out using the Flp recombinase-FRT system as has been described (85). Mikhail Bogdanov and William Dowhan (McGovern Medical School, University of Texas—Houston) provided us with the kind gift of a previously published pssA disruption allele, pss93::kan (72). Strains with the pss93::kan allele were constructed by transforming strains with pDD72GM (86), which carries a wild-type pssA allele, transducing in the pss93::kan allele, and then curing the temperature sensitive pDD72GM plasmid.
TABLE 1.
Strains used in this study
| Strain | Genotype | Source or reference | Plasmids and alleles (source reference) |
|---|---|---|---|
| MG1655 | K-12 F– λ– rph-1 | 87 | |
| AM334 | MG1655 ΔwecA | 26 | |
| AM366 | MG1655 ΔwaaL | 26 | |
| AM395 | MG1655 ΔwzzE ΔwaaL | 26 | |
| AM1357 | MG1655 ΔclsA ΔclsB ΔclsC | This study | |
| AM1453 | MG1655 ΔclsA ΔclsB ΔclsC ΔwaaL::Kanr | This study | |
| AM1358 | MG1655 ΔwzzE ΔwaaL ΔclsA ΔclsB ΔclsC | This study | |
| KM031 | MG1655 pss93::Kanr | This study | pssA93::Kanr allele (72) |
| KM032 | MG1655 ΔwzzE ΔwaaL pss93::Kanr | This study | |
| AM1340 | MG1655 ΔrcsF Δlpp | This study | |
| AM1421 | MG1655 ΔrcsF Δlpp ΔpgsA::Kanr | This study | |
| AM1352 | MG1655 ΔrcsF Δlpp ΔwaaL | This study | |
| AM1452 | MG1655 ΔrcsF Δlpp ΔwaaL ΔpgsA::Kanr | This study | |
| AM1138 | MG1655 pCA24N | 56 | Plasmid is ASKA collection empty vector (88); Cmr |
| AM1144 | MG1655 ΔwzzE ΔwaaL pCA24N | This study | Plasmid is ASKA collection empty vector (88); Cmr |
| AM1220 | MG1655 pCA24N-cdsA | This study | Plasmid (88) |
| AM1286 | MG1655 ΔwzzE ΔwaaL pCA24N-cdsA | This study | |
| AM1326 | MG1655 pCA24N-pssA | This study | Plasmid (88) |
| AM1327 | MG1655 ΔwzzE ΔwaaL pCA24N-pssA | This study | |
| AM1176 | MG1655 pCA24N-psd | This study | Plasmid (88) |
| AM1283 | MG1655 ΔwzzE ΔwaaL pCA24N-psd | This study | |
| AM1324 | MG1655 pCA24N-pgsA | This study | Plasmid (88) |
| AM1325 | MG1655 ΔwzzE ΔwaaL pCA24N-pgsA | This study | |
| AM1174 | MG1655 pCA24N-pgpA | This study | Plasmid (88) |
| AM1282 | MG1655 ΔwzzE ΔwaaL pCA24N-pgpA | This study | |
| AM1328 | MG1655 pCA24N-pgpC | This study | Plasmid (88) |
| AM1329 | MG1655 ΔwzzE ΔwaaL pCA24N-pgpC | This study | |
| AM1330 | MG1655 pCA24N-clsA | This study | Plasmid (88) |
| AM1331 | MG1655 ΔwzzE ΔwaaL pCA24N-clsA | This study | |
| AM1177 | MG1655 pCA24N-clsB | This study | Plasmid (88) |
| AM1284 | MG1655 ΔwzzE ΔwaaL pCA24N-clsB | This study | |
| AM1332 | MG1655 pCA24N-clsC | This study | Plasmid (88) |
| AM1333 | MG1655 ΔwzzE ΔwaaL pCA24N-clsC | This study | |
| KM014 | MG1655 ΔwzzE ΔwaaL pCA24N pJW15 | This study | pJW15 is an empty lux reporter (89); Kanr |
| KM015 | MG1655 ΔwzzE ΔwaaL pCA24N pJW15-Pwec | This study | pJW15-Pwec reporter plasmid (88) |
| KM011 | MG1655 ΔwzzE ΔwaaL pCA24N-cdsA pJW15-Pwec | This study | |
| KM008 | MG1655 ΔwzzE ΔwaaL pCA24N-pssA pJW15-Pwec | This study | |
| KM009 | MG1655 ΔwzzE ΔwaaL pCA24N-psd pJW15-Pwec | This study | |
| KM005 | MG1655 ΔwzzE ΔwaaL pCA24N-pgsA pJW15-Pwec | This study | |
| KM006 | MG1655 ΔwzzE ΔwaaL pCA24N-pgpA pJW15-Pwec | This study | |
| KM012 | MG1655 ΔwzzE ΔwaaL pCA24N-pgpC pJW15-Pwec | This study | |
| KM007 | MG1655 ΔwzzE ΔwaaL pCA24N-clsA pJW15-Pwec | This study | |
| KM013 | MG1655 ΔwzzE ΔwaaL pCA24N-clsB pJW15-Pwec | This study | |
| KM010 | MG1655 ΔwzzE ΔwaaL pCA24N-clsC pJW15-Pwec | This study | |
| AM1225 | MG1655 pJW15 | 56 | |
| AM1455 | MG1655 ΔrcsF Δlpp pJW15 | This study | |
| AM1456 | MG1655 ΔrcsF Δlpp ΔpgsA pJW15 | This study | |
| AM1162 | MG1655 pBAD33 | 56 | Plasmid (65); Cmr |
| AM1158 | MG1655 ΔwzzE ΔwaaL pBAD33 | 56 | |
| AM1425 | MG1655 ΔpgpB ΔpgpA pBAD33-pgpC ΔpgpC | This study | |
| AM1426 | MG1655 ΔwzzE ΔwaaL ΔpgpB ΔpgpA pBAD33-pgpC ΔpgpC | This study | |
| AM1407 | MG1655 pBAD33-pgsA ΔpgsA | This study | |
| AM1408 | MG1655 ΔwzzE ΔwaaL pBAD33-pgsA ΔpgsA | This study | |
| AM1409 | MG1655 ΔrcsF Δlpp pBAD33-pgsA ΔpgsA | This study | |
| AM1412 | MG1655 ΔrcsF Δlpp ΔwzzE ΔwaaL pBAD33-pgsA ΔpgsA | This study | |
| AM1459 | MG1655 ΔrcsF Δlpp ΔwaaL ΔwecA-wzzE pBAD33-pgsA ΔpgsA | This study | ΔwecA-wzzE allele (56) |
| AM1413 | MG1655 ΔclsA ΔclsB pBAD33-clsA ΔclsC | This study | |
| AM1414 | MG1655 ΔwzzE ΔwaaL ΔclsA ΔclsB pBAD33-clsA ΔclsC | This study |
IPTG inducible overexpression constructs were derived from the non-GFP-tagged version of the ASKA collection of E. coli gene overexpression plasmids and were transformed into the indicated strains using standard molecular biology techniques. For construction of PgsA, PgpC, and ClsA depletion strains, pgsA, pgpC, and clsA were cloned into pBAD33 using Gibson Assembly (65). Briefly, pBAD33 was linearized by PCR using the pBAD33 F and R primers (see Table S1). The inserts amplified from MG1655 genomic DNA using the primers listed in Table S1 from 30 bp upstream of the open reading frame (ORF) to the end of the ORF. The PCR fragments were assembled using HiFi Assembly Master Mix (New England Biolabs) according to the manufacturer’s instructions.
Quantification of ECA levels.
ECALPS levels were quantified by staining cells with Alexa Fluor 488-conjugated WGA (Thermo Fisher Scientific) as we have described earlier (56). Briefly, 250-μL aliquots of washed overnight cultures were combined with 10 μg/mL wheat germ agglutinin (WGA) in phosphate-buffered saline (PBS) and then incubated for 10 min at room temperature. Cells were washed twice and resuspended in PBS. The fluorescence (excitation, 485 nm; emission, 519 nm) and the optical density at 600 nm (OD600) were read in a BioTek Synergy H1 plate reader. Fluorescence relative to the OD was calculated.
ECAPG was detected through immunoblot analysis as we have described (26, 56). Briefly, samples from overnight cultures were normalized to equal OD600 and resuspended in BugBuster protein extraction reagent (Millipore Sigma), followed by an equal volume of Laemmli sample buffer (Bio-Rad). Samples were run on 12% acrylamide gels and transferred to nitrocellulose. Blots were probed with α-ECA antibody at a 1:30,000 dilution and a goat α-rabbit-HRP secondary antibody (Prometheus) at a 1:100,000 dilution. α-ECA antibody was kindly provided by Renato Morona (University of Adelaide). Detection was performed using Prosignal Pico ECL (Prometheus) using Prosignal ECL-blotting film (Prometheus).
Luciferase reporter assays.
Activity of the Pwec promoter was assayed with a plasmid-based Pwec:luxCDABE reporter as we have described (56) with minor modifications. Cultures were grown overnight in media without IPTG. For reporter assays, strains were subcultured 1:100 into media with the indicated concentration of IPTG. The luminescence and the OD600 were assayed in technical quadruplicate in a BioTek Synergy H1 plate reader every 3 min for 5 h. The luminescence relative to OD600 was calculated, and technical replicates were averaged. Then, the fold value of each sample to the empty vector control for each time point was calculated.
Depletion growth curves.
For depletion experiments, the indicated strains were grown overnight with arabinose. Then, cultures were washed once in plain Luria-Bertani medium and diluted 1:500 or 1:10,000, as indicated, into media with either arabinose to induce expression from the PBAD plasmid or glucose to repress expression. For strains where the phospholipid was nonessential (i.e., CL, PG in a Δlpp ΔrcsF background), cultures were maintained with chloramphenicol during the course of the growth curve. Cultures were transferred to a 24-well plate and were incubated shaking at 37°C in a BioTek Synergy H1 plate reader. The OD600 was assayed every 5 min for 12 h.
ACKNOWLEDGMENTS
We thank members of the Mitchell lab for productive discussions and for critical reading of our manuscript. We thank Tracy Raivio (University of Alberta) for the pJW15 plasmid and Renato Morona (University of Adelaide) for the ECA antibody. We thank Mikhail Bogdanov and William Dowhan (McGovern Medical School, University of Texas Houston) for providing us with strains carrying a pssA disruption and complementing plasmid.
This work was supported by the National Institute of Allergy and Infectious Diseases under award number R01-AI155915 (to A.M.M.).
Footnotes
Supplemental material is available online only.
Contributor Information
Angela M. Mitchell, Email: amitchell@bio.tamu.edu.
Laurie E. Comstock, University of Chicago
REFERENCES
- 1.Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb Perspect Biol 2:a000414. 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benn G, Mikheyeva IV, Inns PG, Forster JC, Ojkic N, Bortolini C, Ryadnov MG, Kleanthous C, Silhavy TJ, Hoogenboom BW. 2021. Phase separation in the outer membrane of Escherichia coli. Proc Natl Acad Sci USA 118:e2112237118. 10.1073/pnas.2112237118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konovalova A, Silhavy TJ. 2015. Outer membrane lipoprotein biogenesis: Lol is not the end. Philos Trans R Soc B 370:20150030. 10.1098/rstb.2015.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rai AK, Mitchell AM. 2020. Enterobacterial common antigen: synthesis and function of an enigmatic molecule. mBio 11:e01914-20. 10.1128/mBio.01914-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67:593–656. 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henderson JC, Zimmerman SM, Crofts AA, Boll JM, Kuhns LG, Herrera CM, Trent MS. 2016. The power of asymmetry: architecture and assembly of the Gram-negative outer membrane lipid bilayer. Annu Rev Microbiol 70:255–278. 10.1146/annurev-micro-102215-095308. [DOI] [PubMed] [Google Scholar]
- 7.MacNair CR, Brown ED. 2020. Outer membrane disruption overcomes intrinsic, acquired, and spontaneous antibiotic resistance. mBio 11:e01615-20. 10.1128/mBio.01615-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehman KM, Grabowicz M. 2019. Countering gram-negative antibiotic resistance: recent progress in disrupting the outer membrane with novel therapeutics. Antibiotics 8:163. 10.3390/antibiotics8040163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vetterli SU, Zerbe K, Müller M, Urfer M, Mondal M, Wang S-Y, Moehle K, Zerbe O, Vitale A, Pessi G, Eberl L, Wollscheid B, Robinson JA. 2018. Thanatin targets the intermembrane protein complex required for lipopolysaccharide transport in Escherichia coli. Sci Adv 4:eaau2634. 10.1126/sciadv.aau2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srinivas N, Jetter P, Ueberbacher BJ, Werneburg M, Zerbe K, Steinmann J, Van der Meijden B, Bernardini F, Lederer A, Dias RLA, Misson PE, Henze H, Zumbrunn J, Gombert FO, Obrecht D, Hunziker P, Schauer S, Ziegler U, Käch A, Eberl L, Riedel K, DeMarco SJ, Robinson JA. 2010. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science 327:1010–1013. 10.1126/science.1182749. [DOI] [PubMed] [Google Scholar]
- 11.Steenhuis M, Abdallah AM, de Munnik SM, Kuhne S, Sterk G-J, van den Berg van Saparoea B, Westerhausen S, Wagner S, van der Wel NN, Wijtmans M, van Ulsen P, Jong WSP, Luirink J. 2019. Inhibition of autotransporter biogenesis by small molecules. Mol Microbiol 112:81–98. 10.1111/mmi.14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Storek KM, Auerbach MR, Shi H, Garcia NK, Sun D, Nickerson NN, Vij R, Lin Z, Chiang N, Schneider K, Wecksler AT, Skippington E, Nakamura G, Seshasayee D, Koerber JT, Payandeh J, Smith PA, Rutherford ST. 2018. Monoclonal antibody targeting the β-barrel assembly machine of Escherichia coli is bactericidal. Proc Natl Acad Sci USA 115:3692–3697. 10.1073/pnas.1800043115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hart EM, Mitchell AM, Konovalova A, Grabowicz M, Sheng J, Han X, Rodriguez-Rivera FP, Schwaid AG, Malinverni JC, Balibar CJ, Bodea S, Si Q, Wang H, Homsher MF, Painter RE, Ogawa AK, Sutterlin H, Roemer T, Black TA, Rothman DM, Walker SS, Silhavy TJ. 2019. A small-molecule inhibitor of BamA impervious to efflux and the outer membrane permeability barrier. Proc Natl Acad Sci USA 116:21748–21757. 10.1073/pnas.1912345116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Urfer M, Bogdanovic J, Lo Monte F, Moehle K, Zerbe K, Omasits U, Ahrens CH, Pessi G, Eberl L, Robinson JA. 2016. A peptidomimetic antibiotic targets outer membrane proteins and disrupts selectively the outer membrane in Escherichia coli. J Biol Chem 291:1921–1932. 10.1074/jbc.M115.691725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steenhuis M, Corona F, ten Hagen-Jongman CM, Vollmer W, Lambin D, Selhorst P, Klaassen H, Versele M, Chaltin P, Luirink J. 2021. Combining cell envelope stress reporter assays in a screening approach to identify BAM complex inhibitors. ACS Infect Dis 7:2250–2263. 10.1021/acsinfecdis.0c00728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Männel D, Mayer H. 1978. Isolation and chemical characterization of the enterobacterial common antigen. Eur J Biochem 86:361–370. 10.1111/j.1432-1033.1978.tb12318.x. [DOI] [PubMed] [Google Scholar]
- 17.Lugowski C, Romanowska E, Kenne L, Lindberg B. 1983. Identification of a trisaccharide repeating-unit in the enterobacterial common-antigen. Carbohydrate Res 118:173–181. 10.1016/0008-6215(83)88045-8. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt G, Mannel D, Mayer H, Whang HY, Neter E. 1976. Role of a lipopolysaccharide gene for immunogenicity of the enterobacterial common antigen. J Bacteriol 126:579–586. 10.1128/jb.126.2.579-586.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rinno J, Golecki JR, Mayer H. 1980. Localization of enterobacterial common antigen: immunogenic and nonimmunogenic enterobacterial common antigen-containing Escherichia coli. J Bacteriol 141:814–821. 10.1128/jb.141.2.814-821.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuhn HM, Neter E, Mayer H. 1983. Modification of the lipid moiety of the enterobacterial common antigen by the “Pseudomonas factor.” Infect Immun 40:696–700. 10.1128/iai.40.2.696-700.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dell A, Oates J, Lugowski C, Romanowska E, Kenne L, Lindberg B. 1984. The enterobacterial common-antigen, a cyclic polysaccharide. Carbohydr Res 133:95–104. 10.1016/0008-6215(84)85186-1. [DOI] [PubMed] [Google Scholar]
- 22.Acker G, Bitter-Suermann D, Meier-Dieter U, Peters H, Mayer H. 1986. Immunocytochemical localization of enterobacterial common antigen in Escherichia coli and Yersinia enterocolitica cells. J Bacteriol 168:348–356. 10.1128/jb.168.1.348-356.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erbel PJA, Barr K, Gao N, Gerwig GJ, Rick PD, Gardner KH. 2003. Identification and biosynthesis of cyclic enterobacterial common antigen in Escherichia coli. J Bacteriol 185:1995–2004. 10.1128/JB.185.6.1995-2004.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kajimura J, Rahman A, Rick PD. 2005. Assembly of cyclic enterobacterial common antigen in Escherichia coli K-12. J Bacteriol 187:6917–6927. 10.1128/JB.187.20.6917-6927.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuhn H-M, Meier-Dieter U, Mayer H. 1988. ECA, the enterobacterial common antigen. FEMS Microbiol Rev 54:195–222. 10.1111/j.1574-6968.1988.tb02743.x. [DOI] [PubMed] [Google Scholar]
- 26.Mitchell AM, Srikumar T, Silhavy TJ. 2018. Cyclic enterobacterial common antigen maintains the outer membrane permeability barrier of Escherichia coli in a manner controlled by Yhdp. mBio 9:e01321-18. 10.1128/mBio.01321-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao Y, Lee J, Widmalm G, Im W. 2020. Modeling and simulation of bacterial outer membranes with lipopolysaccharides and enterobacterial common antigen. J Phys Chem B 124:5948–5956. 10.1021/acs.jpcb.0c03353. [DOI] [PubMed] [Google Scholar]
- 28.Gao Y, Lee J, Widmalm G, Im W. 2021. Correction to “modeling and simulation of bacterial outer membranes with lipopolysaccharides and enterobacterial common antigen”. J Phys Chem B 125:9384–9385. 10.1021/acs.jpcb.1c06516. [DOI] [PubMed] [Google Scholar]
- 29.Gilbreath JJ, Colvocoresses Dodds J, Rick PD, Soloski MJ, Merrell DS, Metcalf ES. 2012. Enterobacterial common antigen mutants of Salmonella enterica serovar Typhimurium establish a persistent infection and provide protection against subsequent lethal challenge. Infect Immun 80:441–450. 10.1128/IAI.05559-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kunin CM. 1963. Separation, characterization, and biological significance of a common antigen in Enterobacteriaceae. J Exp Med 118:565–586. 10.1084/jem.118.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Minor L, Chalon AM, Veron M. 1972. Detection of a common antigen in Enterobacteriaceae (Kunin antigen) in Yersinia, Levinea, Aeromonas and Vibrio. Ann Inst Pasteur (Paris) 123:761–774. (In French.) [PubMed] [Google Scholar]
- 32.Bottger EC, Jurs M, Barrett T, Wachsmuth K, Metzger S, Bitter-Suermann D. 1987. Qualitative and quantitative determination of enterobacterial common antigen (ECA) with monoclonal antibodies: expression of ECA by two Actinobacillus species. J Clin Microbiol 25:377–382. 10.1128/jcm.25.2.377-382.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bertani B, Ruiz N. 2018. Function and biogenesis of lipopolysaccharides. EcoSal Plus 8. 10.1128/ecosalplus.ESP-0001-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rick PD, Mayer H, Neumeyer BA, Wolski S, Bitter-Suermann D. 1985. Biosynthesis of enterobacterial common antigen. J Bacteriol 162:494–503. 10.1128/jb.162.2.494-503.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barr K, Nunes-Edwards P, Rick PD. 1989. In vitro synthesis of a lipid-linked trisaccharide involved in synthesis of enterobacterial common antigen. J Bacteriol 171:1326–1332. 10.1128/jb.171.3.1326-1332.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rick PD, Barr K, Sankaran K, Kajimura J, Rush JS, Waechter CJ. 2003. Evidence that the wzxE gene of Escherichia coli K-12 encodes a protein involved in the transbilayer movement of a trisaccharide-lipid intermediate in the assembly of enterobacterial common antigen. J Biol Chem 278:16534–16542. 10.1074/jbc.M301750200. [DOI] [PubMed] [Google Scholar]
- 37.Brade H. 1999. Endotoxin in health and disease. CRC Press, Boca Raton, FL. [Google Scholar]
- 38.Barr K, Klena J, Rick PD. 1999. The modality of enterobacterial common antigen polysaccharide chain lengths is regulated by o349 of the wec gene cluster of Escherichia coli K-12. J Bacteriol 181:6564–6568. 10.1128/JB.181.20.6564-6568.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cronan JE, Jr, Rock CO. 2008. Biosynthesis of membrane lipids. EcoSal Plus 3. 10.1128/ecosalplus.3.6.4. [DOI] [PubMed] [Google Scholar]
- 40.Bogdanov M, Pyrshev K, Yesylevskyy S, Ryabichko S, Boiko V, Ivanchenko P, Kiyamova R, Guan Z, Ramseyer C, Dowhan W. 2020. Phospholipid distribution in the cytoplasmic membrane of Gram-negative bacteria is highly asymmetric, dynamic, and cell shape-dependent. Sci Adv 6:eaaz6333. 10.1126/sciadv.aaz6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kessels JM, Ousen H, Van den Bosch H. 1983. Facilitated utilization of endogenously synthesized lysophosphatidic acid by 1-acylglycerophosphate acyltransferase from Escherichia coli. Biochim Biophys Acta 753:227–235. 10.1016/0005-2760(83)90011-5. [DOI] [PubMed] [Google Scholar]
- 42.Carter JR, Jr. 1968. Cytidine triphosphate: phosphatidic acid cytidyltransferase in Escherichia coli. J Lipid Res 9:748–754. 10.1016/S0022-2275(20)42688-4. [DOI] [PubMed] [Google Scholar]
- 43.Gopalakrishnan AS, Chen YC, Temkin M, Dowhan W. 1986. Structure and expression of the gene locus encoding the phosphatidylglycerophosphate synthase of Escherichia coli. J Biol Chem 261:1329–1338. 10.1016/S0021-9258(17)36095-7. [DOI] [PubMed] [Google Scholar]
- 44.Dillon DA, Wu WI, Riedel B, Wissing JB, Dowhan W, Carman GM. 1996. The Escherichia coli pgpB gene encodes for a diacylglycerol pyrophosphate phosphatase activity. J Biol Chem 271:30548–30553. 10.1074/jbc.271.48.30548. [DOI] [PubMed] [Google Scholar]
- 45.Icho T, Raetz CR. 1983. Multiple genes for membrane-bound phosphatases in Escherichia coli and their action on phospholipid precursors. J Bacteriol 153:722–730. 10.1128/jb.153.2.722-730.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu YH, Guan Z, Zhao J, Raetz CR. 2011. Three phosphatidylglycerol-phosphate phosphatases in the inner membrane of Escherichia coli. J Biol Chem 286:5506–5518. 10.1074/jbc.M110.199265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El Ghachi M, Derbise A, Bouhss A, Mengin-Lecreulx D. 2005. Identification of multiple genes encoding membrane proteins with undecaprenyl pyrophosphate phosphatase (UppP) activity in Escherichia coli. J Biol Chem 280:18689–18695. 10.1074/jbc.M412277200. [DOI] [PubMed] [Google Scholar]
- 48.Ota A, Shibuya I, Maruo B, Ishinaga M, Kito M. 1974. An extremely labile phosphatidylserine synthetase of an Escherichia coli mutant with the temperature-sensitive formation of phosphatidylethanolamine. Biochim Biophys Acta 348:449–454. [PubMed] [Google Scholar]
- 49.Hawrot E, Kennedy EP. 1975. Biogenesis of membrane lipids: mutants of Escherichia coli with temperature-sensitive phosphatidylserine decarboxylase. Proc Natl Acad Sci USA 72:1112–1116. 10.1073/pnas.72.3.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan BK, Bogdanov M, Zhao J, Dowhan W, Raetz CR, Guan Z. 2012. Discovery of a cardiolipin synthase utilizing phosphatidylethanolamine and phosphatidylglycerol as the substrates. Proc Natl Acad Sci USA 109:16504–16509. 10.1073/pnas.1212797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nishijima S, Asami Y, Uetake N, Yamagoe S, Ohta A, Shibuya I. 1988. Disruption of the Escherichia coli cls gene responsible for cardiolipin synthesis. J Bacteriol 170:775–780. 10.1128/jb.170.2.775-780.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pluschke G, Hirota Y, Overath P. 1978. Function of phospholipids in Escherichia coli: characterization of a mutant deficient in cardiolipin synthesis. J Biol Chem 253:5048–5055. 10.1016/S0021-9258(17)34655-0. [DOI] [PubMed] [Google Scholar]
- 53.Guo D, Tropp BE. 2000. A second Escherichia coli protein with CL synthase activity. Biochim Biophys Acta 1483:263–274. 10.1016/s1388-1981(99)00193-6. [DOI] [PubMed] [Google Scholar]
- 54.Li C, Tan BK, Zhao J, Guan Z. 2016. In vivo and in vitro synthesis of phosphatidylglycerol by an Escherichia coli cardiolipin synthase. J Biol Chem 291:25144–25153. 10.1074/jbc.M116.762070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dowhan W, Vitrac H, Bogdanov M. 2019. Lipid-assisted membrane protein folding and topogenesis. Protein J 38:274–288. 10.1007/s10930-019-09826-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rai AK, Carr JF, Bautista DE, Wang W, Mitchell AM. 2021. ElyC and cyclic enterobacterial common antigen regulate synthesis of phosphoglyceride-linked enterobacterial common antigen. mBio 12:e02846-21. 10.1128/mBio.02846-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ragolia L, Tropp BE. 1994. The effects of phosphoglycerides on Escherichia coli cardiolipin synthase. Biochim Biophys Acta 1214:323–332. 10.1016/0005-2760(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 58.Rick PD, Hubbard GL, Kitaoka M, Nagaki H, Kinoshita T, Dowd S, Simplaceanu V, Ho C. 1998. Characterization of the lipid-carrier involved in the synthesis of enterobacterial common antigen (ECA) and identification of a novel phosphoglyceride in a mutant of Salmonella Typhimurium defective in ECA synthesis. Glycobiology 8:557–567. 10.1093/glycob/8.6.557. [DOI] [PubMed] [Google Scholar]
- 59.Ikeda M, Wachi M, Jung HK, Ishino F, Matsuhashi M. 1991. The Escherichia coli mraY gene encoding UDP-N-acetylmuramoyl-pentapeptide: undecaprenyl-phosphate phospho-N-acetylmuramoyl-pentapeptide transferase. J Bacteriol 173:1021–1026. 10.1128/jb.173.3.1021-1026.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Danese PN, Oliver GR, Barr K, Bowman GD, Rick PD, Silhavy TJ. 1998. Accumulation of the enterobacterial common antigen lipid II biosynthetic intermediate stimulates degP transcription in Escherichia coli. J Bacteriol 180:5875–5884. 10.1128/JB.180.22.5875-5884.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Castelli ME, Véscovi EG. 2011. The Rcs signal transduction pathway is triggered by enterobacterial common antigen structure alterations in Serratia marcescens. J Bacteriol 193:63–74. 10.1128/JB.00839-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paradis-Bleau C, Kritikos G, Orlova K, Typas A, Bernhardt TG. 2014. A genome-wide screen for bacterial envelope biogenesis mutants identifies a novel factor involved in cell wall precursor metabolism. PLoS Genet 10:e1004056. 10.1371/journal.pgen.1004056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jorgenson MA, Kannan S, Laubacher ME, Young KD. 2016. Dead-end intermediates in the enterobacterial common antigen pathway induce morphological defects in Escherichia coli by competing for undecaprenyl phosphate. Mol Microbiol 100:1–14. 10.1111/mmi.13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eade CR, Wallen TW, Gates CE, Oliverio CL, Scarbrough BA, Reid AJ, Jorgenson MA, Young KD, Troutman JM. 2021. Making the enterobacterial common antigen glycan and measuring its substrate sequestration. ACS Chem Biol 16:691–700. 10.1021/acschembio.0c00983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kikuchi S, Shibuya I, Matsumoto K. 2000. Viability of an Escherichia coli pgsA null mutant lacking detectable phosphatidylglycerol and cardiolipin. J Bacteriol 182:371–376. 10.1128/JB.182.2.371-376.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shiba Y, Yokoyama Y, Aono Y, Kiuchi T, Kusaka J, Matsumoto K, Hara H. 2004. Activation of the Rcs signal transduction system is responsible for the thermosensitive growth defect of an Escherichia coli mutant lacking phosphatidylglycerol and cardiolipin. J Bacteriol 186:6526–6535. 10.1128/JB.186.19.6526-6535.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grabowicz M, Silhavy TJ. 2017. Redefining the essential trafficking pathway for outer membrane lipoproteins. Proc Natl Acad Sci USA 114:4769–4774. 10.1073/pnas.1702248114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sankaran K, Wu HC. 1994. Lipid modification of bacterial prolipoprotein: transfer of diacylglyceryl moiety from phosphatidylglycerol. J Biol Chem 269:19701–19706. 10.1016/S0021-9258(17)32077-X. [DOI] [PubMed] [Google Scholar]
- 70.Yakushi T, Tajima T, Matsuyama S, Tokuda H. 1997. Lethality of the covalent linkage between mislocalized major outer membrane lipoprotein and the peptidoglycan of Escherichia coli. J Bacteriol 179:2857–2862. 10.1128/jb.179.9.2857-2862.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shiba Y, Miyagawa H, Nagahama H, Matsumoto K, Kondo D, Matsuoka S, Matsumoto K, Hara H. 2012. Exploring the relationship between lipoprotein mislocalization and activation of the Rcs signal transduction system in Escherichia coli. Microbiology (Reading) 158:1238–1248. 10.1099/mic.0.056945-0. [DOI] [PubMed] [Google Scholar]
- 72.DeChavigny A, Heacock PN, Dowhan W. 1991. Sequence and inactivation of the pss gene of Escherichia coli: phosphatidylethanolamine may not be essential for cell viability. J Biol Chem 266:5323–5332. 10.1016/S0021-9258(19)67791-4. [DOI] [PubMed] [Google Scholar]
- 73.Hiraoka S, Matsuzaki H, Shibuya I. 1993. Active increase in cardiolipin synthesis in the stationary growth phase and its physiological significance in Escherichia coli. FEBS Lett 336:221–224. 10.1016/0014-5793(93)80807-7. [DOI] [PubMed] [Google Scholar]
- 74.Jiang XE, Tan WB, Shrivastava R, Seow DCS, Chen SL, Guan XL, Chng S-S. 2020. Mutations in enterobacterial common antigen biosynthesis restore outer membrane barrier function in Escherichia coli tol-pal mutants. Mol Microbiol 114:991–1005. 10.1111/mmi.14590. [DOI] [PubMed] [Google Scholar]
- 75.Nishikawa H, Sawasato K, Mori S, Fujikawa K, Nomura K, Shimamoto K, Nishiyama K-I. 2022. Interaction between glycolipid MPIase and proteinaceous factors during protein integration into the cytoplasmic membrane of Escherichia coli. Front Mol Biosci 9:986602. 10.3389/fmolb.2022.986602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nishiyama K-I, Maeda M, Yanagisawa K, Nagase R, Komura H, Iwashita T, Yamagaki T, Kusumoto S, Tokuda H, Shimamoto K. 2012. MPIase is a glycolipozyme essential for membrane protein integration. Nat Commun 3:1260. 10.1038/ncomms2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sawasato K, Sato R, Nishikawa H, Iimura N, Kamemoto Y, Fujikawa K, Yamaguchi T, Kuruma Y, Tamura Y, Endo T, Ueda T, Shimamoto K, Nishiyama K-I. 2019. CdsA is involved in biosynthesis of glycolipid MPIase essential for membrane protein integration in vivo. Sci Rep 9:1372. 10.1038/s41598-018-37809-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moser M, Nagamori S, Huber M, Tokuda H, Nishiyama K-I. 2013. Glycolipozyme MPIase is essential for topology inversion of SecG during preprotein translocation. Proc Natl Acad Sci USA 110:9734–9739. 10.1073/pnas.1303160110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nishiyama K, Ikegami A, Moser M, Schiltz E, Tokuda H, Müller M. 2006. A derivative of lipid A is involved in signal recognition particle/SecYEG-dependent and -independent membrane integrations. J Biol Chem 281:35667–35676. 10.1074/jbc.M608228200. [DOI] [PubMed] [Google Scholar]
- 80.Nomura K, Mori S, Fujikawa K, Osawa T, Tsuda S, Yoshizawa-Kumagaye K, Masuda S, Nishio H, Yoshiya T, Yoda T, Shionyu M, Shirai T, Nishiyama K-I, Shimamoto K. 2022. Role of a bacterial glycolipid in Sec-independent membrane protein insertion. Sci Rep 12:12231. 10.1038/s41598-022-16304-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Endo Y, Shimizu Y, Nishikawa H, Sawasato K, Nishiyama K-I. 2022. Interplay between MPIase, YidC, and PMF during Sec-independent insertion of membrane proteins. Life Sci Alliance 5:e202101162. 10.26508/lsa.202101162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kamemoto Y, Funaba N, Kawakami M, Sawasato K, Kanno K, Suzuki S, Nishikawa H, Sato R, Nishiyama KI. 2020. Biosynthesis of glycolipid MPIase (membrane protein integrase) is independent of the genes for ECA (enterobacterial common antigen). J Gen Appl Microbiol 66:169–174. 10.2323/jgam.2019.05.001. [DOI] [PubMed] [Google Scholar]
- 83.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Silhavy TJ, Berman ML, Enquist LW. 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 85.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bogdanov M, Heacock PN, Dowhan W. 2002. A polytopic membrane protein displays a reversible topology dependent on membrane lipid composition. EMBO J 21:2107–2116. 10.1093/emboj/21.9.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guyer MS, Reed RR, Steitz JA, Low KB. 1981. Identification of a sex-factor-affinity site in Escherichia coli as gamma delta. Cold Spring Harbor Symp Quant Biol 45(Pt 1):135–140. 10.1101/sqb.1981.045.01.022. [DOI] [PubMed] [Google Scholar]
- 88.Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. 2005. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res 12:291–299. 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- 89.MacRitchie DM, Ward JD, Nevesinjac AZ, Raivio TL. 2008. Activation of the Cpx envelope stress response down-regulates expression of several locus of enterocyte effacement-encoded genes in enteropathogenic Escherichia coli. Infect Immun 76:1465–1475. 10.1128/IAI.01265-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S4 and Table S1. Download jb.00403-22-s0001.pdf, PDF file, 1.5 MB (1.5MB, pdf)