

ABSTRACT
The peptidoglycan of mycobacteria has two types of direct cross-links, classical 4-3 cross-links that occur between diaminopimelate (DAP) and alanine residues, and nonclassical 3-3 cross-links that occur between DAP residues on adjacent peptides. The 3-3 cross-links are synthesized by the concerted action of d,d-carboxypeptidases and l,d-transpeptidases (Ldts). Mycobacterial genomes encode several Ldt proteins that can be classified into six classes based upon sequence identity. As a group, the Ldt enzymes are resistant to most β-lactam antibiotics but are susceptible to carbapenem antibiotics, with the exception of LdtC, a class 5 enzyme. In previous work, we showed that loss of LdtC has the greatest effect on the carbapenem susceptibility phenotype of Mycobacterium smegmatis (also known as Mycolicibacterium smegmatis) compared to other ldt deletion mutants. In this work, we show that a M. smegmatis mutant lacking the five ldt genes other than ldtC has a wild-type phenotype with the exception of increased susceptibility to rifampin. In contrast, a mutant lacking all six ldt genes has pleiotropic cell envelope defects, is temperature sensitive, and has increased susceptibility to a variety of antibiotics. These results indicate that LdtC is capable of functioning as the sole l,d-transpeptidase in M. smegmatis and suggest that it may represent a carbapenem-resistant pathway for peptidoglycan biosynthesis.
IMPORTANCE Mycobacteria have several enzymes to catalyze nonclassical 3-3 linkages in the cell wall peptidoglycan. Understanding the biology of these cross-links is important for the development of antibiotic therapies to target peptidoglycan biosynthesis. Our work provides evidence that LdtC can function as the sole enzyme for 3-3 cross-link formation in M. smegmatis and suggests that LdtC may be part of a carbapenem-resistant l,d-transpeptidase pathway.
KEYWORDS: l,d-transpeptidase; mycobacteria; cell wall; peptidoglycan; antibiotic resistance
INTRODUCTION
Despite the decline in the global incidence of tuberculosis (TB), the disease still occurs in every part of the world and has an enormous impact on human health. Current TB treatment involves a prolonged course of multiple antibiotics (1, 2). The rise of drug-resistant TB cases and the prevalence of TB-related HIV deaths has led to the exploration of novel anti-TB therapeutics that target the cell envelope of Mycobacterium tuberculosis.
The mycobacterial cell envelope consists of three covalently linked macromolecules: the mycolic acids, arabinogalactan, and peptidoglycan (PG) (3–5). PG is the anchor for the mycobacterial cell envelope and is composed of a glycan chain with alternating N-acetylglucosamine (GlcNAc) and muramic acid residues, the latter of which are N-acetylated (MurNAc) or N-glycosylated (MurNGc) (3–6). Direct cross-links between pentapeptide chains (l-alanyl-d-isoglutaminyl-meso-diaminopimelate-d-alanyl-d-alanine) attached to the muramyl residues generates the mature structure (3–5). In mycobacteria, up to 80% of the peptides are involved in linkages, the so-called classical 4-3 cross-links and the nonclassical 3-3 cross-links (7–10). Classical 4-3 cross-links are carried out by d,d-transpeptidases, also referred to as penicillin-binding proteins. These enzymes couple the d-alanine residue at the fourth position of the PG peptide with the side chain of the diaminopimelate (DAP) residue at the third position in an adjacent peptide (5, 11, 12). d,d-Transpeptidation reactions are susceptible to inhibition by β-lactam antibiotics, which mimic the terminal ends of the PG peptide substrates (13, 14). 3-3 linkages occur between the DAP residues at the third position of the PG peptides. Nonclassical cross-links are catalyzed by the concerted effort of d,d-carboxypeptidases and l,d-transpeptidases (Ldts) (5). While most bacterial PGs predominantly contain 4-3 cross-links, the PG in mycobacteria primarily contain nonclassical 3-3 cross-links (7, 8, 10, 15–17). The percentages of these linkages appear to be constant during the growth of M. tuberculosis (7) and likely confer increased rigidity to the PG, and therefore increased structural stability, which would be an important function, since the PG anchors the principal mass of the mycobacterial cell envelope.
Biosynthesis of the bacterial PG is the target of β-lactam antibiotics (13); however, many of these drugs are ineffective against mycobacteria in general due to inherent resistance mechanisms, including the production of β-lactamases (2, 18, 19). The predominance of 3-3 cross-links in the mycobacterial PG has also been proposed to limit the effects of β-lactam antibiotics (20, 21). In general, the Ldts are resistant to β-lactam antibiotics with the exception of the carbapenems, which covalently modify the conserved cysteine in the Ldt active site (22–24). Despite the limitations of β-lactam antibiotics in treating M. tuberculosis infections, there has been renewed interest in this modality in combination with β-lactamase inhibitors (25–28).
Mycobacterial genomes contain several ldt genes, depending upon the species. For example, Mycobacterium tuberculosis has five ldt genes, Mycobacterium leprae has four, while Mycobacterium smegmatis (also known as Mycolicibacterium smegmatis) encodes six (29). These Ldt proteins can be grouped into six classes based upon sequence analysis (29, 30). We previously described a series of M. smegmatis strains with single, double, triple, or quadruple insertional or deletion mutations of the ldt genes and their phenotypes (29). The only single gene mutant that had a phenotype was the class 5 ldtC mutant, which displayed increased susceptibility to carbapenem antibiotics. In addition, an M. smegmatis ldtC mutant had a reduction in labeling levels in experiments using Ldt-specific fluorescent probes suggesting a decrease in 3-3 linkages (31).
We have shown that a M. smegmatis double mutant lacking the closely related genes ldtB and ldtF has a 10-fold increase in susceptibility to lysozyme, which cleaves the glycan backbone of the PG, and no changes in antibiotic susceptibility (29). Furthermore, we showed that a triple mutant lacking ldtB, ldtF, and ldtC has moderate morphological changes, increased susceptibility to lysozyme, carbapenems, and increased susceptibility to ampicillin, rifampin, and ethambutol compared to the single ldtC mutant or the double ldtB ldtF mutant (29) and reduced labeling with a 3-3 linkage-specific probe (31). All of these mutant phenotypes could be complemented by the relevant genes from either M. smegmatis or M. tuberculosis, suggesting a conservation of these enzymes between the two species (29). We could find no phenotypical changes in any other ldt mutation combinations. Based on these observations, and the discovery that LdtC, unlike all other Ldt enzymes of M. tuberculosis, is resistant to carbapenem inhibition (22), we hypothesized that LdtC was a key enzyme in M. smegmatis for PG assembly and that it might represent a carbapenem resistance mechanism for 3-3 linkage formation (29). In this study, we report the construction and analysis of a M. smegmatis mutant lacking all ldt genes except ldtC and a mutant lacking all six ldt genes. Our results indicate that LdtC can function by itself in the assembly of the PG of M. smegmatis with little consequence but that loss of all ldt genes results in a strain with multiple phenotypical changes.
RESULTS
Construction of mutant strains.
We used M. smegmatis strain PM2766, which has insertional mutations in the five ldt genes other than ldtC, for allelic exchange with a sacB suicide vector bearing a deletion allele of ldtC, marked with lacZ (plasmid pMP1103). A primary recombinant merodiploid strain, PM2785, with pMP1103 integrated at the ldtC locus, was used to generate the final Δ6 mutant, PM2805, lacking all six ldt genes.
Subsequent attempts to construct a complemented version of the Δ6 mutant after initially identifying phenotypic changes were unsuccessful, as recovery of transformed clones was reduced after electroporation due to increased killing from the electroporation procedure (data not shown). Also, the strain tends to generate large-colony suppressor mutants after prolonged subculture, which makes interpretation of postelectroporation complementation problematic. Hence, we decided to perform a reverse complementation assay by constructing the Δ6 mutant again, but with a complementing plasmid bearing ldtC+ in the strain prior to ldtC deletion. We used strain PM3500, which is the Δ5 primary recombinant merodiploid strain PM2785 bearing the integrated ΔldtC::lacZ sacB suicide vector pMP1103 and transformed with pMP855, a replicative, kanamycin-resistant plasmid bearing the wild-type ldtC gene. We repeated the allelic exchange to generate a new Δ6 mutant bearing the complementing plasmid (strain PM3502, Δ6/ldtC+). This strain was then cured of the complementing plasmid, as described in Materials and Methods, to generate a new Δ6 mutant (PM3507) designated “Δ6 cured.” The original Δ6 mutant, PM2805, had a small-colony morphotype, while the Δ6/ldtC+ complemented strain PM3502 had a normal colony size and the Δ6 cured mutant PM3507 had a small-colony phenotype, as expected (data not shown). A similar reverse complementation procedure was used to test the ability of the M. tuberculosis ldtC gene to complement the M. smegmatis Δ6 mutant. These strains were then compared to each other and to the wild type (WT) in a series of assays to analyze cell envelope biology.
The Δ6 ldt mutant is temperature sensitive for growth and has morphological changes.
Comparison of the growth of the strains showed that the Δ5 mutant grew similarly to the wild type on plates and with similar kinetics in liquid medium (Fig. 1A). In contrast, the Δ6 mutant formed smaller colonies on solid medium and exhibited slower growth in liquid medium (Fig. 1A). The Δ6 mutant was temperature sensitive for growth at 42°C, compared to the Δ5 strain, which grew like the wild type (Fig. 1B and C). This temperature-sensitive phenotype of the Δ6 mutant could be complemented by M. smegmatis ldtC+ in trans (Fig. 1B).
FIG 1.

Growth and temperature sensitivity of the Δ6 ldt strain PM2805. (A) Optical density of broth cultures of the WT strain (PM965; circles), Δ5 mutant (PM2750; squares), Δ6 mutant (PM2805; upward triangles), Δ6/ldtC+ complemented strain (PM3502; downward triangles), and Δ6 plasmid-cured strain (PM3507; diamonds). Significance was determined by calculating area under the curve followed by independent t tests comparing mutant strains to the WT (significance is indicated to the left of the key). Data represent two independent experiments performed in triplicate. ****, P < 0.0001. (B) Spot dilutions of the WT (PM965), Δ5 mutant (PM2750), Δ6 mutant (PM2805), the Δ6/ldtC+ complemented strain PM3502, and the Δ6 plasmid cured strain PM3507 at 37°C (top) and 42°C (bottom). Representative image from three independent experiments. (C) Viability of broth cultures after temperature shift from 37°C to 42°C of the WT strain (PM965; circles), Δ5 mutant (PM2750; squares), and Δ6 mutant (PM2805; triangles). Data represent viable plate counts of samples taken at the time of temperature shift (N0) and every 12 h afterward (N1). The graph is representative of two independent experiments performed in triplicate. ****, P < 0.0001.
The small colony size of the Δ6 mutant prompted us to examine the mutant with scanning electron microscopy (SEM) to visualize whole cells. Early SEM images of the Δ6 mutant showed altered cell morphology compared to the wild type (data not shown), and so the SEM was repeated when all the complemented and control strains were available. The Δ6 mutant cells (Fig. 2E and F) had a rough surface with terminal swellings on some of the cells, in contrast to the wild-type strain (Fig. 2A and B), in which the cells appeared as typical mycobacterial bacilli. The Δ5 cells were morphologically similar to the WT cells (Fig. 2C and D). The morphological phenotype of the Δ6 mutant is complemented in the Δ6/ldtC+ strain, as the cells present as typical thin, rod-shaped bacilli (Fig. 2G and H), comparable to the Δ5 strain, while the Δ6 cured cells appear shortened and rough with bulbous ends (Fig. 2I and J). We used ImageJ to directly measure cell length and found that the Δ6 and Δ6 cured cells were significantly shorter than those of the WT, Δ5, and complemented strains (Fig. 2K). These shortened cell lengths and overall morphological changes are suggestive of abnormal PG synthesis and cell wall maintenance.
FIG 2.
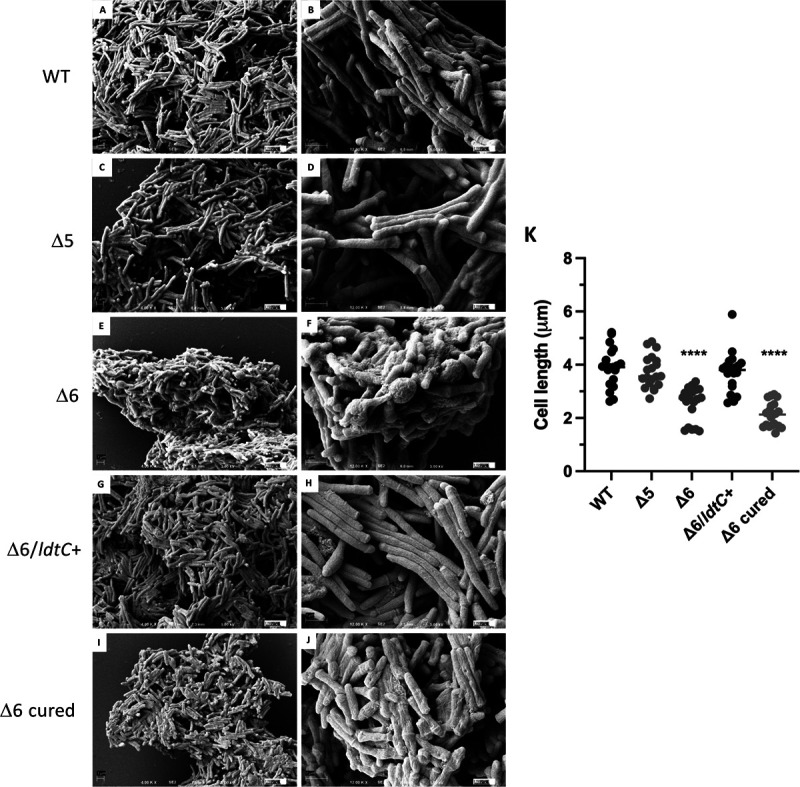
Cellular morphology of the Δ6 ldt mutant. Scanning electron micrographs of (A and B) WT strain PM965, (C and D) Δ5 mutant PM2750, (E and F) Δ6 mutant PM2805, (G and H) Δ6/ldtC+ complemented strain PM3502, and (I and J) Δ6-cured mutant PM3507. Magnifications, ×4,000 (A, C, E, G, and I) and ×12,000 (B, D, F, H, and J). Bar = 1 μm. (K) Cell lengths of ldt mutants (n = 20 for each strain). ****, P < 0.0001.
The Δ6 ldt mutant has increased susceptibility to antibiotics and altered dye binding.
We tested the strains for susceptibility to a variety of antibiotics and chemical agents. As shown in Fig. 3A, the Δ5 mutant had wild-type susceptibility to all antibiotics, with the exception of increased susceptibility to rifampin. The Δ6 mutant, however, had increased susceptibility to all antibiotics tested. We found that the Δ6 mutant could be complemented to the parental phenotype with the M. smegmatis or the M. tuberculosis ldtC+ gene for all three classes of antibiotic susceptibilities tested (Fig. 3B and C).
FIG 3.

Antibiotic susceptibility of the Δ6 ldt mutant by disk diffusion assay. (A) Susceptibility of the WT strain PM965 (gray bars), the Δ5 mutant PM2750 (gray cross-hatched bars), and the Δ6 mutant PM2805 (black bars). (B) Antibiotic susceptibility of the M. smegmatis ldtC gene-complemented Δ6 ldt mutant. The WT strain PM965 (gray bars), Δ5 mutant PM2750 (gray cross-hatched bars), Δ6 mutant PM2805 (black bars), Δ6/ldtC+ complemented strain PM3502 (white bars), and Δ6 cured strain PM3507 (black hatched bars) are shown. (C) Antibiotic susceptibility of the M. tuberculosis ldtC gene-complemented Δ6 ldt mutant. The WT strain PM965 (gray bars), Δ5 mutant PM2750 (gray cross-hatched bars), Δ6 mutant PM2805 (black bars), Δ6/ldtC+ complemented strain PM3502 (white bars), and Δ6 cured strain PM3507 (black hatched bars) are shown. *, P < 0.0001, for Δ6 compared to WT, except for RIF, where the Δ5 strain is compared to the WT and the Δ6 strain is compared to the Δ5 strain. IMI, imipenem; MER, meropenem; ERT, ertapenem; AMP, ampicillin; CFT, ceftriaxone; VAN, vancomycin; EMB, ethambutol; RIF, rifampin. Six repetitions were done for each strain per antibiotic or chemical (either two cultures in triplicate or three cultures in duplicate).
The Δ6 mutant had increased susceptibility to different classes of antibiotics, grew best in liquid medium in which the Tween 80 detergent was replaced with tyloxapol (data not shown), and showed changes in surface characteristics in the SEM images. Therefore, we hypothesized that there were likely changes to the cell envelope beyond the PG. We tested the strains against several dyes and chemicals (malachite green, Congo red, crystal violet, and sodium dodecyl sulfate [SDS] detergent) that interact with the cell envelope. The Δ6 mutant grew more slowly on agar plates with 200 μg/mL Congo red than on plates without and had a darker red color than the Δ5 and wild-type strains. This difference was more pronounced on medium lacking albumin-dextrose-saline supplementation (data not shown). As seen in Fig. 4A, the Δ6 mutant bound more Congo red dye than the wild type or the Δ5 mutant, likely indicating that Δ6 mutant has increased surface hydrophobicity. This phenotype was corrected in the Δ6/ldtC+ complemented strain but not the Δ6 cured strain control, as expected (Fig. 4A). The Δ6 mutant and the Δ6 cured strain control had increased sensitivity to the cell envelope-perturbing detergent SDS compared to the wild type and the Δ5 mutant (Fig. 4B). Additionally, we found no significant difference in SDS sensitivity between the Δ6/ldtC+ complemented strain and the Δ5 mutant (Fig. 4B). There was no change in the sensitivity of the Δ6 mutant to malachite green or crystal violet compared to that of control strains (data not shown).
FIG 4.

Congo red binding and SDS sensitivity of the Δ6 ldt mutant. (A) Congo red dye binding of the WT strain (PM965), the Δ5 mutant (PM2750), the Δ6 mutant (PM2805), the complemented (Δ6/ldtC+) strain (PM3502), and the plasmid-cured Δ6 strain (PM3507), expressed as optical density of dye extracted with acetone per wet weight of cell pellets. *, P = 0.01; **, P = 0.002 (compared to the WT); #, P = 0.004; ##, P = 0.009 (compared to the WT). Each strain was assayed in triplicate, and assays were performed on duplicate sets of cultures. Data from one set are shown. (B) SDS sensitivity by disk diffusion assay of the WT strain (PM965), the Δ5 mutant (PM2750), the Δ6 mutant (PM2805), the complemented (Δ6/ldtC+) strain (PM3502), and the plasmid-cured Δ6 strain (PM3507). ****, P < 0.0001. There was no significant difference between the WT, Δ5, and Δ6/ldtC+ strains. Data represent three independent experiments performed in quadruplicate.
The Δ6 mutant has increased susceptibility to PG-altering agents.
The increased susceptibility of the Δ6 mutant to a variety of antibiotics that affect both PG biosynthesis and other non-PG specific pathways prompted us to examine other treatments that affect only PG. We found that the Δ6 mutant and the cured strain had increased susceptibility to lysozyme, which cleaves the glycan backbone of the mature PG (Fig. 5A), a phenotype that can be complemented by ldtC+ in trans (Fig. 5A; also, see Fig. S1 in the supplemental material). We also found that the Δ6 mutant had increased susceptibility to noncanonical d-amino acid (NCDAA) exposure. NCDAAs can be incorporated into PG precursors by various transpeptidases and carboxypeptidases by exchange reactions utilizing the terminal d-alanine on the PG precursors and the NCDAA. This results in a decrease in PG cross-linking, as the NCDAA does not participate in the cross-linking reaction (32). We conducted viable plating experiments using medium with or without 15 mM d-methionine as the NCDAA and found that the Δ6 mutant and the Δ6 cured strain had increased susceptibility to d-methionine exposure. This was not seen in the Δ5 or the Δ6/ldtC+ complemented mutant (Fig. 5B and Fig. S1).
FIG 5.

Lysozyme and NCDAA sensitivity of the Δ6 ldt mutant. (A) Plate dilutions of the WT strain (PM965), the Δ5 mutant (PM2750), the Δ6 mutant (PM2805), the Δ6/ldtC+ complemented strain (PM3502), and the Δ6 cured strain (PM3507) on medium without lysozyme (top) or with 0.1 mg/mL lysozyme (bottom). (B) Plate dilutions of the WT strain (PM965), the Δ5 mutant (PM2750), the Δ6 mutant (PM2805), the Δ6/ldtC+ complemented strain (PM3502), and the Δ6 cured strain (PM3507) on medium without d-methionine (top) or with 15 mM d-methionine (bottom). Experiments were performed in triplicate. Data from one representative set are shown.
The Δ6 mutant has defective PG architecture.
We analyzed the PG cross-linking in three strains using a method that primarily examines the peptides of the PG but also provides information regarding the degree of transglycosylation, as indicated by the presence of disaccharides composed of MurNAc and GlcNAc. In this technique, the purified mycolyl-PG-arabinogalactan cell wall skeleton is digested with the LysA amidase from mycobacteriophage Ms6 as previously reported (33). The resulting material is then purified via size exclusion and the eluate analyzed by liquid chromatography in-line with mass spectrometry. An in-house database containing calculated monoisotopic masses of possible monomeric, dimeric, and trimeric peptides, including those with amidated carboxylic residues and/or glycine residues (up to a molecular weight of 2 kDa), was used to identify molecular features. The features with a database match were used to identify peptide and degree of cross-linking. The assigned identity of the major peptides was confirmed by tandem mass spectrometry as previously described (33).
As shown in Table 1 and Fig. S2, there were large changes in the cross-linking and overall linkages in the Δ6 mutant compared to the Δ5 mutant and the wild type. In all three strains, the dominant fragments identified were cross-linked dimers. However, these were reduced in abundance in the Δ6 mutant relative to the Δ5 mutant but found in similar abundance in the parental strain. These fragments likely contain both 4-3 and 3-3 cross-links, as it is difficult to distinguish the two types of cross-links by mass spectrometry. Un-cross-linked monomers were present in all three strains but were increased by ~5-fold in the Δ6 mutant. The abundance of trimers was reduced to 0 in the Δ6 mutant, as was that of the identifiable 3-3 cross-linked peptides (dimers and trimers containing multiple tripeptides), as previously reported (33).
TABLE 1.
Classes of muropeptides and disaccharides identified by mass spectral analysis of LysA digested peptidoglycan isolated from ldt mutantsa
| Fragment | %b in: |
||
|---|---|---|---|
| PM965 (WT) | PM2750 (Δ5) | PM2805 (Δ6) | |
| Disaccharides | 20 | 8.7 | 43 |
| Monomers | 4.5 | 5.3 | 25 |
| Dimers | 70 | 90 | 75 |
| Trimers | 25.5 | 3.5 | 0 |
| DAP-DAP-containing cross-linked peptides | 6.7 | 3.6 | 0 |
Cell wall preparations were purified and processed in duplicate for each strain. Each sample was analyzed by mass spectrometry for matches, but only one sample per strain was analyzed in depth.
Values are percentages of total peptides except for disaccharide values, which are percentages of the total detector response. Values were calculated as sum of the indicated class of ions/total percent of detector response for features that could be identified as muropeptides or disaccharides. See Fig. S2 to S4.
Interestingly, the loss of the higher-order cross-linked peptides in the Δ6 mutant was coincident with a large increase in the presence of a previously unidentified feature with an m/z value of 497.20, which was suggestive of a disaccharide composed of MurNAc and GlcNAc and eluted with a retention time similar to that of monomeric muropeptides (Fig. S2). Such a disaccharide would have a calculated monoisotopic mass of 496.190439 Da and a molecular ion, [M+H]+, of 497.197716 Da. This tentative identification was supported by tandem mass spectrometry (MS/MS) analysis (Fig. S3) (34). MS/MS analysis of the ion with m/z of 497.20 resulted in a classic fragmentation pattern where a neutral loss of GlcNAc results in a fragment (Z1) of m/z 276.1083 (calculated); a fragment ion of 276.11 was observed (33). A neutral loss of MurNAc results in a fragment (B1) of m/z 204.0872 (calculated); a fragment ion of 276.09 was observed. In addition, 2 ions (labeled a and b) corresponded to B1 minus H2O and B1 minus 2H2O, respectively. A feature corresponding to MurNGc was also observed (Fig. S2 and S4). MurNAc-GlcNAc/MurNGc-GlcNAc disaccharide values given in Table 1 are relative to the sum of the observed detector response of the disaccharides and muropeptides, relative to either the Δ5 mutant or the parental strain. This observation suggests that the Δ6 mutant is altered in transglycosylation activity as well as transpeptidation.
DISCUSSION
Our previous study examining mutants with either single or multiple deletions of each of the ldt genes in M. smegmatis showed that only certain combinations of mutations yield mutant phenotypes (29). Central to all mutants with a substantial antibiotic phenotype was the loss of the class 5 ldtC gene, suggesting that LdtC is a key enzyme in this pathway. In this work, we tested this hypothesis by constructing an M. smegmatis mutant lacking all ldt genes except ldtC and a mutant lacking all six ldt genes. We found that LdtC is a critical enzyme for M. smegmatis peptidoglycan biosynthesis and is capable of functioning as the sole l,d-transpeptidase. This is not to say that LdtC is the only functional l,d-transpeptidase in M. smegmatis but that under standard laboratory conditions, this enzyme is sufficient.
Our results demonstrate that the Δ5 mutant, which lacks all M. smegmatis Ldts except LdtC, had essentially a WT phenotype in almost all assays but did exhibit increased rifampin susceptibility. This could be attributed to increased permeability of the cell envelope in the mutant, as rifampin is a large, bulky antibiotic. Interestingly, the Δ5 mutant did not have increased susceptibility to carbapenem antibiotics. Others have examined the in vitro activity of purified, recombinant forms of all five M. tuberculosis Ldt enzymes. LdtA, LdtB, LdtC, and LdtE can catalyze 3-3 linkage reactions, LdtD cannot, and all but LdtC can be covalently modified at their active site by carbapenem antibiotics, suggesting that this enzyme is resistant to carbapenems (22). Furthermore, while crystallization studies with LdtC revealed acylation of the protein with meropenem, no binding to various carbapenems was detected by calorimetry (35). These results are consistent with the report that imipenem can bind to purified Ldts of Mycobacterium abscessus, with the exception of LdtMab3, an LdtC ortholog (36). However, another study using fluorescently labeled antibiotics with cell lysates coupled with mass spectrometry showed that LdtC of M. tuberculosis could be labeled with modified meropenem, although this was not confirmed using a deletion mutant (37). The resistance of LdtC to carbapenem inhibition could explain why we see increased carbapenem susceptibility only in the strains lacking ldtC. Our data indicate that, even with the loss of all other Ldts, if LdtC remains intact there is no change in carbapenem resistance, suggesting that perhaps LdtC might represent a carbapenem resistance pathway for 3-3 linkage formation. Taken together, these findings provide insight into the possibility of targeting LdtC in mycobacteria as a potential therapeutic approach.
Some of the increased susceptibility to antibiotics and PG targeting agents seen in the Δ6 mutant is likely due, in part. to increased permeability due to disorganization of the cell envelope. Cellular morphology of Δ6 was altered, with shortened cell lengths and bulges. PG structural analysis showed that Δ6 mutants lost 3-3 cross-links, which is in agreement with a recent study that found that loss of all Ldts in M. smegmatis results in a lack of 3-3 cross-links, numerous cell wall defects, shortened cells, and altered division (38). However, in contrast with this study, which complemented their Ldt null mutant with ldtE and saw partial complementation, our study complemented ldtC with full complementation using the ldtC genes from M. smegmatis or M. tuberculosis. Additionally, it was previously proposed that l,d-transpeptidases are important for rod shape maintenance and that 3-3 cross-links are incorporated at sites of aging cell wall along the lateral body (38). In agreement with this, M. smegmatis and M. tuberculosis are labeled at the septum and the lateral wall with Ldt-specific fluorescent tetrapeptides (31). It is possible that some l,d-transpeptidases are involved with both lateral maintenance and septal growth.
A model of the two PG cross-linking pathways in mycobacteria is shown in Fig. 6. Both pathways utilize the same PG precursor, a disaccharide bearing the PG pentapeptide. This pentapeptide is the substrate for the classical d,d-transpeptidases, which cleave the terminal d-alanine and use the bond energy to link that peptide to the side chain of a DAP residue on another chain, to form a 4-3 linkage. In the 3-3 pathway, a d,d-carboxypeptidase must cleave the terminal d-alanine from the pentapeptide precursor, and the resulting tetrapeptide can be recognized by the l,d-transpeptidases.
FIG 6.

The 4-3 and 3-3 PG cross-linking pathways. Both pathways utilize the same PG precursor, a disaccharide bearing the PG pentapeptide. This pentapeptide is the substrate for the classical d,d-transpeptidases, which cleave off the terminal d-alanine and use the bond energy to link that peptide to the side chain of a DAP residue on another chain, to form a 4-3 linkage. In the 3-3 pathway, a d,d-carboxypeptidase must cleave off the terminal d-alanine from the pentapeptide precursor, and then the resulting tetrapeptide can be recognized by the l,d-transpeptidases (Ldt), which cleave off the terminal d-alanine and use the bond energy to link the DAP residue of the peptide to the side chain of DAP in another chain, thus forming the 3-3 linkage. NAM, N-acylmuramic acid (either N-acetyl or N-glycolyl [MurNAc/MurNGc]); NAG, N-acetylglucosamine (GlcNAc). The image was created with BioRender.com.
There are 3 or 4 d,d-carboxypeptidase genes in mycobacteria, depending upon the species. M. tuberculosis has three unlinked genes (dacB, dacB1, and dacB2), while M. smegmatis has the same three and an additional copy of dacB2 (dacB2b). Notably, there are only two “core” d,d-carboxypeptidases, DacB1 and DacB, found in all mycobacteria. Except for dacB, single-deletion d,d-carboxypeptidases mutants are viable (39, 40).
Baranowski et al. determined that labeled DacB2b localized in areas of aging peptidoglycan in the analysis of their Ldt-null mutant, and they predicted that bleb formation in the mutant may be due to unchecked d,d-endopeptidase activity (38). However, bleb formation was not completely reduced by knockdown of dacB2 expression, indicating involvement of additional d,d-carboxypeptidases in this phenomenon (38). Pandey et al. found that while single dacB2a or dacB2b deletions had no impact on M. smegmatis physiology, the deletion of both genes resulted in a shift from 3-3 cross-linking to 4-3 cross-linking in the PG of the mutant (40). This shift was marked by a relative proportional 9.5-fold decrease in 3-3 cross-links and a 19-fold increase in 4-3 cross-linking (40). The cross-linking shift was also accompanied by increased susceptibility to β-lactam antibiotics and antitubercular therapeutics, as well as a loss of surface glycopeptidolipid, which is consistent with the phenotypes of the Δ6 ldt mutant reported here (39, 40). In contrast to the dacB2a dacB2b double mutant, which had an increase in the relative proportion of 4-3 linkages, the Δ6 mutant reported here has a proportional decrease in 4-3 linkages, suggesting an inability to compensate for loss of 3-3 linkages by increasing the number of 4-3 linkages. It is possible that this is due to d,d-carboxypeptidase activities processing pentapeptide precursors to dead-end tetrapeptide substrates that cannot be used to make 4-3 linkages (Fig. 6). In this view, some of the phenotypes of the Δ6 mutant could be due to the loss of 3-3 linkages and also the inability to compensate by increasing the number of 4-3 linkages, thus resulting in an overall decrease in total PG cross-links. Further experimentation with various dac deletions in the Δ6 mutant should be able to address these questions.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains are listed in Table 2. Escherichia coli was grown at 37°C in Luria-Bertani (LB) broth (Difco/BD Bioscience, San Jose, CA) or on LB agar. M. smegmatis cultures were grown at 37°C or 42°C (as indicated) in Middlebrook 7H9 broth or on 7H10 solid medium (Difco/BD Bioscience), both of which were supplemented with 0.2% glycerol and ADS (0.5% bovine serum albumin fraction V, 0.2% dextrose, and 0.85% NaCl). Middlebrook 7H9 medium was initially supplemented with 0.05% Tween 80 but was subsequently changed to 0.05% tyloxapol, as the Δ6 ldt mutant grew best in tyloxapol-supplemented broth. We used antibiotics at the following concentrations: hygromycin, 100 μg/mL for M. smegmatis and 200 μg/mL for E. coli; kanamycin, 25 μg/mL for M. smegmatis and 50 μg/mL for E. coli; apramycin, 100 μg/mL for both M. smegmatis and E. coli. Bovine serum albumin fraction V and hygromycin were obtained from Roche Applied Science (Indianapolis, IN). All other antibiotics and supplements were obtained from Sigma-Aldrich Chemical (St. Louis, MO).
TABLE 2.
Bacterial strains used in this study
| Strain | Description | Source or reference |
|---|---|---|
| E. coli | ||
| DH10B | F− mcrA Δ(mcrBC-hsdRMS-mrr) [ϕ80dΔlacZΔM15] ΔlacX74 deoR recA1 endA1 araD139 Δ(ara leu)7697 galU galK λ− rpsL nupG | Invitrogen |
| M. smegmatis | ||
| PM965 | ept-1 rpsL4 ΔblaS | 48 |
| PM2110 | PM965 ldtC::res-hyg-res | 29 |
| PM2546 | PM965 ldtB::res-hyg-res ldtF::res ldtC::aacC41 Δ3 strain | 29 |
| PM2705 | PM965 ldtA::aacC41 ldtB::res ldtF::res ldtG::res-hyg-res | 29 |
| PM2727 | PM965 ldtA::aacC41 ldtB::res ldtF::res ldtG::res | This work |
| PM2750 | PM965 ldtA::aacC41 ldtB::res ldtE::res-hyg-res ldtF::res ldtG::res Δ5 strain | This work |
| PM2766 | PM965 ldtA::aacC41 ldtB::res ldtE::res ldtF::res ldtG::res Δ5 strain | This work |
| PM2785 | PM2766 ldtC+::pMP1103 (ΔldtC::lacZ) (primary recombinant) | This work |
| PM2805 | PM2785 ΔldtC::lacZ Δ6 strain (secondary recombinant) | This work |
| PM3500 | PM2785/pMP855 (ldtC+) | This work |
| PM3502 | PM3500 ΔldtC::lacZ pMP855 Δ6/ldtC+ strain | This work |
| PM3507 | PM3502 ΔldtC::lacZ (pMP855 cured) Δ6 (“ldtC+ cured”) | This work |
| PM3679 | PM2785/pMP850 (ldtCMtb+) | |
| PM3575 | PM2785 ΔldtC::lacZ pMP850 Δ6/ldtCMtb+ strain | This work |
| PM3669 | PM3575 ΔldtC::lacZ (pMP850 cured) Δ6 (“ldtCMtb cured”) | This work |
Plasmids and DNA manipulation.
Plasmids used in this study are described in Table 3. DNA manipulations were performed as previously described (41). Qiagen columns (Qiagen, Valencia, CA) were used to prepare plasmids, while the QIAquick gel purification kit (Qiagen) was used to purify DNA fragments. Restriction and DNA modification enzymes were obtained from New England Biolabs (Beverly, MA) or Fermentas (Hanover, MD). We performed plasmid electroporation with M. smegmatis as previously described (42). Oligonucleotide primers were synthesized by Eurofins (Huntsville, AL). PCRs were performed using iProof (Bio-Rad, Hercules, CA) according to the manufacturer, often in conjunction with GC buffer and 3% dimethyl sulfoxide (DMSO). Plasmids were cured from strains by culturing in broth lacking antibiotic for 48 h, plating onto nonselective medium, and then screening for antibiotic-susceptible clones.
TABLE 3.
Plasmids used in this study
| Plasmid | Description | Source or reference |
|---|---|---|
| pJV53 | Kmr recombineering plasmid | 44 |
| pMV261 | Kanr E. coli-Mycobacterium shuttle vector, contains the groEL promoter, ColE1, pAL500 oriM | 49 |
| pYUB657 | Hygr Ampr, sacB allelic exchange vector, ColE1 | 45 |
| pMP850 | pMV261.ldtCMtb+ (Rv0483) complementation plasmid | This work |
| pMP854 | pMV261 γδ resolvase plasmid | 29 |
| pMP855 | pMV261.ldtC+ (MSMEG_0929) complementation plasmid | 29 |
| pMP1103 | pYUB657-based sacB ΔldtC::lacZ allelic exchange plasmid | This work |
Mutant construction.
Strains were constructed in M. smegmatis PM965, which is in the mc2155 lineage but is streptomycin resistant and lacks blaS, encoding the major β-lactamase (29). We started with a previously published strain, PM2705, which has insertional mutations in ldtA (MSMEG_3528), ldtB (MSMEG_4745), ldtF (MSMEG_1322), and ldtG (MSMEG_0674), the latter having a resolvable hygromycin resistance marker (res-hyg-res). The marker was resolved in PM2705 using pMP854, which expressed the γδ resolvase, to yield the hygromycin-sensitive Δ4 mutant PM2727. This strain was then subjected to recombineering with an ldtE::res-hyg-res allele (ldtE is MSMEG_0233) as previously described by us and others (43, 44), yielding the Δ5 mutant PM2750, which was used for another round of res-hyg-res resolution, to yield the hygromycin-susceptible Δ5 mutant PM2766. This strain was used for sacB-mediated allelic exchange with plasmid pMP1103, which bears hygromycin resistance on the vector backbone and an ldtC (MSMEG_0929) deletion allele lacking 598 bp that are replaced with the E. coli lacZ gene expressed by the M. smegmatis mspA promoter (45). This suicide vector was used for allelic exchange at the ldtC locus in PM2766, yielding the primary recombinant PM2785. Subsequent sucrose-mediated counterselection provided the Δ6 mutant PM2805 with the ΔldtC::lacZ allele. Mutants were verified by PCR and Southern blotting.
Reverse complementation was performed by repeating the allelic exchange using strain PM3500, which is the primary ΔldtC::lacZ recombinant strain PM2785 bearing a replicating, kanamycin-resistant ldtC+ plasmid, pMP855 (29), to yield the secondary ΔldtC::lacZ/ldtC+ recombinant complemented (Δ6/ldtC+) strain, PM3502. This strain was cured of the complementing pMP855 plasmid by liquid culture in the absence of kanamycin selection for 48 h and subsequent plate screening for kanamycin sensitivity, to yield the cured control Δ6 mutant, PM3507. A similar strategy was used to test complementation with the M. tuberculosis ldtC gene (Rv0483) using the complementing plasmid pMP850. Mutants were verified by PCR.
Growth curves.
Log-phase broth cultures (optical density at 600 nm [OD600], 0.5 to 0.8) in 7H9 medium were used to inoculate 10 mL 7H9 medium and adjusted to ~0.05 OD600. Cultures were incubated at 37°C with shaking, and OD was measured at 0, 6, 12, 24, and 48 h using a Beckman Du530 Life Science UV/visible-spectrum spectrophotometer. Viable plating was also performed at each time point, with plates incubated at 37°C for 3 days. Temperature sensitivity was assayed by plating and incubation at 37°C and 42°C or by growing liquid cultures at 37°C until late log phase, at which time the culture was shifted to 42°C. Viable plate counts were then determined pre- and post-temperature shift by plating serial dilutions onto agar plates and incubating at 37°C. Assays were performed twice in triplicate.
Susceptibility testing.
Log-phase broth cultures (OD600, 0.5 to 0.8) in 7H9 medium were adjusted to an OD600 of ~0.5. For antibiotic and chemical testing, 200 μL of the suspension was mixed with 3 mL of molten 0.7% Noble agar from Sigma-Aldrich Chemical (St. Louis, MO) and then immediately poured onto 7H10 plates. Once the top agar was solidified, either prepared Sensi-Discs (BD Biosciences) or compound-soaked sterile discs (BD 3 Biosciences) using antibiotics from Sigma-Aldrich Chemical (St. Louis, MO) were placed on the plates. The antibiotics tested were ampicillin (50 μg), rifampin (25 μg), erythromycin (15 μg), vancomycin (30 μg), ceftriaxone (100 μg), imipenem (10 μg), ertapenem (10 μg), meropenem (10 μg), ethambutol (25 μg), isoniazid (5 μg), and ciprofloxacin (50 μg). SDS from EM Science (Gibbstown, NJ) was used at a concentration of 20% in 10-μL volumes on sterile discs. These plates were then placed in the 37°C incubator for 48 h. The zones were measured to the nearest millimeter. Six repetitions were done for each strain per antibiotic or chemical (either two cultures in triplicate or three cultures in duplicate). In the case of SDS testing, three independent experiments were done, each in quadruplicate.
For lysozyme susceptibility testing, cultures were serially spot diluted and plated onto 7H10 plates with or without 100 μg/mL lysozyme (Sigma-Aldrich Chemical, catalog no. L-7651, lot no. 77H7032). For noncanonical d-amino acid sensitivity (NCDAA) testing, cultures were serially spot diluted and plated onto 7H10 plates with or without 15 mM d-methionine. Plates were then incubated at 37°C for 3 days. Lysozyme and NCDAA determinations were done in triplicate, and results from a representative set are shown.
Congo red binding assay.
Congo red binding to cells was done with a previously published method, with a few modifications (46). Strains were streaked onto 7H10 plates lacking ADS enrichment, incubated at 37°C for 3 days, and then incubated at room temperature for 5 days. Cells were scraped off the plates and suspended in phosphate-buffered saline with 0.05% Tween 80 (PBST) in weighed tubes and then pelleted. The PBST was discarded and the cell pellet weight determined before resuspension in 1 mL of acetone. The mixture was incubated on a roller apparatus for 1 h at room temperature and then centrifuged at 12,000 rpm for 5 min to pellet the cells. The OD488 was determined for the acetone supernatant, and the Congo red binding calculated by dividing the OD488 by the wet weight (in grams) of the cell pellet. Each strain was assayed in triplicate, and assays were performed on duplicate sets of cultures.
Electron microscopy.
Scanning electron microscopy (SEM) was performed using M. smegmatis cells from a 50-mL stationary-phase culture grown at 37°C. Cells were recovered by centrifugation and resuspended in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.2) overnight, washed in buffer, postfixed in 1.0% buffered osmium tetroxide, dehydrated in a graded series of ethanol and then hexamethyldisilazane (HMDS). A drop of suspended M. smegmatis in 100% HMDS was allowed to dry on a round glass coverslip mounted on an aluminum stub, which was then sputter coated with gold for imaging using a Zeiss Auriga Supra 40VP field emission SEM at 5 kV. ImageJ was used to measure individual cell lengths. Twenty cells were imaged for each strain.
PG preparation and analysis.
Cells from 2-L cultures grown to mid-log phase were harvested by centrifugation, washed twice with PBS, sonicated (VirSonic 60 with a microtip, 1 min for 10 cycles [60 s on/90 s off, 10 times] on ice), and the centrifuged. The pellet was extracted six times with 2% SDS in PBS at 37°C. Between the fourth and fifth extractions, self-digested pronase was added to a concentration of 0.1 mg/mL, and mixtures were incubated for 1 h at 45°C, followed by 1 h of incubation at 90°C. After all SDS extractions, the preparation was washed five times with PBS, twice with deionized water, and twice with acetone. The final pellets were air dried overnight and then stored at −80°C until further use. PG peptides were isolated from purified MAPc after enzymatic digestion with recombinant LysA amidase, which cleaves peptides from the muramyl moieties in the PG as previously described (47). The reaction mixture was centrifuged and the supernatant applied to a Millipore Ultrafree centrifugal ultrafiltration column with a 5-kDa cutoff.
The subsequent flowthrough, containing PG peptides, was analyzed by an Agilent quadrupole time-of-flight (Q-TOF) mass spectrometer (model 6520) connected up front with an Agilent 1260 series high-pressure liquid chromatography (HPLC) system as previously described (31, 33). The soluble peptides were separated on a Phenomenex HyperClone C18 column (2.0 mm by 150 mm; 5 μm) applying a mobile phase consisting of LC-MS-grade water (solvent A) and methanol (solvent B), both containing 0.1% formic acid, at a flow rate of 0.320 μL/min. A solvent gradient was initiated at 0% B, increasing to 15% over 20 min, increasing to 50% over 10 min, and further increasing to 100% over 7 min, and flow was maintained at this condition for 3 min before returning initial conditions over 5 min. Total analysis time for this analysis was set to 45 min. Column oven temperature was maintained at 40°C throughout the analysis. The eluate was directly introduced into a mass spectrometer equipped with dual electrospray ionization operating in positive mode. Drying gas temperature, drying gas flow, capillary voltage, nebulizer pressure, and fragmentor voltage were set to 325°C, 10.0 L/min, 3,500 V, 45 lb/in2, and 120 V, respectively. The Q-TOF instrument was tuned prior to sample analysis to reveal stability and mass accuracy during analysis (typically < 2 ppm).
The positive-ion MS and tandem MS data were collected using Agilent MassHunter workstation software and processed with the molecular feature extraction algorithm (features with a defined exact mass and retention time) for each sample. A preset minimum value of 500 counts was used to filter out low-abundance molecular features. An in-house database containing calculated monoisotopic masses of possible monomeric, dimeric, and trimeric peptides, including those with amidated carboxylic residues and/or glycine residues (up to a molecular weight of 2 kDa), was used to identify the observed molecular features. The features with a database match were used for identification of peptides with cross-links. The assigned identity of the major peptides was confirmed by tandem mass spectrometry when possible. Cell wall preparations were purified and processed in duplicate for each strain. Each sample was analyzed by mass spectrometry for matches, but only one sample per strain was analyzed in depth.
Statistical analysis.
GraphPad software (GraphPad Software, Inc., La Jolla, CA) was used to analyze data. An unpaired Student's t test was used to compare mutant to WT or to compare an experimental condition to control for a particular strain, as indicated in the figure legends. Alternatively, ordinary one-way analysis of variance (ANOVA) was used.
ACKNOWLEDGMENTS
This work was supported by NIH grants AI073772, AI139058 (M.S.P.), T32 GM68411 (Z.K.F.), and T32 AI007362 (A.N.S.).
We acknowledge Karen Bentley of the University of Rochester Center for Advanced Research Technologies for performing SEM and Lori Wright for assisting with cell wall preparations. We also thank Elaine Smolock for critical reading of the manuscript.
Footnotes
Supplemental material is available online only.
Contributor Information
Martin S. Pavelka, Jr., Email: martin_pavelka@urmc.rochester.edu.
Patricia A. Champion, University of Notre Dame
REFERENCES
- 1.World Health Organization. 2017. Global tuberculosis report.
- 2.Cohen KA, Bishai WR, Pym AS. 2014. Molecular basis of drug resistance in Mycobacterium tuberculosis. Microbiol Spectr 2:MGM2-0036-2013. doi: 10.1128/microbiolspec.MGM2-0036-2013. [DOI] [PubMed] [Google Scholar]
- 3.Alderwick LJ, Harrison J, Lloyd GS, Birch HL. 2015. The mycobacterial cell wall—peptidoglycan and arabinogalactan. Cold Spring Harb Perspect Med 5:a021113. doi: 10.1101/cshperspect.a021113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jankute M, Cox JAG, Harrison J, Besra GS. 2015. Assembly of the mycobacterial cell wall. Annu Rev Microbiol 69:405–423. doi: 10.1146/annurev-micro-091014-104121. [DOI] [PubMed] [Google Scholar]
- 5.Pavelka MS, Jr, Mahapatra S, Crick DC. 2014. Genetics of peptidoglycan biosynthesis. Microbiol Spectr 2:MGM2-0034–2013. doi: 10.1128/microbiolspec.MGM2-0034-2013. [DOI] [PubMed] [Google Scholar]
- 6.Mahapatra S, Scherman H, Brennan PJ, Crick DC. 2005. N glycolylation of the nucleotide precursors of peptidoglycan biosynthesis of Mycobacterium spp. is altered by drug treatment. J Bacteriol 187:2341–2347. doi: 10.1128/JB.187.7.2341-2347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar P, Arora K, Lloyd JR, Lee IY, Nair V, Fischer E, Boshoff HIM, Barry CE. 2012. Meropenem inhibits d,d-carboxypeptidase activity in Mycobacterium tuberculosis. Mol Microbiol 6:367–381. doi: 10.1111/j.1365-2958.2012.08199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lavollay M, Arthur M, Fourgeaud M, Dubost L, Marie A, Veziris N, Blanot D, Gutmann L, Mainardi J-L. 2008. The peptidoglycan of stationary-phase Mycobacterium tuberculosis predominantly contains cross-links generated by l,d-transpeptidation. J Bacteriol 190:4360–4366. doi: 10.1128/JB.00239-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kieser KJ, Rubin EJ. 2014. How sisters grow apart: mycobacterial growth and division. Nat Rev Microbiol 12:550–562. doi: 10.1038/nrmicro3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rimal B, Senzani S, Ealand C, Lamichhane G, Kana B, Kim SJ. 2022. Peptidoglycan compositional analysis of Mycobacterium smegmatis using high-resolution LC-MS. Sci Rep 12:11061. doi: 10.1038/s41598-022-15324-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 12.Sobhanifar S, King DT, Strynadka NC. 2013. Fortifying the wall: synthesis, regulation and degradation of bacterial peptidoglycan. Curr Opin Struct Biol 23:695–703. doi: 10.1016/j.sbi.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 13.Bush K, Bradford PA. 2016. β-Lactams and β-lactamase inhibitors: an overview. Cold Spring Harb Perspect Med 6:a025247. doi: 10.1101/cshperspect.a025247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisher JF, Mobashery S. 2016. β-Lactam resistance mechanisms: Gram-positive bacteria and Mycobacterium tuberculosis. Cold Spring Harb Perspect Med 6:a025221. doi: 10.1101/cshperspect.a025221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lavollay M, Fourgeaud M, Herrmann J-L, Dubost L, Marie A, Gutmann L, Arthur M, Mainardi J-L. 2011. The peptidoglycan of Mycobacterium abscessus is predominantly cross-linked by l,d-transpeptidases. J Bacteriol 193:778–782. doi: 10.1128/JB.00606-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanders AN, Pavelka MS. 2013. Phenotypic analysis of Eschericia coli mutants lacking l,d-transpeptidases. Microbiology (Reading) 159:1842–1852. doi: 10.1099/mic.0.069211-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glauner B, Höltje JV, Schwarz U. 1988. The composition of the murein of Escherichia coli. J Biol Chem 263:10088–10095. doi: 10.1016/S0021-9258(19)81481-3. [DOI] [PubMed] [Google Scholar]
- 18.Flores AR, Parsons LM, Pavelka MS, Jr.. 2005. Genetic analysis of the beta-lactamases of Mycobacterium tuberculosis and Mycobacterium smegmatis and susceptibility to beta-lactam antibiotics. Microbiology (Reading) 151:521–532. doi: 10.1099/mic.0.27629-0. [DOI] [PubMed] [Google Scholar]
- 19.Smith T, Wolff KA, Nguyen L. 2013. Molecular biology of drug resistance in Mycobacterium tuberculosis, p 53–80. In Pieters J, McKinney JD (ed), Pathogenesis of Mycobacterium tuberculosis and its interaction with the host organism. Springer, Berlin, Germany. [Google Scholar]
- 20.Hugonnet JE, Mengin-Lecreulx D, Monton A, den Blaauwen T, Carbonnelle E, Veckerlé C, Brun YV, van Nieuwenhze M, Bouchier C, Tu K, Rice LB, Arthur M. 2016. Factors essential for l,d-transpeptidase-mediated peptidoglycan cross-linking and β-lactam resistance in Escherichia coli. Elife 5:e19469. doi: 10.7554/eLife.19469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Triboulet S, Dubée V, Lecoq L, Bougault C, Mainardi J-L, Rice LB, Ethève-Quelquejeu M, Gutmann L, Marie A, Dubost L, Hugonnet J-E, Simorre J-P, Arthur M. 2013. Kinetic features of l,d-transpeptidase inactivation critical for β-lactam antibacterial activity. PLoS One 8:e67831. doi: 10.1371/journal.pone.0067831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cordillot M, Dubée V, Triboulet S, Dubost L, Marie A, Hugonnet JE, Arthur M, Mainardi JL. 2013. In vitro cross-linking of Mycobacterium tuberculosis peptidoglycan by l,d-transpeptidases and inactivation of these enzymes by carbapenems. Antimicrob Agents Chemother 57:5940–5945. doi: 10.1128/AAC.01663-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar P, Chauhan V, Silva JRA, Lameira J, d'Andrea FB, Li S-G, Ginell SL, Freundlich JS, Alves CN, Bailey S, Cohen KA, Lamichhane G. 2017. Mycobacterium abscessus l,d-transpeptidases are susceptible to inactivation by carbapenems and cephalosporins but not penicillins. Antimicrob Agents Chemother 61:e00866-17. doi: 10.1128/AAC.00866-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mainardi J-L, Hugonnet J-E, Rusconi F, Fourgeaud M, Dubost L, Moumi AN, Delfosse V, Mayer C, Gutmann L, Rice LB, Arthur M. 2007. Unexpected inhibition of peptidoglycan ld-transpeptidase from Enterococcus faecium by the β-lactam imipenem. J Biol Chem 282:30414–30422. doi: 10.1074/jbc.M704286200. [DOI] [PubMed] [Google Scholar]
- 25.Davies Forsman L, Giske CG, Bruchfeld J, Schön T, Juréen P, Ängeby K. 2015. Meropenem-clavulanate has high in vitro activity against multidrug-resistant Mycobacterium tuberculosis. Int J Mycobacteriol 4(Suppl 1):80–81. doi: 10.1016/j.ijmyco.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 26.Diacon AH, van der Merwe L, Barnard M, von Groote-Bidlingmaier F, Lange C, García-Basteiro AL, Sevene E, Ballell L, Barros-Aguirre D. 2016. β-Lactams against tuberculosis—new trick for an old dog? N Engl J Med 375:393–394. doi: 10.1056/NEJMc1513236. [DOI] [PubMed] [Google Scholar]
- 27.Payen MC, De Wit S, Martin C, Sergysels R, Muylle I, Van Laethem Y, Clumeck N. 2012. Clinical use of the meropenem-clavulanate combination for extensively drug-resistant tuberculosis. Int J Tuber Lung Dis 16:558–560. doi: 10.5588/ijtld.11.0414. [DOI] [PubMed] [Google Scholar]
- 28.Payen MC, Muylle I, Vandenberg O, Mathys V, Delforge M, Van den Wijngaert S, Clumeck N, De Wit S. 2018. Meropenem-clavulanate for drug-resistant tuberculosis: a follow-up of relapse-free cases. Int J Tuber Lung Dis 22:34–39. doi: 10.5588/ijtld.17.0352. [DOI] [PubMed] [Google Scholar]
- 29.Sanders AN, Wright LF, Pavelka MS. 2014. Genetic characterization of mycobacterial l,d-transpeptidases. Microbiology (Reading) 160:1795–1806. doi: 10.1099/mic.0.078980-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zandi TA, Marshburn RL, Stateler PK, Brammer Basta LA. 2019. Phylogenetic and biochemical analyses of mycobacterial l,d-transpeptidases reveal a distinct enzyme class that is preferentially acylated by meropenem. ACS Infect Dis 5:2047–2054. doi: 10.1021/acsinfecdis.9b00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pidgeon SE, Apostolos AJ, Nelson JM, Shaku M, Rimal B, Islam MN, Crick DC, Kim SJ, Pavelka MS, Kana BD, Pires MM. 2019. l,d-Transpeptidase specific probe reveals spatial activity of peptidoglycan cross-linking. ACS Chem Biol 14:2185–2196. doi: 10.1021/acschembio.9b00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horcajo P, de Pedro MA, Cava F. 2012. Peptidoglycan plasticity in bacteria: stress-induced peptidoglycan editing by noncanonical D-amino acids. Microb Drug Resist 18:306–313. doi: 10.1089/mdr.2012.0009. [DOI] [PubMed] [Google Scholar]
- 33.Mahapatra S, Piechota C, Gil F, Ma Y, Huang H, Scherman MS, Jones V, Pavelka MS, Jr, Moniz-Pereira J, Pimentel M, McNeil MR, Crick DC. 2013. Mycobacteriophage Ms6 LysA: a peptidoglycan amidase and a useful analytical tool. Appl Environ Microbiol 79:768–773. doi: 10.1128/AEM.02263-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Domon B, Costello CE. 1988. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj J 5:397–409. doi: 10.1007/BF01049915. [DOI] [Google Scholar]
- 35.Brammer Basta LA, Ghosh A, Pan Y, Jakoncic J, Lloyd EP, Townsend CA, Lamichhane G, Bianchet MA. 2015. Loss of a functionally and structurally distinct l,d-transpeptidase, LdtMt5, compromises cell wall integrity in Mycobacterium tuberculosis. J Biol Chem 290:25670–25685. doi: 10.1074/jbc.M115.660753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dousa KM, Kurz SG, Taracila MA, Bonfield T, Bethel CR, Barnes MD, Selvaraju S, Abdelhamed AM, Kreiswirth BN, Boom WH, Kasperbauer SH, Daley CL, Bonomo RA. 2020. Insights into the l,d-transpeptidases and d,d-carboxypeptidase of Mycobacterium abscessus: ceftaroline, imipenem, and novel diazabicyclooctane inhibitors. Antimicrob Agents Chemother 64:e00098-20. doi: 10.1128/AAC.00098-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levine SR, Beatty KE. 2021. Investigating β-lactam drug targets in Mycobacterium tuberculosis using chemical probes. ACS Infect Dis 7:461–470. doi: 10.1021/acsinfecdis.0c00809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baranowski C, Welsh MA, Sham LT, Eskandarian HA, Lim HC, Kieser KJ, Wagner JC, McKinney JD, Fantner GE, Ioerger TR, Walker S, Bernhardt TG, Rubin EJ, Rego EH. 2018. Maturing Mycobacterium smegmatis peptidoglycan requires non-canonical crosslinks to maintain shape. Elife 7:e37516. doi: 10.7554/eLife.37516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ealand CS, Asmal R, Mashigo L, Campbell L, Kana BD. 2019. Characterization of putative dd-carboxypeptidase-encoding genes in Mycobacterium smegmatis. Sci Rep 9:5194. doi: 10.1038/s41598-019-41001-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pandey SD, Pal S, Kumar NG, Bansal A, Mallick S, Ghosh AS. 2018. Two dd-carboxypeptidases from Mycobacterium smegmatis affect cell surface properties through regulation of peptidoglycan cross-linking and glycopeptidolipids. J Bacteriol 200:e00760-17. doi: 10.1128/JB.00760-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ausubel F, Brent R, Kingston R, Moore D, Seidman J, Smith J, Struhl K. 1987. Current Protocols in Molecular Biology. Wiley, New York, NY. [Google Scholar]
- 42.Pavelka MS, Jacobs WR. 1996. Biosynthesis of diaminopimelate, the precursor of lysine and a component of peptidoglycan, is an essential function of Mycobacterium smegmatis. J Bacteriol 178:6496–6507. doi: 10.1128/jb.178.22.6496-6507.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martinelli DJ, Pavelka MS. 2016. The RipA and RipB peptidoglycan endopeptidases are individually nonessential to Mycobacterium smegmatis. J Bacteriol 198:1464–1475. doi: 10.1128/JB.00059-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Kessel JC, Marinelli LJ, Hatfull GF. 2008. Recombineering mycobacteria and their phages. Nat Rev Microbiol 6:851–857. doi: 10.1038/nrmicro2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pavelka MS, Jacobs WR. 1999. Comparison of the construction of unmarked deletion mutations in Mycobacterium smegmatis, Mycobacterium bovis bacillus Calmette-Guérin, and Mycobacterium tuberculosis H37Rv by allelic exchange. J Bacteriol 181:4780–4789. doi: 10.1128/JB.181.16.4780-4789.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cangelosi GA, Palermo CO, Laurent JP, Hamlin AM, Brabant WH. 1999. Colony morphotypes on Congo red agar segregate along species and drug susceptibility lines in the Mycobacterium avium-intracellulare complex. Microbiology (Reading) 145:1317–1324. doi: 10.1099/13500872-145-6-1317. [DOI] [PubMed] [Google Scholar]
- 47.Mahapatra S, Crick DC, McNeil MR, Brennan PJ. 2008. Unique structural features of the peptidoglycan of Mycobacterium leprae. J Bacteriol 190:655–661. doi: 10.1128/JB.00982-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raymond JB, Mahapatra S, Crick DC, Pavelka MS. 2005. Identification of the namH gene, encoding the hydroxylase responsible for the N-glycolylation of the mycobacterial peptidoglycan. J Biol Chem 280:326–333. doi: 10.1074/jbc.M411006200. [DOI] [PubMed] [Google Scholar]
- 49.Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, Snapper SB, Barletta RG, Jacobs WR, Bloom BR. 1991. New use of BCG for recombinant vaccines. Nature 351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S4. Download jb.00424-22-s0001.pdf, PDF file, 0.7 MB (796.9KB, pdf)