

Abstract
Estrogen contributes to the development of breast cancer through estrogen receptor (ER) signaling and by generating genotoxic metabolites that cause oxidative DNA damage. To protect against oxidative stress, cells activate nuclear factor erythroid 2-related factor 2 (Nrf2) and its downstream cytoprotective genes that initiate antioxidant responses and detoxify xenobiotics. Nrf2 activation occurs by inhibiting the protein-protein interaction (PPI) between Nrf2 and its inhibitor Keap1, which otherwise targets Nrf2 for ubiquitination and destruction. In this study, we examined a series of novel direct inhibitors of Keap1-Nrf2 PPI in their role in promoting the availability of Nrf2 for antioxidant activity and attenuating estrogen-mediated responses in breast cancer. ER-positive human breast cancer cells MCF-7 were treated with 17β-estradiol (E2) in the presence or absence of selected Keap1-Nrf2 PPI inhibitors. Keap1-Nrf2 PPI inhibitors suppressed the mRNA and protein levels of estrogen responsive genes induced by E2 exposure, such as PGR. Keap1-Nrf2 PPI inhibitors caused significant activation of Nrf2 target genes. E2 decreased the mRNA and protein level of the Nrf2 target gene NQO1, and the Keap1-Nrf2 PPI inhibitors reversed this effect. The reversal of E2 action by these compounds was not due to binding to ER as ER antagonists. Further, a selected compound attenuated oxidative stress induced by E2, determined by the level of a biomarker 8-oxo-deoxyguanosine. These findings suggest that the Keap1-Nrf2 PPI inhibitors have potent antioxidant activity by activating Nrf2 pathways and inhibit E2-induced gene and protein expression. These compounds may serve as potential chemopreventive agents in estrogen-stimulated breast cancer.
Keywords: Breast cancer, Nrf2, estrogen, Keap1, estrogen receptor
1. Introduction
Estrogen receptor-positive breast cancers represent 65–75% of all breast cancer cases (Burstein, 2020). Estrogen acts as an important mediator in the development of breast cancer. Estrogen receptor (ER) signaling is responsible for cell proliferation and inhibition of apoptosis (Yager and Davidson, 2006). In addition, the metabolites of estrogen cause oxidative damage by forming quinone adducts with DNA (Yager and Davidson, 2006). These two major mechanisms define estrogen as a carcinogen in the development of ER-positive breast cancer. Therefore, targeting estrogen synthesis, signaling and metabolism are key mechanisms in the treatment of ER-positive breast cancer (Burstein, 2020; Jordan, 2006; Smith and Dowsett, 2003).
One way to protect cells from genotoxicity of estrogen and its metabolites is to activate the cellular detoxification mechanism, which involves the action of the nuclear factor erythroid 2-related factor 2 (Nrf2). Nrf2 plays a vital role in regulating antioxidant pathways by activating over 250 target genes in the antioxidant response element (ARE) pathway (Dodson et al., 2019; Yamamoto et al., 2018). Nrf2 target genes, mainly those encoding detoxifying enzymes, are involved in redox regulation, DNA repair and xenobiotic metabolism (Schmidlin et al., 2019). Nrf2 is described as a pleiotropic transcription factor and its role in cancer is context-dependent (Rojo de la Vega et al., 2018). Its action during different stages of cancer development varies from inhibition of carcinogenesis to promotion of cancer progression (Rojo de la Vega et al., 2018; Dodson et al., 2019). Prior to tumor initiation, Nrf2 activation can be useful in combating carcinogenesis via its ability to initiate antioxidant gene transcription, which detoxifies xenobiotics and repairs DNA damage (Rojo de la Vega et al., 2018). As the tumor establishes, high Nrf2 expression renders cells resistant to reactive oxygen species (ROS), genotoxic and metabolic stress, which in turn increases cancer cell proliferation and resistance to radiation and chemotherapies (Cloer et al., 2019). In the context of estrogen-induced carcinogenesis, the Nrf2 system can remove DNA adducts and convert estrogen metabolites to a less toxic form, preventing genomic instability. This is considered as the chemopreventive role of Nrf2, which is the focus of our study.
Nrf2 activation can be achieved by oxidative stress as well as pharmacological interventions. Under basal conditions, Kelch-like ECH associated protein (Keap1) acts as a negative modulator of Nrf2 by binding to Nrf2 at the DLG and ETGE motif of the Neh2 domain as a homodimer (Lee and Hu, 2020). Subsequently, Nrf2 undergoes ubiquitination and is degraded by 26S proteasome, giving rise to a short half-life of 0.33 hours (Cuadrado et al., 2019). However, when cells are exposed to oxidative stress, the interaction between Keap1 and Nrf2 is disrupted. Nrf2 translocates into the nucleus and activates the ARE pathway, which exerts cytoprotective effects against oxidative damage (Lee and Hu, 2020). Compounds have been developed to activate Nrf2 by inhibiting the interaction of Keap1 and Nrf2. Bardoxolone (CDDO, triterpenoid), sulforaphane, and dimethyl fumarate are known Nrf2 activators that have been placed into clinical trials (Dodson et al., 2019). There are other Nrf2 activators under study, including natural products like resveratrol, curcumin, and EGCG (Pouremamali et al., 2022).
To understand the chemoprotective role of Nrf2 activation in mitigating estrogen carcinogenicity, we investigated the effects of novel small molecule inhibitors of Keap1-Nrf2 protein-protein interaction (PPI) in ER-positive breast cancer. Earlier Nrf2 activators are mainly electrophiles, modifying Cys151 of Keap1 and have dose limiting toxicity due to off-target effects (Sivinski et al., 2021). Our Keap1-Nrf2 PPI inhibitors activate Nrf2 by binding noncovalently to the Keap1 Kelch domain, thereby inhibiting the interaction between the Kelch domain and the ETGE or DLG motif on the Nrf2 Neh2 domain (Lee and Hu, 2020). As a result, Nrf2 enters the nucleus and initiates transcription of downstream target genes in the ARE pathway (Lee and Hu, 2020). It has been previously shown that an earlier series of these Keap1-Nrf2 PPI inhibitors activated the Nrf2 system and upregulated Nrf2 target genes and proteins in HEK293 cells (Wen et al., 2015) and mouse BV-2 microglial cells (Abed et al., 2021). However, the effects of the new series of the Keap1-Nrf2 PPI inhibitors on breast cancer and estrogen carcinogenicity have not been examined. In this study, we assessed the ability of the Keap1-Nrf2 PPI inhibitors to mitigate E2-induced estrogen downstream target gene expression and oxidative stress in ER-positive breast cancer via activating the Nrf2 pathway.
2. Material and Methods
2.1. Reagents and Cell Culture
Small-molecule inhibitors (LH1092, LH1093, LH1095, and LH1101) were analogs of 1,4-bis[(4-methoxyphenyl)sulfonamido]naphthalene N,N’-diacetic acid (LH762) and were prepared in-house in a similar fashion as previously reported method by Jiang and coworkers (Jiang et al., 2014; Abed et al., 2021). The Keap1-Nrf2 PPI inhibitors (LH1092, LH1093, LH1095, and LH1101) were dissolved in DMSO and stored as stock solutions (10 mM) at −80°C. 17β-estradiol (E2) was obtained from Sigma-Aldrich (Saint Louis, MO) and dissolved in DMSO. The human breast cancer MCF-7 cell line was obtained from American Type Culture Collection (ATCC). MCF-7 cells were maintained in DMEM/F12 medium containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37°C and 5% CO2. To examine the effects of estrogen stimulation, MCF-7 cells were first seeded in DMEM/F12 medium with 10% FBS. After overnight incubation, the cells were washed with PBS and grown in phenol red-free RPMI medium supplemented with 10% charcoal-stripped FBS containing treatment. Treatment concentrations and conditions are described in respective figure legends.
2.2. PolarScreen™ Green Estrogen Receptor Alpha Competitor Assay
PolarScreen™ Estrogen Receptor Alpha Competitor Assay kit was obtained from Life Technologies Corporation (A15883; Carlsbad, CA). The assay was performed to determine the binding activity of Keap1-Nrf2 PPI inhibitors to estrogen receptor alpha (ERα) following the manufacturer’s protocol with slight modification. A total assay volume of 20 μL was used in 384-well plate format. 2X saturating estradiol solution (20 μM) and test compounds (0, 1, 10, 100 μM) were added to the appropriate wells of the assay plate. The plate was incubated at room temperature for 2 hours before fluorescence was read using a Wallac Victor 3V multilabel microplate reader (PerkinElmer, Shelton, CT) and 485 nm/535 nm filter set. Fluorescence polarization value (mP) was calculated based on the parallel and perpendicular fluorescence intensity (F∥ and F⊥) with respect to the linearly polarized excitation light (Inoyama et al., 2012).
2.3. Quantitative Real-Time PCR Analysis
MCF-7 cells were seeded in 60 mm dishes at a density of 300,000 cells per dish. Cells were treated with 0.1% DMSO, 100 pM E2, 10 μM LH1092 or 100 nM CDDO-IM (a known Nrf2 activator as a positive control) with or without 100 pM E2 for 16 hours before RNA extraction. To assess the activity of all four Keap1-Nrf2 PPI inhibitors, cells were treated with 0.1% DMSO and 100 pM E2 in the presence or absence of 10 μM test compounds (LH1092, LH1093, LH1095 and LH1101) or 100 nM CDDO-IM for 48 hours before RNA extraction. Total RNA was isolated using the TRIzol reagent from Sigma Aldrich (Saint Louis, MO). The cDNA was synthesized from 1 μg of RNA using the High-Capacity cDNA Reverse Transcription Kit from Applied Biosystems (Pleasanton, CA). The mRNA levels of genes of interest were measured by RT-qPCR using TaqMan 2X Universal PCR Master Mix (Applied Biosystems, Pleasanton, CA). The primers, PGR (Hs01556702), CCND1 (Hs00277039), CITED1 (Hs00918445), CTSD (Hs00157205), SERPINA1 (Hs00165475), TFF1 (Hs00907239), HO-1 (Hs01110250_m1), NQO1 (Hs00168547), GCLM (Hs00157694), GPX1 (Hs00829989), GPX2 (Hs01591589), and GAPDH (Hs02758991), were obtained from Applied Biosystems (Pleasanton, CA). The ΔΔCt values were calculated from Ct values compared to a reference gene GAPDH. Results are represented as fold changes compared to the DMSO control group.
2.4. Western Blot Analysis
MCF-7 cells were seeded in 100 mm dishes at a density of 500,000 cells per dish and treated with 0.1% DMSO or 100 pM E2 in the presence or absence of 10 μM LH1092, 10 μM LH1095, or 100 nM CDDO-IM. After 24 hours of treatment, cells were lysed in RIPA buffer containing 1% protease and phosphatase inhibitor. Cell lysates were separated by 10% SDS-PAGE and transferred to a PVDF membrane. The membranes were incubated with primary antibodies at 4°C overnight and with secondary antibodies at room temperature for 1 hour. Primary antibodies against PGR (1:500, 8757), c-MYC (1:1000, 5605), TFF1/pS2 (1:1000, 15571), and Cathepsin D (1:1000, 2284) were from Cell Signaling Technology (Danvers, MA). Primary antibodies against HO-1 (1:200, sc-10789) and NQO1 (1:100, sc-271116) were from Santa Cruz Biotechnology (Dallas, TX). β-actin antibody (1:2000, A1978) was from Sigma Aldrich (Saint Louis, MO). Anti-rabbit and anti-mouse secondary antibodies were from Cell Signaling Technology (Danvers, MA). β-actin was used as a loading control.
2.5. Immunofluorescence Microscopy
To determine the Nrf2 nuclear translocation in response to the test compounds, MCF-7 cells were seeded at 200,000 cells per well in 6-well plates and treated with 0.1% DMSO, 10 μM of LH1092 or LH1095 with or without 1 nM of E2 for 3 hours. To assess the level of oxidative stress changed by the compounds, MCF-7 cells were seeded at 250,000 cells per well in 6-well plates and treated with 0.1% DMSO, 10 μM LH1092 with or without 1 nM E2 or 1 μM etoposide for 48 hours. Cells were fixed using 4% paraformaldehyde for 15 minutes at room temperature. Cells were incubated in PBS containing 0.2% Triton X-100 for 10 minutes for permeabilization, then blocked in PBST with 10% horse serum for 1 hour at room temperature. Cells were incubated with primary antibody against Nrf2 (1:100 dilution, sc-365949, Santa Cruz, Dallas, TX) or 8-oxo-deoxyguanosine (8-oxo-dG, 1:300 dilution, R&D systems, Gaithersburg, MD) at 4°C overnight. After washing with PBS, goat anti-mouse secondary antibody (Alexa Fluor 488, Invitrogen, Carlsbad, CA) was then added to samples and incubated for 1 hour at room temperature. Samples were also treated with TO-PRO-3 dye solution (1:300, Thermo Fisher, Eugene, OR) for 30 minutes at room temperature for nuclear staining. Images were taken with a fluorescence microscope with laser filters at 488 nm (Nrf2 and 8-oxo-dG) and 644 nm (TO-PRO-3). Five fields from each treatment group were imaged. Ten nuclei from each group were randomly selected for analysis. Images were analyzed by measuring the mean gray values of the nuclei using ImageJ (NIH, Bethesda, MD). Data were represented as mean ± SEM of fluorescence intensity.
2.6. Statistical Analysis
The RT-qPCR data were represented as mean ± SEM of fold changes. Ordinary one-way ANOVA followed by Dunnett’s multiple comparisons test was used to compare differences between control and treatment groups. A P value < 0.05 is considered of statistical significance.
3. Results
3.1. Structural modification of a new series of Keap1-Nrf2 PPI inhibitors
Direct inhibitors of Keap1-Nrf2 PPI activate Nrf2 by binding noncovalently to the Keap1 Kelch domain, inhibiting the interaction between the Kelch domain and the ETGE or DLG motif on the Nrf2 Neh2 domain (Lee and Hu, 2020). The series of Keap1-Nrf2 PPI inhibitors used in this study (LH1092, LH1093, LH1095, and LH1101) was synthesized for enhanced activity based on the structure of 1,4-bis[(4-methoxyphenyl)sulfonamido]naphthalene N,N’-diacetic acid (LH762) (Jiang et al., 2014; Abed et al., 2021) (Fig. 1).
Fig. 1.
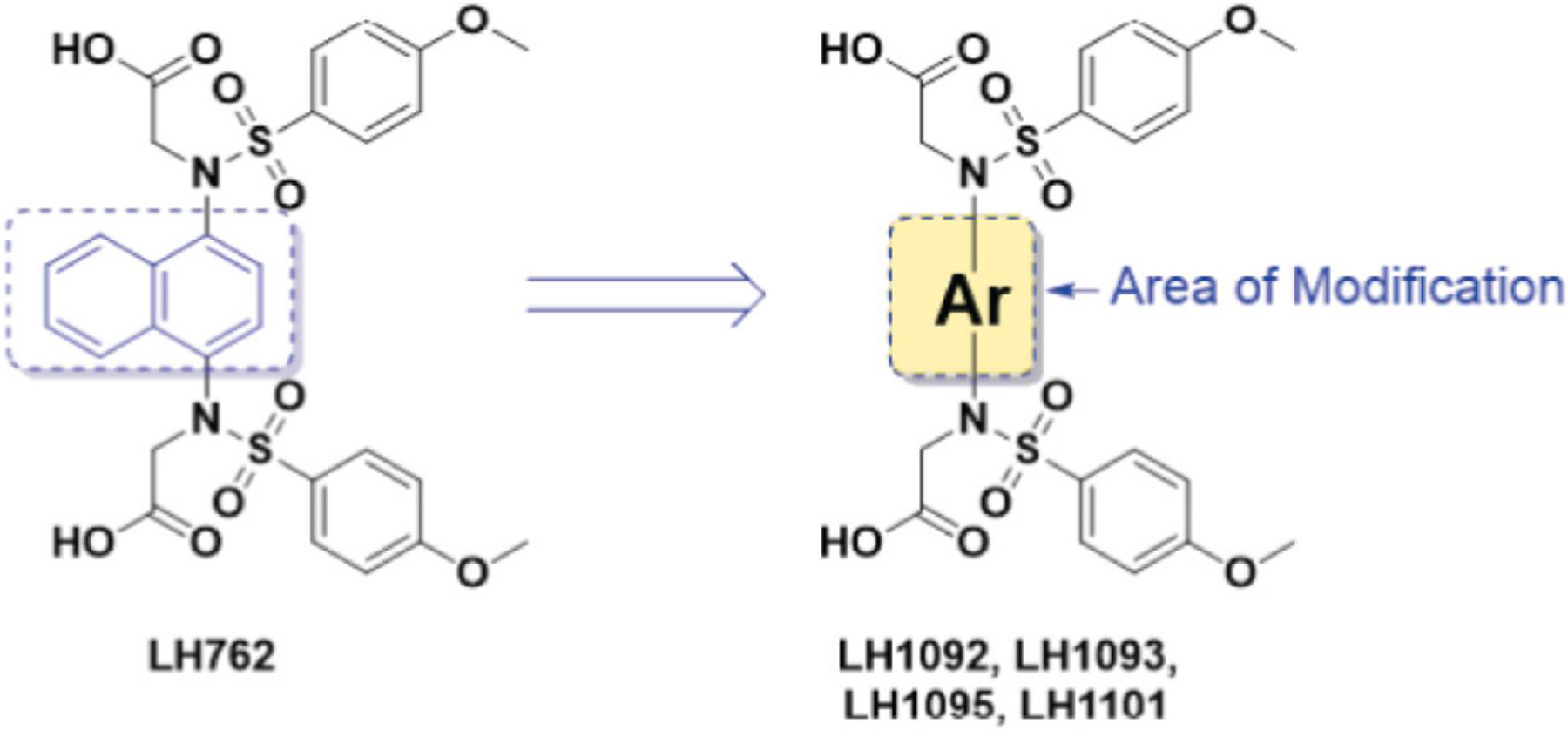
Generic structure of the Keap1-Nrf2 PPI inhibitors used in this study, LH1092, LH1093, LH1095, and LH1101, modified from the structure of 1,4-bis[(4-methoxyphenyl)sulfonamido]naphthalene N,N’-diacetic acid (LH762).
3.2. Keap1-Nrf2 PPI inhibitors downregulate estrogen responsive genes in E2-stimulated MCF-7 cells
Estrogen responsive genes, including progesterone receptor (PGR), cyclin D1 (CCND1), cathepsin D (CTSD), CITED1, SERPINA1, and TFF1, are important players in cell proliferation and cell cycle progression in breast cancer. We treated MCF-7 cells with E2 in the presence of the Keap1-Nrf2 PPI inhibitors and CDDO-IM as a positive control for 16 and 48 hours and examined changes in expression of these estrogen responsive genes. We found that the mRNA level of PGR was significantly elevated by E2, and that Keap1-Nrf2 PPI inhibitors attenuated the E2-stimulated induction at both time points (Fig. 2A and 2B). The mRNA expression of CITED1 and CCND1 increased at 16 hours and returned to baseline at 48 hours in response to E2 (Fig. 2A and 2B). The presence of Keap1-Nrf2 PPI inhibitors downregulated CITED1 and CCND1 expression by about 40% compared to E2 treatment alone at 48 hours (Fig. 2B). The mRNA level of SERPINA1 was increased by E2 at both time points (Fig. 2A and 2B); co-treatment of E2 and LH1092 inhibited this effect of E2 at 16 hours (Fig. 2A). E2 treatment also increased the mRNA levels of CTSD and TFF1, but the Keap1-Nrf2 PPI inhibitors did not have a significant effect on these genes (Fig. 2A and 2B). In addition, we performed Western blot analysis to examine the protein levels of estrogen responsive genes changed by E2 and the Keap1-Nrf2 PPI inhibitors. After 24 hours of treatment, PGR, TFF1/pS2, CTSD and c-MYC protein levels were significantly induced by E2, and this induction was reduced by the selected compounds LH1092 and LH1095 (Fig. 2C).
Fig. 2.
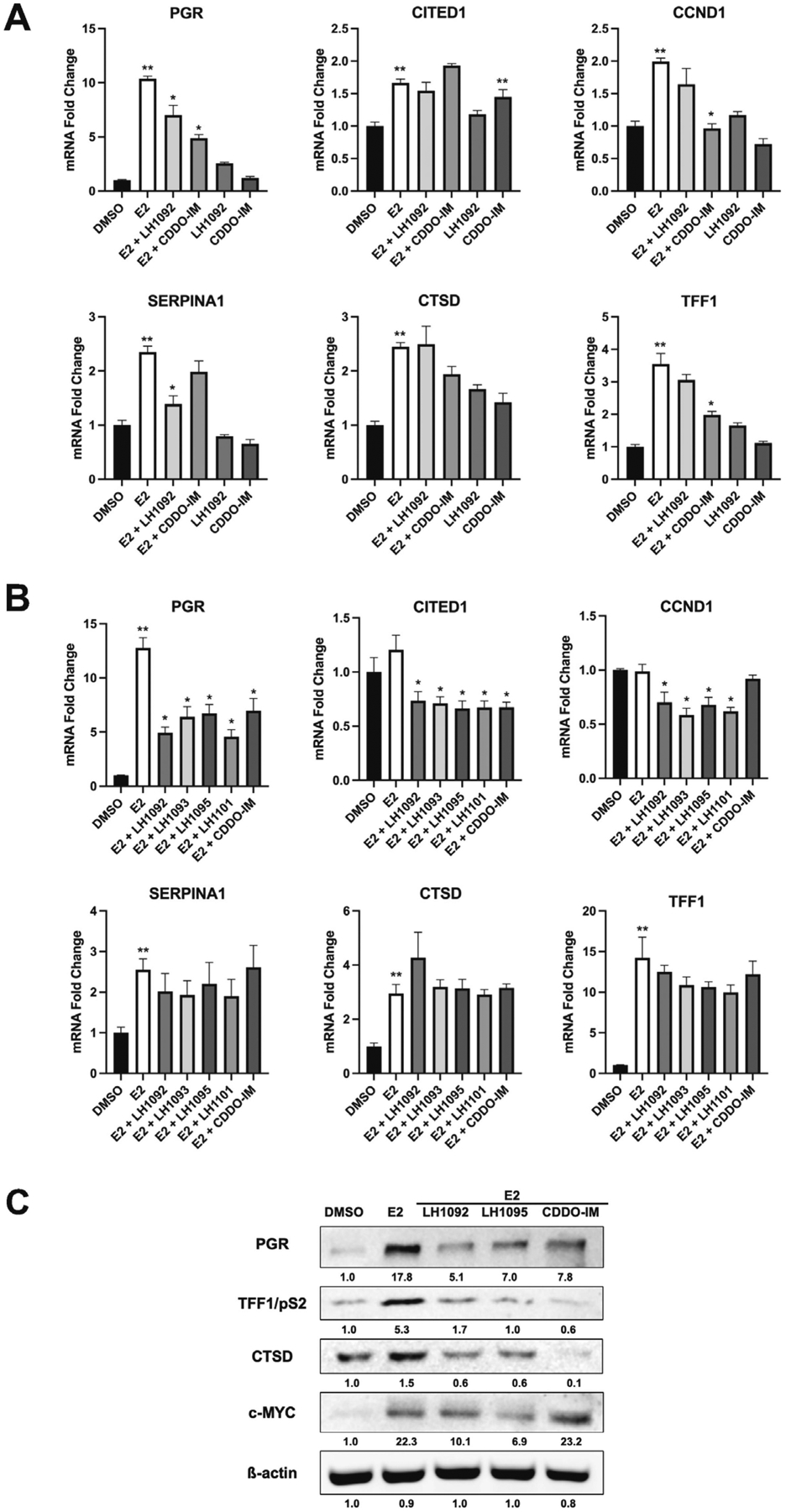
Keap1-Nrf2 PPI inhibitors downregulate estrogen responsive genes in E2-stimulated MCF-7 cells. A. The mRNA expression of estrogen responsive genes at 16 hours after treatment with 10 μM LH1092 or 100 nM CDDO-IM with or without 100 pM E2. B. The mRNA expression of estrogen responsive genes at 48 hours after treatment with 100 pM E2 in the presence or absence of 10 μM test compounds (LH1092, LH1093, LH1095 and LH1101) or 100 nM CDDO-IM. The mRNA levels were analyzed by RT-qPCR. GAPDH was used as an internal control for gene expression. The data are represented as mean ± SEM of mRNA fold changes (n = 6). **p < 0.05, significant difference compared to DMSO control. *p < 0.05, significant difference compared to E2 control. C. MCF-7 cells were treated with 10 μM LH1092, 10 μM LH1095 and 100 nM CDDO-IM in the presence of 100 pM E2 for 24 hours. The protein levels of PGR, TFF1/pS2, CTSD, and c-MYC were determined by Western blot analysis. β-actin was used as a loading control. Quantification of protein levels from the Western blots were analyzed using ImageJ program.
3.3. Keap1-Nrf2 PPI inhibitors do not bind to ERα
Next, we investigated whether the reversal of E2 action by the Keap1-Nrf2 PPI inhibitors is due to ER binding. Ansell et al. showed that the repression of Nrf2 and ARE-dependent gene expression by E2 is achieved through E2-bound ERα (Ansell et al., 2005). Since the Keap1-Nrf2 PPI inhibitors downregulate estrogen responsive genes and relevant protein levels in E2-stimulated MCF-7 cells, we tested the binding activity of the Keap1-Nrf2 PPI inhibitors (LH762, LH1092, LH1093, LH1095 and LH1101) to ERα, using the PolarScreen™ Estrogen Receptor Alpha Competitor Assay. Fluorescence polarization value (mP) was calculated based on the parallel and perpendicular fluorescence intensity (F∥ and F⊥) with respect to the linearly polarized excitation light. The results showed that the Keap1-Nrf2 PPI inhibitors did not bind ERα at any of the concentrations (1, 10, 100 μM) tested (Fig. 3), suggesting that they do not act as ER antagonists.
Fig. 3.
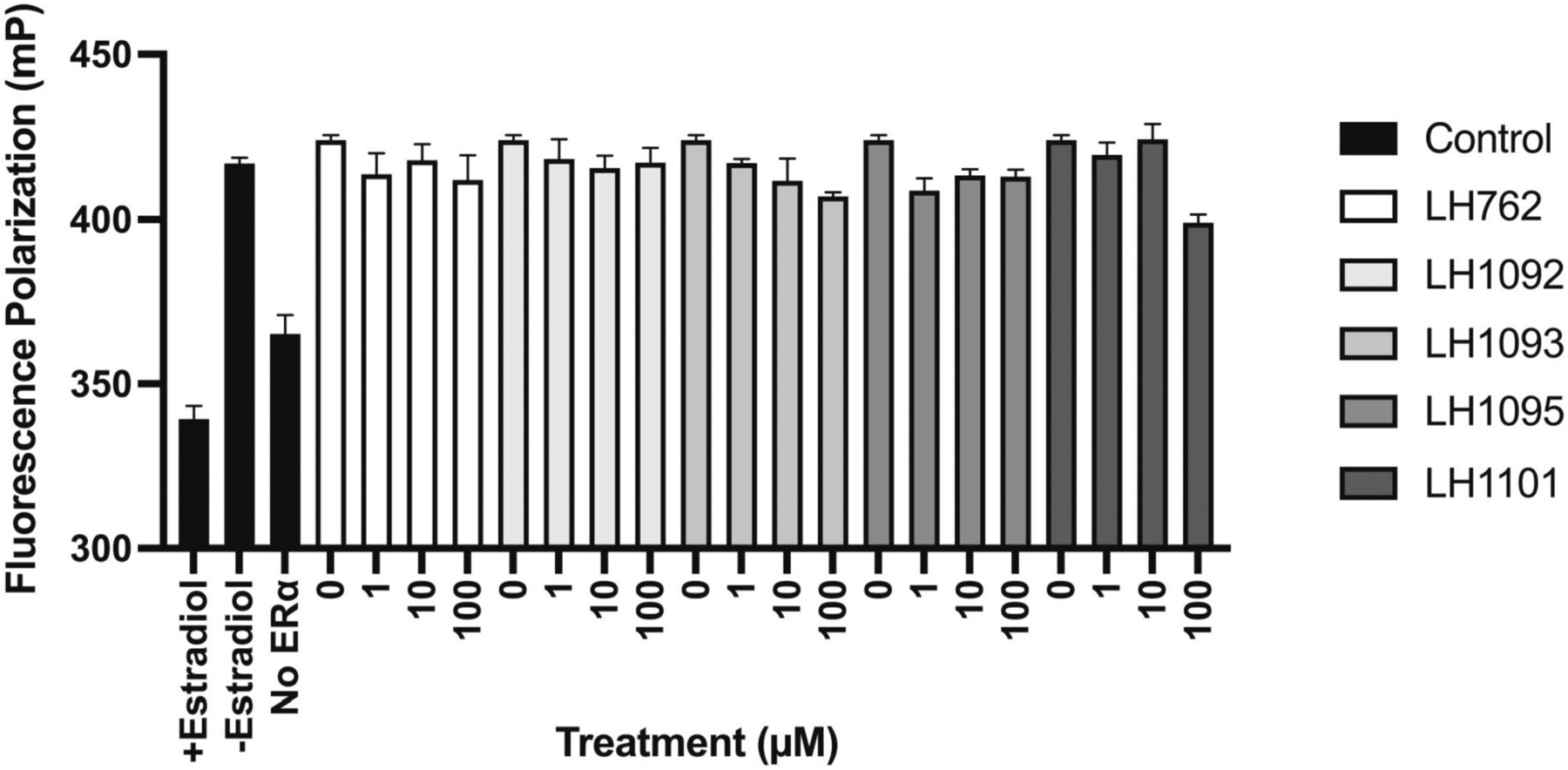
Binding activity of Keap1-Nrf2 PPI inhibitors to estrogen receptor (ER). PolarScreen™ Estrogen Receptor Alpha Competitor Assay kit was obtained from Life Technologies. A total assay volume of 20 μL was used. Estradiol and test compounds (0, 1, 10, 100 μM) were added to the appropriate wells of the assay plate. The plate was incubated at room temperature for 2 hours. Fluorescence polarization value (mP) was calculated based on the parallel and perpendicular fluorescence intensity (F∥ and F⊥) with respect to the linearly polarized excitation light.
3.4. Keap1-Nrf2 PPI inhibitors increase nuclear translocation of Nrf2 in MCF-7 cells
To examine the Nrf2 localization induced by the Keap1-Nrf2 PPI inhibitors, we treated MCF-7 cells with selected compounds LH1092 and LH1095 with or without E2. Immunofluorescence staining was performed at 3 hours to assess the Nrf2 level in the nucleus. LH1092 and LH1095 increased the Nrf2 level (green staining) in the nucleus compared to the DMSO control group regardless of E2 presence (Fig. 4A and 4B). E2 treatment alone did not show significant change in Nrf2 level in the nucleus (Fig. 4A and 4B).
Fig. 4.
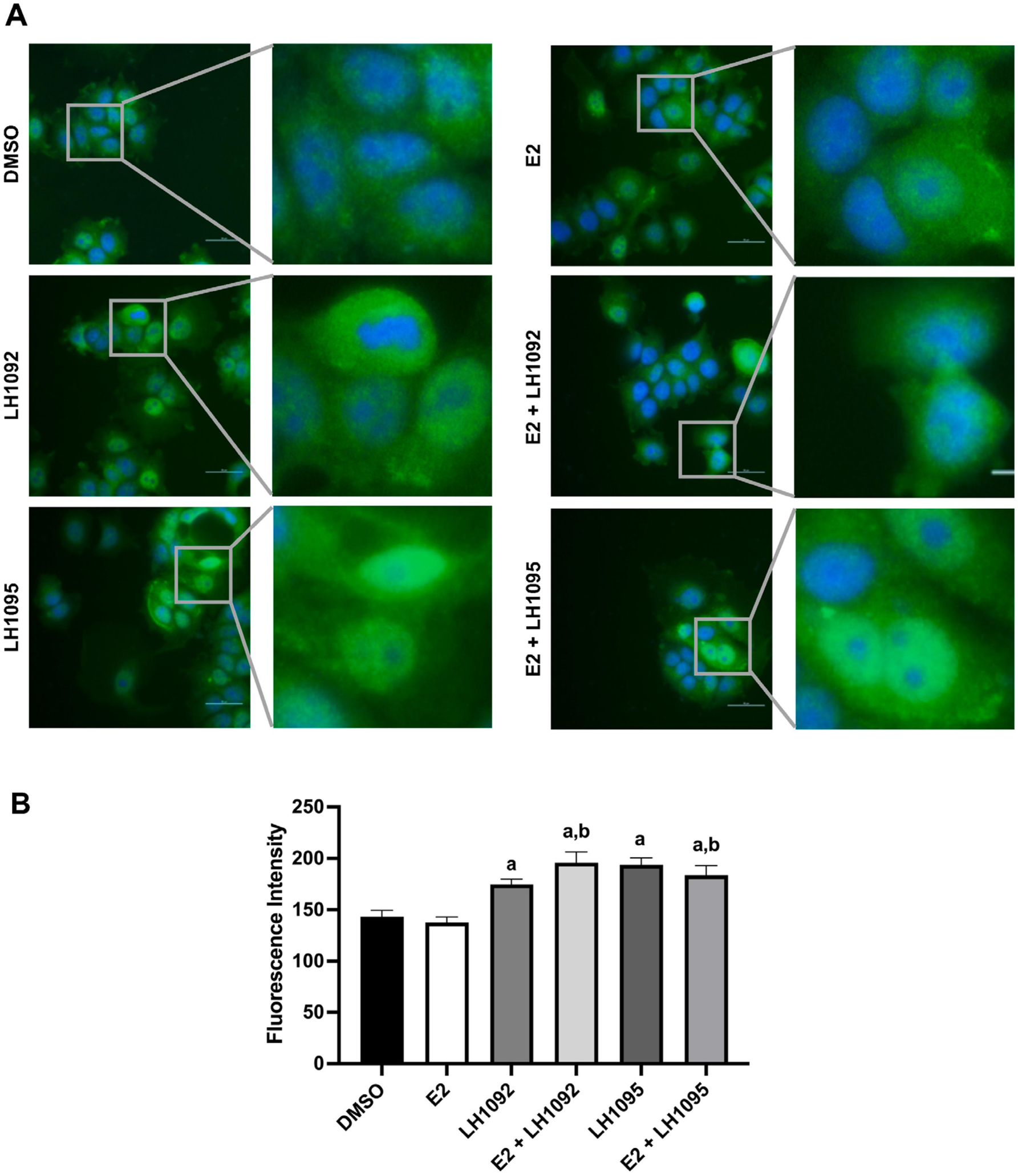
Keap1-Nrf2 PPI inhibitors induce the nuclear translocation of Nrf2 in MCF-7 cells. A. Cells were treated with 10 μM LH1092 and 10 μM LH1095 with or without 1 nM E2 for 3 hours. Cells were fixed and stained for the nuclei (blue) and Nrf2 (green). A fluorescence microscope was used to take the images at 40× magnification. One representative image from each group is shown here along with the expanded view. Scale bar = 50 μm. B. The fluorescence intensity of Nrf2 nucleus staining changed by LH1092 or LH1095 in the presence or absence of E2 is shown. Images were taken by randomly selecting five fields from each treatment group. Two independent experiments were performed. The immunofluorescence staining of Nrf2 in the nuclei was quantified by ImageJ. The data are represented as mean ± SEM of fluorescence intensity. a, p < 0.05, as compared to DMSO. b, p < 0.05, as compared to E2.
3.5. Keap1-Nrf2 PPI inhibitors upregulate Nrf2 target genes in E2-treated MCF-7 cells
As the Nrf2 level is increased in the nucleus in response to the Keap1-Nrf2 PPI inhibitors, we next want to determine if the compounds can activate the Nrf2-ARE pathway in MCF-7 cells exposed to E2. Our results showed that LH1092 upregulated the mRNA expression of Nrf2 target genes (NQO1, HO-1, GCLM and GPX2) regardless of E2 presence at 16 hours (Fig. 5A). The mRNA levels of these genes remained to be elevated by the compounds at 48 hours (Fig. 5B). Notably, GPX2 expression was enhanced by 35-fold in the presence of the Keap1-Nrf2 PPI inhibitors at 48 hours (Fig. 5B). The Keap1-Nrf2 PPI inhibitors had a greater effect than CDDO-IM at inducing the mRNA expression of NQO1, HO-1 and GCLM (Fig. 5A and 5B). It is important to note that E2 downregulated the mRNA and protein level of NQO1; the presence of Keap1-Nrf2 PPI inhibitors or CDDO-IM reversed the effect of E2 by causing an increase in the NQO1 mRNA and protein expression compared to E2 treatment alone (Fig. 5A, 5B and 5C). The change in HO-1 exhibited a different profile from NQO1. We observed a downward trend in HO-1 mRNA level caused by E2, but the difference from DMSO control is not significant (Fig. 5A and 5B). HO-1 protein expression did not significantly change with E2 treatment (Fig. 5D). Treatment of CDDO-IM with E2 had minimal effect to the mRNA and protein level of HO-1 compared to E2 control at the time points we examined (Fig. 5A, 5B and 5C). The mRNA expression of GPX1 did not change in response to the Keap1-Nrf2 PPI inhibitors at 16 and 48 hours (Fig. 5A and 5B). These results demonstrate that the Keap1-Nrf2 PPI inhibitors reverse the E2-mediated repression of specific Nrf2 target genes in a gene-specific manner.
Fig. 5.
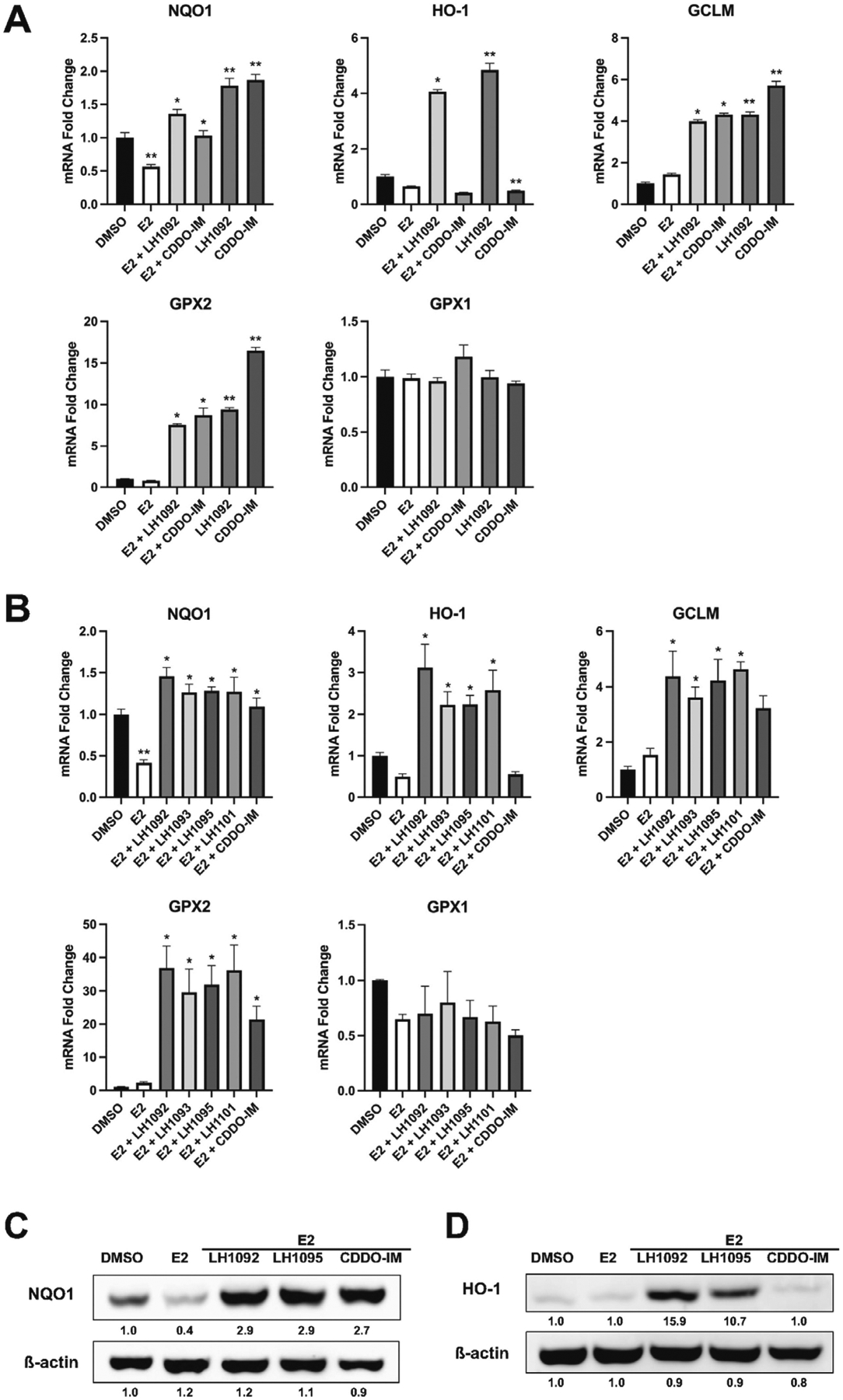
Keap1-Nrf2 PPI inhibitors upregulate Nrf2 target genes in E2-treated MCF-7 cells. A. The mRNA expression of Nrf2 target genes at 16 hours after treatment with 10 μM LH1092 or 100 nM CDDO-IM with or without 100 pM E2. B. The mRNA expression of Nrf2 target genes at 48 hours after treatment with 100 pM E2 in the presence or absence of 10 μM test compounds (LH1092, LH1093, LH1095 and LH1101) or 100 nM CDDO-IM. The mRNA levels were analyzed by RT-qPCR. GAPDH was used as an internal control for gene expression. The data are represented as mean ± SEM of mRNA fold changes (n = 6). **p < 0.05, significant difference compared to DMSO control. *p < 0.05, significant difference compared to E2 control. C. MCF-7 cells were treated with 10 μM LH1092, 10 μM LH1095 and 100 nM CDDO-IM in the presence of 100 pM E2 for 24 hours. The protein levels of NQO1 were determined by Western blot analysis. β-actin was used as a loading control. D. MCF-7 cells were treated with 10 μM LH1092, 10 μM LH1095 or 100 nM CDDO-IM in the presence of 100 pM E2 for 24 hours. The protein levels of HO-1 were determined by Western blot analysis. β-actin was used as a loading control. Quantification of protein levels from the Western blots were analyzed using ImageJ program.
3.6. Keap1-Nrf2 PPI inhibitors modulate oxidative stress caused by E2 in MCF-7 cells
To further investigate the antioxidant property of the Keap1-Nrf2 PPI inhibitors, we treated MCF-7 cells with LH1092 in the presence or absence of E2 or etoposide for 48 hours. Etoposide is a chemotherapeutic drug known to induce oxidative stress and used as a positive control. We have previously shown that E2 and etoposide increased the level of an oxidative stress marker 8-oxo-dG in MCF-7 cells (Bak et al., 2017). Here, we found that E2 and etoposide increased the staining of 8-oxo-dG in the cells compared to the DMSO control group, as indicated by the green fluorescence signals (Fig. 6A). Co-treatment of LH1092 with E2 or etoposide decreased the level of 8-oxo-dG compared to the E2 and etoposide control, respectively (Fig. 6A and 6B). LH1092 treatment alone did not change the level of 8-oxo-dG significantly (Fig. 6A and 6B).
Fig. 6.
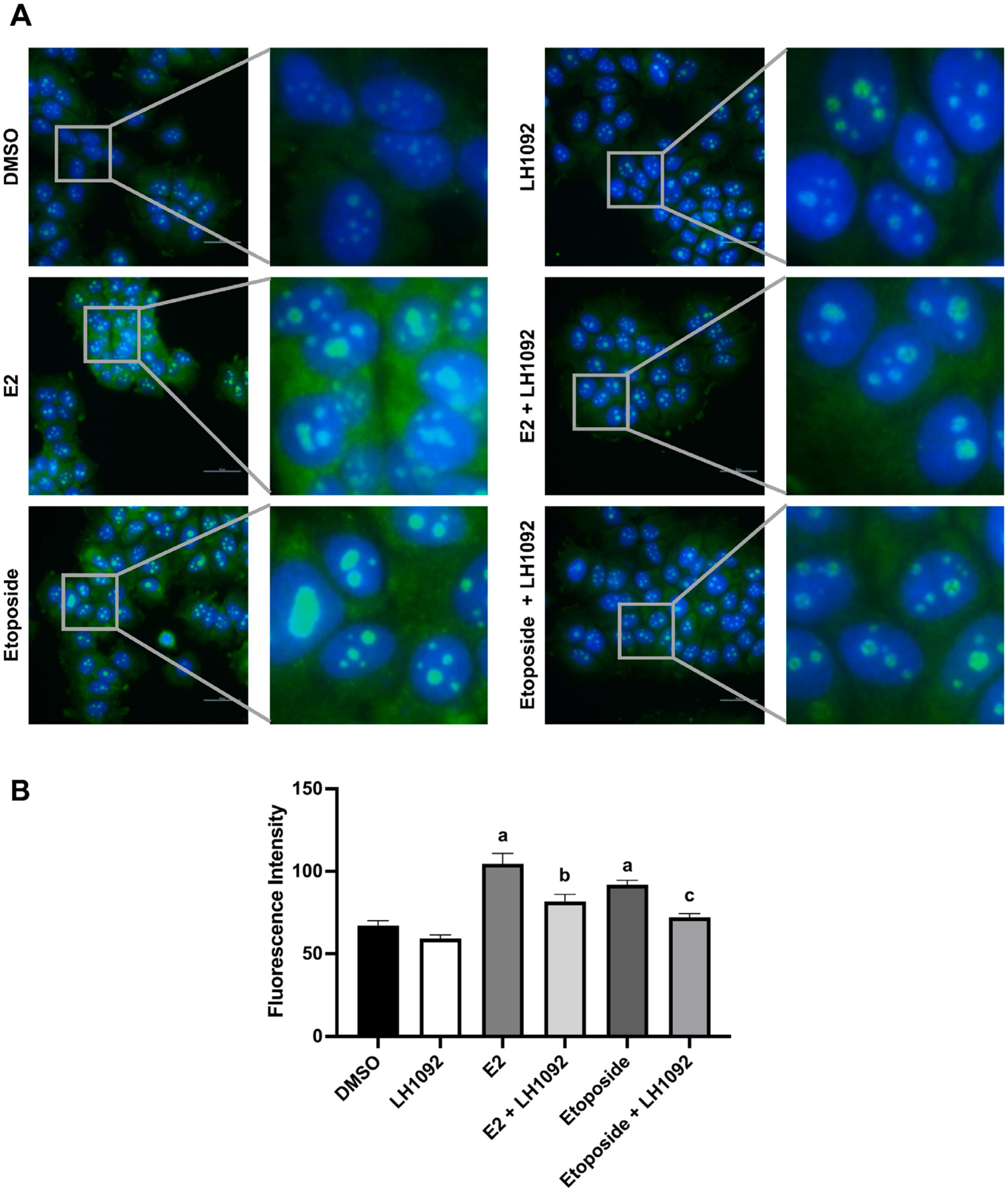
LH1092 attenuates the level of 8-oxo-dG induced by E2 and etoposide in MCF-7 cells. A. Cells were treated with 10 μM LH1092 with or without 1 nM E2 or 1 μM etoposide for 48 hours. Cells were fixed and stained for the nuclei (blue) and 8-oxo-dG (green). A fluorescence microscope was used to take the images at 40× magnification. One representative image from each group is shown here along with the expanded view. Scale bar = 50 μm. B. The immunofluorescence staining of 8-oxo-dG in the nuclei was quantified by ImageJ. Images were taken by randomly selecting five fields from each treatment group. Three independent experiments were performed. The data are represented as mean ± SEM of fluorescence intensity. a, p < 0.05, as compared to DMSO. b, p < 0.05, as compared to E2. c, p < 0.05, as compared to etoposide.
4. Discussion
Nrf2 has been an important target in cancer prevention because of its role in preventing and/or reversing oxidative stress that can promote tumorigenesis (Sporn and Liby, 2012). Both Nrf2 activators and inhibitors have been developed to intervene in different stages of cancer progression (Yamamoto et al., 2018; Rojo de la Vega et al., 2018). We show here that the Keap1-Nrf2 PPI inhibitors induce Nrf2-ARE pathway genes in MCF-7 cells. One of these genes, NQO1, serves as an important modulator of cancer progression. It reduces quinones to hydroquinones as a means of detoxification in cells (Cuadrado et al., 2019). NQO1 deficiency contributes to increased ROS and subsequent genome instability in breast tumors (Fagerholm et al., 2008). In our study, the expression of NQO1 was significantly suppressed by E2, and this suppression was reversed by the Keap1-Nrf2 PPI inhibitors, which implies that the compounds could have cytoprotective effect against E2 oxidative damage to the cells. HO-1, another Nrf2-regulated gene, also has antioxidative properties and its inhibition is related to DNA damage (Chiang et al., 2021). Interestingly, E2 did not significantly change HO-1 expression in MCF-7 cells, but the Keap1-Nrf2 PPI inhibitors increased HO-1 mRNA and protein levels regardless of E2 treatment. Its induction by the Keap1-Nrf2 PPI inhibitors also suggests potential use of these compounds in combating oxidative stress. Our results also indicated an increase in the mRNA level of GCLM, the glutamate cysteine ligase modifier subunit, which participates in the de novo synthesis of glutathione (GSH) (Dodson et al., 2019). Among the Nrf2 target genes examined in this study, GPX2 (glutathione peroxidase 2) displayed the highest mRNA induction by the Keap1-Nrf2 PPI inhibitors at 48 hours. GPX2 uses GSH to reduce organic hydroperoxides and hydrogen peroxide (H2O2) in redox regulation, which is a crucial mechanism in Nrf2-mediated chemoprevention (Dodson et al., 2019). Low GPX2 mRNA level in human breast cancer leads to poor patient survival and oncogenic signaling (Ren et al., 2022), suggesting that the Keap1-Nrf2 PPI inhibitors could inhibit breast cancer via GPX2-mediated mechanisms. Induction of these cytoprotective genes catalyzes the detoxification process and contributes to chemoprevention by preventing DNA damage from carcinogens such as E2 (Abed et al., 2015).
Current literature is divergent regarding E2 regulation of Nrf2-mediated transcription of target genes. Ansell et al. found that low levels of E2 can repress Nrf2-related phase II enzymes, GST and QR in mice (Ansell et al., 2004). Further, Yao et al. demonstrated that the inhibition of Nrf2 target gene NQO1 is caused by ERα and SIRT1 recruited by E2 at the NQO1 promoter (Yao et al., 2010). On the other hand, Gorrini et al. demonstrated that a high dose of E2 at 10 nM induces Nrf2 and its related antioxidant genes, including GCLM, HMOX1, and NQO1, in BRCA1-deficient cells through the PI3K-AKT pathway (Gorrini et al., 2014). Wu et al. showed that a high dose of E2 upregulates Nrf2 activity by increasing ARE-Luc activity and HO-1 mRNA expression through the PI3K/GSK-3β pathway in MCF-7 cells (Wu et al., 2014). These contradictory data regarding the regulation of Nrf2 activity by E2 could be attributed to ligand- and cell-specific factors as well as the varied levels of estrogen exposure (Ansell et al., 2005; Wu et al., 2014).
Regarding the modulation of E2-induced effects, the Keap1-Nrf2 PPI inhibitors downregulated the mRNA levels of estrogen responsive genes, including PGR, CITED1 and CCND1, as well as the protein levels of PGR, TFF1/pS2, CTSD and c-MYC, compared to E2 control. Progesterone receptor (PGR) is a direct target of ER signaling. Its inhibition by the compounds suggests that Nrf2 activation may interact with ER signaling by inhibiting estrogen response element (ERE). The changes in the mRNA expression of estrogen responsive genes are time dependent. E2 increased the mRNA levels of CITED1 and CCND1 at 16 hours and returned the levels to the baseline at 48 hours. The mRNA expression of CITED1 and CCND1 did not change by the LH1092 treatment at 16 hours but decreased at 48 hours. CITED1 is a transcriptional coregulator that enhances sensitivity to E2 in MCF-7 cells (Yahata et al., 2001). CCND1, cyclin D1, is an oncogene overexpressed in breast cancer. Inhibition of these estrogen responsive genes indicate that the Keap1-Nrf2 PPI inhibitors may attenuate E2 responses from estrogen receptor signaling.
Although we do not yet know the precise mechanism by which the Keap1-Nrf2 PPI inhibitors exert these inhibitory effects on E2 signaling, we did find that the compounds themselves did not bind directly to ER. The compounds activate and increase nuclear translocation of Nrf2 in MCF-7 cells regardless of E2 presence. LH1092 attenuated oxidative stress induced by E2 and etoposide, as shown by decreasing the level of 8-oxo-dG produced in MCF-7 cells, and these results indicate potential antioxidant effect of the Keap1-Nrf2 PPI inhibitors against oxidative stress. A proposed mechanism of activation of the Nrf2-ARE pathway and inhibition of ER signaling pathway by the Keap1-Nrf2 PPI inhibitors is depicted in Fig. 7. Our data demonstrate that the Keap1-Nrf2 PPI inhibitors mitigate E2-induced estrogen downstream target gene expression and oxidative stress in ER-positive breast cancer via activating the Nrf2 pathway. These findings warrant further validation in a physiologically relevant estrogen-induced in vivo model of ER-positive breast cancer. Taken together, our study suggests that the Keap1-Nrf2 PPI inhibitors may block E2-mediated effects in ER-positive breast cancer and could serve as agents useful in preventing development or progression of these tumors.
Fig. 7.
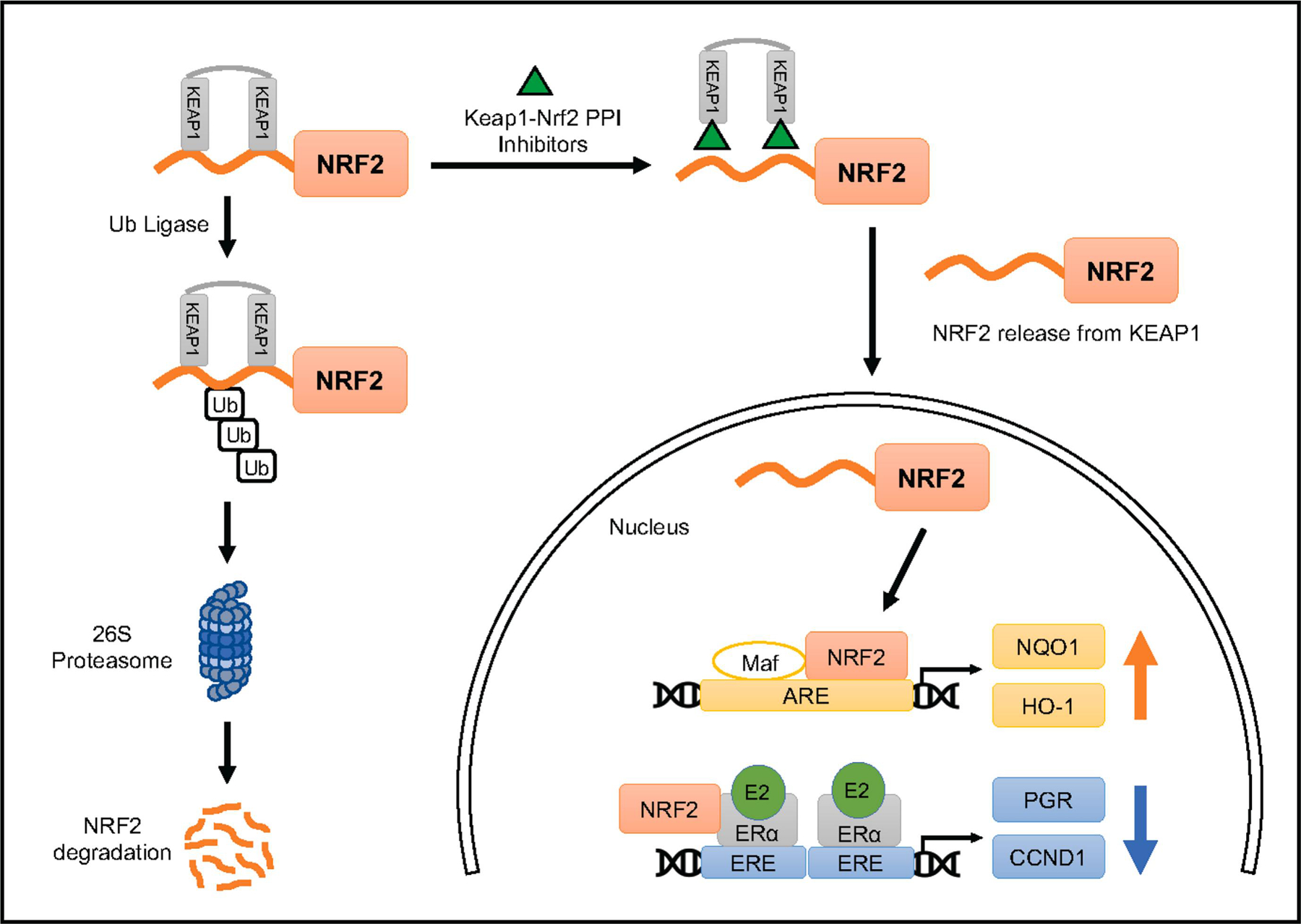
Schematic diagram showing the proposed mechanism of activation of the Nrf2 pathway by Keap1-Nrf2 PPI inhibitors and Nrf2 interaction with ER signaling. Under basal conditions, Nrf2 binds to Keap1 homodimer and is ubiquitinated by a ubiquitin ligase complex and undergoes degradation through 26S proteasome. Use of direct inhibitors of the Keap1-Nrf2 protein-protein interaction allows Nrf2 to translocate to the nucleus and initiate the transcription of antioxidant response element (ARE)-related genes. A proposed mechanism is depicted here that Nrf2 activated by the Keap1-Nrf2 PPI inhibitors interacts with ERα and inhibits ER signaling.
Supplementary Material
Highlights.
The direct inhibitors of Keap1-Nrf2 protein-protein interaction activate the Nrf2 pathway.
The compounds reverse estrogen action in gene and protein expression in MCF-7 cells.
The compounds reduce estrogen responsive gene and protein levels, such as PGR.
Selected compound mitigates estrogen-induced oxidative stress in ER-positive MCF-7 cells.
Acknowledgements
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding
This work was supported by the National Center for Complementary and Integrative Health of the National Institutes of Health [R01 AT007036], the National Institute of Environmental Health Sciences grant [ES005022, R25 ES020721], National Cancer Institute [R03 CA259650, R01 CA133791], Rutgers TechAdvance Grant [TA2019-0300], Charles and Johanna Busch Memorial Fund at Rutgers University and the New Jersey Health Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT authorship contribution statement
Tingying Xie: Methodology, Formal analysis, Investigation, Conceptualization, Data curation, Writing- original draft, Visualization. Husam Zahid: Methodology, Investigation, Conceptualization, Resources, Writing- original draft, Writing- review and editing, Visualization. Ahmed R. Ali: Methodology, Investigation, Conceptualization, Resources, Writing- original draft, Writing- review and editing, Visualization. Ryan Joyce: Methodology, Formal analysis, Investigation, Conceptualization, Resources, Data curation, Writing- original draft, Writing- review and editing, Visualization. Ge Yang: Methodology, Resources, Writing- review and editing. Cassandra Winz: Methodology, Resources, Writing- review and editing. Yicong Le: Methodology, Resources, Writing- review and editing. Renping Zhou: Methodology, Resources, Writing- review and editing. Philip Furmanski: Methodology, Formal analysis, Investigation, Conceptualization, Writing- review and editing, Supervision. Longqin Hu: Methodology, Formal analysis, Investigation, Conceptualization, Resources, Data curation, Writing- review and editing, Visualization, Supervision, Project administration, Funding acquisition. Nanjoo Suh: Methodology, Investigation, Conceptualization, Resources, Data curation, Writing-original draft, Writing- review and editing, Visualization, Supervision, Project administration, Funding acquisition.
Conflict of Interest Statement: Some of the authors are inventors of a provisional patent application. Rutgers University is in the process of filing with the US Patent and Trademark Office on small molecule direct inhibitors of Keap1-Nrf2 interaction.
Declaration of interests
Longqin Hu has patent pending to Rutgers University. Ahmed R. Ali has patent pending to Rutgers University.
References
- Abed DA, Goldstein M, Albanyan H, Jin H, & Hu L (2015). Discovery of direct inhibitors of Keap1–Nrf2 protein–protein interaction as potential therapeutic and preventive agents. Acta Pharmaceutica Sinica B, 5(4), 285–299. 10.1016/j.apsb.2015.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abed DA, Lee S, Wen X, Ali AR, Mangipudy V, Aleksunes LM, & Hu L (2021). Optimization of 1,4-bis(arylsulfonamido)naphthalene-N,N’-diacetic acids as inhibitors of Keap1-Nrf2 protein-protein interaction to suppress neuroinflammation. Bioorganic & Medicinal Chemistry, 44, 116300. 10.1016/j.bmc.2021.116300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansell PJ, Espinosa-Nicholas C, Curran EM, Judy BM, Philips BJ, Hannink M, & Lubahn DB (2004). In Vitro and in Vivo Regulation of Antioxidant Response Element-Dependent Gene Expression by Estrogens. Endocrinology (Philadelphia), 145(1), 311–317. 10.1210/en.2003-0817 [DOI] [PubMed] [Google Scholar]
- Ansell PJ, Lo S−, Newton LG, Espinosa-Nicholas C, Zhang DD, Liu J−, Hannink M, & Lubahn DB (2005). Repression of cancer protective genes by 17β-estradiol: Ligand-dependent interaction between human Nrf2 and estrogen receptor α. Molecular and Cellular Endocrinology, 243(1), 27–34. 10.1016/j.mce.2005.08.002 [DOI] [PubMed] [Google Scholar]
- Bak MJ, Das Gupta S, Wahler J, Lee HJ, Li X, Lee M, Yang CS, & Suh N (2017). Inhibitory Effects of γ- and δ-Tocopherols on Estrogen-Stimulated Breast Cancer In Vitro and In Vivo. Cancer Prevention Research (Philadelphia, Pa.), 10(3), 188–197. 10.1158/1940-6207.CAPR-16-0223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein HJ (2020). Systemic Therapy for Estrogen Receptor–Positive, HER2-Negative Breast Cancer. The New England Journal of Medicine, 383(26), 2557–2570. 10.1056/NEJMra1307118 [DOI] [PubMed] [Google Scholar]
- Chiang S, Chen S, & Chang L (2021). The Role of HO-1 and Its Crosstalk with Oxidative Stress in Cancer Cell Survival. Cells (Basel, Switzerland), 10(9), 2401. 10.3390/cells10092401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemons M, & Goss P (2001). Estrogen and the Risk of Breast Cancer. N Engl J Med, 344(4), 276–285. 10.1056/NEJM200101253440407 [DOI] [PubMed] [Google Scholar]
- Cloer EW, Goldfarb D, Schrank TP, Weissman BE, & Major MB (2019). NRF2 Activation in Cancer: From DNA to Protein. Cancer Research (Chicago, Ill.), 79(5), 889–898. 10.1158/0008-5472.CAN-18-2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A, Rojo AI, Wells G, Hayes JD, Cousin SP, Rumsey WL, Attucks OC, Franklin S, Levonen A, Kensler TW, & Dinkova-Kostova AT (2019). Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nature Reviews. Drug Discovery, 18(4), 295–317. 10.1038/s41573-018-0008-x [DOI] [PubMed] [Google Scholar]
- Dodson M, de la Vega Montserrat Rojo, Cholanians AB, Schmidlin CJ, Chapman E, & Zhang DD (2019). Modulating NRF2 in Disease: Timing Is Everything. Annual Review of Pharmacology and Toxicology, 59(1), 555–575. 10.1146/annurev-pharmtox-010818-021856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerholm R, Hofstetter B, Tommiska J, Aaltonen K, Vrtel R, Syrjaekoski K, Kallioniemi A, Kilpivaara O, Mannermaa A, Kosma V, Uusitupa M, Eskelinen M, Kataja V, Aittomaeki K, Von Smitten K, Heikkilae P, Lukas J, Holli K, Bartkova J, … Nevanlinna H (2008). NAD(P)H:quinone oxidoreductase 1 NQO1 super(2) genotype (P187S) is a strong prognostic and predictive factor in breast cancer. Nature Genetics, 40(7), 844–853. 10.1038/ng.155 [DOI] [PubMed] [Google Scholar]
- Gorrini C, Gang BP, Bassi C, Wakeham A, Baniasadi SP, Hao Z, Li WY, Cescon DW, Li Y, Molyneux S, Penrod N, Lupien M, Schmidt EE, Stambolic V, Gauthier ML, & Mak TW (2014). Estrogen controls the survival of BRCA1-deficient cells via a PI3K–NRF2-regulated pathway. Proceedings of the National Academy of Sciences - PNAS, 111(12), 4472–4477. 10.1073/pnas.1324136111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoyama D, Chen Y, Huang X, Beamer LJ, Kong AT, & Hu L (2012). Optimization of Fluorescently Labeled Nrf2 Peptide Probes and the Development of a Fluorescence Polarization Assay for the Discovery of Inhibitors of Keap1-Nrf2 Interaction. Journal of Biomolecular Screening, 17(4), 435–447. 10.1177/1087057111430124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Lu M, Xu L, Yang T, Xi M, Xu X, Guo X, Zhang X, You Q, & Sun H (2014). Discovery of Potent Keap1–Nrf2 Protein–Protein Interaction Inhibitor Based on Molecular Binding Determinants Analysis. Journal of Medicinal Chemistry, 57(6), 2736–2745. 10.1021/jm5000529 [DOI] [PubMed] [Google Scholar]
- Jordan VC (2006). Tamoxifen (ICI46,474) as a targeted therapy to treat and prevent breast cancer. British Journal of Pharmacology, 147(S1), S269–S276. 10.1038/sj.bjp.0706399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, & Hu L (2020). Nrf2 activation through the inhibition of Keap1–Nrf2 protein–protein interaction. Medicinal Chemistry Research, 29(5), 846–867. 10.1007/s00044-020-02539-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouremamali F, Pouremamali A, Dadashpour M, Soozangar N, & Jeddi F (2022). An update of Nrf2 activators and inhibitors in cancer prevention/promotion. Cell Communication and Signaling, 20(1), 100. 10.1186/s12964-022-00906-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, & Kensler TW (2001). Sensitivity to Carcinogenesis is Increased and Chemoprotective Efficacy of Enzyme Inducers is Lost in nrf2 Transcription Factor-Deficient Mice. Proceedings of the National Academy of Sciences - PNAS, 98(6), 3410–3415. 10.1073/pnas.051618798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Liang H, Galbo J, Phillip M, Dharmaratne M, Kulkarni AS, Fard AT, Aoun ML, Martinez-Lopez N, Suyama K, Benard O, Zheng W, Liu Y, Albanese J, Zheng D, Mar JC, Singh R, Prystowsky MB, Norton L, & Hazan RB (2022). Redox signaling by glutathione peroxidase 2 links vascular modulation to metabolic plasticity of breast cancer. Proceedings of the National Academy of Sciences - PNAS, 119(8), 1. 10.1073/pnas.2107266119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo de la Vega Montserrat, Chapman E, & Zhang DD (2018). NRF2 and the Hallmarks of Cancer. Cancer Cell, 34(1), 21–43. 10.1016/j.ccell.2018.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidlin CJ, Dodson MB, Madhavan L, & Zhang DD (2019). Redox regulation by NRF2 in aging and disease. Free Radical Biology & Medicine, 134, 702–707. 10.1016/j.freeradbiomed.2019.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivinski J, Zhang DD, & Chapman E (2021). Targeting NRF2 to treat cancer. Seminars in Cancer Biology, 76, 61–73. 10.1016/j.semcancer.2021.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith IE, & Dowsett M (2003). Aromatase Inhibitors in Breast Cancer. The New England Journal of Medicine, 348(24), 2431–2442. 10.1056/NEJMra023246 [DOI] [PubMed] [Google Scholar]
- Sporn MB, & Liby KT (2012). NRF2 and cancer: the good, the bad and the importance of context. Nature Reviews. Cancer, 12(8), 564–571. 10.1038/nrc3278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Thorne G, Hu L, Joy MS, & Aleksunes LM (2015). Activation of NRF2 Signaling in HEK293 Cells by a First-in-Class Direct KEAP1-NRF2 Inhibitor. Journal of Biochemical and Molecular Toxicology, 29(6), 261–266. 10.1002/jbt.21693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Williams D, Walter GA, Thompson WE, & Sidell N (2014). Estrogen increases Nrf2 activity through activation of the PI3K pathway in MCF-7 breast cancer cells. Experimental Cell Research, 328(2), 351–360. 10.1016/j.yexcr.2014.08.030 [DOI] [PubMed] [Google Scholar]
- Yager JD, & Davidson NE (2006). Estrogen Carcinogenesis in Breast Cancer. The New England Journal of Medicine, 354(3), 270–282. 10.1056/NEJMra050776 [DOI] [PubMed] [Google Scholar]
- Yahata T, Shao W, Endoh H, Hur J, Coser KR, Sun H, Ueda Y, Kato S, Isselbacher KJ, Brown M, & Shioda T (2001). Selective coactivation of estrogen-dependent transcription by CITED1 CBP/p300-binding protein. Genes & Development, 15(19), 2598–2612. 10.1101/gad.906301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kensler TW, & Motohashi H (2018). The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiological Reviews, 98(3), 1169–1203. 10.1152/physrev.00023.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Brodie AMH, Davidson NE, Kensler TW, & Zhou Q (2010). Inhibition of estrogen signaling activates the NRF2 pathway in breast cancer. Breast Cancer Research and Treatment, 124(2), 585–591. 10.1007/s10549-010-1023-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.