

Abstract
Background and Objectives
Nemaline myopathy (NM) is a genetically heterogeneous inherited myopathy related with at least 12 genes, whereas pathogenic variants in NEB gene are the most common genetic cause. The clinical spectrum of NM caused by NEB pathogenic variants (NM-NEB) is very broad, ranging from mild to severe presentations manifesting with generalized weakness, as well as respiratory and bulbar involvement. There is currently not enough data regarding the progression of the disease. In this study, we present a genotypic and phenotypic spectrum of 33 patients with NM caused by NEB variants (NM-NEB) classified according to age groups and the use of ventilatory support. We focused on interventional support, genotype-phenotype correlation, and association between respiratory, bulbar, and motor systems in groups of patients stratified by age and by the use of ventilatory support (VS).
Methods
Clinical and genetic data from patients with NM-NEB followed up in one specialized center were collected through regular consultations. Patients were evaluated regarding motor, bulbar, and respiratory functions.
Results
Thirty-three patients with NM-NEB were evaluated consisting of 15 females and 18 males with an average age of 18 (±12) years and a median of 17 (±11) years. 32% of patients with NM-NEB used a G tube, 35% were not able to walk without support, and 55% needed VS. Scoliosis and dysphagia were more common among patients who used VS. Described for the first time, half of the patients presented tongue atrophy in a triple furrow pattern, and the presence of the atrophy was associated with dysphagia. Comparing the patients grouped by age, we found that, proportionally, older patients had more scoliosis and respiratory dysfunction than younger groups, suggesting the progression of the disease in these domains. In addition to that, we showed that VS use was associated with scoliosis and dysphagia.
Discussion
NM-NEB is a very debilitating disease. There is an association between scoliosis and respiratory dysfunction while patients using VS have more often scoliosis than the no-VS group. Triple furrow tongue atrophy is a novel and frequent finding, which is directly associated with dysphagia. Grouping patients by age suggested disease stability in motor and swallow function, but a progression in respiratory dysfunction and skeletal deformities. All observations are relevant in the management care of patients with NM.
Nemaline myopathy (NM) is a genetically heterogeneous inherited myopathy and constitutes one of the most common forms of congenital myopathy. It is defined histologically by the presence of nemaline bodies in muscle biopsy.1 It is caused by at least 11 genes coding thin filaments or related proteins reported to date.
NEB and ACTA1 are the genes responsible for most of the cases, followed by TPM3, TPM2, KLHL40, KLHL41, KBTKD13, CFL2, LMOD3, TNNT1, and MYPN.1-4 NEB is one of the largest genes in the human genome and encodes nebulin, one of the biggest structural proteins in the skeletal muscle. The clinical spectrum of NM caused by NEB pathogenic variants (NM-NEB) is very broad, ranging from severe to mild presentations. In addition to histologically defined NM, pathogenic variants in NEB may cause core-rod myopathy, distal nebulin myopathy without nemaline rods, lethal multiple pterygium syndrome, and a dominant distal nemaline/cap myopathy.5,6 More recently, a dominantly inherited distal myopathy and a recessive congenital asymmetric distal myopathy with hemifacial weakness were associated with large deletions in NEB, further expanding its clinical spectrum.5,7
On purely clinical grounds, NM can be classified into groups of relative severity according to the age of symptoms' onset and degree of muscle weakness: (1) severe congenital NM, with patients lacking spontaneous movements and manifesting respiratory insufficiency at birth; (2) intermediate congenital NM: patients who were moving and breathing at birth but did not achieve of loose independent ambulation and/or developed respiratory failure in the first years of life; (3) typical congenital NM: patients with congenital onset and delayed milestones followed by a slowly progressive course; (4) childhood/juvenile-onset form: first symptoms occurring in the late first or second decade of life after independent ambulation; (5) adult-onset NM: weakness developing usually after 20 years of age; and (6) other forms of NM with unusual associated features, such as cardiomyopathy and ophthalmoplegia.8 Recently, a new classification was proposed pointing to the difficulties related with the classification system because the progression into childhood is one of the features distinguishing the groups.1
Nebulin is a giant muscle protein that is fundamental in the sarcomeric structure. The clinical severity of NM caused by NEB pathogenic recessive variants is wide and seems to be correlated to the amount and size of expressed protein in the muscle tissue and the preservation of actin and tropomyosin binding sites.9 Most causative variants result from frameshift premature stop codons and splicing changes. Missense variants are rare and are considered pathogenic when functionally accessed or when predicted to interfere in the actin or tropomyosin-nebulin interaction sites. These variants are expected to be less disruptive and cause milder presentations.6,10 However, the effect of these mutations is hard to predict.
Despite the clinical variability, most patients present generalized muscle weakness with a predominant facial, neck flexor, respiratory, and bulbar involvement. The respiratory dysfunction is usually disproportional to the limb weakness and might occur early in childhood as an insidious symptom leading to the need of serial and specialized evaluations.1 There are not enough systematic data about the correlation between the most frequent symptoms among patients with NM. Recently, a cross-sectional study aiming to access the respiratory function of patients with NM demonstrated the low correlation between the forced vital capacity and the motor function measure scores.11 The association between the respiratory involvement with dysphagia and spine deformities, also very frequent in NM, has not been reported.
There is also insufficient data defining the natural history of NM-NEB. Despite its previous consideration as a stable disease, a recent cross-sectional study with 18 individuals with NM-NEB from a cohort of 57 patients with NM suggested a slowly progressive course affecting respiratory, motor, and bulbar systems. The same study also showed 58% of the patients requiring interventional support in at least one of those affected systems, illustrating how debilitating this disease can be.12-14
In this study, we present the mutational and phenotypic spectrum in a cohort of 33 Brazilian patients with NM-NEB focusing on interventional support, genotype-phenotype correlation, and association between respiratory, bulbar, and motor functions in groups of patients stratified by age and the use of ventilatory support (VS). In addition, we described the triple furrow tongue sign as a novel clinical finding, which might be valuable in the evaluation of bulbar involvement of the patients. This study adds valuable insights about the progression of NM-NEB disease and management care.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by the Clinical Hospital of Universidade de São Paulo Ethical Committe. Written consent was obtained from the legal guardians or the patient if appropriate, and patient assent was obtained when possible.
Patients
Thirty-three patients with NM caused by NEB variants were seen at the Department of Neurology of FMUSP from 2011 to 2022. Patients participated in the study after signing the informed consent. The study was approved by a local ethical committee. Clinical, genetical, and histologic data were retrospectively collected from patients' medical records. The first author saw and examined all patients.
Motor Function and Skeletal Deformities Assessment
Direct muscle strength of 4 limbs and neck was measured with the Medical Research Council grading. Facial weakness, ocular movements, and the presence of scoliosis, rigid spine, and articular contractions were tested in all patients.
Patients were stratified according to their motor function into 4 groups according to their maximum motor skills: (1) none (i.e., patients who were not able to sit or roll); (2) sit with or without support; (3) stand and/or walk few steps; and (4) walk independently.15
Respiratory Function
Data of VS included the age that patients started using VS devices, time spent in ventilation, type of dispositive used, and the presence or absence of tracheostomy (TQ). According to the total time the patient remained in ventilation, we stratified it as continuous when the patient used VS for more than 22 h/d and as intermittent when the patient used VS nightly and an extra few hours during daytime (ranging from 15 to 22 h/d) and nighttime only. We also classified VS as noninvasive VS (NIVS) or invasive VS (IVS) when the patient had TQ.
Swallow and Bulbar Function
Patients were stratified according to their swallow function using the Neuromuscular Disease Swallowing Status Scale (NdSSS) (Wada et al., 2015). This is an 8-stage scale that considered food intake: (1) tube feeding only with saliva suctioning in the oral cavity necessary; (2) tube feeding only without suctioning needed; (3) tube feeding mainly with occasional/fun oral intake; (4) oral feeding only with supplemental nutrients exclusively, such as enteral solution; (5) oral feeding only with easy-to-swallow food and supplemental nutrients; (6) oral feeding only with easy-to-swallow food exclusively; (7) totally oral fed with some food restrictions; and (8) oral fed only with no restrictions.16
Genetic Testing
Molecular studies were performed through commercial genetic panels (8 patients) and research WES (25 patients) according to availability. The genetic panel included all the main genes associated with congenital myopathies. All genes associated with NM were included and well covered. Variants were annotated for the transcript NEB:ENST00000397345.8 using ANNOVAR (ANNOtate VARiation) software17 and Ensembl Variant Predictor18 and then filtered using custom R-scrips. Filtered variants were rare or absent in control population databases (gnomAD exome and gnomAD Genome) and absent in homozygous individuals in controls.19 To detect whether the variant was previously reported, we used the literature in addition to ClinVar and Leiden Open Variation Database. The in silico predictors used were Polyphen, Genomic Evolutionary Rate Profiling score, and Combined Annotation Dependent Depletion score for missense variants and Splice AI for splice site variants. Owing to the main mechanism associated with NEB variants, we considered frameshift variants, premature stop-gain and splicing variants, inframe indels, and exon deletion as predicted loss-of-function alleles. Rare missense variants were included in the analysis if previously reported or had functional assays performed or if they were rare and predicted to be damaging.
Statistical Analysis
The normality assumption for continuous variables was checked using histograms and normality plot. Continuous variables were presented as mean (SD) or median and interquartile range and discrete and categorical variables as counts and percentages. Univariate analysis was performed to calculate the prevalence and describe the characteristics of the patients. Non-normally distributed variables were associated with categorical variables using the Dunn test of multiple comparisons using rank sums and Bonferroni as a post hoc analysis when appropriate. Odds ratio was calculated as a measure of association between the independent and dependent variables using contingency tables and the Mantel-Haenszel χ2 test, and 95% CI and p value were calculated. All tests were 2-sided, and a p value of 0.05 was considered as the threshold for significance. The Stata software was used for statistical analysis STATA 16.0 MP (STATA®, Statistical Data Analysis, Statacorp, LLC, TX) and R-packages.
Data Availability
Deidentified data will be shared on request with any qualified investigator.
Results
General Clinical Characteristics
Our cohort consisted of 33 patients, 15 females and 18 males, from 30 families harboring NEB variants. They presented an average age of 18 (±12) years and a median of 17 (±11) years, ranging from 2 to 59 years. Two families reported consanguinity. Muscle biopsy was performed in 26/33 patients, and nemaline rods were confirmed in all of them. Seven patients were diagnosed through the molecular test only, and muscle histology was not performed. Demographics are presented in Figure 1. Clinical findings are summarized in eTable 1, links.lww.com/NXG/A576.
Figure 1. Demographics and Main Characteristics of the Cohort.
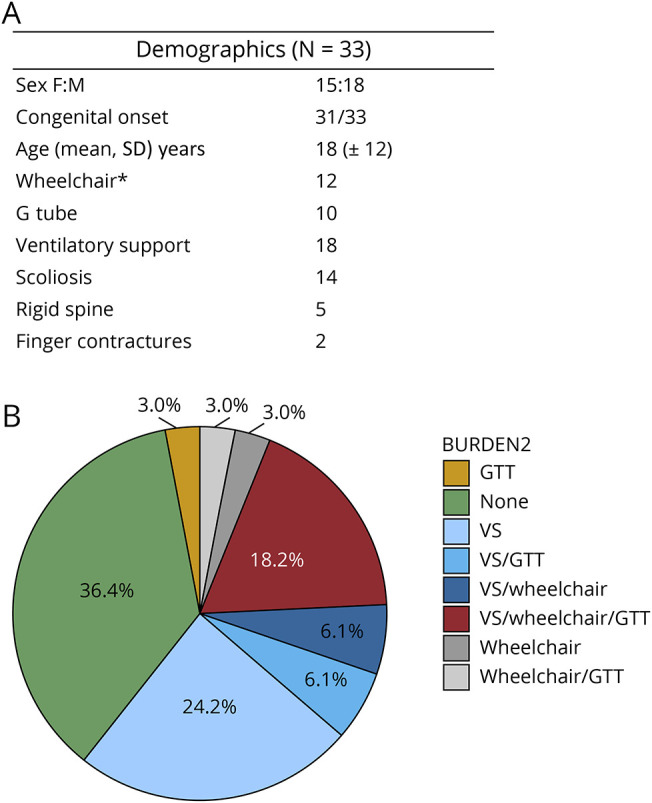
Demographics table (A) with the main characteristics of the cohort and a pie chart (B) illustrating the need for support in the main involved domains (motor, respiratory, and bulbar). Only 36% of patients did not need interventional support in the respiratory, motor, or bulbar systems and 18% of them needed support in all 3 systems.
Most of the patients presented congenital onset, and only 2 patients (6%) had the first symptoms during childhood. At the final evaluation, most of the patients (55%) were able to walk independently, 4 patients (12%) were able to walk only with support, 3 patients (9%) could stand only and help with transfers, 4 patients (12%) could sit unsupported, and 3 patients were not able to sit or roll or they had head support (age of 2, 2.6, and 0.7 years). Of the walking patients, only 2 were able to climb stairs unsupported, 12 were able to climb stairs with unilateral support and 5 needed bilateral support. Three patients lost their gait at ages of 13, 15 and 20 years (P12, P21, and P26), respectively.
All patients presented with a high arched palate and 2 patients with childhood onset had less pronounced facial weakness. In addition, tongue atrophy with a triple furrow sign was detected in 12 patients of the 23 accessed for this sign (Figure 2, A–D). Among those, the atrophy was mild in 6 patients and was pronounced in 6 others.
Figure 2. Triple Furrow Sign in Nemaline Myopathy Caused by NEB Variants.

Pronounced tongue atrophy with the triple furrow sign in 4 patients in A, B, C, and D.
The presence of tongue atrophy was statistically associated with the scores in the NdSSS (p = 0.006) (Figure 3). Patients with more pronounced tongue atrophy also had the tendency to be most dysphagic, followed by those with mild tongue atrophy and those with an absence of atrophy.
Figure 3. Association Between Tongue Atrophy and Swallow Function in NM-NEB Represented by NdSSS Scores.
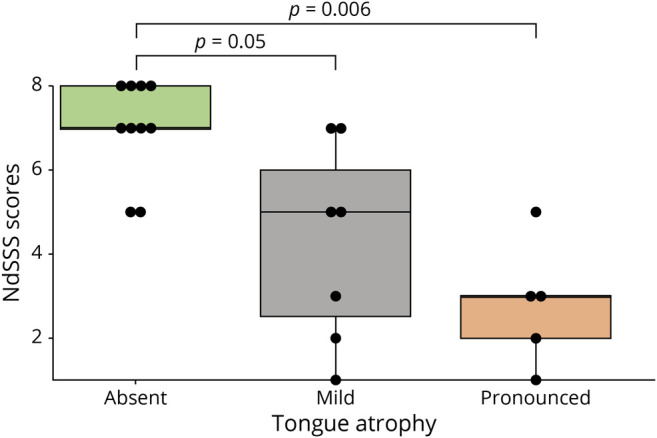
Box plot showing the comparison of the NdSSS scores and the presence of tongue atrophy. There was significant association between tongue atrophy and swallow function. Patients with a trophic tongue had the highest scores of NdSSS, followed by those with mild tongue atrophy and those with pronounced tongue atrophy, which presented with the lowest scores on the swallow scale.
Skeletal deformities were found in 63% of the cohort, excluding 3 patients who were younger than 3 years and who were too young to manifest this complication. Scoliosis was the most frequent deformity and was found in 14 (46%) of the patients, followed by rigid spine present in 5 (16%) and finger contractures in 2 (6%) patients (Figure 4, A–D). The current mean age of patients with spine deformities was 24 (±12) years compared with 12 (±5) years of patients without spine deformities. Four patients had scoliosis correction surgery (15% of patients who were older than 10 years). Two patients (P14.1 and P14.2) had severe thoracic deformities and kyphoscoliosis (Figure 4E), and they had indication for spine surgery, but due to clinical/respiratory conditions, the procedure was suspended by the orthopedic team.
Figure 4. General Characteristics of the Cohort.

Finger contractures (A and B) were found in patients with NM-NEB. Scoliosis and thoracic deformities were present in most of the patients, but pectus carinatum and kyphoscoliosis (C) were found only in 2 siblings. (D) Prominent cervical flexor weakness in a patient with a childhood-onset form despite presenting with no facial or proximal weakness.
Ventilatory Support
In the evaluation, 55% of the patients were using VS. Five patients (16%) used it continuously. Of them, 4 had tracheostomy and used ventilator. P25 refused to have TQ, although it was recommended, and used bilevel positive airway pressure (BiPAP) continuously with a facial mask during the final evaluation. Six patients were in intermittent VS; all of them used BiPAP, but 2 had TQ. Six patients used BiPAP at night only, and only 1 of them had TQ (P31). Patients started VS at a mean age of 9 (±6) years, and the mean age of the 16 patients who were not in VS was 18 (±4) years. Among the patients who were using VS, 6 started VS in their first days of life (neonatal-VS group). Most of the others started VS during childhood (mean age of 15 [±9] years) (later-VS group). Two patients required transient VS in infancy in the setting of additional contributors to respiratory compromise. P30 needed early but temporary VS. She was born at 32 weeks, twin brother was not affected, and she was intubated neonatally, but was off the ventilator in 2 months. At the last evaluation, she was 7 months of age, did not use VS, and was developing with motor gains. On the contrary, P31 was born well and needed VS few months later. She had an uneventful neonatal period, but developed with failure to thrive, recurrent vomits, and recurrent pulmonary infections. At the age of 18 months, she developed a severe pulmonary infection, and she was intubated, tracheostomized, and gastrostomized. After the GT, she was not having recurrent infections, and 1 month after the procedures, she improved her ventilatory function, needed VS only while sleeping, and improved her weight gain. At the final evaluation, she was 8 years of age, and she still had TQ but was using BiPAP only at night.
Comparison between the 15 patients who used VS (VS-group) with the 18 who did not use VS (no-VS group) is presented in Figure 5A. There was a significant difference in the presence of scoliosis, tongue atrophy, and lower NdSSS scores (NdSSS <6 and NdSSS >7). Patients from no-VS group presented less frequently scoliosis and tongue atrophy and had higher scores of NdSSS. There was a tendency of significance comparing the presence of gastrostomy tube (GT). Only 2 patients from the no-VS group had GT compared with 8 patients from the no-VS group. The use of wheelchair, maximum motor function, and presence of rigid spine and finger contractures were not different in these 2 groups.
Figure 5. Ventilatory Support Characterization.

(A) Main characteristics of patients who used ventilatory support vs those who did not use ventilatory support during evaluation. (B) Time-to-event analysis to the ventilatory support demonstrates that most of the patients start the ventilatory support before the age of 20 years and that, at approximately 40 years of age, less than 15% of the patients are not going to need ventilatory assistance. *In this analysis, we included only patients older than 3 years. **In this analysis, we excluded patients who were not accessed for tongue atrophy.
The neonatal-VS group's age in the final evaluation ranged from 2 to 12 years. All of them developed motor gains, but 3 patients (P1, P2, and P6) were still in IVS (mean age of 4 ± 3 years), 1 patient (P5) used NIVS (BiPAP) intermittently at the age of 11 years, and 1 patient (P11) used NIVS (BiPAP) nighttime only at the age of 13 years. They also presented frequently severe and early spine deformities; 2 of them were able to acquire gait, but lost it still in their first decade.
Comparison between the neonatal-VS group and later-VS group showed that patients who started VS neonatally were also more severe in the motor domain (all of them needed a wheelchair), all of them had severe scoliosis, and 90% had GT. However, the difference was not statistically significant.
Progression of the Disease
To gather insights of the natural history of the disease, we stratified patients according to the group of ages that could be relevant in the disease progression: (1) infants (younger than 3 years, n = 3); (2) children (aged 4–12 years, n = 6); and (3) teenagers (aged 13–20 years, n = 13) and (4) adults (older than 21 years, n = 10). The first group had only 3 individuals and was excluded from this analysis.
The swallow function and maximum motor function were similar in the different age groups. In the respiratory domain, we observed a decrease in the proportion of patients who did not use any VS and an increase in the proportion of patients who presented with scoliosis. Most of the patients aged 13–20 years (65%) did not use VS when compared with 20% of the group comprising those older than 20 years. In addition, 60% of patients aged 4–12 years did not have skeletal deformities when compared with 40% in the group comprising those aged 13–20 years. All patients older than 21 years had skeletal deformities, whereas most of them presented with scoliosis (Figure 6, A–D).
Figure 6. Progression of the Disease.

Clinical manifestations separated by age groups. Bar plot showing patients categorized by age groups according to maximum motor function (A) type of ventilatory support (B), the presence of spine deformities (C) and the use of ventilatory support (D). This analysis showed stability of the disease in the bulbar and motor domains and a progressive course in the respiratory domain and among the skeletal deformities.
Genetic Analysis
A DNA study was performed through exome sequencing in 25 patients and commercial neuromuscular panel in 8 patients. Data are included in eTable 2, links.lww.com/NXG/A576. Most of the patients (26, 80%) presented 2 pathogenic/likely pathogenic (P/LP) variants; 4 of them had a (P/LP) variant in homozygous state and 22 presented 2 monoallelic P/LP variants in trans state. Four of the patients presented 1 pathogenic variant in trans with a likely pathogenic missense variant (12%). Therefore, those 26 patients were classified as definite NM-NEB. Two patients presented 1 P/LP variant in trans with a novel missense variant classified as variant of unknown significance, and 5 patients presented only 1 P/LP. Therefore, we classified those 7 patients (20%) as probable NM-NEB.
All variants were classified according to the American College of Medical Genetics guidelines,20 and details about the variant annotation and classification are summarized in eTable 2, links.lww.com/NXG/A576. Of the 45 variants found, 22 were previously reported in the literature or ClinVar in association to NM, and 23 were novel. Most of the variants were predicted to truncate the protein. Twelve variants were in the splice site region, 16 were frameshift variants, 8 presented a nucleotide change that created a premature stop codon, and 6 were rare missenses; of those, 4 were previously reported in association to congenital myopathy. Two variants were exon deletion/duplication, and 1 was an inframe indel. Seven variants were recurrent. The variant c.19944G > A (p.Ser6648=) was present in 3 patients (P2, P25, and P31) in trans with another pathogenic variant. All of them were severely affected. P2 was ventilator dependent since her first weeks of life, and P25 was able to stand and change in transfers but was also in 24-hour Bipap ventilation due to a severe restrictive pulmonary involvement. The variant c.5343 + 5G > A was found in 5 patients. It was found in a homozygous state in P29 and as a compound heterozygosity in P9, P19, P26, and P27. This variant is not present in gnomAD and was previously reported in patients with NM. The variants c.24840_24841del (p.Arg8281Glyfs) and c.9723 + 1G > A were novel and shared in a heterozygous state in 2 patients each.
Discussion
We presented clinical and genetic findings in 33 patients with NM caused by NEB variants, representing one of the largest cohorts reported thus far. These data provided a novel clinical finding in this group of patients and provided important insights into the disease progression among age groups.
Previous studies have shown a high disease burden regarding NM in general comprehending 3 main domains: bulbar, respiratory, and motor functions.13,14,21 In a cross-sectional study, Amburgey et al. presented data from 57 patients with NM, whereas 20 were caused by NEB pathogenic variants. Considering interventional support in this study, 32% of the NM-NEB used GT, 35% used wheelchair, and 55% used VS. In the cohort presented in this study, we observed similar results, where 25% of the patients used a GT, 46% of them were not able to walk, and 55% used VS. In general, 36% of patients did not require any interventional support, 30% needed support in 1 domain, 15% in 2 domains, and 18% needed interventional support in all 3 domains.
Time-to-event analysis to the VS demonstrated that 50% of patients started the VS before 20 years. At 40 years of age, 85% of patients needed ventilatory assistance. According to the age that patients started VS, we noticed 3 distinct groups of patients: the ones who started VS neonatally (neonatal-VS), the ones who started VS later in childhood (later-VS), and the ones who did not use VS at the moment of the final evaluation (no-VS). We observed differences in the disease progression among those groups that could be relevant for management care. Despite the low number of observations, the neonatal-VS patients were more severely affected in all domains. Two were still too young for us to make any assumptions. The remaining 3 developed severe kyphoscoliosis while still in their first decade of life and maintained severe ventilatory dysfunction. Two of them acquired their gait but were able to walk for a few steps with bilateral assistance device. This finding suggested that requiring VS neonatally might be an important severity marker. But careful attention is needed for other factors influencing the early use of VS in childhood such as the prematurity described in P30.
The comparison between patients using VS with patients not using VS showed that both were similar regarding their maximum motor function and wheelchair use. However, dysphagia, tongue atrophy, and scoliosis were more frequent in patients using VS. It is hard to infer whether presenting one of those features might aggravate the ventilatory dysfunction or whether they reflect the axial involvement that led to the weakness in the respiratory muscles commonly found in NM. But we should look carefully in patients presenting ventilatory dysfunction, spine deformities, and/or dysphagia because they might be connected. P31 had a typical presentation but was tracheostomized and started on IVS early in childhood after successive respiratory infections because of broncoaspiration, which exemplifies how careful should be the dysphagia assessment in these patients.
Orofacial and bulbar muscles are commonly involved in patients with NM. Our data agreed with this statement and demonstrated that although most of the patients were orally fed, only 30% mentioned they were able to eat all kinds of food. The remaining patients explained they needed to adapt their food and/or take supplements to get the nutrition they need. The cause of dysphagia in NM is not completely understood. However, it seems to result from a combination between orofacial weakness and orofacial deformities such as high-arched palate, bad dental occlusion, and the inability to close the mouth, also presented among most of the patients in this cohort. A detailed assessment of the bulbar function is preferable for more consistent conclusions and is ongoing.
We described the tongue atrophy in a triple furrow pattern as a novel clinical finding presented by several patients in this cohort. The atrophy was more pronounced in patients with lower scores in the NdSSS, suggesting a direct association between the tongue atrophy and dysphagia. This observation added weight to the orofacial muscular involvement for NM-NEB and pointed to tongue weakness as a relevant and clinically accessible factor for dysphagia.
Despite not having a large prospective study, NM has been considered a relatively stable disease.13,14,22 It has been recently suggested that scoliosis and motor function might progress over time but that respiratory function and walk support were stable.14 Comparing clinical manifestations among patients grouped by age, we agreed that they were relatively stable in the bulbar and motor domains and that skeletal deformities might present a progressive character because all patients in the adult group had spine deformities when compared with 60% of the teenagers and 25% of the children. However, we showed that respiratory function might also present a progressive character. There was a decrease in the proportion of no-VS patients, decreasing from 65% in the teenager group to 20% among the adults. A prospective study would be necessary to gather stronger conclusions; but we believe that respiratory dysfunction and skeletal deformities are slowly progressive and that a short-period natural history study might not be enough to catch the changes those patients might present.
Agreeing with previous publications, we did not identify any genotype-phenotype correlation.6,22 Most of the patients had biallelic pathogenic variants in NEB gene defining it as the causal gene among those patients. In 20% of patients, we found only 1 P/LP variant (NEB-probable) despite the efforts to gather copy number variation (CNV) and exon duplications and deletions. All of them required muscle biopsy to confirm NM diagnosis. Causative variants, including CNVs, are known to be distributed throughout the whole gene, and gain or losses into the repeated region might also be pathogenic and represent a big challenge for short-read sequencing techniques.9 For this reason, we believed that there is a second variant in the patients who presented only 1 pathogenic variant, and a deep investigation using specific CNV pipelines and RNA sequencing or the whole-genome sequencing might be necessary to define the association. Most P/LP variants identified in the cohort resulted in frameshift, premature stop codon creation, or splicing changes that caused truncation or deletion in the protein as previously described. Missense variants appear to be rare and require further investigation to presume any effect in the protein; for this reason, we did not include them as strong candidates unless they were previously reported in patients with NM.6,23 Two patients presented 1 pathogenic variant in combination to a missense of unknown significance. We considered it as NEB-probable because more studies are required to verify the effect of this missense.
Our study had several limitations. A prospective study design would be preferable for natural history or disease progression considerations and to avoid survivorship bias that might be a confounder in cross-sectional studies. We were not able to perform functional measures in all patients. To address that, a new study accessing functional measures, respiratory function, and swallow function is on the way. However, considering that NM is a rare disease and that severe patients are usually not willing to participate in long follow-up studies, we added valuable data for different age groups, which could be translated into information about the progression of the disease. We showed a triple furrow atrophy in tongue as a novel clinical sign, which is associated with dysphagia. This easily assessed finding is very valuable in the evaluation of bulbar involvement in patients. Finally, we demonstrated that scoliosis, tongue atrophy, and dysphagia were more frequent in patients who require VS and that scoliosis and respiratory function presented a slowly progressive character. These findings could help clinician in the diagnosis and care of NM caused by NEB mutations.
Glossary
- BiPAP
bilevel positive airway pressure
- CNV
copy number variantion
- GT
gastrostomy tube
- IVS
invasive VS
- NdSSS
Neuromuscular Disease Swallowing Status Scale
- NIVS
noninvasive VS
- NM
nemaline myopathy
- P/LP
pathogenic/likely pathogenic
- TQ
tracheostomy
- VS
ventilatory support
Appendix. Authors

Study Funding
The authors report no targeted funding.
Disclosure
C.A.M. Moreno, M.C. Artilheiro, A.T.Q.S.M. Fonseca, C.G. Camelo, G.C. Medeiros, F.C. Sassi, C.R.F Andrade, S. Donkervoort, A.M.S. Silva, L.D. Junior, O.L. Abath-Neto, U.C. Reed, C.G. Bonnemann, and E. Zanoteli report no disclosures relevant to the manuscript. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Sewry CA, Laitila JM, Wallgren-Pettersson C. Nemaline myopathies: a current view. J Muscle Res Cell Motil. 2019;40(2):111-126. doi: 10.1007/s10974-019-09519-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallgren-Pettersson C, Sewry CA, Nowak KJ, Laing NG. Nemaline myopathies. Semin Pediatr Neurol. 2011;18(4):230-238. doi: 10.1016/j.spen.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Sandaradura SA, Bournazos A, Mallawaarachchi A, et al. Nemaline myopathy and distal arthrogryposis associated with an autosomal recessive TNNT3 splice variant. Hum Mutat. 2018;39(3):383-388. doi: 10.1002/humu.23385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miyatake S, Mitsuhashi S, Hayashi YK, et al. Biallelic mutations in MYPN, encoding myopalladin, are associated with childhood-onset, slowly progressive nemaline myopathy. Am J Hum Genet. 2017;100(1):169-178. doi: 10.1016/j.ajhg.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiiski KJ, Lehtokari VL, Vihola AK, et al. Dominantly inherited distal nemaline/cap myopathy caused by a large deletion in the nebulin gene. Neuromuscul Disord. 2019;29(2):97-107. doi: 10.1016/j.nmd.2018.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Lehtokari VL, Kiiski K, Sandaradura SA, et al. Mutation update: the spectra of nebulin variants and associated myopathies. Hum Mutat. 2014;35(12):1418-1426. doi: 10.1002/humu.22693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sagath L, Lehtokari VL, Välipakka S, et al. Congenital asymmetric distal myopathy with hemifacial weakness caused by a heterozygous large de novo mosaic deletion in nebulin. Neuromuscul Disord. 2021;31(6):539-545. doi: 10.1016/j.nmd.2021.03.006. [DOI] [PubMed] [Google Scholar]
- 8.Wallgren-Pettersson C, Laing NG. Report of the 70th ENMC international workshop: nemaline myopathy, 11-13 June 1999, Naarden, The Netherlands. Neuromuscul Disord. 2000;10(4-5):299-306. doi: 10.1016/s0960-8966(99)00129-7. [DOI] [PubMed] [Google Scholar]
- 9.Pelin K. Nebulin: size matters for optimal muscle function. J Gen Physiol. 2021;153(3):e202012848. doi: 10.1085/jgp.202012848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuen M, Ottenheijm CAC. Nebulin: big protein with big responsibilities. J Muscle Res Cell Motil. 2020;41(1):103-124. doi: 10.1007/s10974-019-09565-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Kleef ESB, van Doorn JLM, Gaytant MA, et al. Respiratory muscle function in patients with nemaline myopathy. Neuromuscul Disord. 2022;32(8):654-663. doi: 10.1016/j.nmd.2022.06.009. [DOI] [PubMed] [Google Scholar]
- 12.Lehtokari VL, Pelin K, Sandbacka M, et al. Identification of 45 novel mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Hum Mutat. 2006;27(9):946-956. doi: 10.1002/humu.20370. [DOI] [PubMed] [Google Scholar]
- 13.Colombo I, Scoto M, Manzur AY, et al. Congenital myopathies: natural history of a large pediatric cohort. Neurology. 2015;84(1):28-35. doi: 10.1212/wnl.0000000000001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amburgey K, Acker M, Saeed S, et al. A cross-sectional study of nemaline myopathy. Neurology. 2021;96(10):e1425-e1436. doi: 10.1212/wnl.0000000000011458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wijnhoven TM, de Onis M, Onyango AW, et al. Assessment of gross motor development in the WHO multicentre growth reference study. Food Nutr Bull. 2004;25(1 suppl 1):S37-S45. doi: 10.1177/15648265040251s106. [DOI] [PubMed] [Google Scholar]
- 16.Wada A, Kawakami M, Liu M, et al. Development of a new scale for dysphagia in patients with progressive neuromuscular diseases: the Neuromuscular Disease Swallowing Status Scale (NdSSS). J Neurol. 2015;262(10):2225-2231. doi: 10.1007/s00415-015-7836-y. [DOI] [PubMed] [Google Scholar]
- 17.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLaren W, Gil L, Hunt SE, et al. The Ensembl variant effect predictor. Genome Biol. 2016;17(1):122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lahuerta Pueyo C, Aibar Arregui MÁ, Gracia Gutierrez A, Bueno Juana E, Menao Guillén S. Estimating the prevalence of allelic variants in the transthyretin gene by analysing large-scale sequencing data. Eur J Hum Genet. 2019;27(5):783-791. doi: 10.1038/s41431-019-0337-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryan MM, Schnell C, Strickland CD, et al. Nemaline myopathy: a clinical study of 143 cases. Ann Neurol. 2001;50(3):312-320. doi: 10.1002/ana.1080. [DOI] [PubMed] [Google Scholar]
- 22.Malfatti E, Romero NB. Nemaline myopathies: state of the art. Rev Neurol (Paris). 2016;172(10):614-619. doi: 10.1016/j.neurol.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 23.Lehtokari VL, Pelin K, Herczegfalvi A, et al. Nemaline myopathy caused by mutations in the nebulin gene may present as a distal myopathy. Neuromuscul Disord. 2011;21(8):556-562. doi: 10.1016/j.nmd.2011.05.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Deidentified data will be shared on request with any qualified investigator.