

Abstract
Building on our previous work on ibrutinib-based reversible covalent Bruton’s tyrosine kinase (BTK) PROTACs, we explored a different irreversible BTK inhibitor poseltinib as the BTK binder for PROTAC development. Different from ibrutinib, converting the irreversible cysteine reacting acrylamide group of poseltinib to a reversible covalent cyano-acrylamide group dramatically decreases the binding affinity to BTK by over 700 folds. Interestingly, one of the reversible covalent BTK PROTACs based on poseltinib with a rigid linker, dubbed as PS-RC-1, is highly potent (IC50 = ~10 nM) in Mino cells but not in other mantle cell lymphoma (MCL) cell lines, such as Jeko-1 and Rec-R cells. We showed that PS-RC-1 potently induces degradation of IKZF1 and IKZF3 but not BTK or GSPT1, accounting for its toxicity in Mino cells. We further decreased the molecular size of PS-RC-1 by shrinking the BTK binding moiety and developed PS-2 as a potent BTK and IKZF1/3 triple degrader with high specificity.
1. Introduction
Bruton’s Tyrosine Kinase (BTK) is a Tec-family tyrosine kinase present in all blood cells except for T cells and natural killer cells (Lindvall et al., 2005). Overexpression of BTK is associated with various B cell malignancies, including mantle cell lymphoma (MCL) and chronic lymphocytic leukemia (CLL). BTK is a clinically proven target to attenuate BCR signaling and induce cell death in these cancer cells (Cinar et al., 2013). Several BTK kinase inhibitors have been approved to treat B cell malignancies (Wang et al., 2013; Woyach et al., 2014; Estupi ñá n et al., 2021; Wen et al., 2021). Not only serving as a kinase, BTK can also enhance antigen receptor-induced calcium influx in a kinase-independent manner (Saito et al., 2003; Middendorp et al., 2003). Therefore, developing BTK degraders to abolish both kinase-dependent and -independent functions attracts significant interests in academia and industry (Buhimschi et al., 2018; Ronen et al., 2020; Guo et al., 2020; Tinworth et al., 2019; Sun et al., 2019; Yang et al., 2021; Zorba et al., 2018; Dobrovolsky et al., 2019).
A proteolysis targeting chimera (PROTAC) is a heterobifunctional molecule that can bind both a targeted protein and an E3 ubiquitin ligase to facilitate the formation of a ternary complex, leading to ubiquitination and ultimate degradation of the target protein (Burslem and Crews, 2017). Taking advantage of the PROTAC technology, we recently developed a reversible covalent BTK PROTAC RC-1 with a high target occupancy, serving as a dual functional inhibitor and degrader (Guo et al., 2020). Additionally, we serendipitously discovered that cyano-acrylamide-based reversible covalent chemistry can significantly enhance the intracellular accumulation and target engagement of PRO-TACs (Guo et al., 2020). To develop an orally available BTK degrader, we explore a different irreversible BTK inhibitor poseltinib as the PROTAC warhead in this study, which has a smaller molecular weight than ibrutinib. To our surprise, converting the irreversible cysteine reacting acrylamide group of poseltinib to a reversible covalent cyano-acrylamide group decreases the binding affinity to BTK by over 700 folds. Interestingly, the reversible covalent BTK PROTAC based on poseltinib, dubbed as PS-RC-1, is highly potent (IC50 = ~ 10 nM) in Mino cells but not in other MCL cell lines, such as Jeko-1 and Rec-R cells. Considering the common neosubstrates for Cereblon, we find that PS-RC-1 potently induces degradation of IKZF1 and IKZF3 but not BTK or GSPT1. We further decreased the molecular size of PS-RC-1 by shrinking the BTK binding moiety and developed PS-2 as a potent BTK and IKZF1/3 triple degrader.
2. Materials and methods
2.1. Cell line engineering and culture
HEK-293T17 cells were engineered to stably express nLuc-fused to the C-terminal of BTK or IKZF1 or IKZF3 via lentivectors. Briefly, BTK/IKZF1/IKZF3 expression constructs (DNASU: Cat. No. HsCD00514411, HsCD00512327, and HsCD00513042) were Gateway cloned into pLenti6.2-ccdB-Nanoluc vector (Addgene: Cat. No. 87078), which were co-transfected into HEK-293T cells with lentiviral envelope protein construct pCMV-VSV-G (Addgene, Cat. No. 8454) and lentiviral packaging plasmid (Addgene, Cat. No. 8454) for viral production. Transfections were carried out using calcium phosphate-mediated transfection method (Promega, Cat. No. E1200). Virus was harvested at 48 h and 72 h post transfection and transfection was performed in the presence of 8 μg/mL of polybrene. HEK-293T17 cells were treated with the concentrated viral particles. Following lentiviral transduction, stable cell lines were selected by 7.5 μg/mL of blasticidin. Successful establishment of stable cell lines were confirmed with Nano-Glo Luciferase Assay (Promega, Cat. No. N1110).
HEK-293T17, Mino,Jeko-1, Ramos, and A20 were obtained from the American Type Culture Collection (ATCC, Cat. No. CRL-11268, CRL-3000,CRL3006, CRL1596, and TIB-208). MOLM14 and Rec-R cells were shared by Dr. Orla Conneely at Baylor College of Medicine and Dr. Michael L. Wang at MD Anderson Cancer Center, respectively.
All cell lines were cultured in RPMI-1640 medium (Thermo Fisher Scientific, Cat. No. MT10040CV), except engineered HEK-293T17-BTK/IKZF1/IKZF3-nLuc cell lines, which were cultured in DMEM medium (Thermo Fisher Scientific, Cat. No. MT10013CV). All media contained 10% fetal bovine serum (GE Healthcare, Cat. No. SH30071.03) plus 1% Pen/Strep (Thermo Fisher Scientific, Cat. No. 15140163). All cells were grown in a humidified incubator at 37 °C with 5% CO2.
2.2. Immunoblotting
Mino cells were seeded into the wells of six-well plates at the density of 5 × 105 cells per mL in 2 mL of complete RPMI-1640 culture medium. After overnight adaptation, cells were treated with serially diluted compounds (from 1,000–1.6 nM, 5-fold dilution) for 24 h. After treatment, whole cell lysates for immunoblotting were prepared by pelleting Mino cells at 4 °C and 200× g for 5 min. The resulting cell pellets were washed once with ice-cold PBS and lysed in 1 × RIPA lysis buffer (Alfa Aesar, Cat. No. J62524) supplemented with protease and phosphatase inhibitor cocktail (Thermo Fisher, Cat. No.78430). Lysates were centrifuged at 15,000 × g for 10 min at 4 °C and protein concentrations were assessed using BCA assay (Pierce, Cat. No.23225). Same amounts of protein (30 μg) for each sample were loaded onto sodium dodecyl sulfate-polyacrylamide gel, separated by electrophoresis (Bio-Rad) at 120 V for 1.5 h and transferred to PVDF membrane using a Transblot Turbo system (Bio-Rad). After blocking for 1 h at room temperature in 1% BSA-TBST, the membranes were immunoblotted with the specified primary antibodies at the dilution of 1:1000 in TBST (Cell Signaling Technology: anti-BTK Cat. No.8547, anti-IKZF1 Cat. No.14859, anti-IKZF3 Cat. No.15103, anti-β-actin Cat. No.4570, Proteintech: anti-GSPT1, Cat. No.10763-1-AP) overnight at 4 °C and the HRP-conjugated secondary antibodies (Cell Signaling, Cat. No.7074, 1:1000 in TBST) for 1 h at room temperature. Imaging was performed using the ECL Prime chemiluminescent Western blot detection reagents (Kindle Biosciences, Cat. No. R1100) by visualization of the blots with an Imager (Kindle Biosciences, Cat. No. D1001). All western blots were subsequently processed and quantified with Imager software ImageJ and protein level was normalized to the β-actin loading controls.
2.3. Nanoluc-based endpoint and protein degradation kinetics assays
For endpoint assays, engineered HEK-293T17 cells were plated in white opaque 96-well plate (Thermo Fisher) at a density of 4,000 cells per well in either 100 μL of Opti-MEM (4 h treatment) or 100 μL of DMEM whole medium (24 h treatment). The nanoluc activity was determined using the furimazine substrate (Promega, Cat. No. N1110). Briefly, after 4 h incubation of indicated compounds, cells were directly lysed by addition of 100 μL of nanoluciferase detection solution, containing lysis buffer and furimazine (1/50 dilution). For 24 h treatment group, the old medium was replaced with 100 μL fresh opti-MEM, followed by the addition of equal volume of lysis buffer plus furimazine (1/50 dilution). The endpoint luminescence was measured with a microplate reader (BioTek Synergy H1).
For protein degradation kinetics assays, HEK-293T17 cells stably expressing nLuc fusion proteins were plated in white opaque 96-well plates (Thermo Fisher) at a density of 4,000 cells per well in 100 μL of Opti-MEM. Cells were allowed to attach overnight and treated with 40 nM or 200 nM of indicated compounds plus endurazine (1:2000, Promega, Cat. No. N2570) and extracellular nanoluc inhibitor (1:2000, Promega, Cat. No. N2162), after which the real-time luminescence was measured with a microplate reader (BioTek Synergy H1) immediately every 5 min for 24 h. The kinetics data were fitted in the “one phase decay” model in Prism 9 to calculate the half-life, defined as time to reach 50% of maximal protein degradation.
2.4. Cell viability assays
Mino cells were plated in 96-well plates (Corning, Cat. No.3598) at a density of 6,000 cells per well in 100 μL of medium. Cells were treated the next day with indicated compounds in a five-fold dilution series (from 1000 to 0.64 nM), followed by 72 h incubation at 37 °C with 5% CO2. Cell viabilities were measured using the Alarma Blue assay by adding prewarmed Resazuerin sodium (Sigma, Cat. No. 199303) solution (1 mg mL−1 in PBS) in an amount equal to 10% of the volume in the well. Four hours after incubation, fluorescence signals were measured with a BioTek Synergy H1 microplate reader at excitation/emission 544/590 nm from top with a gain of 60. The EC50 values were calculated using GraphPad Prism 9.3 (GraphPad Software, La Jolla, CA) with the nonlinear fitting model of “[Inhibitor] vs. response – Variable slope (four parameters)”. The IC50 values, defined as compound concentrations that reduce cell viabilities by 50%, were extrapolated based on the nonlinear fitting model and reported in figure panels.
2.5. TR-FRET biochemical binding assays
Time-resolved fluorescence resonance energy transfer (TR-FRET) assay was carried out to evaluate the binding of indicated compounds and BTK by competition with a BODIPY-FL labeled BTK tracer (Fig. 1, BTK TR-FRET tracer). The assay was performed in 20 μL assay buffer (50 mM Tris, pH7.5, 0.1% Triton X-100, 0.01% BSA, and 1 mM DTT) with 0.3 nM Tb-anti-His (Cisbio, Cat.No. 61HI2TLA), 2 nM His-BTK (Signal-Chem, Cat. No. B10–10H-10), 150 nM BTK-BODIPY tracer and serial diluted compounds (10,000–0.64 nM, 5-fold dilutions) in opaque 384-well plates. Unless specified otherwise, all assays were performed in triplicates. The assay mixtures were incubated at room temperature in dark for 120 min, unless specified otherwise, and the signals were collected using a BioTek Synergy H1 microplate reader to measure the fluorescence emission ratio (I520 nm/I490 nm) of each well, using a 340-nm excitation filter, a 100-μs delay, and a 200-μs integration time. Raw data from the plate reader were used directly for analysis. The curve-fitting software GraphPad Prism 9 was used to generate graphs and curves and to determine IC50 values.
Fig. 1.
Chemical structures of compounds used in this study.
2.6. Proteomics
Two million of Mino cells were treated with DMSO or 200 nM of PS-2 for 4 h in biological triplicates and cells were harvested by centrifugation (200× g, 4 °C, 5 min). Lysis buffer (100 mM Triethylammonium bicarbonate (TEAB, Thermo Fisher Scientific, Cat. No. 90114), 2% SDS) was added to the cell pellets and homogenized with a microtip sonicator (Branson) to achieve a cell lysate with a protein concentration of ~2 mg mL−1. Protein concentration was determined using BCA assay (Pierce, Cat. No.23225) and normalized to 1 mg mL−1. One hundred μg of protein for each sample were reduced and alkylated with 10 mM Tris(2-carboxyethyl) phosphine hydrochloride (TCEP, Sigma, Cat. No. C4706) and 17 mM iodoacetamide (IAA, Sigma, Cat. No. 16125-5G), followed by digestion with trypsin (1:20, enzyme: protein, Thermo Fisher Scientific, Cat. No.90058) for 16 h shaking at 37 °C. Tandem mass tag (TMT) reagents (Thermo Fisher Scientific, Cat. No. A44520) were dissolved in anhydrous acetonitrile (ACN, Sigma, Cat. No. 271004) according to manufacture instructions, 0.08 mg of label reagent was used for 10 μg of protein digest. The 16-plex labeling reactions were performed for 1 h at room temperature and the reaction was quenched by the addition of 5% hydroxylamine solution for 15 min at room temperature. The sample channels were combined at a 1:1:1:1:1:1 ratio, desalted using peptide desalting columns (Thermo Fisher Cat. No. 89852). After drying down in a speed vacuum overnight, the combined sample was resuspended in 1% formic acid and subjected to Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA).
3. Results
3.1. Development of poseltinib based reversible covalent BTK PROTACs
Based on a molecular docking modeling for poseltinib in the ATP binding pocket of BTK, we found that the piperazine moiety is solvent exposed and chosen as the exit vector to develop BTK PROTACs. Following our and others’ work on BTK PROTACs, we choose Cereblon as the targeted E3 ligase (Buhimschi et al., 2018; Ronen et al., 2020; Guo et al., 2020; Tinworth et al., 2019; Sun et al., 2019; Yang et al., 2021; Zorba et al., 2018; Dobrovolsky et al., 2019). A series of reversible covalent BTK PROTACs with different linkers are synthesized (Fig. 1). PS-RC-1 has a rigid linker, while PS-RC-2, PS-RC-3 and PS-RC-4 share flexible PEG linkers with different lengths. To test whether the four PROTACs can induce BTK degradation, we treated Mino cells with PS-RC-1, PS-RC-2, PS-RC-3 and PS-RC-4 for 24 h, followed by western blotting to quantify the total BTK levels. To our surprise, none of the tested PROTACs induce appreciable BTK degradation, while the positive control compound DD-03-171 developed by the Gray group (Dobrovolsky et al., 2019) potently induces BTK degradation (Fig. 2a-e, Table 1). To corroborate with the Western blot results, we tested these compounds in HEK-293 cells stably expressing a BTK-Nano-Luciferase (nLuc) fusion protein as a reporter and found similar results (Fig. 2f, Table 1).
Fig. 2.
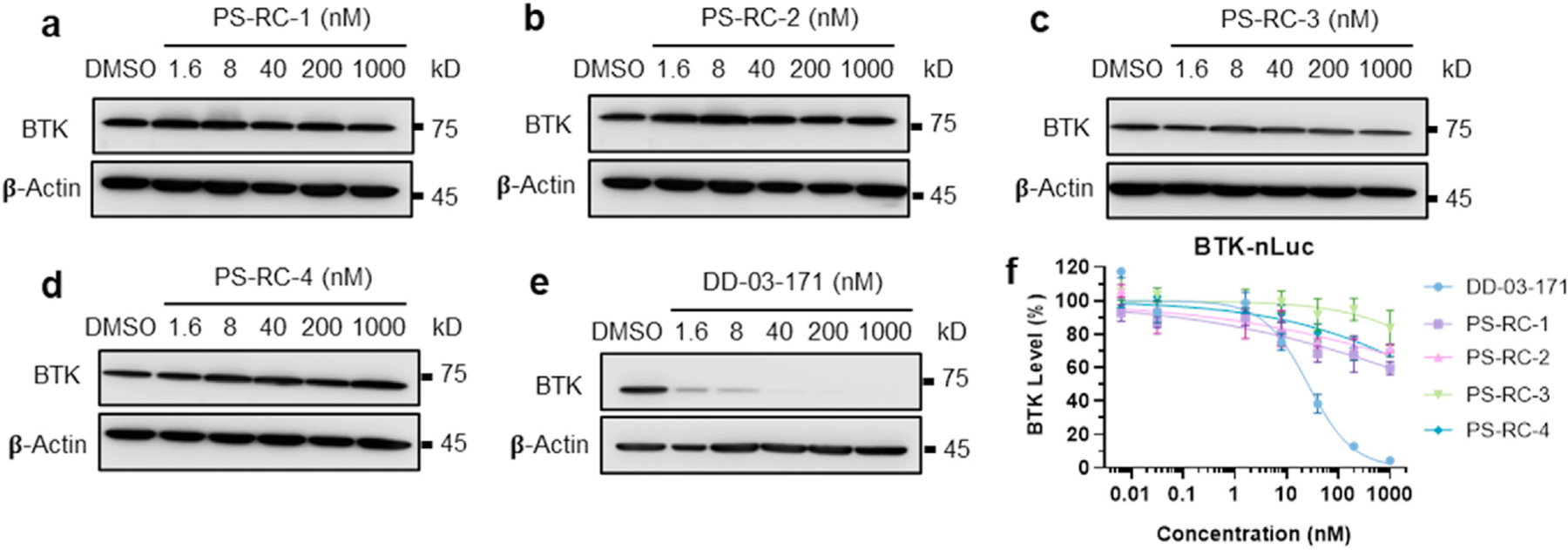
Poseltinib-based reversible covalent BTK PROTACs cannot induce BTK degradation in cells. a-e. Mino cells were treated with indicated compounds at 0, 1.6, 8, 40, 200, and 1000 nM for 24 h, followed by Western blotting for BTK. PS-RC-1, PS-RC-2, PS-RC-3, and PS-RC-4 are poseltinib-based reversible covalent BTK PROTACs. DD-03-171 is a BTK degrader developed by the Gray group and used as a positive control. f. HEK-293T cells stably expressing a BTK-nLuc fusion protein were treated with indicated compounds (same as in a-e) for 24 h. The BTK degradation was determined by evaluating luminescence signals of NanoLuc. The DC50 (concentration of PROTACs required to achieve 50% degradation of the target protein) and Dmax (maximum level of target protein can be degraded by PROTACs) values obtained through this assay are listed in Table 1.
Table 1.
Degradation of BTK induced by reversible covalent BTK PROTACs.
| Compounds | BTK-nLuc |
BTK WB |
BTK binding |
||
|---|---|---|---|---|---|
| DC50 (nM) | Dmax (%) | DC50 (nM) | Dmax (%) | IC50 (nM) | |
| DD-03-171 | 25.9 | 96 | 0.9 | 99 | – |
| PS-RC-1 | >1,000 | 40 | >1,000 | – | >10,000 |
| PS-RC-2 | >1,000 | 30 | >1,000 | – | >10,000 |
| PS-RC-3 | >1,000 | 16 | >1,000 | – | 4,367 |
| PS-RC-4 | >1,000 | 30 | >1,000 | – | 6,614 |
| Poseltinib | – | – | – | – | 1.21 |
| PS-RC-Ctrl | – | – | – | – | 869 |
We next examined the potencies of PS-RC-1, PS-RC-2, PS-RC-3 and PS-RC-4 in inhibiting cell growth with poseltinib as a positive control (Fig. 3a-b). In MOLM-14 cells, an acute monocytic leukemia (AML) cell line, the IC50 value of poseltinib is 2.9 μM, while all the reversible covalent BTK PROTACs do not show any appreciable inhibitory effects in the same cells at 10 μM, the highest concentration tested (Fig. 3a). Interestingly, in Mino cells, a mantle cell lymphoma (MCL) cell line, PS-RC-1 is highly potent with an IC50 value of 12 nM. In contrast, PS-RC-2, PS-RC-3 and PS-RC-4 barely inhibit 50% of cell growth at 10 μM in Mino cells (Fig. 3b). To test whether the potent inhibitory effect of PS-RC-1 is MCL cell specific, we tested its potency in two additional MCL cell lines, Jeko-1 and Rec-R, but found that these two MCL cells poorly respond to PS-RC-1 (Fig. 3c). Based on these data, we postulate that the growth inhibitory effect of PS-RC-1 in Mino cells is not due to BTK degradation but related to a specific cell signaling pathway in this cell line.
Fig. 3.
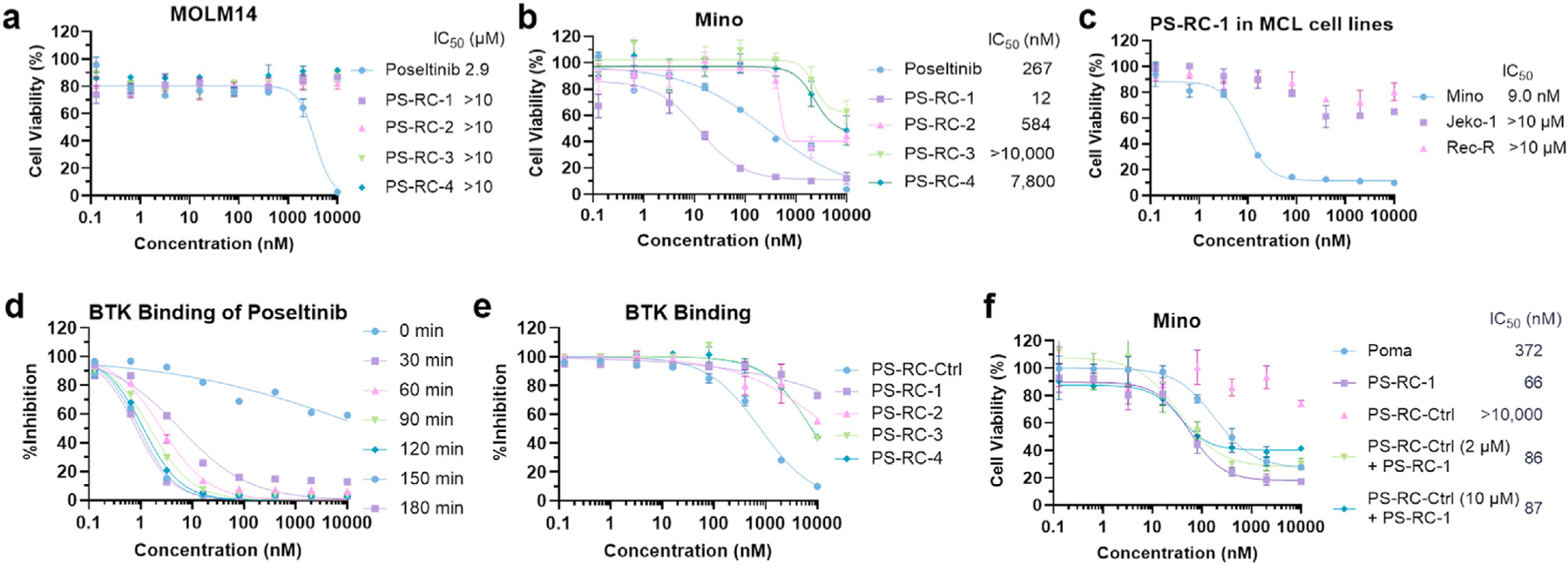
Toxicities of poseltinib-based BTK PROTACs in cells and their binding affinities to BTK. a-b. MOLM14 and Mino cells were treated with serially diluted poseltinib and PS-RC-1 to PS-RC-4 for 72 h, followed by Alarma Blue assay to quantify the cell viabilities. c. Three MCL cell lines, including Mino, Jeko-1, and Rec-R cells, were treated with serially diluted PS-RC-1 for 72 h, followed by Alarma Blue assay to quantify the cell viabilities. d. TR-FRET based binding kinetics assay between poseltinib and BTK. Serial dilutions of poseltinib mixed with 2 nM of His-BTK, 0.3 nM Tb-anti-His, and 150 nM of BTK-BODIPY tracer. e. BTK binding affinity assays for poseltinib-based PROTACs (PS-RC-1 to PS-RC-4), following the same protocol as described in d. After 2 h incubation, TR-FRET signals were measured. The IC50 values were listed in Table 1 f. PS-RC-1 serves as a molecular glue to inhibit growth in Mino cells. Mino cells were pre-treated with a large excess of PS-RC-Ctrl (2 μM or 10 μM), followed by PS-RC-1 incubation for 72 h. The cell viabilities were quantified using an Alarma Blue assay. For cell viability assays, data represent mean ± SD (n = 3) and the IC50 values are defined as compound concentrations that reduce cell viabilities by 50%. For BTK binding assays, data represent mean ± SD (n = 3) and the IC50 values are defined as compound concentrations that reduce tracer binding by 50%. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.2. Biochemical binding affinities of reversible covalent PROTACs to BTK
To measure the binding affinities between PROTACs and BTK, we developed a time-resolved fluorescence resonance energy transfer (TR-FRET) competition binding assay following previous studies (Asami et al., 2015; Yu et al., 2018). A BTK tracer was synthesized by conjugating an ibrutinib-based reversible noncovalent binder with the BODIPY-FL dye (Fig. 1). In a saturation binding assay, it was shown that the BTK tracer has a Kd value of 155 nM to the recombinant BTK protein. In the presence of the BTK tracer, competition assays were performed to measure the binding affinities between BTK and its inhibitors and PROTACs. As an irreversible covalent inhibitor, poseltinib is highly potent with an IC50 value of 1.1 nM for 2 h of incubation (Fig. 3d). Interestingly, substituting the acrylamide in poseltinib with a reversible covalent cyano-acrylamide group (i.e. PS-RC-Ctrl) dramatically decreases the binding affinity to BTK by > 700 folds (IC50 1.2 nM vs 869 nM, Table 1). Converting the reversible covalent cyano-acrylamide containing poseltinib to PROTACs further decreases their binding affinities to BTK (IC50 > 7.5 μM, Fig. 3e, Table 1). The very weak BTK binding affinities for PS-RC-1, PS-RC-2, PS-RC-3 and PS-RC-4 explain their minimal BTK degradation effects in cells and further support our hypothesis that the observed growth inhibitory effect of PS-RC-1 in Mino cells is unrelated to the BTK pathway.
3.3. Examining off-target effects of CRBN engaged PROTACs
CC-885 and CC-90009, two derivatives of lenalidomide, potently degrade the cell cycle regulator and translation termination factor GSPT1 as a neo-substrate for Cereblon (Hansen et al., 2021; Matyskiela et al., 2016). The groups of Winter, Wang, Yao and Tang reported GSPT1 as a common off-target for phthalimide-based PROTACs (Yang et al., 2019, 2021; Ishoey et al., 2018; Wang et al., 2021). Additionally, depending on the linker position on thalidomide, the CRBN engaged PROTACs can also degrade IKZF1 and IKZF3 (Guo et al., 2020; Dobrovolsky et al., 2019; Bricelj et al., 2021; Nguyen et al., 2021). To test these three off-target degradations, we treated Mino cells with PS-RC-1 at different concentrations for 24 h, followed by Western blotting. PS-RC-1 does not degrade GSPT1 (Fig. 4a) but potently degrades IKZF3 (DC50 = 44 nM, Fig. 4b and Table 2) and IKZF1 to a lesser extent (DC50 802 nM, Fig. 4b and Table 2). Immunomodulatory drugs (IMiDs), such as pomalidomide and lenalidomide, have been explored to treat relapsed or refractory mantle-cell lymphoma (Wang et al., 2012), presumably due to its degradation effect of IKZF1/3. We treated Mino, Jeko-1 and Rec-R cells with pomalidomide and found that only Mino cells are responsive to pomalidomide (IC50 = 329 nM, Fig. 3d), which is less potent than PS-RC-1 (IC50 = 9.0 nM, Fig. 3c) in the same cell line. Comparing the IKZF3 degradation efficiency, PS-RC-1 is more potent than pomalidomide (DC50 3.6 vs 14.3 nM (nLuc assay), DC50 44 vs 54 nM (Western blot), Table 2).
Fig. 4.
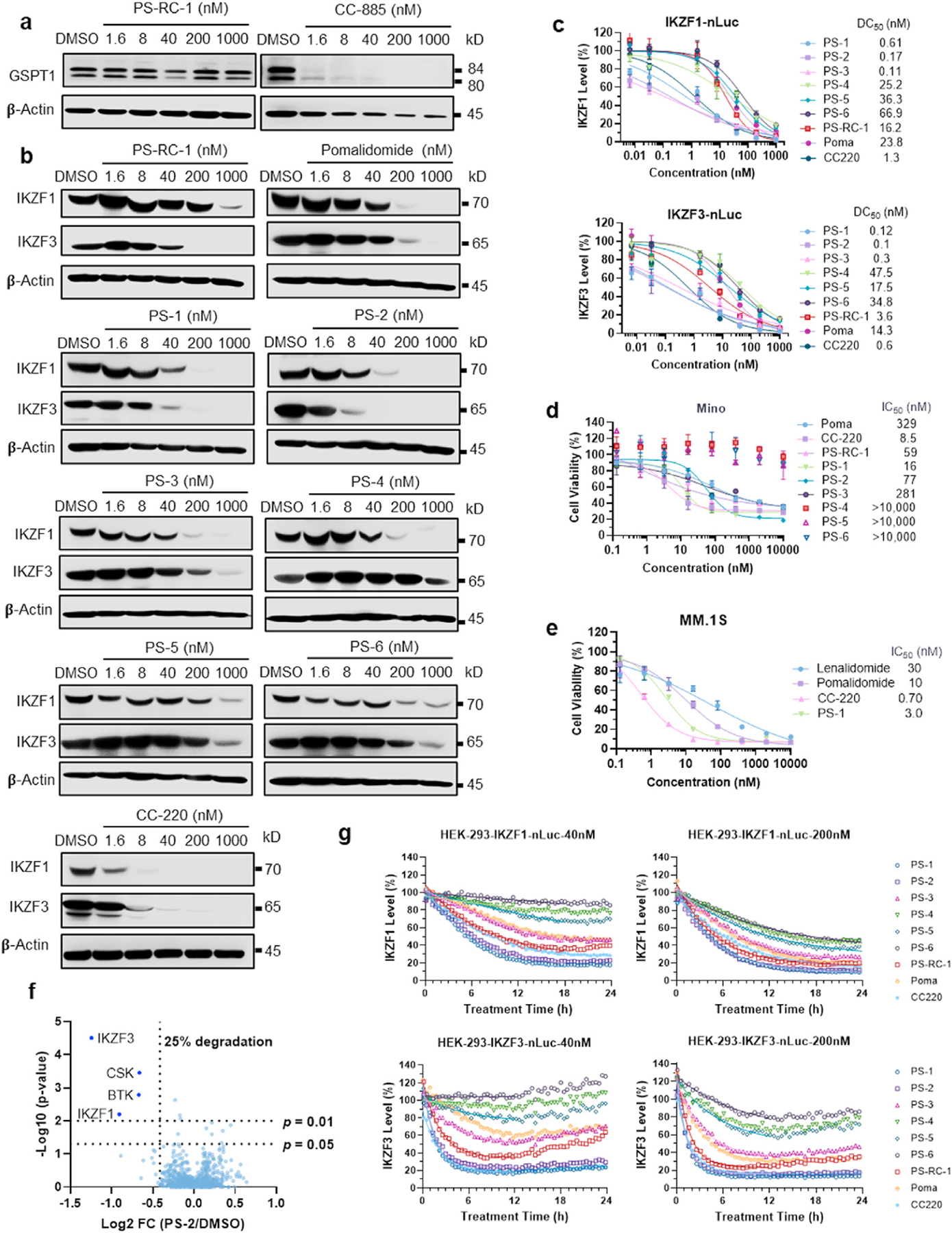
PS-1 and PS-2 potently degrades IKZF1 and IKZF3 and showed significant toxicity in Mino cells. a. Mino cells were treated with PS-RC-1 or CC-885 at 0, 1.6, 8, 40, 200, and 1000 nM for 24 h, followed by Western blotting for GSPT1. In contrast to the potent GSPT1 degrader CC-885, PS-RC-1 does not degrade GSPT1. b. Mino cells were treated with indicated compounds at 0, 1.6, 8, 40, 200, and 1000 nM for 24 h, followed by Western blotting for IKZF1 and IKZF3. c. HEK-293T-IKZF1/IKZF3-nLuc cells were treated with indicated compounds for 24 h and the degradation of IKZF1/IKZF3 were determined by measuring Nanoluc luminescence signals. Data represent mean ± SD (n = 3) and the DC50 values are defined as compound concentrations that reduce nLuc signals by 50%. d-e. Mino and MM.1S cells were treated with serially diluted indicated compounds for 72 and 120 h, respectively, followed by Alarma Blue assay to quantify the cell viabilities. For cell viability assays, data represent mean ± SD (n = 3 and the IC50 values are defined as compound concentrations that reduce cell viabilities by 50%. f. TMT-based quantitative proteome analysis of protein expression changes from PS-2 (200 nM) treatment for 4 h in Mino cells. Volcano plot shows protein abundance (log2) as a function of significance level (log10). The vertical dotted line denotes 25% reduction of protein levels, whereas the horizontal dotted lines marks p = 0.01 and p = 0.05 statistical significance thresholds. Downregulated proteins with statistical significance are found in the upper left quadrant of the plots. Dataset represents an average of n = 3 replicates. A total of 2,543 proteins were identified, and only the ones with at least one uniquely identified peptide are plotted. g. Degradation kinetics of IKZF1 and IKZF3. HEK-293T-IKZF1/IKZF3-nLuc cells were treated with indicated compounds, followed by immediate measurements of Nanoluc luminescence signals every 5 min for 24 h. The degradation kinetics were fitted into a mono-exponential decay model. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Table 2.
Degradation of IKZF1 and IKZF3 induced by PS-RC-1 and its analogs.
| Compounds | IKZF1-nLuc Endpoint |
IKZF1-nLuc Degradation Kinetics |
IKZF1-WB Endpoint |
IKZF3-nLuc Endpoint |
IKZF3-nLuc Degradation Kinetics |
IKZF3-WB |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DC50 (nM) | Dmax (%) | 40 nM Compounds |
200 nM Compounds |
DC50 (nM) | Dmax (%) | DC50 (nM) | Dmax (%) | 40 nM Compounds |
200 nM Compounds |
DC50 (nM) | Dmax (%) | |||||
| t1/2 (h) |
Dmax (%) |
t1/2 (h) |
Dmax (%) |
t1/2 (h) |
Dmax (%) |
t1/2 (h) |
Dmax (%) |
|||||||||
| PS-RC-1 | 16 | 95 | 5.4 | 60 | 3.4 | 80 | 802 | 62 | 3.6 | 91 | 1.6 | 43 | 1.0 | 65 | 44 | 92 |
| Pomalidomide | 24 | 90 | 15 | 53 | 5.5 | 80 | 42 | 96 | 14 | 94 | 5.1 | 52 | 1.9 | 64 | 54 | 96 |
| CC220 | 1.3 | 92 | 7.2 | 72 | 5.1 | 82 | 3.1 | 95 | 0.6 | 96 | 1.5 | 80 | 1.2 | 84 | 3.8 | 98 |
| PS-1 | 0.6 | 98 | 4.3 | 83 | 3.4 | 90 | 44 | 99 | 0.1 | 98 | 1.2 | 79 | 0.9 | 88 | 39 | 95 |
| PS-2 | 0.2 | 97 | 4.9 | 80 | 3.4 | 89 | 28 | 99 | 0.1 | 98 | 1.2 | 75 | 0.9 | 84 | 2.5 | 99 |
| PS-3 | 0.1 | 94 | 15 | 52 | 5.6 | 75 | 91 | 90 | 0.3 | 94 | 2.8 | 45 | 1.9 | 55 | 250 | 78 |
| PS-4 | 25 | 82 | 23 | 20 | 8.3 | 56 | 146 | 86 | 48 | 85 | >24 | 12 | >24 | 20 | >1,000 | 15 |
| PS-5 | 36 | 85 | >24 | 33 | 7.2 | 63 | 464 | 64 | 18 | 87 | >24 | 23 | >24 | 27 | >1,000 | 45 |
| PS-6 | 67 | 80 | >24 | 12 | 8.4 | 50 | 384 | 65 | 35 | 85 | >24 | – | >24 | 12 | 301 | 70 |
It is possible that reversible covalent poseltinib (PS-RC-Ctrl) may bind other off-targets, contributing to the observed cell growth inhibition effect of PS-RC-1 in Mino cells. We found that PS-RC-Ctrl has no appreciable effect on cell growth in Mino cells. To test whether the phenotype of PS-RC-1 in Mino cells is due to its bivalent structure-based protein degradation or a monovalent molecular glue, we pretreated Mino cells with a large excess of PS-RC-Ctrl (2 and 10 μM) for 2 h to prevent ternary complex formation with PS-RC-1, followed by PS-RC-1 treatment for 24 h. We did not observe appreciable IC50 shift for PS-RC-1 in Mino cells upon PS-RC-Ctrl pre-treatment, suggesting that the reversible covalent poseltinib moiety in PS-RC-1 does not significantly contribute to the observed toxicity in Mino cells (Fig. 3f). Therefore, we concluded that PS-RC-1 is a potent IKZF1/3 molecular glue degrader.
3.4. Optimization of IKZF1/3 degrader
As PS-RC-1 degrades IKZF1/3 instead of BTK, we were curious whether we can further shrink the molecular size by removing the BTK binding moiety. A series of PS-RC-1 analogs, PS-1, PS-2, PS-3, PS4, PS-5 and PS-6 (Fig. 1), were developed by maintaining the pomalidomide group while systemically shrinking the reversible covalent poseltinib moiety to explore the structure-activity relationship. We tested the IKZF1 and IKZF3 degradation potencies for these compounds in both HEK-293 cells stably expressing nLuc labeled IKZF1 and IKZF3 and in Mino cells using Western blotting. It is interesting to note that PS-RC-1, PS-1, and PS-2 have similar potencies to induce IKZF1 and IKZF3 degradation. PS-3, a PS-2 analog without the phenoxyl group, PS-4, PS-5 and PS-6 show significant reduction in the degradation potencies and preferential degradation of IKZF1 over IKZF3, similar to pomalidomide. Based on the Western blot in Mino cells, we found that among these analogs, PS-2 is the most potent to degrade IKZF1 and IKZF3 (DC50 27.8 nM and 2.5 nM), but less potent than CC-220 (DC50 3.1 nM and 3.8 nM, Fig. 4b and Table 2), an IMiD developed by Celgene. PS-RC-1, PS-1, and PS-2 inhibits Mino cell growth with IC50 values of 59, 16 and 77 nM, respectively (Fig. 4d).
IMiDs are commonly used to treat multiple myeloma (MM). We further tested PS-1 in MM.1S cells, an MM cell line. As shown in Fig. 4e, PS-1 is more potent than pomalidomide and lenalidomide in inhibiting the growth of MM.1S cells (IC50 = 3.0 nM).
Next, we performed tandem mass tagging (TMT)-based quantitative proteomic profiling to evaluate the degradation specificity of PS-2. PS-2 was chosen for the proteomics study because it is the most potent to induce degradation of IKZF1 and IKZF3 in Mino cells. To avoid the potential secondary effects induced by PS-2 treatment, Mino cells were treated with 200 nM of PS-2 for 4 h. Quantitative proteomics employing TMT chemical labeling coupled with LC/MS/MS enabled the detection of >2,500 unique proteins without fractionation (Fig. 4f). Consistent with Western blots (Fig. 4b), PS-2 induces >60% degradation of IKZF3 and ~50% degradation of IKZF1 after 4-h treatment, indicating the selectivity of PS-2 as a IKZF1/IKZF3 degrader. We also observed two additional downregulated targets, BTK and CSK, suggesting that downsized poseltinib can still bind to these two kinases. We further validated that PS-2, but not PS-1 or PS-3, potently induces BTK degradation in Mino cells using Western blotting (Fig. 5), corroborating the proteomics data. No other proteins are downregulated by PS-2 for at least 25% with statistical significance, indicating that PS-2 degrades IKZF1/3 with high specificity in Mino cells.
Fig. 5.

PS-2 induces BTK degradation in cells. Mino cells were treated with indicated compounds at 0, 1.6, 8, 40, 200, and 1000 nM for 24 h, Ramos and A20 cells were incubated with PS-1, PS-2, PS-3, and DD-03-171 at 40, 200, and 1000 nM for 24 h, followed by Western blotting for BTK. CC-220 is a IKZF1/3 degrader developed by Celgene and used for comparison. In contrast to PS-1, PS-3 and CC-220, PS-2 potently induces BTK degradation in Mino cells. In Ramos and A20 cell lines, PS-1 and PS-3 elicit very weak or no BTK degradation, while PS-2 induces potent BTK degradation that is comparable to DD-03-171.
Compared with PS-1, the BTK binding moiety of PS-2 lacks the furan ring fused to pyrimidine, which might be flexible enough to allow formation of a more stable ternary complex with BTK and CRBN in cells. In contrast, PS-3, whose warhead is further reduced compared with PS-2, loses its ability to function as either a BTK degrader or a potent IKZF1/3 molecular glue. To confirm this observation, we also tested BTK degradation induced by PS-1, PS-2, and PS-3 in both a human B lymphocyte cell line Ramos and mouse BALB/c B cell lymphoma line A20, with DD-03-171 from the Gray group as a positive control (Fig. 5). Consistent with the results observed in Mino cells, only PS-2, but not PS-1 or PS-3, can induce potent BTK degradation in Ramos and A20 cells with a potency comparable or better than DD-03-171.
We further examined the degradation kinetics of IKZF1 and IKZF3 using the HEK-293 cells stably expressing IKZF1-nLuc or IKZF3-nLuc. PS-1 and PS-2 induce the fastest degradation of IKZF1 and IKZF3 with degradation half-lives of ~2.8 and ~0.8 h, respectively (Fig. 4g and Table 2), which is faster than pomalidomide (4.9 h and 1.9 h for IKZF1 and IKZF3, respectively) and generally consistent with the potency trends measured by endpoint nLuc assays and Western blotting.
4. Discussion
In our previous study, we developed an ibrutinib-based reversible covalent BTK PROTAC, RC-1, which can potently degrade BTK, IKZF1 and IKZF3 with high intracellular accumulation and target engagement (Guo et al., 2020). The motivation of this study is to further improve reversible covalent BTK PROTACs to achieve oral availability. When designing poseltinib-based reversible covalent PROTACs, we docked poseltinib to the available BTK crystal structure (PDB: 5P9I) and found the predicted binding pose of poseltinib aligned well with those of spebrutinib and DD-03-171. Therefore, we chose the pyrimidine group in poseltinib as the linker attachment point, which is consistent with DD-03-171, the BTK degrader developed by the Gray group. It should be noted that the linker attachment point for our ibrutinib-based BTK PROTAC is on the covalent reactive group. For future studies, it would be interesting to take poseltinib or other BTK binders to explore two different exit vectors and compare their potencies.
Replacing the acrylamide in poseltinib with a reversible covalent cyano-acrylamide group unexpectedly decreases the binding affinity to BTK by > 700 folds. This may be due to the steric clash between BTK and the t-butyl group at the β-position of the cyano-acrylamide group. Smaller substituents at the β-position of the cyano-acrylamide group may alleviate the steric clash but render the Michael acceptor to non-specifically react with thiols, the millimolar level of intracellular glutathione, in particular (Krishnan et al., 2014; Bradshaw et al., 2015; Jiang et al., 2015, 2017, 2019; Chen et al., 2017). The t-butyl group at the β-position of the cyano-acrylamide group ensures the Michael acceptor has a millimolar Kd when reacting with glutathione. For future work, it would be interesting to explore other substituents at the β-position of the Michael acceptor with small molecular sizes, such as halogens, to reduce its reactivity towards nucleophiles.
Additionally, it is very interesting to note that compared with PS-2, the furan ring in PS-1 precludes it from degrading BTK. We speculate that when forming the BTK-PROTAC-CRBN ternary complexes, some side chains around the ATP binding pocket of BTK might need to reorient to accommodate CRBN binding. The smaller size of PS-2 might provide a larger wiggle room for the BTK side chains. To test this hypothesis, ternary complex binding assays and/or x-ray co-crystallization of the BTK-PROTAC-CRBN ternary complex would need to be performed. This could be an interesting example of how a modification in the target recognition element but not in the linker profoundly influences targeted protein degradation. We could further explore PS-2 analogs to develop BTK, IKZF1 and IKZF3 triple degraders.
We developed HEK-293 cells stably expressing targeted protein-nLuc fusions driven by a CMV promoter for high throughput screening of degraders. We infected HEK-293 cells with different MOIs and selected the clones that can achieve similar sensitivity to known degraders compared with the Western blot quantification. This assay is useful when screening a large number of degraders. However, the major caveat is that the expression level of the targeted protein-nLuc fusion is controlled by the CMV promoter instead of its endogenous promoter. Sometimes we observed discrepancies for protein levels between the nLuc based assay and Western blotting. For example, PS-5 and PS-6 appear to be potent IKZF3 degraders in the nLuc assay, but do not achieve appreciable degradation of IKZF3 on Western blots (Table 2 and Fig. 3). This may be because the degradation of IKZF3 triggers its compensation at the transcription level through its endogenous promoter. However, the CMV-driven nLuc assay cannot reflect the compensation effect. Ideally, nLuc should be endogenously knocked in to the target gene, although this process has low efficiency and is time consuming due to the growth of single cell clones.
Although the design of PROTACs seems to be modular, the linkers play critical roles in both the biological and physicochemical properties of PROTACs, usually dubbed as linkerology (Khan et al., 2020). In hematological cancer applications, it is important to continuously suppress BTK activities in patients. Therefore, it would be ideal to develop BTK degraders as orally available agents from the translational point of view. Based on the disclosed structures of PROTACs in clinical trials, both ARV-110, an androgen receptor PROTAC, and ARV-471, an estrogen receptor PROTAC, have highly rigid linkers. The Shaomeng Wang group reported an orally available androgen receptor degrader ARD-2128, which also has a rigid linker (Han et al., 2021). Pike et al. analyzed the oral bioavailability of PROTACs across a diverse set of projects at AstraZeneca and found that high oral bioavailability (>30%) can be achieved among approximately one third of CRBN-targeted PROTACs, while VHL-targeted PROTACs have no more than 3% of oral bioavailability (Pike et al., 2020). Therefore, we focused our efforts primarily on CRBN-targeted BTK PROTACs with rigid linkers. Recently, the Chaudhary group reported a systematic study regarding how the linker structures and the attachment points on imides affect the off-targets (Nguyen et al., 2021). PS-3 and PS-4 achieved substantial IKZF1 degradation at 200 nM, while PS-5 and PS-6 are less potent at the same concentration, suggesting that the amino-phenyl group in PS-4 contributes binding to IKZF1. PS-RC-1, PS-1 and PS-2 all potently degrade both IKZF1 and IKZF3 at 40 nM, while PS-3, PS-4, PS-5 and PS-6 do not degrade IKZF3 at the same concentration, suggesting that the phenoxyl group in PS-2 plays an important role to bind to IKZF3. Thalidomide, lenalidomide, and pomalidomide induce rapid degradation of IKZF1 and IKZF3 by recruiting them to the Cereblon E3 ubiquitin ligase through a Cys2-His2 (C2H2) zinc finger domain that is required for their degradation (Sievers et al., 2018). To better understand the structure-activity relationship, more analogs of PS-RC-1 should be developed and tested for IKZF1/3 degradation. Additionally, structural biology and MD simulations can also help to elucidate the detailed binding mode between the molecular glues and IKZF1/3.
Supplementary Material
Acknowledgments
The research was supported in part by National Institute of Health (R01-GM115622 and R01-CA250503 to J.W.), the Welch Foundation (Q1912 to M.C.W.), Howard Hughes Medical Institute (to M.C.W.), and the Michael E. DeBakey, M.D., Professorship in Pharmacology (to J.W.). Additionally, we thank Drs. Danette Daniels, Matt Robers, and Jim Vasta at Promega for their support and advice on Nanoluc based assays and the Protein & Monoclonal Antibody Production Core at Baylor College of Medicine. We also appreciate Reviewer 1’s constructive suggestions.
Footnotes
Declaration of competing interest
J.W. is the co-founder of CoActigon Inc. and Chemical Biology Probes LLC.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.crchbi.2022.100029.
References
- Asami T, Kawahata W, Sawa M, 2015. TR-FRET binding assay targeting unactivated form of Bruton’s tyrosine kinase. Bioorg. Med. Chem. Lett 25, 2033–2036. [DOI] [PubMed] [Google Scholar]
- Bradshaw JM, et al. , 2015. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol 11, 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricelj A, et al. , 2021. Influence of linker attachment points on the stability and neosubstrate degradation of cereblon ligands. ACS Med. Chem. Lett 12, 1733–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhimschi AD, et al. , 2018. Targeting the C481S ibrutinib-resistance mutation in Bruton’s tyrosine kinase using PROTAC-mediated degradation. Biochemistry 57, 3564–3575. [DOI] [PubMed] [Google Scholar]
- Burslem GM, Crews CM, 2017. Small-molecule modulation of protein homeostasis. Chem. Rev 117, 11269–11301. [DOI] [PubMed] [Google Scholar]
- Chen J, et al. , 2017. Reversible reaction-based fluorescent probe for real-time imaging of glutathione dynamics in mitochondria. ACS Sens 2, 1257–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinar M, et al. , 2013. Bruton tyrosine kinase is commonly overexpressed in mantle cell lymphoma and its attenuation by Ibrutinib induces apoptosis. Leuk. Res 37, 1271–1277. [DOI] [PubMed] [Google Scholar]
- Dobrovolsky D, et al. , 2019. Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood 133, 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estupiñán HY, Berglöf A, Zain R, Smith CIE, 2021. Comparative analysis of BTK inhibitors and mechanisms underlying adverse effects. Front. Cell Dev. Biol 9, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W-H, et al. , 2020. Enhancing intracellular accumulation and target engagement of PROTACs with reversible covalent chemistry. Nat. Commun 11, 4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, et al. , 2021. Strategies toward discovery of potent and orally bioavailable proteolysis targeting chimera degraders of androgen receptor for the treatment of prostate cancer. J. Med. Chem 64, 12831–12854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JD, et al. , 2021. CC-90009: a cereblon E3 ligase modulating drug that promotes selective degradation of GSPT1 for the treatment of acute myeloid leukemia. J. Med. Chem 64, 1835–1843. [DOI] [PubMed] [Google Scholar]
- Ishoey M, et al. , 2018. Translation termination factor GSPT1 is a phenotypically relevant off-target of heterobifunctional phthalimide degraders. ACS Chem. Biol 13, 553–560. [DOI] [PubMed] [Google Scholar]
- Jiang X, et al. , 2015. Quantitative imaging of glutathione in live cells using a reversible reaction-based ratiometric fluorescent probe. ACS Chem. Biol 10, 864–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, et al. , 2017. Quantitative real-time imaging of glutathione. Nat. Commun 8, 16087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, et al. , 2019. Quantitative real-time imaging of glutathione with subcellular resolution. Antioxidants Redox Signal 30, 1900–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, et al. , 2020. PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene 39, 4909–4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan S, et al. , 2014. Design of reversible, cysteine-targeted Michael acceptors guided by kinetic and computational analysis. J. Am. Chem. Soc 136, 12624–12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindvall JM, et al. , 2005. Bruton’s tyrosine kinase: cell biology, sequence conservation, mutation spectrum, siRNA modifications, and expression profiling. Immunol. Rev 203, 200–215. [DOI] [PubMed] [Google Scholar]
- Matyskiela ME, et al. , 2016. A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nature 535, 252–257. [DOI] [PubMed] [Google Scholar]
- Middendorp S, Dingjan GM, Maas A, Dahlenborg K, Hendriks RW, 2003. Function of Bruton’s tyrosine kinase during B cell development is partially independent of its catalytic activity. J. Immunol 171, 5988–5996. [DOI] [PubMed] [Google Scholar]
- Nguyen TM, et al. , 2021. Proteolysis targeting chimeras with reduced off-targets. bioRxiv 11 (18), 468552. 10.1101/2021.11.18.468552, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike A, Williamson B, Harlfinger S, Martin S, McGinnity DF, 2020. Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: a drug metabolism and pharmacokinetics perspective. Drug Discov Today 25, 1793–1800. [DOI] [PubMed] [Google Scholar]
- Ronen G, et al. , 2020. Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs [DOI] [PMC free article] [PubMed]
- Saito K, et al. , 2003. BTK regulates PtdIns-4,5-P2 synthesis: importance for calcium signaling and PI3K activity. Immunity 19, 669–678. [DOI] [PubMed] [Google Scholar]
- Sievers QL, et al. , 2018. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 362, eaat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, et al. , 2019. Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia 10.1038/s41375-019-0440-x. [DOI] [PubMed]
- Tinworth CP, et al. , 2019. PROTAC-mediated degradation of Bruton’s tyrosine kinase is inhibited by covalent binding. ACS Chem. Biol 14, 342–347. [DOI] [PubMed] [Google Scholar]
- Wang M, et al. , 2012. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol 10.1016/s1470-2045(12)70200-0. [DOI] [PubMed]
- Wang ML, et al. , 2013. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med 369, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, et al. , 2021. Development of MDM2 degraders based on ligands derived from Ugi reactions: lessons and discoveries. Eur. J. Med. Chem 219, 113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen T, Wang J, Shi Y, Qian H, Liu P, 2021. Inhibitors targeting Bruton’s tyrosine kinase in cancers: drug development advances. Leukemia 35, 312–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woyach JA, et al. , 2014. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib . N. Engl. J. Med 370, 2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, et al. , 2019. Simple structural modifications converting a bona fide MDM2 PROTAC degrader into a molecular glue molecule: a cautionary tale in the design of PROTAC degraders. J. Med. Chem 62, 9471–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, et al. , 2021. Merging PROTAC and molecular glue for degrading BTK and GSPT1 proteins concurrently. Cell Res 1–4. 10.1038/s41422-021-00533-6. [DOI] [PMC free article] [PubMed]
- Yu H, et al. , 2018. Homogeneous BTK occupancy assay for pharmacodynamic assessment of tirabrutinib (GS-4059/ONO-4059) target engagement. Slas Discov 23, 919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorba A, et al. , 2018. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc Natl Acad Sci U A 115, E7285–E7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.