

Abstract
Autophagy is a conserved metabolic pathway that is central to many diseases. Recently, there has been a lot of interest in targeting autophagy with small molecule inhibitors as a possible therapeutic strategy. However, many of the compounds used for autophagy are non-selective. Here, we explored the inhibition of autophagy in pancreatic cancer cells using established selective small molecule inhibitors and discovered an unexpected link between the autophagy pathway and progression through the cell cycle. Our findings revealed that treatments with inhibitors that have different autophagy pathway targets block cell replication and activate other metabolic pathways to compensate for the blockade in autophagy. An unbiased screen looking for known drugs that might synergize with autophagy inhibition revealed new combination treatments that might provide a blueprint for therapeutic approaches to pancreatic cancer. The drugs quizartinib and THZ1 showed a strong synergistic effect in pancreatic cells with autophagy inhibition.
Graphical Abstract:
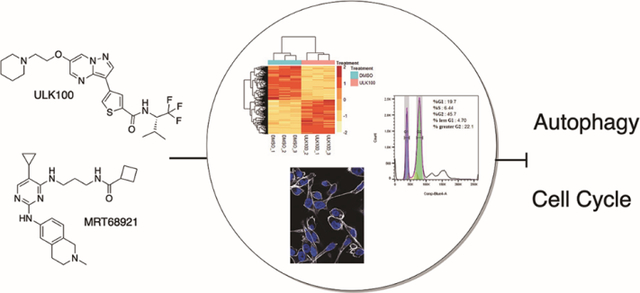
Introduction
Pancreatic cancer is the most lethal form of cancer in the United States with poor treatment options.1 While great progress has been made in treating several cancers through strategies like the targeting of oncogenic kinases, outcomes of pancreatic cancer treatment have not seen any real improvement. In part, this is due to the fact that the driving oncogene in pancreatic ductal adenocarcinoma (PDAC, the predominant form of the disease) is KRAS, which was thought to be undruggable, except for the G12C mutation that has recently been successfully targeted.2 Therefore, additional approaches are needed to find new vulnerabilities and develop novel therapeutic strategies. One key characteristic of cancer that has not been readily exploited is the altered metabolic state of tumors.3–5 For over 80 years, it has been known that tumor cells consume massive amounts of glucose to undergo glycolysis to produce lactate rather than undergo oxidative phosphorylation.6 A consequence of these metabolic changes, known as the Warburg Effect,7,8 is that cancer cells are particularly vulnerable to changes in nutrient availability.9 For example, tumors face a shortage of glucose prior to angiogenesis.10 Targeting starvation-response pathways may therefore be a way to selectively treat cancer cells. Altered cellular energy regulation is one of the major novel hallmarks of cancer and an emerging therapeutic target.11
A major cellular mechanism for responding to starvation is autophagy, a process in which cellular components are degraded for energy and building blocks like amino acids. Autophagy is initiated under starvation when a key kinase called Unc-51 like autophagy activating kinase (ULK1) gets activated by AMPK and mTOR (positively and negatively, respectively) (Figure 1A).12,13 Upon activation, ULK1 forms the ULK1 complex14,15 (ULK1/2, FIP200, ATG13, and ATG101), which in turn activates the VPS34 complex (VPS34, VPS15, Beclin1, and autophagy-related gene (ATG)14L, or UVRAG), which then activates a cascade of reactions leading to the formation of the double membrane structures called autophagophores.16,17 Autophagophores form around cellular components, from proteins to whole organelles14,18 and fuse with lysosomes leading to the degradation of its components. Autophagy is a fundamental process that plays a particularly important role in cancer.19–21 In pancreatic cancer, it has been shown to be an essential source of amino acids for the metabolic needs of the tumor cells and occurs at an extremely high basal level compared to normal cells.22,23 In other cases, it has been shown to be used by tumors to resist chemotherapy.24–26
Figure 1. Autophagy Pathway Inhibitors.
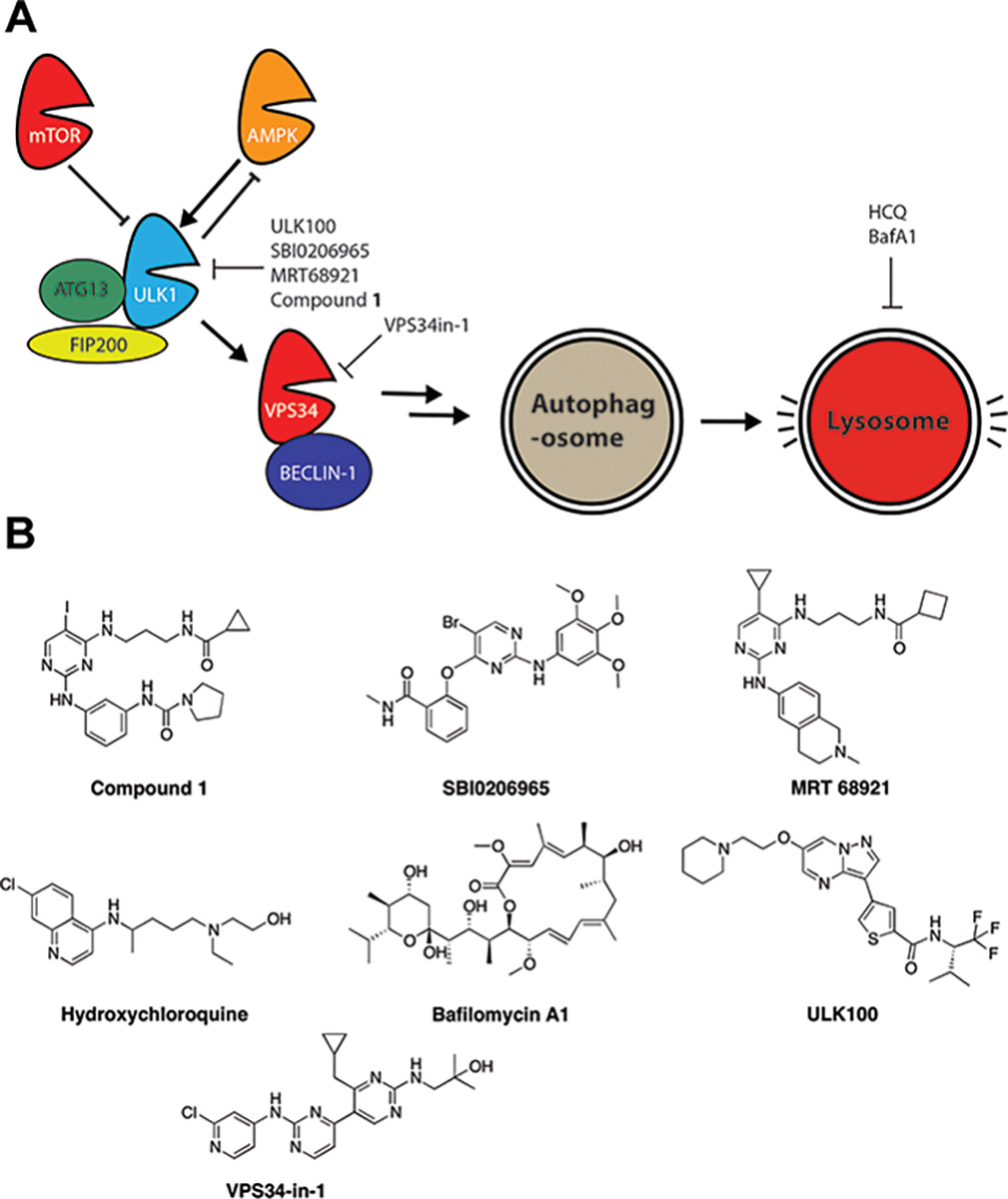
A) Schematic of the pathway that activates autophagic degradation in the lysosome, starting with activation of ULK1. Not all complex members are shown, for clarity. B) Structures of autophagy inhibitors in the literature and used in this study. The compounds target different stages of the autophagy pathway and have different selectivities towards their targets.
A number of recent studies have shown that pancreatic cancer cells require autophagy for growth27,28 and autophagy activation is correlated with poor clinical prognosis.29 Compared to other malignancies, pancreatic cancer, and other Ras-dependent malignancies, are particularly dependent on nutrients. By producing glutamine, for example, autophagy promotes the survival of the tumor under hypoxic and nutrient-poor conditions.27,30–33 Therefore, autophagy is attractive as a new target in Ras-driven pancreatic cancer. Autophagy has not been properly evaluated as a target because selective inhibitors did not exist. Typical compounds that modulate autophagy in cell culture models, including rapamycin and bafilomycin, have broad cellular effects beyond the autophagy pathway.
Recent studies showed a clinical benefit obtained by combining known tumor therapies like MEK inhibition with an autophagy-modulating drug like chloroquine or hydroxychloroquine (HCQ), a compound that targets all lysosomes, not just autolysosomes.34,35 While those results are exciting, the underlying mechanisms of synergy are unclear due to the nonselective nature of autophagy inhibitors used in those studies. Therefore, we studied the known autophagy drugs to examine their vulnerabilities and possible synergistic targets.
If autophagy inhibitors are to be used to block cancer cell growth, it is important to understand their mechanism. It is likely that different compounds with different scaffolds and targets, including off-targets, could work differently on blocking growth. We recently reported Compound 1, a nanomolar inhibitor against ULK1, which we co-crystallized with the kinase.36 Two other ULK1 inhibitors were reported, SBI-020695 and MRT68921 (Figure 1B).37,38 Of these three, MRT68921 had the best selectivity, showing a preference for ULK1, although there was significant inhibition of AMPK and 70% inhibition of Aurora B at 1 μM.38 VPS34-in1 is an inhibitor of the lipid kinase VPS34 (encoded by the PIK3C3 gene), which is downstream of the ULK1 complex. VPS34-in1 has no direct activity against Aurora kinases, which is predictable since lipid kinases are more divergent from protein kinases and their inhibitors are often more selective.39,40 Lastly, ULK100 and its related analog ULK101 were recently reported as a novel scaffold that inhibits ULK1 potently and potentially more selectively41 (Figure 1). It has to be noted that most of these inhibitors have other important kinases as off-targets, including the family of Aurora kinases (Aurora kinase A, B and C) that are required for mitosis.42 SBI-020695 and MRT68921 both inhibit Aurora A,43 therefore, using these inhibitors complicates the interpretation of data concerning autophagy inhibition and the cell cycle. According to the profiling done by Martin et al.41, SBI-020695 was more potent against Aurora kinase A and Aurora kinase B than ULK1 itself. Inhibition of Aurora kinases as an off-target is a concern as it causes cell cycle arrest regardless of the intended target. ULK100 was the most selective against ULK1 over Aurora kinases. ULK101 inhibits Aurora kinase more than ULK100 but inhibits AMPK less. Therefore, ULK101 could have uses in other contexts, such as distinguishing between the role of ULK1 and AMPK inhibition. In summary, ULK100 and VPS34-in1 are the most selective compounds at inhibiting autophagy while sparing Aurora kinases.
Others have previously described a connection between autophagy and the cell cycle, focusing mostly on the effects the cell cycle has on autophagy regulation.44 It has also previously been shown that knockdown of the VPS34 complex components leads to cytokinesis arrest, likely through depletion of PtdIns(3)P. Nevertheless, this has not been seen with other autophagy components like ULK1, and it is unclear if it is due to the autophagy role of these proteins or their non-autophagic function, or due to their catalytic or scaffolding functions.45,46 Therefore, interrogating different arms of the autophagic machinery with selective compounds will help shed light on the complex interplay between autophagy and the cell cycle. While others have noted the link between the cell cycle and autophagy, it is difficult to interpret this data because many compounds used are non-selective. Using the most selective compounds, which spare Aurora kinases, we sought to uncover the cellular effects of blocking autophagy at different stages of the pathway.
Results and Discussion
Selective inhibitors of autophagy still show inhibition of Aurora kinase signaling
Because we suspected that many autophagy inhibitors interfere with Aurora kinases, we investigated the effect of these compounds on mitosis. Visualizing nuclei by DAPI incorporation, we examined cells after autophagy inhibitor treatment, looking for signs of cell cycle disruption. Polyploidy is a hallmark of severe cell cycle disruption and has been observed from selective inhibition of both Aurora A and Aurora B.47,48 As seen in Figure 2, we can see polyploid and enlarged nuclei with many of the compounds that are used as autophagy inhibitors. The compounds with known Aurora inhibition showed strong polyploidy in this assay, including compound 1, SBI-020695 and MRT68921. However, VPS34in1, ULK100, hydroxychloroquine (HCQ), and bafilomycin A1 showed minimal to no polyploidy induction, suggesting that they are good compounds to study the connection between autophagy and the cell cycle, and that autophagy inhibition alone does not cause polyploidy. AMG-900, a potent selective pan Aurora inhibitor,49 was used as a control compound and showed a strong polyploidy phenotype.
Figure 2. Cell cycle perturbations from autophagy inhibition seen by confocal microscopy after DAPI incorporation.
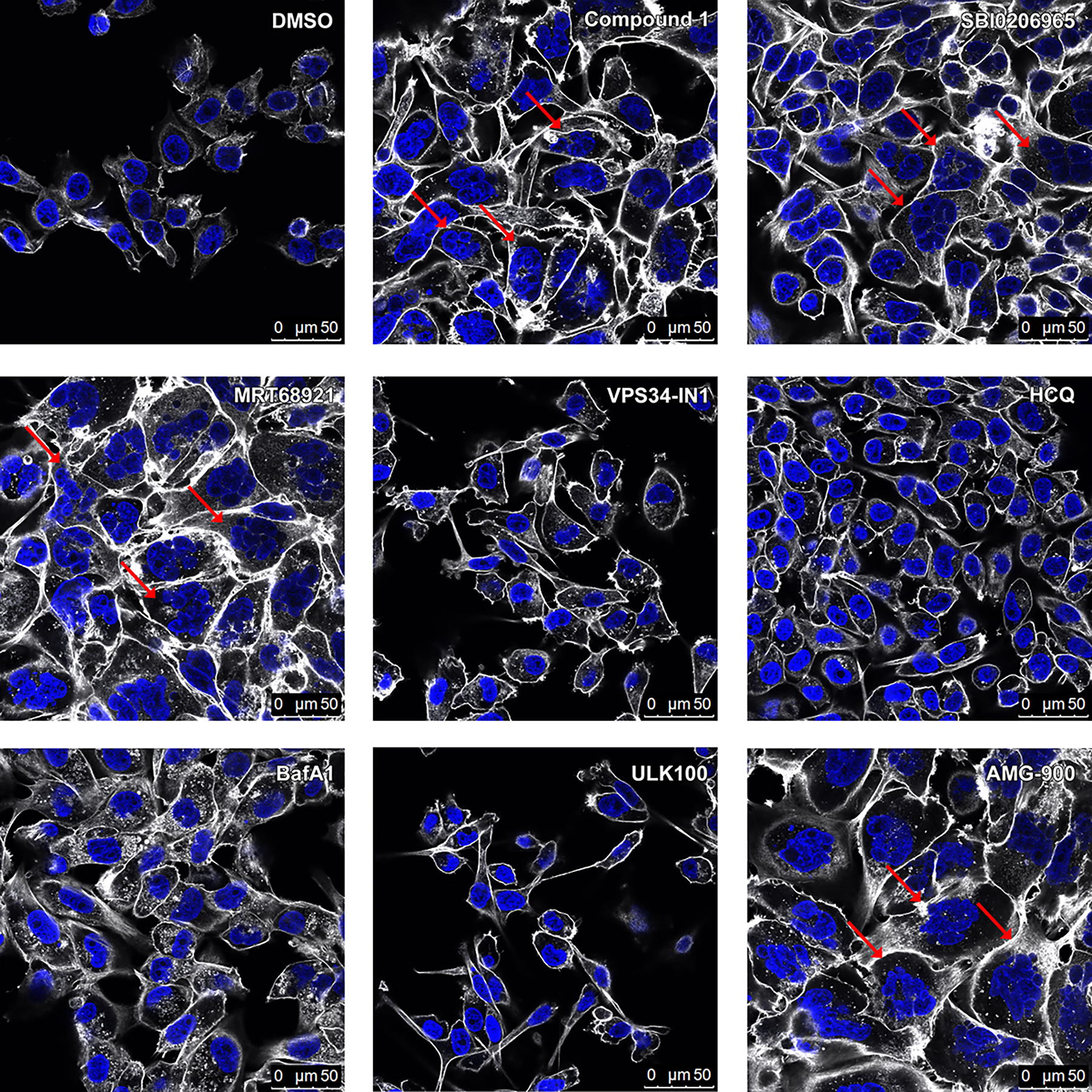
Pa 8988T cells were treated with compounds at the following concentrations for 48 hours: Compound 1: 1 μM; SBI0206965: 2 μM; MRT68921: 1 μM; VPS34in-1: 1 μM; Hydroxychloroquine (HCQ): 5 μM; Bafilomycin A1 (BafA1): 10 nM; ULK100: 1 μM; AMG-900: 250 nM. Red arrows depict some of the cells with increased DNA content compared to vehicle treatment. DAPI is shown in blue, Phalloidin-647 in white.
Polyploidy is one phenotype of cell cycle disruption. We next looked for additional readouts of cell cycle alteration using two different pancreatic cancer cell lines (PANC-1 and 8988T), as some cell lines have different sensitivities to autophagy inhibition. We explored the effect of known autophagy inhibitors by western blotting, using phospho-Histone 3 at Serine 10 (H3 S10) as a readout for Aurora signaling, total Aurora A as an additional marker for cell cycle regulation, and LC3 II formation (the processed lipidated form of LC3 I) as a readout for autophagy inhibition.
As seen in Figure 3A and Figure 3B, the compound with known Aurora activity, MRT68921, had strong effects on phospho-H3 activity. Surprisingly, however, compounds with no Aurora activity, including ULK100, VPS34-in1, and HCQ, all had a dose-dependent inhibitory effect on both H3 S10 phosphorylation and Aurora A protein levels. We tested another inhibitor of VPS34 with a different scaffold, SAR405, to ensure that it was not an off-target artifact of VPS34-in1. While less potent than VPS34-in1 in this assay, SAR405 showed the same dose-dependent trend in interfering with the cell cycle markers. This shows that inhibition of autophagy at different stages of the autophagic pathway with selective inhibitors can indirectly interfere with Aurora kinases, independent of off-target effects of the less selective autophagy inhibitors that bind to and inhibit Aurora kinases directly. The dose-response also extended to autophagy inhibition, with the build-up of LC3 I and LC3 II observed for the early (ULK1, VPS34) and late (HCQ) autophagy inhibitors (replicates and quantification shown in Figure S1). Because these are different scaffolds that target different parts of the autophagy pathway, this suggests that full inhibition of autophagy at the higher doses of compounds leads to maximal disruption of the cell cycle, regardless of which point in the pathway is targets.
Figure 3. Mechanisms and biological changes from autophagy inhibition.
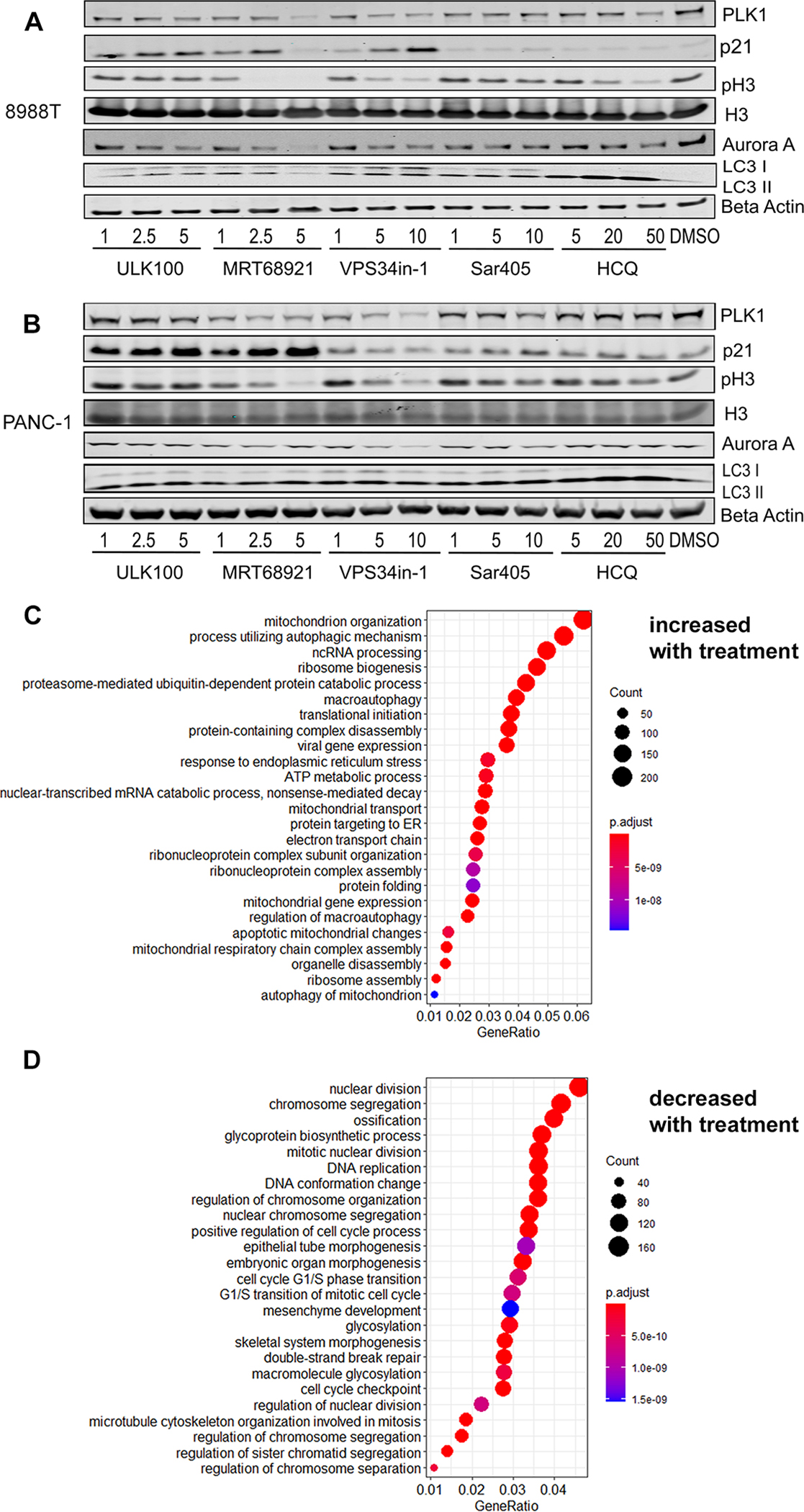
A) 8988T cells were treated for 24 hours with the compounds at the indicated concentrations (μM) and the protein lysate blotted with the indicated antibodies. B) The same compounds were used in PANC-1 cells. C-D) GO-enrichment analysis of RNA-Seq data revealed a list of biological processes that are upregulated (C) or downregulated (D) in PANC-1 cells after inhibition of autophagy with VPS34in-1.
RNA-Seq data reveals strong blockade of cell division upon inhibition of autophagy
To get an unbiased global view of the cellular changes that occur upon autophagy inhibition, we performed RNA-Seq on PANC-1 cells after treatment with either ULK100, VPS34-in1, or DMSO vehicle, in triplicates. We used the high doses of the autophagy inhibitors from the western blot experiments to ensure full inhibition of autophagy. Principal component analysis (PCA) confirmed robust reproducibility among the replicates and showed substantially diverse global expression patterns among treatments with the two autophagy inhibitors (Figure S2). Gene Ontology (GO) enrichment analysis on genes that were significantly upregulated (Figure 3C) or downregulated (Figure 3D) upon autophagy inhibition with VPSin34in1 or with ULK100 (Figure S3) showed a common regulation of genes with both inhibitors, even though they target different stages of the autophagy pathway. Strikingly, there was a significant downregulation of genes involved in cell division and DNA replication. This is consistent with our discovery of the downregulation of Aurora kinase A protein levels and a decrease in phospho-H3. Indeed, both Aurora A and Aurora B were highly significantly decreased at the RNA level (AURKA, AURKB, Figure S4). Conversely, one of the main upregulated processes was autophagy itself, showing that these cells have a compensatory mechanism for increasing autophagy despite the inhibition of an essential step in the process. For example, VPS34 and ULK1 inhibition led to a significant increase in ULK1 expression. In both cases, we also saw a dramatic increase in RNA levels for MAP1LC3A (the gene that encodes LC3) (Figure S4) which agrees with our observation by western blot of increased LC3-I and LC3-II after ULK1 or VPS34 inhibition (Figure 3A and B). An increase of LC3-II is typically an effect of lysosomal inhibitors that block LC3-II degradation (see HCQ treatment in Figure 3A and B), whereas early-stage inhibitors should only increase LC3-I levels. Thus, upon autophagy inhibition the cells are compensating by increasing the amount of LC3 that is expressed and translated, further complicating the interpretation of autophagic flux by looking at LC3 levels only.
While examining key regulators of autophagy and the cell cycle, we detected many other significant alterations upon drug treatment, showing the coordinated effort to regulate these pathways. After autophagy inhibition, other autophagy genes had increased expressions, including OPTN, ATG2A, ATG13, and TMEM41B (Figure S4). Conversely, several genes that regulate the cell cycle had significantly decreased expression after treatment with either of the two autophagy inhibitors, including BUB1, BUB1B, PCNA, and CDK1 (Figure S4). Other important genes had significant changes in expression but were different for the two inhibitors, suggesting that there are specific differences in inhibiting ULK1 and VPS34, both of which have additional cellular roles outside of autophagy. These genes include TP53, KRAS, and MYC. Lastly, we note that the gene that had the most decrease in expression was collagen (COL1A1), which has been observed before50, but was striking nonetheless and might have implications for more complex tumor models. We explored further cell cycle genes in our RNA-Seq data and many were significantly altered upon autophagy inhibition, most of which indicated an arrest in cell cycle progression (Figure S5). The totality of these RNA changes suggests a broad decrease in cell-cycle progression gene expression upon autophagy inhibition. Subsequently, we explored some of these cell cycle targets by western blot, to see if the changes in mRNA levels correlated with changes in protein levels in the two pancreatic cancer cell lines (Figure 3A and B). Correlating with the RNA-Seq result, PLK1 was reduced in a dose-dependent manner in both cell lines with inhibition of ULK1 or VPS34. Additionally, p21 (CDKN1A) showed a dose-dependent increase in expression upon inhibition of ULK1, which mirrored the results in the RNA-Seq data. Interestingly, in the PANC-1 cells, VPS34 inhibition did not increase p21 levels, which agrees with the RNA-Seq data that shows VPS34 inhibition instead induced the closely related CDKN1B gene. In summary, using two different kinase inhibitors that do not inhibit Aurora kinases directly, we found numerous autophagy genes that respond to autophagy inhibition by a significant increase in expression, and abundant cell cycle genes that decrease in expression in response to autophagy inhibition, including Aurora A and Aurora B.
Cell Cycle Analysis after autophagy inhibition
To learn more about the connection between autophagy and the cell cycle, we performed cell cycle analysis by flow cytometry (FACS) after propidium iodide (PI) staining of cells treated with the inhibitors of different stages of autophagy (Table 1 and Figure S6). The effects of the compounds clustered into two different categories based on their selectivity. Compound 1, MRT68921, and SBI-020695 increased the amount of cells arrested in the G2 phase, indicating G2/M checkpoint failure and polyploidy, similar to what is reported with Aurora kinase inhibition.51 This is likely due to the off-target inhibition of Aurora kinase by these less selective compounds. All other autophagy inhibitors increased the percentage of cells arrested in the G1 phase, which is consistent with the RNA-Seq data of an increase in CDKN1A and CDKN1B and a decrease in CDK2 (Figure S5), which can block the G1-S transition.52 This was previously observed for Bafilomycin A1.53 The fact that these inhibitors all have the same buildup of cells in the G1 phase suggests it is a general autophagy phenomenon since they are targeting different stages of the autophagic pathway (initiation, nucleation, and degradation). In agreement with the polyploidy that we observed by DAPI visualization (Figure 2), PI analysis confirmed that treatments with SBI-020695, Compound 1 and MRT68921 increase the DNA content of the cells shown as >G2 in Table 1 and Supplementary Figure 6.
Table 1.
Cell cycle changes from autophagy inhibitors.
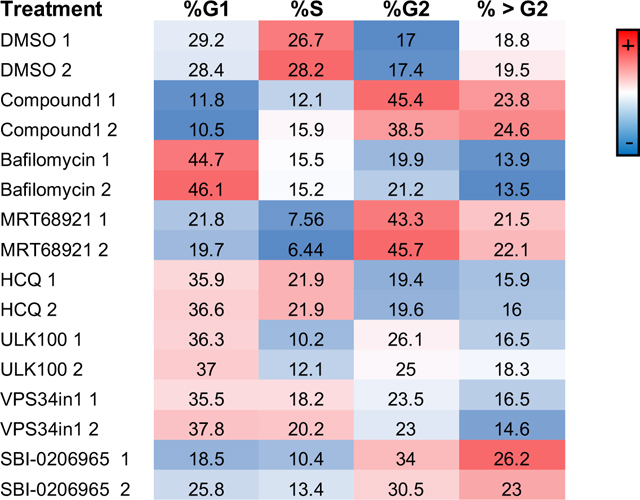
|
Cells were treated with each compound in duplicates, harvested, treated with propidium iodide (PI) and analyzed by flow cytometry. The percentage of cells in each cell cycle phase is shown and color coded by abundance. The compounds were applied at the same concentrations as in the microscopy images (Fig. 2) for 24 hours. >G2 shows the percentage of cells that have an excessive DNA content and therefore are likely polyploid.
Screen for synergy leads to discovery of two novel targets
Using this information, we set out to find synergistic inhibitors of pancreatic cancer cells with autophagy inhibition. Because of the high levels of autophagy in certain cancer types like pancreatic cancer, and the benefits of drug synergy, it is possible that combining chemotherapy with autophagy inhibitors will be more potent and less toxic than single drug regimens.54 There has been great interest in combining non-specific autophagy inhibitors like chloroquine or HCQ with known chemotherapy agents to improve the potency and ward off resistance. In fact, there are several ongoing clinical trials for these combination treatments. However, besides the non-specificity of the autophagy compounds, the mechanism of synergy is unclear. Therefore, we first performed a screen using a commercial kinase inhibitor library with and without VPS34-in1 in PANC-1 cells. Surprisingly, instead of seeing traditionally associated synergistic compounds like trametinib, the top hits were multitargeting receptor tyrosine kinase inhibitors, with ceritinib and quizartinib being the top hits. Quizartinib, which is an FLT3 inhibitor, was the most potent in combination, seen in multiple pancreatic cancer cell lines (PANC1 and MIA-Paca2). Treating these cell lines with the compounds in combination showed an increase in cleaved PARP (Figure S7), suggesting a synergistic effect with autophagy inhibition. Since quizartinib had a stronger effect at a lower dose than ceritinib, we focused on this compound. As seen in Figure 4A, quizartinib alone had almost no effect on cell viability in MiaPaca-2 cells. However, in combination with low doses of VPS34in1, there was a significant decrease in cell viability. Subsequently, we did a full dose-response matrix to measure drug synergy and saw a large synergistic effect over most concentration ranges by Loewe analysis (Figure 4B). Subsequently, we did a screen of PANC-1 cells with the kinase inhibitor library with and without ULK100, to look for compounds that were only toxic in the presence of ULK100. We found several interesting candidates, but the most potent was THZ1, a CDK7 inhibitor. This was exciting, because of the connection between the cell cycle and autophagy, and it suggested that it might be possible to take advantage of this novel mechanism. Indeed, when we treated cells with varying doses of ULK100 and THZ1, we saw a strong synergistic effect, as evidenced by the Loewe score (Figure 4C).55 This suggests that combining autophagy inhibitors with cell cycle inhibitors like THZ1 could be more selective and efficacious than cell cycle inhibitors alone.
Figure 4. Synergistic inhibition of pancreatic cancer cell growth with autophagy inhibitors and known drugs.
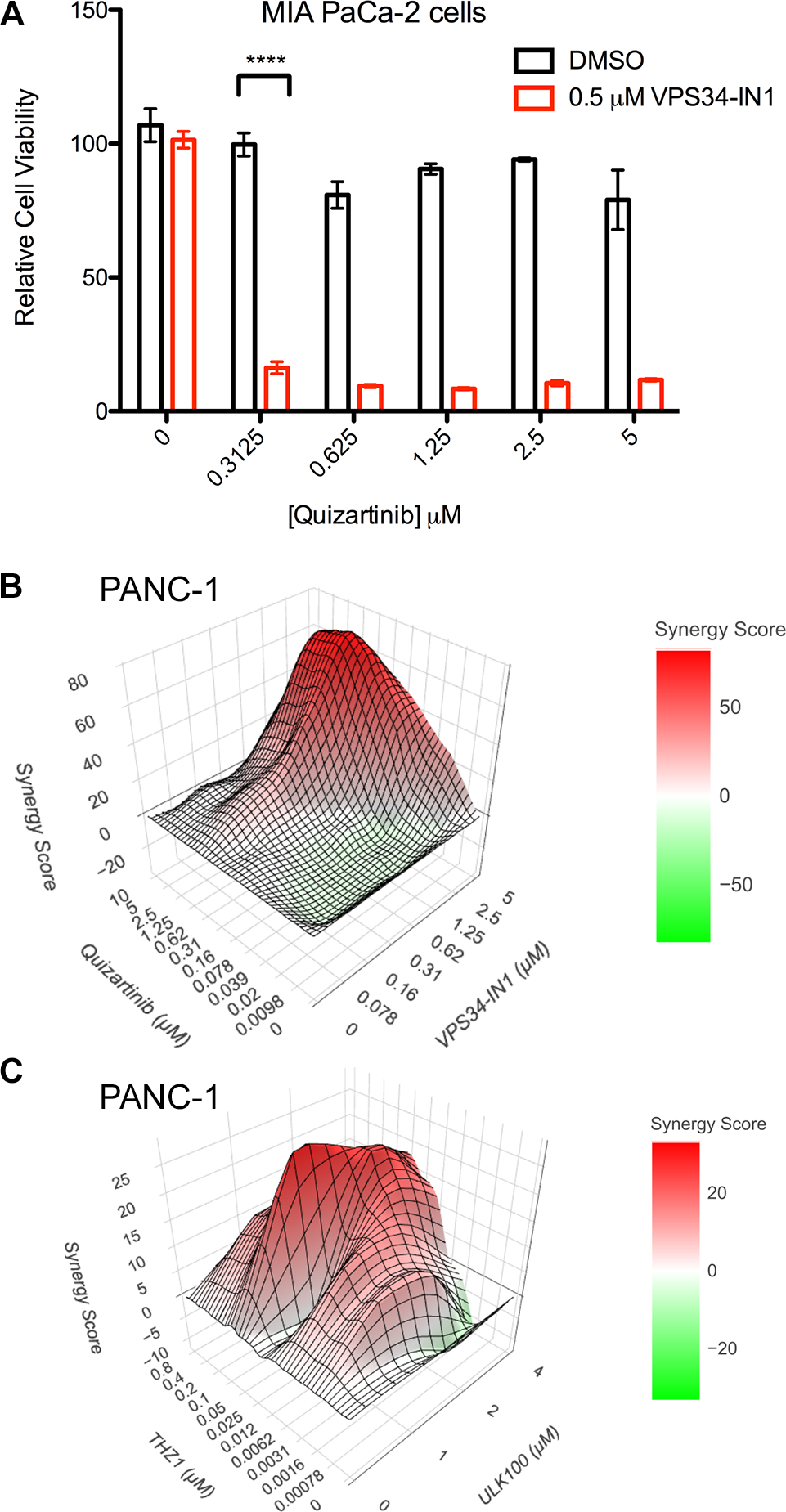
A) Cell viability of MIA PaCa-2 cells after treatment with either quizartinib, VPS34in-1 or both. Error bars show SD, n=3. B) Synergy plot of PANC-1 cells treated with quizartinib and VPS34in-1, using a treatment matrix with the indicated concentrations. The graph shows the contours of the Loewe synergy score, calculated using SynergyFinderPlus. C) Synergy plot of PANC-1 cells treated with ULK100 and THZ-1, treated at the indicated concentrations with the Loewe score graphed with synergism shown in red.
The synergy between quizartinib and VPS34in-1 was particularly surprising. Quizartinib is a selective FLT3 inhibitor that was investigated to treat AML.56 There were no reports of FLT3 expression in pancreatic cells, which our RNA-Seq data confirmed (Figure S8). We also looked at kinome profiling of quizartinib to hypothesize other targets and identified several candidates that were shown in vitro to be inhibited by quizartinib 56. These could potentially be the targets of quizartinib in these cells, or perhaps inhibiting some combination of them has a polypharmacological effect that induces autophagy and leads to cell death when that escape mechanism is blocked. We examined the 12 kinases that are reported to be most inhibited by quizartinib in vitro. Of those 12, only DDR1, and potentially RET, were expressed at levels that were sufficient to deem them as plausible targets of the drug in these cells. The other kinases, including FLT3, were barely expressed with or without autophagy inhibitor treatment. Intriguingly, DDR1, the most expressed of all of these is also known to be regulated by collagen,57 suggesting that autophagy inhibition is altering signaling of this receptor.
For ULK100, THZ1, a CDK7 inhibitor, was the most potent synergistic compound that we identified in our screen. This suggested that, because of the natural feedback between the cell cycle and autophagy, inhibition of both pathways could be beneficial as a treatment strategy. The mechanism needs to be further evaluated, but in these pancreatic cell lines, which we chose because of their known dependence on autophagy and high autophagic flux, it is possible that the cells block division as a survival mechanism until autophagy is restored and can provide sufficient energy.
We have found that there are two mechanisms linking autophagy inhibition to cell cycle aberrations. First, since ULK1 and Aurora kinase share many similarities in the ATP binding pocket,43 many autophagy inhibitors are also direct inhibitors of Aurora kinase. These compounds should not be used for cellular experiments studying autophagy, as their dual pharmacology effects will make the interpretation of experimental results difficult. Second, we find that other selective autophagy compounds also interfere with the cell cycle, primarily at the G1/S transition, but also inhibit histone H3 S10 phosphorylation. The mechanism appears to be at the transcriptional level and involves numerous changes to genes involved in cell-cycle progression. ULK100 and VPS34in-1 and related compounds are in this class. These compounds are useful autophagy probes and can be potentially synergistic with other drug mechanisms in pancreatic cancer cells. While chloroquine is being widely investigated in combination with other compounds, we suggest that these other selective compounds might provide more straightforward mechanisms of action. Furthermore, the synergy between autophagy inhibition and other pathways is more complex than previously thought, so unbiased screens might be necessary for identifying synergistic drug combinations.
Supplementary Material
Acknowledgements
This work was supported by R01DK118946 to M.J.W., the Mallinckrodt Foundation and N.I.H K22CA201103 and R35GM124838 to M.B.L. We would also like to thank the Flow Cytometry core at Mount Sinai for assistance with the FACS experiments.
Footnotes
Competing interests
The authors declare no competing interests.
Supporting Information
Supplementary figures including further RNA-Seq analysis and cell-cycle analysis, full methods, and supplementary references.
Data
RNA-Seq data was deposited to the GEO database with accession number GSE206261.
References
- 1.Kleeff J et al. Pancreatic cancer. Nat Rev Dis Primers 2, 16022 (2016). 10.1038/nrdp.2016.22 [DOI] [PubMed] [Google Scholar]
- 2.Eser S, Schnieke A, Schneider G & Saur D Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 111, 817–822 (2014). 10.1038/bjc.2014.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vander Heiden MG, Cantley LC & Thompson CB Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009). 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White-Gilbertson S, Kurtz DT & Voelkel-Johnson C The role of protein synthesis in cell cycling and cancer. Molecular Oncology 3, 402–408 (2009). 10.1016/j.molonc.2009.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dang CV Links between metabolism and cancer. Genes & Development 26, 877–890 (2012). 10.1101/gad.189365.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warburg OH, Dickens F & Berlin (Germany). Kaiser Wilhelm-Institut für Biologie. The metabolism of tumours; investigations from the Kaiser Wilhelm institute for biology, Berlin-Dahlem. (Constable & co. ltd., 1930). [Google Scholar]
- 7.Warburg O On respiratory impairment in cancer cells. Science 124, 269–270 (1956). [PubMed] [Google Scholar]
- 8.Warburg O On the origin of cancer cells. Science 123, 309–314 (1956). [DOI] [PubMed] [Google Scholar]
- 9.Jones RG & Thompson CB Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes & Development 23, 537–548 (2009). 10.1101/gad.1756509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagy JA, Chang S-H, Dvorak AM & Dvorak HF Why are tumour blood vessels abnormal and why is it important to know? British Journal of Cancer 100, 865–869 (2009). 10.1038/sj.bjc.6604929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 12.Egan DF et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461 (2011). 10.1126/science.1196371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim J, Kundu M, Viollet B & Guan KL AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13, 132–141 (2011). 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong PM, Puente C, Ganley IG & Jiang X The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 9, 124–137 (2013). 10.4161/auto.23323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stjepanovic G et al. Assembly and dynamics of the autophagy-initiating Atg1 complex. Proc Natl Acad Sci U S A 111, 12793–12798 (2014). 10.1073/pnas.1407214111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan EY, Kir S & Tooze SA siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem 282, 25464–25474 (2007). 10.1074/jbc.M703663200 [DOI] [PubMed] [Google Scholar]
- 17.Mizushima N, Yoshimori T & Ohsumi Y The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27, 107–132 (2011). 10.1146/annurev-cellbio-092910-154005 [DOI] [PubMed] [Google Scholar]
- 18.Mizushima N, Levine B, Cuervo AM & Klionsky DJ Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 (2008). 10.1038/nature06639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathew R, Karantza-Wadsworth V & White E Role of autophagy in cancer. Nat Rev Cancer 7, 961–967 (2007). 10.1038/nrc2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rubinsztein DC, Codogno P & Levine B Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11, 709–730 (2012). 10.1038/nrd3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang X, Overholtzer M & Thompson CB Autophagy in cellular metabolism and cancer. J. Clin. Invest. 125, 47–54 (2015). 10.1172/JCI73942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang S et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729 (2011). 10.1101/gad.2016111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.White E Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 12, 401–410 (2012). 10.1038/nrc3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu YL, Jahangiri A, Delay M & Aghi MK Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. Cancer Res. 72, 4294–4299 (2012). 10.1158/0008-5472.CAN-12-1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu L et al. Induction of autophagy counteracts the anticancer effect of cisplatin in human esophageal cancer cells with acquired drug resistance. Cancer Lett 355, 34–45 (2014). 10.1016/j.canlet.2014.09.020 [DOI] [PubMed] [Google Scholar]
- 26.Fan QW et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal 3, ra81 (2010). 10.1126/scisignal.2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang S et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 25, 717–729 (2011). 10.1101/gad.2016111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perera RM et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365 (2015). 10.1038/nature14587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujii S et al. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci 99, 1813–1819 (2008). 10.1111/j.1349-7006.2008.00893.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Son J et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 (2013). 10.1038/nature12040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perera RM & Bardeesy N Pancreatic Cancer Metabolism: Breaking It Down to Build It Back Up. Cancer Discov 5, 1247–1261 (2015). 10.1158/2159-8290.CD-15-0671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo JY et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25, 460–470 (2011). 10.1101/gad.2016311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lock R et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 22, 165–178 (2011). 10.1091/mbc.E10-06-0500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bryant KL et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med 25, 628–640 (2019). 10.1038/s41591-019-0368-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kinsey CG et al. Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med 25, 620–627 (2019). 10.1038/s41591-019-0367-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lazarus MB & Shokat KM Discovery and structure of a new inhibitor scaffold of the autophagy initiating kinase ULK1. Bioorg Med Chem 23, 5483–5488 (2015). 10.1016/j.bmc.2015.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Egan DF et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell 59, 285–297 (2015). 10.1016/j.molcel.2015.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petherick KJ et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem 290, 11376–11383 (2015). 10.1074/jbc.C114.627778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bago R et al. Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem J 463, 413–427 (2014). 10.1042/BJ20140889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whitmarsh-Everiss T & Laraia L Small molecule probes for targeting autophagy. Nat Chem Biol 17, 653–664 (2021). 10.1038/s41589-021-00768-9 [DOI] [PubMed] [Google Scholar]
- 41.Martin KR et al. A Potent and Selective ULK1 Inhibitor Suppresses Autophagy and Sensitizes Cancer Cells to Nutrient Stress. iScience 8, 74–84 (2018). 10.1016/j.isci.2018.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willems E et al. The functional diversity of Aurora kinases: a comprehensive review. Cell Div 13, 7 (2018). 10.1186/s13008-018-0040-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chaikuad A et al. Conservation of structure, function and inhibitor binding in UNC-51-like kinase 1 and 2 (ULK1/2). Biochem J 476, 875–887 (2019). 10.1042/BCJ20190038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mathiassen SG, De Zio D & Cecconi F Autophagy and the Cell Cycle: A Complex Landscape. Front Oncol 7, 51 (2017). 10.3389/fonc.2017.00051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Joo JH et al. The Noncanonical Role of ULK/ATG1 in ER-to-Golgi Trafficking Is Essential for Cellular Homeostasis. Mol Cell 62, 491–506 (2016). 10.1016/j.molcel.2016.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galluzzi L & Green DR Autophagy-Independent Functions of the Autophagy Machinery. Cell 177, 1682–1699 (2019). 10.1016/j.cell.2019.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nair JS, Ho AL & Schwartz GK The induction of polyploidy or apoptosis by the Aurora A kinase inhibitor MK8745 is p53-dependent. Cell Cycle 11, 807–817 (2012). 10.4161/cc.11.4.19323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nair JS et al. Aurora B kinase regulates the postmitotic endoreduplication checkpoint via phosphorylation of the retinoblastoma protein at serine 780. Mol Biol Cell 20, 2218–2228 (2009). 10.1091/mbc.E08-08-0885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Payton M et al. Preclinical evaluation of AMG 900, a novel potent and highly selective pan-aurora kinase inhibitor with activity in taxane-resistant tumor cell lines. Cancer Res 70, 9846–9854 (2010). 10.1158/0008-5472.CAN-10-3001 [DOI] [PubMed] [Google Scholar]
- 50.Li CX et al. Inhibiting autophagy promotes collagen degradation by regulating matrix metalloproteinases in pancreatic stellate cells. Life Sci 208, 276–283 (2018). 10.1016/j.lfs.2018.07.049 [DOI] [PubMed] [Google Scholar]
- 51.Ryu J, Pyo J, Lee CW & Kim JE An Aurora kinase inhibitor, AMG900, inhibits glioblastoma cell proliferation by disrupting mitotic progression. Cancer Med 7, 5589–5603 (2018). 10.1002/cam4.1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsai LH, Lees E, Faha B, Harlow E & Riabowol K The cdk2 kinase is required for the G1-to-S transition in mammalian cells. Oncogene 8, 1593–1602 (1993). [PubMed] [Google Scholar]
- 53.Yuan N et al. Bafilomycin A1 targets both autophagy and apoptosis pathways in pediatric B-cell acute lymphoblastic leukemia. Haematologica 100, 345–356 (2015). 10.3324/haematol.2014.113324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lehar J et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol 27, 659–666 (2009). 10.1038/nbt.1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ianevski A, Giri AK & Aittokallio T SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res 48, W488–W493 (2020). 10.1093/nar/gkaa216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kampa-Schittenhelm KM et al. Quizartinib (AC220) is a potent second generation class III tyrosine kinase inhibitor that displays a distinct inhibition profile against mutant-FLT3, -PDGFRA and -KIT isoforms. Mol Cancer 12, 19 (2013). 10.1186/1476-4598-12-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Juskaite V, Corcoran DS & Leitinger B Collagen induces activation of DDR1 through lateral dimer association and phosphorylation between dimers. Elife 6 (2017). 10.7554/eLife.25716 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.