

Abstract
COPD is a heterogeneous disease with multiple clinical phenotypes. COPD endotypes can be determined by different expressions of hypoxia-inducible factors (HIFs), which, in combination with individual susceptibility and environmental factors, may cause predominant airway or vascular changes in the lung. The pulmonary vascular phenotype is relatively rare among COPD patients and characterised by out-of-proportion pulmonary hypertension (PH) and low diffusing capacity of the lung for carbon monoxide, but only mild-to-moderate airway obstruction. Its histologic feature, severe remodelling of the small pulmonary arteries, can be mediated by HIF-2 overexpression in experimental PH models. HIF-2 is not only involved in the vascular remodelling but also in the parenchyma destruction. Endothelial cells from human emphysema lungs express reduced HIF-2α levels, and the deletion of pulmonary endothelial Hif-2α leads to emphysema in mice. This means that both upregulation and downregulation of HIF-2 have adverse effects and that HIF-2 may represent a molecular “switch” between the development of the vascular and airway phenotypes in COPD. The mechanisms of HIF-2 dysregulation in the lung are only partly understood. HIF-2 levels may be controlled by NAD(P)H oxidases via iron- and redox-dependent mechanisms. A better understanding of these mechanisms may lead to the development of new therapeutic targets.
Short abstract
Upregulation of endothelial HIF-2 may represent a new mechanism of the vascular phenotype in COPD, whereas downregulation of HIF-2 may cause damage of airways and lung parenchyma. HIF-2 may provide an important tool to achieve precision medicine in COPD. https://bit.ly/3UByVmR
Introduction
COPD is one of the major causes of chronic morbidity and one of the top three causes of death worldwide [1]. Due to an aging population and continued exposure to risk factors (e.g. cigarette smoke and air pollution), the medical and social burdens of COPD are progressively increasing. Being very divergent clinically, COPD is also heterogeneous with respect to the severity of airway, pulmonary parenchyma, lung vessel and systemic vasculature involvement. Therefore, a better understanding of the pathobiology of the disease is crucial for developing novel therapeutic approaches targeting key molecular “hubs” in different lung cells.
Pulmonary vascular involvement, eventually resulting in pulmonary hypertension (PH), is a typical feature of COPD. PH due to COPD (COPD-PH) occurs in a significant proportion of patients, may lead to right heart failure and is characterised by a very poor prognosis [2]. Most patients with COPD-PH belong to group 3 PH (associated with lung diseases and/or hypoxia) [3]. A subgroup of COPD patients present with “out-of-proportion” (severe) pre-capillary PH, significantly reduced diffusion capacity of the lung (DLCO) and relatively low oxygen and carbon dioxide partial pressures in the systemic blood, but only moderate airflow limitation. This specific pattern has been proposed to represent the “pulmonary vascular phenotype” [4].
The pathologic mechanisms leading to pulmonary vascular alterations in COPD are complex and include lung hyperinflation due to emphysema, left ventricular failure, increased pulmonary vascular stiffness, and vasoconstriction. The latter, on the one hand, is driven by direct effects of tobacco smoke on the vascular wall with the development of chronic smoking-induced endothelial dysfunction and, on the other hand, by hypoxic pulmonary vasoconstriction (HPV) with remodelling of the small pulmonary arteries [5]. HPV is a specific compensatory response to alveolar hypoxia aiming at redistributing blood flow from the less oxygenated alveoli to the better-oxygenated ones. However, in chronic lung diseases, this physiological response may result in PH. The loss of pulmonary capillaries (at least, radiographically) and small arterioles (distal pruning) with or without emphysema may also contribute to the development of PH, and a decreased DLCO may be an indicator for this particular pathology [6].
Hypoxia-inducible factors (HIFs) play not only a pivotal role in the pathogenesis of COPD-PH, but also in physiologic lung development, including angiogenesis and repair [7, 8]. In this review, we focus on HIF-related mechanisms in COPD and COPD-PH, with an emphasis on the possible role of HIF-2α in the structural lung cells for the pulmonary vascular phenotype. We hope this will contribute to a better understanding of COPD pathogenesis and the development of new biomarkers and therapeutic targets.
Methods
In this review, we aimed to combine the recent achievements in lung cell phenotyping and interactions, based on state-of-art transcriptomic methods, and recent advances in the understanding of hypoxic signalling in the lung in order to elucidate possible molecular determinants of the vascular phenotype in COPD. We relied on our own experience and the published literature. We performed searches of articles published in PubMed (https://pubmed.ncbi.nlm.nih.gov/) up to 31 July 2022 in the English language using the search terms (including MeSH and free-text search terms) “vascular phenotype”, “vascular remodeling”, “hypoxic signaling”, “hypoxia-inducible factors”, “HIF-2” AND “COPD” OR “LUNG”. We assessed the eligibility of the identified articles by screening the titles and abstracts and thus narrowed down the number of articles used for the review.
Heterogeneity and complexity of COPD
COPD is a conventional umbrella term that, according to Global Initiative for Chronic Obstructive Pulmonary Disease (GOLD), covers different pathological conditions characterised by persistent respiratory symptoms and airflow limitation due to airway and/or alveolar abnormalities usually caused by exposure to noxious particles/gases and influenced by host factors including genetic predisposition and abnormal lung development [9]. A time-dependent interaction between environmental factors (exposome) and genetic predisposition define an individual's susceptibility and the predominant involvement of different cell populations (epithelial, endothelial and/or immune) in the lung, thus leading to different pathologic traits and the development of particular endo- and phenotypes.
While cigarette smoking is considered a major environmental factor associated with 90% of deaths from COPD in developed countries, the other risk factors that determine the development of particular phenotypes remain poorly understood [10]. Recently, genome-wide association studies have identified many candidate genes for disease development, including, for instance, HO-1 (hemoxygenase-1), IREB2 (iron regulatory binding protein-2), HHIP (hedgehog-interacting protein), RAGE (receptor for advanced glycosylation end products), CHRNA3/5 (cholinergic nicotine receptor alpha 3/5), VEGFR2/KDR (vascular endothelial growth factor receptor-2), MMP-12 (matrix metalloproteinase-12) and others [11, 12].
Regarding COPD endotyping (molecular characterisation), many pathological mechanisms of the disease have been described, including alterations in the innate and acquired immune responses, enhanced oxidative stress, abnormal tissue repair, accelerated cellular senescence, impaired apoptosis and defective efferocytosis [13], and, yet, only congenital α1-antitrypsin deficiency is considered as an established COPD endotype [14]. According to Petersen et al. [15], different COPD endotypes may be derived from different populations of the susceptible lung cells exposed to the environmental factors over time. Following this idea, the cellular targets of the endotypes range from predominant epithelial (airway and parenchyma) to predominant endothelial (pulmonary vasculature) damage. The particular molecular pathways underlying the different susceptibilities of the lung cell populations in the COPD pathogenesis are not fully understood.
Vascular component of the disease
Pulmonary vascular involvement in COPD has been extensively studied [4, 16, 17]. We will focus on pre-capillary PH, pulmonary vascular disease and the pulmonary vascular phenotype in COPD.
The vascular component of COPD-PH is haemodynamically characterised by a decreased pulmonary vascular compliance and an increased pulmonary vascular resistance (PVR), which may be due to chronic hypoxic vasoconstriction, pulmonary artery remodelling and loss of the microvasculature [18]. Pulmonary microvascular blood flow measured using gadolinium-enhanced magnetic resonance imaging was reduced even in early COPD, including lung regions without emphysematous changes [19]. Pre-capillary PH has been defined as a mean pulmonary arterial pressure (mPAP) >20 mmHg, pulmonary artery wedge pressure ≤15 mmHg and PVR ≥3 Wood units [20]. COPD-PH must be confirmed by right heart catheterisation and there is evidence that PH is present in a substantial proportion of COPD patients with a prevalence between 30% and 70% [18]. mPAP may increase at a rate of 1 mmHg year−1; however, with large individual differences [21, 22], and usually manifests at the advanced stages of COPD. Actually, 90% of GOLD stage IV patients have mPAP >20 mmHg [22]. However, only 1–4% of moderate-to-severe COPD patients develop severe PH (mPAP >35 mmHg), partly associated with mild airflow obstruction, reflecting the vascular phenotype [2, 23]. The first analytical identification of this phenotype succeeded, employing a cluster analysis in a French cohort of patients with severe COPD [24].
Based on pathologic studies of lung specimens, pulmonary vascular remodelling in COPD is characterised by narrowing of small arteries (with a diameter <500 μm) due to intimal and medial thickening, as well as neomuscularisation of small arterioles [18]. Pulmonary artery remodelling also includes changes in the adventitia, including infiltration with inflammatory cells [25, 26].
While chronic HPV may lead to medial thickening of the pulmonary arteries and neomuscularisation of small arterioles, with no or minimal intimal remodelling (thus resembling the lesions observed in highlanders) [27], changes in the intima of the pulmonary arteries indicate the development of endothelial dysfunction, with areas of endothelial cell (EC) denudation and intercellular detachment, as well as so-called “neointimal lesions” that encompass the proliferation of cells that possess features of both endothelial and smooth muscle cells (formerly myofibroblasts), with the deposition of elastic and collagen fibres [17, 28]. These complex intimal changes were suggested to be a result of multiple “hits”, including not only hypoxia, but also toxic effects of airborne particles/gases [16]. Therefore, a combination of at least two types of pulmonary vascular remodelling in COPD – HPV-induced and cigarette smoking-related – may underlie the structural alterations in pulmonary arteries [29].
Vanishing capillary syndrome has been suggested as a hypothesis for the development of the vascular phenotype in COPD, explaining severe PH in association with significantly lowered DLCO and mild-to-moderate airway obstruction [30, 31]. Under chronic hypoxia, in contrast to distinctive angiogenesis in the systemic circulation, human lung capillary rarefaction has been described [32]. However, enhanced angiogenesis with increased bronchial wall vascularity was found in the small airways of COPD patients with moderate bronchial obstruction [33]. Therefore, microvascular alterations in COPD may vary in the different vascular beds (bronchial versus pulmonary) and depend on the particular phenotype.
Bunel et al. [34] found that patients with severe COPD-PH were characterised by more distinctive neomuscularisation of small arterioles and loss of alveolar capillaries, compared to less severe PH. In general, vascular pruning and capillary rarefaction can be explained by vascular regression due to migration and/or apoptosis of ECs [35]. In accordance with the latter, inhibition of EC apoptosis prevented the development of PH in monocrotaline and SU5416 (sugen) animal models [36]. Alternatively, vessel loss may also result from so-called “intussusceptive vascular pruning”, being a consequence of significant blood flow limitation in capillaries and/or downregulation of VEGF signalling in ECs [37, 38]. Therefore, in COPD, severe PH may result from a different pathological mechanism than the pulmonary arterial remodelling in mild-to-moderate PH.
Interestingly, in contrast to the loss of parenchymal blood capillaries, there was an increased lymphatic microvessel density in COPD lung parenchyma [39]. Therefore, dysregulated blood capillary and lymphangiogenesis may represent important components of the vascular remodelling in COPD. For instance, mutations of VEGFR2/KDR were associated with a specific phenotype of idiopathic pulmonary arterial hypertension that is reminiscent of the vascular phenotype in COPD including severe PH and low DLCO [12, 40, 41]. Further studies are required to explore the cellular and molecular mechanisms of vascular remodelling in COPD, taking into account unique features of the lung physiology such as the dual circulation, a particularly oxygen-rich environment and an ever-moving matrix [42].
HIF as a key molecular “hub” in lung physiology and COPD
HIF is a heterodimeric transcription factor that consists of a constitutively expressed HIF-1β subunit and an O2-regulated HIF-α subunit (HIF-1α or HIF-2α): under normoxia, the HIF-α subunit is hydroxylated by PHD (prolyl hydroxylase) and degraded by proteasome via the von Hippel–Lindau (VHL) ubiquitylation complex [43]. HIFs play a key role in lung development and homeostasis, including repair and angiogenesis [7, 44]. HIF-1α is expressed and stabilised by hypoxia in more-or-less all lung cells, whereas HIF-2α expression is restricted to the endothelium, alveolar epithelial type 2 cells (AEC2), and several types of bronchial epithelial cells [45]. Interestingly, in the lungs, during fetal development, HIF-1α protein is abundant in the branching epithelium, whereas HIF-2α protein is found in both epithelium and mesenchymal structures that are important for the vessel formation [44]. Also, it is generally believed that HIF-1α drives the early stages of angiogenesis, whereas HIF-2α is required for maturation of the vascular network [46]. Thus, in vascular biology, HIF-2α has been considered a central regulator of physiological and pathological angiogenic phenotypes [8].
Hif-2α+/– mice (haploinsufficient for HIF-2α) are characterised by augmented carotid body sensitivity to hypoxia, irregular breathing and systemic hypertension [47], but they are completely protected from hypoxic PH [48]. Dai et al. [49] confirmed this and showed that HIF-2α overexpressing mice developed very severe PH. On the parenchymal level, HIF-2α may play a different role, as recently revealed by Pasupneti et al. [50]: human emphysema lung ECs expressed reduced HIF-2α levels and EC-specific deletion of Hif-2α in the mouse model led to emphysema development, whereas its overexpression prevented emphysema after SU5416 exposure. Considering this and the fact that HIF-2 controls the main target genes that are involved in the vascular remodelling in PH, HIF-2 may represent a molecular “switch” between the development of the vascular phenotype that is associated with well-ventilated but poorly perfused lung and the nonvascular (airway) phenotypes characterised by poor ventilation–perfusion matching and severe hypoxemia and hypercapnia.
The next sections provide an overview of the main intercellular interactions within lung parenchyma, small pulmonary arteries and small airways, with an emphasis on the role of the HIF system in homeostatic signalling and pathogenesis of the disease.
Structure and role of HIFs in the alveolar-capillary unit (ACU)
To date, transcriptomic methods have revealed 58 molecular cell types in human healthy lung: 15 epithelial, nine endothelial, nine stromal and 25 immune populations [51]. At the level of the ACU, an intimate cooperation of lung cells and a precise regulation of their interactions is essential for oxygen transport into the arterial blood. At least 10 different cell types are involved in the spatial and temporal organisation of the ACU (figures 1 and 2). Mapping of the ligand-receptor signalling interactions within the unit predicts hundreds of communications among neighbouring cells [51].
FIGURE 1.

Schematic representation of paracrine interactions within the alveolar-capillary unit of the lung acinus: focus on the microvasculature. An alveolar capillary consists of “respiratory” and “homeostatic” compartments. The first one provides gas exchange and is composed of alveolar epithelial type 1 cells, aerocytes (aCap) and a thin basal membrane between them. The second one consists of closely connected general capillary endothelial cells (gCap) and pericytes that both may contribute to structural stability, regeneration and vascular tone. Crosstalk between gCap and pericytes is mediated by angiopoietin-1 (Ang-1), endothelin (ET)-1, platelet-derived growth factor (PDGF) and fibroblast growth factor-2 (FGF2). Aerocytes are unique microvascular endothelial cells with a large surface designed for effective gas diffusion. They produce apelin that is antagonistic to ET-1 released by gCAP. In addition, a subset of gCap cells represents progenitors for both gCap and aCap. The main pro-survival and mitogenic signals for endothelial cells within the alveolar-capillary unit are vascular endothelial growth factor (VEGF) and Ang-1, produced by alveolar epithelial type 1 cells and pericytes, respectively. Secretion of most of the angiocrine factors is hypoxia-inducible factor (HIF)-dependent: HIF-2α is a predominant form in pericytes and gCAP, while HIF-1α is predominantly expressed in alveolar epithelial type 1 cells. HIF isoforms in aerocytes and factors that may affect alveolar epithelial cells have not been described. HGF: hepatocyte growth factor.
FIGURE 2.

Schematic representation of paracrine interactions within the alveolar-capillary unit of the lung: focus on the alveolar epithelium. Alveolar epithelial type 2 (AEC2) cells play a central role in homeostasis as progenitor cells for alveolar epithelial type 1 cells (blue dashed arrows) and producers of surfactant. AEC2 cells are regulated by multiple paracrine interactions between neighbouring cells. The main pro-survival signal is mediated by hepatocyte growth factor (HGF), produced by lipofibroblasts (alveolar fibroblasts) and pericytes in a hypoxia-inducible factor (HIF)-2-dependent manner. Additionally, fibroblasts secrete interleukin-6 (IL-6) and fibroblast growth factor (FGF)-7. In turn, AEC2 cells produce platelet-derived growth factor (PDGF) that regulates fibroblast and pericyte activity. Capillary endothelial cells secrete matrix metalloproteinase-14 (MMP-14), which increases the bioavailability of the cryptic ligands for the epidermal growth factor receptor on AEC2, thus promoting regenerative alveologenesis. On the other hand, AEC2 cells constitutively release prostaglandin E2 (PGE2), promoting endothelial barrier integrity. Alveolar macrophages and bone marrow-derived monocytes can be the source of additional factors affecting AEC2 function and proliferative activity (cytokines, growth factors). In turn, AEC2 cells regulate the pool of alveolar macrophages by granulocyte–monocyte colony stimulating factor (GM-CSF). HIF-2α is the predominant isoform in AEC2 and, probably, pericytes, while fibroblasts express both HIF-1α and HIF-2α. S1P: sphingosine-1-posphate; TSP-1: thrombospondin-1.
Microvascular ECs
HIFs are crucial factors for ECs. The latter play a pivotal role in lung antenatal development, post-natal homeostasis and tissue regeneration by releasing paracrine (“angiocrine”) cytokines, growth factors and metabolites [52]. Transcriptomic studies have discovered a mosaic pattern and stable functional compartmentalisation of two distinct molecular phenotypes of microvascular ECs within the ACU: general capillary (gCap) ECs and so-called “aerocyte” capillary (aCap) ECs [53, 54]. Compared to aCap ECs, gCap ECs have different transcription profiles and functions producing vasoactive substances (endothelin (ET)-1, NO and prostaglandin I2) and closely interacting with pericytes [55]. In addition, gCap ECs are considered to be progenitor cells for all microvascular ECs in the lung [56]. Furthermore, network analysis predicted gCap ECs to be a major source of CXCL-12 (C-X-C motif chemokine ligand-12) in the ACU of COPD lungs [57]. CXCL-12 is controlled by HIFs and mediates the recruitment of bone marrow derived angiogenic cells [58]. In turn, aCap ECs are specialised for gas exchange, having a large surface area which is five times greater than that of gCap ECs [53, 59].
Two types of pulmonary microvascular ECs have reciprocal regulation: aCap ECs produce apelin that serves as a ligand for the apelin receptor displayed by gCap ECs; in turn, gCap ECs are a source of ET-1 and VEGF, which both act through cognate receptors on aCap ECs (endothelin receptor B (ETB) and VEGFR2/KDR, respectively), thus forming a feedback loop in vascular tone regulation [60]. Apelin expression is controlled by HIF-1α and, in chronic hypoxia, apelin-null mice developed severe PH with loss of pulmonary microvasculature [61]. In turn, HIF-2α may play a specific role in vessel integrity by regulating expression of VEGF (alongside with HIF-1α), VEGF receptor-1 (Flt-1), VEGFR2 and Tie2 (tyrosine kinase receptor for angiopoietins) in ECs [62, 63]. Being one of the leading survival factors for ECs, VEGF also regulates antenatal alveologenesis and post-natal maintenance of the alveolar structure [64]. Alveolar epithelial type (AEC) 1 but not AEC2 cells have recently been recognised as a near-exclusive source of VEGF during the late stages of lung development and repair [65]. Despite HIF-dependent upregulation of VEGF expression in chronic hypoxia, its levels were reduced in the epithelial lining fluid from patients with emphysema [66, 67]. The latter corresponds with decreased levels of HIF-2α in severe COPD. However, contrary to emphysema, VEGF levels were increased in the sputum of COPD patients with predominant bronchitis [68], reflecting different phenotypes.
Single-cell connectomic analysis revealed that microvascular ECs received signals not only from neighbouring cells in the alveolar niche, but also from remote ones. For instance, both types of microvascular ECs expressed cognate receptors for calcitonin gene-related peptide (CGRP), which is mainly excreted by pulmonary neuroendocrine cells (PNECs) in the airways [54, 69]. CGRP is a potent pulmonary vasodilator neuropeptide, but its level was increased in rats with hypoxia-induced PH, probably reflecting a compensatory upregulation [70]. In addition, CGRP promoted angiogenesis via regulation of the HIF-1α/VEGF axis in ECs [71].
Pericytes
Pericytes regulate lung tissue growth and homeostasis via the coordination of alveolar epithelial and ECs, mainly through paracrine release of hepatocyte growth factor (HGF) and angiopoetin-1 (figure 1) [72]. In severe COPD, uncoupling of pericytes from the microvasculature, as well as their migration towards the larger pulmonary arteries, are important pathogenic factors of emphysema and PH [73]. Interestingly, knockout of prolyl hydroxylase-2 (PHD2) causing an increase of HIF-2α in pulmonary artery ECs increased pericyte recruitment and muscularisation of small arterioles [74]. In turn, pericyte uncoupling from ECs may be facilitated by angiopoietin-2, which is upregulated in ECs by hypoxia during COPD exacerbations [75].
In mice treated with lipopolysaccharide, knockout of sirtuin-3 resulted in a significant reduction of pericyte/EC coverage due to upregulation of angiopoietin-2, but downregulation of HIF-2α/Notch3 and Tie-2 expression [76]. Another animal model confirmed the development of severe PH with pulmonary pericyte differentiation into contractile cells and subsequent muscularisation of distal arterioles after SU5416 exposure followed by 3 weeks of hypoxia [77]. Later, Yuan et al. [78] identified Wnt5a (wingless/integrated-5) as a key regulator of pulmonary pericyte/EC interaction and showed that reduced production of Wnt5 by microvascular ECs resulted in decreased migration and polarisation of pericytes towards ECs, resulting in the development of persistent PH in mice. Intriguingly, HIF-2α drove Wnt5a expression in multiple duodenal organoid models [79], but it is not known if the same mechanism is relevant for the pulmonary microvascular ECs.
Reduced expression of HGF associated with fewer ECs and pericytes was noted in the lungs of Hif-2α-deficient mice and in the lungs from COPD patients, suggesting HIF-2-dependent pro-survival signalling between ECs, pericytes and alveolar epithelial cells [50, 66].
Alveolar epithelial cells
Being thin and flat, covering up to 95% of the gas exchange surface, AEC1 cells form intimate associations with aCap ECs, AEC2 cells and mesenchymal cells [65]. AEC1 cells are the source of ligands not only for VEGF, but also for Wnt (wingless/integrated-1), Shh (Sonic hedgehog) and PDGF (platelet-derived growth factor) pathways. This makes AEC1 cells an important hub of intercellular communications within the ACU in lung morphogenesis and homeostasis [80]. AEC1 cells may also be an important source of ET-3 that, being a selective ligand for ETB on ECs, causes vasodilation and has antiapoptotic effects [81].
AEC2 cells play a central role for surfactant production and as progenitor cells for AEC1 cells (figure 2) [57, 82, 83]. Interestingly, HIF-2α contributes to AEC2 maturation during antenatal development encoding important proteins involved in phospholipid metabolism and surfactant production [84]. Self-renewal of AEC2 cells is controlled by ECs via the MMP-14/epidermal growth factor receptor pathway [85], and by intimately connected platelet-derived growth factor receptor α-positive fibroblasts via bone morphogenetic protein (BMP), fibroblast growth factor (FGF) 7 and Wnt paracrine signalling [56, 82, 86]. Differentiation of AEC2 cells into AEC1 cells is regulated by sphingosine-1-phosphate (S1P) released by ECs [87]. In addition to its general growth-like factor properties, S1P may act as a canonical activator of HIF-2α expression (via the Akt/mammalian target of rapamycin (mTOR) pathway), as it was shown in multiple cancer cell lines [88].
It is worth mentioning that in the human lung, respiratory airway secretory (RAS) cells in the respiratory bronchioles may also act as progenitors for AEC2 cells. RAS cell differentiation into AEC2 cells is regulated by Notch and Wnt signalling that is impaired in COPD [89]. The role of HIFs in RAS cells is not understood.
Immune cells
Chronic inflammation plays a pivotal role in both COPD and hypoxic PH [90]. In turn, HIF pathways in multiple immune cell types are context-specific and can significantly affect inflammatory process [91, 92]. The role of leukocyte-specific HIF isoforms in pulmonary inflammation has been investigated [93, 94], suggesting that HIF-2α regulates essential inflammatory functions of immune cells [95].
In murine macrophages, HIF-2α regulated the expression of pro-inflammatory cytokines in vitro [96], arginase [97] and polarisation into the M2 phenotype [98], although the latter was not observed in the model of aseptic tissue damage [99]. It is unclear whether HIF-2α dysregulation contributes to alveolar macrophage dysfunction in COPD. Expression of HIF-2α in blood neutrophils was elevated in patients with chronic inflammatory diseases and, in murine neutrophils, HIF-2α appeared to increase cell survival, thus promoting persistent neutrophilic inflammation [100]. In natural killer (NK) cells, HIF-2 limits cellular cytotoxicity [101]. In dendritic cells, HIF-1 has an anti-inflammatory function, while little is known about the role of HIF-2 [91]. The role of HIF-2 in adaptive immunity is yet to be fully elucidated. To date, it was shown that HIF-2α enhanced T-helper cell 9 development, inhibited regulatory T-cell induction [102] and played a substantial role in antigen-specific IgM expression by B-cells [103].
Role of HIFs in small pulmonary arteries
In all vessels, HIF signalling has a strong impact on vascular wall permeability, angiogenesis and repair after injury, although each vascular bed may respond differently due to distinct differences between HIF-1α and HIF-2α expression [104]. In animal models, endothelial-specific deletion of HIF-2α increased lung vascular permeability in acute lung injury and activation of HIF-2α by inhibition of PHD2 suppressed vascular leakiness [105]. Interestingly, knockout of lymphatic endothelial Hif-2α also led to increased lymphatic leakage and lymphedema due to loss of Tie2 tonic activation [106].
HIFs control the expression of the main target genes that play a key role in the vascular remodelling in PH (figure 3) [107]. For instance, mutations that stabilise HIF-2α protein lead to hereditary erythrocytosis and elevated mPAP even under normoxic conditions [108]. In multiple animal experiments, hypoxic activation of endothelial HIF-2α initiated vascular cell proliferation and recruitment of inflammatory cells in pulmonary arteries in the early stages of PH and, conversely, knockdown of Hif-2α or inhibition of HIF-2α significantly reduced the development of hypoxia-induced PH [109]. Previously, beneficial effects of HIF-2α inhibition on the development of PH were shown in different animal models [49]. Although HIF-1α plays an important role for HPV in pulmonary artery smooth muscle cells (PASMCs) and adventitial fibroblasts [110], endothelial HIF-2α appears to be the predominant factor in the pathogenesis of COPD-PH.
FIGURE 3.

Schematic representation of the hypoxia-inducible factor (HIF)-dependent mechanisms of pulmonary hypertension: focus on small pulmonary arteries. In endothelial cells (ECs), chronic hypoxia causes predominant activation of HIF-2α and subsequent production of vasoconstrictive and mitogenic factors. In turn, HIF-1α is the predominant factor in the smooth muscle cells (SMCs). Among HIF-2α target genes in ECs, C-X-C motif chemokine ligand-12 (CXCL-12), arginase-2, thrombospondin-1 (TSP-1), resistin-like molecule (RELM) and endothelin-1 (ET-1) play important roles in vascular remodelling. CXCL-12 stimulates proliferation (red dashed arrow) and expansion of SMCs and acts as a chemoattractant for immune cells. Arginase-2 limits NO bioavailability causing vasoconstriction. TSP-1 suppresses both the NO and the vascular endothelial growth factor signalling pathways (antiangiogenic effect) and activates transforming growth factor-β (pro-fibrotic effect). RELM also stimulates the proliferation of SMCs. ET-1 is a strong vasoconstrictor and activator of HIF-1α expression in SMCs (feedforward loop). Platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF) are controlled by HIF-1α and secreted by adventitial fibroblasts, in which activation of HIF-1α induces expression of angiotensin-converting enzyme (ACE) that, in turn, causes angiotensin receptor-1 (AT1) stimulation with angiotensin II and subsequent additional upregulation of HIF-1α expression (positive feedback loop). Simultaneously, in fibroblasts, HIF-2α activates nuclear factor of activated T-lymphocytes (NFAT), stimulating fibroblast proliferation (violet dashed arrow) contributing to perivascular fibrosis.
Several mechanisms have been proposed to explain the role of endothelial HIF-2α in the development of pulmonary vascular remodelling and PH, including upregulation of ET-1, CXCL-12, arginase-2, thrombospondin-1 (TSP-1), hypoxia-induced mitogenic factor (HIMF)/resistin-like molecule (RELM) and intercellular adhesion molecule 1 (ICAM1), and downregulation of the apelin receptor [111]. Endothelial- and smooth muscle cell-derived CXCL-12 promotes proliferation of PASMCs, increases pericyte coverage, acts as a chemoattractant for the immune cells and stimulates production of HIMF/RELM [112]. Arginase-2 decreases the level of endothelial NO disrupting the NO-dependent mechanism of vascular relaxation [113]. TSP-1 suppresses both NO and VEGF signalling pathways and activates transforming growth factor-β [114]. HIMF/RELM induces the proliferation of smooth muscle cells by triggering the release of HMGB1 (high mobility group box-1) from pulmonary arterial ECs which, in turn, suppresses BMPR2 (bone morphogenetic protein receptor-2) [115]. Several HIF-dependent mechanisms of hypoxic PH have also been reported in PASMCs, where HIF-1α is the main isoform [116].
Regarding the adventitial fibroblasts in PA, HIF-2α but not HIF-1α activates NFAT (nuclear factor of activated T-lymphocytes) signalling, thus promoting fibroblast proliferation [117]. In turn, HIF-1α regulates expression of PDGF, FGF, angiotensin-converting enzyme (ACE) and AT1 (angiotensin receptor-1), thus promoting proliferation of PASMCs [118].
Both HIF-1α and HIF-2α play a role in the recruitment of inflammatory cells in PA but the mechanisms are quite different. While, in PASMCs, HIF-1α is upregulated by HIMF/RELM, promoting activation of monocytes and interleukin-6 secretion in macrophages and fibroblasts [119], HIF-2α orchestrates the transactivation of ICAM1 in ECs, thus promoting recruitment of inflammatory cells [120].
Role of HIFs in small airways
Cellular and functional heterogeneity of the bronchial epithelium has been described elsewhere [121]. Here, we focus on the HIF signalling in the cells that play an important role in the maintenance and regeneration of the airways.
Airway basal cells are key modulators of lung homeostasis and epithelial regeneration due to their stem cell properties and ability to self-renew and differentiate into most of other cell types including goblet, ciliated and club cells, ionocytes, and PNECs [122]. PNECs are epithelial cells that have many characteristics of neurons, including oxygen sensing and production of CGRP, serotonin and gamma-aminobutyric acid [123]. Hypoxia leads to a proliferation of PNECs due to differentiation of the basal stem cells and this process is driven by stabilisation of HIF-1α, but not HIF-2α [124]. Besides stimulation of type 2 innate lymphoid cells, CGRP is a neuropeptide with angiogenic and vasomotor functions [70].
Club cells secrete protective proteins and proliferate under hypoxic conditions, thus providing protection and maintenance of the bronchial epithelium [125]. In contrast to basal cells and PNECs, the hypoxic response of club cells requires HIF-2α stabilisation that leads to the upregulation of FoxM1 and HIMF/RELM [126]. HIMF/RELM has antiapoptotic effects and is upregulated in bronchial epithelial and AEC2 cells during lung alveolarisation [127]. Intriguingly, under hypoxia, HIMF/RELM may be also produced by pulmonary vascular cells, being a more potent vasoconstrictor than ET-1 or angiotensin II [128].
An additional factor that may play a local cytoprotective role in the lung is erythropoietin; it is expressed at very low levels in bronchial epithelial and AEC2 cells in mice [129]. Considering that HIF-2α is an exclusive regulator of erythropoietin expression in the kidney and liver [130, 131], it is tempting to suggest that the same mechanism of regulation applies in the lung. Thus, HIF-2α upregulation in AEC2 and bronchial epithelial cells would have a beneficial effect on lung function in the case of COPD patients with mild-to-moderate emphysema and/or bronchitis.
Mechanisms of HIF-2α (dys)regulation in COPD
Cellular adaptation to hypoxia through HIFs has been extensively studied and described elsewhere [132]. Mechanisms of HIF regulation include transcriptional (hormones, cytokines and epigenetics), translational (iron-regulatory proteins, mTORC1/2 and calcitriol) and post-translational (O2-dependent and O2-independent stabilisation) mechanisms [133]. To date, several distinct post-translational modifications of HIF-1α and HIF-2α have been identified, including hydroxylation, phosphorylation, acetylation, methylation and nitrosylation [134]. In addition, regulatory mechanisms of the HIF pathway via ubiquitin and nonubiquitin degradation systems are emerging [135]. Here, we will focus on the pathways that may play important role in up- or downregulation of HIF-2α in COPD.
Currently there is no explanation why pulmonary ECs, AEC2 cells and club cells rely so much more on HIF-2α as compared to HIF-1α in the rest of the lung cells [136]. Interestingly, in the lung tissue, HIF-2α was detectable at higher oxygen levels as compared to other organs, whereas HIF-1α was undetectable even at 6% ambient oxygen concentration [45]. This suggests an important role for nonhypoxic HIF-2α activation and/or stabilisation in the lung. For instance, S1P is an oxygen-independent regulator of HIFs (via intracellular iron accumulation and ceramide production) [137]. In addition, inflammatory cytokines, adenosine, intracellular ascorbate and even microbe-derived products (siderophores and short-chain fatty acids) modulate activity of HIFs in both normoxia and hypoxia [138]. Interestingly, decreased Hif-2α expression was observed in chronically cigarette smoke exposed mice [139]. However, the particular mechanisms of HIF-2α downregulation after cigarette smoke exposure are not well understood, as well as the presence of a similar effect in humans.
HIF activation is also modulated by intracellular iron. Iron acts as a cofactor for PHDs, and HIF-2α translation is controlled by the iron regulatory proteins-1/2 (IRP1/2) that function as HIF-2α repressors [140]. This explains why Irp1−/– animals developed PH, having an increased HIF-2α protein level in pulmonary artery ECs, whereas genetic variants near IREB2 (encoding IRP2) were a risk factor for pulmonary artery enlargement and PH in COPD [141, 142]. This suggests that, in the lung, there are multiple nonhypoxic, but iron-dependent pathways upregulating HIF-2α. In turn, HIFs directly trans-activate several iron-related genes, including the transferrin receptor, heme oxygenase and coeruloplasmin [143], and HIF-2 regulates the expression of frataxin (mitochondrial aconitase chaperone) which assembles iron–sulphur clusters [144]. The mechanisms of abnormal iron homeostasis in COPD have recently been reviewed by Cloonan et al. [145].
HIFs can be modified by reactive oxygen species (ROS) in a direct and indirect fashion, but oxidation of the cysteine residues (direct redox effect) is only present in the DNA-binding domain of HIF-2α and not HIF-1α [146]. Whether this HIF-2α feature is cell-type specific or universal is unknown. The indirect effects of ROS are mediated via modulation of PHDs, FIH (factor inhibiting HIF), redox-sensitive kinases and phosphatases [147]. In particular, ROS are known to activate the extracellular signal-regulated protein kinase-1/2 (ERK1/2) that phosphorylates HIF-2α, thus controlling its nucleus shuttling and transcriptional activity [148].
NAD(P)H oxidases (Nox) are important oxygen sensors and source of ROS in the cells [149]. In COPD, Nox may be an intriguing clue to the possible mechanism of HIF-2α dysregulation and its role as a “switch” between phenotypes. Thus, Nox2-deficient mice developed spontaneous emphysema [150]. Nox subunit NOXO1 (Nox organiser-1) was upregulated in both Nox2−/– animals and wild-type mice with cigarette smoke-induced emphysema, as well as in human COPD, whereas Noxo1−/– mice were protected from cigarette smoke-induced emphysema and PH with right heart hypertrophy. However, mice had an increased vascular muscularisation after 8 months of cigarette smoke exposure [151]. Nagaraj et al. [152] showed that p22phox, an essential subunit of several Nox [153], was significantly reduced in patients with severe COPD as compared to controls, but was preserved in the subgroup with severely increased mPAP and lowered DLCO. Furthermore, downregulation of p22phox expression inhibited Akt phosphorylation and decreased HIF-2α translation [154] while p22phox stabilisation promoted hypoxic PH in mice [155]. In addition, cancer research studies showed that p22phox-dependent Nox1 and Nox4 were essential for HIF-2α expression and activity in VHL-deficient cells [156]. Considering all these facts, we hypothesise that development of the vascular phenotype in COPD may be mediated by stabilisation of several Nox isoforms via iron- and redox-dependent factors, controlling endothelial HIF-2α expression and activity (figure 4).
FIGURE 4.
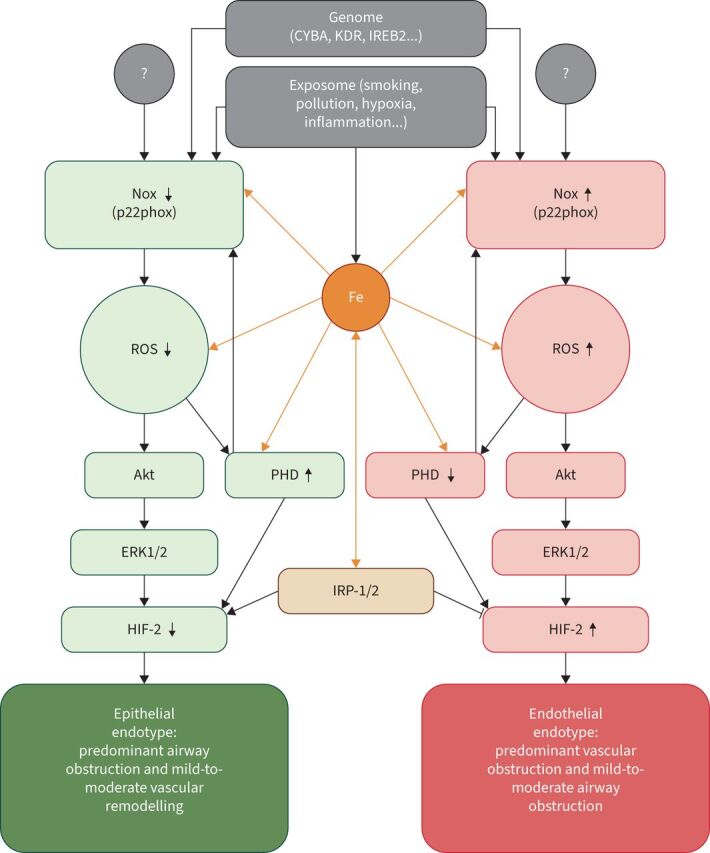
Schematic of the hypothetic mechanism of hypoxia-inducible factor-2α (HIF-2α) (dys)regulation in COPD. NAD(P)H oxidase (Nox) stabilisation (including the p22phox subunit) increases production of reactive oxygen species (ROS) that deactivate prolyl hydroxylase (PHD), but activate the Akt-kinase (Akt)/extracellular signal-regulated kinase-1/2 (ERK1/2) pathway resulting in endothelial HIF-2α upregulation and development of the endothelial endotype. The latter may be associated with the vascular phenotype in COPD. In contrast, reduced expression and/or activity of Nox leads to HIF-2α downregulation and development of the epithelial endotype that may encompass nonvascular phenotypes (bronchitis or emphysema). Since PHD and Nox (p22phox) are iron-dependent enzymes, the intracellular iron level modifies the production of ROS, PHD activity and regulates translation of HIF-2α via iron regulatory protein-1/2 (IRP-1/2). Hypoxia, cytokines and environmental factors affect the level and/or activity of Nox. CYBA: gene encoding p22phox; IREB2: gene encoding IRP-2; KDR: gene encoding vascular endothelial growth factor receptor 2.
Points for clinical practice
In COPD, upregulation of HIF-2 in the lung may increase the risk of development of the vascular phenotype.
Cell-specific HIF-2 surrogate readouts (related endothelial, alveolar type 2 epithelial or bronchial epithelial biomarkers) may provide a clue towards better molecular characterisation of the individual COPD patients and their management.
Questions for future research
Will HIF-2 augmentation be beneficial for COPD patients with loss of airway structures?
Will HIF-2 suppression be beneficial for COPD patients with the pulmonary vascular phenotype?
Conclusion and future perspectives
The HIF system plays a critical role in the pulmonary development and homeostasis. Cell-specific expression of HIF-2α is predominant in ECs, AEC2 cells and club cells, and mediates unique functions in the lung. In COPD, upregulation of HIF-2 in ECs may lead to predominant pulmonary vascular remodelling and severe PH, but preserved bronchial and parenchymal structures – due to the pro-surviving functions of HIF-2 in AEC2 and bronchial club cells. In contrast, downregulation of HIF-2 in AEC2 and bronchial club cells may lead to parenchymal degeneration and loss of small airways, but more preserved vascular structures due to beneficial effects in pulmonary arterial ECs. These effects of up- or downregulation of HIF-2α may explain, at a molecular level, why different phenotypes of COPD develop.
Figure 5 illustrates the putative role of HIF-2 as a “switch” between the vascular and nonvascular phenotypes in COPD. This hypothesis suggests that HIF-2-modulating therapies may affect both the vascular and airway system of the lung. However, it will be important to consider the timing, cell specificity and off-target side effects of such therapies. Nevertheless, potential HIF-2 surrogate readouts may have an important diagnostic meaning in COPD endotyping, establishing the “endotype – biomarker – precision medicine” strategy and identifying new treatable targets.
FIGURE 5.

Hypothesis of hypoxia-inducible factor-2 (HIF-2) as a “switch” between the vascular and nonvascular phenotypes in COPD. HIF-2 might represent a molecular “switch” between progression into a vascular or nonvascular phenotype. Under the influence of multiple exogenous and endogenous factors, maladaptive upregulation of HIF-2 in endothelial cells, alveolar type 2 cells and some types of bronchial cells may result in predominant vascular involvement, with pronounced remodelling and severe (out-of-proportion) pulmonary hypertension, but relatively preserved airways and alveolar-capillary units of the lung (the vascular phenotype). Loss of adaptive HIF-2 upregulation (HIF-2 downregulation) in the mentioned cells leads to the loss of alveoli and/or small bronchioles, but mild-to-moderate vascular remodelling and proportionate pulmonary hypertension.
Acknowledgements
Imaginative figures were created with BioRender.com.
Provenance: Submitted article, peer reviewed.
Conflicts of interest: The authors have no conflicts of interest to disclose.
Support statement: This research was funded in whole, or in part, by the Austrian Science Fund (FWF) DOC 129-B (RESPImmun Doctoral Program). For the purpose of open access, the authors have applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
References
- 1.Halpin DMG, Celli BR, Criner GJ, et al. The GOLD Summit on chronic obstructive pulmonary disease in low- and middle-income countries. Int J Tuberc Lung Dis 2019; 23: 1131–1141. doi: 10.5588/ijtld.19.0397 [DOI] [PubMed] [Google Scholar]
- 2.Kovacs G, Agusti A, Barbera JA, et al. Pulmonary vascular involvement in chronic obstructive pulmonary disease. Is there a pulmonary vascular phenotype? Am J Respir Crit Care Med 2018; 198: 1000–1011. doi: 10.1164/rccm.201801-0095PP [DOI] [PubMed] [Google Scholar]
- 3.Galie N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J 2015; 46: 903–975. doi: 10.1183/13993003.01032-2015 [DOI] [PubMed] [Google Scholar]
- 4.Hoeper MM, Humbert M, Souza R, et al. A global view of pulmonary hypertension. Lancet Respir Med 2016; 4: 306–322. doi: 10.1016/S2213-2600(15)00543-3 [DOI] [PubMed] [Google Scholar]
- 5.El-Mahdy MA, Abdelghany TM, Hemann C, et al. Chronic cigarette smoke exposure triggers a vicious cycle of leukocyte and endothelial-mediated oxidant stress that results in vascular dysfunction. Am J Physiol Heart Circ Physiol 2020; 319: H51–H65. doi: 10.1152/ajpheart.00657.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weatherald J, Montani D, Humbert M. Seeing the forest for the (arterial) tree: vascular pruning and the chronic obstructive pulmonary disease pulmonary vascular phenotype. Am J Respir Crit Care Med 2019; 200: 406–408. doi: 10.1164/rccm.201901-0248ED [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pullamsetti SS, Mamazhakypov A, Weissmann N, et al. Hypoxia-inducible factor signaling in pulmonary hypertension. J Clin Invest 2020; 130: 5638–5651. doi: 10.1172/JCI137558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Befani C, Liakos P. The role of hypoxia-inducible factor-2 alpha in angiogenesis. J Cell Physiol 2018; 233: 9087–9098. doi: 10.1002/jcp.26805 [DOI] [PubMed] [Google Scholar]
- 9.Halpin DMG, Criner GJ, Papi A, et al. Global initiative for the diagnosis, management, and prevention of chronic obstructive lung disease. The 2020 GOLD science committee report on COVID-19 and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2021; 203: 24–36. doi: 10.1164/rccm.202009-3533SO [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siroux V, Agier L, Slama R. The exposome concept: a challenge and a potential driver for environmental health research. Eur Respir Rev 2016; 25: 124–129. doi: 10.1183/16000617.0034-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho MH, McDonald ML, Zhou X, et al. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir Med 2014; 2: 214–225. doi: 10.1016/S2213-2600(14)70002-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swietlik EM, Greene D, Zhu N, et al. Bayesian inference associates rare KDR variants with specific phenotypes in pulmonary arterial hypertension. Circ Genom Precis Med 2020; 14: e0031551. doi: 10.1161/CIRCGEN.120.003155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agusti A, Vestbo J. Current controversies and future perspectives in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2011; 184: 507–513. doi: 10.1164/rccm.201103-0405PP [DOI] [PubMed] [Google Scholar]
- 14.Agustí A, Celli B, Faner R. What does endotyping mean for treatment in chronic obstructive pulmonary disease? Lancet 2017; 390: 980–987. doi: 10.1016/S0140-6736(17)32136-0 [DOI] [PubMed] [Google Scholar]
- 15.Petersen H, Vazquez Guillamet R, Meek P, et al. Early endotyping: a chance for intervention in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2018; 59: 13–17. doi: 10.1165/rcmb.2018-0002PS [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakao S, Voelkel NF, Tatsumi K. The vascular bed in COPD: pulmonary hypertension and pulmonary vascular alterations. Eur Respir Rev 2014; 23: 350–355. doi: 10.1183/09059180.00007913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blanco I, Piccari L, Barbera JA. Pulmonary vasculature in COPD: the silent component. Respirology 2016; 21: 984–994. doi: 10.1111/resp.12772 [DOI] [PubMed] [Google Scholar]
- 18.Minai OA, Chaouat A, Adnot S. Pulmonary hypertension in COPD: epidemiology, significance, and management: pulmonary vascular disease: the global perspective. Chest 2010; 137: Suppl. 6, 39S–51S. doi: 10.1378/chest.10-0087 [DOI] [PubMed] [Google Scholar]
- 19.Hueper K, Vogel-Claussen J, Parikh MA, et al. Pulmonary microvascular blood flow in mild chronic obstructive pulmonary disease and emphysema. The MESA COPD study. Am J Respir Crit Care Med 2015; 192: 570–580. doi: 10.1164/rccm.201411-2120OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53: 1801913. doi: 10.1183/13993003.01913-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nathan SD, Barbera JA, Gaine SP, et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J 2019; 53: 1801914. doi: 10.1183/13993003.01914-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kessler R, Faller M, Weitzenblum E, et al. ‘Natural history’ of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am J Respir Crit Care Med 2001; 164: 219–224. doi: 10.1164/ajrccm.164.2.2006129 [DOI] [PubMed] [Google Scholar]
- 23.Kovacs G, Avian A, Bachmaier G, et al. Severe pulmonary hypertension in COPD: impact on survival and diagnostic approach. Chest 2022; 162: 202–212. doi: 10.1016/j.chest.2022.01.031 [DOI] [PubMed] [Google Scholar]
- 24.Dauriat G, Reynaud-Gaubert M, Cottin V, et al. Severe pulmonary hypertension associated with chronic obstructive pulmonary disease: a prospective French multicenter cohort. J Heart Lung Transplant 2021; 40: 1009–1018. doi: 10.1016/j.healun.2021.04.021 [DOI] [PubMed] [Google Scholar]
- 25.Peinado VI, Barbera JA, Abate P, et al. Inflammatory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999; 159: 1605–1611. doi: 10.1164/ajrccm.159.5.9807059 [DOI] [PubMed] [Google Scholar]
- 26.Vergadi E, Chang MS, Lee C, et al. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation 2011; 123: 1986–1995. doi: 10.1161/CIRCULATIONAHA.110.978627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tuder RM. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res 2017; 367: 643–649. doi: 10.1007/s00441-016-2539-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santos S, Peinado VI, Ramirez J, et al. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J 2002; 19: 632–638. doi: 10.1183/09031936.02.00245902 [DOI] [PubMed] [Google Scholar]
- 29.Gredic M, Blanco I, Kovacs G, et al. Pulmonary hypertension in chronic obstructive pulmonary disease. Br J Pharmacol 2021; 178: 132–151. doi: 10.1111/bph.14979 [DOI] [PubMed] [Google Scholar]
- 30.Chaouat A, Bugnet AS, Kadaoui N, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005; 172: 189–194. doi: 10.1164/rccm.200401-006OC [DOI] [PubMed] [Google Scholar]
- 31.Pistenmaa CL, Nardelli P, Ash SY, et al. Pulmonary arterial pruning and longitudinal change in percent emphysema and lung function: the genetic epidemiology of COPD study. Chest 2021; 160: 470–480. doi: 10.1016/j.chest.2021.01.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hopkins N, McLoughlin P. The structural basis of pulmonary hypertension in chronic lung disease: remodelling, rarefaction or angiogenesis? J Anat 2002; 201: 335–348. doi: 10.1046/j.1469-7580.2002.00096.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hashimoto M, Tanaka H, Abe S. Quantitative analysis of bronchial wall vascularity in the medium and small airways of patients with asthma and COPD. Chest 2005; 127: 965–972. doi: 10.1378/chest.127.3.965 [DOI] [PubMed] [Google Scholar]
- 34.Bunel V, Guyard A, Dauriat G, et al. Pulmonary arterial histologic lesions in patients with COPD with severe pulmonary hypertension. Chest 2019; 156: 33–44. doi: 10.1016/j.chest.2019.02.333 [DOI] [PubMed] [Google Scholar]
- 35.Korn C, Augustin HG. Mechanisms of vessel pruning and regression. Dev Cell 2015; 34: 5–17. doi: 10.1016/j.devcel.2015.06.004 [DOI] [PubMed] [Google Scholar]
- 36.Jurasz P, Courtman D, Babaie S, et al. Role of apoptosis in pulmonary hypertension: from experimental models to clinical trials. Pharmacol Ther 2010; 126: 1–8. doi: 10.1016/j.pharmthera.2009.12.006 [DOI] [PubMed] [Google Scholar]
- 37.Mentzer SJ, Konerding MA. Intussusceptive angiogenesis: expansion and remodeling of microvascular networks. Angiogenesis 2014; 17: 499–509. doi: 10.1007/s10456-014-9428-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hlushchuk R, Ehrbar M, Reichmuth P, et al. Decrease in VEGF expression induces intussusceptive vascular pruning. Arterioscler Thromb Vasc Biol 2011; 31: 2836–2844. doi: 10.1161/ATVBAHA.111.231811 [DOI] [PubMed] [Google Scholar]
- 39.Kropski JA, Richmond BW, Gaskill CF, et al. Deregulated angiogenesis in chronic lung diseases: a possible role for lung mesenchymal progenitor cells (2017 Grover Conference Series). Pulm Circ 2018; 8: 2045893217739807. doi: 10.1177/2045893217739807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eyries M, Montani D, Girerd B, et al. Familial pulmonary arterial hypertension by KDR heterozygous loss of function. Eur Respir J 2020; 55: 1902165. doi: 10.1183/13993003.02165-2019 [DOI] [PubMed] [Google Scholar]
- 41.Kasahara Y, Tuder RM, Taraseviciene-Stewart L, et al. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 2000; 106: 1311–1319. doi: 10.1172/JCI10259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eldridge L, Wagner EM. Angiogenesis in the lung. J Physiol 2019; 597: 1023–1032. doi: 10.1113/JP275860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Epstein AC, Gleadle JM, McNeill LA, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001; 107: 43–54. doi: 10.1016/S0092-8674(01)00507-4 [DOI] [PubMed] [Google Scholar]
- 44.Shimoda LA, Semenza GL. HIF and the lung: role of hypoxia-inducible factors in pulmonary development and disease. Am J Respir Crit Care Med 2011; 183: 152–156. doi: 10.1164/rccm.201009-1393PP [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wiesener MS, Jurgensen JS, Rosenberger C, et al. Widespread hypoxia-inducible expression of HIF-2α in distinct cell populations of different organs. FASEB J 2003; 17: 271–273. doi: 10.1096/fj.02-0445fje [DOI] [PubMed] [Google Scholar]
- 46.Koh MY, Powis G. Passing the baton: the HIF switch. Trends Biochem Sci 2012; 37: 364–372. doi: 10.1016/j.tibs.2012.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng YJ, Nanduri J, Khan SA, et al. Hypoxia-inducible factor 2α (HIF-2α) heterozygous-null mice exhibit exaggerated carotid body sensitivity to hypoxia, breathing instability, and hypertension. Proc Natl Acad Sci USA 2011; 108: 3065–3070. doi: 10.1073/pnas.1100064108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brusselmans K, Compernolle V, Tjwa M, et al. Heterozygous deficiency of hypoxia-inducible factor-2α protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest 2003; 111: 1519–1527. doi: 10.1172/JCI15496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dai Z, Zhu MM, Peng Y, et al. Therapeutic targeting of vascular remodeling and right heart failure in pulmonary arterial hypertension with a HIF-2aα inhibitor. Am J Respir Crit Care Med 2018; 198: 1423–1434. doi: 10.1164/rccm.201710-2079OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pasupneti S, Tian W, Tu AB, et al. Endothelial HIF-2α as a key endogenous mediator preventing emphysema. Am J Respir Crit Care Med 2020; 202: 983–995. doi: 10.1164/rccm.202001-0078OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Travaglini KJ, Nabhan AN, Penland L, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020; 587: 619–625. doi: 10.1038/s41586-020-2922-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Augustin HG, Koh GY. Organotypic vasculature: from descriptive heterogeneity to functional pathophysiology. Science 2017; 357: eaal2379. doi: 10.1126/science.aal2379 [DOI] [PubMed] [Google Scholar]
- 53.Gillich A, Zhang F, Farmer CG, et al. Capillary cell-type specialization in the alveolus. Nature 2020; 586: 785–789. doi: 10.1038/s41586-020-2822-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schupp JC, Adams TS, Cosme C,Jr, et al. Integrated single-cell atlas of endothelial cells of the human lung. Circulation 2021; 144: 286–302. doi: 10.1161/CIRCULATIONAHA.120.052318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kruger-Genge A, Blocki A, Franke RP, et al. Vascular endothelial cell biology: an update. Int J Mol Sci 2019; 20: 4411. doi: 10.3390/ijms20184411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu H, Tang N. Stem cells in pulmonary alveolar regeneration. Development 2021; 148: dev193458. doi: 10.1242/dev.193458 [DOI] [PubMed] [Google Scholar]
- 57.Sauler M, McDonough JE, Adams TS, et al. Characterization of the COPD alveolar niche using single-cell RNA sequencing. Nat Commun 2022; 13: 494. doi: 10.1038/s41467-022-28062-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Samanta D, Prabhakar NR, Semenza GL. Systems biology of oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med 2017; 9: e1382. doi: 10.1002/wsbm.1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weibel ER. Lung morphometry: the link between structure and function. Cell Tissue Res 2017; 367: 413–426. doi: 10.1007/s00441-016-2541-4 [DOI] [PubMed] [Google Scholar]
- 60.Hennigs JK, Matuszcak C, Trepel M, et al. Vascular endothelial cells: heterogeneity and targeting approaches. Cells 2021; 10: 2712. doi: 10.3390/cells10102712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chandra SM, Razavi H, Kim J, et al. Disruption of the apelin-APJ system worsens hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol 2011; 31: 814–820. doi: 10.1161/ATVBAHA.110.219980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Skuli N, Liu L, Runge A, et al. Endothelial deletion of hypoxia-inducible factor-2α (HIF-2α) alters vascular function and tumor angiogenesis. Blood 2009; 114: 469–477. doi: 10.1182/blood-2008-12-193581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duan LJ, Zhang-Benoit Y, Fong GH. Endothelium-intrinsic requirement for Hif-2α during vascular development. Circulation 2005; 111: 2227–2232. doi: 10.1161/01.CIR.0000163580.98098.A3 [DOI] [PubMed] [Google Scholar]
- 64.Yun EJ, Lorizio W, Seedorf G, et al. VEGF and endothelium-derived retinoic acid regulate lung vascular and alveolar development. Am J Physiol Lung Cell Mol Physiol 2016; 310: L287–L298. doi: 10.1152/ajplung.00229.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang J, Hernandez BJ, Martinez Alanis D, et al. The development and plasticity of alveolar type 1 cells. Development 2016; 143: 54–65. doi: 10.1242/dev.130005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanazawa H, Tochino Y, Asai K, et al. Simultaneous assessment of hepatocyte growth factor and vascular endothelial growth factor in epithelial lining fluid from patients with COPD. Chest 2014; 146: 1159–1165. doi: 10.1378/chest.14-0373 [DOI] [PubMed] [Google Scholar]
- 67.Voelkel NF, Vandivier RW, Tuder RM. Vascular endothelial growth factor in the lung. Am J Physiol Lung Cell Mol Physiol 2006; 290: L209–L221. doi: 10.1152/ajplung.00185.2005 [DOI] [PubMed] [Google Scholar]
- 68.Kanazawa H, Asai K, Hirata K, et al. Possible effects of vascular endothelial growth factor in the pathogenesis of chronic obstructive pulmonary disease. Am J Med 2003; 114: 354–358. doi: 10.1016/S0002-9343(02)01562-0 [DOI] [PubMed] [Google Scholar]
- 69.Raredon MSB, Adams TS, Suhail Y, et al. Single-cell connectomic analysis of adult mammalian lungs. Sci Adv 2019; 5: eaaw3851. doi: 10.1126/sciadv.aaw3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lo CCW, Moosavi SM, Bubb KJ. The regulation of pulmonary vascular tone by neuropeptides and the implications for pulmonary hypertension. Front Physiol 2018; 9: 1167. doi: 10.3389/fphys.2018.01167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guo Y, Zhang Q, Chen H, et al. Overexpression of calcitonin gene-related peptide protects mouse cerebral microvascular endothelial cells from high-glucose-induced damage via ERK/HIF-1/VEGF signaling. J Physiol Sci 2019; 69: 939–952. doi: 10.1007/s12576-019-00708-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kato K, Dieguez-Hurtado R, Park DY, et al. Pulmonary pericytes regulate lung morphogenesis. Nat Commun 2018; 9: 2448. doi: 10.1038/s41467-018-04913-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Birbrair A. Pericyte biology: development, homeostasis, and disease. Adv Exp Med Biol 2018; 1109: 1–3. doi: 10.1007/978-3-030-02601-1_1 [DOI] [PubMed] [Google Scholar]
- 74.Wang S, Zeng H, Xie XJ, et al. Loss of prolyl hydroxylase domain protein 2 in vascular endothelium increases pericyte coverage and promotes pulmonary arterial remodeling. Oncotarget 2016; 7: 58848–58861. doi: 10.18632/oncotarget.11585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pichiule P, Chavez JC, LaManna JC. Hypoxic regulation of angiopoietin-2 expression in endothelial cells. J Biol Chem 2004; 279: 12171–12180. doi: 10.1074/jbc.M305146200 [DOI] [PubMed] [Google Scholar]
- 76.Zeng H, He X, Tuo QH, et al. LPS causes pericyte loss and microvascular dysfunction via disruption of Sirt3/angiopoietins/Tie-2 and HIF-2α/Notch3 pathways. Sci Rep 2016; 6: 20931. doi: 10.1038/srep20931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bordenave J, Tu L, Berrebeh N, et al. Lineage tracing reveals the dynamic contribution of pericytes to the blood vessel remodeling in pulmonary hypertension. Arterioscler Thromb Vasc Biol 2020; 40: 766–782. doi: 10.1161/ATVBAHA.119.313715 [DOI] [PubMed] [Google Scholar]
- 78.Yuan K, Shamskhou EA, Orcholski ME, et al. Loss of endothelium-derived wnt5a is associated with reduced pericyte recruitment and small vessel loss in pulmonary arterial hypertension. Circulation 2019; 139: 1710–1724. doi: 10.1161/CIRCULATIONAHA.118.037642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Garcia Garcia CJ, Acevedo Diaz AC, Kumari N, et al. HIF2 Regulates Intestinal Wnt5a Expression. Front Oncol 2021; 11: 769385. doi: 10.3389/fonc.2021.769385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zepp JA, Morley MP, Loebel C, et al. Genomic, epigenomic, and biophysical cues controlling the emergence of the lung alveolus. Science 2021; 371: eabc3172. doi: 10.1126/science.abc3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Montani D, Souza R, Binkert C, et al. Endothelin-1/endothelin-3 ratio: a potential prognostic factor of pulmonary arterial hypertension. Chest 2007; 131: 101–108. doi: 10.1378/chest.06-0682 [DOI] [PubMed] [Google Scholar]
- 82.Barkauskas CE, Cronce MJ, Rackley CR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest 2013; 123: 3025–3036. doi: 10.1172/JCI68782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zacharias WJ, Frank DB, Zepp JA, et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018; 555: 251–255. doi: 10.1038/nature25786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huang Y, Kempen MB, Munck AB, et al. Hypoxia-inducible factor 2α plays a critical role in the formation of alveoli and surfactant. Am J Respir Cell Mol Biol 2012; 46: 224–232. doi: 10.1165/rcmb.2011-0024OC [DOI] [PubMed] [Google Scholar]
- 85.Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014; 507: 190–194. doi: 10.1038/nature12930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zepp JA, Zacharias WJ, Frank DB, et al. Distinct mesenchymal lineages and niches promote epithelial self-renewal and myofibrogenesis in the lung. Cell 2017; 170: 1134–1148.e10. doi: 10.1016/j.cell.2017.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen Q, Rehman J, Chan M, et al. Angiocrine sphingosine-1-phosphate activation of S1PR2-YAP signaling axis in alveolar type II cells is essential for lung repair. Cell Rep 2020; 31: 107828. doi: 10.1016/j.celrep.2020.107828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bouquerel P, Gstalder C, Muller D, et al. Essential role for SphK1/S1P signaling to regulate hypoxia-inducible factor 2α expression and activity in cancer. Oncogenesis 2016; 5: e209. doi: 10.1038/oncsis.2016.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Basil MC, Cardenas-Diaz FL, Kathiriya JJ, et al. Human distal airways contain a multipotent secretory cell that can regenerate alveoli. Nature 2022; 604: 120–126. doi: 10.1038/s41586-022-04552-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pugliese SC, Poth JM, Fini MA, et al. The role of inflammation in hypoxic pulmonary hypertension: from cellular mechanisms to clinical phenotypes. Am J Physiol Lung Cell Mol Physiol 2015; 308: L229–L252. doi: 10.1152/ajplung.00238.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Taylor CT, Doherty G, Fallon PG, et al. Hypoxia-dependent regulation of inflammatory pathways in immune cells. J Clin Invest 2016; 126: 3716–3724. doi: 10.1172/JCI84433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Watts ER, Walmsley SR. Inflammation and hypoxia: HIF and PHD isoform selectivity. Trends Mol Med 2019; 25: 33–46. doi: 10.1016/j.molmed.2018.10.006 [DOI] [PubMed] [Google Scholar]
- 93.Whyte MK, Walmsley SR. The regulation of pulmonary inflammation by the hypoxia-inducible factor-hydroxylase oxygen-sensing pathway. Ann Am Thorac Soc 2014; 11: Suppl. 5, S271–S276. doi: 10.1513/AnnalsATS.201403-108AW [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Evans CE. Hypoxia-inducible factor signaling in inflammatory lung injury and repair. Cells 2022; 11: 183. doi: 10.3390/cells11020183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Palazon A, Goldrath AW, Nizet V, et al. HIF transcription factors, inflammation, and immunity. Immunity 2014; 41: 518–528. doi: 10.1016/j.immuni.2014.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Imtiyaz HZ, Williams EP, Hickey MM, et al. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest 2010; 120: 2699–2714. doi: 10.1172/JCI39506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lin N, Simon MC. Hypoxia-inducible factors: key regulators of myeloid cells during inflammation. J Clin Invest 2016; 126: 3661–3671. doi: 10.1172/JCI84426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Colgan SP, Furuta GT, Taylor CT. Hypoxia and innate immunity: keeping up with the HIFsters. Annu Rev Immunol 2020; 38: 341–363. doi: 10.1146/annurev-immunol-100819-121537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gondin J, Theret M, Duhamel G, et al. Myeloid HIFs are dispensable for resolution of inflammation during skeletal muscle regeneration. J Immunol 2015; 194: 3389–3399. doi: 10.4049/jimmunol.1401420 [DOI] [PubMed] [Google Scholar]
- 100.Thompson AA, Elks PM, Marriott HM, et al. Hypoxia-inducible factor 2α regulates key neutrophil functions in humans, mice, and zebrafish. Blood 2014; 123: 366–376. doi: 10.1182/blood-2013-05-500207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang J, Han C, Dai H, et al. Hypoxia-inducible factor-2α limits natural killer T cell cytotoxicity in renal ischemia/reperfusion injury. J Am Soc Nephrol 2016; 27: 92–106. doi: 10.1681/ASN.2014121248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Singh Y, Garden OA, Lang F, et al. MicroRNAs regulate T-cell production of interleukin-9 and identify hypoxia-inducible factor-2α as an important regulator of T helper 9 and regulatory T-cell differentiation. Immunology 2016; 149: 74–86. doi: 10.1111/imm.12631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.McGettrick AF, O'Neill LAJ. The role of HIF in immunity and inflammation. Cell Metab 2020; 32: 524–536. doi: 10.1016/j.cmet.2020.08.002 [DOI] [PubMed] [Google Scholar]
- 104.Marsboom G, Rehman J. Hypoxia signaling in vascular homeostasis. Physiology 2018; 33: 328–337. 10.1152/physiol.00018.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gong H, Rehman J, Tang H, et al. HIF2α signaling inhibits adherens junctional disruption in acute lung injury. J Clin Invest 2015; 125: 652–664. doi: 10.1172/JCI77701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jiang X, Tian W, Granucci EJ, et al. Decreased lymphatic HIF-2α accentuates lymphatic remodeling in lymphedema. J Clin Invest 2020; 130: 5562–5575. doi: 10.1172/JCI136164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med 2011; 365: 537–547. doi: 10.1056/NEJMra1011165 [DOI] [PubMed] [Google Scholar]
- 108.Formenti F, Beer PA, Croft QP, et al. Cardiopulmonary function in two human disorders of the hypoxia-inducible factor (HIF) pathway: von Hippel–Lindau disease and HIF-2α gain-of-function mutation. FASEB J 2011; 25: 2001–2011. doi: 10.1096/fj.10-177378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hu CJ, Poth JM, Zhang H, et al. Suppression of HIF2 signalling attenuates the initiation of hypoxia-induced pulmonary hypertension. Eur Respir J 2019; 54: 1900378. doi: 10.1183/13993003.00378-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Waypa GB, Schumacker PT. Roles of HIF1 and HIF2 in pulmonary hypertension: it all depends on the context. Eur Respir J 2019; 54: 1901929. doi: 10.1183/13993003.01929-2019 [DOI] [PubMed] [Google Scholar]
- 111.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 2006; 99: 675–691. doi: 10.1161/01.RES.0000243584.45145.3f [DOI] [PubMed] [Google Scholar]
- 112.Dai Z, Li M, Wharton J, et al. Prolyl-4 hydroxylase 2 (PHD2) deficiency in endothelial cells and hematopoietic cells induces obliterative vascular remodeling and severe pulmonary arterial hypertension in mice and humans through hypoxia-inducible factor-2α. Circulation 2016; 133: 2447–2458. doi: 10.1161/CIRCULATIONAHA.116.021494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cowburn AS, Crosby A, Macias D, et al. HIF2α-arginase axis is essential for the development of pulmonary hypertension. Proc Natl Acad Sci U S A 2016; 113: 8801–8806. doi: 10.1073/pnas.1602978113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Labrousse-Arias D, Castillo-Gonzalez R, Rogers NM, et al. HIF-2α-mediated induction of pulmonary thrombospondin-1 contributes to hypoxia-driven vascular remodelling and vasoconstriction. Cardiovasc Res 2016; 109: 115–130. doi: 10.1093/cvr/cvv243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lin Q, Fan C, Gomez-Arroyo J, et al. HIMF (hypoxia-induced mitogenic factor) signaling mediates the HMGB1 (high mobility group box 1)-dependent endothelial and smooth muscle cell crosstalk in pulmonary hypertension. Arterioscler Thromb Vasc Biol 2019; 39: 2505–2519. doi: 10.1161/ATVBAHA.119.312907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Stenmark KR, Frid MG, Graham BB, et al. Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension. Cardiovasc Res 2018; 114: 551–564. doi: 10.1093/cvr/cvy004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Senavirathna LK, Huang C, Yang X, et al. Hypoxia induces pulmonary fibroblast proliferation through NFAT signaling. Sci Rep 2018; 8: 2709. doi: 10.1038/s41598-018-21073-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Krick S, Hanze J, Eul B, et al. Hypoxia-driven proliferation of human pulmonary artery fibroblasts: cross-talk between HIF-1α and an autocrine angiotensin system. FASEB J 2005; 19: 857–859. doi: 10.1096/fj.04-2890fje [DOI] [PubMed] [Google Scholar]
- 119.Johns RA, Takimoto E, Meuchel LW, et al. Hypoxia-inducible factor 1α is a critical downstream mediator for hypoxia-induced mitogenic factor (FIZZ1/RELMα)-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol 2016; 36: 134–144. doi: 10.1161/ATVBAHA.115.306710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang S, Wang Y, Liu C, et al. EPAS1 (endothelial PAS domain protein 1) orchestrates transactivation of endothelial ICAM1 (intercellular adhesion molecule 1) by small nucleolar RNA host gene 5 (SNHG5) to promote hypoxic pulmonary hypertension. Hypertension 2021; 78: 1080–1091. doi: 10.1161/HYPERTENSIONAHA.121.16949 [DOI] [PubMed] [Google Scholar]
- 121.Davis JD, Wypych TP. Cellular and functional heterogeneity of the airway epithelium. Mucosal Immunol 2021; 14: 978–990. doi: 10.1038/s41385-020-00370-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rock JR, Randell SH, Hogan BL. Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis Model Mech 2010; 3: 545–556. doi: 10.1242/dmm.006031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Noguchi M, Furukawa KT, Morimoto M. Pulmonary neuroendocrine cells: physiology, tissue homeostasis and disease. Dis Model Mech 2020; 13: dmm046920. doi: 10.1242/dmm.046920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shivaraju M, Chitta UK, Grange RMH, et al. Airway stem cells sense hypoxia and differentiate into protective solitary neuroendocrine cells. Science 2021; 371: 52–57. doi: 10.1126/science.aba0629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Laucho-Contreras ME, Polverino F, Gupta K, et al. Protective role for club cell secretory protein-16 (CC16) in the development of COPD. Eur Respir J 2015; 45: 1544–1556. doi: 10.1183/09031936.00134214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Torres-Capelli M, Marsboom G, Li QO, et al. Role of Hif2α oxygen sensing pathway in bronchial epithelial club cell proliferation. Sci Rep 2016; 6: 25357. doi: 10.1038/srep25357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wagner KF, Hellberg AK, Balenger S, et al. Hypoxia-induced mitogenic factor has antiapoptotic action and is upregulated in the developing lung: coexpression with hypoxia-inducible factor-2α. Am J Respir Cell Mol Biol 2004; 31: 276–282. doi: 10.1165/rcmb.2003-0319OC [DOI] [PubMed] [Google Scholar]
- 128.Teng X, Li D, Champion HC, et al. FIZZ1/RELMα, a novel hypoxia-induced mitogenic factor in lung with vasoconstrictive and angiogenic properties. Circ Res 2003; 92: 1065–1067. doi: 10.1161/01.RES.0000073999.07698.33 [DOI] [PubMed] [Google Scholar]
- 129.Haine L, Yegen CH, Marchant D, et al. Cytoprotective effects of erythropoietin: what about the lung? Biomed Pharmacother 2021; 139: 111547. doi: 10.1016/j.biopha.2021.111547 [DOI] [PubMed] [Google Scholar]
- 130.Rankin EB, Biju MP, Liu Q, et al. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest 2007; 117: 1068–1077. doi: 10.1172/JCI30117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kobayashi H, Liu Q, Binns TC, et al. Distinct subpopulations of FOXD1 stroma-derived cells regulate renal erythropoietin. J Clin Invest 2016; 126: 1926–1938. doi: 10.1172/JCI83551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol 2020; 21: 268–283. doi: 10.1038/s41580-020-0227-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Choudhry H, Harris AL. Advances in hypoxia-inducible factor biology. Cell Metab 2018; 27: 281–298. doi: 10.1016/j.cmet.2017.10.005 [DOI] [PubMed] [Google Scholar]
- 134.Daly LA, Brownridge PJ, Batie M, et al. Oxygen-dependent changes in binding partners and post-translational modifications regulate the abundance and activity of HIF-1α/2α. Sci Signal 2021; 14: eabf6685. doi: 10.1126/scisignal.abf6685 [DOI] [PubMed] [Google Scholar]
- 135.Gunter J, Ruiz-Serrano A, Pickel C, et al. The functional interplay between the HIF pathway and the ubiquitin system – more than a one-way road. Exp Cell Res 2017; 356: 152–159. doi: 10.1016/j.yexcr.2017.03.027 [DOI] [PubMed] [Google Scholar]
- 136.Bartoszewski R, Moszynska A, Serocki M, et al. Primary endothelial cell-specific regulation of hypoxia-inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J 2019; 33: 7929–7941. doi: 10.1096/fj.201802650RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ottolenghi S, Zulueta A, Caretti A. Iron and sphingolipids as common players of (Mal)adaptation to hypoxia in pulmonary diseases. Int J Mol Sci 2020; 21: 307. doi: 10.3390/ijms21010307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Malkov MI, Lee CT, Taylor CT. Regulation of the hypoxia-inducible factor (HIF) by pro-inflammatory cytokines. Cells 2021; 10: 2340. doi: 10.3390/cells10092340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yoo S, Takikawa S, Geraghty P, et al. Integrative analysis of DNA methylation and gene expression data identifies EPAS1 as a key regulator of COPD. PLoS Genet 2015; 11: e1004898. doi: 10.1371/journal.pgen.1004898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hirota K. An intimate crosstalk between iron homeostasis and oxygen metabolism regulated by the hypoxia-inducible factors (HIFs). Free Radic Biol Med 2019; 133: 118–129. doi: 10.1016/j.freeradbiomed.2018.07.018 [DOI] [PubMed] [Google Scholar]
- 141.Lee JH, Cho MH, Hersh CP, et al. IREB2 and GALC are associated with pulmonary artery enlargement in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2015; 52: 365–376. doi: 10.1165/rcmb.2014-0210OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ghosh MC, Zhang DL, Jeong SY, et al. Deletion of iron regulatory protein 1 causes polycythemia and pulmonary hypertension in mice through translational derepression of HIF2α. Cell Metab 2013; 17: 271–281. doi: 10.1016/j.cmet.2012.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Peyssonnaux C, Nizet V, Johnson RS. Role of the hypoxia inducible factors HIF in iron metabolism. Cell Cycle 2008; 7: 28–32. doi: 10.4161/cc.7.1.5145 [DOI] [PubMed] [Google Scholar]
- 144.Oktay Y, Dioum E, Matsuzaki S, et al. Hypoxia-inducible factor 2α regulates expression of the mitochondrial aconitase chaperone protein frataxin. J Biol Chem 2007; 282: 11750–11756. doi: 10.1074/jbc.M611133200 [DOI] [PubMed] [Google Scholar]
- 145.Cloonan SM, Mumby S, Adcock IM, et al. The ‘iron’-y of iron overload and iron deficiency in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2017; 196: 1103–1112. doi: 10.1164/rccm.201702-0311PP [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lando D, Pongratz I, Poellinger L, et al. A redox mechanism controls differential DNA binding activities of hypoxia-inducible factor (HIF) 1α and the HIF-like factor. J Biol Chem 2000; 275: 4618–4627. doi: 10.1074/jbc.275.7.4618 [DOI] [PubMed] [Google Scholar]
- 147.Kietzmann T, Gorlach A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin Cell Dev Biol 2005; 16: 474–486. doi: 10.1016/j.semcdb.2005.03.010 [DOI] [PubMed] [Google Scholar]
- 148.Gkotinakou IM, Befani C, Simos G, et al. ERK1/2 phosphorylates HIF-2α and regulates its activity by controlling its CRM1-dependent nuclear shuttling. J Cell Sci 2019; 132: jcs225698. doi: 10.1242/jcs.225698 [DOI] [PubMed] [Google Scholar]
- 149.Wolin MS, Ahmad M, Gupte SA. Oxidant and redox signaling in vascular oxygen sensing mechanisms: basic concepts, current controversies, and potential importance of cytosolic NADPH. Am J Physiol Lung Cell Mol Physiol 2005; 289: L159–L173. doi: 10.1152/ajplung.00060.2005 [DOI] [PubMed] [Google Scholar]
- 150.Kassim SY, Fu X, Liles WC, et al. NADPH oxidase restrains the matrix metalloproteinase activity of macrophages. J Biol Chem 2005; 280: 30201–30205. doi: 10.1074/jbc.M503292200 [DOI] [PubMed] [Google Scholar]
- 151.Seimetz M, Sommer N, Bednorz M, et al. NADPH oxidase subunit NOXO1 is a target for emphysema treatment in COPD. Nat Metab 2020; 2: 532–546. doi: 10.1038/s42255-020-0215-8 [DOI] [PubMed] [Google Scholar]
- 152.Nagaraj C, Tabeling C, Nagy BM, et al. Hypoxic vascular response and ventilation/perfusion matching in end-stage COPD may depend on p22phox. Eur Respir J 2017; 50: 1601651. doi: 10.1183/13993003.01651-2016 [DOI] [PubMed] [Google Scholar]
- 153.Rastogi R, Geng X, Li F, et al. NOX activation by subunit interaction and underlying mechanisms in disease. Front Cell Neurosci 2016; 10: 301. doi: 10.3389/fncel.2016.00301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Block K, Gorin Y, Hoover P, et al. NAD(P)H oxidases regulate HIF-2α protein expression. J Biol Chem 2007; 282: 8019–8026. doi: 10.1074/jbc.M611569200 [DOI] [PubMed] [Google Scholar]
- 155.Zhang Z, Trautz B, Kracun D, et al. Stabilization of p22phox by hypoxia promotes pulmonary hypertension. Antioxid Redox Signal 2019; 30: 56–73. doi: 10.1089/ars.2017.7482 [DOI] [PubMed] [Google Scholar]
- 156.Maranchie JK, Zhan Y. Nox4 is critical for hypoxia-inducible factor 2-α transcriptional activity in von Hippel–Lindau-deficient renal cell carcinoma. Cancer Res 2005; 65: 9190–9193. doi: 10.1158/0008-5472.CAN-05-2105 [DOI] [PMC free article] [PubMed] [Google Scholar]