

Abstract
Severe COVID-19 is often associated with detrimental elevations in blood glucose, but the mechanisms are poorly understood. In this issue of Nature Metabolism, Wan et al. identify GP73 as a SARS-CoV-2-induced secreted factor that enhances hepatic glucose production and is necessary for SARS-CoV-2-associated glucose abnormalities.
The maintenance of glucose homeostasis in healthy individuals reflects a tightly controlled balance between whole-body glucose production and glucose utilization that is governed by a regulated network of circulating hormones and metabolites1. Acute and critical illness disrupts this balance and provokes hyperglycaemia due to the heightened production of pro-inflammatory mediators and counter-regulatory hormones that antagonize insulin action and stimulate excessive hepatic glucose production (HGP)2. The pathophysiology of hyperglycaemia in the context of severe viral infection has become even more significant due to the COVID-19 pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Indeed, SARS-CoV-2 infection is often associated with elevations in blood glucose that impact disease severity and clinical outcomes3, but the mechanism(s) underlying COVID-associated dysglycaemia in vivo, the target tissue(s) involved and the chronicity of disrupted glycaemic control following resolution of acute illness are poorly understood. In this issue of Nature Metabolism, Wan and colleagues4 fill an important gap in the field by identifying a new function for Golgi protein 73 (GP73) as a stress-induced secreted factor that elevates blood glucose and is required for SARS-CoV-2 associated dysglycaemia (Fig. 1).
Fig. 1 |. GP73 is a stress-induced secreted factor that increases hepatic glucose production, impairs whole-body glucose utilization and contributes to SARS-CoV-2-associated dysglycaemia.
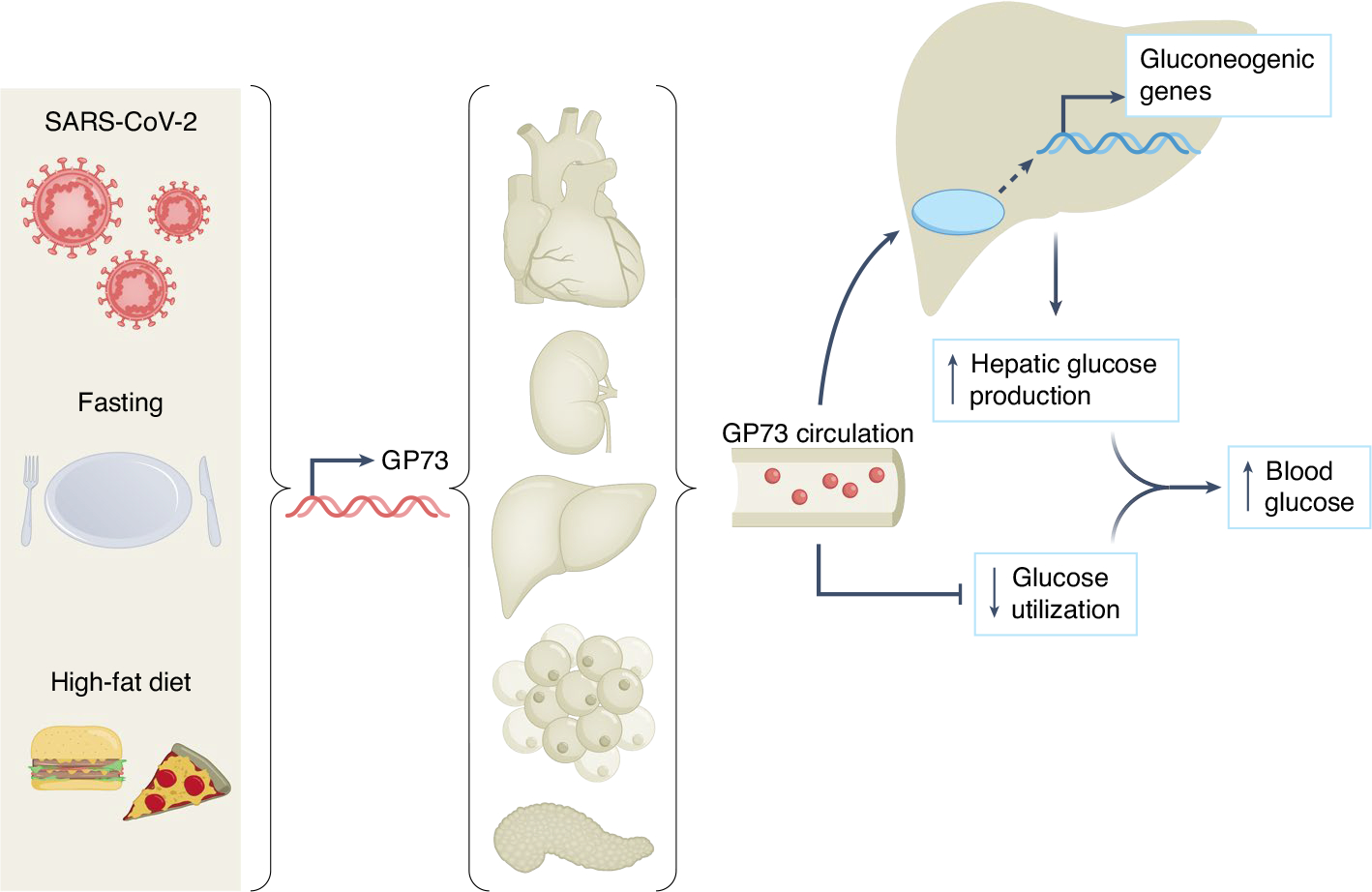
Different forms of metabolic stress, including SARS-CoV-2 infection, fasting or high-fat diet feeding, induce the expression and secretion of GP73 from a variety of tissues depending on the context. Circulating GP73 travels to the liver, where it activates PKA and its downstream signalling molecules, increases the expression of gluconeogenic genes and augments hepatic glucose release. GP73 also impairs insulin-mediated glucose utilization, which, when combined with the above, elevates blood glucose.
GP73 was initially characterized as a Golgi-localized protein that is induced in response to viral infection5. Later studies showed that both viral and non-viral liver diseases enhance the production and secretion of GP73, which may serve as a serological marker for these conditions6. Here, Wan and colleagues4 found that GP73 is induced in multiple metabolic tissues in mice upon fasting or upon high-fat diet feeding and is coordinately elevated in the circulation of mice and humans in these contexts. To assess the acute metabolic effect of circulating GP73, Wan et al.4 injected mice with a purified recombinant form of the secreted protein (rmGP73). They observed a rapid (within 15 minutes) and sharp increase in blood glucose and insulin that was abolished by administration of a GP73-specific antibody. Of note, the rapid hyperglycaemic effect of rmGP73 occurred in the absence of corresponding changes in counter-regulatory hormones (that is, glucagon, catecholamines and glucocorticoids) or liver glycogen levels, thereby arguing against a temporal effect on hepatic glycogen breakdown. Intriguingly, neutralization of endogenous circulating GP73 also lowered fasting glucose in mice, supporting a physiologic role for this factor in the maintenance of glucose homeostasis during fasting.
An increase in blood glucose may reflect either enhanced HGP or decreased glucose utilization (or both). To determine which of these processes was affected by rmGP73, Wan et al.4 performed nutrient tolerance tests in mice 2 hours after injection with phosphate-buffered saline or rmGP73. They found that rmGP73 provoked significantly greater glycaemic excursions in response to glucose, pyruvate and alanine. In the case of pyruvate, this effect was eliminated by antibody-mediated neutralization of GP73. Although these data suggest that rmGP73 enhances the ability of the liver to convert gluconeogenic substrates to glucose within 2 hours, it is unclear whether increased gluconeogenesis explains the rapid (within 15 minutes) hyperglycaemic effect of rmGP73. Furthermore, differences in glucose tolerance may reflect differences in pancreatic islet hormone secretion and action, glucose effectiveness and/or glucose utilization. To address some of these possibilities, the authors then performed hyperinsulinemic–euglycaemic clamps in GP73-null mice infused with physiologic levels of rmGP73. During the basal period, mice treated with rmGP73 exhibited significantly greater HGP than vehicle-treated controls. During the clamp, the glucose infusion rate required to maintain euglycaemia was markedly lower in rmGP73-treated mice due to impairments in insulin-mediated suppression of HGP and stimulation of glucose disposal, suggesting that circulating GP73 induces hyperglycaemia by augmenting HGP and impairing glucose utilization.
To address the mechanistic basis of these findings, the authors treated primary mouse hepatocytes with rmGP73 and observed a dose- and time-dependent increase in glucose release in association with increased levels of intracellular cAMP, activation of protein kinase A (PKA) and expression of key gluconeogenic genes (namely, Pcx, Pck1 and G6pc). Furthermore, global phosphoproteomics and kinase enrichment analyses of primary mouse hepatocytes treated with rmGP73 or glucagon revealed a marked shift toward activation of PKA and its downstream signalling molecules, indicating that GP73 and glucagon have overlapping functions at the signalling level.
Since GP73 expression was shown previously7 to be elevated in the liver of patients who died of COVID-19, the authors hypothesized that SARS-CoV-2 may have direct effects on hepatic GP73. To test this, they infected cultured human liver cells with SARS-CoV-2 and observed a robust dose- and time-dependent increase in GP73 expression and secretion, along with increased PKA activation, gluconeogenic gene expression and glucose release. Importantly, these effects were blocked by a GP73-specific antibody. A key finding of the paper was identification of an elevation in circulating GP73 in patients with COVID-19 that averaged twofold and positively correlated with their blood glucose levels and disease severity. Indeed, exposure of human liver cells to medium conditioned with GP73-enriched serum from COVID-19 patients led to heightened activation of PKA and induction of gluconeogenic gene expression. These effects were blocked by antibody-mediated neutralization of GP73, suggesting that circulating GP73 is a pathogenic mediator of COVID-associated hyperglycaemia.
Finally, to test the relevance of their findings in vivo, the authors infected mice with a mouse-adapted strain of SARS-CoV-2, MASCp6, and observed significantly higher fasting blood glucose levels and gluconeogenic gene expression compared to those in mock-infected controls. Importantly, these hyperglycaemic effects were abolished in GP73 knockout animals or by pre-treatment with a GP73-specific antibody, providing compelling evidence in a pre-clinical model that SARS-CoV-2-induced dysglycaemia is GP73-dependent. However, transcriptional regulation of gluconeogenic genes cannot explain the rapid hyperglycaemic effect of rmGP73 noted earlier, thereby suggesting that other mechanisms are also involved.
The contribution by Wan and colleagues4 is timely and important as it advances our understanding of the mechanism(s) underlying COVID-associated dysglycaemia. In addition, their findings shed light on a novel function for GP73 as a glucagon-like ‘hormone’ that activates gluconeogenic signalling molecules and participates in the regulation of glucose homeostasis. Caveats and questions that warrant further investigation include the following. (1) What is the receptor for GP73? (2) Do the metabolic effects of exogenously administered rmGP73 in healthy animals reflect the function of the endogenously secreted protein in a pathophysiologic context? (3) Are GP73-dependent disruptions in glucose homeostasis transient and reversible following resolution of COVID-19? (4) How does GP73 affect insulin action in extra-hepatic tissues? (5) And how is GP73 expression and secretion regulated in such disparate contexts? Future studies are needed to address these questions and others, and to clarify the relevance of these findings to humans with COVID-19 and/or non-COVID-related illnesses.
Acknowledgements
Work by the author is supported by DK117147 and DK20593 (Vanderbilt Diabetes Research and Training Center).
Footnotes
Competing interests
The author declares no competing interests.
References
- 1.Cherrington AD Diabetes 48, 1198–1214 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Jivanji CJ, Asrani VM, Windsor JA & Petrov MS Mayo Clin. Proc. 92, 762–773 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Khunti K et al. Diabetes Care 44, 2645–2655 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wan L et al. Nat. Metab. 10.1038/s42255-021-00508-2 (2021). [DOI]
- 5.Kladney RD et al. Gene 249, 53–65 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xia Y et al. Cell Prolif. 52, e12538 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nie X et al. Cell 184, 775–91.e14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]