

Abstract
Anti-PD-1 (aPD1) immunotherapy reinvigorates CD8 T cell responses in cancer patients. In addition to tumor-infiltrating T cells, PD-1 is also expressed by other immune cells including follicular helper CD4 T cells (Tfh) involved in germinal center (GC) responses. Little is known, however, about the effects of anti-PD-1 immunotherapy on non-cancer immune responses in humans. To investigate this question, we examined the impact of aPD1 immunotherapy for cancer on the Tfh-B cell axis responding to unrelated viral antigens. Following influenza vaccination, a subset of adults receiving aPD1 had more robust circulating Tfh (cTfh) responses than adults not receiving immunotherapy, and cTfh responses correlated with plasmablasts. PD-1 pathway blockade also resulted in transcriptional signatures of increased cellular proliferation in cTfh and responding B cells compared to controls. Moreover, aPD1 therapy was associated with a higher seroconversion rate after immunization, although some differences in antibody glycosylation and affinity were evident at baseline. Furthermore, a subset of participants with robust changes in cTfh were enriched for previous (or future) immune related adverse events (irAEs) associated with immunotherapy. These latter observations suggest an underlying change in the Tfh-B cell and GC axis in a subset of immunotherapy patients that may predispose to autoreactivity and also highlight analytical vaccination as an approach that may reveal underlying immune predisposition to adverse events. Together, these results demonstrate dynamic effects of aPD1 therapy on influenza vaccine responses and provide a framework for dissecting the impact of immune modulating therapies on overall immune health.
One sentence summary:
Anti-PD1 immunotherapy dynamically affects influenza vaccine-induced immune responses.
Cancer immunotherapy has revolutionized our ability to target and redirect immune system activity to treat malignancies. Programmed Death-1 (PD-1, CD279) expressed on T cells during activation and exhaustion is the most common target of cancer immunotherapy1. Blockade of PD-1 signaling using anti-PD1 (aPD1) or aPD-L1 immunotherapy has been associated with improved survival or cure in several different types of malignancies, in part due to the reinvigoration of exhausted CD8 T cells2. However, PD-1 is expressed by other immune cells including T follicular helper CD4 cells (Tfh) that provide help to B cells in the germinal center (GC) thereby enabling affinity-matured, long-lived antibody responses3,4. Indeed, Tfh cells in secondary lymphoid tissue have high PD-1 expression in humans3,5–7. Little is known, however, about the effects of aPD1 immunotherapy on Tfh or on related events including the outcomes of Tfh-B cell interactions and GC-dependent immune responses. Moreover, approximately 50% of patients on aPD1 immunotherapy develop immune-related adverse events (irAE)8–11. At least some of these irAEs have been postulated to be linked to autoreactive humoral responses12–14, but precise mechanisms remain poorly defined. Thus, there is a need to better understand the immunological effects of aPD1 on parts of the immune system not directly related to the on-tumor effects.
The main goal of immunotherapy for cancer or other diseases is to treat the tumor or affected tissue or system. As a result, the impact of immunotherapies on immune responses unrelated to disease have received less attention. For example, the Tfh-B cell axis is central for vaccine responses, yet the effects of aPD1 therapy on vaccine-induced Tfh and humoral responses have not been extensively examined in humans. Indeed, examining immune responses to vaccine antigens in the setting of PD-1 blockade may provide insights into the function of PD-1 in the “normal” immune system and potentially reveal new opportunities to improve vaccination or understand the role of the PD-1 pathway in other diseases. A small number of studies have evaluated influenza vaccines, mainly for safety or serology in the setting of aPD1 immunotherapy15–20. However, no information exists on how perturbing the PD-1 pathway in humans impacts Tfh responses to vaccination or the Tfh-B cell axis responsible for generating high quality antibodies. To examine these questions, we used influenza vaccination in human cancer patients to directly investigate the effects of aPD1 immunotherapy on Tfh and B cell responses to non-cancer antigens. Specifically, we examined responses of the circulating subset of PD-1+ CXCR5+ CD4 cells, termed circulating Tfh (cTfh), that share phenotypic and functional characteristics of lymphoid Tfh 5,21–25. These studies demonstrate that aPD1 treatment was associated with more robust vaccine-induced cTfh responses and increased proliferation of responding cTfh and B cells including antibody secreting cells (ASC). Mechanistically, this increased cTfh and ASC proliferation was coupled to reduced transcriptional signatures of cytokine signaling including IL-2/STAT5 in cTfh and TNF/NFkB in ASC. These aPD1-induced changes in vaccine responding cTfh and B cells were associated with greater increase in titer but lower affinity and lower sialylation of neutralizing influenza-specific antibodies compared to those not receiving immunotherapy. Increases in serum CXCL13 following vaccination in the presence of aPD1 implicated altered GC activity. Finally, although vaccination does not provoke immunotherapy associated complications 17, a subset of participants with robust changes in cTfh were enriched for future (or previous) immune related adverse events (irAEs) associated with immunotherapy, suggesting an underlying change in Tfh-B cell, humoral and/or GC activity. This latter observation points to a potential common signature or baseline immune state altered by PD-1 that impacts vaccination and autoreactivity. Moreover, these studies highlight the potential to use analytical vaccination to detect these changes, possibly identifying patients with higher likelihood for irAEs. Together, these data demonstrate that anti-PD1 immunotherapy has broad effects on the Tfh-B cell axis with major implications for vaccination, humoral responses and changes in baseline immune health.
Results
aPD1 is associated with increased cTfh and plasmablast activity following influenza vaccination
Anti-PD-1 (aPD1) immunotherapy with nivolumab or pembrolizumab is given over months or even years 1 with the intention of enhancing the immune response to cancer. The effects of long-term disruption of the PD-1 pathway on immune responses to other antigens including humoral responses to vaccines that involve PD-1-expressing Tfh remain poorly understood. To address this question, we enrolled adults with renal cell carcinoma or urothelial carcinoma who were receiving immunotherapy (Cohort 1, Supplemental Table 1) and were due to receive seasonal inactivated influenza vaccine. Participants were divided into two groups: non-aPD1-based therapy (n=10, median age=65) or aPD1-based therapy (n=29, median age=71.5). In a second, independently-generated cohort at a different institution, we enrolled adults with melanoma receiving immunotherapy (n=30, median age=61.5) and healthy adults not receiving immunotherapy (n=27, median age=33) (Cohort 2, Supplemental Table 2). Participants received influenza vaccination on the same day as one of their maintenance immunotherapy infusions (median of cycle 12 of aPD1 for Cohort 1 and cycle 7 for Cohort 2; Extended Data Fig. 1a,b). Blood was drawn on the day of vaccination (baseline), one week later, and again 3–6 weeks after vaccination (late) (Extended Data Fig. 1a), as has been done previously21,22,25,26.
In healthy adults, CXCR5+ PD-1+ cTfh expressing the activation markers Inducible Costimulator (ICOS, CD278) and CD38 expand one week after influenza vaccination and this population contains influenza-specific CD4 T cells25. We therefore first asked whether aPD1 immunotherapy alters the cTfh response to the vaccine. To avoid bias of using PD-1 itself in detection of cTfh due to treatment with therapeutic anti-PD-1 antibodies, we used a broader definition of non-naive CXCR5+ CD4 T cells that coexpressed ICOS and CD38 (ICOS+CD38+ cTfh) to identify responding cTfh in this study (Extended Data Fig. 1c-e). Following vaccination in Cohort 1, there was a 2.6-fold induction of ICOS+CD38+ cTfh in cancer patients receiving aPD1 compared to a 1.1-fold change in cancer patients on therapies other than aPD1 (P=0.013; Wilcoxon test, Fig. 1b), demonstrating that aPD1 therapy is associated with augmented cTfh responses to influenza vaccination.
Figure 1. aPD1 treatment is associated with enhanced cTfh, B cell and GC responses following influenza vaccination.
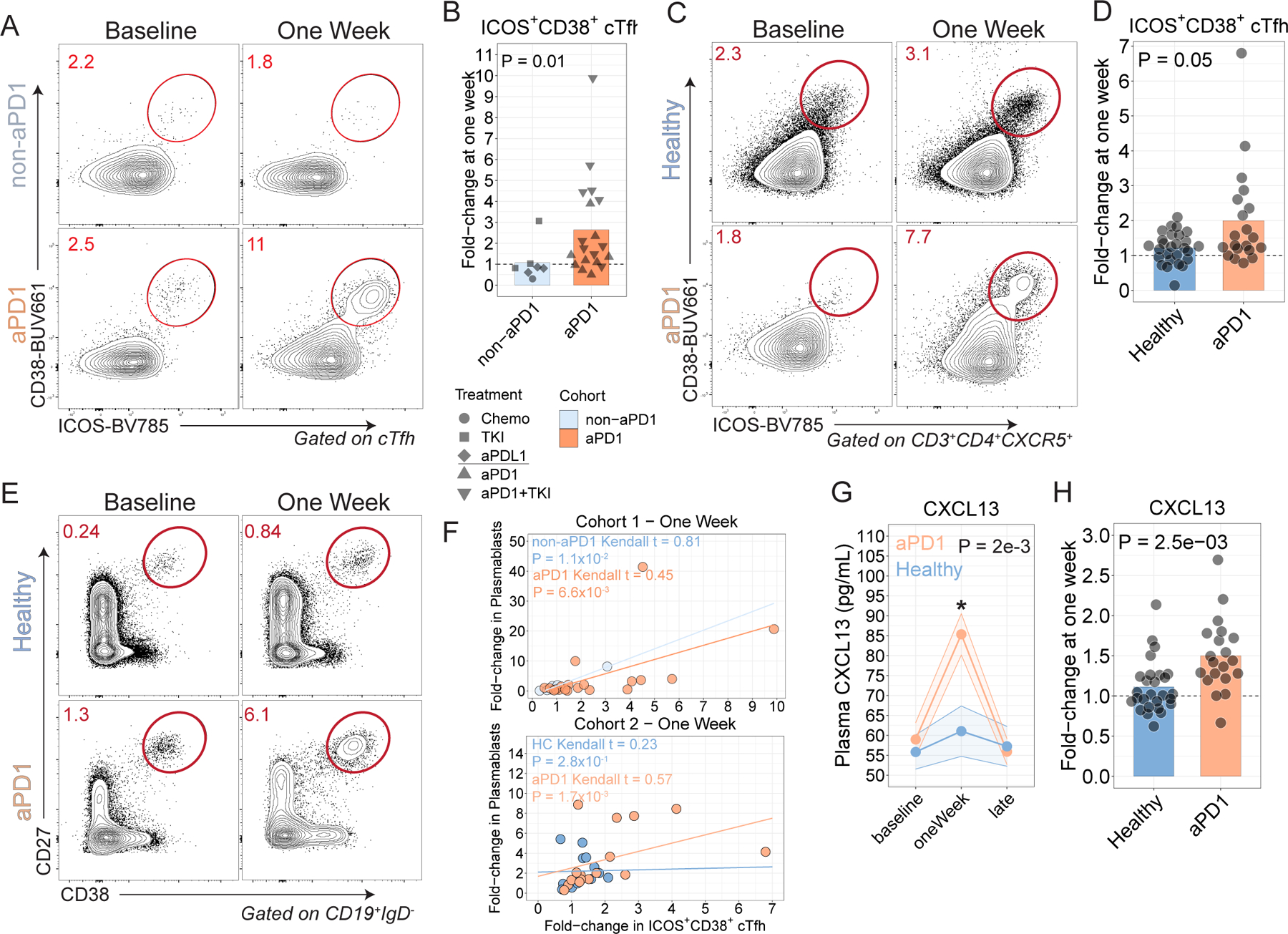
A. cTfh were profiled for expression of ICOS and CD38 at baseline and at one week after influenza vaccine in Cohort 1. B. ICOS+CD38+ cTfh fold-change at one week in Cohort 1 (P=0.01; Wilcoxon test). C. Patients with melanoma were recruited in Cohort 2 and profiled following influenza vaccination. cTfh responses shown at baseline and one week. D. ICOS+CD38+ cTfh fold-change at one week compared to baseline in Cohort 2 (P=0.05; Wilcoxon test). E. Plasmablast responses after influenza vaccination in Cohort 2. F. Correlation between Plasmablasts and ICOS+CD38+ cTfh at one week for Cohorts 1 and 2. G. Plasma CXCL13 over time (P=2x10−3 for aPD1 oneWeek compared to baseline; two-way ANOVA with Tukey’s post-test). H. Plasma CXCL13 fold-change at one week in Cohort 2 (P=2.5x10−3; two-sample t-test).
We examined an independent cohort of melanoma patients receiving aPD1 immunotherapy (Cohort 2, Supplemental Table 1). Again, aPD1 therapy was associated with more robust increases in cTfh responses compared to healthy control participants one week after vaccination (P=0.05, Wilcoxon test), with responses returning to baseline by the late time point (Fig. 1c,d; Extended Data Fig. 1f-h). We also confirmed that using PD-1 in the definition of cTfh also resulted in similar observations (Extended Data Fig. 1i-k). The increase in the cTfh response was not associated with more recent initiation of aPD1 therapy (Extended Data Fig. 1l) and was unlikely to be due to age-related differences between participants (Extended Data Fig. 1m,n). Further subsetting based on CCR6 and CXCR3, as described previously23, did not identify differences between treatment groups (Extended Data Fig. 1o). Finally, we considered other regulators of the B cell response. In particular, T follicular regulatory cells (Tfr) phenotypically resemble Tfh but express Foxp3 and can regulate Tfh-B cell interactions27. There was a greater decrease in the frequency of circulating Tfr in the aPD-1 treated participants than in the healthy adults in Cohort 2 (Extended Data Fig. 1p-r). However, this change could reflect dilution of cTfr by the ICOS+CD38+ CXCR5+ cTfh or perhaps due to altered kinetics. Given the structure of the study, it was not possible to assess cTfh and cTfr responses at different time points. Nonetheless, these data indicate that aPD1 treatment was associated with an augmented numerical response of cTfh in a subset of patients one week after influenza vaccination.
We next asked if the B cell response to vaccination was affected by aPD1 therapy. Although the plasmablast frequency was not higher in the aPD1 group following vaccination, there was a subset of aPD1 participants with robust induction of plasmablasts one week after vaccination, (Fig. 1e, Extended Data Fig. 2a-d). Correlations have been observed previously between plasmablast and cTfh responses to influenza vaccination22,25. Here, the plasmablast response correlated with ICOS+CD38+ cTfh responses one week after vaccination more strongly in the aPD1 group in both Cohorts than with either non-aPD1 or healthy control participants (Fig. 1f). These data suggest that aPD1 therapy alters Tfh-B cell interactions, likely in secondary lymphoid tissues, resulting in higher frequencies of both cTfh and plasmablasts in the peripheral blood following vaccination.
One biomarker of GC activity in lymphoid tissue is plasma CXCL1328. We observed weak induction of plasma CXCL13 in healthy adults one week after influenza vaccination (Fig. 1g), consistent with previous studies showing that influenza vaccination induced only modest changes in this chemokine detectable in blood thought to be related to the relatively weak immune response to this vaccine28. In contrast, however, aPD1-treated participants had substantial induction of plasma CXCL13 one week after vaccine compared to baseline in both Cohorts (Fig. 1g,h; Extended Data Fig. 2e-g). Although this CXCL13 in the blood did not correlate strongly with the cTfh or plasmablast response, there was a correlation with later antibody responses (Extended Data Fig. 2h). Collectively, these data reveal a robust enhancement of vaccine-induced cTfh, plasmablast and likely GC activity in the presence of aPD1 therapy.
aPD1 is associated with altered antibody responses
Humoral responses to influenza vaccination are typically measured by an increase in hemagglutinin inhibitory (HAI) antibody titers, a correlate of protection following vaccination29. Thus, we first investigated whether aPD1 therapy impacted the induction of HAI titers. We focused on Cohort 2 because of the larger number of individuals available for analysis.
We first examined the fold-change in the HAI antibody titer at the 3–6 week time point compared to the baseline titer. Indeed, across all three strains of influenza included in the vaccine, neutralizing antibody titers increased by a median of 4-fold in the presence of aPD1 compared to 2-fold in the absence of aPD1 (Fig. 2a and Extended Data Fig. 3a). Most aPD1-treated adults had antibody titers of >1:40 following vaccination and were thus seroprotected, similar to healthy adults, indicating no clinical deficiency in the outcome of vaccination in this setting (Fig. 2b). The absolute HAI titer was similar between the aPD1 and non-aPD1 at the late time point, and difference in fold-induction reflected slightly lower baseline HAI titers in the aPD1 group than in the non-aPD1 group (Fig. 2c, Extended Data Fig. 3b). These data are consistent with a previous study showing increases in influenza-specific antibodies in patients vaccinated while on aPD1 therapy15. In Cohort 1, there were similar trends, but these differences in HAI titers in Cohort 1 did not reach statistical significance, perhaps because of a smaller number of participants, more varied treatment regimens, or the effects of prior cancer therapy (Extended Data Fig. 3c,d). In this setting, correlations between circulating Tfh or B cell responses and HAI titers were not apparent (Extended Data Fig. 3e). The differences in antibody observed in Cohort 2 were unlikely to be due to the differences in age between the two groups, since independent analyses did not reveal age-associated differences in influenza-specific neutralizing antibodies (Extended Fig. 3f). Together, these data show that the increase in cTfh, plasmablast and CXCL13 responses were associated with quantitatively increased antibody responses in the setting of aPD1 compared to participants not receiving aPD1 therapy.
Figure 2. aPD1 is associated with quantitatively similar but qualitatively worse anti-HA antibodies.
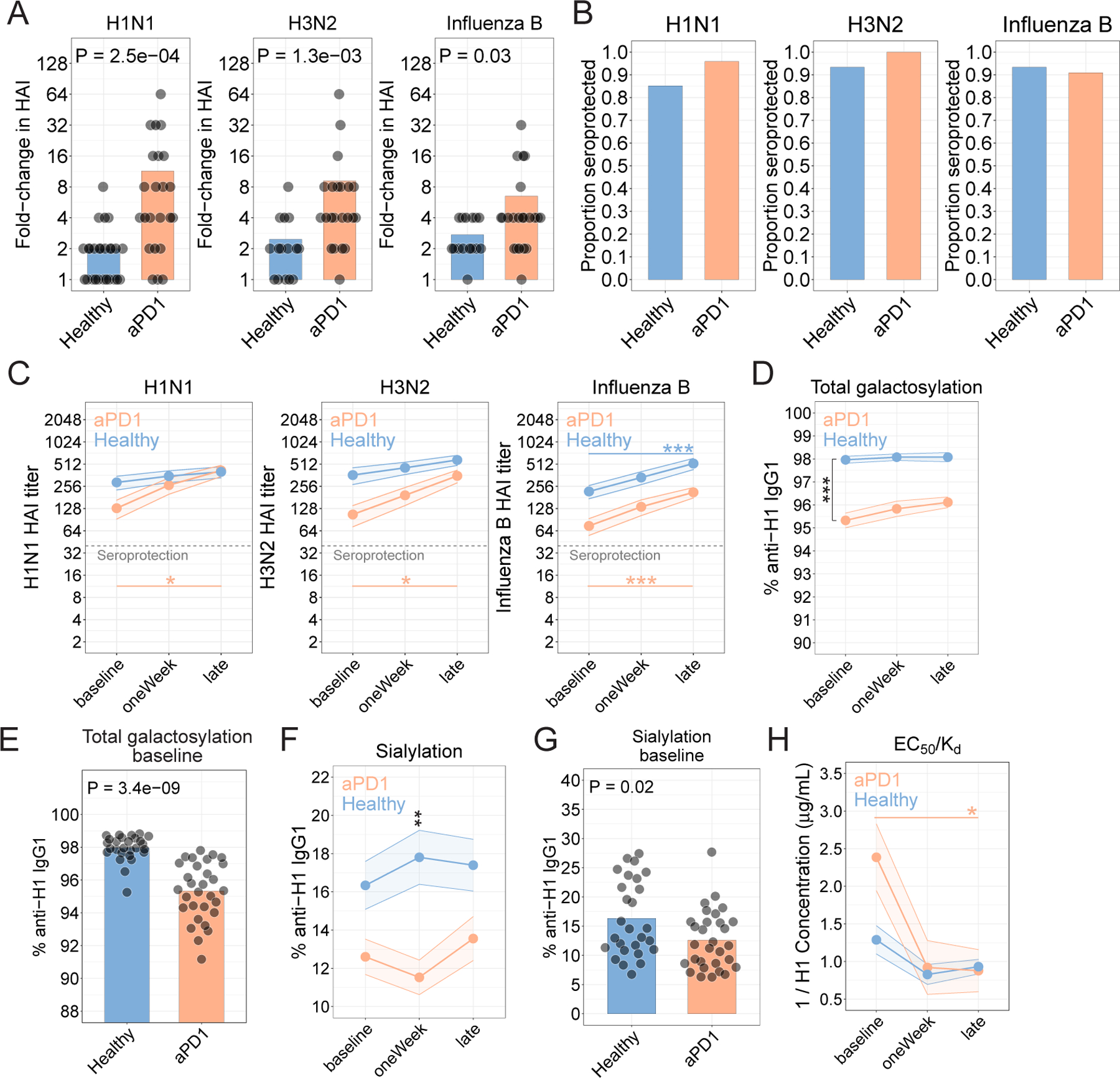
A. HAI as fold-change at the late time point compared to baseline for each strain. Nominal P values from t-test comparisons are shown. B. Seroprotection for each strain, shown as the proportion of participants who achieved an HAI titer of 1:40 or higher 21–42 days after vaccination. C. Hemagglutinin inhibition titers determined for the H1N1, H3N2, and influenza B strains. (*P<0.05, **P<0.01, ***P<0.001; two-way ANOVA with Tukey’s). D. Proportion of anti-H1 IgG1 antibodies galactosylated (***P=1.8x10−12; two-way ANOVA with Tukey’s; comparison of aPD1 vs Healthy at baseline). E. Proportion anti-H1 antibodies galactosylated at baseline (P=3.4x10−9; t-test). F. Sialylation for anti-H1 IgG1 antibodies (**P=7.1x10−3; two-way ANOVA with Tukey’s; comparison of aPD1 vs Healthy at oneWeek). G. Proportion anti-H1 antibodies sialylated at baseline (P=0.02; t-test). H. Affinity determined as EC50/Kd (*P=0.02; two-way ANOVA with Tukey’s; comparison of aPD1 at baseline vs late).
Antibodies provide protection by being present at sufficient quantities, and by undergoing qualitative improvements during the T cell-dependent phase of induction in lymphoid tissues. However, because the data above revealed a quantitative impact of aPD1 on induction of antibody following vaccination, but endpoint HAI titers were similar, we next wanted to investigate potential qualitative effects of aPD1 treatment on antibody responses. For example, antibody effector function is dictated by two important components of the antibody Fc fragment, both the amino acid sequence (e.g. IgG subclass IgG1–4) and the composition of the conserved N-linked glycan at asparagine position 29730. We previously demonstrated that changes to anti-HA glycoforms, in particular increased total sialylation of influenza-specific IgG, drove B cell affinity selection and determined the efficacy of influenza vaccination31. We therefore investigated how disruption of the PD-1 pathway impacted antibody glycosylation and subsequent affinity. We focused on the antibody response to hemagglutinin (H1), the primary target of the antibody response in vaccinated individuals32. IgG subclass and glycan distribution of antigen-specific IgG was evaluated using mass spectrometry31. Overall, anti-H1 antibodies were enriched for the IgG1 subclass, with less IgG2 and IgG3/4 in the setting of aPD1 compared to controls (Extended Data Fig. 4a, P=9x10−3, two-way ANOVA with Sidak’s post-test). We next investigated how aPD1 treatment impacted antibody glycosylation. Afucosylation of antibodies results in enhanced antibody-dependent cytotoxicity30, but was not affected by aPD1 treatment (Extended Data Fig. 4b). Sialylation and galactosylation are two additional Fc glycosylation events that can regulate antibody function. Sialylated antibodies conferred improved protection to influenza in vivo31 and galactosylation is a prerequisite for sialylation30. Indeed, sialylation and galactosylation of anti-H1 antibodies was correlated for both aPD1 treated and non-treated groups (Extended Data Fig. 4c). However, the aPD1-treated patients had lower total galactosylation (Fig. 2d,e) and sialylation (Fig. 2f,g) of anti-H1 antibodies than non-aPD1 treated participants at baseline and after vaccination, and these observations were consistent across all Ig subclasses (Extended Data Fig. 4d-f). In Cohort 1, aPD1-treated participants had lower galactosylation and sialylation of anti-H1 antibodies at baseline (Extended Data Fig. 4g-j). In previous studies, we have found similar changes in glycosylation between antigen-specific and total antibodies 31,33,34. We therefore focused here on anti-H1 antibodies because these are a major mechanism of vaccine induced protection. However, we observed similar changes in glycosylation patterns on anti-H1 antibodies of all antibody subclasses examined suggesting a broad impact on antibody glycosylation (Extended Data Fig. 4d,f). Altogether, these data identify an effect of aPD1 therapy on sialylation of anti-H1 antibodies at baseline and after vaccination, effects that might be predicted to impact the quality of influenza-specific antibodies.
To test whether altered antibody glycosylation in the setting of aPD1 treatment had functional consequences, we next examined antibody affinity. Sialylation of antigen-specific IgG and immune complexes contributes to affinity maturation through FcγRIIb-mediated modulation of GC B cell selection favoring higher affinity clones31. Thus, lower sialylation in the context of aPD1 was predicted to impact subsequent antibody affinity. Indeed, baseline antibody affinity was lower in patients on aPD1 compared to Healthy controls (Fig. 2h, Extended Data Fig. 4k). Using a second assay that estimates affinity based on binding to low- versus high-density antigen31, we confirmed that aPD1-treated participants had lower affinity IgG1 at baseline (Extended Data Fig. 4l-n). Collectively, these data identify both quantitative and qualitative changes in antibody responses to influenza vaccine associated with blockade of PD-1. Because some of these changes were present prior to yearly vaccination, these data suggest perhaps an underlying impact of aPD1 therapy on influenza specific immune memory, in addition to the effects on the responses provoked by the vaccination studied here.
Given these changes in antibody, we next investigated whether aPD1 treatment was associated with changes in B cell subsets that responded to vaccination including antibody secreting cells (ASC, gated as CD19+IgD−CD71+CD20lo) and activated B cells (ABC, gated as CD19+IgD−CD71+CD20hi)35. Even before vaccination, aPD1 therapy was associated with a higher frequency of circulating ASC and reciprocally reduced ABC frequencies (Extended Data Fig. 4o,p). However, whereas ABC only weakly correlated with ICOS+CD38+ cTfh, at baseline and after vaccination, ASC frequencies were positively correlated with ICOS+CD38+ Tfh in aPD1 patients before and after vaccination (Extended Data Fig. 4q,r). Taken together, these data indicate that aPD1 treatment is associated with alterations in B cell subsets at baseline and in vaccine-induced antibody quantity and quality. Moreover, these alterations prior to vaccination suggest that treatment with aPD1 may alter the baseline Tfh-B cell and humoral immune set point in these individuals.
aPD1 therapy is associated with transcriptional changes in cTfh and B cells
To further interrogate the effects of aPD1 on immune responses to influenza vaccination, we performed transcriptional profiling on sorted ICOS+CD38+ cTfh, ABC, ASC, or naïve B cells (gated as CD19+IgD+CD27lo) from participants in Cohort 2. Sample distribution in tSNE space was driven primarily by cell subset and minimally by treatment or timing relative to vaccination (Extended Data Fig. 5a-e). However, at one week post vaccination, transcriptional differences were readily apparent in ICOS+CD38+ cTfh from aPD1-treated adults compared to ICOS+CD38+ cTfh from healthy adults including upregulation of genes such as MKI67 and ESPL1, indicating recent proliferation36, and lower expression of genes including IFI44L, OASL, and TNFRSF1B (Fig. 3a and Supplemental Table 5), suggesting reduced response to interferon signaling37,38. A similar upregulation of ESPL1, MKI67, and other cell cycle genes has also been observed in aPD-1 induced “burned-out” CD8 T cells in cancer patients39. Indeed, gene ontology (GO) term enrichment highlighted proliferation and cell cycle in the transcriptional signatures of ICOS+CD38+ cTfh from aPD1-treated participants following vaccination compared to leukocyte activation, migration, as well as cytokine production and signaling in the control group (Fig. 3b and Extended Data Fig. 5f). Indeed, Ki67 protein expression one week after vaccination correlated with the fold-change in the ICOS+CD38+ cTfh (Extended Data Fig. 5g), supporting the notion that aPD1 therapy was associated with an enhanced proliferative cTfh response after immunization.
Figure 3. aPD1 is associated with cellular proliferation and reduced IL-2/STAT5 signaling.
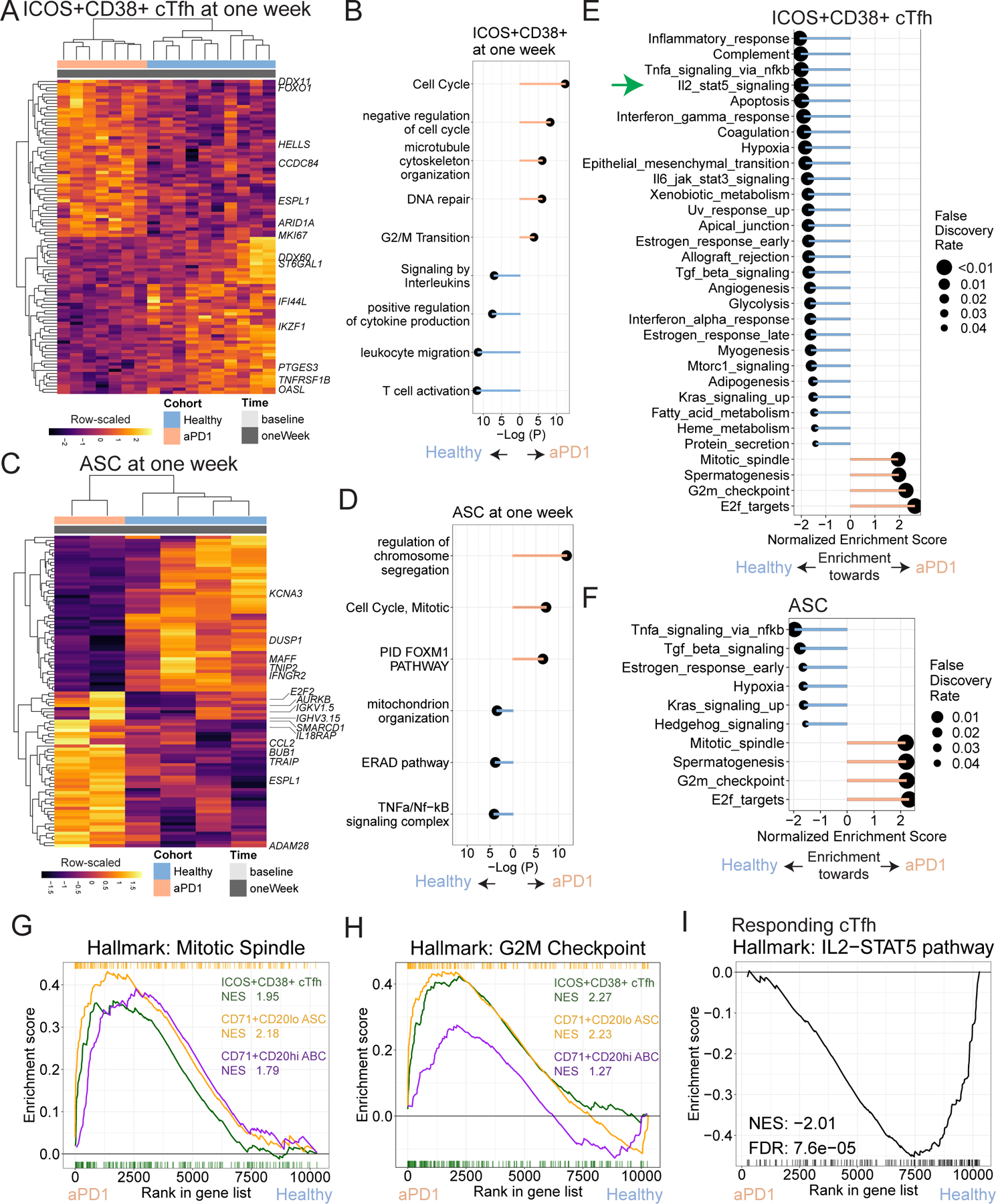
A. Top and bottom 50 genes from differential expression analysis of ICOS+CD38+ cTfh at one week after vaccination. B. Top and bottom 50 genes from differential expression analysis of ASC at one week after vaccination. C-D. Gene ontology analysis shown for genes enriched in aPD1 (peach) and those enriched in Healthy (blue) for ICOS+CD38+ cTfh (C) and ASC (D). E. Gene set enrichment analysis (GSEA) for MSigDB Hallmark gene sets used to compare ICOS+CD38+ cTfh at one week for aPD1 and Healthy. Positive enrichment scores denote enrichment towards the aPD1 cohort. F. GSEA for Hallmark gene sets for ASC at one week for aPD1 and Healthy. Positive enrichment scores denote enrichment towards the aPD1 cohort. G. GSEA shown for the Mitotic Spindle gene set for ICOS+CD38+ cTfh, ASC, and ABC at one week after vaccine. Normalized enrichment scores (NES) shown. H. GSEA for the G2M checkpoint gene set with NES is shown. I. GSEA plot shown for the IL2/STAT-5 pathway in the MSigDB Hallmark database for ICOS+CD38+ cTfh.
Similarly, ASC from vaccinated aPD1-treated participants upregulated genes such as AURKB, BUB1, and ESPL1, consistent with greater proliferation relative to ASC from healthy adults (Fig. 3c and Extended Data Fig. 5h), and had terms for “cell cycle” and “mitotic cell division” by gene ontology (Fig. 3d and Extended Data Fig. 5i). ASC from the aPD1 group also had higher expression of CCL2 and IL18RAP, but lower expression of IFNGR2, DUSP1, and KCNA3. Moreover, upregulation of ARID1A in ICOS+CD38+ cTfh and SMARCD1 in ASC from the aPD1 group also suggested a potential impact on SWI/SNF complex use in these cells responding to vaccination in the absence of normal PD-1 signaling. Although there were other differences at baseline in aPD-1-treated versus health subject activated cTfh and ASC, these proliferation-related genes were not observed prior to vaccination (Extended Data Fig. 5i,j).
We next performed gene set enrichment analysis (GSEA)40 for both cTfh and ASC to identify pathways altered due to aPD1 at one week after vaccination. For the ICOS+CD38+ cTfh, we identified four gene sets that were enriched in the aPD1 participants, all of which were associated with proliferation, whereas 27 gene sets were enriched in the control (Fig. 3e). Indeed, enrichment of gene sets associated with proliferation were also identified in the ASC and ABC subsets of aPD1 participants one week after vaccination (Fig. 3f-h, Extended Data Fig. 6a,b). These data indicate that aPD1 drove greater activation and proliferation of cTfh and B cell populations during the vaccine response.
In addition to the genes and pathways upregulated in the presence of aPD1, genes and pathways downregulated or that failed to be induced following influenza vaccination in aPD1 patients were also of interest. Indeed, aPD1 was associated with relative reduction in several pathways of direct relevance for cTfh biology in vaccine responding ICOS+CD38+ cTfh. First, the STAT5/Blimp-1 axis is a negative regulator of Tfh differentiation and function, in part through antagonism of the Tfh-driving transcription factor Bcl641,42. Here, aPD1 treatment was associated with downregulation of IL2/STAT5 signaling in activated cTfh (Fig. 3i) consistent with the enhanced cTfh response in a subset of aPD1 patients. Moreover, enrichment for the IL2/STAT5 gene set was negatively correlated with ICOS+CD38+ cTfh frequency in aPD1 patients but not healthy controls, consistent with a regulatory effect of this pathway on Tfh activation and/or expansion that is revealed by aPD1 treatment (Extended Data Fig. 6c). Other Tfh-regulating pathways, such as TGFβ signaling and Apoptosis, were also downregulated in the setting of aPD1 treatment (Fig. 3e). Downregulation of regulatory signaling pathways such as IL2/STAT5 signaling that normally impede Tfh activity in the setting of aPD1 may help explain the greater influenza vaccine-induced cTfh and antibody responses in the setting of aPD1 (Fig. 2).
A second relevant transcriptional imprint of aPD1 treatment was downregulation of genes involved in the TNF/NFkB pathway. These changes were of interest at least in part because we have recently found that TNF and NFkB signaling are regulators of cTfh biology43. In the setting of aPD1 treatment, signatures of TNF/NFkB related genes in ICOS+CD38+ cTFH were highly biased to the control participants (Fig. 3e) suggesting that aPD1 treatment impaired efficient use of TNF/NFkB signaling in vaccine responding activated cTfh. A similar picture emerged for ASC (Fig. 3f) suggesting a coordinated set of biological pathways impacted by aPD1 therapy for both cTfh and ASC. Thus, these data indicate that aPD1 may alter the humoral, cTfh and likely GC responses by dysregulating control of proliferation and altered sensing of key cytokine circuits needed for proper Tfh control of humoral immunity.
cTfh responses are predictive of immune-related adverse events
A major limitation of current checkpoint blockade in cancer immunotherapy is the development of irAE12,13. Although the underlying mechanisms have been unclear, one possibility is dysregulated GC-dependent immune responses including CD4 T cells (e.g. Tfh) and antibody responses44,45. Thus, we hypothesized that differences in Tfh biology during aPD1 therapy may offer insights into the mechanisms of irAE.
To test this idea, we focused on participants in Cohort 2 where aPD1 was used as monotherapy, and considered irAE irrespective of timing to influenza vaccination (Extended Data Fig. 7a, Supplemental Table 6). We considered whether mRNA profiling of ICOS+CD38+ cTfh at baseline could identify characteristics of those who had an irAE versus those who did not have any irAE. Patients with irAEs had slightly more transcripts for genes associated with cellular activation, such as PDCD1 and ICOS, relative to patients who did not have irAE though these differences were not statistically significant (Extended Data Fig. 7b,c). However, higher expression of ICOS protein and PD-1 protein, using αIgG4, as a proxy for PD-12, was indeed observed in aPD1-treated adults with irAE than in aPD1-treated adults who did not have irAE (Extended Data Fig. 7d-f). Transcriptional analysis of ICOS+CD38+ cTfh revealed 18 genes upregulated in activated cTfh from patients with irAE, including KIF2C, MKI67, and BIRC5 compared to patients without irAE (Fig. 4a, Extended Data Fig. 7g, Supplemental Table 7). Consistent with these proliferation-associated genes, gene ontology analysis also revealed enrichment for terms related to cell cycle and cellular activation in patients with irAEs (Extended Data Fig. 7h). Other genes of potential interest were also revealed including the cytokine IL32, the splicing factor XAB2, and CD52, the target of alemtuzumab used to treat chronic lymphocytic leukemia and multiple sclerosis46, that were all more highly expressed in activated cTfh from aPD1-treated adults with irAEs. In contrast, higher ITGA5, CD82 and FOS were found in cTfh from aPD1-treated adults without irAEs. These data indicated that, prior to influenza vaccination, ICOS+CD38+ cTfh in aPD1-treated adults with irAE were more activated than in aPD1-treated adults without irAE and may engage additional biologically relevant pathways.
Figure 4. ICOS+CD38+ cTfh activation state is associated with development of irAE.
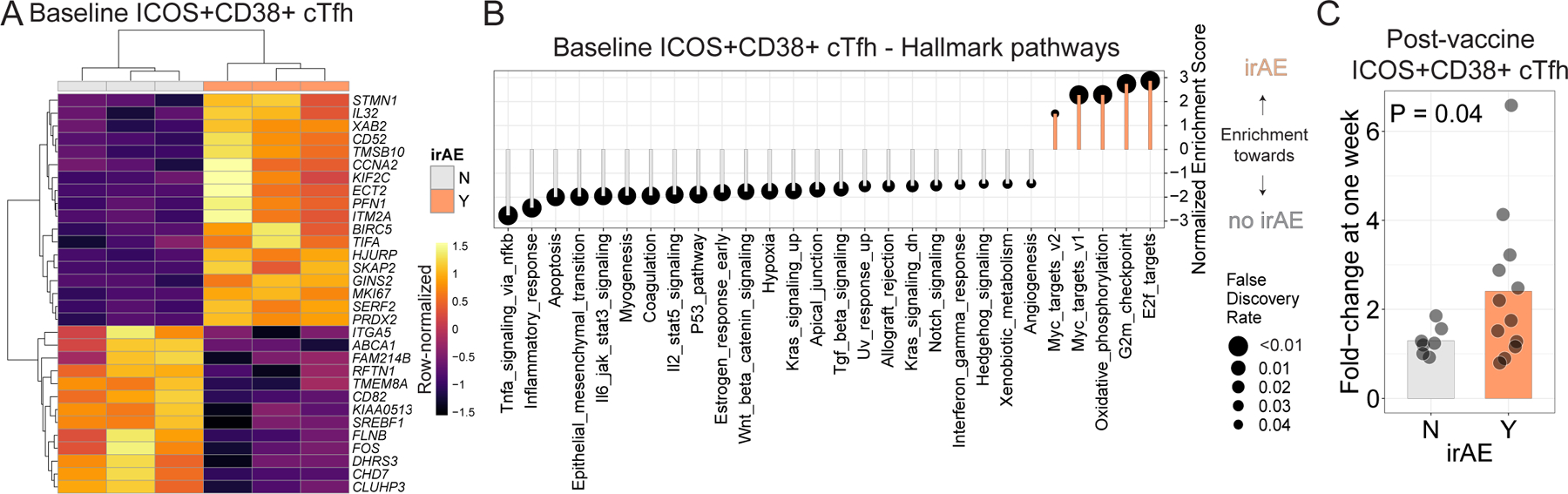
A. aPD1 participants in Cohort 2 were subgrouped based on irAE (Y=Yes, N=No). Differential expression analysis was performed on ICOS+CD38+ cTfh at baseline, and all genes with Padj < 0.05 are shown. B. GSEA for Hallmark gene sets in the setting of irAE. C. Fold-change in cTfh responses after influenza vaccine, calculated as the frequency at one week relative to baseline.
GSEA analysis identified enrichment of G2M checkpoint and E2F targets in ICOS+CD38+ cTfh at baseline from participants with irAE compared to those without irAE (Fig. 4B), further supporting the notion that ICOS+CD38+ cTfh from patients with irAE were more activated, even prior to influenza vaccination, compared to patients without irAE. This observation is consistent with other reports that identified T cell activation as predictors of irAE47. In addition to proliferation-associated pathways multiple other irAE associated biologic pathway changes in cTfh were revealed, including downregulation of IL2/STAT5, TNF/NFkB, IL6/JAK/STAT, IFN𝛾 and TGFβ signaling, consistent with the results above. Taken together, these data support the notion of an aPD1-mediated re-wiring of cTfh biology in patients with irAE that includes increased activation and proliferation concomitant with blunting of cytokine pathway signaling.
These observations provoked the hypothesis that the post-influenza vaccine cTfh response might distinguish aPD1 patients with irAEs from those without irAEs because of an underlying change in the global setpoint of Tfh regulation. To test this idea, we examined the magnitude of the ICOS+CD38+ cTfh increase one week following influenza vaccination in patients who did, or did not, have irAEs. Indeed, in patients with irAEs, influenza vaccination had a more robust ICOS+CD38+ cTfh response compared to patients without irAE (~2.4-fold compared to 1.3-fold; Fig. 4c). A similar trend was seen in patients in Cohort 1 who were receiving aPD1 monotherapy (Extended Data Fig. 7i). No difference was observed in the frequency of ICOS+CD38+ cTfh prior to vaccination (Extended Data Fig. 7j), suggesting a key role for the vaccine-induced perturbation in revealing this biology. Moreover, we did not observe increases in titers of anti-nuclear or anti-dsDNA autoantibodies with aPD1 therapy or in the setting of irAE (Extended Data Fig. 7k,l). Thus, these observations suggest that aPD1 therapy alters Tfh-B cell and likely underlying GC biology, at least in a subset of patients and that these changes can alter the magnitude and quality of the response to vaccination. The alteration in this Tfh-B cell and GC axis correlates with propensity for aPD1-associated irAEs and perhaps points to underlying rewiring of Tfh and GC biology contributing to these outcomes that may be more sensitive than general autoantibody measurements such as anti-nuclear or anti-dsDNA antibodies. Thus, using influenza vaccination as an “analytical probe” of underlying changes in immune function revealed a proliferatively enhanced, but biologically altered, response to vaccination that also identified participants predisposed to irAEs.
Discussion
Checkpoint blockade therapies including anti-PD-1 antibodies are now a major tool in the arsenal for the treatment of cancer. A significant amount of effort has focused on the in vivo effects of aPD1 therapy on the CD8 T cell response1. However, as evidenced by the global effects of PD-1 deficiency48 and the immune-related complications of aPD1 therapy, better understanding of the mechanisms of aPD1 therapy on additional immune cell types is needed. Here, we exploited influenza vaccine-induced immune responses to evaluate the effects of aPD1 on the Tfh-B cell axis. We identified evidence of increased early germinal center activation that was associated with proliferation in cTfh and responding B cells. Moreover, transcriptional profiling of ICOS+CD38+ cTfh following vaccination identified key altered signaling pathways including reduced IL2/STAT5 signaling in the setting of aPD1, and these changes were associated with quantitatively robust but qualitatively impaired antibody responses to vaccination. Finally, we demonstrated that cTfh from participants with irAE had evidence of hyperresponsiveness compared to those without irAE.
Mechanistic studies of the in vivo effects of immunomodulators such as aPD1 are challenging given limits on our ability to study immune mechanisms in humans. In previous studies, we established ICOS+CD38+ cTfh as a circulating cellular biosensor in the setting of aging49, given that many cTfh are recent emigrants from lymphoid tissue50,51. Thus, platforms that employ an immunologic stimulus, particularly one widely used in routine clinical care, offer a window of opportunity to elucidate the in vivo effects of therapeutics on the Tfh-B cell axis. Indeed, little is known about the protective effects of vaccines used clinically in cancer patients receiving immunologically-relevant therapies. In this study, aPD1 therapy was associated with augmented proliferative responses in vaccine-responding cTfh and B cell populations, based on increased circulating cell frequency, differential expression of cell cycle genes, and the coordinated expression of genes associated with the Mitotic Spindle and G2M Checkpoint using gene-set enrichment analysis. Moreover, influenza vaccination in the presence of aPD-1 augmented the induction of antibody, though antibody levels achieved became similar to Healthy adults. However, the antibodies generated in aPD-1-treated patients had reduced sialylation, a change that has implications for regulation of germinal center responses and antibody affinity maturation31. Although it is currently assumed that influenza vaccination offers similar efficacy in patients receiving aPD1 compared to patients not receiving checkpoint inhibitors, further studies will be needed to determine whether alternative vaccination strategies, such as use of adjuvants, high-dose vaccine or perhaps mRNA-based influenza vaccines, are needed to optimally protect patients.
It is worth noting that some differences observed between aPD-1 treated patients and controls were present prior to vaccination, for example in sialylation of antibodies. It is not clear whether these findings reflect differences between participant groups, or possible effects of aPD1 on other biology, such as reduction in longevity of bone marrow plasma cells. Indeed, PD-L1 is expressed on plasma cells52 but the PD-1 signaling pathway, and the effects of PD-1 blockade, have not been extensively studied in the bone marrow. One distinct aspect of using influenza vaccine for these studies is that the response to influenza vaccination likely mainly involves a recall response of previously-primed memory CD4 T cells and B cells, with perhaps a smaller newly-primed response depending on the year and strains of influenza virus in the vaccine53. Indeed, we previously observed recall responses in cTfh following successive annual vaccination25. It is worth noting that the impact of aPD1 may differ for primary versus secondary responses54 and deconvolving the primary versus recall responses in humans is complex, especially at 1 week following vaccination when both primary and recall responses would both already be clonally expanded. Thus, future studies, perhaps using antigens not previously encountered (e.g. SARS-CoV-2 mRNA vaccines) should help further dissect the role of aPD1 on pre-existing versus newly-primed CD4 T cell and B cell responses.
A major gap in our knowledge is the inability to predict cancer treatment success or failure at the time of initiation of immunotherapy. Accurately predicting these responses requires better understanding of the mechanisms by which immunotherapies work. We identified increased Tfh and B cell responses to vaccination in the setting of aPD1, and our findings are consistent with other reports of aPD1 inducing Tfh proliferation55. Although evidence supports a major role for aPD1 augmenting antitumor immunity via cytotoxic CD8 T cells2, recent studies have also suggested a link between the Tfh-B cell axis and efficacy of immunotherapy56–59. Here, we enrolled participants who were receiving immunotherapy in the form of single-agent aPD1 therapy (e.g. pembrolizumab or nivolumab). This strategy has two implications. First, the immune response to initiation of aPD1 therapy is dynamic and typically occurs within the first 1–2 cycles. Thus, the patients studied here were biased towards those in whom loss of PD-1 signaling was more established, allowing the setting of treatment-induced steady-state PD-1 deficiency to be assessed. Second, because these patients on average had received 7 or more cycles of immunotherapy corresponding to 4–6 months since initiation, they were likely enriched for participants who were having a successful clinical response. This latter effect was advantageous in allowing us to study reasonably healthy patients, but is also worth noting since perhaps not all cancer patients receiving aPD1 therapy will respond the same way to vaccines. Whether use of such analytical vaccination as a measure of immune fitness will aid in determining outcomes of cancer therapy is unclear, but these data suggest that immuno-monitoring of the Tfh-B cell axis during cancer therapy could provide additional insights about overall immune health.
Immune-related adverse events lead to morbidity and premature discontinuation of therapy but are not well-understood. Here, we found that patients with irAE had evidence of greater sensitivity to aPD1 in ICOS+CD38+ cTfh, compared to patients without irAE, all of whom were receiving aPD1. We also found that patients with irAE also had more robust influenza vaccine-induced cTfh responses relative to patients without irAE. Indeed, there is additional evidence from other studies for enhanced T cell activation in the setting of irAE47. Patients with irAE following immune checkpoint blockade often have improved anticancer responses and better survival60–62 suggesting broad re-wiring of immunity by aPD1 immunotherapy that impacts responses as diverse as those targeting tumor and self antigens. Moreover, a recent study highlighted the utility of CD4 T cell profiling as predictive for subsequent irAE and these studies identified an association between abundance of memory CD4 T cells,TCR diversity and severe irAEs63. Although the mechanisms for the increased sensitivity of cTfh and the germinal center-dependent immune responses to aPD1 identified here are not fully understood, these data are consistent with previous work identifying cTfh as sensitive readouts of underlying changes in overall immune system status43. Indeed, in the setting of immunotherapy, we hypothesize that coordination of germinal center-dependent vaccine responses may serve as a proxy for understanding broader immune response regulation in the context of aPD1 therapy. The coordinated response provoked by vaccination, however, may allow a temporally-synchronized immune response and provide a high-resolution window into the aPD1-dependent changes in underlying immune biology. Thus, the use of influenza vaccine as an analytical probe of broader immune function on aPD1 therapy revealed insights into underlying immune set point, specifically for immune features and pathways that have a role in germinal center-dependent immune responses that may include both responses to vaccination and autoimmunity..
In summary, these studies not only reveal a role for treatment with aPD1 to reset the immune landscape beyond anti-cancer responses in some patients, but also suggest potential of PD1 pathway manipulation in anti-vaccine responses, and point to activated cTfh including in the context of an analytical vaccine to identify patients at risk of irAE. The data presented here demonstrate the utility of immunoprofiling studies during a vaccine-induced perturbation as a means of uncovering mechanisms of immunomodulator therapy, which will have major implications for design and use of immunomodulator therapy.
Materials and Methods
Human subjects
Participants were recruited and consented between 2015 – 2019 at the Memorial Sloan Kettering Cancer Center (Cohort 1) and at the University of Pennsylvania (Cohort 2), in accordance with the Institutional Review Boards of each institution, also described in NCT03346772 and NCT03315975. Participants were eligible if they required influenza vaccine for routine care and had not received influenza vaccine in the prior 6 months; they were excluded if they had contraindications to influenza vaccine, or needed immunosuppressive medications (i.e. corticosteroids). Participants received influenza vaccine on the same day as their scheduled dose of aPD1. In Cohort 1, participants under age 65 received Flulaval quadrivalent vaccine (GlaxoSmithKline), whereas participants over age 65 received Fluzone High-Dose quadrivalent vaccine (Sanofi-Pasteur). In Cohort 2, Healthy adults received Fluzone quadrivalent vaccine (GlaxoSmithKline) and aPD1 adults received quadrivalent vaccine (manufacturer information unavailable).
Peripheral venous blood was drawn at baseline (day 0), at one week (days 7–10), and late (21–28 days) after seasonal inactivated influenza vaccination. Blood was collected into heparinized tubes. Plasma was isolated by centrifugation, and PBMC were isolated using Ficoll-Paque PLUS (GE Healthcare) or Sepmate (Stem Cell) tubes. Participants in influenza vaccine studies in the setting of aging were described previously21.
Flow cytometry
PBMC were stained for surface proteins for 20 minutes at room temperature. Permeabilization was performed using the Intracellular Fixation/Permeabilization Concentrate and Diluent kit (ThermoFisher) for 20 minutes at room temperature. Intracellular staining was performed for 60 minutes at room temperature. Antibodies and clones are described in Supplemental Table 3. Cells were resuspended in 1% para-formaldehyde until acquisition on a BD Symphony cytometer. Fluorescence-minus-one controls were performed in pilot studies.
RNAseq processing
PBMC were sorted on a BD Aria II cell sorter, followed by total RNA extraction by RNeasy Micro Plus kit (Qiagen) into DNA LoBind tubes (Eppendorf). Samples were transferred to a skirted twin.tec 96-well PCR plate (Eppendorf), followed by polyA amplification with SMART-Seq HT Kit (Clontech) according to manufacturer instructions. Libraries were prepared using the Nextera XT Library Preparation kit (Illumina) using the protocol modification recommended by the Clontech kit. Libraries were indexed with a unique dual indexing strategy (IDT for Illumina Nextera DNA Set A, Illumina) to account for index hopping. Sequencing was performed on an Illumina Novaseq 6000, using 51 + 10 + 10 + 51 run geometry. FASTQ files were trimmed with Trimmomatic (version 0.32) and assessed for quality with FastQC. Libraries were aligned using STAR (version 2.5.2a) and normalized by PORT (https://github.com/itmat/Normalization/, version 0.8.5) against the GRCh38 reference assembly of the human genome. One of the 89 resultant libraries was excluded from further analysis due to poor sequencing (10-fold fewer reads than next least sample).
RNAseq analysis
Variance-stabilizing transformation and differential expression were performed using DESeq2 (version 1.28.1) based on genes with at least 20 counts in at least 25% of all libraries, using the R environment (version 4.0.2). Plots were made by ggplot2 (version 3.3.2). Heatmap coloring was based on the “inferno” color-scheme from the viridis library. t-SNE maps were produced using Rtsne (version 0.15) from the variance-stabilized counts data. Gene ontology was performed using Metascape (https://metascape.com). Gene set enrichment analyses were performed with at least 10000 permutations of pre-ranked GSEA (http://software.broadinstitute.org/gsea/downloads.jsp), and gene set variation analysis64 was performed with the GSVA library (version 1.36.2).
Influenza-specific antibodies
The two influenza A vaccine strains of the 2014/2015 seasonal influenza vaccine, A/California/7/2009 (H1N1) pdm09-like virus and A/Victoria/361/2011 (H3N2)-like virus, were obtained from the Centers for Disease Control and Prevention (Atlanta, GA). Influenza B strain B/Brisbane/33/2008 was obtained from BEI Bioresources (NR-42006). Assays to detect hemagglutinin inhibition assay (HAI) antibody (Ab) titers were performed as previously31. Infectious virus was used for neutralizing Ab assays or inactivated by β-propiolactone for H1N1/California-, H3N2/Victoria-, or B/Victoria specific binding Ab ELISA assays. Nunc Maxisorp plates (Nunc) were coated with 10 mg/mL influenza A/H1N1/California, A/H3N2/Victoria, or B/Brisbane/33/2008 virus along with isotype standards for IgG (Athens Research and Technology) in bicarbonate buffer overnight at 4°C. Plates were blocked with 3% BSA in PBS and incubated with heat-inactivated sera of young and elderly adults. Abs were detected using alkaline phosphatase-conjugated mouse anti-human IgG (Southern Biotechnology).
ELISAs
HA ELISAs were performed as described previously31. Negative control sera values were subtracted from readings given by test samples. Serum antibody 50% effective concentration (EC50) levels were determined by nonlinear regression analysis of the serially diluted serum OD values, calculated GraphPad Prism. The EC50 takes into account both the concentration (maximum binding) and affinity of antibodies (e.g. the slope of the binding curve) and is an accurate measure of total binding which generally correlates with the endpoint titer65. For hi-lo affinity ELISAs, sera diluted 1:200 were incubated on plates coated with 1ug/ml HA protein (low density) or 6 mg/ml HA (high density). The affinity of HA-specific IgG was expressed as a ratio of binding to low-density:high-density HA-coated plates, as previously31.
Anti-nuclear antibody (ANA) and anti-double stranded DNA (dsDNA) screen
Anti-nuclear antibodies were detected by ELISA (BioVIsion). Here, diluted patient sera (1:21) was added to the ELISA plate coated with purified nuclear antigens. The enzyme conjugate was then added to all wells and incubated for 20 minutes at room temperature. Following removal and washing three times with wash buffer, 100 ul of TMB substrate was added and allowed to incubate for 10 minutes prior to adding 100 ul of stop solution. Optical density of the plates was measured at 450 nm. Antibody indices were calculated per manufacturer recommendations and values >0.9 were considered positive. Similarly, anti-dsDNA antibodies were detected by quantitative ELISA (Eagle Biosciences). Briefly, the antibodies of the calibrators, control and diluted patient were incubated with dsDNA immobilized on the solid phase of microtiter plates. Following an incubation period of 60 min at room temperature (RT), unbound sample components were removed, and plates were washed three times. The bound IgG antibody complexes were detected using anti-human-IgG conjugated to horseradish peroxidase (HRP) for 30 minutes at room temperature. 100 ul of TMBsolution was added to each of the wells and allowed to incubate for 10 minutes at room temperature prior to the addition of 100 ul of stop solution. The optical density (OD) of the solution was measured at 450 nm. A standard curve was established by plotting the antibody concentrations of the calibrators (x-axis) and their corresponding OD values (y-axis) measured. The antibody concentration of each specimen was then interpolated from the standard curve.
Fc Glycan Analysis
IgGs were isolated from serum by protein G purification. HA-specific IgGs were isolated on agarose resin (Pierce) coupled to HA protein. Total anti-H1 HA IgGs were captured on Cal/09-coupled resin. IgG Fc-associated glycans were analyzed by mass spectrometry following tryptic digestion of purified IgG or on-bead IgG.
Protein samples (10 µg) enriched by Protein G beads either in-solution or on-beads in 50mM Tris buffer pH 8.0 were denatured with 6M guanidine-HCl, reduced with 10 mM dithiothreitol (DTT) at 56°C for 45 minutes and alkylated with 60mM iodoacetamide for 1 hour at room temperature in the dark. Excess iodoacetamide was quenched with 20mM dithiothreitol for 30 minutes at room temperature. The samples were diluted to a final volume of 200µL with 50mM ammonium bicarbonate to reduce the Guanidine HCL concentration to less than 1M. The samples were digested by adding trypsin (0.5–1µg, Promega, Madison, WI) at a 1:20 ratio (w/w) and incubated at 37ºC for 16 hours. All samples were desalted using solid phase extraction (SPE) on Sep-Pak Cartridges (Waters, Milford, MA) and the eluted tryptic peptides were evaporated to dryness in a Speedvac SC110 (Thermo Savant, Milford, MA) before analysis.
The nanoLC-MS/MS analysis for characterization of glycosylation sites was performed on an UltiMate3000 nanoLC (Dionex, Sunnyvale, CA) coupled with a hybrid triple quadrupole linear ion trap mass spectrometer, the 4000 Q Trap (AB SCIEX, Framingham, MA). The digested samples were reconstituted in 50 µl of 0.2% formic acid, of which 2–3 µl were injected with an autosampler onto a PepMap C18 trap column (5 µm, 300 µm x 5 mm, Dionex) with 0.1% FA at 20 mL/min for 1 minute, then separated on a PepMap C18 RP nano column (3 µm, 75 µm x 15 cm, Dionex) using a 60-minute gradient of 10% to 35% ACN in 0.1% FA at 300 nL/min, followed by a 3-minute ramp to 95% ACN-0.1% FA and a 5-minute hold at 95% ACN-0.1% FA.
MS data acquisition was performed using Analyst 1.4.2 software (Applied Biosystems) for precursor ion scan triggered information dependent acquisition (IDA) analysis and enhanced MS-based IDA analysis66,67. The precursor ion scan of the oxonium ion (HexNAc+ at m/z 204.08) was monitored at a step size of 0.2 Da cross a mass range of m/z 400 to 1600 for detecting glycopeptides containing N-acetylhexosamine unit. The nanospray voltage was 1.9 kV, and was used in positive ion mode for all experiments. The declustering potential was set at 50 eV and nitrogen as collision gas. In IDA analysis, after each precursor ion scan or EMS scan, and enhanced resolution scan, the two to three highest intensity ions with multiple charge states were selected for tandem MS (MS/MS) with rolling collision energy applied for detected ions based on different charge states and m/z values. All acquired MS/MS spectra from EMS-IDA were subjected to Mascot database search. All acquired MS/MS spectra for detected glycopeptides ions by precursor ion scanning were manually inspected and interpreted with Analyst 1.4.2 and BioAnalysis 1.4 software (Applied Biosystems). The peak areas of detected precursor ions were determined by extracted chromatogram (XIC) at each specific m/z representing glycopeptides isoforms. The relative quantitations of the sugar glycan isoforms of N-linked peptide ions was carried out based on precursor ion peak areas under assumption that all sugar glycan isoforms linked to the same core peptide have identical or a similar ionization efficiency.
Statistics
Statistical analyses were performed with R (version 4.0.2) using the rstatix library (version 0.6.0) or Prism 8 (GraphPad). Data was compared using Student’s t test, paired t test, Wilcoxon test, one-way or two-way Analysis of Variance (ANOVA) with Tukey post-hoc analysis, non-parametric ANOVA with Friedman’s post-test, or Fisher’s Exact test, as indicated. All tests were performed as two-tailed tests with α = 0.05.
Data and code
Transcriptional profiling data reported here are available at the Gene Expression Omnibus (GEO accession GSE179487). R scripts used in analyses and figure generation are available on Github (https://github.com/Sedmic/PembroFluVac).
Extended Data
Extended Data Figure 1.
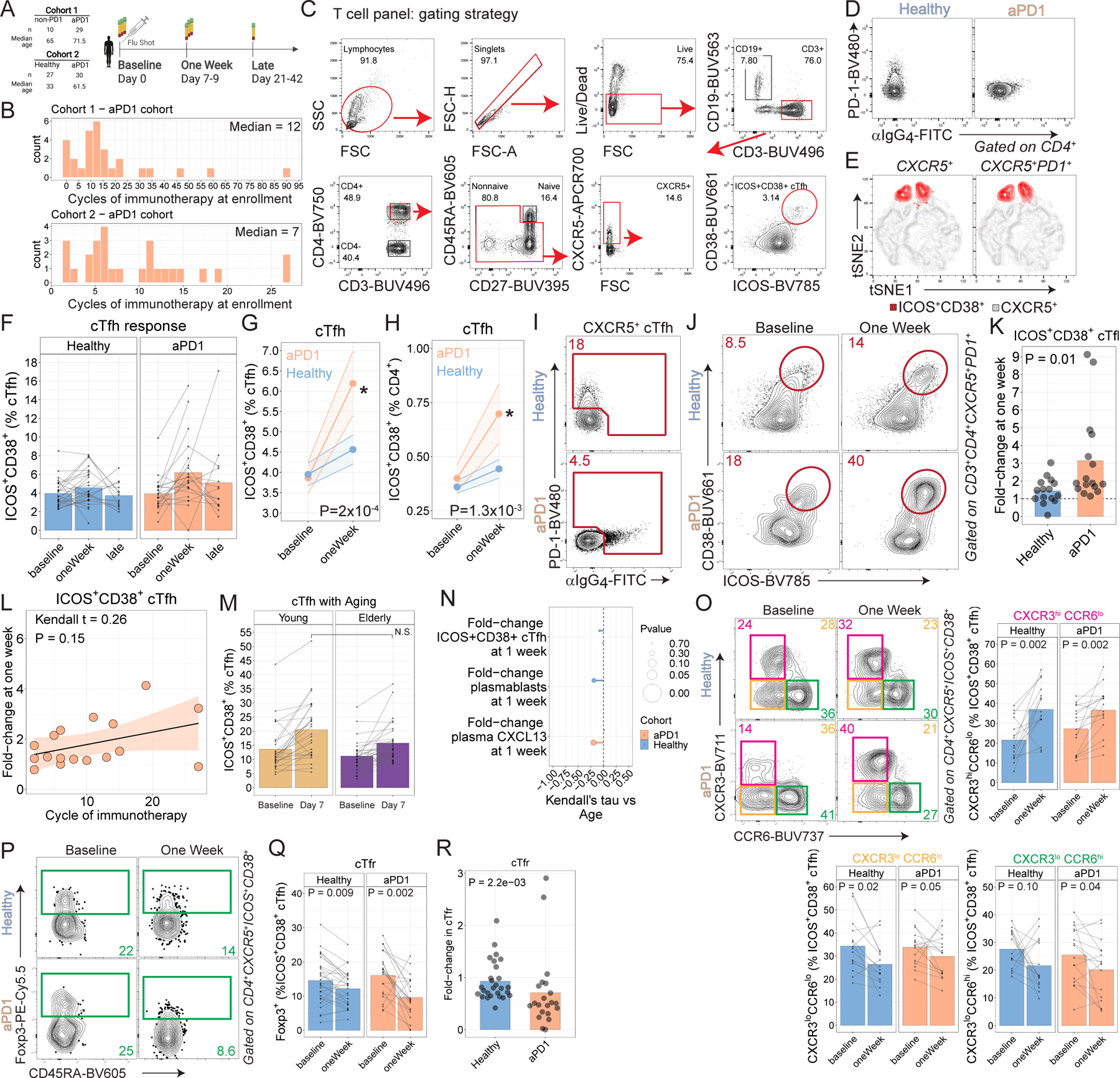
A. Study schematic. B. Histogram of cycle of immunotherapy shown for each cohort. C. Gating strategy shown for CD4 T cells. D. aPD1 binding to cells demonstrated by absence of PD-1 staining and presence of α-human IgG4 staining. E. tSNE representation showing gating on ICOS+CD38+ either directly on CXCR5+ CD4 T cells (left) or after first gating on PD-1+ (right). F. ICOS+CD38+ cTfh responses over time. Repeat measurements from the same subjects shown as connected lines. G. ICOS+CD38+ responses shown as % cTfh (P = 2x10−4 for aPD1 oneWeek compared to baseline; paired two-way ANOVA with Sidak’s correction). H. ICOS+CD38+ cTfh responses shown as % CD4 (P=1.3x10−3 for aPD1 at one week compared to baseline; paired two-way ANOVA with Sidak’s correction). I. PD-1 expression among CXCR5+ CD4 T cells was identified by either direct staining for PD-1 or by indirect staining for α-human IgG4 staining. J. ICOS+CD38+ coexpression shown after gating on CXCR5+PD-1+ as identified in panel I. K. Fold-change in ICOS+CD38+ cTfh shown after gating on PD-1 as in panel J. L. cTfh responses vs cycle of immunotherapy for the melanoma cohort (Kendall t=0.26; P=0.15). M. An independent cohort of young and elderly adults without cancer was assessed for cTfh responses following influenza vaccination. N. Correlations between the cellular responses and chronological age shown for Cohort 2. O. ICOS+CD38+ cTfh were analyzed for expression of CXCR3 and CCR6. P-values on summary plots from paired t-test comparisons. P. T follicular regulatory (Tfr) cells were identified among ICOS+CD38+ cTfh based on expression of Foxp3. Q. Summary plots for Tfr in Cohort 2, gated on ICOS+CD38+ cTfh. R. Fold-change in cTfr at one week relative to baseline for Cohort 2 (P=2.2x10−3; Wilcoxon test).
Extended Data Figure 2.
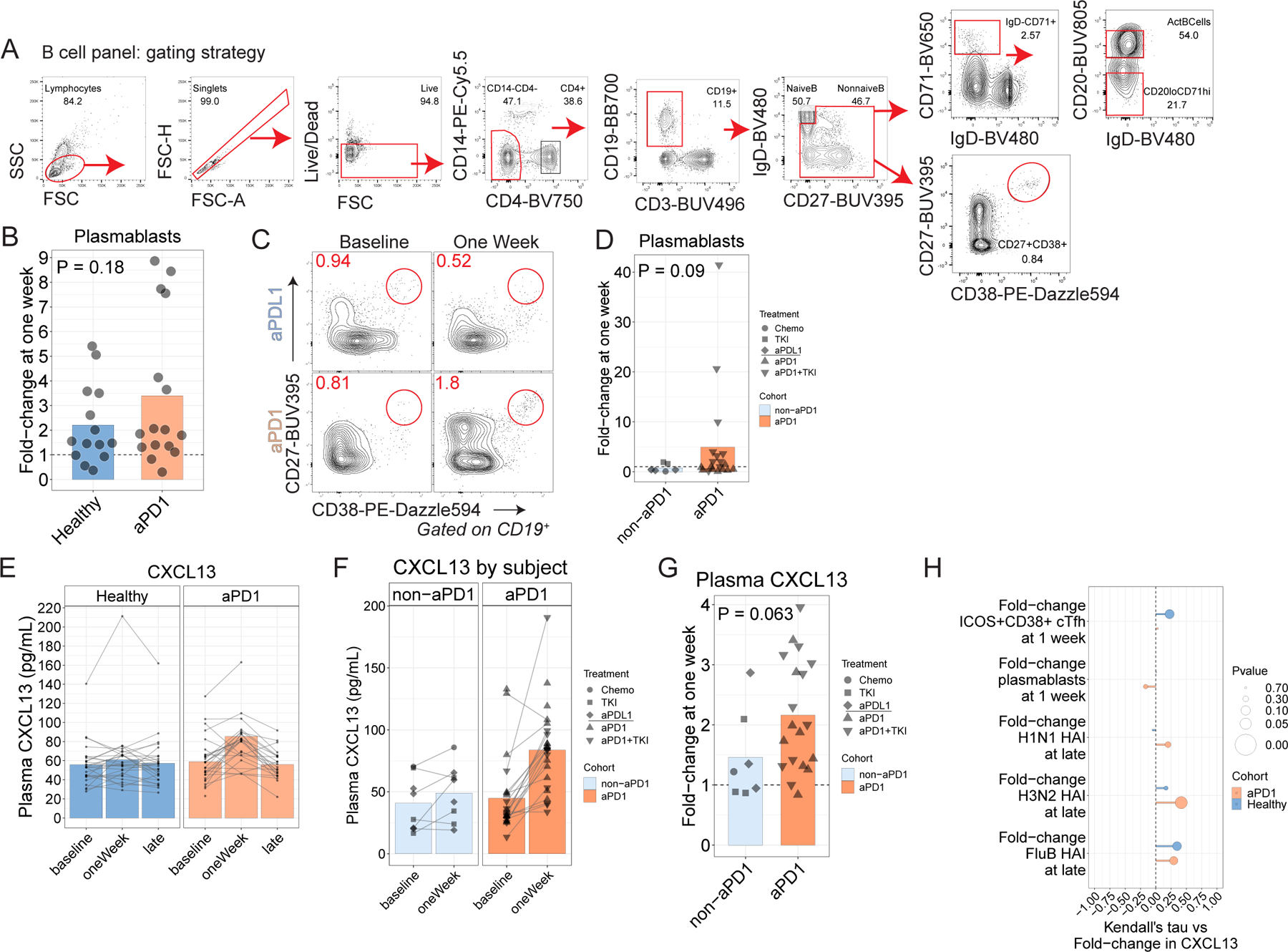
A. Gating strategy for B cell analysis. B. Plasmablast fold-change at one week compared to baseline in Cohort 1 (P=0.18; t-test). C. Plasmablast responses, gated as CD27++CD38++ nonnaive CD19+ lymphocytes in Cohort 1. D. Plasmablast responses shown as fold-change (P=0.09, t-test). N. Fold-change in plasmablast responses at one week compared to fold-change in cTfh responses at one week in Cohort 1. O. Plasma CXCL13 for individual subjects in Cohort 2. P. Plasma CXCL13 in Cohort 1. Q. Plasma CXCL13 fold-change in Cohort 1 (P=0.06; Wilcoxon test).
Extended Data Figure 3.
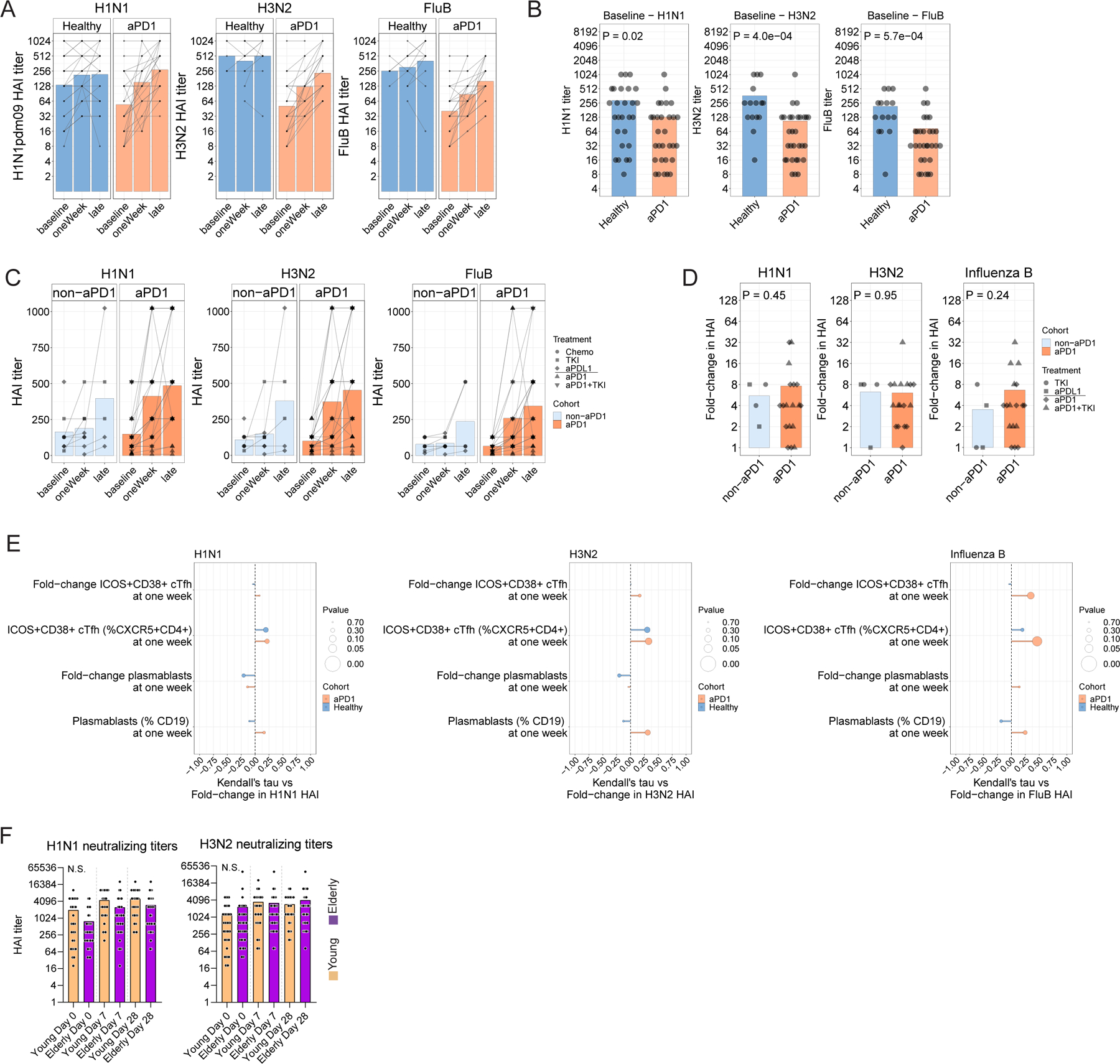
A. Hemagglutinin-inhibitory titers (HAI) for three influenza virus strains for Cohort 2. Connected lines indicate repeated measurements from the same individual over time. B. Direct comparison of HAI titers per strain at the time of influenza vaccination for Cohort 2. B. HAI titers over time for Cohort 1. Connected lines indicate repeated measurements form the same individual over time. D. Seroconversion as a fold-change at the late time point relative to baseline for Cohort 1. E. Kendall correlations shown for the cellular responses shown versus the fold-change in HAI titer for each viral strain for Cohort 2. F. HAI titers were determined in an independent cohort of young and elderly adults without cancer for H1N1 (left) and H3N2 (right) strains.
Extended Data Figure 4.
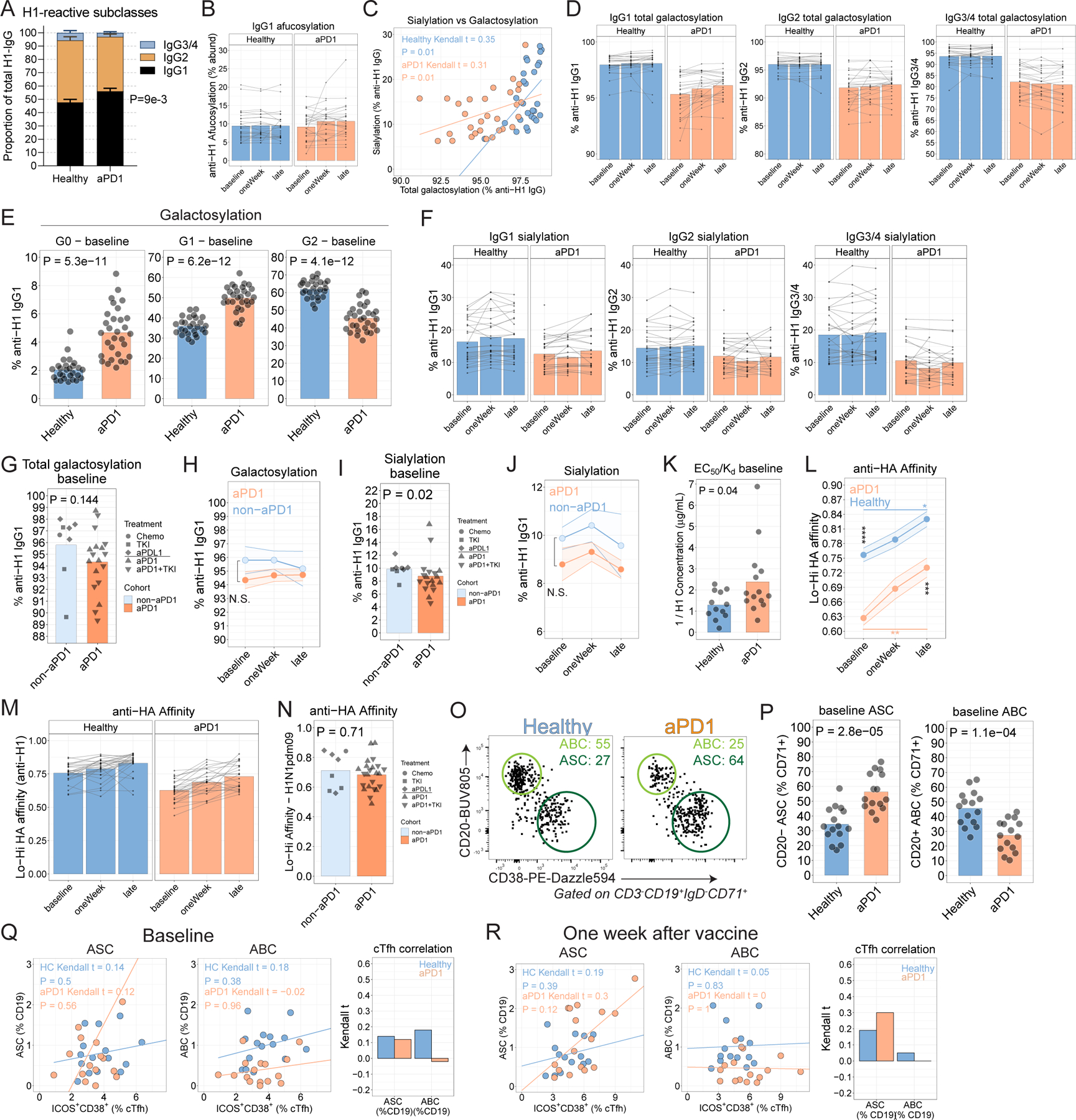
A. Plasma levels of IgG subclasses (P=9.6x10−3 for IgG1 between Healthy and aPD1; two-way ANOVA with Sidak’s correction). B. Proportion of afucosylated anti-H1 IgG1 antibodies. C. Kendall correlation between sialylated anti-HA IgG1 antibodies and proportion of galactosylated anti-H1 IgG1 antibodies for Cohort 2. D. Proportion of total (G1 + G2) galactosylated anti-H1 IgG1 antibodies by Ig subclass. E. Galactosylation states shown for no galactosylation (left), one galactose residue (middle), or two galactose residues (right). P values from Wilcoxon test. F. Sialylated anti-H1 IgG1 antibodies by subclass. G. Total galactosylation for anti-H1 IgG1 antibodies at baseline in Cohort 1. H. Total galactosylation for anti-H1 IgG1 antibodies over time in Cohort 1. I. Sialylation of anti-H1 IgG1 antibodies at baseline in Cohort 1. J. Sialylation for anti-H1 IgG1 antibodies over time in Cohort 1. K. Anti-HA affinity at baseline. L. Anti-HA affinity for Cohort 2, as determined by ELISA and calculated as a binding ratio of low-density to high-density of plate-bound HA protein (*P < 0.05, **P<0.01, ***P<0.001, two-way ANOVA with Tukey’s). M. Anti-HA affinity over time. N. EC50/Kd affinity at baseline for Cohort 2 for H1N1. P value from t-test. O. Examples of ASC and ABC at baseline. P. Baseline ASC frequencies (P=2.8x10−5; t-test) and ABC frequencies by cohort (P=1.1x10−4; t-test). Q. Kendall correlations at baseline between ICOS+CD38+ cTfh for ASC (left) or ABC (middle) for Cohort 2. Bar graph shows Kendall correlation coefficients. R. Kendall correlations at one week after vaccine between ICOS+CD38+ cTfh for ASC (left) or ABC (middle) for Cohort 2. Bar graph shows Kendall correlation coefficients.
Extended Data Figure 5.
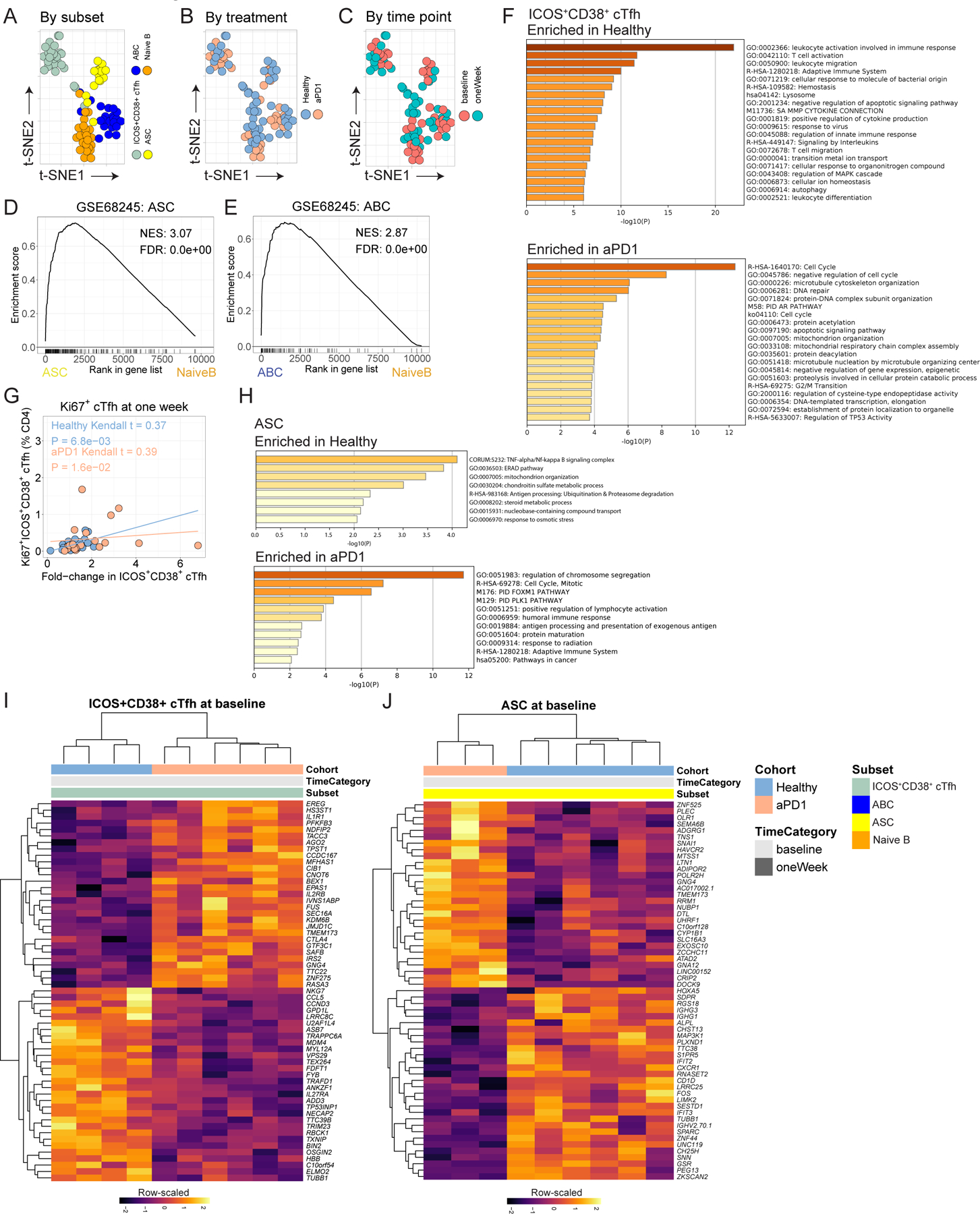
A. tSNE for cellular subsets evaluated by bulk RNAseq. B. tSNE recolored by treatment. C. tSNE plot recolored by time point. D. ASC transcriptional profile validated against GSE68245. E. ABC transcriptional profile validated against GSE68245. F. Gene ontology for differentially expressed genes at p<0.01 for ICOS+CD38+ cTfh for Healthy (top) or aPD1 (bottom). H. Gene ontology for differentially expressed genes at p<0.01 for ASC for Healthy (top) or aPD1 (bottom). I. Differential expression analysis of ICOS+CD38+ cTfh at baseline. J. Differential expression analysis of ASC at baseline.
Extended Data Figure 6.
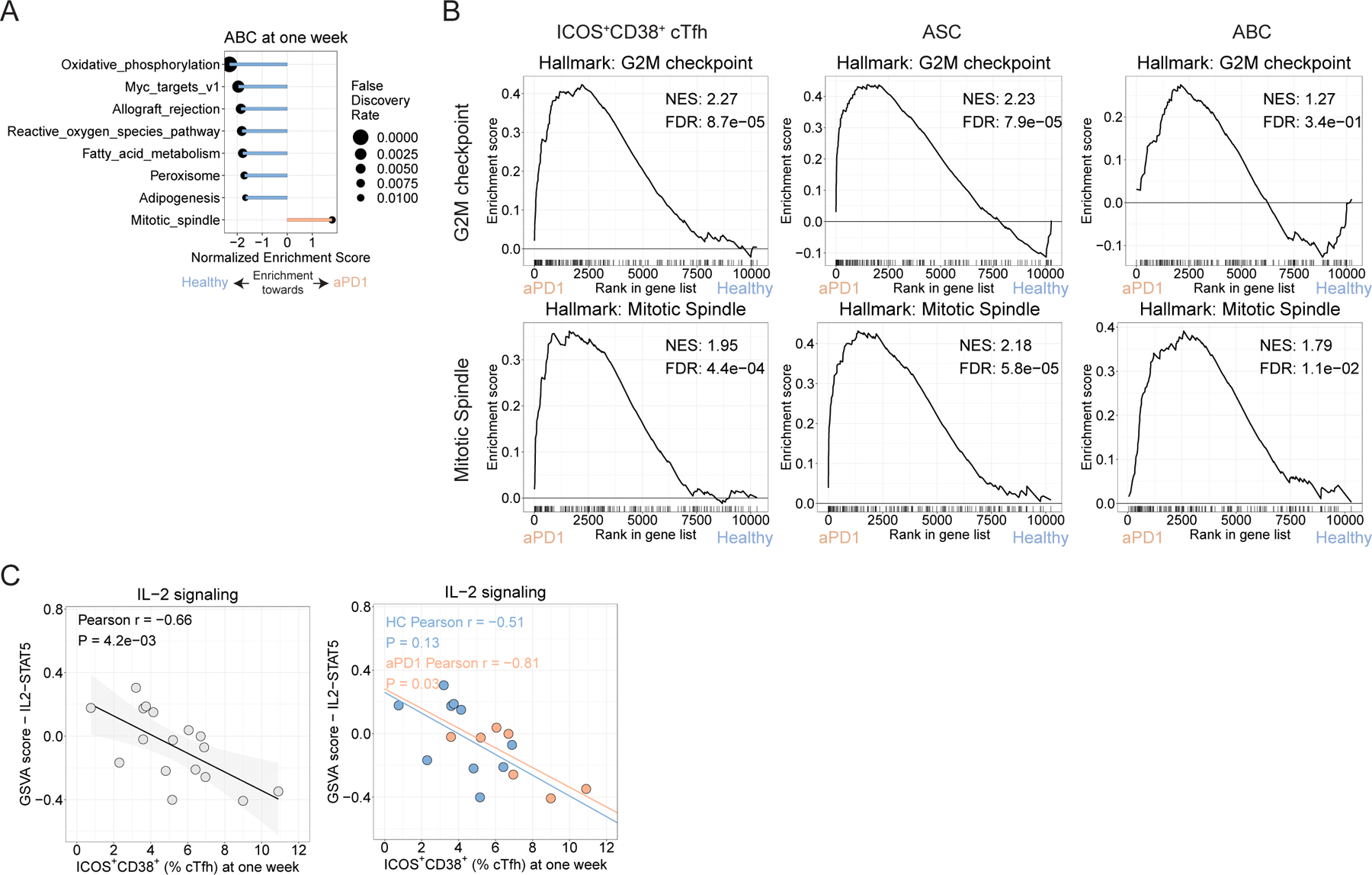
A. GSEA for Hallmark gene sets for ABC for FDR <0.05. B. GSEA plots for the G2M checkpoints and Mitotic Spindle gene sets. C. Pearson correlation for ICOS+CD38+ cTfh frequency at one week versus the GSVA score for the IL2/STAT5 gene set for ICOS+CD38+ cTfh at one week for the full study (left) and split by treatment group (right).
Extended Data Figure 7.
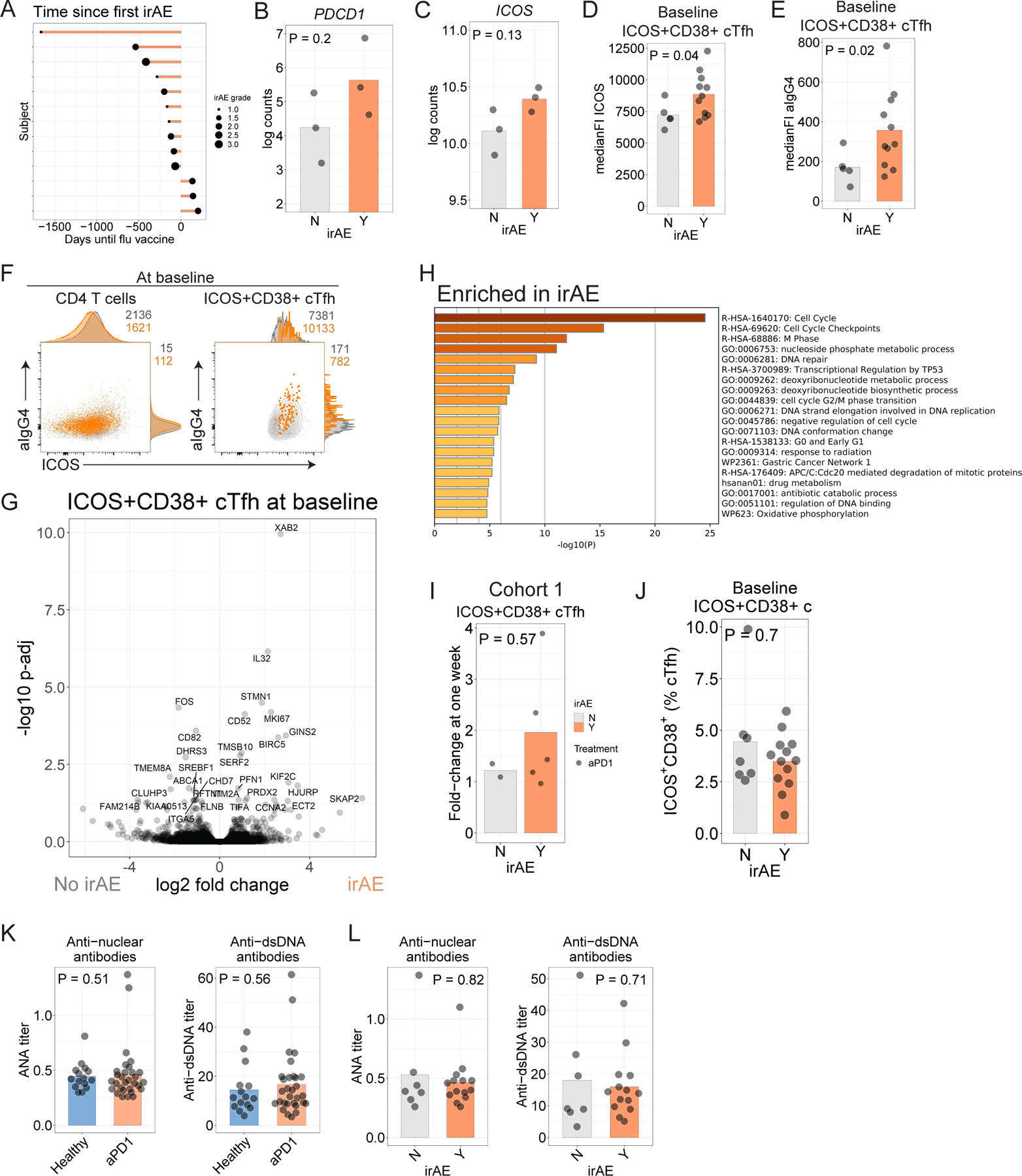
A. Time to development of irAE in Cohort 2. B-C. Log-transformed counts for PDCD1 (B) and ICOS (C). P values from t-test comparison. D. ICOS protein in ICOS+CD38+ cTfh before influenza vaccination in Cohort 2. E. αIgG4 staining in ICOS+CD38+ cTfh before influenza vaccination in Cohort 2. F. αIgG4 and ICOS protein in total CD4 (left) or ICOS+CD38+ cTfh (right) for participants who had irAE (orange) versus those who did not have irAE (grey). G. Volcano plot of genes differentially expressed in aPD1 cohort with respect to irAE. Genes labeled where Padj < 0.05. H. Gene ontology for genes differentially expressed at nominal P<0.05 for ICOS+CD38+ cTfh at baseline in participants in Cohort 2 who developed irAE. I. Fold-change in ICOS+CD38+ cTfh at one week compared to pre-vaccination baseline in Cohort 1. J. Baseline ICOS+CD38+ cTfh frequency among who developed irAE in Cohort 2. K. Plasma levels of antinuclear antibodies (ANA, left) and anti-dsDNA antibodies (right) were assessed at baseline in Cohort 2. L. Plasma ANA and anti-dsDNA antibodies compared among aPD1 participants split by irAE status.
Supplementary Material
Acknowledgements
We would like to thank the participants who volunteered for our studies, as well as members of the Wherry and Ravetch labs who provided critical feedback in this project. Thanks to James Pippin and Struan Grant (Children’s Hospital of Philadelphia) for assistance with sequencing. We would also like to thank the Flow Cytometry Core and the Human Immunology Core at the University of Pennsylvania for their advice and assistance in these experiments. This work was supported by NIH grants AI114852, AI082630, AI158617, and AG047773 to R.S.H, the Pediatric Infectious Diseases Society Fellowship Award, the National Center for Advancing Translational Sciences of the National Institutes of Health under award number KL2TR001879 to L.A.V. This work was supported by NIH grant K08 CA230157 and the Parker Bridge Scholar Award for ACH. Blood collection was supported by SPORE in Skin Cancer grant P50-CA174523.
This work was also supported by NIH grants AI155577, AI108545, AI112521, AI082630, AI201085, AI149680 and AI117950 to E.J.W. the U.S. Broad Agency Announcement HHSN272201100018C (to H.C.J.E. and E.J.W.) and the Allen Institute for Immunology (E.J.W.). We thank Dr. Kenneth Schmader and Sue Doyle for their help in influenza vaccine studies. E.J.W. is also supported by the Parker Institute for Cancer Immunotherapy which supports the cancer immunology program at UPenn. Additionally, research reported in this publication was supported by the National Cancer Institute and the National Institute Of Allergy and Infectious Diseases of the National Institutes of Health under Award Numbers U19AI142737, R35CA196620, K08CA248966, R01AI145870, P30CA008748, and P50CA221745. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The project described was also supported in part by grant #UL1TR001866 from the National Center for Advancing Translational Sciences (NCATS, National Institutes of Health (NIH) Clinical and Translational Science Award (CTSA) program. The following reagent was obtained through BEI Resources, NIAID, NIH: Influenza B Virus, B/Brisbane/33/2008 (Victoria Lineage), NR-42006.
Footnotes
Competing financial interests
EJW is an advisor for Merck, Marengo, Janssen, Related Sciences, Synthekine, and Surface Oncology. EJW is a founder of Surface Oncology, Danger Bio, and Arsenal Biosciences. EJW has a patent on the PD1 pathway. A.C.H is a consultant for Immunai and received research funding from BMS. T.C.M. has had advisory roles with Merck, Bristol-Myers Squibb, and OncoSec. D.B. reports consulting fees from Bristol Myers Squibb, Merck, Genentech‐Roche, AstraZeneca, and Pfizer and institutional research support from Merck, Genentech‐Roche, AstraZeneca, Novartis, and Bristol‐Myers Squibb. S.F. reports consulting fees from Merck; institutional research support from AstraZeneca and Genentech/Roche; stock and other ownership interest in Urogen, Allogene Therapeutics, Neogene Therapeutics, Kronos Bio, Iconovir, and Vida Ventures. C.H.L. served in a consultancy or advisory role to Amgen, Bristol-Myer Squibb, Exelixis, Merck, Pfizer, EMD Serono, and Eisai, and also received research funding from Bristol-Myers Squibb, Calithera, Eisai, Exelixis, Eli Lilly, Merck, and Pfizer. R.M. served in a consultancy or advisory role for Pfizer, Novartis, Merck, Genentech/Roche, Eisai and Exelixis, and received research funding from Bristol-Myers Squibb, Merck, Pfizer, Genentech/Roche, Eisai, Exelixis, and Novartis outside of the submitted work. M.V. consulting or advisory role: Alexion Pharmaceuticals, Calithera Biosciences, Exelixis, GlaxoSmithKline, Natera, Novartis, Pfizer; research funding: Bristol-Myers Squibb, Genentech/Roche, Pfizer; personal fees: Novartis, Takeda; travel fees: Novartis, Takeda, Eisai, AstraZeneca; honoraria: Novartis, outside of the submitted work.
References
- 1.Waldman AD, Fritz JM & Lenardo MJ A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol 20, 651–668 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang AC et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crotty S T follicular helper cell differentiation, function, and roles in disease. Immunity 41, 529–542 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crotty S T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 50, 1132–1148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Locci M et al. Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity 39, 1–12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crotty S Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol 29, 621–663 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Rasheed A-U, Rahn H-P, Sallusto F, Lipp M & Müller G Follicular B helper T cell activity is confined to CXCR5(hi)ICOS(hi) CD4 T cells and is independent of CD57 expression. Eur. J. Immunol 36, 1892–1903 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Freeman-Keller M et al. Nivolumab in Resected and Unresectable Metastatic Melanoma: Characteristics of Immune-Related Adverse Events and Association with Outcomes. Clin. Cancer Res 22, 886–894 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haratani K et al. Association of Immune-Related Adverse Events With Nivolumab Efficacy in Non-Small-Cell Lung Cancer. JAMA Oncol 4, 374–378 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishihara H et al. Association between immune-related adverse events and prognosis in patients with metastatic renal cell carcinoma treated with nivolumab. Urol. Oncol. 37, 355.e21–355.e29 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Verzoni E et al. Real-world efficacy and safety of nivolumab in previously-treated metastatic renal cell carcinoma, and association between immune-related adverse events and survival: the Italian expanded access program. J Immunother Cancer 7, 99 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kennedy LB & Salama AKS A Review of Immune-Mediated Adverse Events in Melanoma. Oncol Ther 7, 101–120 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinmann SC & Pisetsky DS Mechanisms of immune-related adverse events during the treatment of cancer with immune checkpoint inhibitors. Rheumatology 58, vii59–vii67 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willsmore ZN et al. B Cells in Patients With Melanoma: Implications for Treatment With Checkpoint Inhibitor Antibodies. Front. Immunol 11, 622442 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Läubli H et al. Influenza vaccination of cancer patients during PD-1 blockade induces serological protection but may raise the risk for immune-related adverse events. J Immunother Cancer 6, 40 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wijn DH et al. Influenza vaccination in patients with lung cancer receiving anti-programmed death receptor 1 immunotherapy does not induce immune-related adverse events. Eur. J. Cancer 104, 182–187 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Chong CR et al. Safety of Inactivated Influenza Vaccine in Cancer Patients Receiving Immune Checkpoint Inhibitors (ICI). Clin. Infect. Dis (2019) doi: 10.1093/cid/ciz202. [DOI] [PMC free article] [PubMed]
- 18.Failing JJ et al. Safety of Influenza Vaccine in Patients With Cancer Receiving Pembrolizumab. JCO Oncol Pract 16, e573–e580 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bersanelli M et al. INfluenza Vaccine Indication During therapy with Immune checkpoint inhibitors: a transversal challenge. The INVIDIa study. Immunotherapy 10, 1229–1239 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Bayle A et al. Immunogenicity and safety of influenza vaccination in cancer patients receiving checkpoint inhibitors targeting PD-1 or PD-L1. Ann. Oncol 31, 959–961 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Herati RS et al. Circulating CXCR5+PD-1+ response predicts influenza vaccine antibody responses in young adults but not elderly adults. J. Immunol 193, 3528–3537 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bentebibel S-E et al. Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci. Transl. Med 5, 176ra32–176ra32 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morita R et al. Human Blood CXCR5 + CD4 + T Cells Are Counterparts of T Follicular Cells and Contain Specific Subsets that Differentially Support Antibody Secretion. Immunity 34, 108–121 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Obeng-Adjei N et al. Circulating Th1-Cell-type Tfh Cells that Exhibit Impaired B Cell Help Are Preferentially Activated during Acute Malaria in Children. Cell Rep 13, 425–439 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herati RS et al. Successive annual influenza vaccination induces a recurrent oligoclonotypic memory response in circulating T follicular helper cells. Sci Immunol 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wrammert J et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453, 667–671 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sage PT & Sharpe AH T follicular regulatory cells. Immunol. Rev 271, 246–259 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Havenar-Daughton C et al. CXCL13 is a plasma biomarker of germinal center activity. Proceedings of the National Academy of Sciences 201520112–201520112 (2016) doi: 10.1073/pnas.1520112113. [DOI] [PMC free article] [PubMed]
- 29.Cox RJ Correlates of protection to influenza virus, where do we go from here? Hum. Vaccin. Immunother 9, 405–408 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang TT & Ravetch JV Functional diversification of IgGs through Fc glycosylation. J. Clin. Invest 129, 3492–3498 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang TT et al. Anti-HA Glycoforms Drive B Cell Affinity Selection and Determine Influenza Vaccine Efficacy. Cell 162, 160–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krammer F The human antibody response to influenza A virus infection and vaccination. Nat. Rev. Immunol 19, 383–397 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Wang TT et al. IgG antibodies to dengue enhanced for FcγRIIIA binding determine disease severity. Science 355, 395–398 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bournazos S et al. Antibody fucosylation predicts disease severity in secondary dengue infection. Science 372, 1102–1105 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellebedy AH et al. Defining antigen-specific plasmablast and memory B cell subsets in human blood after viral infection or vaccination. Nat. Immunol 17, 1226–1234 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun Y et al. Separase is recruited to mitotic chromosomes to dissolve sister chromatid cohesion in a DNA-dependent manner. Cell 137, 123–132 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeDiego ML, Martinez-Sobrido L & Topham DJ Novel Functions of IFI44L as a Feedback Regulator of Host Antiviral Responses. J. Virol 93, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang E & Li MMH All About the RNA: Interferon-Stimulated Genes That Interfere With Viral RNA Processes. Front. Immunol 11, 605024 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanmamed MF et al. A Burned-Out CD8+ T-cell Subset Expands in the Tumor Microenvironment and Curbs Cancer Immunotherapy. Cancer Discov 11, 1700–1715 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnston RJ, Choi YS, Diamond J. a., Yang J. a. & Crotty S STAT5 is a potent negative regulator of TFH cell differentiation. J. Exp. Med 209, 243–250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnston RJ et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325, 1006–1010 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herati RS et al. Vaccine-induced ICOS+CD38+ circulating Tfh are sensitive biosensors of age-related changes in inflammatory pathways. Cell Reports Medicine 0, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinov T et al. Programmed Death-1 Restrains the Germinal Center in Type 1 Diabetes. J. Immunol 203, 844–852 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Das R et al. Early B cell changes predict autoimmunity following combination immune checkpoint blockade. J. Clin. Invest 128, 715–720 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gallo P, Centonze D & Marrosu MG Alemtuzumab for multiple sclerosis: the new concept of immunomodulation. Multiple Sclerosis and Demyelinating Disorders 2, 1–11 (2017). [Google Scholar]
- 47.Jing Y et al. Multi-omics prediction of immune-related adverse events during checkpoint immunotherapy. Nat. Commun 11, 4946 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Good-Jacobson KL et al. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat. Immunol 11, 535–542 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herati RS et al. Vaccine-induced ICOS+CD38+ cTfh are sensitive biosensors of age-related changes in inflammatory pathways. bioRxiv 711911–711911 (2019) doi: 10.1101/711911. [DOI] [PMC free article] [PubMed]
- 50.Vella LA et al. T follicular helper cells in human efferent lymph retain lymphoid characteristics. J. Clin. Invest 129, 3185–3200 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sage PT et al. Circulating T follicular regulatory and helper cells have memory-like properties. J. Clin. Invest 124, 5191–5204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yousef S et al. Immunomodulatory molecule PD-L1 is expressed on malignant plasma cells and myeloma-propagating pre-plasma cells in the bone marrow of multiple myeloma patients. Blood Cancer J 5, e285 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turner JS et al. Human germinal centres engage memory and naive B cells after influenza vaccination. Nature 586, 127–132 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi J et al. PD-1 Controls Follicular T Helper Cell Positioning and Function. Immunity 49, 264–274.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Satpathy AT et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol 37, 925–936 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Helmink BA et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cabrita R et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020). [DOI] [PubMed] [Google Scholar]
- 58.Petitprez F et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560 (2020). [DOI] [PubMed] [Google Scholar]
- 59.Hollern DP et al. B Cells and T Follicular Helper Cells Mediate Response to Checkpoint Inhibitors in High Mutation Burden Mouse Models of Breast Cancer. Cell 179, 1191–1206.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sato K et al. Correlation between immune-related adverse events and efficacy in non-small cell lung cancer treated with nivolumab. Lung Cancer 115, 71–74 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Grangeon M et al. Association Between Immune-related Adverse Events and Efficacy of Immune Checkpoint Inhibitors in Non-small-cell Lung Cancer. Clin. Lung Cancer 20, 201–207 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Ricciuti B et al. Impact of immune-related adverse events on survival in patients with advanced non-small cell lung cancer treated with nivolumab: long-term outcomes from a multi-institutional analysis. J. Cancer Res. Clin. Oncol 145, 479–485 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Lozano AX et al. T cell characteristics associated with toxicity to immune checkpoint blockade in patients with melanoma. Nat. Med (2022) doi: 10.1038/s41591-021-01623-z. [DOI] [PMC free article] [PubMed]
- 64.Hänzelmann S, Castelo R & Guinney J GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Andrews SF et al. High preexisting serological antibody levels correlate with diversification of the influenza vaccine response. J. Virol 89, 3308–3317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang S & Williamson BL Characterization of protein glycosylation using chip-based nanoelectrospray with precursor ion scanning quadrupole linear ion trap mass spectrometry. J. Biomol. Tech 16, 209–219 (2005). [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang S et al. Comparative characterization of the glycosylation profiles of an influenza hemagglutinin produced in plant and insect hosts. Proteomics 12, 1269–1288 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.