

Abstract
To elucidate principles operating in native biological systems and to develop novel biotechnologies, synthetic biology aims to build and integrate synthetic gene circuits within native transcriptional networks. The utility of synthetic gene circuits for cell engineering relies on the ability to control the expression of all constituent transgene components. Transgene silencing, defined as the loss of expression over time, persists as an obstacle for engineering primary cells and stem cells with transgenic cargos. In this review, we highlight the challenge that transgene silencing poses to the robust engineering of mammalian cells, outline potential molecular mechanisms of silencing, and present approaches for preventing transgene silencing. We conclude with a perspective identifying future research directions for improving the performance of synthetic gene circuits.
eTOC
Transgene silencing, defined as the loss of transgene expression over time, persists as an obstacle for engineering mammalian cells and limits various applications of mammalian cell-based biotechnology. Diverse mechanisms contribute to transgene silencing, suggesting a variety of strategies may be needed to overcome these mechanisms. Furthermore, benchmarking of genetic components will be integral for designing and achieving roust transgene expression in mammalian cells.
Graphical Abstract
Introduction
Genome engineering within mammalian cells enables the stable expression of transgenes to support the design and implementation of custom genetic programs across a wide range of biotechnology applications 1–9. Engineered cells must retain control over the expression of transgenes over many cell generations. However, stably integrated transgenes often experience silencing, or diminished expression over time, thus limiting the use of engineered cells for applications that require weeks or more of expression 10. In the context of synthetic gene circuits, silencing interferes with circuit regulation, limiting the translation of engineered gene circuits for therapeutic and other applications. In this perspective, we highlight the challenge that transgene silencing poses to the robust engineering of mammalian cells, along with opportunities to mitigate this phenomenon.
Transgene silencing appears conserved across diverse organisms 11–13. Host cell identity, sequence of the integrated transgene(s), its location of integration, and gene delivery methods all putatively contribute to the rate and degree of transgene silencing. Silencing can manifest as an all-or-nothing phenomenon in which a portion of cells do not express the transgene. Often the proportion of engineered cells that express the transgene decreases over time in culture 14–17 (Figure 1A). In some cases, transgene silencing can be observed as a decrease in transgene expression levels in individual cells 18,19, and it often appears as a heritable change passed down through cell generations 14,15,20.
Figure 1. Definition and impact of transgene silencing in mammalian cells.
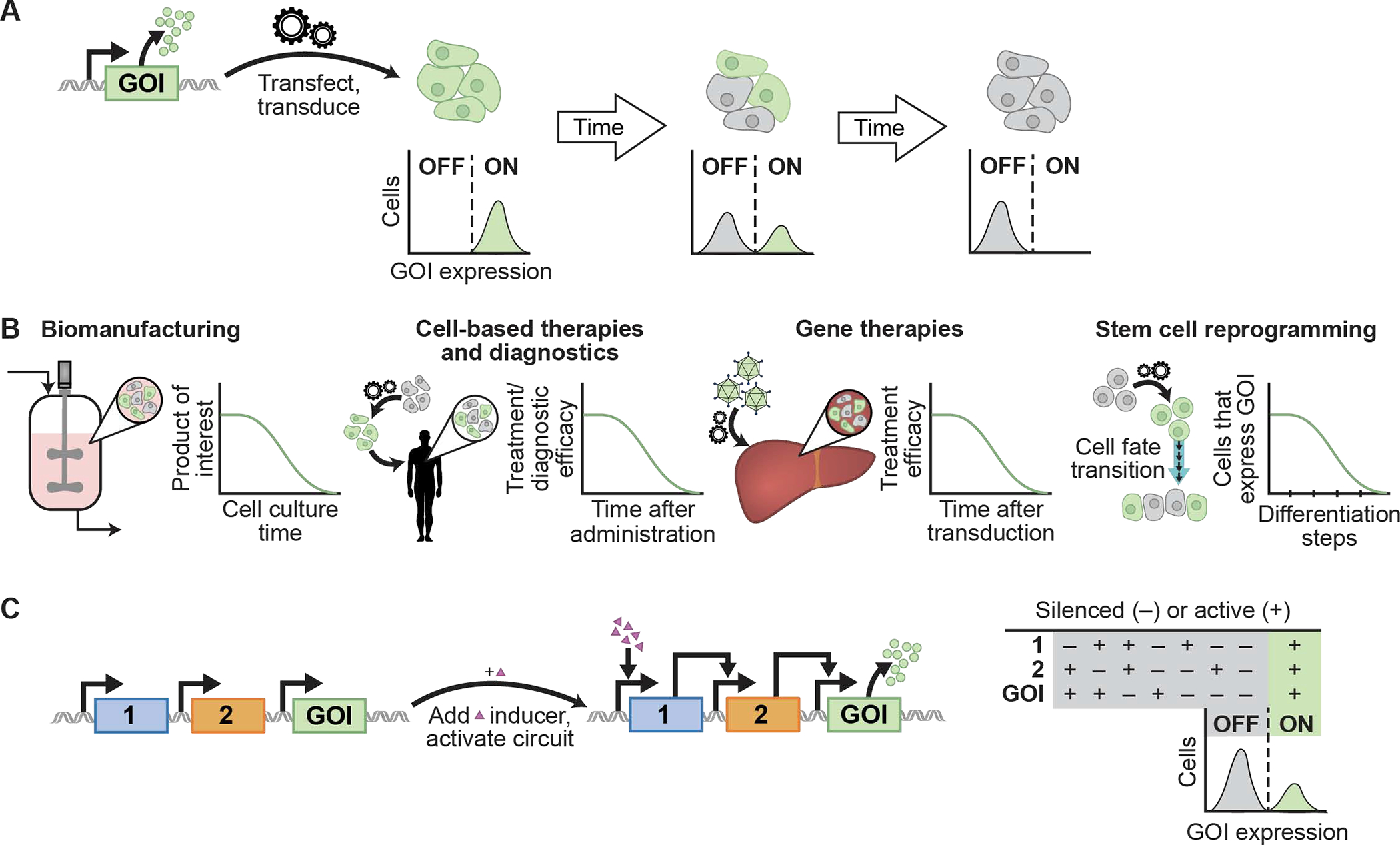
A. Mammalian cells engineered to express a transgene often undergo silencing. A variety of host-cell mechanisms contribute to transgene silencing which correlates with changes in chromatin structure at the site of the integrated transgene. Over time, transgene silencing generates a proportion of the engineered cell population that does not express the gene of interest (GOI). Transgene silencing is often observed as a bimodal distribution of cells that express the transgene (are in the ON state) or do not express the transgene (are in the OFF state) as shown 14,15. Transgene silencing may also be observed as a decrease in the relative levels of transgene expression rather than a complete loss of expression. B. Diverse applications in biotechnology rely on stable expression of transgenes in engineered mammalian cells. In biomanufacturing, silencing of mammalian cells engineered to produce a product of interest results in a decrease in product produced over time 21,22. Similarly, silencing of theranostic circuits in mammalian cells engineered ex vivo or engineered in vivo via gene therapies leads to waning efficacy over time 23. In cellular reprogramming and differentiation, cells engineered to express a gene or circuit of interest often undergo silencing as they change cell fates. In particular, differentiation of induced pluripotent stem cells into mature cell types often generates the desired cell type with a low proportion of cells that retain expression of the GOI 47. C. Gene circuits often require robust expression of multiple transgenes. Silencing of any individual transgene may limit the performance of stably integrated genetic circuits 24. In the example shown, a cascade of inducible transgenes regulates expression of the GOI. Silencing of any of the transgenes will result in failure to express the GOI.
Transgene silencing represents a bottleneck for many mammalian cell-based biotechnology applications (Figure 1B). For instance, in industrial cell lines such as Chinese hamster ovary (CHO) cells, or human embryonic kidney (HEK) 293 cells, the silencing of integrated transgenes reduces the long-term production yield for biopharmaceutical manufacturing 21,22. Similarly, silencing of sense-and-respond theranostic circuits lead to waning efficacy over time 23–25. Additionally, more complex synthetic gene circuits with multiple transgenes may be more susceptible to performance failure as silencing of any individual transgene renders the whole circuit nonfunctional (Figure 1C). Furthermore, silencing can spread to neighboring genes through direct and indirect effects, resulting in silencing compounding over time 24,26. Moreover, loss of expression of transgenes delivered via retroviruses has been well-documented in reprogramming, which may inhibit cell-fate transitions and lead to partially reprogrammed cells 27–30. Thus, transgene silencing is a critical challenge to understand and overcome for effective cell-based technologies.
In this perspective, we provide an overview of known mechanisms for transgene silencing, provide practical guidelines on how to avoid transgene silencing, and offer a look into future efforts that can further expand our understanding and improve our ability to control transgene expression in synthetic gene circuits. In addition, we suggest that future publications include discussions of observed cases of transgene silencing to help move toward more predictable and reliable cell reprogramming (Box 1).
Box 1. Publishing transgene silencing - making the invisible discoverable.
A large swath of valuable information generated by routine cell engineering goes unreported due to the lack of a system to share findings that are typically not incentivized for publication. Customarily, only the best-performing transgenic lines or clones take center stage in the final manuscript. We encourage scientists to include in figures, supplemental data, or materials and methods the frequencies of transgene silencing (e.g. rates of expressing and non-expressing clones). For example, a succinct description of “transgene performance” accompanied by tabulation of sub-optimal and misbehaving clones will not only provide discoverable data for meta-analyses, but also give authors an opportunity to highlight the magnitude of effort behind their work. For this reporting, we recommend inclusion of: cell type, special culturing conditions (if applicable), promoter type, transfection/transduction vector, number of passages since delivery when the transgene exhibited undesirable behavior, a description of the misregulation, and whether attempts were made to alleviate the silencing with their outcomes. Collectively, this reporting will help move the field forward to predictably and successfully engineer cells.
MECHANISMS OF TRANSGENE SILENCING
Cells rely on transcriptional regulation to tune gene expression, respond to environmental stressors, and generate phenotypic diversity in complex multicellular organisms. Epigenetic regulation complements dynamic transcriptional control. Through the deposition, recognition, and erasure of covalent modifications to DNA and histones, epigenetic regulation confers stable memory and hysteresis within biological systems. Epigenetic regulation harmonizes with transcriptional control including the assembly of the preinitiation complex, double-stranded DNA melting at the promoter, initiation, or elongation 31. In all, these can be affected by DNA modifications and influence the structure of the local chromatin and the protein-DNA complexes surrounding the gene of interest. Synthetic gene circuits must contend with native epigenetic and transcriptional regulatory mechanisms, which may support or impede transgene expression. As silencing often correlates with specific chromatin modifications profiles of transgenes, epigenetic regulation putatively supports and reinforces transgene silencing. Therefore, an understanding of the epigenetic mechanisms that silence transgenic cargoes in mammalian cells may facilitate the design of more robust genetic circuits and engineered transcriptional programs 32.
Cells engineered with multi-component circuits rely on the tight regulation of multiple transcriptional units to control cellular behavior. For example, cell-based therapies might employ circuits composed of multiple transgenic cassettes encoding biosensing and signal processing functions. Hence, one malfunctioning unit, or cassette, could result in the breakdown of the entire circuit (Figure 1C). In one study, a genetic circuit consisting of four transcription cassettes in HEK293T cells was silenced at an estimated rate of ~2% of the population per week 24, whereas a more rapid shutdown of an 8 kb circuit was observed after approximately three weeks of culture in mouse embryonic stem cells 25. Notably, in both examples the DNA circuits were integrated at genomic safe-harbor sites. While safe-harbor sites provide genomic regions that support transgene integration without adversely affecting normal cellular functions, cassettes integrated at these sites are still subject to silencing. These results highlight the need for a better understanding of how genomic context and composition of the synthetic gene circuit can influence transgene silencing.
How do cells identify transgenes for silencing?
Over time, cells selectively silence integrated transgenes while maintaining endogenous genes at homeostatic levels 33,34. Given that silencing appears to selectively impact transgenic elements, how do cells distinguish transgenes from other genomic elements to generate specific profiles of silencing? As recruitment of chromatin-modifying enzymes likely serves an essential step in epigenetic silencing, cells may recruit these enzymes through mechanisms that are dependent and independent of the transgene sequence. In sequence-dependent mechanisms, interactions at the exact site of transgene integration may prime transgenes for selective silencing. This may include protein recognition of specific DNA motifs, such as CpG islands, and the subsequent formation of complexes with chromatin-modifying activity 35. Additionally, sequence-dependent formation of DNA, RNA, or hybrid structures may recruit chromatin-modifying enzymes. For instance, GC-rich sequences can induce G-quadruplexes, R-loops, and other DNA structures that may contribute to RNA polymerase (RNAP) stalling and recruitment of chromatin modifiers through factors that directly recognize these structures 36,37. Furthermore, terminal repeat sequences that enable transposon-based insertion of transgenes into the genome can trigger RNAi-mediated silencing 38. As the particular mechanism may vary based on the sequence of the transgene, we expect that interventions may show different efficacy across transgenic cargos.
Alternatively, sequence-independent silencing may result from passive loss of transcriptional activity which may be influenced by local genomic context effects. For instance, activity of chromatin modifiers near the locus of transgene integration may contribute to nonspecific silencing through spreading of heterochromatin. Additionally, chromatin-modifying enzymes may broadly survey the genome, actively silencing genes through reversible, transient modification 39. Nascent chromatin remains inaccessible and transcriptionally inactive following DNA replication, and transcriptional reactivation is required to regain accessibility 40. Potentially, gene activity is reestablished for endogenous genes through selective transcriptional reactivation by combinations of endogenous transcription factors after DNA replication. Lacking such mechanisms, transgenes may remain nonspecifically silenced.
In principle, sequence-dependent and -independent mechanisms may combine to induce transgene silencing. Whether recruited in a sequence-dependent manner or not, chromatin modifying enzymes may prevent expression of synthetic circuits through heterochromatin formation, DNA methylation and post-translational modifications (PTMs) to histones. Processes that lead to silencing may be induced at the site of transgene integration via direct recruitment of chromatin regulators, encroachment of heterochromatin, activation of viral and transposon defense systems, or proliferation-mediated processes. Below, we discuss these mechanisms and their relation to transgene silencing.
Proliferation-associated processes can mediate silencing
Transgene silencing increases over time in proliferating cells and correlates with the number of cell divisions. Both the fraction of cells that express the transgene and the mean expression level of marker-positive cells can decrease over time and with cell division 24,27,41. Putatively, processes linked to the cell cycle provide a mechanism that enhances transgene silencing.
Transgene silencing may be accelerated by the inherent antagonism between transcription and the DNA replication necessary for proliferation, each increasing torsional strain and steric interference on the DNA polymer 42,43. These processes can lead to the accumulation of positive and negative supercoiling (over- or under-wound DNA, respectively), which can in turn promote the formation of structures such as R-loops, interactions between DNA and nascent RNA 44,45. R-loops have been shown to alter the binding of chromatin remodelers 36, suggesting a potential mechanism by which persistent changes in gene expression could arise (Figure 2A). Indeed, overexpression of transcription factors in reprogramming induces markers of genomic stress including increased negative DNA supercoiling, R-loop formation, and DNA replication fork stalling 27. Thus, collisions between the transcription and replication machinery in proliferating cells may contribute to transgene silencing.
Figure 2. Mechanisms of transgene silencing.
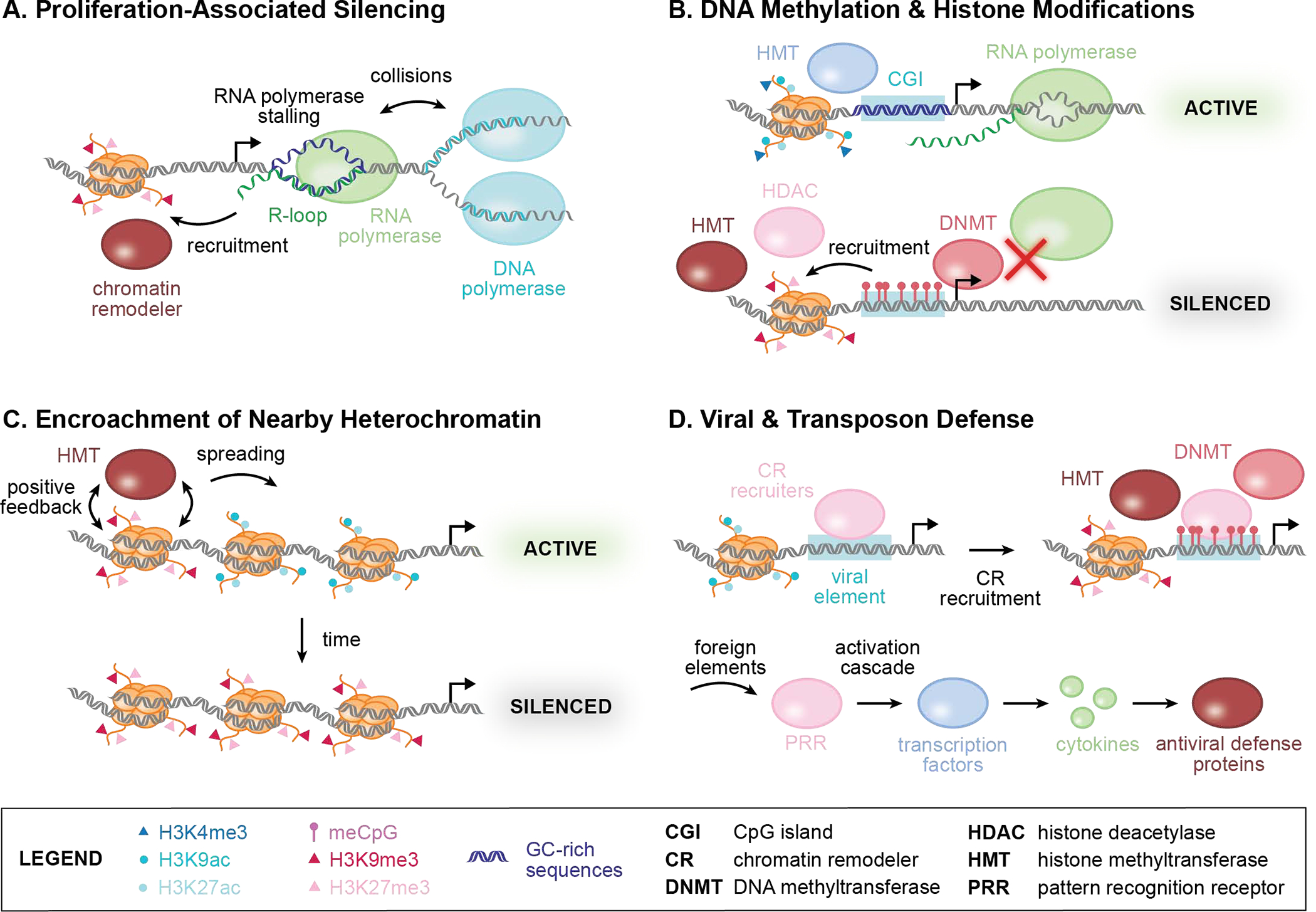
A. Proliferation may contribute to gene silencing via antagonism between transcription and replication machinery. Increased strain on the DNA and collisions between RNA polymerases and DNA polymerases can promote the formation of structures such as R-loops, which can alter binding of chromatin remodelers and thus reshape epigenetic states at the sites of transgene integration. B. DNA methylation and histone modifications are associated with gene silencing. Top: Hypomethylated CpG islands (CGIs) can recruit histone methyltransferases (HMTs) and accrue H3K4me3, associated with active transcription by RNA polymerases. High GC content at promoter CGIs is correlated with resistance to silencing. Bottom: Hypermethylation of CpGs (meCpG) by DNA methyltransferases (DNMTs) can recruit histone deacetylases (HDACs) and subsequently HMTs, replacing active H3K9ac and H3K27ac marks with repressive H3K9me3 and H3K27me3 marks. Competition between transcriptional machinery and DNMTs may reinforce the association of DNA methylation and gene expression states (i.e., hypomethylation/active, hypermethylation/silenced). C. Heterochromatin spreading to neighboring regions may silence transgenes. H3K9me3 and meCpG can spread to neighboring genes via positive feedback supported by HMT recruitment to methylated sites, establishing a repressive chromatin state at nearby integrated synthetic circuits. D. Endogenous pathways that recognize viral and transposon elements may suppress transgene expression. Top: Proteins that recognize viral DNA sequences, such as LTRs, recruit chromatin remodelers (CRs), including HDACs, HMTs, and DNMTs. Bottom: Recognition of foreign elements, such as unmethylated CpGs, by pattern recognition receptors (PRRs) activates transcription factors involved in innate immunity (e.g., NFκB). These factors promote cytokine production and lead to upregulation of antiviral defense proteins, which may result in transgene silencing.
Proliferation and silencing are intimately linked in the process of stem cell reprogramming and differentiation. For example, proliferation has been shown to promote cellular reprogramming to induce pluripotent cells and to neurons 27,46. The loss of transgene expression delivered via retroviruses is well-documented in reprogramming, and this loss of expression may inhibit cell-fate transitions and lead to partially reprogrammed cells 27–30. On the other hand, proliferation can also drive transgene silencing while simultaneously increasing the probability that a cell will reprogram 27,46. A tradeoff between transgene expression and proliferation emerges, leaving a narrow window of time for reprogramming. Notably, in a recent study, cells that sustain high transgene expression while undergoing rapid proliferation reprogrammed to neurons at high rates and display increased functional maturity 27. While the loss of transgene expression may induce heterogeneity and reduce efficiency, the loss of reprogramming factors may improve differentiation of pluripotent stem cells to new cell fates 30. Silencing of transgenes has been observed after they have been placed in various safe harbor loci during human pluripotent stem cell (hPSC) and mouse embryonic stem cell (mESC) differentiation into various lineages 47–50. In some cases, transgene expression could be maintained by the introduction of a flanking chromatin insulator derived from the chicken β-globulin hypersensitivity site 4 (cHS4) 47. In fibroblast conversion to induced pluripotent stem cells, Velychko et al found that retroviral silencing varied based on the reprogramming factors used and that silencing could occur early in the reprogramming process, even before the loss of fibroblast identity 28. In particular, inclusion of cMyc, which drives proliferation, increased transgene silencing. In reprogramming to neurons, loss of expression of retroviral transgenes occurs at higher rates in hyperproliferative cells 27. In addition to these proliferation-associated phenomena, silencing also occurs in post-mitotic or slowly dividing cells 23,51. Overall, silencing is often enhanced by proliferation but does not require proliferation.
DNA methylation contributes to stable silencing
Cytosine methylation at CpG motifs contributes to epigenetic silencing (Figure 2B). The distribution of CpG dinucleotides delineates DNA with different states of methylation and plays a key role in epigenetic regulation 52. When interspersed across genomic tracks including introns and exons, CpGs are canonically methylated and may contribute to transgene silencing. On the other hand, when CpGs cluster at promoters and enhancers to form CpG islands (CGIs), they are often hypomethylated 53.
Methyltransferases establish and maintain CGI methylation. During embryogenesis, DNA methyltransferase 3 Alpha, Beta, and Like (DNMT3A, DNMT3B, and DNMT3L) establish CpG methylation which is maintained during DNA replication by DNA methyltransferase 1 (DNMT1) 54–57. Understanding CGI methylation and transcriptional repression may guide strategies to mitigate CpG-mediated transgene silencing 58–61. In addition to a direct contribution to epigenetic silencing, DNA methylation can be involved in the recruitment of H3K9me3 to nucleosomes, which contributes to the formation of heterochromatin 62. Heterochromatin is associated with limited DNA accessibility, nuclear reorganization, and silenced transcription, as discussed further below 63,64.
CGIs associate with ubiquitously expressed genes, but not with tissue-specific genes 65. The presence of CpGs on promoters significantly impacts the silencing of downstream genes 55,57,66–71. Furthermore, CGI hypomethylation and active transcription may reinforce one another through competition between transcription and methylase complexes, causing active promoters to remain hypomethylated and inactive genes to accrue methylation 65,72.
Native and synthetic CGIs may confer specific patterns of DNA methylation. Endogenous promoters ectopically inserted into the β-globin locus of mouse embryonic stem cells exhibited CGI methylation patterns that resemble their native counterparts 73. Similarly, synthetic elements comprising CGIs and bacterial sequences recapitulate expected patterns of methylation 73. However, CGIs do not effectively shield promoters from methylation if positioned beyond 100–200bp from the transcription start site 61,68,74. Synthetic promoter-less CGIs may recruit histone methyltransferases and accrue H3K4me3, but the capacity to remain CpG-hypomethylated requires both high CpG density and high GC content, suggesting that AT-rich sequences act as DNA methyltransferase docking sites 71. Overall, DNA CpG methylation is implicated in transcriptional silencing, whereas high GC-content CGIs associated with transcription sites remain hypomethylated.
Heterochromatin-associated histone modifications are found at silenced transgenes
Histone modifications play a central role in epigenetic gene silencing through constitutive heterochromatin formation 75,76. Heterochromatin is characterized by regions with relatively high nucleosomal density, which may impede transcription initiation. DNA methylation can initiate formation of heterochromatin through the recruitment of histone deacetylase enzymes (HDACs), which remove histone acetylation, a feature typically associated with transcriptionally active chromatin (Figure 2B). Importantly, treatment of cells with the HDAC inhibitor sodium butyrate can restore inducible gene activation in mouse embryonic stem and transformed mammalian breast cancer cells 25,77. To induce durable silencing, deacetylated histones are subsequently trimethylated by histone methyltransferases 78–80. Silencing is facilitated by nuclear proteins such as heterochromatin protein-1 (HP1) which promote heterochromatin maintenance 81. Targeted inhibitions of these processes at sites of transgene integration may prevent local heterochromatin formation without the global epigenetic perturbations induced by broad chemical inhibitors, potentially offering an approach to mitigate silencing due to heterochromatin formation.
Spreading of heterochromatin silences proximal regions at the locus of integration
Although heterochromatin formation may initiate focally, proximal regions may be silenced through spreading of heterochromatin (Figure 2C). Silencing of proximal regions was identified in Drosophila and termed position effect variegation in 1930 by Muller et al. 82,83. Although this phenomenon was initially described with genomic rearrangement of endogenous genetic elements, integration of transgenic payloads mimics these phenomena 84–86. Encroachment of the surrounding heterochromatin can disrupt transcription. Spreading of H3K9me3 propagates via a feedback loop of chromatin regulators 75,87,88. Combined with DNA methylation, spreading of H3K9me3 leads to heterochromatization and transcriptional repression. In the case of an integrated transgene heterochromatization of a nearby gene can spread to, and silence, the transgene 26. Furthermore, spreading of heterochromatin has been observed using multicopy transcription arrays in hamster, mouse, and Drosophila cells showing that gene silencing correlates with appearance of repressive chromatin at transgene arrays 89–91.
Epigenetic silencing of transgenes depends on the specific locus and genomic context of integration 92,93. Integration within topological associated domains (TADs) may impact transgene activity through TAD-specific determinants of chromatin state 94. For example, H3K9me3 often spreads throughout a TAD 95. Notably, integration near centromeres may influence the epigenetic silencing of nearby transgenes. In cases when random integration methods are used to engineer synthetic genetic circuits in mammalian cells, wide variability in gene expression and epigenetic silencing may result.
Site-specific genome engineering methods can be utilized to integrate transgenes at so-called safe-harbor loci, yet there remain associated complexities that require further examination. For example, silencing of safe-harbor loci is well-documented 25,47,48,96. Furthermore, transgene insertion can alter the local chromatin state in a locus-specific manner and affect transgene expression 32. A key open challenge is understanding why transgene insertion into safe-harbor loci confers stable, consistent gene expression in some scenarios (e.g. cell types, transgene sequences, and insertion conditions) but not others. For a comprehensive review of silencing of transgenes in safe harbor sites and discussions of criteria for identifying genomic safe harbors, we direct readers to the following perspective 97.
Viral and transposon defense systems contribute to transgene silencing
Viral vectors and transposon systems provide powerful tools to integrate transgenic cargo into mammalian cells with high efficiency, but they confer specific challenges in maintaining transgene activity. Key gene therapy delivery agents such as lentiviral and gammaretroviral vectors are subject to transcriptional silencing upon integration into the mammalian genome 98–100. Mechanistically, the silencing of lentiviral vectors is often associated with promoter methylation, especially during differentiation of stem cells 101. Viral promoters appear to be more prone to epigenetic silencing compared to endogenous promoters 101, highlighting the need to choose an appropriate promoter for clinical gene delivery when viral vectors are used for transgene delivery. For example, DNA methylation and silencing was observed in murine hematopoietic stem cells (HSCs) following Moloney murine leukemia virus (MoMuLV)-based retroviral transduction in vivo following serial transplantation 102. In these studies, both murine stem cell virus (MSCV) and human immunodeficiency virus type 1 (HIV-1) virus led to DNA methyltransferase activity independent of silencing in transgenic mice, murine embryonic stem cells, and Drosophila 103.
As a defensive adaptation against pathogens and transposon-mediated genomic instability, mammalian cells use epigenetic regulation mechanisms to specifically identify and repress virally integrated transgenes 104. Mammalian hosts possess dedicated machinery that detects proviral sequences and recruits histone-modification complexes that mediate transcriptional repression (Figure 2D). One such proviral sequence is the primer binding site, an 18-bp element residing near the 5’ long terminal repeat (LTR) from which viral reverse transcription is initiated. The LTR sequence varies across viruses and is complementary to ribosomal tRNAs, allowing retroviruses to hijack tRNAs and prime reverse transcription of the minus strand 105,106. Reciprocally, the host cell can use this site as a target for transcriptional repression. For example, pluripotent cells strongly repress gene expression from MoMuLV 107–109. Biochemical analyses have shown that ZFP809 and TRIM28 (KAP1) bind the primer binding site and form a complex 110,111 that recruits the H3K9 methyltransferase SETDB1 112,113 and components of the NuRD histone deacetylase complex 114. Accordingly, ZFP809 and TRIM28 are enriched in an endogenous retrovirus sequence spanning the LTR, 5’UTR and beginning of gag, and are essential for H3K9me3 deposition, histone deacetylation, and repression of proviral genomes 113,115,116. Additional native proteins have been implicated in reinforcing this epigenetic repression complex by acting as a scaffold for SETDB1 and NuRD components 117. In this way, endogenous proteins recognize viral DNA motifs and induce epigenetic changes in a sequence-dependent manner.
Viral LTRs serve as prominent targets for CpG methylation. The DNMT-binding scaffold protein Daxx mediates repression of invading viruses and contributes to maintenance of LTR methylation 118. Additionally, methylation of endogenous proviruses is facilitated by the ZFP809-TRIM28-SETDB1 complex 119. Combining TRIM28 knockout with 5-azacytidine-induced CpG demethylation increases provirus transcriptional reactivation stronger than either treatment alone 115. However, SETDB1 knockout reactivates endogenous retroviruses without affecting CpG methylation, and SETDB1-mediated H3K9me3 deposition is unaffected by co-deletion of Dnmt3a/Dnmt3b/Dnmt1 113. Hence, it appears that DNMT-Daxx-mediated CpG methylation and SETDB1-NuRD histone modifications act synergistically to robustly ensure retrovirus and transposon repression. It therefore seems plausible that engineering of viral vectors, e.g. modifying the ZFP809 recognition sequence in the LTR, could result in stealth variants that are less susceptible to epigenetic silencing.
Another defense mechanism involves the recognition of non-self macromolecules carrying pathogen-associated molecular patterns (PAMPs). Recognition of PAMPs by host-expressed pattern recognition receptors (PRRs) triggers an innate immune signaling reaction 120,121. The Toll-like receptor (TLR) 9 recognizes bacterial and viral DNA lacking methylated CpGs, triggering activation of NFκB and resulting in the production of cytokines in dendritic cells and macrophages 122,123. An shRNA screen of baculovirus-infected A549 cells identified genes of the TLR, interferon, and interleukin families that silence transgene expression 124,125. In another example of immune-mediated transgene silencing, influenza and Sendai virus infections of macrophages trigger IFN-α-mediated upregulation of TLR1–3 and TLR7 126. IFN-α reduces histone acetylation and transcription of hepatitis B viral mini chromosomes in HepG2 cells 127, and upregulates Daxx in HeLa cells 118. PAMP-PRR reactions thus convert infected cells into cytokine hubs that signal an upregulation of repressive barriers against invading transgenes (Figure 2C). To ensure transgene expression, these reactions should be avoided through careful consideration and engineering of delivery vectors.
PRACTICAL GUIDELINES FOR AVOIDING TRANSGENE SILENCING
Loss of transgene expression compounds with the myriad other challenges of cell engineering. The field needs improved methodological guidelines and data sharing of successes and failures alike across diverse systems to identify common, useful tools and frameworks. Here, we propose practical guidelines for stable engineering of mammalian cells.
Choice of promoter influences the probability of transgene silencing
Promoters vary in their transcriptional activities and sensitivity to epigenetic silencing. Transgene expression is dependent on multiple factors that vary across genetically engineered clones (e.g. loci of integration, copy number, and target cell), which can obscure the role of the promoter in silencing. Thus, it is important to test the effect of the promoter while controlling for genomic context, for instance by comparing multiple clones with the transgene cassette integrated in the same location, differing only in the selected promoter. To our knowledge, there has not been a comprehensive comparison that systematically evaluated the long-term activity of all promoters commonly used in mammalian synthetic biology. Here, we surveyed the literature to assess promoter performance in terms of expression levels and stable activity over time in the context of their respective experimental details (Table 1).
Table 1.
Summary of published studies on the effect of promoter choice in transgene yields (“Levels”) and sustained expression (“Stability”) over culture time.
| Transgene Delivery Method | Host Cell Type | Transgene Expression | Locus of Integration | Clone Information | Top Three Promoters |
Reference | |
|---|---|---|---|---|---|---|---|
| Levels | Stability | ||||||
|
| |||||||
| Lentiviral vector | Various | Stable | Random | Polyclonal | CAG hEF1α CMV | n.d. | Qin et al 2010 |
| Lipofectamine 2000 | HEK-293F | Stable | Random or Episomal | Polyclonal | CAG hEF1α CMV | CAG hEF1α CMV | Dou et al 2021 |
| Lipofectamine 2000 | CHO-K1 | Stable | Random or Episomal | Clonal lines | hEF1α CMV CAG | Only hEF1α tested | X. Wang et al 2017 |
| Lentiviral vector | mESC (J1) | Transient | Random | Polyclonal | hEF1α CAG hPGK1 | Only hEF1α tested | Hong et al 2007 |
| Lentiviral vector | mESC (JM8.N4) | Stable | Random | Polyclonal | n.d. | hPGK hEF1α CMV | Herbst et al 2012 |
| φC31 integrase | mESC (IDG26.10–3) | Stable | Rosa26 | Clonal lines | CAG hUbC hEF1α | n.d. | Chen et al 2011 |
| Electroporation | mESC (E14Ju09) | Stable | Random | Clonal lines | CAG; mPGK1 | n.d. | Malaguti et al 2022 |
| Lentiviral vector | hESC (SA121; Hues-4) | Stable | Random | Polyclonal | hEF1α hACTB PGK | hACTB PGK EF1α | Norrman et al 2010 |
For stability in stem cells, the list refers to robust transgene expression over prolonged stem cell maintenance (for differentiation, please see text).
Abbreviations: CAG: CMV early enhancer/β-actin synthetic hybrid promoter; CMV: cytomegalovirus promoter; EF1α: elongation factor 1-alpha promoter; PGK1: phosphoglycerate kinase 1 promoter; UbC: polyubiquitin-C promoter. m or h indicate mouse or human promoter origin and cells respectively; n.d.: not determined.
Inducible systems provide extra safety by offering the ability to turn on and off expression of a transgene using external control, such as the addition of a small molecule, light, or other user-imposed or cell-sensed stimuli. Regulation by inducible promoters allows stably integrated transgenes to be left in the inactive or OFF state for periods of time before induction. These periods of inactivity correlate with an increase in the proportion of cells that do not respond to induction. This phenomenon has been documented for tetracycline-inducible promoter systems, where continuous induction or higher basal activity of the promoter results in less silencing compared to versions with tightly regulated OFF states 77,128. More recently, inducible expression systems have been developed that resist silencing over longer time periods compared to a tetracycline-inducible promoter, employing constitutive transgene transcription with post-transcriptional regulation to mitigate silencing 129.
Viral promoters such as cytomegalovirus (CMV), spleen focus-forming virus (SFFV), and Rous sarcoma virus (RSV) undergo CpG methylation resulting in silencing of the transgene within a few cell divisions, however, this silencing can be alleviated with 5-azacytidine, a DNA methyltransferase inhibitor, to partially restore the transgene expression 22,101,130,131. A gene driven by the RSV promoter has also been shown to be silenced by polycomb repression complex 2 (PRC2) in CHO cells 26. However, silencing of the cassette containing RSV and a downstream transgene, can be prevented by proximal cHS4 insulators, perhaps due to increased local histone acetylation and protection of the promoter from DNA methylation 132,133. In mESCs, transgenes containing LTR and SV40 promoters can also be acutely silenced 134–136. However, unlike in pluripotent stem cells, 5-azacytidine may not rescue transgene expression, as has been observed in undifferentiated pluripotent stem cells 25. Additionally, while episome disappearance and provirus methylation occurred seven to ten days after infection, the viral cassette was silenced prior to that point, supporting a DNA methylation-independent mechanism of provirus silencing in pluripotent stem cells 108,109. This mechanism appears to be histone methylation (H3K9me3 and H4K20me3) catalyzed by SETDB1 in complex with TRIM28 and ZFP809. A mutation in the LTR-associated primer binding site, demonstrated improved long-term transgene expression in bone marrow cells transplanted into irradiated recipient mice 137. Together, these data show that viral promoters alone should be avoided for long-term transgene expression due to their propensity to be silenced, but engineered variants show promise.
Comparison of various promoters in their ability to drive GFP expression via lentiviral transduction of murine and human cell lines has shown that EF1α and CAG promoters consistently produce high fluorescence intensities; CAG exhibited the least variation between transductions, whereas the CMV promoter demonstrated fluorescent variability depending on the host cellular context (e.g. HEK-293T and human MRC5 fibroblasts) 138. Similarly, high CMV activity in HEK-293T cells is corroborated by transient transfections when transgenes are not permanently integrated into the genome 139. Non-viral Lipofectamine-mediated transgene delivery into HEK-293F cells showed that the top three promoters driving highest yields were, in descending order, CAG, EF1α and CMV, and the ranking remained consistent over time 140. Therefore, the EF1α and CAG promoters are commonly used in workhorse mammalian cells because they seem to be the most suitable for long-term expression of high transgene levels.
The choice of promoter in stem cells appears to be more complicated. For example, EF1α, CAG and PGK promoters all have been successfully used to drive long-term transgene expression in undifferentiated mESCs 101,141,142. However, their performance is influenced by context-specific factors such as viral elements or locus of integration 141,143. With respect to differentiation, EF1α promoter activity has been observed to suffer the least silencing during early-stage embryoid body differentiation 101,141, however, during neuronal maturation, the CMV promoter outperformed EF1α and CAG promoters in mESC-derived neurons, illustrating lineage-dependent promoter performance 141. In hESCs transduced with lentiviral vectors, fluorescence intensity was highest when the EF1α, β-actin, and PGK promoters were used to drive GFP expression 33. Moreover, promoters can exhibit different behaviors based on the type of silencing considered. For example, β-actin remained the most active during long-term hESC maintenance, however, during differentiation it drove the highest overall GFP intensity. In contrast, EF1α retained the largest percentage of GFP positive cells and overall activity across a number of lineage markers 33. Overall, EF1α, CAG, and β-actin promoters efficiently drive transgene expression in stem cells, however, it is important to consider the context of reprogramming stem cells and whether the goal is for long-term stem cell maintenance, differentiation, or both.
Additionally, beyond stability of the mean expression level, consideration of the expression dynamics may also be relevant for some applications. Future work to characterize these parameters of common promoters across contexts is therefore needed. Ultimately, promoter choice should be considered in concert with other factors including insulating elements, locus of integration, and cell type.
Insulators can block transgene silencing
One commonly adopted strategy to counteract transgene silencing is to include insulating DNA elements in the expression cassette. Two types of DNA insulator functions exist: barrier activity that blocks the spreading of heterochromatin from nearby repressive regions, and enhancer-blocking activity that prevents enhancer-dependent gene activation 144. In the context of mitigating transgene silencing, the prevention of heterochromatin spreading is important. Various insulating elements have been reported and tested in mammalian cells, including the prototypic insulator, cHS4 145,146, scaffold/matrix attachment regions (S/MAR) 147, ubiquitous chromatin opening elements (UCOE) 148,149 and human tRNA gene tDNA 150 (Table 2). Readers are directed to other reviews for more detailed discussions on this topic 151–153. These barrier elements typically function by recruiting proteins (e.g., histone modifying enzymes, chromatin remodelers) that prevent the spreading of repressive heterochromatin and thus establish a local transcriptionally permissive environment 151. More specifically, the core region of the cHS4 insulator has protein binding sites for VEZF1, CTCF and USF1/2, which protect against DNA methylation, help form chromosomal loops, and recruit histone modifying enzymes associated with active expression states, respectively 154. Accumulating evidence has shown the significant role that chromatin insulators play in regulating the 3D genome architecture 155. For instance, the binding of CTCF, the primary insulator protein in mammals, is essential to establish the boundaries of TADs 156. The role of CTCF as an enhancer blocker has been well characterized, leading to the discovery of highly potent enhancer-blocking insulators from the high affinity CTCF-binding sites in the human genome 157. Interestingly, genome-wide analysis of CTCF-binding sites in chromatin barrier regions indicate that CTCF may also play an important role in the barrier activity of insulators 158. Hence, future studies on chromatin boundary regions will likely contribute to the discovery of novel insulators beneficial for mammalian synthetic biology.
Table 2.
Summary of commonly used insulators in mammalian cells.
| Insulator | Size (bp) | Source | Chromosome coordinate | Reference |
|---|---|---|---|---|
| cHS4 | 1200 (full) 250 (core) | Chicken beta-globin locus | Chr1:197,298,879–197,300,081 (galGal6) | 145 |
| A2-UCOE | 1500 | Human HNRPA2B1-CBX3 locus | Chr7:26,239,804–26,241,504 (hg19) | 262 |
| CBX3-UCOE | 700 | Human HNRPA2B1-CBX3 locus | Chr7:26,240,735–26,241,449 (hg19) | 149 |
| MAR 1–68 | 3630 | Human Chromosome 1 intergenic between SPATA6 and AGBL4 | Chr1:48,947,776–48,951,409 (hg19) | 147 |
| tDNA | 1200 | Human tRNA genes | Chr17: 7,963,112–7,964,183 (hg19) | 150 |
Insulator elements are described in terms of their size and genomic source. cHS4: chicken hypersensitive site 4. UCOE: ubiquitous chromatin opening element. MAR: matrix attachment region. tDNA: tRNA gene.
The insulating DNA elements identified so far face various challenges that limit their use in mammalian cell engineering. Incorporating the cHS4 sequence into the transgene cassette can significantly reduce the titer of packaged lentivirus carrying the transgene 159,160. In addition, the relatively large size of S/MAR elements (e.g. the S/MAR 1–68 element is ~3.6kb) renders them unfavorable when using vectors with limited cargo capability. Although UCOE-type insulators have been shown to prevent silencing when used with numerous promoters in stem cells 149, their potential bidirectional promoter activity may lead to transcriptional activation of nearby genes upon integration, which poses a safety concern in gene therapy 161. Interestingly, a recent study screened candidate UCOEs with various truncations, demonstrating their potential to function as barrier-type insulators without intrinsic promoter activity 162. However, there is a lack of systematic comparison of the barrier activity of different insulating elements under the same conditions (e.g., cell line, chromosome context, and copy number). Therefore, both direct comparison of existing insulators within the same context and identification of other novel insulators with better features (e.g. compact size, broad tissue compatibility, and no intrinsic promoter activity) would be beneficial to the synthetic biology community. Overall, the choice of promoter and insulator combined with exclusion of transcriptional repression target elements is important to stabilize high levels of transgene expression (Figure 3A).
Figure 3. Design considerations for preventing transgene silencing via selection of genetic elements, locus, and cell type of interest.
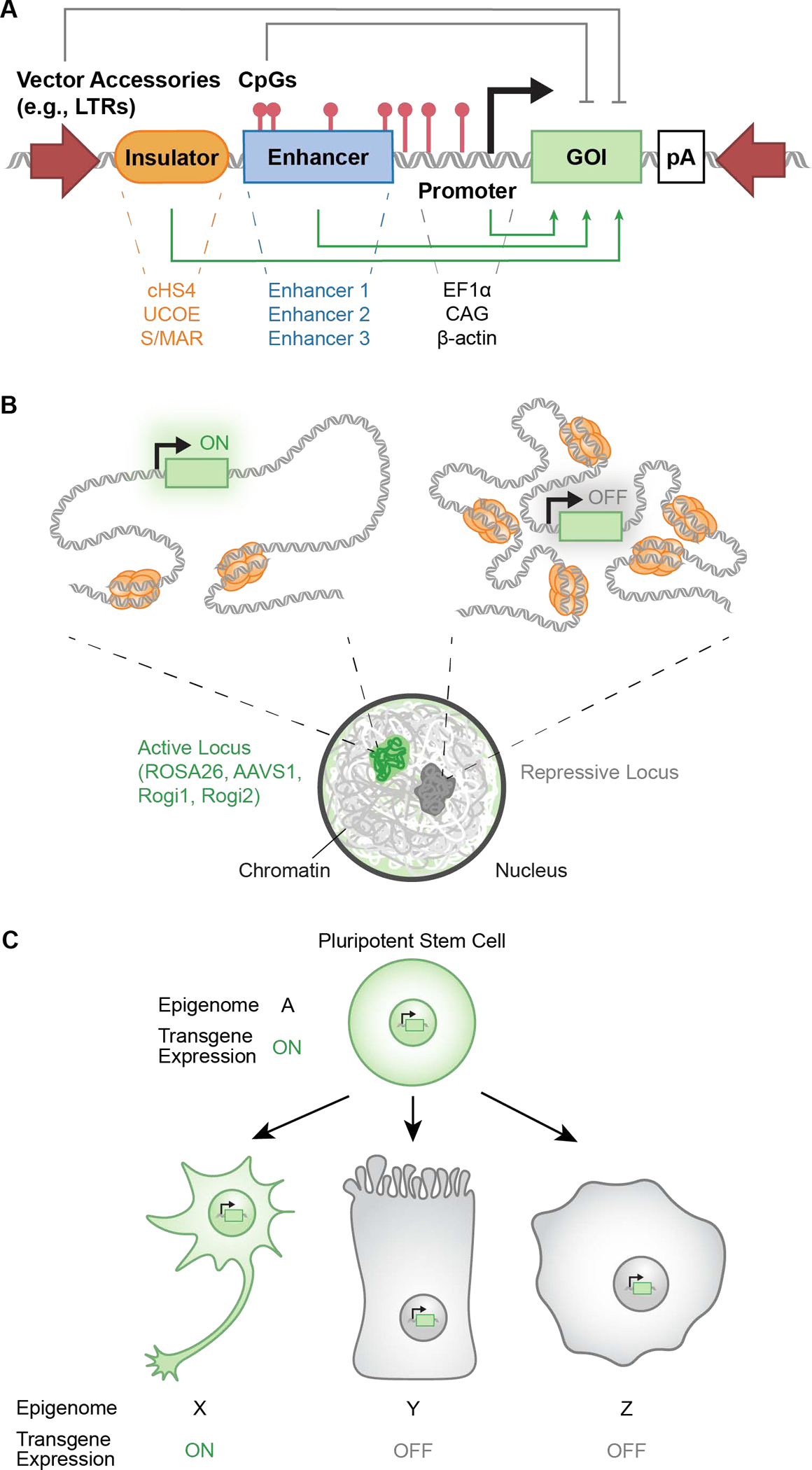
A. Genetic elements including insulators, promoters, and the combination of activating and repressive elements regulates gene expression. Promoters, enhancers, insulators, and CpG islands facilitate continuous gene expression. Elements such as low-density CpGs among GC-poor regions and viral sequences such as long-terminal repeats act as targets for transcriptional repression. Stability of transgene expression can be improved through the inclusion of activating elements, exclusion of repressive elements, and sequence-specific parameter optimization. B. Transgene circuit integration into heterochromatic, repressive genomic loci increases the likelihood of silencing. Targeted integration of transgenes into genomic safe harbors that remain ubiquitously euchromatic may reduce silencing. C. Stem cell differentiation induces genome-wide changes across CpG methylation, histone modification, and chromatin remodeling landscapes. Transgene expression depends on the epigenome of the differentiated lineage; expression might be safeguarded through CpG islands, tissue-specific enhancers, and transgene integration into ubiquitously open genomic safe harbors. Abbreviations: Gene of interest (GOI); Poly-adenylation signal (pA); Long terminal repeat (LTR).
Genomic locus of integration affects stable transgene expression
One major driver of transgene silencing and the instability of expression over time is the local repressive environment of the integration site in the genome. Viral vectors (e.g., retroviruses or lentiviruses) or transposase/transposon systems (e.g. piggyBac or Sleeping Beauty transposase) can deliver synthetic DNA cargo into the mammalian genome with high efficiency in a semi-random manner, as different vectors and systems have their own integration biases 163,164. However, due to the uncontrolled integration, there is often minimal to no regulation over the integrant copy number or the integration site(s), which may result in concatemer-induced epigenetic silencing 21 or silencing caused by the existence or spreading of local repressive chromatin at the integration site 165. More importantly, because transgenes exhibit different levels of expression when integrated into different chromosomal sites, random insertion is often unfavorable when systematic comparison or characterization of multiple DNA elements (promoter, enhancer, insulator, etc.) is desired.
One strategy to avoid these drawbacks associated with uncontrolled integration is to insert transgenic DNA at a predefined, transcription-permissible locus in the genome. Often the empirically determined genomic safe-harbor regions were chosen for this purpose 166. Currently, the popular choices of safe harbor loci include AAVS1, CCR5, and hRosa26 in the human genome, and Rosa26 and Hipp11 in the mouse genome. These commonly adopted safe harbor loci have been validated in various models including human iPSCs and ESCs, human CD34+ T cells, HEK293T cells, CHO cells as well as transgenic mice 167–171. Interestingly, a recent study by Aznauryan et al. identified two novel safe harbor sites (Rogi1 and Rogi2) that are capable of stable and safe expression of transgenes 172. These two sites were first tested in HEK293T and Jurkat cells for long-term transgene expression, and were further validated in primary T cells and dermal fibroblasts, thus offering more target choices in the human genome (Figure 3B). However, safe-harbor regions may remain vulnerable to epigenetic silencing for reasons discussed above. Potentially, a combination of strategies may most effectively reduce the probability of transgene silencing.
These loci can be targeted with programmable genome editing tools such as the CRISPR-Cas9 system to achieve targeted insertion of relatively short DNA sequences (e.g., single gene cassettes) with high efficiency. However, considering that synthetic gene circuits typically consist of multiple transcription units, the inevitably large size (e.g. greater than 15 kb) of the circuit makes it challenging for CRISPR-based genomic insertion 25,173,174. A serine integrase, on the other hand, is capable of integrating large DNA cargos with high specificity in mammalian cells 25,173,174. Recently, the serine integrase-based landing-pad strategy has been widely adopted for various applications involving large DNA constructs, with examples including the rapid prototyping of synthetic DNA circuits 175, the parallel assessment of large human gene variants library 176, and the integration of up to nine copies (~100 kb) of a monoclonal antibody-expressing gene cassette to improve antibody production in mammalian cells 177. Although serine integrases can be advantageous in their high specificity and large cargo capability, one caveat is that they require a landing pad (namely, an att recognition site) to be previously inserted at the chosen site to create a chassis cell line, which limits the ability to multiplex such a strategy. Therefore, recent development in novel genome editing tools combining CRISPR and integrases for targeted insertion of large DNA sequences 178 as well as the discovery of novel integrases with better activities at both landing pads and directly targeting the human genome 179, could enable the synthetic biology community to more rapidly test locations in the genome to characterize synthetic gene circuits in mammalian cells.
Cell-type choice influences the stability of transgene expression
It is important not to assume that the transgene-silencing factors discussed here are present at the same levels in all cell types. Data downloaded from the Human Protein Atlas (HPA) show that one or more transgene-silencing–associated factors are expressed at high levels in cell lines that are often used as test beds for cell engineering, and levels vary across cell lines (Table 3). The wealth of available epigenomic data for these widely used cell lines (e.g., ENCODE 180 and 4DN, https://www.4dnucleome.org/cell-lines.html) should be leveraged to investigate context-dependent transgene behavior. For instance, chromatin immunoprecipitation (ChIP) signals can be used to compare levels of transcriptional silencing or activating chromatin features at genomically mapped safe harbor loci. Additionally, RNA-seq data can be used to identify factors that are expressed at high levels, and their impact on transgene silencing can be tested by genetic knock-down or chemical inhibition. For example, high HDAC1 expression may contribute to transgene silencing in T cells, as indicated by the HPA data for Jurkat cells. In T cells that showed lentiviral and retroviral transgene silencing after four weeks of passaging, treatment with HDAC inhibitors was used to restore transgene expression 181. Future work could similarly identify context-specific methods to mitigate transgene silencing.
Table 3.
Expression levels (normalized transcripts per million, TPM) from public RNA-seq data (Human Protein Atlas, proteinatlas.org 263 for key mediators of transgenes silencing.
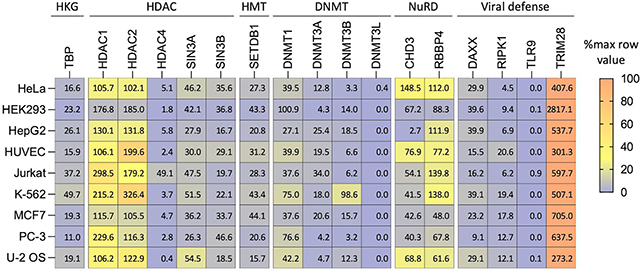
|
HKG: housekeeping gene (shown for comparison), HDAC: histone deacetylase, HMT: histone H3K9 methyltransferase, DNMT: DNA methyltransferase, NuRD: nucleosome remodeling and deacetylase complex.
For cancer-derived cell lines components of the SWItch/Sucrose Non-Fermentable (SWI/SNF) ATP-dependent chromatin remodeling complex frequently show loss of function mutations or low expression 182,183. This needs to be taken into consideration when attempting to modify histone marks via HDAC inhibition or epigenome editing to restore transgene expression because SWI/SNF is often required to act in concert with chromatin modifications such as histone acetylation to activate transcription.
Pluripotent stem cells allow for unlimited self-renewal and the ability to contribute to all germ-layers that give rise to the adult body. The pluripotent state entails unique epigenetic properties as unveiled through MoMuLV infection experiments of mouse embryos and pluripotent cells 107–109. First, ESCs express Zfp809 that mediates Setdb1-catalyzed H3K9 and H4K20 methylation of retroviral sequences through the Zfp809-Trm28-Setb1 complex, allowing the cells to efficiently repress expression of transgenes delivered and integrated through viral vectors 110,111,113,184. Second, mouse preimplantation stem cells abundantly express tRNA-derived fragments that inhibit translation of retroviral transcripts by competing for the primer binding site 185. Third, mouse embryos carrying a human β-globin gene regulated by a Cre-excisable methylation-resistant CGI methylate and silence the transgene only if the island is excised before implantation 61. These findings illustrate that pluripotent cells possess unique mechanisms that could silence transgene expression, and exit from pluripotency concomitant to implantation involves de novo methylation that is associated with transcriptional silencing 54,55.
During mouse development, high-density CpG promoters and CGIs are resistant to de novo methylation, most of which occurs during implantation at the E4.5-E5.5 transition catalyzed by DNMT3A and DNMT3B 55. Given the comparability of naïve pluripotent ESCs to preimplantation E4.5 epiblast cells 186, it is conceivable that differentiation recapitulates passage through the developmental stage of implantation and the surge of de novo DNA methylation. Indeed, mESCs exiting naïve pluripotency exhibited increased DNMT3A/DNMT3B expression and genome-wide CpG methylation after 24 hours of differentiation triggered by PD0325901/CHIR99021 withdrawal from the media (with notable resistance of CGI promoters), although no correlation between promoter methylation and respective gene expression was determined 187. Similarly, differentiating mESCs accrue DNMT3A and DNMT3B-dependent CpG methylation in the Oct4 promoter (curiously reduction of Oct4 mRNA preceded methylation 188, which has been described as a non-CpG promoter 68. High-density CpG and CGI promoters might provide candidates for safeguarding promoter activity during pluripotent stem cell differentiation.
Differentiation encompasses dynamic chromatin state changes, with different loci changing from an open to a closed chromatin state and vice versa. This can lead to silencing of randomly integrated transgenes in a promoter-independent and locus-dependent manner. Constitutively active loci allowing ubiquitous transgene expression have been identified to tackle this problem: the Rosa26 locus in the mouse genome 189, and AAVS1, CCR5 and Rosa26 in the human genome 97,167. It will be interesting to see how the two newly identified human genomic safe harbors 172 fare in ensuring ubiquitous transgene expression during human stem cell differentiation. Altogether, global changes in DNA methylation and chromatin states are critical factors of transgene activity during stem cell differentiations and reprogramming (Figure 3C). CGIs and safe harbors may provide solutions to these barriers.
Avoiding nutrient limitations supports active transgene expression
Engineered cell lines may encounter signals from the microenvironment that induce dramatic shifts in metabolic states that could impact epigenetic regulation of transgenes. A pool of metabolites that become depleted or replenished in response to environmental cues also provides substrates for the chromatin modification machinery. For instance, the free pool of acetyl-coA, the sole substrate for acetylation of histones in transcriptionally active chromatin, is heavily regulated by signals linked to nutrient availability and is primarily derived from extracellular glucose levels. Exposure to fatty acids or insulin can increase lipid storage and synthesis 190–192. In yeast, when glucose becomes unavailable and cells enter the stationary phase, lipid synthesis outcompetes histone acetyltransferases (HATs) for acetyl-coA, and histone acetylation levels decrease 193,194, which favors the formation of closed chromatin. Human cancer cells show increased lipogenesis and broad reprogramming of gene expression in response to signals from adipocytes 195–197. Thus, low levels of glucose in cell culture media could lead to transgene silencing through decreased availability of acetyl-coA, as well as high levels of fatty acids or insulin. Lactate can play a similar role to acetylation through lactylation of histones. Evidence thus far of this novel histone mark supports that lactylation promotes maintenance of active genes 198,199, so low levels of lactate could result in silencing. However, a delicate balance must be struck as high levels of lactate generally reduce cell growth and protein production 200. Furthermore, inhibition of histone demethylases by D-2-hydroxyglutarate (D2HG), an “oncometabolite” produced by mutated IDH1/2, has been implicated in gene silencing in cancer cells, and has been mechanistically linked to elevated H3K39me3 and gene silencing in yeast 201. Taken together, these observations suggest the importance of careful growth medium design and feeding strategies in order to reduce transgene silencing, focusing on providing sufficient glucose and reducing flux through the D2HG pathway.
PROSPECTS FOR THE FUTURE
Despite the identification of abundant potential mechanisms and diverse strategies for mitigating transgene silencing, silencing persists as a challenge for cellular engineering, highlighting the importance of new descriptive studies and novel strategies for stable transgene expression. Basic research into the biology of silencing could elucidate its molecular and physical basis, identify the responsible host genes and pathways, and inform new strategies to address this challenge. Here, we propose future research directions that could propel the field of mammalian synthetic biology past the current challenges of transgene silencing. This research includes the application of publicly available data to investigate silencing at the epigenetic level or identify silencing-resistant promoters, CRISPR-based screens to identify genes associated with silencing and massively parallel reporter assays (MPRAs) to evaluate new circuit components to prevent silencing. Finally, alternative engineering solutions could be further developed to mitigate transgene silencing, including non-integrating methods of stable expression, post-transcriptional and translational regulation, and epigenetic modifying circuits.
Mapping chromatin modifications in the transgene and at the integration loci
Mapping chromatin modifications both at endogenous integration loci and in silenced transgenes will be useful for better understanding the characteristics of effective safe-harbor loci as well as the mechanisms by which transgenes are epigenetically silenced (Figure 4A). Investigating chromatin modifications at integration sites across different cell types of interest could help identify any differences that affect silencing of a transgene upon integration. Several databases including ENCODE 180, 4DN 202 and Human Cell Atlas 203 contain data on chromatin modifications (ChIP-seq, CUT&RUN), chromatin accessibility (ATAC-seq) and gene expression (RNA-seq) in both human cell lines and primary cells. Using this epigenetic information to better characterize existing safe harbor loci will also aid the discovery of new integration sites that are less prone to transgene silencing. Similar epigenetic profiling of silenced transgenes for different chromatin modifications such as DNA methylation, H3K27me3, H3K9me3, and H3K27ac will provide insights into how transgenes are silenced, including which chromatin complexes are involved and how transgenes are recognized or potentially targeted for silencing by the cell. Mapping these chromatin modifications will help inform larger screens to determine genes that are responsible for transgene silencing.
Figure 4. Technologies being applied to better understand or manipulate transgene silencing.
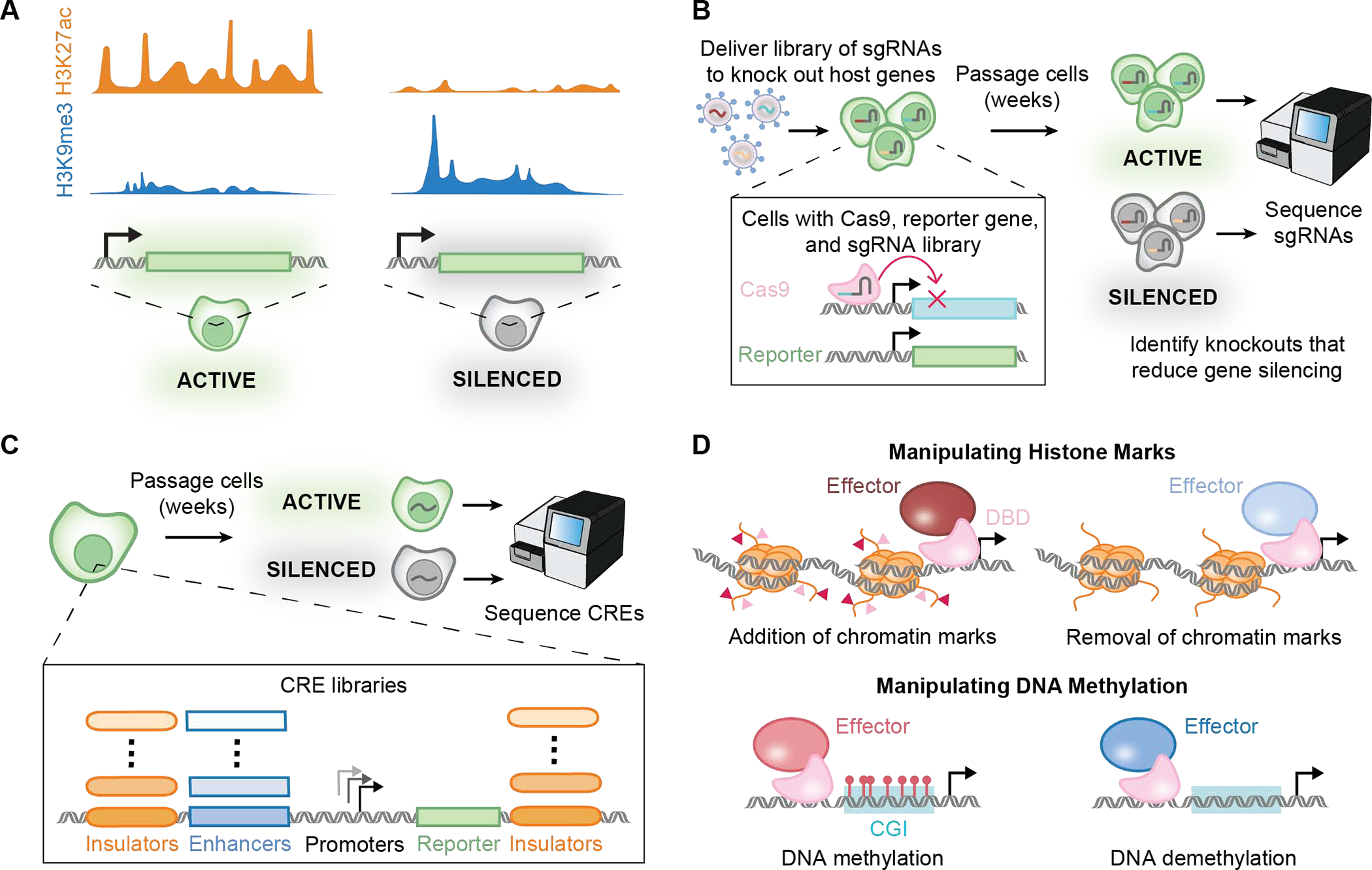
A. Measurement of chromatin marks and DNA methylation can inform the epigenetic state of a synthetic genetic element as well as the epigenetic modifications involved. The measurements can be performed using techniques such as ChIP-Seq. B. CRISPR screens can identify genetic dependencies for transgene silencing. A reporter gene is engineered in cells that also express Cas9. A library for sgRNAs is delivered to the cells. A cell receives a unique sgRNA that targets and knocks out a unique endogenous gene. The cells are passaged, and time is allowed for gene silencing to occur. Sequencing of the gRNAs is performed to identify knockouts that reduce epigenetic silencing. C. Massively parallel reporter assays (MPRAs) enable systematic identification of cis-regulatory elements (CREs), including promoters, enhancers, and insulators that can maintain transgene expression and prevent silencing. A library of CREs regulating a reporter gene is installed in a population of cells, the cells are passaged to allow time for gene silencing to occur, the cells are separated by reporter level, and the CREs are sequenced to identify library elements that are enriched in the population of cells with maintained gene expression. D. Synthetic chromatin regulators can be engineered by fusing DNA-binding domains (DBDs) to epigenetic modifying effectors, such as enzymes that catalyze specific additions or removal of methylation on histones or DNA. This and other technologies such as ChIP-qPCR, CUT&RUN, methylated DNA immunoprecipitation, and bisulfite conversion enable synthetic biologists to understand the effect of different epigenetic effectors and to manipulate epigenetic silencing.
Identifying constitutive endogenous promoters by harnessing publicly available data
Identification of additional stable constitutive promoters could also be useful for maintaining transgene expression, as these elements may be silencing-resistant. This could be accomplished by scanning the mammalian transcriptomes for ubiquitously expressed housekeeping genes, mapping their respective promoters in the genome, and utilizing highly conserved candidates for stable transgene expression. The FANTOM5 database lists such housekeeping gene promoters for mouse and human cells, making it an attractive tool for identifying species-conserved stable promoter sequences 204. Indeed, the ubiquitous-uniform promoter category contains β-actin and elongation factor-family genes (e.g. EF1α), along with p53 and members of the ribonucleoprotein processing machinery. On the other hand, promoters of non-coding RNAs showed the least cross-species conservation, while non-TATA and CGI-based promoters demonstrated non-ubiquitous expression 204. CGIs are thought to confer resistance to DNA methylation-dependent gene silencing, so the non-ubiquitous expression associated with CGI promoters possibly reflects differential methylation: most CGIs are ubiquitously unmethylated, about 25% are ubiquitously methylated, and a few thousand exhibit tissue-specific methylation 68. Therefore, the subcategory of housekeeper-associated unmethylated CGIs might hold attractive candidates for safeguarding transgene expression. Alternatively, novel CGI-promoter hybrid combinations could create synthetic promoters with desired properties. In support of this notion, fusion of the CGI of theCHO-K1 β-actin promoter to the CMV promoter improved long-term transgene expression and antibody production yields in CHO-K1 cells, compared to the original CMV promoter 205. A high-throughput synthetic biology approach utilizing databases listing CGIs, transcription start sites 68 and mammalian promoters 204 could facilitate the computational design and genetic engineering of novel CGI-promoter variants optimized to ensure stable transgene expression.
CRISPR screens to map genes responsible for transgene silencing
CRISPR screens can provide a better understanding of mechanisms that have evolved to avoid silencing and will inspire new synthetic biology strategies 206–208. Endogenous cellular pathways are responsible for the silencing of transgenic payloads. Therefore, an identification of such pathways could facilitate the prevention of these silencing mechanisms. In order to map the cellular pathways involved in transgene silencing, one could perform high-throughput loss-of-function screens, using gene-perturbation strategies to avoid unintended consequences associated with the use of small-molecule inhibitors, such as effects on native gene expression and potential cellular toxicity 209,210. Pooled CRISPR screens are a promising strategy to identify the host genes required for silencing (Figure 4B). One could use a genome-wide or epigenetics-focused sgRNA library to knockout host genes and then measure sgRNA enrichments in cells where a synthetic gene circuit is silenced versus maintained. Identification of the required host genes could be immediately useful. CRISPR screens have been used to identify genes involved with drug resistance, influenza A virus infections, and cellular reprogramming 211–213. For example, one could generate stable knockout or knockdown cell lines that are silencing-incompetent, or transiently inhibit the host silencing machinery with small molecules or siRNAs to prevent or reverse silencing 125,214–216
Aside from the practical outcomes of stable transgene expression, a deeper investigation of these host silencing pathways could expand our understanding of how the host machinery evolved to silence synthetic genes. Additionally, studies of the cGAS-STING sensing 217, and chromatinization of episomal transgenes indicate that DNA (e.g., linear DNA donor template for CRISPR editing and plasmid DNA used in Sleeping Beauty transposon system) can be sensed by the cell and modified prior to integration. Therefore, CRISPR screens prior to, and post, integration can provide insights into the pathways involved in silencing. One possible outcome is the identification of new mechanisms that have evolved to defend the cell against other foreign DNA including viruses and transposable elements.
Massively parallel reporter assays (MPRAs) as a method to find novel elements and understand their optimal deployment
Advances in technologies for genetic screening, epigenomics, and synthetic gene circuit design are creating new opportunities to characterize and prevent transcriptional silencing in mammalian cells. Furthermore, high-throughput screening of endogenous genetic elements could facilitate the rational design of new construct components and aid in the understanding of epigenetic regulation with the ability to rapidly screen synthetic gene circuit stability after integration into the genome. Massively parallel reporter assays (MPRAs) including CapSTARR-seq, a high-throughput method to quantify enhancer activity 218,219 and Functional Identification of Regulatory Elements Within Accessible Chromatin (FIREWACh) 220 for mammalian cells have primarily been used to measure enhancer and promoter activity; however, most studies probe enhancers with the same minimal promoter that is decoupled from genomic locus as most reporters are not integrated into the genome 221,222. Chromosomal domains have long been shown to affect transgene expression 223, and lentivirus based MPRA has shown that genomically integrated reporters have different expression than their episomal counterparts 224. A method termed Thousands of Reporters Integrated in Parallel (TRIP), allows high-throughput investigation of the influence of genomic integration loci on transgene expression 165. Analysis of two promoters, mPGK and tet-Off, at 27,000 loci in mESCs showed 1,000-fold variation in expression levels, where chromatin state is related to expression level and lamina-associated domains attenuated transcription, while nearby enhancers increased expression 165. Additionally, MPRA of enhancers have also shown cell-type specificity 225. While there are numerous screens on the effect of enhancer-promoter interactions, promoters and insulators have been less well characterized. Screening human promoters at AAVS1 safe harbor locus in K562 cells found that core promoters drive unidirectional transcription 226. Screening more broadly across loci, promoter activity scales across regions of integration suggesting that integration context provides a factor over the promoter-intrinsic properties 227. However, there remain many human promoters to characterize for their variance in expression levels in different cell types and for their propensity of transgene silencing (Table 3). Using MPRAs to better characterize promoters in different genomic contexts and cell types would help identify which loci and promoter combinations in specific cell types reduce transgene silencing, as well as help further understand the mechanisms that drive silencing (Figure 4C). In addition to testing different cell types, promoters should also be tested in different cell states such as under various metabolic conditions or in the presence of immune simulation. High-throughput screening may facilitate identification of novel insulator elements that resist transgene silencing. When encoded proximal to transgenes, existing insulators such as UCOE and cHS4 promote stable transgene expression. Candidate UCOE elements vary in their ability to limit transgene silencing and depend upon the choice of the promoter 162. Further efforts to screen and characterize diverse insulators will help add more reliable insulators to the synthetic biology toolbox.
New genetic elements should be benchmarked against current gold-standard regulatory elements to define their effectiveness. As noted above, genetic elements function differently between cell types and lines, so the performance of these elements will likely require a systematic comparison within the relevant cellular context. MPRAs have immense potential to discover new parts and optimize circuit configuration, but researchers ought to begin standardizing currently available genetic elements. For instance, alternate sequences of related genetic elements such as EF1α and EFS promoters can have drastically different performance and properties 228. Therefore, a systematic comparison of currently available elements is a necessary step towards standardization of best practices to inform optimal construction, enhance circuit robustness, and minimize systemic inefficiencies.
Inspiration from evolved solutions to transgene silencing
Viruses have evolved many mechanisms to avoid identification and silencing of viral elements. Therefore, there is significant potential in repurposing evolved viral defense mechanisms to design novel stable transgene expression strategies. For instance, incorporation of the S/MAR element was shown to enhance nuclear transport of transfected episomal DNA 229. Engineering of this phenomenon may eventually yield robust expression of transgenes and synthetic circuits. As another example, viral covalently closed circular DNA (cccDNA) persistence is a hallmark of Hepatitis B virus (HBV) infection and there are non-integrated HBV elements that are believed to interact with host chromatin related proteins to regulate viral gene transcription. Additionally, the persistence of these foreign elements is believed to be the cause of relapse after viral infection clearance 230. The production of recombinant cccDNA has been employed in the search for drugs to treat and remove these persistent HBV elements 231. Although these tools will need to be further explored and engineered, recombined cccDNA may be leveraged for stable transgene expression. Another viral defense mechanism involves the suppressors of RNA silencing (VSRs), a mechanism used by viruses to interfere with host RNA interference following infection of plant, insect, and mammalian cells 232 Engineering elements inspired by these and newly discovered viral defense mechanisms have the potential to result in the design of robust genetic circuits.
Managing silencing in extrachromosomal vectors
Our understanding of the challenges associated with silencing in extrachromosomal vectors is informed in part by substantial experience with adeno-associated virus (AAV) vectors, which exist as episomal DNA. Adeno-associated viruses (AAVs) have attracted a significant amount of attention for use as a gene therapy vector to deliver DNA in vivo because they can have low immunogenicity and low rates of insertional mutagenesis 233. Nevertheless, two early-phase clinical trials of gene therapy for inherited vision loss reported only short-term vision improvement following the treatment of patients with recombinant AAV 234,235. Although the underlying mechanism of the decline in improved vision in the long-term remains unclear, transgene silencing was proposed as a potential cause in one of the studies 234. The hypothesis that transgene silencing was the major cause of the poor robustness of AAV gene therapy approaches has yet to be proven, especially since cellular turnover and immune responses could also play a significant role. Determining the mechanism(s) involved in the durability of AAV gene therapy in vivo presents a challenge, and this challenge becomes increasingly complicated as studies focus on more complex and therapeutically relevant systems. For example, a preclinical study of liver-targeted AAV found a strong correlation between liver vector DNA copy number and transgene protein expression level in mice, but that there was very little protein expression from non-human primate liver despite DNA levels of approximately 1–100 vector copies per cell. The authors proposed vector silencing as one possible culprit 236. A recent preprint describes a study on the loss of AAV transgene expression in the primate liver that used in situ hybridization and found a disproportionate loss of transgene RNA relative to DNA over time 23. At day 14 there was high transgene expression and AAV DNA was found dispersed throughout the nucleus, whereas by day 77, the expression was largely lost and AAV DNA was found in a few distinct foci, which may be transcriptionally inactive. Since AAV rarely integrates into the genome and is not rapidly diluted in non-dividing cells, the use of AAV is viewed as one of the safest and most practical approaches for gene therapies 233. However, with a positional bias towards transcriptionally active regions 237–239, it has been proposed that the few integration events may drive what is left of transgene expression after the virus wanes through cell divisions. Further, SETDB1, the H3K9 methyltransferase, has been identified by several groups as a host factor that can reduce both the percentage of transgene-expressing cells and the level of expression among those transduced cells when using AAV, lentivirus, and adenovirus delivery methods 240–242. These findings further implicate chromatin-mediated transgene silencing as a mechanism with significant influence over long-term AAV expression. In summary, the loss of AAV expression is an important area of active investigation, and transgene silencing of gene therapy vectors may be a major barrier to achieving long-term high treatment efficacy.
Introducing artificial chromosomes may avoid silencing mechanisms inherent to integration in endogenous loci. Human artificial chromosome (HACs) are a potential solution to limitations regarding transgene size limits, positional regulation effects and silencing associated with viral elements 243,244. So far, the complexity of assembling HACs has limited their construction as well as their benchmarking with other transgene delivery methods 245. Ongoing efforts have resulted in more streamline assembly and delivery of HACs into human T cells and iPSCs 246. However, transcriptional silencing does occur on current-generation HACs. Thus, while bottom-up engineering may provide silencing-resistant HACs in the future, further characterization is required for HACs to become a viable method for preventing transgene silencing.
Post-transcriptional and post-translational mechanisms as alternative regulatory strategies
Genetic circuit designs that robustly resist epigenetic silencing may not be compatible with common methods of transcriptional control, meaning alternative regulatory strategies are needed. For instance, synthetic circuits that are exclusively composed of active promoters may silence less than transcriptionally regulated circuits. In order to achieve the regulatory function of the genetic circuit, post-transcriptional or post-translational mechanisms could be employed instead. Post-transcriptional control can be achieved by including regulatory elements in untranslated regions of mRNA. These parts include microRNA and microRNA binding sites, RNA binding protein motifs, and ribozyme switches. For a more detailed review on this topic, see 247. Recently, CRISPR Cas-binding motifs 129,248 and toehold switches 249 have also been used to engineer post-transcriptional or translational control. Alternatively, regulation can be implemented at the post-translational level using protein domains responsive to external inputs (such as small molecule- or light-inducible degrons) or by engineering protein-protein interactions. Complex logic has been achieved entirely post-translationally using proteases 250–252. Because these circuits act independently from transcription, they are compatible with an array of promoters and expression methods. Thus, post-transcriptional and post-translational regulatory strategies could facilitate the decoupling of functional modules from transcriptional components that resist epigenetic silencing, allowing each to be optimized separately.
Engineered epigenetic modulation to counteract silencing
Synthetic biology tools for epigenetic modulation can be potentially used in genetic circuits to directly counteract epigenetic silencing. Previously developed tools include engineered synthetic chromatin regulators, which typically consist of a DNA binding domain fused to an epigenetic effector domain 253. The DNA binding domain can be programmable, including Zinc-finger proteins, TALEs and CRISPR proteins 254,255. This enables epigenetic modifications to be targeted to synthetic genetic circuits, such as removal of silencing modifications (e.g., demethylases) or addition of activating modifications (e.g. acetyltransferases) 256,257 (Figure 4D). Besides using these approaches to study and modulate epigenetic silencing 14, engineered synthetic chromatin regulators can be used as part of synthetic gene circuits to counteract transgene silencing. A potential approach involves the use of feedback, which is already employed in synthetic gene circuits and natural epigenetic regulatory pathways. For instance, in a positive autoregulatory synthetic gene circuit, synthetic chromatin regulators could be used to constitutively remove repressive epigenetic marks and maintain an open chromatin structure or maintain epigenetic marks associated with active transcription. This approach can be complemented with control theoretic systems biology strategies that mathematically model and study the role of feedback in epigenetics 258,259. In another approach, a circuit could employ mechanisms to detect epigenetic silencing and subsequently activate an effector to remove the repressive marks. In this case, VP64-based transcriptional activators have been engineered to specifically bind repression-associated histone marks such as H3K27me3 260 and ChIP data from the epithelial cell line, U2OS, suggest a change in chromatin state from silenced to active perhaps through Mediator recruitment (MED25, MED17) 261. To use this approach for transgenes, a DNA sequence recognition module would need to be incorporated to achieve transgene-specific regulation and avoid off-target activation elsewhere in the genome. Finally, synthetic chromatin regulators can be used to engineer efficient transitions between closed and open chromatin states. Linking the establishment of an epigenetic state to an input of interest would enable the use of epigenetic memory as a form of information storage.
Conclusion
In conclusion, although transgene silencing poses significant challenges for mammalian cell engineering, efforts are underway to elucidate the molecular mechanisms responsible for this phenomenon and develop solutions to mitigate it. The field of mammalian synthetic biology can overcome the challenge of transgene silencing by sharing silencing data with current elements discovering new regulatory elements and delivery approaches identifying and intervening with problematic pathways, and employing additional layers of transcriptional and post-transcriptional regulation. The design of more robust tools for mammalian cell engineering will undoubtedly accelerate the fields of cell and gene therapy, biomanufacturing, and basic biology research.
ACKNOWLEDGMENTS
Funding for this work was supported in part by the National Institutes of Health grants 1DP2CA250006-01 (T.L.D.), 1R01GM129011 (W.W.W.), R01EB029483 (W.W.W.), 1R01EB026510 (J.N.L), R21EB030772 (I.B.H.), R35GM143532 (I.B.H), R35GM143033 (K.E.G), R35GM138256 (L.M.), 4K00DK126120-03 (J.T.), R01EB029483 (A.S.K.), R35GM128947 (L.B.), R01EB030946 (R.W.), R01EB025256 (R.W.), 1UM1HG009402 (H.Z.), U54DK107965 (H.Z.), R21CA232244 (K.A.H.), 1RC2DK120535-01A1 (J.J.C.), and 1 U01 DK 127420- 01 (M.B.E.), the National Science Foundation grants CBET-2034495 (L.M.), CBET-2145528 (L.M.), 2141064 (K.S.L.), EF-1921677 (A.S.K) and EF-2021552 under subaward UWSC10142 (M.B.E.). Further support was also provided by the Biotechnology and Biological Sciences Research Council (BBSRC) BB/S006206/1 (K.P.) and BB/M018040/1 (S.J.R.), ElectroGene 785800 (M.F.), the SNF (M.F.), the Paul Allen Foundation (W.W.W.), AOFSR FA9550-22-1-0316 (K.E.G.), the Wellcome Sanger Institute LEAP 21-275 (L.M.), the Parker Institute for Cancer Immunotherapy (Y.Y.C.), the W.H. Coulter Department of Biomedical Engineering at Emory University (C.S.), and the DoD Vannevar Bush Faculty Fellowship N00014-20-1-2825 (A.S.K.). M.B.E. is a Howard Hughes Medical Institute Investigator.
Glossary
- Transgene
A gene that is delivered and expressed in a cell to produce a desired phenotype. A transgene can refer to a native gene that is introduced artificially (for example, for high expression), a gene from another organism, or an entirely synthetic gene. A transgene is often implemented as a transcriptional unit with other components that regulate its expression such as a promoter, a Kozak consensus sequence, and poly-adenylation signal
- Transgene silencing
Loss or down-regulation of expression of a transgene in a cell despite its encoding DNA remaining present in the cell’s nucleus
- Hysteresis
a property or phenomenon in a system, where the state of the system depends on its history or prior events
- Cassette
A single unit consisting of a transgene that has yet to be integrated into a genome
- Synthetic gene circuit or genetic program
An assembly of cassettes that encode RNA or protein molecules that interact with each other to perform one or more biological functions
- Transcriptional unit
The DNA sequence necessary to produce a single, unified RNA transcript. The transcriptional unit supports expression of one gene or unit of genes. A transcriptional unit often includes a promoter sequence, coding sequence and poly-adenylation sequence. Transcriptional units may include additional elements within the coding sequence such as a splice site or other regulatory elements within the untranslated regions (UTRs)
- Safe-harbor site or safe-harbor locus
A locus in the genome with an open chromatin state that is amenable for stable transgene expression without adversely affecting normal cellular functions (e.g. the activation of nearby oncogenes)
- Insulator
DNA elements that serve as barriers to transcriptional and epigenetic regulation of surrounding genes. Specifically, DNA insulators can exhibit barrier activity to block the spreading of heterochromatin or repressive epigenetic modifications. Alternatively, DNA insulators may function as enhancer blockers to prevent the acting of enhancers on the promoters of neighboring genes. Certain DNA insulators such as the prototypic cHS4 exhibit both functions
- Enhancer
DNA elements that interact with target promoters to amplify initiation of transcription
- Epigenetic effector
A protein that can modulate the addition or removal of epigenetic modifications on histones or DNA
- Poly-adenylation signal (pA)
DNA sequence that signals the transcription complex to poly-adenylate the RNA being transcribed. This stabilizes RNA molecules by preventing their degradation to enable their export from the nucleus to the cytoplasm to be translated into protein by ribosomes
- Long terminal repeat (LTR)
One of a pair of DNA sequences that form a retrotransposon, retrovirus or previrus. LTRs flank retroviral genomes and are common in lentiviral vectors and lentiviral integrations
- CpG islands (CGIs)
Large DNA segments with high content of CpGs, particularly as compared to other regions of DNA
- H3K9me3
A heterochromatin associated mark that is associated with the downregulation of nearby genes
- Viral vectors
Engineered or modified viruses that serve as a vehicle for efficient delivery of nucleic acids to cells. Examples include lentiviral vectors and adeno-associated virus (AAV) vectors
- Topologically associating domains (TADs)
TADs are self-interacting chromosomal domains identified by chromosome conformation capture technologies. DNA sequences located within the same TAD interact more frequently with each other than with DNA sequences outside the TAD. TADs are proposed as the fundamental regulatory units of the genome three-dimensional architecture
- Serine Integrases
Single protein systems, derived from mobile genetic elements, that can integrate a large DNA sequence into the genome by mediating recombination between attachment sites (DNA motifs of ~30 bp) on the genome and donor. Examples include the BxB1, PhiC31, and Pa01 large serine recombinases
- Transposon systems
Systems for transgene integration in the genome that utilize transposition as a mechanism for genomic integration. Examples include the piggyBac system and the sleeping beauty system
- EF1α and EFS promoters
EF1α is the promoter sequence derived from the human EEF1A1 gene that expresses the alpha subunit of eukaryotic elongation factor 1. EF1α is known as one of the strong promoters in various mammalian cell lines. The EFS promoter is the short, intronless form of the EF1α promoter
- cHS4 and cHS4 core insulators
The cHS4 (chicken hypersensitive site 4) insulator is the prototypical chromatin insulator derived from the chicken β-like globin gene cluster. It possesses both the enhancer-blocking and barrier activities and has been adopted for transgene insulation in various mammalian cell lines. The cHS4 core usually refers to the 5’ 250 bp of the full-length cHS4 insulator
Footnotes
DECLARATION OF INTERESTS
J.T. and L.B. acknowledge outside interest in Stylus Medicine.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xie M, and Fussenegger M (2018). Designing cell function: assembly of synthetic gene circuits for cell biology applications. Nat. Rev. Mol. Cell Biol. 19, 507–525. [DOI] [PubMed] [Google Scholar]
- 2.Kitada T, DiAndreth B, Teague B, and Weiss R (2018). Programming gene and engineered-cell therapies with synthetic biology. Science 359. 10.1126/science.aad1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kis Z, Pereira HS, Homma T, Pedrigi RM, and Krams R (2015). Mammalian synthetic biology: emerging medical applications. J. R. Soc. Interface 12. 10.1098/rsif.2014.1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lienert F, Lohmueller JJ, Garg A, and Silver PA (2014). Synthetic biology in mammalian cells: next generation research tools and therapeutics. Nat. Rev. Mol. Cell Biol. 15, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson MB, March AR, and Morsut L (2017). Engineering multicellular systems: using synthetic biology to control tissue self-organization. Curr Opin Biomed Eng 4, 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang NB, Beitz AM, and Galloway K (2020). Engineering cell fate: Applying synthetic biology to cellular reprogramming. Current Opinion in Systems Biology 24, 18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacDonald IC, and Deans TL (2016). Tools and applications in synthetic biology. Adv. Drug Deliv. Rev. 105, 20–34. [DOI] [PubMed] [Google Scholar]
- 8.Deans TL, Cantor CR, and Collins JJ (2007). A tunable genetic switch based on RNAi and repressor proteins for regulating gene expression in mammalian cells. Cell 130, 363–372. [DOI] [PubMed] [Google Scholar]
- 9.Keung AJ, Joung JK, Khalil AS, and Collins JJ (2015). Chromatin regulation at the frontier of synthetic biology. Nat. Rev. Genet. 16, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.E. O, Subkhankulova T, and Tolmachov T (2013). Silencing of transgene expression: A gene therapy perspective. In Gene Therapy - Tools and Potential Applications (InTech; ). [Google Scholar]
- 11.Brachmann CB, Sherman JM, Devine SE, Cameron EE, Pillus L, and Boeke JD (1995). The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 9, 2888–2902. [DOI] [PubMed] [Google Scholar]
- 12.Duan B, Ding P, Navarre WW, Liu J, and Xia B (2021). Xenogeneic Silencing and Bacterial Genome Evolution: Mechanisms for DNA Recognition Imply Multifaceted Roles of Xenogeneic Silencers. Mol. Biol. Evol. 38, 4135–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gartenberg MR, and Smith JS (2016). The Nuts and Bolts of Transcriptionally Silent Chromatin in Saccharomyces cerevisiae. Genetics 203, 1563–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bintu L, Yong J, Antebi YE, McCue K, Kazuki Y, Uno N, Oshimura M, and Elowitz MB (2016). Dynamics of epigenetic regulation at the single-cell level. Science 351, 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hathaway NA, Bell O, Hodges C, Miller EL, Neel DS, and Crabtree GR (2012). Dynamics and memory of heterochromatin in living cells. Cell 149, 1447–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Felsenfeld G (1992). Chromatin as an essential part of the transcriptional mechanim. Nature 355, 219–224. [DOI] [PubMed] [Google Scholar]
- 17.Behe M, and Felsenfeld G (1981). Effects of methylation on a synthetic polynucleotide: the B--Z transition in poly(dG-m5dC).poly(dG-m5dC). Proc. Natl. Acad. Sci. U. S. A. 78, 1619–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uckelmann M, and Davidovich C (2022). An added layer of repression for human genes. Nature 604, 41–42. [DOI] [PubMed] [Google Scholar]
- 19.Tchasovnikarova IA, Timms RT, Matheson NJ, Wals K, Antrobus R, Göttgens B, Dougan G, Dawson MA, and Lehner PJ (2015). GENE SILENCING. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 348, 1481–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sutherland HG, Kearns M, Morgan HD, Headley AP, Morris C, Martin DI, and Whitelaw E (2000). Reactivation of heritably silenced gene expression in mice. Mamm. Genome 11, 347–355. [DOI] [PubMed] [Google Scholar]
- 21.Chusainow J, Yang YS, Yeo JHM, Toh PC, Asvadi P, Wong NSC, and Yap MGS (2009). A study of monoclonal antibody-producing CHO cell lines: what makes a stable high producer? Biotechnol. Bioeng. 102, 1182–1196. [DOI] [PubMed] [Google Scholar]
- 22.Yang Y, Mariati, Chusainow J, and Yap MGS. (2010). DNA methylation contributes to loss in productivity of monoclonal antibody-producing CHO cell lines. J. Biotechnol. 147, 180–185. [DOI] [PubMed] [Google Scholar]
- 23.Greig JA, Breton C, Martins KM, Zhu Y, He Z, White J, Bell P, Wang L, and Wilson JM (2022). Loss of transgene expression limits liver gene therapy in primates. bioRxiv, 2022.03.24.485675. 10.1101/2022.03.24.485675. [DOI] [Google Scholar]
- 24.Zimak J, Wagoner ZW, Nelson N, Waechtler B, Schlosser H, Kopecky M, Wu J, and Zhao W (2021). Epigenetic silencing directs expression heterogeneity of stably integrated multi-transcript unit genetic circuits. Sci. Rep. 11, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fitzgerald M, Livingston M, Gibbs C, and Deans TL (2020). Rosa26 docking sites for investigating genetic circuit silencing in stem cells. Synth. Biol. 5. 10.1093/synbio/ysaa014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lensch S, Herschl MH, Ludwig CH, Sinha J, Hinks MM, Mukund A, Fujimori T, and Bintu L (2021). Dynamic spreading of chromatin-mediated gene silencing and reactivation between neighboring genes in single cells. bioRxiv, 2021.11.04.467237. 10.1101/2021.11.04.467237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Babos KN, Galloway KE, Kisler K, Zitting M, Li Y, Shi Y, Quintino B, Chow RH, Zlokovic BV, and Ichida JK (2019). Mitigating Antagonism between Transcription and Proliferation Allows Near-Deterministic Cellular Reprogramming. Cell Stem Cell 25, 486–500.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Velychko S, Adachi K, Kim K-P, Hou Y, MacCarthy CM, Wu G, and Schöler HR (2019). Excluding Oct4 from Yamanaka Cocktail Unleashes the Developmental Potential of iPSCs. Cell Stem Cell 25, 737–753.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okada M, and Yoneda Y (2011). The timing of retroviral silencing correlates with the quality of induced pluripotent stem cell lines. Biochim. Biophys. Acta 1810, 226–235. [DOI] [PubMed] [Google Scholar]
- 30.Hu X, Wu Q, Zhang J, Kim J, Chen X, Hartman AA, Eastman AE, Park I-H, and Guo S (2021). Reprogramming progressive cells display low CAG promoter activity. Stem Cells 39, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernández-Tornero C (2018). RNA polymerase I activation and hibernation: unique mechanisms for unique genes. Transcription 9, 248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lombardo A, Cesana D, Genovese P, Di Stefano B, Provasi E, Colombo DF, Neri M, Magnani Z, Cantore A, Lo Riso P, et al. (2011). Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat. Methods 8, 861–869. [DOI] [PubMed] [Google Scholar]
- 33.Norrman K, Fischer Y, Bonnamy B, Wolfhagen Sand F, Ravassard P, and Semb H (2010). Quantitative comparison of constitutive promoters in human ES cells. PLoS One 5, e12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfaff N, Lachmann N, Ackermann M, Kohlscheen S, Brendel C, Maetzig T, Niemann H, Antoniou MN, Grez M, Schambach A, et al. (2013). A ubiquitous chromatin opening element prevents transgene silencing in pluripotent stem cells and their differentiated progeny. Stem Cells 31, 488–499. [DOI] [PubMed] [Google Scholar]
- 35.Smith E, and Shilatifard A (2010). The chromatin signaling pathway: diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol. Cell 40, 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uruci S, Lo CSY, Wheeler D, and Taneja N (2021). R-Loops and Its Chro-Mates: The Strange Case of Dr. Jekyll and Mr. Hyde. Int. J. Mol. Sci. 22. 10.3390/ijms22168850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reina C, and Cavalieri V (2020). Epigenetic Modulation of Chromatin States and Gene Expression by G-Quadruplex Structures. Int. J. Mol. Sci. 21. 10.3390/ijms21114172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rauschhuber C, and Ehrhardt A (2012). RNA interference is responsible for reduction of transgene expression after Sleeping Beauty transposase mediated somatic integration. PLoS One 7, e35389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davidovich C, Zheng L, Goodrich KJ, and Cech TR (2013). Promiscuous RNA binding by Polycomb repressive complex 2. Nat. Struct. Mol. Biol. 20, 1250–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stewart-Morgan KR, Reverón-Gómez N, and Groth A (2019). Transcription Restart Establishes Chromatin Accessibility after DNA Replication. Mol. Cell 75, 408–414. [DOI] [PubMed] [Google Scholar]
- 41.Zopf CJ, Quinn K, Zeidman J, and Maheshri N (2013). Cell-cycle dependence of transcription dominates noise in gene expression. PLoS Comput. Biol. 9, e1003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kotsantis P, Silva LM, Irmscher S, Jones RM, Folkes L, Gromak N, and Petermann E (2016). Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 7, 13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keszthelyi A, Minchell NE, and Baxter J (2016). The Causes and Consequences of Topological Stress during DNA Replication. Genes 7. 10.3390/genes7120134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, and Li X (2011). R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 25, 2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manzo SG, Hartono SR, Sanz LA, Marinello J, De Biasi S, Cossarizza A, Capranico G, and Chedin F (2018). DNA Topoisomerase I differentially modulates R-loops across the human genome. Genome Biol. 19, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo S, Zi X, Schulz VP, Cheng J, Zhong M, Koochaki SHJ, Megyola CM, Pan X, Heydari K, Weissman SM, et al. (2014). Nonstochastic reprogramming from a privileged somatic cell state. Cell 156, 649–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ordovás L, Boon R, Pistoni M, Chen Y, Wolfs E, Guo W, Sambathkumar R, Bobis-Wozowicz S, Helsen N, Vanhove J, et al. (2015). Efficient Recombinase-Mediated Cassette Exchange in hPSCs to Study the Hepatocyte Lineage Reveals AAVS1 Locus-Mediated Transgene Inhibition. Stem Cell Reports 5, 918–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhagwan JR, Collins E, Mosqueira D, Bakar M, Johnson BB, Thompson A, Smith JGW, and Denning C (2019). Variable expression and silencing of CRISPR-Cas9 targeted transgenes identifies the AAVS1 locus as not an entirely safe harbour. F1000Res. 8, 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beard C, Hochedlinger K, Plath K, Wutz A, and Jaenisch R (2006). Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis 44, 23–28. [DOI] [PubMed] [Google Scholar]
- 50.Haenebalcke L, Goossens S, Naessens M, Kruse N, Farhang Ghahremani M, Bartunkova S, Haigh K, Pieters T, Dierickx P, Drogat B, et al. (2013). Efficient ROSA26-based conditional and/or inducible transgenesis using RMCE-compatible F1 hybrid mouse embryonic stem cells. Stem Cell Rev Rep 9, 774–785. [DOI] [PubMed] [Google Scholar]
- 51.Pelascini LPL, Janssen JM, and Gonçalves MAFV (2013). Histone deacetylase inhibition activates transgene expression from integration-defective lentiviral vectors in dividing and non-dividing cells. Hum. Gene Ther. 24, 78–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lyko F (2018). The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 19, 81–92. [DOI] [PubMed] [Google Scholar]
- 53.Hughes AL, Kelley JR, and Klose RJ (2020). Understanding the interplay between CpG island-associated gene promoters and H3K4 methylation. Biochim. Biophys. Acta Gene Regul. Mech. 1863, 194567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reik W, Dean W, and Walter J (2001). Epigenetic reprogramming in mammalian development. Science 293, 1089–1093. [DOI] [PubMed] [Google Scholar]
- 55.Auclair G, Guibert S, Bender A, and Weber M (2014). Ontogeny of CpG island methylation and specificity of DNMT3 methyltransferases during embryonic development in the mouse. Genome Biol. 15, 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Veland N, Lu Y, Hardikar S, Gaddis S, Zeng Y, Liu B, Estecio MR, Takata Y, Lin K, Tomida MW, et al. (2019). DNMT3L facilitates DNA methylation partly by maintaining DNMT3A stability in mouse embryonic stem cells. Nucleic Acids Res. 47, 152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ooi SKT, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin S-P, Allis CD, et al. (2007). DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Busslinger M, Hurst J, and Flavell RA (1983). DNA methylation and the regulation of globin gene expression. Cell 34, 197–206. [DOI] [PubMed] [Google Scholar]
- 59.Kruczek I, and Doerfler W (1983). Expression of the chloramphenicol acetyltransferase gene in mammalian cells under the control of adenovirus type 12 promoters: effect of promoter methylation on gene expression. Proc. Natl. Acad. Sci. U. S. A. 80, 7586–7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Keshet I, Yisraeli J, and Cedar H (1985). Effect of regional DNA methylation on gene expression. Proc. Natl. Acad. Sci. U. S. A. 82, 2560–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Siegfried Z, Eden S, Mendelsohn M, Feng X, Tsuberi BZ, and Cedar H (1999). DNA methylation represses transcription in vivo. Nat. Genet. 22, 203–206. [DOI] [PubMed] [Google Scholar]
- 62.Rose NR, and Klose RJ (2014). Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta 1839, 1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elgin SCR, and Reuter G (2013). Position-effect variegation, heterochromatin formation, and gene silencing in Drosophila. Cold Spring Harb. Perspect. Biol. 5, a017780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Richards EJ, and Elgin SCR (2002). Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell 108, 489–500. [DOI] [PubMed] [Google Scholar]
- 65.Bird AP (1986). CpG-rich islands and the function of DNA methylation. Nature 321, 209–213. [DOI] [PubMed] [Google Scholar]
- 66.Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, and Schübeler D (2007). Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 39, 457–466. [DOI] [PubMed] [Google Scholar]
- 67.Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr ARW, James KD, Turner DJ, Smith C, Harrison DJ, Andrews R, and Bird AP (2010). Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 6, e1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Straussman R, Nejman D, Roberts D, Steinfeld I, Blum B, Benvenisty N, Simon I, Yakhini Z, and Cedar H (2009). Developmental programming of CpG island methylation profiles in the human genome. Nat. Struct. Mol. Biol. 16, 564–571. [DOI] [PubMed] [Google Scholar]
- 69.Clouaire T, Webb S, Skene P, Illingworth R, Kerr A, Andrews R, Lee J-H, Skalnik D, and Bird A (2012). Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 26, 1714–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thomson JP, Skene PJ, Selfridge J, Clouaire T, Guy J, Webb S, Kerr ARW, Deaton A, Andrews R, James KD, et al. (2010). CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 464, 1082–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wachter E, Quante T, Merusi C, Arczewska A, Stewart F, Webb S, and Bird A (2014). Synthetic CpG islands reveal DNA sequence determinants of chromatin structure. Elife 3, e03397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Turker MS (2002). Gene silencing in mammalian cells and the spread of DNA methylation. Oncogene 21, 5388–5393. [DOI] [PubMed] [Google Scholar]
- 73.Lienert F, Wirbelauer C, Som I, Dean A, Mohn F, and Schübeler D (2011). Identification of genetic elements that autonomously determine DNA methylation states. Nat. Genet. 43, 1091–1097. [DOI] [PubMed] [Google Scholar]
- 74.Brandeis M, Frank D, Keshet I, Siegfried Z, Mendelsohn M, Nemes A, Temper V, Razin A, and Cedar H (1994). Sp1 elements protect a CpG island from de novo methylation. Nature 371, 435–438. [DOI] [PubMed] [Google Scholar]
- 75.Saksouk N, Simboeck E, and Déjardin J (2015). Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kouzarides T (2007). Chromatin modifications and their function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- 77.Yu Y, Lowy MM, and Elble RC (2016). Tet-On lentiviral transductants lose inducibility when silenced for extended intervals in mammary epithelial cells. Metab Eng Commun 3, 64–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cedar H, and Bergman Y (2009). Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 10, 295–304. [DOI] [PubMed] [Google Scholar]
- 79.Müller MM, Fierz B, Bittova L, Liszczak G, and Muir TW (2016). A two-state activation mechanism controls the histone methyltransferase Suv39h1. Nat. Chem. Biol. 12, 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maison C, Bailly D, Quivy J-P, and Almouzni G (2016). The methyltransferase Suv39h1 links the SUMO pathway to HP1α marking at pericentric heterochromatin. Nat. Commun. 7, 12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maison C, and Almouzni G (2004). HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 5, 296–304. [DOI] [PubMed] [Google Scholar]
- 82.Muller HJ (1930). Types of visible variations induced by X-rays inDrosophila. J. Genet. 22, 299–334. [Google Scholar]
- 83.Henikoff S, and Dreesen TD (1989). Trans-inactivation of the Drosophila brown gene: evidence for transcriptional repression and somatic pairing dependence. Proc. Natl. Acad. Sci. U. S. A. 86, 6704–6708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sun F-L, Haynes K, Simpson CL, Lee SD, Collins L, Wuller J, Eissenberg JC, and Elgin SCR (2004). cis-Acting determinants of heterochromatin formation on Drosophila melanogaster chromosome four. Mol. Cell. Biol. 24, 8210–8220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Haynes KA, Caudy AA, Collins L, and Elgin SCR (2006). Element 1360 and RNAi components contribute to HP1-dependent silencing of a pericentric reporter. Curr. Biol. 16, 2222–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Riddle NC, Leung W, Haynes KA, Granok H, Wuller J, and Elgin SCR (2008). An investigation of heterochromatin domains on the fourth chromosome of Drosophila melanogaster. Genetics 178, 1177–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Villaseñor R, and Baubec T (2021). Regulatory mechanisms governing chromatin organization and function. Curr. Opin. Cell Biol. 70, 10–17. [DOI] [PubMed] [Google Scholar]
- 88.Becker JS, Nicetto D, and Zaret KS (2016). H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet. 32, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang F, Koyama N, Nishida H, Haraguchi T, Reith W, and Tsukamoto T (2006). The assembly and maintenance of heterochromatin initiated by transgene repeats are independent of the RNA interference pathway in mammalian cells. Mol. Cell. Biol. 26, 4028–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Calero-Nieto FJ, Bert AG, and Cockerill PN (2010). Transcription-dependent silencing of inducible convergent transgenes in transgenic mice. Epigenetics Chromatin 3, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sabl JF, and Henikoff S (1996). Copy number and orientation determine the susceptibility of a gene to silencing by nearby heterochromatin in Drosophila. Genetics 142, 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barrett CM, McCracken R, Elmer J, and Haynes KA (2020). Components from the Human c-myb Transcriptional Regulation System Reactivate Epigenetically Repressed Transgenes. Int. J. Mol. Sci. 21. 10.3390/ijms21020530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Szabo Q, Bantignies F, and Cavalli G (2019). Principles of genome folding into topologically associating domains. Sci Adv 5, eaaw1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Penagos-Puig A, and Furlan-Magaril M (2020). Heterochromatin as an Important Driver of Genome Organization. Front Cell Dev Biol 8, 579137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Klatt D, Cheng E, Hoffmann D, Santilli G, Thrasher AJ, Brendel C, and Schambach A (2020). Differential Transgene Silencing of Myeloid-Specific Promoters in the AAVS1 Safe Harbor Locus of Induced Pluripotent Stem Cell-Derived Myeloid Cells. Hum. Gene Ther. 31, 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sadelain M, Papapetrou EP, and Bushman FD (2011). Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 12, 51–58. [DOI] [PubMed] [Google Scholar]
- 98.Ellis J (2005). Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum. Gene Ther. 16, 1241–1246. [DOI] [PubMed] [Google Scholar]
- 99.Chang AH, Stephan MT, and Sadelain M (2006). Stem cell-derived erythroid cells mediate long-term systemic protein delivery. Nat. Biotechnol. 24, 1017–1021. [DOI] [PubMed] [Google Scholar]
- 100.Mok HP, Javed S, and Lever A (2007). Stable gene expression occurs from a minority of integrated HIV-1-based vectors: transcriptional silencing is present in the majority. Gene Ther. 14, 741–751. [DOI] [PubMed] [Google Scholar]
- 101.Herbst F, Ball CR, Tuorto F, Nowrouzi A, Wang W, Zavidij O, Dieter SM, Fessler S, van der Hoeven F, Kloz U, et al. (2012). Extensive methylation of promoter sequences silences lentiviral transgene expression during stem cell differentiation in vivo. Mol. Ther. 20, 1014–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Challita PM, and Kohn DB (1994). Lack of expression from a retroviral vector after transduction of murine hematopoietic stem cells is associated with methylation in vivo. Proc. Natl. Acad. Sci. U. S. A. 91, 2567–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pannell D, Osborne CS, Yao S, Sukonnik T, Pasceri P, Karaiskakis A, Okano M, Li E, Lipshitz HD, and Ellis J (2000). Retrovirus vector silencing is de novo methylase independent and marked by a repressive histone code. EMBO J. 19, 5884–5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Suzuki MM, and Bird A (2008). DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 9, 465–476. [DOI] [PubMed] [Google Scholar]
- 105.Berkhout B (1997). The primer binding site on the RNA genome of human and simian immunodeficiency viruses is flanked by an upstream hairpin structure. Nucleic Acids Res. 25, 4013–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li X, Mak J, Arts EJ, Gu Z, Kleiman L, Wainberg MA, and Parniak MA (1994). Effects of alterations of primer-binding site sequences on human immunodeficiency virus type 1 replication. J. Virol. 68, 6198–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jähner D, Stuhlmann H, Stewart CL, Harbers K, Löhler J, Simon I, and Jaenisch R (1982). De novo methylation and expression of retroviral genomes during mouse embryogenesis. Nature 298, 623–628. [DOI] [PubMed] [Google Scholar]
- 108.Gautsch JW, and Wilson MC (1983). Delayed de novo methylation in teratocarcinoma suggests additional tissue-specific mechanisms for controlling gene expression. Nature 301, 32–37. [DOI] [PubMed] [Google Scholar]
- 109.Niwa O, Yokota Y, Ishida H, and Sugahara T (1983). Independent mechanisms involved in suppression of the Moloney leukemia virus genome during differentiation of murine teratocarcinoma cells. Cell 32, 1105–1113. [DOI] [PubMed] [Google Scholar]
- 110.Wolf D, and Goff SP (2009). Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 458, 1201–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wolf D, and Goff SP (2007). TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell 131, 46–57. [DOI] [PubMed] [Google Scholar]
- 112.Schultz DC, Ayyanathan K, Negorev D, Maul GG, and Rauscher FJ 3rd (2002). SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 16, 919–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, Tachibana M, Lorincz MC, and Shinkai Y (2010). Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464, 927–931. [DOI] [PubMed] [Google Scholar]
- 114.Schultz DC, Friedman JR, and Rauscher FJ 3rd (2001). Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev. 15, 428–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard-Liesching H, Verp S, Marquis J, et al. (2010). KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463, 237–240. [DOI] [PubMed] [Google Scholar]
- 116.Wolf G, Yang P, Füchtbauer AC, Füchtbauer E-M, Silva AM, Park C, Wu W, Nielsen AL, Pedersen FS, and Macfarlan TS (2015). The KRAB zinc finger protein ZFP809 is required to initiate epigenetic silencing of endogenous retroviruses. Genes Dev. 29, 538–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yang BX, El Farran CA, Guo HC, Yu T, Fang HT, Wang HF, Schlesinger S, Seah YFS, Goh GYL, Neo SP, et al. (2015). Systematic identification of factors for provirus silencing in embryonic stem cells. Cell 163, 230–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shalginskikh N, Poleshko A, Skalka AM, and Katz RA (2013). Retroviral DNA methylation and epigenetic repression are mediated by the antiviral host protein Daxx. J. Virol. 87, 2137–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rowe HM, Friedli M, Offner S, Verp S, Mesnard D, Marquis J, Aktas T, and Trono D (2013). De novo DNA methylation of endogenous retroviruses is shaped by KRAB-ZFPs/KAP1 and ESET. Development 140, 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Akira S, Uematsu S, and Takeuchi O (2006). Pathogen recognition and innate immunity. Cell 124, 783–801. [DOI] [PubMed] [Google Scholar]
- 121.Chan YK, and Gack MU (2016). Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 14, 360–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, et al. (2000). A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745. [DOI] [PubMed] [Google Scholar]
- 123.Bauer S, Kirschning CJ, Häcker H, Redecke V, Hausmann S, Akira S, Wagner H, and Lipford GB (2001). Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. U. S. A. 98, 9237–9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang C-H, Naik NG, Liao L-L, Wei S-C, and Chao Y-C (2017). Global Screening of Antiviral Genes that Suppress Baculovirus Transgene Expression in Mammalian Cells. Mol Ther Methods Clin Dev 6, 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Herold MJ, van den Brandt J, Seibler J, and Reichardt HM (2008). Inducible and reversible gene silencing by stable integration of an shRNA-encoding lentivirus in transgenic rats. Proc. Natl. Acad. Sci. U. S. A. 105, 18507–18512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Miettinen M, Sareneva T, Julkunen I, and Matikainen S (2001). IFNs activate toll-like receptor gene expression in viral infections. Genes Immun. 2, 349–355. [DOI] [PubMed] [Google Scholar]
- 127.Belloni L, Allweiss L, Guerrieri F, Pediconi N, Volz T, Pollicino T, Petersen J, Raimondo G, Dandri M, and Levrero M (2012). IFN-α inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J. Clin. Invest. 122, 529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhu P, Aller MI, Baron U, Cambridge S, Bausen M, Herb J, Sawinski J, Cetin A, Osten P, Nelson ML, et al. (2007). Silencing and un-silencing of tetracycline-controlled genes in neurons. PLoS One 2, e533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.DiAndreth B, Wauford N, Hu E, Palacios S, and Weiss R (2022). PERSIST platform provides programmable RNA regulation using CRISPR endoRNases. Nat. Commun. 13, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bednarik DP, Cook JA, and Pitha PM (1990). Inactivation of the HIV LTR by DNA CpG methylation: evidence for a role in latency. EMBO J. 9, 1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Garrison BS, Yant SR, Mikkelsen JG, and Kay MA (2007). Postintegrative gene silencing within the Sleeping Beauty transposition system. Mol. Cell. Biol. 27, 8824–8833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mutskov VJ, Farrell CM, Wade PA, Wolffe AP, and Felsenfeld G (2002). The barrier function of an insulator couples high histone acetylation levels with specific protection of promoter DNA from methylation. Genes Dev. 16, 1540–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhao H, and Dean A (2004). An insulator blocks spreading of histone acetylation and interferes with RNA polymerase II transfer between an enhancer and gene. Nucleic Acids Res. 32, 4903–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stewart CL, Vanek M, and Wagner EF (1985). Expression of foreign genes from retroviral vectors in mouse teratocarcinoma chimaeras. EMBO J. 4, 3701–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wagner EF, Vanek M, and Vennström B (1985). Transfer of genes into embryonal carcinoma cells by retrovirus infection: efficient expression from an internal promoter. EMBO J. 4, 663–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Stewart CL, Schuetze S, Vanek M, and Wagner EF (1987). Expression of retroviral vectors in transgenic mice obtained by embryo infection. EMBO J. 6, 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rivière I, Brose K, and Mulligan RC (1995). Effects of retroviral vector design on expression of human adenosine deaminase in murine bone marrow transplant recipients engrafted with genetically modified cells. Proc. Natl. Acad. Sci. U. S. A. 92, 6733–6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Qin JY, Zhang L, Clift KL, Hulur I, Xiang AP, Ren B-Z, and Lahn BT (2010). Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PLoS One 5, e10611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ede C, Chen X, Lin M-Y, and Chen YY (2016). Quantitative Analyses of Core Promoters Enable Precise Engineering of Regulated Gene Expression in Mammalian Cells. ACS Synth. Biol. 5, 395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dou Y, Lin Y, Wang T-Y, Wang X-Y, Jia Y-L, and Zhao C-P (2021). The CAG promoter maintains high-level transgene expression in HEK293 cells. FEBS Open Bio 11, 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hong S, Hwang D-Y, Yoon S, Isacson O, Ramezani A, Hawley RG, and Kim K-S (2007). Functional analysis of various promoters in lentiviral vectors at different stages of in vitro differentiation of mouse embryonic stem cells. Mol. Ther. 15, 1630–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Malaguti M, Portero Migueles R, Annoh J, Sadurska D, Blin G, and Lowell S (2022). SyNPL: Synthetic Notch pluripotent cell lines to monitor and manipulate cell interactions in vitro and in vivo. Development 149. 10.1242/dev.200226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chen C-M, Krohn J, Bhattacharya S, and Davies B (2011). A comparison of exogenous promoter activity at the ROSA26 locus using a ΦiC31 integrase mediated cassette exchange approach in mouse ES cells. PLoS One 6, e23376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Emery DW (2011). The use of chromatin insulators to improve the expression and safety of integrating gene transfer vectors. Hum. Gene Ther. 22, 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chung JH, Whiteley M, and Felsenfeld G (1993). A 5’ element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell 74, 505–514. [DOI] [PubMed] [Google Scholar]
- 146.Rincón-Arano H, Furlan-Magaril M, and Recillas-Targa F (2007). Protection against telomeric position effects by the chicken cHS4 beta-globin insulator. Proc. Natl. Acad. Sci. U. S. A. 104, 14044–14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Girod P-A, Nguyen D-Q, Calabrese D, Puttini S, Grandjean M, Martinet D, Regamey A, Saugy D, Beckmann JS, Bucher P, et al. (2007). Genome-wide prediction of matrix attachment regions that increase gene expression in mammalian cells. Nat. Methods 4, 747–753. [DOI] [PubMed] [Google Scholar]
- 148.Antoniou M, Harland L, Mustoe T, Williams S, Holdstock J, Yague E, Mulcahy T, Griffiths M, Edwards S, Ioannou PA, et al. (2003). Transgenes encompassing dual-promoter CpG islands from the human TBP and HNRPA2B1 loci are resistant to heterochromatin-mediated silencing. Genomics 82, 269–279. [DOI] [PubMed] [Google Scholar]
- 149.Müller-Kuller U, Ackermann M, Kolodziej S, Brendel C, Fritsch J, Lachmann N, Kunkel H, Lausen J, Schambach A, Moritz T, et al. (2015). A minimal ubiquitous chromatin opening element (UCOE) effectively prevents silencing of juxtaposed heterologous promoters by epigenetic remodeling in multipotent and pluripotent stem cells. Nucleic Acids Res. 43, 1577–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Raab JR, Chiu J, Zhu J, Katzman S, Kurukuti S, Wade PA, Haussler D, and Kamakaka RT (2012). Human tRNA genes function as chromatin insulators. EMBO J. 31, 330–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Barkess G, and West AG (2012). Chromatin insulator elements: establishing barriers to set heterochromatin boundaries. Epigenomics 4, 67–80. [DOI] [PubMed] [Google Scholar]
- 152.Guo X, Wang C, and Wang T-Y (2020). Chromatin-modifying elements for recombinant protein production in mammalian cell systems. Crit. Rev. Biotechnol. 40, 1035–1043. [DOI] [PubMed] [Google Scholar]
- 153.Gaszner M, and Felsenfeld G (2006). Insulators: exploiting transcriptional and epigenetic mechanisms. Nat. Rev. Genet. 7, 703–713. [DOI] [PubMed] [Google Scholar]
- 154.Raab JR, and Kamakaka RT (2010). Insulators and promoters: closer than we think. Nat. Rev. Genet. 11, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Phillips-Cremins JE, and Corces VG (2013). Chromatin insulators: linking genome organization to cellular function. Mol. Cell 50, 461–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kentepozidou E, Aitken SJ, Feig C, Stefflova K, Ibarra-Soria X, Odom DT, Roller M, and Flicek P (2020). Clustered CTCF binding is an evolutionary mechanism to maintain topologically associating domains. Genome Biol. 21, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Liu M, Maurano MT, Wang H, Qi H, Song C-Z, Navas PA, Emery DW, Stamatoyannopoulos JA, and Stamatoyannopoulos G (2015). Genomic discovery of potent chromatin insulators for human gene therapy. Nat. Biotechnol. 33, 198–203. [DOI] [PubMed] [Google Scholar]
- 158.Cuddapah S, Jothi R, Schones DE, Roh T-Y, Cui K, and Zhao K (2009). Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 19, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nielsen TT, Jakobsson J, Rosenqvist N, and Lundberg C (2009). Incorporating double copies of a chromatin insulator into lentiviral vectors results in less viral integrants. BMC Biotechnol. 9, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Urbinati F, Arumugam P, Higashimoto T, Perumbeti A, Mitts K, Xia P, and Malik P (2009). Mechanism of reduction in titers from lentivirus vectors carrying large inserts in the 3’LTR. Mol. Ther. 17, 1527–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yi Y, Noh MJ, and Lee KH (2011). Current advances in retroviral gene therapy. Curr. Gene Ther. 11, 218–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rudina SS, and Smolke CD (2019). A Novel Chromatin-Opening Element for Stable Long-term Transgene Expression. bioRxiv, 626713. 10.1101/626713. [DOI] [Google Scholar]
- 163.Mitchell RS, Beitzel BF, Schroder ARW, Shinn P, Chen H, Berry CC, Ecker JR, and Bushman FD (2004). Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2, E234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Liang Q, Kong J, Stalker J, and Bradley A (2009). Chromosomal mobilization and reintegration of Sleeping Beauty and PiggyBac transposons. Genesis 47, 404–408. [DOI] [PubMed] [Google Scholar]
- 165.Akhtar W, de Jong J, Pindyurin AV, Pagie L, Meuleman W, de Ridder J, Berns A, Wessels LFA, van Lohuizen M, and van Steensel B (2013). Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell 154, 914–927. [DOI] [PubMed] [Google Scholar]
- 166.Papapetrou EP, and Schambach A (2016). Gene Insertion Into Genomic Safe Harbors for Human Gene Therapy. Mol. Ther. 24, 678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Irion S, Luche H, Gadue P, Fehling HJ, Kennedy M, and Keller G (2007). Identification and targeting of the ROSA26 locus in human embryonic stem cells. Nat. Biotechnol. 25, 1477–1482. [DOI] [PubMed] [Google Scholar]
- 168.Hong SG, Yada RC, Choi K, Carpentier A, Liang TJ, Merling RK, Sweeney CL, Malech HL, Jung M, Corat MAF, et al. (2017). Rhesus iPSC Safe Harbor Gene-Editing Platform for Stable Expression of Transgenes in Differentiated Cells of All Germ Layers. Mol. Ther. 25, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee Y-L, et al. (2008). Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 26, 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chi X, Zheng Q, Jiang R, Chen-Tsai RY, and Kong L-J (2019). A system for site-specific integration of transgenes in mammalian cells. PLoS One 14, e0219842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tasic B, Hippenmeyer S, Wang C, Gamboa M, Zong H, Chen-Tsai Y, and Luo L (2011). Site-specific integrase-mediated transgenesis in mice via pronuclear injection. Proc. Natl. Acad. Sci. U. S. A. 108, 7902–7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Aznauryan E, Yermanos A, Kinzina E, Devaux A, Kapetanovic E, Milanova D, Church GM, and Reddy ST (2022). Discovery and validation of human genomic safe harbor sites for gene and cell therapies. Cell Rep Methods 2, 100154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhang M, Yang C, Tasan I, and Zhao H (2021). Expanding the Potential of Mammalian Genome Engineering via Targeted DNA Integration. ACS Synth. Biol. 10, 429–446. [DOI] [PubMed] [Google Scholar]
- 174.Jusiak B, Jagtap K, Gaidukov L, Duportet X, Bandara K, Chu J, Zhang L, Weiss R, and Lu TK (2019). Comparison of Integrases Identifies Bxb1-GA Mutant as the Most Efficient Site-Specific Integrase System in Mammalian Cells. ACS Synth. Biol. 8, 16–24. [DOI] [PubMed] [Google Scholar]
- 175.Duportet X, Wroblewska L, Guye P, Li Y, Eyquem J, Rieders J, Rimchala T, Batt G, and Weiss R (2014). A platform for rapid prototyping of synthetic gene networks in mammalian cells. Nucleic Acids Res. 42, 13440–13451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Matreyek KA, Stephany JJ, and Fowler DM (2017). A platform for functional assessment of large variant libraries in mammalian cells. Nucleic Acids Res. 45, e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gaidukov L, Wroblewska L, Teague B, Nelson T, Zhang X, Liu Y, Jagtap K, Mamo S, Tseng WA, Lowe A, et al. (2018). A multi-landing pad DNA integration platform for mammalian cell engineering. Nucleic Acids Res. 46, 4072–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ioannidi EI, Yarnall MTN, Schmitt-Ulms C, Krajeski RN, Lim J, Villiger L, Zhou W, Jiang K, Roberts N, Zhang L, et al. (2021). Drag-and-drop genome insertion without DNA cleavage with CRISPR-directed integrases. bioRxiv, 2021.11.01.466786. 10.1101/2021.11.01.466786. [DOI] [Google Scholar]
- 179.Durrant MG, Fanton A, Tycko J, Hinks M, Chandrasekaran SS, Perry NT, Schaepe J, Du PP, Lotfy P, Bassik MC, et al. (2021). Large-scale discovery of recombinases for integrating DNA into the human genome. bioRxiv, 2021.11.05.467528. 10.1101/2021.11.05.467528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Moore TV, Scurti GM, DeJong M, Wang S-Y, Dalheim AV, Wagner CR, Hutchens KA, Speiser JJ, Godellas CV, Fountain C, et al. (2021). HDAC inhibition prevents transgene expression downregulation and loss-of-function in T-cell-receptor-transduced T cells. Mol Ther Oncolytics 20, 352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Peinado P, Andrades A, Cuadros M, Rodriguez MI, Coira IF, Garcia DJ, Álvarez-Perez JC, Baliñas-Gavira C, Arenas AM, Patiño-Mercau JR, et al. (2020). Comprehensive Analysis of SWI/SNF Inactivation in Lung Adenocarcinoma Cell Models. Cancers 12. 10.3390/cancers12123712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Decristofaro MF, Betz BL, Rorie CJ, Reisman DN, Wang W, and Weissman BE (2001). Characterization of SWI/SNF protein expression in human breast cancer cell lines and other malignancies. J. Cell. Physiol. 186, 136–145. [DOI] [PubMed] [Google Scholar]
- 184.Karimi MM, Goyal P, Maksakova IA, Bilenky M, Leung D, Tang JX, Shinkai Y, Mager DL, Jones S, Hirst M, et al. (2011). DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8, 676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Schorn AJ, Gutbrod MJ, LeBlanc C, and Martienssen R (2017). LTR-Retrotransposon Control by tRNA-Derived Small RNAs. Cell 170, 61–71.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Boroviak T, Loos R, Bertone P, Smith A, and Nichols J (2014). The ability of inner-cell-mass cells to self-renew as embryonic stem cells is acquired following epiblast specification. Nat. Cell Biol. 16, 516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kalkan T, Olova N, Roode M, Mulas C, Lee HJ, Nett I, Marks H, Walker R, Stunnenberg HG, Lilley KS, et al. (2017). Tracking the embryonic stem cell transition from ground state pluripotency. Development 144, 1221–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Athanasiadou R, de Sousa D, Myant K, Merusi C, Stancheva I, and Bird A (2010). Targeting of de novo DNA methylation throughout the Oct-4 gene regulatory region in differentiating embryonic stem cells. PLoS One 5, e9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Soriano P (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70–71. [DOI] [PubMed] [Google Scholar]
- 190.Hao J-W, Wang J, Guo H, Zhao Y-Y, Sun H-H, Li Y-F, Lai X-Y, Zhao N, Wang X, Xie C, et al. (2020). CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat. Commun. 11, 4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Manning BD, and Cantley LC (2007). AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.DeBose-Boyd RA, and Ye J (2018). SREBPs in Lipid Metabolism, Insulin Signaling, and Beyond. Trends Biochem. Sci. 43, 358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zhang M, Galdieri L, and Vancura A (2013). The yeast AMPK homolog SNF1 regulates acetyl coenzyme A homeostasis and histone acetylation. Mol. Cell. Biol. 33, 4701–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Galdieri L, and Vancura A (2012). Acetyl-CoA carboxylase regulates global histone acetylation. J. Biol. Chem. 287, 23865–23876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Koundouros N, and Poulogiannis G (2020). Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 122, 4–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Micallef P, Wu Y, Bauzá-Thorbrügge M, Chanclón B, Vujičić M, Peris E, Ek CJ, and Wernstedt Asterholm I (2021). Adipose Tissue-Breast Cancer Crosstalk Leads to Increased Tumor Lipogenesis Associated with Enhanced Tumor Growth. Int. J. Mol. Sci. 22. 10.3390/ijms222111881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nickel A, Blücher C, Kadri OA, Schwagarus N, Müller S, Schaab M, Thiery J, Burkhardt R, and Stadler SC (2018). Adipocytes induce distinct gene expression profiles in mammary tumor cells and enhance inflammatory signaling in invasive breast cancer cells. Sci. Rep. 8, 9482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al. (2019). Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yu J, Chai P, Xie M, Ge S, Ruan J, Fan X, and Jia R (2021). Histone lactylation drives oncogenesis by facilitating m6A reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 22, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Torres M, Altamirano C, and Dickson AJ (2018). Process and metabolic engineering perspectives of lactate production in mammalian cell cultures. Curr. Opin. Chem. Eng. 22, 184–190. [Google Scholar]
- 201.Janke R, Iavarone AT, and Rine J (2017). Oncometabolite D-2-Hydroxyglutarate enhances gene silencing through inhibition of specific H3K36 histone demethylases. Elife 6. 10.7554/eLife.22451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Dekker J, Belmont AS, Guttman M, Leshyk VO, Lis JT, Lomvardas S, Mirny LA, O’Shea CC, Park PJ, Ren B, et al. (2017). The 4D nucleome project. Nature 549, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, Bodenmiller B, Campbell P, Carninci P, Clatworthy M, et al. (2017). Science forum: the human cell atlas. Elife 6, e27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.FANTOM Consortium and the RIKEN PMI and CLST (DGT), Forrest ARR, Kawaji H, Rehli M, Baillie JK, de Hoon MJL, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, et al. (2014). A promoter-level mammalian expression atlas. Nature 507, 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zúñiga RA, Gutiérrez-González M, Collazo N, Sotelo PH, Ribeiro CH, Altamirano C, Lorenzo C, Aguillón JC, and Molina MC (2019). Development of a new promoter to avoid the silencing of genes in the production of recombinant antibodies in chinese hamster ovary cells. J. Biol. Eng. 13, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kelly M, Mandel RJ, Nguyen M, and Trono D (1998). A third-generation lentivirus vector with a conditional packaging system. Journal of. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wang D, Tai PWL, and Gao G (2019). Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 18, 358–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wolf G, Greenberg D, and Macfarlan TS (2015). Spotting the enemy within: Targeted silencing of foreign DNA in mammalian genomes by the Krüppel-associated box zinc finger protein family. Mob. DNA 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Liu H, Wang J, He T, Becker S, Zhang G, Li D, and Ma X (2018). Butyrate: A Double-Edged Sword for Health? Adv. Nutr. 9, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Prasad KN, and Sinha PK (1976). Effect of sodium butyrate on mammalian cells in culture: a review. In Vitro 12, 125–132. [DOI] [PubMed] [Google Scholar]
- 211.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Li B, Clohisey SM, Chia BS, Wang B, Cui A, Eisenhaure T, Schweitzer LD, Hoover P, Parkinson NJ, Nachshon A, et al. (2020). Genome-wide CRISPR screen identifies host dependency factors for influenza A virus infection. Nat. Commun. 11, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Yang J, Rajan SS, Friedrich MJ, Lan G, Zou X, Ponstingl H, Garyfallos DA, Liu P, Bradley A, and Metzakopian E (2019). Genome-Scale CRISPRa Screen Identifies Novel Factors for Cellular Reprogramming. Stem Cell Reports 12, 757–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Chung S, Quarmby V, Gao X, Ying Y, Lin L, Reed C, Fong C, Lau W, Qiu ZJ, Shen A, et al. (2012). Quantitative evaluation of fucose reducing effects in a humanized antibody on Fcγ receptor binding and antibody-dependent cell-mediated cytotoxicity activities. MAbs 4, 326–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Grav LM, la Cour Karottki KJ, Lee JS, and Kildegaard HF (2017). Application of CRISPR/Cas9 Genome Editing to Improve Recombinant Protein Production in CHO Cells. Methods Mol. Biol. 1603, 101–118. [DOI] [PubMed] [Google Scholar]
- 216.Martínez MA (2009). Progress in the therapeutic applications of siRNAs against HIV-1. Methods Mol. Biol. 487, 343–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Decout A, Katz JD, Venkatraman S, and Ablasser A (2021). The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 21, 548–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Vanhille L, Griffon A, Maqbool MA, Zacarias-Cabeza J, Dao LTM, Fernandez N, Ballester B, Andrau JC, and Spicuglia S (2015). High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq. Nat. Commun. 6, 6905. [DOI] [PubMed] [Google Scholar]
- 219.Sheng T, Ho SWT, Ooi WF, Xu C, Xing M, Padmanabhan N, Huang KK, Ma L, Ray M, Guo YA, et al. (2021). Integrative epigenomic and high-throughput functional enhancer profiling reveals determinants of enhancer heterogeneity in gastric cancer. Genome Med. 13, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Murtha M, Tokcaer-Keskin Z, Tang Z, Strino F, Chen X, Wang Y, Xi X, Basilico C, Brown S, Bonneau R, et al. (2014). FIREWACh: high-throughput functional detection of transcriptional regulatory modules in mammalian cells. Nat. Methods 11, 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Babbitt CC, Markstein M, and Gray JM (2015). Recent advances in functional assays of transcriptional enhancers. Genomics 106, 137–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Andersson R, and Sandelin A (2020). Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 21, 71–87. [DOI] [PubMed] [Google Scholar]
- 223.Gierman HJ, Indemans MHG, Koster J, Goetze S, Seppen J, Geerts D, van Driel R, and Versteeg R (2007). Domain-wide regulation of gene expression in the human genome. Genome Res. 17, 1286–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Inoue F, Kircher M, Martin B, Cooper GM, Witten DM, McManus MT, Ahituv N, and Shendure J (2017). A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res. 27, 38–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kheradpour P, Ernst J, Melnikov A, Rogov P, Wang L, Zhang X, Alston J, Mikkelsen TS, and Kellis M (2013). Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 23, 800–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Weingarten-Gabbay S, Nir R, Lubliner S, Sharon E, Kalma Y, Weinberger A, and Segal E (2019). Systematic interrogation of human promoters. Genome Res. 29, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Hong CKY, and Cohen BA (2022). Genomic environments scale the activities of diverse core promoters. Genome Res. 32, 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Yaguchi M, Ohashi Y, Tsubota T, Sato A, Koyano KW, Wang N, and Miyashita Y (2013). Characterization of the properties of seven promoters in the motor cortex of rats and monkeys after lentiviral vector-mediated gene transfer. Hum. Gene Ther. Methods 24, 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Hagedorn C, Antoniou MN, and Lipps HJ (2013). Genomic cis-acting Sequences Improve Expression and Establishment of a Nonviral Vector. Mol. Ther. Nucleic Acids 2, e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Allweiss L, and Dandri M (2017). The Role of cccDNA in HBV Maintenance. Viruses 9. 10.3390/v9060156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Guo X, Chen P, Hou X, Xu W, Wang D, Wang T-Y, Zhang L, Zheng G, Gao Z-L, He C-Y, et al. (2016). The recombined cccDNA produced using minicircle technology mimicked HBV genome in structure and function closely. Sci. Rep. 6, 25552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Li F, and Ding S-W (2006). Virus counterdefense: diverse strategies for evading the RNA-silencing immunity. Annu. Rev. Microbiol. 60, 503–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Naso MF, Tomkowicz B, Perry WL 3rd, and Strohl WR (2017). Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 31, 317–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Jacobson SG, Cideciyan AV, Roman AJ, Sumaroka A, Schwartz SB, Heon E, and Hauswirth WW (2015). Improvement and decline in vision with gene therapy in childhood blindness. N. Engl. J. Med. 372, 1920–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bainbridge JWB, Mehat MS, Sundaram V, Robbie SJ, Barker SE, Ripamonti C, Georgiadis A, Mowat FM, Beattie SG, Gardner PJ, et al. (2015). Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med. 372, 1887–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hurlbut GD, Ziegler RJ, Nietupski JB, Foley JW, Woodworth LA, Meyers E, Bercury SD, Pande NN, Souza DW, Bree MP, et al. (2010). Preexisting immunity and low expression in primates highlight translational challenges for liver-directed AAV8-mediated gene therapy. Mol. Ther. 18, 1983–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Nakai H, Iwaki Y, Kay MA, and Couto LB (1999). Isolation of recombinant adeno-associated virus vector-cellular DNA junctions from mouse liver. J. Virol. 73, 5438–5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Nakai H, Montini E, Fuess S, Storm TA, Grompe M, and Kay MA (2003). AAV serotype 2 vectors preferentially integrate into active genes in mice. Nat. Genet. 34, 297–302. [DOI] [PubMed] [Google Scholar]
- 239.Miller DG, Trobridge GD, Petek LM, Jacobs MA, Kaul R, and Russell DW (2005). Large-scale analysis of adeno-associated virus vector integration sites in normal human cells. J. Virol. 79, 11434–11442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Ngo AM, and Puschnik AS (2022). Genome-scale analysis of cellular restriction factors that inhibit transgene expression from adeno-associated virus vectors. bioRxiv, 2022.07.13.499963. 10.1101/2022.07.13.499963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Smith-Moore S, Neil SJD, Fraefel C, Linden RM, Bollen M, Rowe HM, and Henckaerts E (2018). Adeno-associated virus Rep proteins antagonize phosphatase PP1 to counteract KAP1 repression of the latent viral genome. Proc. Natl. Acad. Sci. U. S. A. 115, E3529–E3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Das A, Vijayan M, Walton EM, Stafford VG, Fiflis DN, and Asokan A (2022). Epigenetic Silencing of Recombinant Adeno-associated Virus Genomes by NP220 and the HUSH Complex. J. Virol. 96, e0203921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kazuki Y, and Oshimura M (2011). Human artificial chromosomes for gene delivery and the development of animal models. Mol. Ther. 19, 1591–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Harrington JJ, Van Bokkelen G, Mays RW, Gustashaw K, and Willard HF (1997). Formation of de novo centromeres and construction of first-generation human artificial microchromosomes. Nat. Genet. 15, 345–355. [DOI] [PubMed] [Google Scholar]
- 245.Kouprina N, Earnshaw WC, Masumoto H, and Larionov V (2013). A new generation of human artificial chromosomes for functional genomics and gene therapy. Cell. Mol. Life Sci. 70, 1135–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Kazuki Y, Uno N, Abe S, Kajitani N, Kazuki K, Yakura Y, Sawada C, Takata S, Sugawara M, Nagashima Y, et al. (2021). Engineering of human induced pluripotent stem cells via human artificial chromosome vectors for cell therapy and disease modeling. Mol. Ther. Nucleic Acids 23, 629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Pardi ML, Wu J, Kawasaki S, and Saito H (2022). Synthetic RNA-based post-transcriptional expression control methods and genetic circuits. Adv. Drug Deliv. Rev. 184, 114196. [DOI] [PubMed] [Google Scholar]
- 248.Kawasaki S, Ono H, Hirosawa M, Kuwabara T, and Saito H (2021). Programmable mammalian translational modulators by CRISPR-associated proteins. bioRxiv, 2021.09.17.460758. 10.1101/2021.09.17.460758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Zhao EM, Mao AS, de Puig H, Zhang K, Tippens ND, Tan X, Ran FA, Han I, Nguyen PQ, Chory EJ, et al. (2022). RNA-responsive elements for eukaryotic translational control. Nat. Biotechnol. 40, 539–545. [DOI] [PubMed] [Google Scholar]
- 250.Gao XJ, Chong LS, Kim MS, and Elowitz MB (2018). Programmable protein circuits in living cells. Science 361, 1252–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Chen Z, Linton JM, Zhu R, and Elowitz MB (2022). A synthetic protein-level neural network in mammalian cells. bioRxiv, 2022.07.10.499405. 10.1101/2022.07.10.499405. [DOI] [Google Scholar]
- 252.Fink T, Lonzarić J, Praznik A, Plaper T, Merljak E, Leben K, Jerala N, Lebar T, Strmšek Ž, Lapenta F, et al. (2019). Design of fast proteolysis-based signaling and logic circuits in mammalian cells. Nat. Chem. Biol. 15, 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Thakore PI, Black JB, Hilton IB, and Gersbach CA (2016). Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat. Methods 13, 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Brocken DJW, Tark-Dame M, and Dame RT (2018). dCas9: A Versatile Tool for Epigenome Editing. Curr. Issues Mol. Biol. 26, 15–32. [DOI] [PubMed] [Google Scholar]
- 255.Waryah CB, Moses C, Arooj M, and Blancafort P (2018). Zinc Fingers, TALEs, and CRISPR Systems: A Comparison of Tools for Epigenome Editing. Methods Mol. Biol. 1767, 19–63. [DOI] [PubMed] [Google Scholar]
- 256.Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, and Maehr R (2015). Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 12, 401–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, and Gersbach CA (2015). Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 33, 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Bruno S, Williams RJ, and Del Vecchio D Epigenetic cell memory: The gene’s inner chromatin modification circuit. 10.1101/2022.02.02.476953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Bruno S, Williams RJ, and Del Vecchio D (2022). Epigenetic cell memory: The gene’s inner chromatin modification circuit. PLoS Comput. Biol. 18, e1009961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Tekel SJ, Vargas DA, Song L, LaBaer J, Caplan MR, and Haynes KA (2018). Tandem Histone-Binding Domains Enhance the Activity of a Synthetic Chromatin Effector. ACS Synth. Biol. 7, 842–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Nyer DB, Daer RM, Vargas D, Hom C, and Haynes KA (2017). Regulation of cancer epigenomes with a histone-binding synthetic transcription factor. NPJ Genom Med 2. 10.1038/s41525-016-0002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Williams S, Mustoe T, Mulcahy T, Griffiths M, Simpson D, Antoniou M, Irvine A, Mountain A, and Crombie R (2005). CpG-island fragments from the HNRPA2B1/CBX3 genomic locus reduce silencing and enhance transgene expression from the hCMV promoter/enhancer in mammalian cells. BMC Biotechnol. 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Karlsson M, Zhang C, Méar L, Zhong W, Digre A, Katona B, Sjöstedt E, Butler L, Odeberg J, Dusart P, et al. (2021). A single-cell type transcriptomics map of human tissues. Sci Adv 7. 10.1126/sciadv.abh2169. [DOI] [PMC free article] [PubMed] [Google Scholar]