

Abstract

Photocatalysis has become a prominent tool in the arsenal of organic chemists to develop and (re)imagine transformations. However, only a handful of versatile organic photocatalysts (PCs) are available, hampering the discovery of new reactivities. Here, we report the design and complete physicochemical characterization of 9-aryl dihydroacridines (9ADA) and 12-aryl dihydrobenzoacridines (12ADBA) as strong reducing organic PCs. Punctual structural variations modulate their molecular orbital distributions and unlock locally or charge-transfer (CT) excited states. The PCs presenting a locally excited state showed better performances in photoredox defunctionalization processes (yields up to 92%), whereas the PCs featuring a CT excited state produced promising results in atom transfer radical polymerization under visible light (up to 1.21 Đ, and 98% I*). Unlike all the PC classes reported so far, 9ADA and 12ADBA feature a free NH group that enables a catalytic multisite proton-coupled electron transfer (MS-PCET) mechanism. This manifold allows the reduction of redox-inert substrates including aryl, alkyl halides, azides, phosphate and ammonium salts (Ered up to −2.83 vs SCE) under single-photon excitation. We anticipate that these new PCs will open new mechanistic manifolds in the field of photocatalysis by allowing access to previously inaccessible radical intermediates under one-photon excitation.
Introduction
Over the past decades, the design and development of new catalysts has been a driving force for synthetic chemists to enable new reaction manifolds toward compounds of high interest in fine chemical synthesis.1 In this context, the current development of photocatalysis has set previously inaccessible mechanistic vistas,2 while allowing operation under mild and sustainable reaction conditions.3 In particular, the identification and use of simple and purely organic molecules as photocatalysts (PCs) has become a major goal in the chemical community.4 Organic PCs present clear advantages with respect to their metal counterparts (e.g., Ru- or Ir-complexes), including (i) their higher accessibility and sustainability,3c (ii) their overall lower price,4 and (iii) a higher tunability of their scaffolds,5 which enables (iv) a more direct assessment of the structure–property relationships.6 Likewise the metal-to-ligand charge transfer (MLCT) in metal complexes, organic PCs can access a charge transfer (CT) excited state. The CT excited state occurs when the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) are spatially separated in the molecule,5c and involves a directional electron movement from the HOMO to the LUMO. The new electron distribution grants access to more stable excited states that have an extended lifetime (from ns to μs) as well as more balanced redox potentials.3c In this context, highly reducing organic PCs play a crucial role.6,7 These molecules are highly efficient for the activation of alkyl and aryl halides, ketones, and aldehydes, as well as for the generation of polymers under mild metal-free conditions, thus overcoming metal-contamination issues.4,7
The preliminary findings in this area have involved the use of phenothiazine (PTH) 1 (Figure 1),8 which is still one of the PCs of choice for thermodynamically challenging reductive processes. Nevertheless, the low visible-light absorption of PTH, its poor tunability, and inefficiency under atom transfer radical polymerization (ATRP) processes have encouraged the community to identify different scaffolds.6b In that respect, structural motifs such as 2 and 3 have been recently reported to serve as convenient alternatives to PTH (Figure 1). These scaffolds merge high reducing power with a CT character, rendering their excited-state lifetime longer and their redox properties rationally tunable. Recently, dihydrophenazines 2 have shown their potential in light-driven metal-free polymerization processes.9 However, the widespread use of these known PC variants is often hampered by tedious synthetic routes, poor solubility, or low visible-light absorption. More importantly, the classes of PCs 1–3 share a moderate versatility when moving from classical oxidative quenching processes to ATRP and vice versa. Hence, the identification of novel structural classes capable of switching from the functionalization of small molecules to the synthesis of polymers, while unlocking previously inaccessible reactivity, is a formidable challenge. With this aim in mind, we turned our attention to 9,10-dihydroacridine scaffolds that are typically used in material chemistry as electron-donor moieties.10 Until recently, their use as photocatalysts has been largely underdeveloped, which might be explained by the challenges associated with their synthesis and their tunability. However, we recently solved this issue by devising a simple protocol to access those compounds in a single step from common alkyne and diarylamine precursors.11 We herein document the rational design and structural refinements of two new classes of highly reducing purely organic PCs incorporating a 9,10-dihydroacridine scaffold (Figure 1, 4 and 5). We performed complete physicochemical characterization of various scaffolds, while delineating structure–property relationships. Their synthetic potential is illustrated in thermodynamically challenging reductive photoredox processes. Furthermore, we disclose a new mechanistic pathway based on a catalytic proton-coupled electron transfer (PCET) mechanism that relies on the crucial activity of the free NH group, a key structural signature of these molecules. This reaction manifold is currently not possible to access by the previous PC classes (1–3) as they lack the essential H-bond donor moiety. Lastly, the use of our catalysts in metal-free controlled radical polymerization processes under visible-light was investigated.
Figure 1.

Highly reducing PCs previously reported and our new classes of organic PCs disclosed.
Results and Discussion
Photophysical Characterization
By assessing the previously reported classes of reducing PCs, we questioned whether the presence of an aryl ring on the N atom was essential to reach a CT excited state. Our investigation was guided by the idea that having a free NH group would allow new types of reactivity. We first considered the 9-aryl dihydroacridine (9ADA) scaffold 4a–c bearing electronically varied functional groups (H, CF3, CN; Figure 2a), in which the exocyclic aromatic ring could potentially accommodate the LUMO. Indeed, DFT calculations at the B3LYP/6-311++G** level of theory confirmed our hypothesis and revealed the possibility of accessing a CT excited state (Figure 2a, bottom). Time dependent DFT calculations were performed to verify which MOs are actually involved in the formation of the lowest excited state of each PC (see Table S7 in the SI). In the case of compound 4a, the lowest excited state primarily involves a transition between HOMO and LUMO+1 MOs, which are both located on the tricyclic core, while the LUMO is located on the external aryl ring (Figure 2a, bottom). This MO distribution suggests access to a CT excited state while allowing the exploitation of the free amino group. Based on previous precedents,4−6,10 the CF3 and CN groups were selected to evaluate the impact of an increased electron-withdrawing (EW) character of the exocyclic aryl ring on the MOs, and later on the overall photoredox properties of the molecules.
Figure 2.

Absorption and emission profiles of (a) 9ADA PCs 4a–c, (b) 2,7-OMeADA PCs 6a–c, and (c) 12ADBA PCs 5a–d. All the experimental data were collected in MeCN (see SI, Section F). Selected frontier MOs and relative calculated energies of PCs bearing an unsubstituted Ph ring at position 9 or 12 (4a, 6a, and 5a) and PCs substituted with a p-CN group (4c, 6c, and 5d). The LUMO+1 levels are depicted by lighter color bars.
By adding the CN group, we computed a LUMO-lowering and a lowest excited state primarily involving a transition between HOMO and LUMO. These data suggest the CT character of the lowest excited state of 4c. With respect to the experimental data, it strongly increases the CT character within 4c. The absorption slightly increases along the series, with 4c tailing to the visible region (Figure 2a, abs.). On the other hand, the emission profile is significantly red-shifted, with the λmax passing from 362 to 418 and 484 nm (Figure 2a, em.). We next evaluated a HOMO-raising strategy by introducing an electron-donating group (EDG, OMe) at position 2 and 7 of the original scaffold 4 (Figure 2b, bottom). As expected, this class of molecules behaves similarly to 4 while showing a red-shifted absorption, tailing up to 390 nm for 6c. Additionally, the emission is red-shifted, passing from 362 nm for 6a, up to 543 nm for 6c. Over the two series of PCs 4 and 6 (Figure 2a and 2b), the addition of EWGs at the exocyclic aryl group (H to CF3 and CN) resulted in a higher Stokes shift, passing from 77 to 204 nm for PCs 4, and from 24 to 201 nm for PCs 6 (vide infraTable 1). This behavior indicates an increased CT excited state character. It is noteworthy that the emission profile of 6c and in part also the one of 4c presented two maxima, which can be attributed to two alternative excited configurations: a local S1 excited configuration and a S1 CT excited configuration.12 Indeed, the solvatochromic properties of PC 6c were experimentally observed by dissolving this PC in solvent having an increased polarity (Figure 3).13 These analyses indicate that the photochemical properties of these molecules (4a–c and 6a–c) can be simply tuned by modifying the substitution pattern of the exocyclic aryl ring. In fact, a mere H-to-CN replacement confers increased visible-light absorption and red-shifted emission, while highly impacting the PC’s lifetime and redox properties (vide infra).
Table 1. Summary of the Excited- and Ground-State Photoredox Propertiesa.

All potentials were measured in MeCN. Values are reported in V versus SCE (see SI, Sections A and F). The colored bars qualitatively indicate the presence of a CT excited state–the color intensity is based on Stokes shift and QY.
Figure 3.

Photograph of the PC 6c dissolved in solvents with increasing polarity, as indicated by their increasing Reichardt parameter (ET(30) in kcal/mol). From left to right: n-hexane (ET(30) = 31.0 kcal/mol), toluene (ET(30) = 33.9 kcal/mol), ethyl acetate (ET(30) = 38.1 kcal/mol), tetrahydrofuran (ET(30) = 37.4 kcal/mol), dichloromethane (ET(30) = 40.7 kcal/mol), acetone (ET(30) = 42.2 kcal/mol), acetonitrile (ET(30) = 45.6 kcal/mol), and dimethyl sulfoxide (ET(30) = 45.1 kcal/mol).
While computationally investigating alternative structural variants and maintaining the free NH group, we considered the 12-aryl dihydrobenzoacridine scaffold (12ADBA) 5. In the case of this family of PCs, the calculated LUMO is placed at a different position depending on the presence or absence of an EWG on the exocyclic aryl ring (Figure 2c, bottom). In the presence of a H, OMe, or CF3 group (5a–5c), the LUMO lies onto the naphthalene within the PC’s core, whereas, in the case of CN-substituted PC 5d, it is placed on the exocyclic ring similarly to the other PC’s classes. As a result, a different substitution pattern was used to deliberately alter the LUMO distribution within the scaffolds and thus their photochemical properties. Thanks to the extended conjugation, all PCs 5 have an enhanced visible-light absorption with respect to the other classes of PCs 4 and 6 (Figure 2c, top). The emission profiles are almost identical for PCs 5a–c while an alternative spectroscopic signature is observed for 5d, as suggested by the alternative LUMO distribution. The simple addition of a CN group also results in a significant variation of the MO’s distribution which has a major impact on the photochemical properties of this molecule.
Characterization of the PCs Redox Properties
We continued the structure–property relationship study by analyzing the cyclic voltammetry (CV) of our PCs to define their ground and excited state redox potentials (Figure 4). As predicted by DFT calculations, the PCs 4a–c showed Eox within a narrow range of potentials spanning from 0.79 to 0.86 V vs SCE. However, the cathodic sweeps revealed the presence of two different peaks. Such behavior has been already observed for structurally related systems.
Figure 4.

Normalized cyclic voltammograms of PCs 4a–c, 6a–c, and 5a–d. Potentials are reported in V vs SCE. Normalized cyclic voltammograms of PCs 6a–c (solid traces) compared with the normalized cyclic voltammogram of PC 4a (dashed trace).
The second peak was attributed to the presence of electroactive dimerization products in solution deriving from the corresponding radical cations reacting at position 2 and/or 7.14 Introducing substituents at such positions inhibited the dimerization pathway. Indeed, the CVs of PCs 5a–d and 6a–c showed a completely reversible behavior with a single cathodic peak. In turn, the more electron-rich PCs 6a–c have less positive Eox than both 4a–c and 5a–d (Figure 4), spanning from 0.44 to 0.49 V vs SCE. The presence of OMe groups on the dihydroacridine scaffold results in a HOMO-raising, that facilitates the single electron oxidation by 0.3 V. Although being characterized by a red-shifted absorption, PCs 5a–d showed Eox very close to those of 4a–c, spanning from 0.74 to 0.80 V vs SCE. Indeed, insights from the calculated HOMO revealed negligible alteration through the addition of the fused phenyl ring. In the case of PC 5a, the CV analysis under oxidative scan shows a reversible one-electron wave (ΔEp = 74 mV, ipa/ipc = 0.99 at 0.1 V/s scan rate, Figure 5), while the plot of the anodic peak current vs v1/2 provided a linear relationship, indicating a diffusion-controlled process.15
Figure 5.
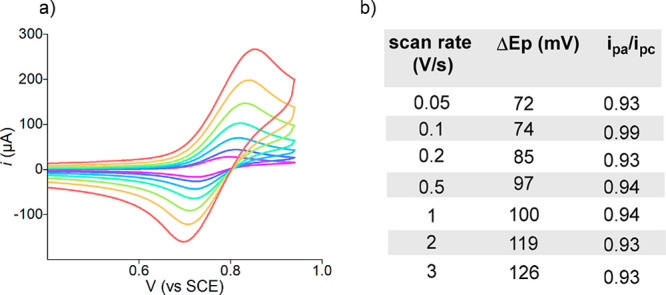
(a) Cyclic voltammograms of 5a recorded at increasing scan rates. (b) Selected experimental values of ΔEp and ipa/ipc at different scan rate (see Section E in the SI).
It is important to stress how punctual structural modifications on the scaffolds 4–6 can be used to impart a substantial variation of the redox properties while still maintaining a highly reducing character, with the Eox spanning from 0.86 to 0.44 for 4c and 6a, respectively. This feature is not always possible or easy to access by the currently available highly reducing organic PCs (e.g., 1–3).
We next calculated excited state redox potentials across all the developed PCs. All the E*ox values are highly negative, spanning from −2.31 to −2.91 eV. These data are summarized together with the photochemical and redox properties in Table 1.
Evaluation of the Excited State Charge Transfer Character
We then completed the assessment of the PCs properties by looking at the ability of the diverse molecules to access a CT excited state. We thus measured the luminescence quantum yield (QY) and the excited state lifetimes (τ). As summarized in Table 1 (green box), the QY decreases over the series of PCs 4 and 6, passing from 11% to 3% for PCs 4, and from 8% to 0.7% for PCs 6, according to the increased CT character. On the other hand, τ increases from 4.2 to 14.3 ns for PCs 4a → 4c, and from 3.3 to 4.2 ns for PCs 6a → 6c. These trends, together with the highly increased Stokes shift—which exceeds 200 nm for PCs 4c and 6c—clearly indicate that PCs 4b, 4c, and 6c can access a CT excited state. A different behavior was observed for the 12ADBA series. Specifically, PCs 5a–c have moderate CT character, with QY and τ remaining to similar levels for these scaffolds. On the other hand, PC 5d showed an increased Stokes shift of 81 nm, a reduced QY of 6%, and a slightly extended τ of 9.4 nm, indicating a more significant CT character. Remarkably, selecting the suitable substitution pattern allows not only control of the photochemical properties but also access to CT character with longer τ and more balanced redox potentials.
Exploitation of the NH Group To Accessing PCET Manifolds
Having established structure–property relationships, we next sought to use the free NH group to explore new types of reactivity that were challenging with the previously reported photocatalytic systems. We reasoned that, in the presence of a suitable base associated in a hydrogen-bonding network with the PC, the reduction of the substrate by the PC* could occur through a PCET mechanism, with definite kinetic and thermodynamic benefits.16 Apart from the possibility of opening new mechanistic perspectives, a PCET would allow the reduction of redox-inert substrates by extending the redox window of the PC.16a,16b The oxidation potential of the PC* is expressed in eq 1 (where E0,0 is the energy gap between the lowest vibrational levels of the excited and the ground states) and refers to the semi reaction shown in eq 2.
| 1 |
| 2 |
In the presence of a base B capable of promoting a PCET with the PC, the involved semi reaction turns into eq 3, that can be considered as the sum of eqs 2, 4, and 5:
| 3 |
| 4 |
| 5 |
The reduction potential from eq 3 can be expressed in eq 6, and it is thus dependent on the potential of the PCNH•+/PCNH* couple and on the difference in acidity of PCNH•+ and BH+:
| 6 |
with 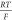 = 0.0257 V.
= 0.0257 V.
The oxidation potential of the PCNH*/B couple expressed in eq 6 can be in principle adjusted depending on the strength of the base. In particular, the stronger the base (pKa(BH+) higher), the more negative the oxidation potential, and the higher the reducing power of the PC*/B couple is. With the aim of evaluating the benefits of a PCET vs a classical ET manifold, we started testing the ability of the PCs to engage in reductive dehalogenation processes (Figure 6a). These types of photoreactions require an extremely reducing PC excited state. In fact, they classically proceed either under strong UV-light irradiation,17 or by relying on the photoexcitation of specific radical anions.18 While in previous reports, PTH 1 was used under UV-light irradiation (<380 nm),17 we compared the reaction performance of the designed PCs under visible light (427 nm) using both kinetics and final yields (see Section G of the SI, and Figure 6). The best results were obtained with PCs 5a-c that outperformed all the other 9ADA variants 4 and 6, as well as PTH 1 – the previous PC of choice for this reaction, that furnished the product in 52% yield (see SI, Section G). Besides, the CT character does not infer any advantage in terms of reactivity. We finally wondered which of the excited forms of the PCs (S1 vs T1) was engaged under the reductive process. We thus performed the control experiments in the presence of two well-known triplet quenchers. The reaction of 5a in the presence of O2 (air) or diazabicyclo[2.2.2]octane (DABCO) furnished the product 8 in nearly identical yield, 81% and 82%, respectively, ruling out the hypothesis of a T1-based reactivity of the PCs.
Figure 6.
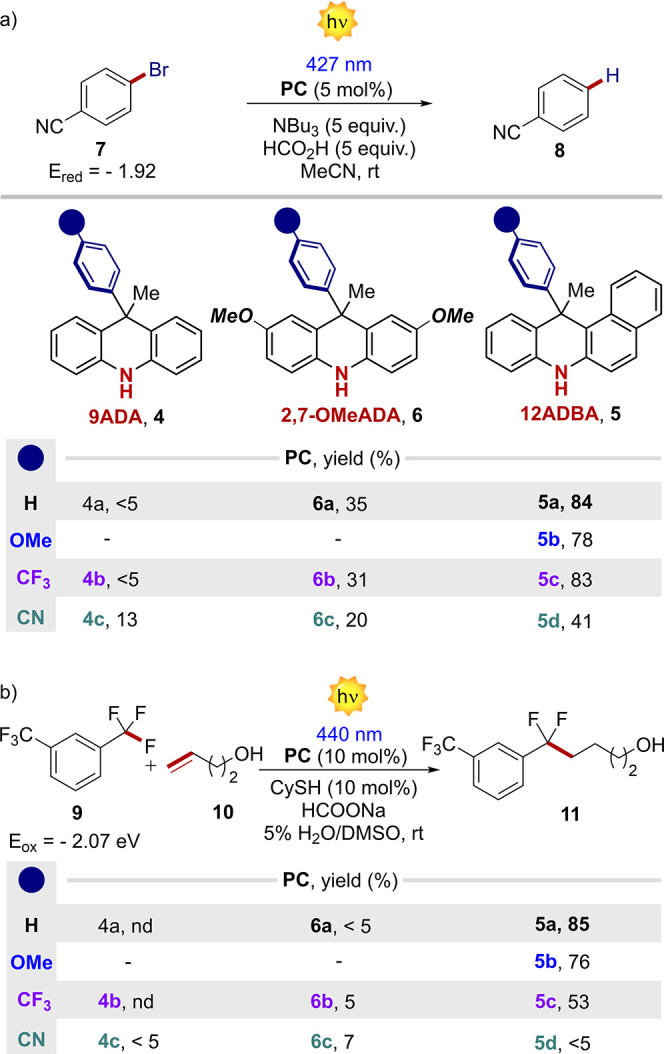
(a) Photocatalytic dehalogenation reaction. (b) Defluorinative arene alkylation. Reaction time 6 h. Yields were determined by GC-FID and 19F-NMR (see SI, Section G).
Of note, apart from previous mechanistic studies,18 the activity of a XAT-based manifold under the reported reaction conditions is unlikely due to the highly reducing nature of the PCs and to the presence of a stoichiometric amount of acid. To confirm this finding, we selected a second photoreaction to evaluate the PCs under the ET reaction manifold, namely the photoreduction of trifluoromethylarenes (Figure 6b),13 employing 1,3-bistrifluoromethylbenzene 9 (Ered = −2.07 V vs SCE)14 as a model substrate with olefin 10 (for the reported mechanism see Section G of SI). It has been recently reported that only extremely reducing PCs such as PTH 1 could promote this challenging process under energetic light irradiation (380–400 nm).13,15 We decided to screen all the PCs in our possession under a less energetic 440 nm irradiation, while comparing their kinetic profiles at the initial stage of the reaction (see Section G of the SI). The analysis of kinetic profiles showed the best performances with PCs 5a, 5b, and 5c. In the case of PCs 5a and 5b, the target product 11 was obtained in 85% and 76% yield in 6 h, respectively. As a comparison, PTH 1 reached 87% yield after 24 h under a more energetic irradiation.
Having identified 5a as the best PC under the classical ET manifold, we subsequently evaluated the feasibility of a PCET mechanism. In this case, we targeted the photoreductive dehalogenation of the thermodynamically challenging aryl chloride 12 (Ered = −2.08 vs SCE), comparing the reaction outcome in the presence of different bases (pKa in the range 23.4–26.0 in MeCN) at a short reaction time (2 h). The reaction in the presence of NBu3 proved to be highly inefficient (9% yield), while no reaction was observed without a base. The addition of a base engaging in H-bonding showed improved performances (Figure 7a). MeTBD was identified as the best base, delivering the product in 27% yield. In agreement with the involvement of a PCET mechanism (Figure 7c), a competent HAT donor (e.g., γ-terpinene 13) was added to the reaction mixture. Under these conditions, the process ended up being faster and more selective. The diverse bases showed a trend with respect to their pKa. TBD, the only solid base along the series, was the only exception, likely due to its inferior solubility. The best result was obtained when MeTBD was used, providing product 8 in 40% yield after only 2 h with a virtually perfect mass balance. In this case, the control experiments also point to the operation of an S1 excited state, as the reactions in the presence of O2 or DABCO resulted in 31% and 36% yields, respectively.
Figure 7.

(a) Base screening. (b) Cyclic voltammetry of 5a in the presence and absence of MeTBD in MeCN, V vs SCE. (c) Proposed reaction manifold. (d) 1H NMR titration experiment revealing the presence of a H-bonding complex. Reactions performed at 0.2 mmol scale, [10]0 = 0.1 M. * TBD resulted partially insoluble under the reaction conditions (see SI, Section H).
Finally, an extended reaction time with MeTBD (12 h) led to 8 in 65% yield. We next attempted to rationalize the obtained results. Consistent with eq 3, the cyclic voltammetry of 5a in the presence of MeTBD showed an anodic wave peaking at 0.32 V vs SCE, thus being anticipated by 0.44 V (Figure 7b). This experiment suggests an interaction between the PC and the base, and demonstrates that the oxidation of PC 5a is anticipated of 0.44 V in the presence of MeTBD. Based on this experimental evidence, we can assume that when the base is added to the reaction mixture, an alternative mechanistic pathway operates, in which the PC is preorganized into a H-bonded complex with MeTBD (Figure 7c). To get insight into the nature of such PC-base interaction, we performed 1H NMR titrations. Those experiments, in the presence of increasing amounts of MeTBD, clearly indicate the existence of an H-bonding complex between the base and the PC (Figure 7d). An alternative deprotonation process was ruled out due to the pKa of the involved species (MeTBD = 25.4, pKa5a = 32, vide infra), and the different 1H NMR trace of the K-salt of 5a (see SI, Sections D and J). Following this mechanistic proposal (Figure 7c), upon light irradiation, the PC-MeTBD complex reaches an electronically excited state that takes part in a multisite PCET with chlorobenzonitrile 12. While the electron is transferred to 12, the proton moves to the MeTBD, resulting in the generation of (i) the aryl radical, (ii) the protonated MeTBD, and (iii) the PC N-centered radical 16. At this juncture, a HAT donor is necessary to close the catalytic cycle, delivering the final product 8 while restoring the PC. This last step was confirmed by the detection of p-cymene (15), deriving from the aromatization of γ-terpinene, in the reaction crude by both 1H NMR and GC-FID. The quantum yield of the reaction was found to be 0.15, corroborating our mechanistic hypothesis, ruling out at the same time an alternative XAT process (see Section H of the SI). DFT calculations further supported the experimental data, computing a ground-state H-bonding complex of PC 5a and MeTBD with a NH···N distance of 1.9 Å (Figure 8, left). In agreement with the experimental data, when one electron is removed from the complex, the proton moves to the base with the formation of the N-centered radical 16 (Figure 8, right). The spin-density map reveals that the radical is mainly localized on the N atom. DFT calculations were also performed to estimate the pKa values for 5a and its radical cation 5a•+. We used a relative determination method, based on the experimental pKa of the base and the difference in energy of the optimized adducts, where the proton is deliberately located on the PC or on the base (see SI, Section H).
Figure 8.
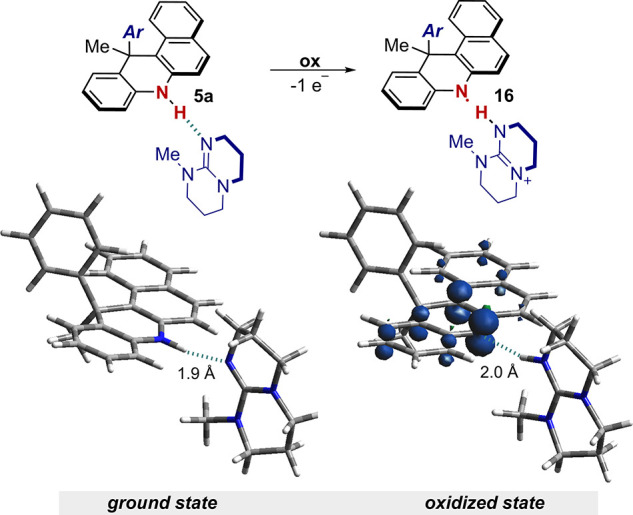
Calculated H-bonding complexes of the PC 5a with MeTBD at the ground (left), and oxidized state (right) with spin-density map. Structures, energies and electronic features of the complexes were calculated at B3LYP/6-311++G** level of theory (see SI, Section H).
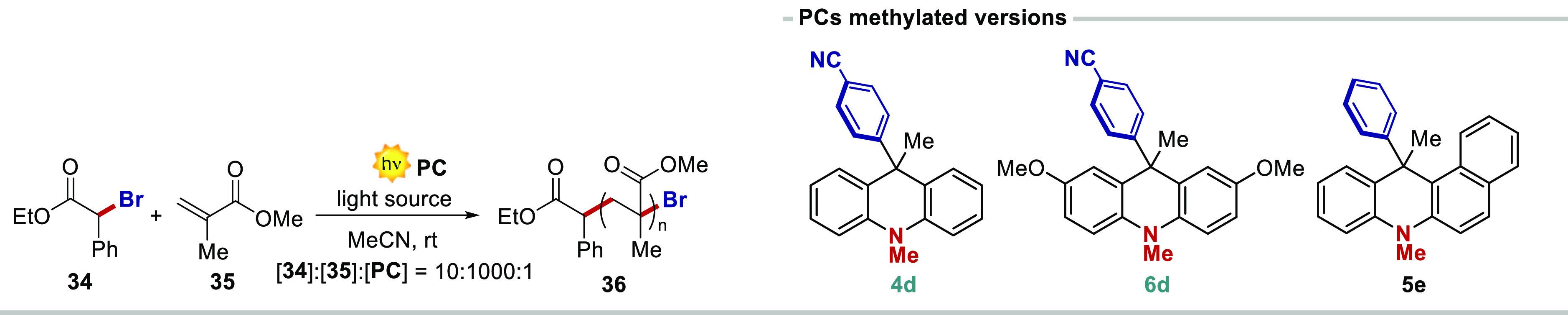
This estimation led to a pKa of 32 and 17 for 5a and 5a•+, respectively, thus in agreement with a proton shift during the PCET. Having designed an alternative manifold to the classical ET, we then investigated the generality of the PCET manifold with respect to the ET approach for the defunctionalization of structurally diverse substrates. In this regard, we synthesized the methylated PC 5e, for which a PCET is not possible. From there, we selected a series of compounds 17-33 of diverse chemical nature that were all characterized by a very negative Ered, approaching −2.8 eV vs SCE (in MeCN). Remarkably, without the need of any optimization process, aryl and benzyl halides, as well as azides were found to be suitable substrates, with yields ranging from 40 to 60%. In contrast, under the ET manifold, the corresponding defunctionalized products were obtained, almost in all the cases, in inferior yields (Figure 9). Phosphonates and phosphates were more reluctant substrates, although the PCET manifold delivered the corresponding products in up to 37% yield. Lastly, the ability of defunctionalize alkyl ammonium salts was explored. Here the PCET manifold demonstrated his superior generality and synthetic potential with yields up to 71%. In several cases, under ET, we observed a significant decrease of the efficiency, ammonium salts 28, 29, 31 and 32 proving completely unreactive. Remarkably, the PCET manifold also demonstrated its versatility in the presence of difunctionalized substrates (31), amino acids derivatives (32) and hydroxyl-group-free compounds (33). It has to be stressed that the reduction of these molecules (26-33) usually require photoelectrochemical synthesis or a multiphoton excitation process.19 An alternative XAT-based strategy is also not possible.18 Here, we just exploited the structural feature of the PCs under a single-photon excitation. While 5a can still afford the defunctionalized products even beyond its redox potentials (E*ox = −2.37) through a PCET, the N-methylated PC 5e—working by ET—loses the ability to promote photoreductions when approaching its redox potential limit. These results indicate that, by accessing a catalytic PCET manifold, 5a has key kinetic and thermodynamic benefits.
Figure 9.

Generality and limits of the PCET manifold (base = MeTBD) and comparison with the classical ET process (base = NBu3). Yields determined by GC-FID or 1H NMR analysis of the crude reaction mixture; isolated yields are reported in parentheses (see SI, Section G). *The reported value refers to the conversion of the substrate.
PCs for Photoinduced Organocatalyzed Atom Transfer Radical Polymerization (O-ATRP)
As mentioned above, highly reducing PCs have found important applications in organocatalyzed atom transfer radical polymerization (O-ATRP).9 We thus decided to assess the ability of our PCs in this type of industrially relevant transformation. As model substrates, we selected ethyl α-bromophenylacetate (BPA) 34 as the initiator and methyl methacrylate (MMA) 35 as the monomer. The efficient use of the developed PCs under ET or PCET manifolds did not guarantee their applicability under ATRP. This is because the reaction manifold in ATRP is significantly different from ET or PCET processes (Figure 10). Upon light excitation, the PC reaches an electronically excited state that is responsible for the activation step by single-electron reduction of the alkyl radical (chain or 28).
Figure 10.

O-ATRP and reported reaction manifold.
This ET, which requires a high reducing power, leads to the formation of the corresponding radical 37 and the PC•+. A delicate interlay should be balanced between the stability of the PC•+ and its ability to oxidize the propagating radical 38 or the Br anion, to efficiently deactivate the propagation process and deliver a controlled polymerization. PCs 4–6 were subsequently tested as catalysts in photoinduced organocatalyzed atom transfer radical polymerization (O-ATRP) of MMA 35 in acetonitrile (MeCN) as a solvent (Table 2). All the structurally different PCs were able to trigger the polymerization of MMA. An important parameter in the evaluation of the PCs performances in O-ATRP is the molecular weight dispersity of the polymer chains Đ (Đ = Mw/Mn, where Mw is the mass-average molecular weight and Mn is the number-average molecular weight).9 Ideally, the Đ value should be as close as possible to 1 to obtain a virtually monodispersed polymer. Another relevant parameter is the initiation efficiency (I*) that corresponds to Mn (theoretical)/Mn (experimental), where the Mn,exp is the experimentally measured number-average molecular weight.9 In this case the optimal value is 100%, where the PC exerts the highest control over the polymerization process, with a perfect balance on the activation and deactivation steps. PCs 4a–c provided poly(methyl methacrylate) (PMMA) 36 with Đ between 1.33 and 1.59 (entries 1–4). In the case of 4c, the polymerization proceeded more rapidly than with the structurally related 4a–b. This was presumably due to the superior CT character of this particular PC and a corresponding longer excited state lifetime. Of note, while PCs 4a–b displayed moderate to low initiator efficiency (I* = Mn,theo/Mn,exp × 100%, where Mn,theo is the theoretical and Mn,exp the experimentally measured number-average molecular weight),9 the compound 4c exhibited an I* of 113%. Given the red-shifted light absorption of 4c, we also evaluated its performance as PC for O-ATRP under visible light irradiation at 400 nm (entry 4). Under these conditions, O-ATRP of MMA catalyzed by 4c showed improved control, reaching Đ = 1.33, albeit with a lower I* (70%). This behavior could be attributed to an inferior catalytic activity of the PC, thus exerting lower control on the MW.6c Since all PCs of the series 6a–c (entries 5–8) featured a red-shifted absorption with respect to 4a–c, they provided relatively high MMA conversion after only 2 h of irradiation (76–86%) with high I*, which approached 100% for 6a (98%) and 6c (96%). However, Đ remained moderately low, reaching 1.38 for PC 6c (entry 8).
Table 2. Results of the Polymerization of MMA in Batcha.
| Entry | PC | Wavelength (nm) | Polymerization time (h) | Conversion (%) | Mw (kDa) | Mn (kDa) | Đ | I* (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 4a | 390 | 9 | >98 | 19.8 | 12.6 | 1.59 | 78 |
| 2 | 4b | 390 | 7 | 85 | 18.7 | 13.4 | 1.40 | 63 |
| 3 | 4c | 390 | 2 | 95 | 13.1 | 8.4 | 1.56 | 113 |
| 4 | 4c | 400 | 4 | 98 | 18.5 | 13.9 | 1.33 | 70 |
| 5 | 6a | 390 | 2 | 84 | 12.1 | 8.6 | 1.41 | 98 |
| 6 | 6b | 390 | 2 | 76 | 13.8 | 9.6 | 1.43 | 79 |
| 7 | 6c | 390 | 2 | 86 | 13.2 | 9.0 | 1.46 | 96 |
| 8 | 6c | 400 | 4 | 81 | 17.4 | 12.6 | 1.38 | 64 |
| 9 | 5a | 427 | 6 | 86 | 10.8 | 6.9 | 1.52 | 124 |
| 10 | 5b | 427 | 6 | 81 | 25.8 | 13.2 | 1.96 | 61 |
| 11 | 5c | 427 | 6 | >98 | 13.1 | 8.2 | 1.57 | 119 |
| 12 | 5d | 427 | 6 | >98 | 12.4 | 8.0 | 1.55 | 122 |
| 13 | 4d | 400 | 4 | 76 | 19.8 | 13.1 | 1.51 | 58 |
| 14 | 6d | 400 | 4 | 91 | 16.3 | 12.2 | 1.34 | 74 |
| 15 | 5e | 427 | 6 | >98 | 16.2 | 12.2 | 1.44 | 80 |
Reaction were performed in MeCN with a ratio [MMA]:[EBP]:[PC] = 1000:10:1, [MMA]:[MeCN] = 1:1 (v/v) (see SI, Section G).
The activity of PCs 5a–5d for O-ATRP of MMA was subsequently evaluated by using 427 nm irradiation. 5c and 5d provided full MMA conversion in 6 h, while for 5a and 5b the reaction was relatively slower. PCs 5a, 5c, and 5d all provided similar control on Đ and exhibited similarly high I*. In turn, 5b stood out in the series by showing a poor I* (61%) and providing rather polydisperse PMMA (Đ = 1.96). Control experiments performed with the N-methylated scaffolds of 4c, 6c, and 5a (entries 13–15) resulted in poor control over O-ATRP, highlighting the benefit of the free NH group within the PC structure for the controlled synthesis of PMMA.20 In order to test the temporal control over O-ATRP, we performed a series of experiments in which the reaction mixture was exposed to a pulsed irradiation (400 nm, 30 min per cycle). As it can be observed in Figure 11a, using PCs 4c, 6c, and 5a, polymerization occurred only during irradiation, while it reversibly stopped when the reaction solution was kept in the dark. Hence, O-ATRP could be efficiently controlled by switching on and off the light source, enabling excellent control over chain growth by using irradiation as a trigger.7,21 Among the PCs, 5a performed the best, in terms of both initial polymerization rate and conversion over 2 h. The result could be ascribed to a more efficient visible photon harvesting of 5a with respect to 4c and 6c (Figure 2).
Figure 11.

(a) On/off studies performed at 400 nm with PCs 4c, 6c, and 5a. (b) In-flow O-ATRP process (see SI, Section G).
Finally, we evaluated the performance of 5a as PC for O-ATRP under flow conditions (Figure 11b). During a flow process the irradiation is more efficient and homogeneous, permitting a more effective excitation of the PC and activation of the alkyl bromide.22 In addition, under these conditions, it is possible to obtain an increased amount of polymer just by parallelization or numbering-up strategy.23 In the present study, a simple syringe pump connected to a 3D-printed flow reactor was employed as a flow reactor (see SI, Section G). We scaled-up the polymerization using 28.2 mmol of the monomer. Remarkably, in a relatively short reaction time (RT) of 4 h, and under visible-light irradiation (427 nm), 86% of MMA conversion with Đ = 1.21 was obtained, although a relatively low I* was recorded (see Table 2, entry 9 for comparison). These results further highlight the potential and versatility of the newly developed PCs, and their applicability in O-ATRP, especially when it is performed in continuous flow.
Conclusions
In conclusion, we have reported free-NH 9-aryl (9ADA) and 12-aryl dihydroacridines (12ADBA), two novel classes of strongly reducing organic PCs engaging under both catalytic PCET and ATRP processes. Based on UV–vis absorption, emission, excited-state lifetime, quantum yield, cyclic voltammetry, and DFT calculations, we have assessed their complete structure–property relationships. The key structural features of these PCs allow access to a CT excited state even in the absence of the common N–Ar moiety, which is present in all the other reported PCs classes (Figure 1). Here, the free NH group is engaging in an H-bonding interaction with a competent base (e.g., MeTBD), expanding the PC’s redox limits to enable the activation of redox-inert alkyl and aryl halides as well as azides, and aryl and alkyl ammonium salts (yields up 71%). The disclosed PCET-based reactivity has been investigated by CV and 1H NMR titration along with computational data. Finally, we also evaluated the potential use of these new PCs in catalyzing O-ATRP processes. Across the diverse PC structures, we determined that the 9ADA family was more performant, delivering the polymer under controlled conditions in up to 1.21 Đ, and 98% I* (Table 2). Importantly, the PCs accessing a CT excited state (longer τ, and more balanced redox potentials) gave better results under ATRP processes, while the PCs accessing a locally excited state (shorter τ, and higher E*ox) performed better for the activation of thermodynamically challenging substrates. Here, we have shown that previously inaccessible mechanistic vistas can be opened by using the free NH group, without losing the ability of catalyzing O-ATRP processes. We thus foresee a broad utilization of these versatile purely organic PCs for the activation of redox-inert substrates under synthetic and material chemistry settings.
Acknowledgments
This work was supported by MUR (Ministero dell’Università) PRIN 2020927WY3_002, (European Research Council) ERC-Starting Grant 2021 SYNPHOCAT 101040025 (L.D.); the CariParo Foundation Synergy–Progetti di Eccellenza 2018 (A.S.); and the Interdisciplinary Thematic Institute ITI-CSC via the IdEx Unistra (ANR-10-IDEX-0002) within the program Investissement d’Avenir (D.L.). D.L. thanks the CNRS. D.L. thank Dr. Shengdong Wang and Prof. Vincent Gandon for initial studies. Prof. Marco Fantin is gratefully acknowledged for insightful discussions, and Mr. Carlos Pavón Regaña, for technical support during the polymerization experiments.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c11364.
Experimental procedures, characterization data, and NMR spectra (PDF)
The authors declare no competing financial interest.
This paper was published on January 6, 2023. The colors in Figure 4 have been corrected and the paper was re-posted on January 9, 2023.
Supplementary Material
References
- a Vega-Peñaloza A.; Paria S.; Bonchio M.; Dell’Amico L.; Companyó X. Profiling the Privileges of Pyrrolidine-Based Catalysts in Asymmetric Synthesis: From Polar to Light-Driven Radical Chemistry. ACS Catal. 2019, 9, 6058–6072. 10.1021/acscatal.9b01556. [DOI] [Google Scholar]; b Douglas J. J.; Sevrin M. J.; Stephenson C. R. J. Visible Light Photocatalysis: Applications and New Disconnections in the Synthesis of Pharmaceutical Agents. Org. Process Res. Dev. 2016, 20, 1134–1147. 10.1021/acs.oprd.6b00125. [DOI] [Google Scholar]; c Bogdos M. K.; Pinard E.; Murphy J. A. Applications of Organocatalysed Visible-Light Photoredox Reactions for Medicinal Chemistry. Beilstein J. Org. Chem. 2018, 14, 2035–2064. 10.3762/bjoc.14.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzetti L.; Crisenza G. E. M.; Melchiorre P. Mechanistic Studies in Photocatalysis. Angew. Chem., Int. Ed. 2019, 58, 3730–3747. 10.1002/anie.201809984. [DOI] [PubMed] [Google Scholar]
- a Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shaw M. H.; Twilton J.; MacMillan D. W. C. Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81 (16), 6898–6926. 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bortolato T.; Cuadros S.; Simionato G.; Dell’Amico L. The Advent and Development of Organophotoredox Catalysis. Chem. Commun. 2022, 58, 1263–1283. 10.1039/D1CC05850A. [DOI] [PubMed] [Google Scholar]
- a Nicewicz D. A.; Nguyen T. M. Recent Applications of Organic Dyes as Photoredox Catalysts in Organic Synthesis. ACS Catal. 2014, 4, 355–360. 10.1021/cs400956a. [DOI] [Google Scholar]; b Romero N. A.; Nicewicz D. A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]; c Bortolato T.; Dyguda M.; Vega-Peñaloza A.; Dell’Amico L. Properties and Synthetic Performances of Phenylamino Cyanoarenes under One-Photon Excitation Manifolds. Synthesis 2022, 54, 3409–3413. 10.1055/a-1776-0929. [DOI] [Google Scholar]; d Cuadros S.; Rosso C.; Barison G.; Costa P.; Bonchio M.; Prato M.; Dell’Amico L.; Filippini G. Unveiling the Synthetic Potential of Substituted Phenols as Fully Recyclable Organophotoredox Catalysts for the Iodosulfonylation of Olefins. ACS Catal. 2022, 12, 4290–4295. 10.1021/acscatal.2c00565. [DOI] [Google Scholar]
- a Speckmeier E.; Fischer T. G.; Zeitler K. A Toolbox Approach to Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor-Acceptor Cyanoarenes. J. Am. Chem. Soc. 2018, 140, 15353–15365. 10.1021/jacs.8b08933. [DOI] [PubMed] [Google Scholar]; b Elliott L. D.; Kayal S.; George M. W.; Booker-Milburn K. Rational Design of Triplet Sensitizers for the Transfer of Excited State Photochemistry from UV to Visible. J. Am. Chem. Soc. 2020, 142, 14947–14956. 10.1021/jacs.0c05069. [DOI] [PubMed] [Google Scholar]; c Vega-Peñaloza A.; Mateos J.; Companyó X.; Escudero-Casao M.; Dell’Amico L. A Rational Approach to Organo-Photocatalysis. Novel Designs and Structure-Property-Relationships. Angew. Chem., Int. Ed. 2021, 133, 1096–1111. 10.1002/ange.202006416. [DOI] [PubMed] [Google Scholar]
- a Mateos J.; Rigodanza F.; Vega-Peñaloza A.; Sartorel A.; Natali M.; Bortolato T.; Pelosi G.; Companyó X.; Bonchio M.; Dell’Amico L. Naphthochromenones: Organic Bimodal Photocatalysts Engaging in Both Oxidative and Reductive Quenching Processes. Angew. Chem., Int. Ed. 2020, 59, 1302–1312. 10.1002/anie.201912455. [DOI] [PubMed] [Google Scholar]; b Wu C.; Corrigan N.; Lim C.-H.; Liu W.; Miyake G.; Boyer C. Rational Design of Photocatalysts for Controlled Polymerization: Effect of Structures on Photocatalytic Activities. Chem. Rev. 2022, 122, 5476–5518. 10.1021/acs.chemrev.1c00409. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Mateos J.; Cuadros S.; Vega-Peñaloza A.; Dell’Amico L. Unlocking the Synthetic Potential of Light-Excited Aryl Ketones – Applications in Direct Photochemistry and Photoredox Catalysis. Synlett 2022, 33, 116–128. 10.1055/a-1403-4613. [DOI] [Google Scholar]; d McCarthy B. G.; Pearson R. M.; Lim C.; Sartor S. M.; Damrauer N. H.; Miyake G. M. Structure – Property Relationships for Tailoring Phenoxazines as Reducing Photoredox Catalysts. J. Am. Chem. Soc. 2018, 140, 5088–5101. 10.1021/jacs.7b12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin D. A.; Miyake G. M. Photoinduced Organocatalyzed Atom Transfer Radical Polymerization (O-ATRP): Precision Polymer Synthesis Using Organic Photoredox Catalysis. Chem. Rev. 2022, 122, 1830–1874. 10.1021/acs.chemrev.1c00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treat N. J.; Sprafke H.; Kramer J. W.; Clark P. G.; Barton B. E.; De Alaniz J. R.; Fors B. P.; Hawker C. J. Metal-Free Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2014, 136, 16096–16101. 10.1021/ja510389m. [DOI] [PubMed] [Google Scholar]
- a Theriot J. C.; Musgrave C. B.; Miyake G. M. Organocatalyzed Atom Transfer Radical Polymerization Driven by Visible Light. Science 2016, 352 (6289), 1082–1086. 10.1126/science.aaf3935. [DOI] [PubMed] [Google Scholar]; b Buss B. L.; Lim C.-H.; Miyake G. Dimethyl Dihydroacridines as Photocatalysts in Organocatalyzed Atom Transfer Radical Polymerization of Acrylate Monomers. Angew. Chem., Int. Ed. 2020, 59, 3209–3217. 10.1002/anie.201910828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected applications, see:; a Park M. S.; Lee J. Y. Indolo Acridine-Based Hole-Transport Materials for Phosphorescent OLEDs with Over 20% External Quantum Efficiency in Deep Blue and Green. Chem. Mater. 2011, 23, 4338–4343. 10.1021/cm201634w. [DOI] [Google Scholar]; b Méhes G.; Nomura H.; Zhang Q.; Nakagawa T.; Adachi C. Enhanced Electroluminescence Efficiency in a Spiro-Acridine Derivative through Thermally Activated Delayed Fluorescence. Angew. Chem., Int. Ed. 2012, 51, 11311–11315. 10.1002/anie.201206289. [DOI] [PubMed] [Google Scholar]; c Kim M.; Lee J. Y. Improved Power Efficiency in Deep Blue Phosphorescent Organic Light-Emitting Diodes Using an Acridine Core Based Hole Transport Material. Org. Electron. 2012, 13, 1245–1249. 10.1016/j.orgel.2012.03.040. [DOI] [Google Scholar]; d Zhang Q.; Li B.; Huang S.; Nomura H.; Tanaka H.; Adachi C. Efficient Blue Organic Light-Emitting Diodes Employing Thermally Activated Delayed Fluorescence. Nat. Photonics 2014, 8, 326–332. 10.1038/nphoton.2014.12. [DOI] [Google Scholar]; e Zhang Y.-X.; Zhang L.; Cui L.-S.; Gao C.-H.; Chen H.; Li Q.; Jiang Z.-Q.; Liao L.-S. Control of Conjugation Degree via Position Engineering to Highly Efficient Phosphorescent Host Materials. Org. Lett. 2014, 16, 3748–3751. 10.1021/ol501603b. [DOI] [PubMed] [Google Scholar]; f Liu X.-Y.; Liang F.; Yuan Y.; Jiang Z.-Q.; Liao L.-S. Utilizing 9,10-Dihydroacridine and Pyrazine-Containing Donor-Acceptor Host Materials for Highly Efficient Red Phosphorescent Organic Light-Emitting Diodes. J. Mater. Chem. C 2016, 4, 7869–7874. 10.1039/C6TC02180H. [DOI] [Google Scholar]; g Zhang L.; Zhang Y.-X.; Hu Y.; Shi X.-B.; Jiang Z.-Q.; Wang Z.-K.; Liao L. S. Highly Efficient Blue Phosphorescent Organic Light-Emitting Diodes Employing a Host Material with Small Bandgap. ACS Appl. Mater. Interfaces 2016, 8, 16186–16191. 10.1021/acsami.6b01304. [DOI] [PubMed] [Google Scholar]; h Zeng W.; Lai H.-Y.; Lee W.-K.; Jiao M.; Shiu Y.-J.; Zhong C.; Gong S.; Zhou T.; Xie G.; Sarma M.; Wong K.-T.; Wu C.-C.; Yang C. Achieving Nearly 30% External Quantum Efficiency for Orange-Red Organic Light Emitting Diodes by Employing Thermally Activated Delayed Fluorescence Emitters Composed of 1,8-Naphthalimide-Acridine Hybrids. Adv. Mater. 2017, 29, 1704961. [DOI] [PubMed] [Google Scholar]; i Yu L.; Wu Z.; Xie G.; Zeng W.; Ma D.; Yang C. Molecular Design to Regulate the Photophysical Properties of Multifunctional TADF Emitters Towards High-Performance TADF-Based OLEDs with EQES up to 22.4% and Small Efficiency Roll-Offs. Chem. Sci. 2018, 9, 1385–1391. 10.1039/C7SC04669C. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Liu X.-Y.; Ma Y.-Y.; Zhang W.; Song B.; Ding L.; Fung M.-K.; Fan J. A Novel Linking Strategy of Using 9,10-Dihydroacridines to Construct Efficient Host Materials for Red Phosphorescent Organic Light-Emitting Diodes. Chem.—Eur. J. 2018, 24, 11755–11762. 10.1002/chem.201802030. [DOI] [PubMed] [Google Scholar]; k Wada Y.; Kubo S.; Kaji H. Adamantyl Substitution Strategy for Realizing Solution-Processable Thermally Stable Deep-Blue Thermally Activated Delayed Fluorescence Materials. Adv. Mater. 2018, 30, 1705641. 10.1002/adma.201705641. [DOI] [PubMed] [Google Scholar]; l Yang Y.; Wang S.; Zhu Y.; Wang Y.; Zhan H.; Cheng Y. Thermally Activated Delayed Fluorescence Conjugated Polymers with Backbone-Donor/Pendant-Acceptor Architecture for Nondoped OLEDs with High External Quantum Efficiency and Low Roll-Off. Adv. Funct. Mater. 2018, 28, 1706916. 10.1002/adfm.201706916. [DOI] [Google Scholar]; m Sartor S. M.; McCarthy B. G.; Pearson R. M.; Miyake G. M.; Damrauer N. H. Exploiting Charge-Transfer States for Maximizing Intersystem Crossing Yields in Organic Photoredox Catalysts. J. Am. Chem. Soc. 2018, 140, 4778–4781. 10.1021/jacs.8b01001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Force G.; Carpentier J.-F.; Sarazin Y.; Bour C.; Gandon V.; Lebœuf D. Modular Synthesis of 9,10-Dihydroacridines through an Ortho-C Alkenylation/Hydroarylation Sequence between Anilines and Aryl Alkynes in Hexafluoroisopropanol. Org. Lett. 2021, 23, 2565–2570. 10.1021/acs.orglett.1c00487. [DOI] [PubMed] [Google Scholar]
- Romero N. A.; Nicewicz D. A. Mechanistic Insight into the Photoredox Catalysis of Anti-Markovnikov Alkene Hydrofunctionalization Reactions. J. Am. Chem. Soc. 2014, 136, 17024–17035. 10.1021/ja506228u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt C. Solvatochromic Dyes as Solvent Polarity Indicators. Chem. Rev. 1994, 94 (8), 2319–23582. 10.1021/cr00032a005. [DOI] [Google Scholar]
- a Andrew T. L.; Swager T. M. Detection of Explosives via Photolytic Cleavage of Nitroesters and Nitramines. J. Org. Chem. 2011, 76, 2976–2993. 10.1021/jo200280c. [DOI] [PubMed] [Google Scholar]; b Liu Y.; Chen Q.; Tong Y.; Ma Y. 9,9-Dimethyl Dihydroacridine-Based Organic Photocatalyst for Atom Transfer Radical Polymerization from Modifying “Unstable” Electron Donor. Macromolecules 2020, 53, 7053–7062. 10.1021/acs.macromol.0c00377. [DOI] [Google Scholar]
- Bard A. J.; Faulkner L. R.. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley & Sons: 2001. [Google Scholar]
- a Agarwal R. G.; Coste S. C.; Groff B. D.; Heuer A. M.; Noh H.; Parada G. A.; Wise C. F.; Nichols E. M.; Warren J. J.; Mayer J. M. Free Energies of Proton-Coupled Electron Transfer Reagents and Their Applications. Chem. Rev. 2022, 122, 1–49. 10.1021/acs.chemrev.1c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gentry E. C.; Knowles R. R. Synthetic Applications of Proton-Coupled Electron Transfer. Acc. Chem. Res. 2016, 49, 1546–1556. 10.1021/acs.accounts.6b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Murray P. R. D.; Cox J. H.; Chiappini N. D.; Roos C. B.; McLoughlin E. A.; Hejna B. G.; Nguyen S. T.; Ripberger H. H.; Ganley J. M.; Tsui E.; et al. Photochemical and Electrochemical Applications of Proton-Coupled Electron Transfer in Organic Synthesis. Chem. Rev. 2022, 122, 2017–2291. 10.1021/acs.chemrev.1c00374. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Warren J. J.; Tronic T. A.; Mayer J. M. Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev. 2010, 110, 6961–7001. 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Gupta N.; Linschitz H. Hydrogen-Bonding and Protonation Effects in Electrochemistry of Quinones in Aprotic Solvents. J. Am. Chem. Soc. 1997, 119, 6384–6391. 10.1021/ja970028j. [DOI] [Google Scholar]
- Discekici E. H.; Treat N. J.; Poelma S. O.; Mattson K. M.; Hudson Z. M.; Luo Y.; Hawker C. J.; De Alaniz J. R. A Highly Reducing Metal-Free Photoredox Catalyst: Design and Application in Radical Dehalogenations. Chem. Commun. 2015, 51, 11705–11708. 10.1039/C5CC04677G. [DOI] [PubMed] [Google Scholar]
- a Constantin T.; Julia F.; Leonori D. Applications of Halogen-Atom Transfer (XAT) for the Generation of Carbon Radicals in Synthetic Photochemistry and Photocatalysis. Chem. Rev. 2022, 122, 2292–2352. 10.1021/acs.chemrev.1c00558. [DOI] [PubMed] [Google Scholar]; b Ruffoni A.; Mykura R. C.; Bietti M.; Leonori D. The Interplay of Polar Effects in Controlling the Selectivity of Radical Reactions. Nat. Synth. 2022, 1, 682–695. 10.1038/s44160-022-00108-2. [DOI] [Google Scholar]
- a Ghosh I.; Ghosh T.; Bardagi J. I.; König B. Reduction of aryl halides by consecutive visible light-induced electron transfer processes.. Science 2014, 346, 725–728. 10.1126/science.1258232. [DOI] [PubMed] [Google Scholar]; b Kim H.; Kim H.; Lambert T. H.; Lin S. Reductive Electrophotocatalysis: Merging Electricity and Light to Achieve Extreme Reduction Potentials. J. Am. Chem. Soc. 2020, 142, 2087–2092. 10.1021/jacs.9b10678. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cowper N. G. W.; Chernowsky C. P.; Williams O. P.; Wickens Z. K. Potent Reductants via Electron-Primed Photoredox Catalysis: Unlocking Aryl Chlorides for Radical Coupling. J. Am. Chem. Soc. 2020, 142, 2093–2099. 10.1021/jacs.9b12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the kinetic analysis of the polymerization process performed on PC 5a see Section G of the SI.
- a Sifri R. J.; Ma Y.; Fors B. P. Photoredox Catalysis in Photocontrolled Cationic Polymerizations of Vinyl Ethers. Acc. Chem. Res. 2022, 55, 1960–1971. 10.1021/acs.accounts.2c00252. [DOI] [PubMed] [Google Scholar]; b Dadashi-Silab S.; Pan X.; Matyjaszewski K. Phenyl Benzo[b]phenothiazines as a Visible Light Photoredox Catalyst for Metal-Free Atom Transfer Radical Polymerization. Chem.—Eur. J. 2017, 23, 5972–5977. 10.1002/chem.201605574. [DOI] [PubMed] [Google Scholar]
- a Buglioni L.; Raymenants F.; Slattery A.; Zondag S. D. A.; Noël T. Technological Innovations in Photochemistry for Organic Synthesis: Flow Chemistry, High-Throughput Experimentation, Scale-up, and Photoelectrochemistry. Chem. Rev. 2022, 122, 2752–2906. 10.1021/acs.chemrev.1c00332. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mateos J.; Cherubini-Celli A.; Carofiglio T.; Bonchio M.; Marino N.; Companyó X.; Dell’Amico L. A Microfluidic Photoreactor Enables 2-Methylbenzophenone Light-Driven Reactions with Superior Performance. Chem. Commun. 2018, 54, 6820–6823. 10.1039/C8CC01373J. [DOI] [PubMed] [Google Scholar]
- a Hartman R. L.; McMullen J. P.; Jensen K. F. Deciding Whether To Go with the Flow: Evaluating the Merits of Flow Reactors for Synthesis. Angew. Chem., Int. Ed. 2011, 50, 7502–7519. 10.1002/anie.201004637. [DOI] [PubMed] [Google Scholar]; b Buss B. L.; Miyake G. M. Photoinduced Controlled Radical Polymerizations Performed in Flow: Methods, Products, and Opportunities. Chem. Mater. 2018, 30, 3931–3942. 10.1021/acs.chemmater.8b01359. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Junkers T.; Wenn B. Continuous Photoflow Synthesis of Precision Polymers. React. Chem. Eng. 2016, 1, 60–64. 10.1039/C5RE00042D. [DOI] [Google Scholar]; d Tucker J. W.; Zhang Y.; Jamison T. F.; Stephenson C. R. J. Visible-Light Photoredox Catalysis in Flow. Angew. Chem., Int. Ed. 2012, 51, 4144–4147. 10.1002/anie.201200961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.