

Abstract
Neuropeptide Y (NPY), a 36 amino acid peptide, is widely expressed in the mammalian brain. Changes in NPY levels in different brain regions and plasma have been described in several neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, Huntington's disease, Amyotrophic Lateral Sclerosis, and Machado-Joseph disease. The changes in NPY levels may reflect the attempt to set up an endogenous neuroprotective mechanism to counteract the degenerative process. Accumulating evidence indicates that NPY can function as an anti-apoptotic, anti-inflammatory, and pro-phagocytic agent, which may be used effectively to halt or to slow down the progression of the disease. In this review, we will focus on the neuroprotective roles of NPY in several neuropathological conditions, with a particular focus on the anti-inflammatory properties of NPY.
Keywords: Neuropeptide Y, neurodegenerative disease, neuroprotection, neuroinflammation, apoptosis, autophagy
1. INTRODUCTION
1.1. Neuroprotective Effects of Neuropeptide Y
Neuropeptide Y (NPY), a 36 amino acid peptide, is widely expressed in the mammalian brain [1]. NPY is involved in many physiological and pathological processes such as feeding behavior, anxiety, seizure, memory, and pain [2]. The NPY acts through 5 different G protein-coupled receptors designated as Y1, Y2, Y3, Y5, and Y6 receptors that are widely expressed during development and adulthood [3]. High levels of NPY mRNAs are found in numerous brain regions, such as the striatum and the dentate gyrus [1]. In the striatum, NPY is expressed by medium-size GABAergic neurons that receive both direct dopaminergic inputs from the substantia nigra and glutamatergic inputs from the cortex.
The levels of the NPY are altered in several neurodegenerative disorders, such as Alzheimer's disease, Parkinson's disease, Huntington's disease, Amyotrophic Lateral Sclerosis, and Machado-Joseph disease. The changes in NPY levels may reflect the development of endogenous neuroprotective mechanisms to counteract the degenerative process. Therefore, NPY could be a potential neuroprotective agent for the treatment of neurodegenerative disease. Indeed, several studies have demonstrated, both in vitro and in vivo, a neuroprotective action of exogenous NPY in several models of excitotoxicity and neuroinflammation, as well as in various neurodegenerative pathologies [4]. In vitro, the neuroprotective effects of NPY have been revealed with respect to several neurotoxicity models. For example, NPY has a neuroprotective effect on organotypic cultures of rat and mouse hippocampus exposed to excitotoxic lesions [5] and on retinal cells against ecstasy-induced toxicity [6]. Similarly, C-terminal fragments of NPY protected human neuron cultures from the neurotoxic effect of the peptide β-amyloid [7]. In vivo, in the nigrostriatal system, methamphetamine-induced toxicity in the striatum is reduced by intracerebroventricular injection of NPY [8]. In humans, in epilepsy, an increase in the number of NPY-expressing interneurons has been observed, and, in the animal model of this condition, an increase in the mRNA of this peptide has been shown in many brain regions. Collectively, these observations highlight the neuroprotective potential of NPY for the treatment of neurodegenerative disease (Table 1).
Table 1.
Alteration of neuropeptide Y (NPY) levels in neurodegenerative diseases.
| Neurodegenerative Disease | Changes of NPY | Regions of Changes | References |
|---|---|---|---|
| Parkinson Disease | *Increase of NPY neurons number in animal models and in PD patients. | *Striatum Caudate nucleus and putamen of PD. |
[21, 22] |
| *Neuroprotective role of NPY in vitro and in vivo in animal PD models. | *Substantia nigra and striatum of rodent PD models. | [23, 24] | |
| Alzheimer Disease | *Decrease of the number of hippocampal NPY cells in animal models and in AD patients *Alteration of number of NPY receptors. |
*Hippocampal and cortical regions of AD transgenic mouse models. | [27-30] |
| *Changes of NPY plasma and cerebrospinal fluid levels in AD patients. | *Plasma and cerebro-spinalfluid of AD patients. |
[31, 32] | |
| *Neuroprotective role of NPY in vitro and in vivo in animal AD models. | *Prefrontal cortex and hippocampus of AD mouse models. | [7, 33] | |
| Huntington Disease | *Increase of NPY neurons in the brain of HD patient. | *Striatum of HD patient. | [34] |
| *Neuroprotective role of NPY in an animal HD model. | *Cortex and striatum of HD mouse model. | [35, 36] | |
| Amyotrophic Lateral Sclerosis | *Increase of NPY levels in animal models and in ALS patients *Neuroprotection of NPY in ALS. |
*Motor cortex of ALS mice and blood of ALS patients. | [39-41] |
| Machado-Joseph Disease | *Decrease of NPY levels in animal model and in MJD patients *Neuroprotection of NPY in MJD. |
*Cerebellum and striatum of MJD transgenic mice model and cerebellar extracts of MJD patients. | [4, 44, 45] |
Abbreviations: NPY, Neuropeptide Y; BDNF, Brain-Derived Neurotrophic Factor; ALS, Amyotrophic Lateral Sclerosis; AD, Alzheimer's Disease; PD, Parkinson’s Disease; HD, Huntington's Disease; ALS, Amyotrophic Lateral Sclerosis; MJD, Machado-Joseph Disease; MS, Multiple Sclerosis; Meth, Methamphetamine.
2. NEUROPROTECTIVE EFFECTS OF NPY IN PARKINSON'S DISEASE
Parkinson’s disease (PD) is the second most common neurodegenerative disease, affecting about 1% of the population over the age of 60 [9]. Clinically, PD is associated with motor impairments, including bradykinesia, akinesia, rigidity, resting tremor, and gait disturbance [10, 11]. PD is characterized by the progressive and relatively selective loss of dopaminergic neurons in the substantia pars compacta (SN) and the presence of misfolded and aggregated α-synuclein inclusion named Lewy bodies [12, 13]. Degeneration of dopaminergic neurons leads to dopamine depletion in the striatum. The evolution of PD is progressive, and motor disturbances only appear when 50 to 70% of the dopaminergic neurons of the SNpc have degenerated [14-17]. Despite the lack of curative treatment, current symptomatic treatments provide good relief for motor disorders. Pharmacological treatment based on L-Dopa, a dopamine precursor molecule, allows dopamine supplementation in the striatum and has beneficial effects on motor activity [18]. However, the effectiveness of this treatment diminishes due to the progression of the neurodegenerative process, and L-Dopa causes very disabling side effects in the long term. The search for therapies to prevent the loss of dopaminergic neurons or their replacement [19, 20] is a crucial issue in regenerative medicine today.
In PD, the loss of dopaminergic neurons induces adaptive changes in the expression of NPY proteins and NPY mRNA. Thus, an increase in the number of NPY neurons in the striatum was observed in an animal model of PD [21]. Patients with PD have a greater number of cells expressing NPY mRNA in the caudate-putamen compared to healthy subjects [22]. The changes in NPY or NPY mRNA expression observed in PD, as well as in other neurodegenerative diseases, may reflect the development of endogenous neuroprotective mechanisms to counteract the degenerative process. Several studies have indicated a neuroprotective role for NPY in vitro and in vivo. NPY has been shown to protect dopaminergic cells in vitro from neurotoxicity of 6-hydroxydopamine (6-OHDA) [23]. In an animal model of PD, striatal injection of NPY prevented the nigrostriatal pathway from degeneration, as shown by anatomical and molecular studies. In addition, HPLC analysis revealed that NPY treatment in 6-OHDA lesioned mice results in higher levels of striatal dopamine and DOPAC compared to lesioned but untreated animals. Interestingly, this neuroprotective effect is accompanied by a significant improvement in motor function. The use of agonists and antagonists specific for Y1 or Y2 receptors and transgenic mice knockout for one of these receptors led the conclusion that the neuroprotective effect of NPY is preferentially mediated via Y2 receptors and may involve activation of the mitogen-activated protein kinase and Akt pathways and modified the expression of neither BDNF nor GDNF, suggesting a direct action of the neuropeptide on dopaminergic neurons [23]. The neuroprotective effect of NPY has also been confirmed in the 6-OHDA rat model of PD. Indeed, Pain et al. have shown that NPY induced a neuroprotective effect on microglia and caused inflammation, as observed by the specific binding of a ligand on the receptor translocator protein localized on microglial elements in the substantia nigra and striatum [24].
3. NEUROPROTECTIVE EFFECTS OF NPY IN ALZHEIMER'S DISEASE
Alzheimer's disease (AD) is an age-related neurodegenerative disorder characterized by cognitive dysfunction, memory impairment, and behavioral issues [25]. Neuropathological hallmarks of AD are extracellular amyloid-beta (Aβ) protein aggregation and intraneuronal protein clusters of hyperphosphorylated Tau protein (neurofibrillary tangles) [26]. Several studies have reported the alteration in NPY levels in different brain regions and in plasma in AD, suggesting the implication of NPY on the pathophysiology of the disease. The number of hippocampal NPY cells was significantly decreased in presymptomatic transgenic mouse model AD, and this decrease coincides with the early impairment of neuronal network activity [27]. Moreover, the expressions of NPY mRNAs were also decreased in the hippocampal and cortical regions of a transgenic mouse model of AD [28]. In the brain of patients with AD, the levels of NPY are significantly reduced in the hippocampal regions [29], and this reduction is associated with an alteration of the number of NPY receptors [30]. Moreover, there is a correlation between NPY levels in plasma and cerebrospinal fluid and stage of diseases severity; indeed, NPY plasma and cerebrospinal fluid levels are reduced with the progression of AD [31, 32]. Interestingly, several in vivo and in vitro studies have demonstrated that NPY has neuroprotective effects in AD. The administration of C-terminal NPY fragments in the brains of amyloid precursor protein transgenic mice ameliorated the neurodegenerative pathology [7]. Moreover, in vitro, the amidated C-terminal NPY fragments protected human neuronal cells from the neurotoxic effects of Aβ [7]. In the amyloid-β mouse model of AD, a single intracerebroventricular administration of NPY prevents depressive-like behavior, spatial memory impairments, and oxidative stress following amyloid-β administration in mice [33]. In this model, pre-treatment with a selective Y2 receptor antagonist abolished the protective effects of NPY on spatial memory [33].
4. NEUROPROTECTIVE EFFECTS OF NPY IN HUNTINGTON'S DISEASE
Huntington's disease (HD) is an inherited neurodegenerative disorder characterized by progressive neuronal dysfunction and cell loss, especially striatal GABAergic neurons, generating motor, cognitive and affective disturbances [15]. Furthermore, striatal neurons that express neuropeptide Y are preferentially spared in HD. The number of striatal GABAergic neurons co-expressing NPY is increased in the brain of HD patients [34], suggesting that NPY may be of therapeutic interest in patients with HD. Interestingly, a single ICV injection of NPY in R6/2mice, a transgenic mouse model of HD, increased survival time through reduced weight loss and a beneficial effect on motor function [35]. In addition, the degree of cerebral and striatal atrophy was reduced following NPY treatment [35]. More recently, Fatoba et al. have shown that intranasal administration of NPY to R6/2 mice model improved motor function, reduced inflammation, and increased BDNF expression through the activation of Y2 receptors [36].
5. NEUROPROTECTIVE EFFECTS OF NPY IN AMYOTROPHIC LATERAL SCLEROSIS
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by the progressive loss of motor neurons in the brain and spinal cord, leading to muscle weakness and paralysis [37, 38]. Extensive evidence indicates that the NPY may play a role in ALS pathology. Numerous studies reported the changes in the levels of NPY in ALS patients and as well as in rodent models of ALS. An increased number of NPY+ cells were found in the motor cortex of ALS transgenic mice carrying the G93A mutation on the superoxide dismutase 1 gene at the end-stage [39]. Increased levels of NPY were found in the blood of ALS patients [40]. Interestingly, increased levels of NPY correlated with shorter disease duration [40], indicating a role of this peptide in neuronal injury and disease progression. It has been suggested that NPY, through the activation of Y receptor pathways, could modulate several pathways implicated in ALS such as motor neuron hyperexcitability, glutamatergic excitotoxicity, oxidative stress, endoplasmic reticulum stress, neuroinflammation, and autophagy [41].
6. NEUROPROTECTIVE EFFECTS OF NPY IN MACHADO-JOSEPH DISEASE
Machado-Joseph disease (MJD), also known as Spinocerebellar Ataxia 3, is the second most common polyglutamine neurodegenerative disorder associated with neurodegeneration of brain regions, including the cerebellum and striatum [42]. MJD is caused by the expansion of CAG repeats within the ATXN3 gene and is characterized by progressive impairment of motor coordination [43]. The levels of NPY were reduced in both post-mortem cerebellar extracts of MJD patients and in striata and cerebella of transgenic mice model of MJD. Interestingly, overexpression of NPY in transgenic mice model of MJD ameliorated the motor coordination, preserved cerebellar structure, and reduced striatal neuronal dysfunction. Additionally, NPY overexpression increased the levels of brain-derived neurotrophic factors and reduced neuroinflammation associated with MJD [4, 44]. Collectively, these data suggest that NPY is a potential therapeutic strategy for MJD. More recently, Duarte-Neves et al. also demonstrated that intranasal administration of NPY improved motor and balance performance of MJD transgenic mice and reduced cerebellar neuropathology [45].
7. MECHANISMS RESPONSIBLE FOR THE NEUROPROTECTIVE EFFECTS OF NPY
Many neurodegenerative diseases, including Parkinson's disease, Alzheimer's disease, and Huntington's disease, occur as a result of neuronal loss arising from numerous cellular and molecular events such as inflammation and apoptosis. NPY exerts its neuroprotective effects by modulating some of the mechanisms involved in neurodegenerative processes and that are common to several neurodegenerative diseases. Accumulating evidence indicates that NPY can function as an anti-apoptotic, anti-inflammatory, and pro-phagocytic agent, which may be used effectively to halt or to slow down the progression of disease (Fig. 1).
Fig. (1).
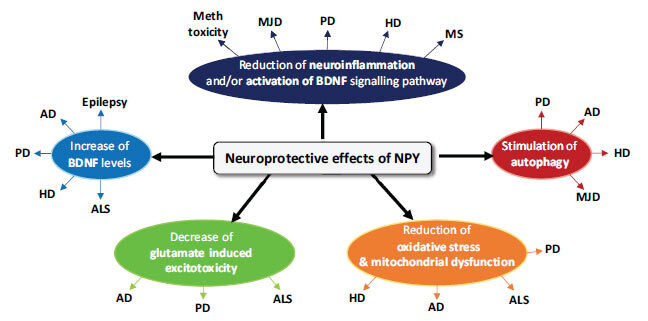
Neuroprotective effects of NPY. The neuroprotective effects of NPY in neurodegenerative diseases include reduction of neuroinflammation, increase of BDNF level, stimulation of autophagy, antiapoptotic effect by reduction of oxidative stress and mitochondrial dysfunction, decrease of glutamate-induced excitotoxicity. NPY, neuropeptide Y; BDNF, Brain-derived Neurotrophic Factor; ALS, Amyotrophic Lateral Sclerosis; AD, Alzheimer's Disease; PD, Parkinson’s Disease; HD, Huntington's Disease; ALS, Amyotrophic Lateral Sclerosis; MJD, Machado-Joseph Disease; MS, Multiple Sclerosis; Meth, Methamphetamine.
7.1. NPY Decreases Glutamate-induced Excitotoxicity
Glutamate is the main excitatory neurotransmitter in the central nervous system (CNS). Excitotoxicity is a process of neuronal death caused by the overactivation of glutamate receptors. Several studies have shown that NPY can exert neuroprotective effects against excitotoxicity induced by glutamate in multiple brain regions such as the hippocampus and striatum [5, 23, 46-48]. Similarly, NPY is implicated in the neuroprotective role against necrotic and apoptotic cell death induced by glutamate in rat retinal cells both in culture and in situ in the retina [49]. In PD, loss of nigral dopaminergic neurons and the subsequent striatal depletion of dopamine leads to the overactivation of glutamatergic projections from the subthalamic nucleus to the basal ganglia output nuclei. The implication of glutamatergic excitotoxicity in nigrostriatal degeneration provides potential targets for therapeutic intervention in PD. Interestingly, the glutamate receptor antagonist improves motor impairment in Parkinson's patients. Some studies suggest that NPY is a neuroprotective agent against glutamate excitotoxicity. Indeed, NPY has been shown in vitro to protect cells of the hippocampus, cortex, and retina against glutamate excitotoxicity by activating Y2 and Y5 receptors. These neuroprotective effects of NPY against glutamate excitotoxicity have also been demonstrated in rodent models, reducing cell death in both the hippocampus and retina. In ALS, an increase in the levels of glutamate in the cerebral spinal fluid of patients has been shown [50, 51]. As NPY decreases glutamate-induced excitotoxicity in other pathological conditions, one can expect a neuroprotective action of NPY in ALS.
7.2. NPY Reduces Neuroinflammation
Evidence from PD patients indicates that neuroinflammation, associated with microglia activation, may contribute to the development of the disease [52]. Several studies have demonstrated a relationship between dopaminergic neuron death and microglial activation in PD patients, both postmortem and in vivo, using molecular imaging methods. A cascade of events is involved in neuroinflammation processes in PD, including activation of microglia and increased secretion of cytokines [53]. Several clinical studies have shown an increased level of inflammatory enzymes, such as cyclo-oxygenase-2, in DA neurons of the postmortem PD brain and in a mouse model of PD [54]. Activated microglial cells intensively express a mitochondrial protein, the translocator protein, which is a sensitive biomarker associated with neuroinflammation. Recent work suggests a direct effect of NPY on neuroinflammatory processes [24]. In an animal model of PD, obtained by injection of 6-OHDA, treatment with NPY allows protection of dopaminergic neurons by decreasing the microglial reaction. Indeed, injured animals treated with NPY show a decrease in the binding of [3H]-PK11195, a TSPO ligand, in the striatum and black matter, compared to injured and untreated animals.
In HD, intranasal NPY reduced induction of proinflammatory cytokine and inflammatory mediators in R6/2 mice [36].
In the Machado-Joseph Disease, overexpressing NPY in the striatum and in the cerebellum of two different MJD mouse models reduced the mRNA levels of IL-6 and prevented the mutant ataxin-3-induced increase of microglial immunoreactivity [44]. Moreover, overexpression of the NPY induced up-regulation of brain-derived neurotrophic factor (BDNF), and it has been shown that BDNF participates in the regulation of neuroinflammation by reducing astrocytosis and proinflammatory cytokines levels [55, 56]. Therefore, NPY could directly restrain the exacerbation of the inflammatory response via the activation of the BDNF signalling pathway. NPY was also observed to protect hippocampal cells against METH-induced toxicity through the release of BDNF from microglia, which has been shown to attenuate neuroinflammation by reducing astrocytosis and pro-inflammatory cytokine production [57].
Anti-inflammatory effects of NPY were also investigated in neuroimmune diseases. Multiple sclerosis (MS) is a chronic autoimmune disease of the CNS characterized by multifocal inflammation in the brain, extensive demyelination, axonal loss, and gliosis. It was found that the levels of NPY were decreased in the cerebrospinal fluid of MS patients [58]. The suppressive role of exogenous NPY has been demonstrated in experimental autoimmune encephalomyelitis, an animal model of MS [59].
The mechanisms underlying NPY-mediated anti-inflammatory effects in neurodegenerative disorders may involve suppression of excessive inflammatory response and/or activation of the BDNF signalling pathway.
7.3. Interplay between NPY and BDNF
Brain-derived neurotrophic factor (BDNF) is a member of the neurotrophin family of growth factors that plays a crucial role in neuronal survival, growth, differentiation, and neurogenesis [60]. The concentration of BDNF is reduced in the blood and brain of patients in several pathological conditions and neurodegenerative diseases, including epilepsy, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis [61]. During the development of epilepsy, BDNF is up-regulated in different brain areas, including the hippocampus. Several studies have shown that, in animal models of epilepsy, infusion of BDNF into the hippocampus can attenuate the development of epilepsy, and this effect is mediated by an increase in the expression of neuropeptide Y [62, 63]. In patients with epilepsy, the level of NPY is increased in the cortex, and this increase is correlated with the BDNF level [64]. Exposure of mouse organotypic hippocampal cultures to a glutamate receptor agonist evoked a significant increase in BDNF. Toxicity mediated by AMPA receptor activation could be prevented by neuroprotective pathways activated by NPY receptors, which can be impacted by BDNF released by microglia and neurons [57].
Regarding Alzheimer's disease, it has been reported that the neuropeptide Y protects cultured rat cortical neurons exposed to amyloid-beta. The neuroprotective effects of NPY are mediated by alteration of the expression of microRNA, small non-coding DNA fragments involved in BDNF tuning expression. Treatment with NPY decreased microRNA expression and increased BDNF mRNA and protein expression [65].
The interplay between NPY and BDNF was also investigated in Huntington's disease. In R6/2 mice model Huntington's disease, NPY induced a decrease in aggregated mutant huntingtin and mediated increase in dopamine and cAMP-regulated phosphoprotein, 32kDa, BDNF, and activated extracellular signal-regulated protein kinases levels [36].
7.4. NPY Stimulates Autophagy
Autophagy is a cellular process responsible for removing protein aggregates and damaged organelles. Many neurodegenerative diseases are associated with an accumulation of undegraded and aggregated proteins as well as autophagic dysfunction. Indeed, protein misfolding and aggregation cause neurofibrillary tangles and amyloid β plaques in Alzheimer's disease [66]. These α-synuclein aggregates constitute Lewy bodies in Parkinson's disease [67] and expanded polyglutamine protein aggregates in polyglutamine diseases, such as Huntington's disease and Machado-Joseph Disease [68-70]. Increasing evidence suggests that autophagy defects are involved in PD pathogenesis. Aberrant autophagy activity is identified in the substantia nigra of patients with PD [71]. Recent studies have shown that modulation of autophagy could be an interesting therapeutic strategy for these diseases. NPY has been shown to exert a neuroprotective effect on the striatum and cerebellum of two mouse models of spinocerebellar ataxia type 3. It has been suggested that this action could be related to NPY stimulation of autophagy.
Aveleira and collaborators showed that NPY not only induces autophagy, increasing the number of autophagosomes, but also increases autophagic flux in hypothalamic neurons [72, 73]. In both hypothalamic neuronal in vitro models, rat hypothalamic neural cell primary cultures, and a mouse hypothalamic cell line, NPY1R (neuropeptide Y receptor Y1), NPY2R, or NPY5R antagonists were able to block autophagy induction. Therefore, their results show that NPY induces autophagic flux through NPY1R and NPY5R activation, and this involves a concerted action of the PI3K, MAPK1/3, and PKA pathways [73].
Because hypothalamic autophagy and NPY levels decrease with aging and NPY regulates autophagy in the hypothalamus, Aveleira et al. [72] suggest that modulation of NPY levels may be manipulated to produce protective effects against hypothalamic impairments associated with age.
7.5. NPY Reduces Oxidative Stress and Mitochondrial Dysfunction
Activation of the apoptosis signalling pathway is associated with oxidative stress and reactive oxidative species (ROS) production [74]. In this line, Lee et al. have shown that NPY was able to decrease key pro-apoptotic caspases (caspases 3 and 4) activation [75]. Indeed, NPY could attenuate oxidative stress through inhibition of ROS via Y1 and Y2 receptor modulation [76].
In the case of neurodegenerative disease, NPY protects mitochondria against oxidative damage by inhibiting nitric oxide production [33, 77].
In ALS, oxidative damage is dependent on NO-induced progressive motor neuron death by inhibition of mitochondrial respiration, increased glutamate release, and excitotoxicity [78].
The production of mitochondrial superoxide contributed to cellular damage. However, it has been reported that the exposure of neurons to a low and temporary level of superoxide has a neuroprotective effect. This neuroprotective effect induced by a subtoxic increase in oxidative stress has been termed preconditioning hormesis [79-81]. In this line, the hormesis mechanism may account for the observed protective effects of NPY, which could be hormesis. Several studies demonstrated that hormesis dietary phytochemicals might improve health and extend lifespan through mild elevation of ROS [82]. It has been reported that low concentrations of natural polyphenols generate moderate stress that prolongs lifespan in animal models of PD [83, 84]. Taken together, these observations suggest that hormesis effects could be exploited to prevent the emergence of various neurodegenerative disorders and to slow down the aging process [85].
7.6. Concluding Remarks and Perspectives for NPY in Clinical Therapeutics
Neuropeptide Y is widely expressed in the brain, and its level of expression is increased in several neuropathological conditions. Accumulating evidence indicates that NPY can function as an anti-apoptotic, anti-inflammatory, and pro-phagocytic agent, which may be used effectively to halt or to slow down the progression of the disease.
However, given the multiple physiological functions that are regulated by NPY, its potential use as a therapeutic agent in the clinic is not without critical aspects. To restrain the action of NPY, the development of specific agonists or antagonists targeting NPY receptors is attractive in clinical settings. Over the past years, several agonists and antagonists against Y receptors have been developed and tested as research tools and in clinical trials [86]. However, to date, these drug leads have not yet been translated into clinical tools.
Another important issue concerning the use of NPY as a therapeutic agent is its mode of administration. Indeed, NPY has a poor capacity to cross the blood-brain barrier, a low oral bioavailability, and a short half-life. Previous studies have demonstrated the feasibility of intranasal delivery of NPY in rodents [45] and in humans [87]. It has been shown that following intranasal administration, NPY reaches several brain regions along olfactory and trigeminal pathways that innervate the nasal cavity [87].
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
ACKNOWLEDGEMENTS
Declared none.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Adrian T.E., Allen J.M., Bloom S.R., Ghatei M.A., Rossor M.N., Roberts G.W., Crow T.J., Tatemoto K., Polak J.M. Neuropeptide Y distribution in human brain. Nature. 1983;306(5943):584–586. doi: 10.1038/306584a0. [DOI] [PubMed] [Google Scholar]
- 2.Wettstein J.G., Earley B., Junien J.L. Central nervous system pharmacology of neuropeptide Y. Pharmacol. Ther. 1995;65(3):397–414. doi: 10.1016/0163-7258(95)98598-K. [DOI] [PubMed] [Google Scholar]
- 3.Silva A.P., Cavadas C., Grouzmann E. Neuropeptide Y and its receptors as potential therapeutic drug targets. Clin. Chim. Acta. 2002;326(1-2):3–25. doi: 10.1016/S0009-8981(02)00301-7. [DOI] [PubMed] [Google Scholar]
- 4.Duarte-Neves J., Pereira de Almeida L., Cavadas C., Neuropeptide Y., Neuropeptide Y. NPY) as a therapeutic target for neurodegenerative diseases. Neurobiol. Dis. 2016;95:210–224. doi: 10.1016/j.nbd.2016.07.022. [DOI] [PubMed] [Google Scholar]
- 5.Silva A.P., Pinheiro P.S., Carvalho A.P., Carvalho C.M., Jakobsen B., Zimmer J., Malva J.O. Activation of neuropeptide Y receptors is neuroprotective against excitotoxicity in organotypic hippocampal slice cultures. FASEB J. 2003;17(9):1118–1120. doi: 10.1096/fj.02-0885fje. [DOI] [PubMed] [Google Scholar]
- 6.Alvaro A.R., Martins J., Costa A.C., Fernandes E., Carvalho F., Ambrósio A.F., Cavadas C. Neuropeptide Y protects retinal neural cells against cell death induced by ecstasy. Neuroscience. 2008;152(1):97–105. doi: 10.1016/j.neuroscience.2007.12.027. [DOI] [PubMed] [Google Scholar]
- 7.Rose J.B., Crews L., Rockenstein E., Adame A., Mante M., Hersh L.B., Gage F.H., Spencer B., Potkar R., Marr R.A., Masliah E. Neuropeptide Y fragments derived from neprilysin processing are neuroprotective in a transgenic model of Alzheimer’s disease. J. Neurosci. 2009;29(4):1115–1125. doi: 10.1523/JNEUROSCI.4220-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thiriet N., Deng X., Solinas M., Ladenheim B., Curtis W., Goldberg S.R., Palmiter R.D., Cadet J.L. Neuropeptide Y protects against methamphetamine-induced neuronal apoptosis in the mouse striatum. J. Neurosci. 2005;25(22):5273–5279. doi: 10.1523/JNEUROSCI.4893-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pringsheim T., Jette N., Frolkis A., Steeves T.D.L. The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov. Disord. 2014;29(13):1583–1590. doi: 10.1002/mds.25945. [DOI] [PubMed] [Google Scholar]
- 10.Fahn S. Description of Parkinson’s disease as a clinical syndrome. Ann. N. Y. Acad. Sci. 2003;991:1–14. doi: 10.1111/j.1749-6632.2003.tb07458.x. [DOI] [PubMed] [Google Scholar]
- 11.Jankovic J. Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry. 2008;79(4):368–376. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- 12.Spillantini M.G., Schmidt M.L., Lee V.M., Trojanowski J.Q., Jakes R., Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 13.Poewe W., Seppi K., Tanner C.M., Halliday G.M., Brundin P., Volkmann J., Schrag A-E., Lang A.E. Parkinson disease. Nat. Rev. Dis. Primers. 2017;3:17013. doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 14.Marsden C.D. Parkinson’s disease. Lancet. 1990;335(8695):948–952. doi: 10.1016/0140-6736(90)91006-V. [DOI] [PubMed] [Google Scholar]
- 15.Ross G.W., Petrovitch H., Abbott R.D., Nelson J., Markesbery W., Davis D., Hardman J., Launer L., Masaki K., Tanner C.M., White L.R. Parkinsonian signs and substantia nigra neuron density in decendents elders without PD. Ann. Neurol. 2004;56(4):532–539. doi: 10.1002/ana.20226. [DOI] [PubMed] [Google Scholar]
- 16.Lang A.E., Lozano A.M. Parkinson’s disease. First of two parts. N. Engl. J. Med. 1998;339(15):1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- 17.Dauer W., Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39(6):889–909. doi: 10.1016/S0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 18.Bergman H., Wichmann T., DeLong M.R. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249(4975):1436–1438. doi: 10.1126/science.2402638. [DOI] [PubMed] [Google Scholar]
- 19.Gaillard A., Decressac M., Frappé I., Fernagut P.O., Prestoz L., Besnard S., Jaber M. Anatomical and functional reconstruction of the nigrostriatal pathway by intranigral transplants. Neurobiol. Dis. 2009;35(3):477–488. doi: 10.1016/j.nbd.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 20.Gaillard A., Jaber M. Rewiring the brain with cell transplantation in Parkinson’s disease. Trends Neurosci. 2011;34(3):124–133. doi: 10.1016/j.tins.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Ma Y., Zhan M., OuYang L., Li Y., Chen S., Wu J., Chen J., Luo C., Lei W. The effects of unilateral 6-OHDA lesion in medial forebrain bundle on the motor, cognitive dysfunctions and vulnerability of different striatal interneuron types in rats. Behav. Brain Res. 2014;266:37–45. doi: 10.1016/j.bbr.2014.02.039. [DOI] [PubMed] [Google Scholar]
- 22.Cannizzaro C., Tel B.C., Rose S., Zeng B-Y., Jenner P. Increased neuropeptide Y mRNA expression in striatum in Parkinson’s disease. Brain Res. Mol. Brain Res. 2003;110(2):169–176. doi: 10.1016/S0169-328X(02)00555-7. [DOI] [PubMed] [Google Scholar]
- 23.Decressac M., Pain S., Chabeauti P-Y., Frangeul L., Thiriet N., Herzog H., Vergote J., Chalon S., Jaber M., Gaillard A. Neuroprotection by neuropeptide Y in cell and animal models of Parkinson’s disease. Neurobiol. Aging. 2012;33(9):2125–2137. doi: 10.1016/j.neurobiolaging.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 24.Pain S., Vergote J., Gulhan Z., Bodard S., Chalon S., Gaillard A. Inflammatory process in Parkinson disease: neuroprotection by neuropeptide Y. Fundam. Clin. Pharmacol. 2019;33(5):544–548. doi: 10.1111/fcp.12464. [DOI] [PubMed] [Google Scholar]
- 25.Burns A., Iliffe S. Alzheimer’s disease. BMJ. 2009;338:b158. doi: 10.1136/bmj.b158. [DOI] [PubMed] [Google Scholar]
- 26.Tiraboschi P., Hansen L.A., Thal L.J., Corey-Bloom J. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 2004;62(11):1984–1989. doi: 10.1212/01.WNL.0000129697.01779.0A. [DOI] [PubMed] [Google Scholar]
- 27.Mahar I., Albuquerque M.S., Mondragon-Rodriguez S., Cavanagh C., Davoli M.A., Chabot J-G., Williams S., Mechawar N., Quirion R., Krantic S. Phenotypic alterations in hippocampal NPY- and PV-expressing interneurons in a presymptomatic transgenic mouse model of Alzheimer’s disease. Front. Aging Neurosci. 2017;8:327. doi: 10.3389/fnagi.2016.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramos B., Baglietto-Vargas D., del Rio J.C., Moreno-Gonzalez I., Santa-Maria C., Jimenez S., Caballero C., Lopez-Tellez J.F., Khan Z.U., Ruano D., Gutierrez A., Vitorica J. Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1xAPP transgenic model of Alzheimer’s disease. Neurobiol. Aging. 2006;27(11):1658–1672. doi: 10.1016/j.neurobiolaging.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 29.Kowall N.W., Beal M.F. Cortical somatostatin, neuropeptide Y, and NADPH diaphorase neurons: normal anatomy and alterations in Alzheimer’s disease. Ann. Neurol. 1988;23(2):105–114. doi: 10.1002/ana.410230202. [DOI] [PubMed] [Google Scholar]
- 30.Martel J.C., Alagar R., Robitaille Y., Quirion R. Neuropeptide Y receptor binding sites in human brain. Possible alteration in Alzheimer’s disease. Brain Res. 1990;519(1-2):228–235. doi: 10.1016/0006-8993(90)90082-M. [DOI] [PubMed] [Google Scholar]
- 31.Alom J., Galard R., Catalan R., Castellanos J.M., Schwartz S., Tolosa E. Cerebrospinal fluid neuropeptide Y in Alzheimer’s disease. Eur. Neurol. 1990;30(4):207–210. doi: 10.1159/000117347. [DOI] [PubMed] [Google Scholar]
- 32.Koide S., Onishi H., Hashimoto H., Kai T., Yamagami S. Plasma neuropeptide Y is reduced in patients with Alzheimer’s disease. Neurosci. Lett. 1995;198(2):149–151. doi: 10.1016/0304-3940(95)11973-Z. [DOI] [PubMed] [Google Scholar]
- 33.dos Santos V.V., Santos D.B., Lach G., Rodrigues A.L.S., Farina M., De Lima T.C.M., Prediger R.D., Neuropeptide Y., Neuropeptide Y. NPY) prevents depressive-like behavior, spatial memory deficits and oxidative stress following amyloid-β (Aβ(1-40)) administration in mice. Behav. Brain Res. 2013;244:107–115. doi: 10.1016/j.bbr.2013.01.039. [DOI] [PubMed] [Google Scholar]
- 34.Dawbarn D., De Quidt M.E., Emson P.C. Survival of basal ganglia neuropeptide Y-somatostatin neurones in Huntington’s disease. Brain Res. 1985;340(2):251–260. doi: 10.1016/0006-8993(85)90921-7. [DOI] [PubMed] [Google Scholar]
- 35.Decressac M., Wright B., Tyers P., Gaillard A., Barker R.A. Neuropeptide Y modifies the disease course in the R6/2 transgenic model of Huntington’s disease. Exp. Neurol. 2010;226(1):24–32. doi: 10.1016/j.expneurol.2010.07.022. [DOI] [PubMed] [Google Scholar]
- 36.Fatoba O., Kloster E., Reick C., Saft C., Gold R., Epplen J.T., Arning L., Ellrichmann G. Activation of NPY-Y2 receptors ameliorates disease pathology in the R6/2 mouse and PC12 cell models of Huntington’s disease. Exp. Neurol. 2018;302:112–128. doi: 10.1016/j.expneurol.2018.01.001. [DOI] [PubMed] [Google Scholar]
- 37.Chiò A., Logroscino G., Traynor B.J., Collins J., Simeone J.C., Goldstein L.A., White L.A. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41(2):118–130. doi: 10.1159/000351153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor J.P., Brown R.H., Jr, Cleveland D.W. Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clark R.M., Blizzard C.A., Young K.M., King A.E., Dickson T.C. Calretinin and Neuropeptide Y interneurons are differentially altered in the motor cortex of the SOD1G93A mouse model of ALS. Sci. Rep. 2017;7:44461. doi: 10.1038/srep44461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahmed R.M., Phan K., Highton-Williamson E., Strikwerda-Brown C., Caga J., Ramsey E., Zoing M., Devenney E., Kim W.S., Hodges J.R., Piguet O., Halliday G.M., Kiernan M.C. Eating peptides: biomarkers of neurodegeneration in amyotrophic lateral sclerosis and frontotemporal dementia. Ann. Clin. Transl. Neurol. 2019;6(3):486–495. doi: 10.1002/acn3.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clark C.M., Clark R.M., Hoyle J.A., Dickson T.C. Pathogenic or protective? Neuropeptide Y in amyotrophic lateral sclerosis. J. Neurochem. 2021;156(3):273–289. doi: 10.1111/jnc.15125. [DOI] [PubMed] [Google Scholar]
- 42.Paulson H.L. Dominantly inherited ataxias: lessons learned from Machado-Joseph disease/spinocerebellar ataxia type 3. Semin. Neurol. 2007;27(2):133–142. doi: 10.1055/s-2007-971172. [DOI] [PubMed] [Google Scholar]
- 43.Kawaguchi Y., Okamoto T., Taniwaki M., Aizawa M., Inoue M., Katayama S., Kawakami H., Nakamura S., Nishimura M., Akiguchi I. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 1994;8(3):221–228. doi: 10.1038/ng1194-221. [DOI] [PubMed] [Google Scholar]
- 44.Duarte-Neves J., Gonçalves N., Cunha-Santos J., Simões A.T., den Dunnen W.F.A., Hirai H., Kügler S., Cavadas C., Pereira de Almeida L. Neuropeptide Y mitigates neuropathology and motor deficits in mouse models of Machado-Joseph disease. Hum. Mol. Genet. 2015;24(19):5451–5463. doi: 10.1093/hmg/ddv271. [DOI] [PubMed] [Google Scholar]
- 45.Duarte-Neves J., Cavadas C., Pereira de Almeida L., Neuropeptide Y., Neuropeptide Y. NPY) intranasal delivery alleviates Machado-Joseph disease. Sci. Rep. 2021;11(1):3345. doi: 10.1038/s41598-021-82339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silva A.P., Xapelli S., Grouzmann E., Cavadas C. The putative neuroprotective role of neuropeptide Y in the central nervous system. Curr. Drug Targets CNS Neurol. Disord. 2005;4(4):331–347. doi: 10.2174/1568007054546153. [DOI] [PubMed] [Google Scholar]
- 47.Smiałowska M., Domin H., Zieba B., Koźniewska E., Michalik R., Piotrowski P., Kajta M. Neuroprotective effects of neuropeptide Y-Y2 and Y5 receptor agonists in vitro and in vivo. Neuropeptides. 2009;43(3):235–249. doi: 10.1016/j.npep.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 48.Xapelli S., Silva A.P., Ferreira R., Malva J.O. Neuropeptide Y can rescue neurons from cell death following the application of an excitotoxic insult with kainate in rat organotypic hippocampal slice cultures. Peptides. 2007;28(2):288–294. doi: 10.1016/j.peptides.2006.09.031. [DOI] [PubMed] [Google Scholar]
- 49.Santos-Carvalho A., Elvas F., Alvaro A.R., Ambrósio A.F., Cavadas C. Neuropeptide Y receptors activation protects rat retinal neural cells against necrotic and apoptotic cell death induced by glutamate. Cell Death Dis. 2013;4:e636. doi: 10.1038/cddis.2013.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rothstein J.D., Tsai G., Kuncl R.W., Clawson L., Cornblath D.R., Drachman D.B., Pestronk A., Stauch B.L., Coyle J.T. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann. Neurol. 1990;28(1):18–25. doi: 10.1002/ana.410280106. [DOI] [PubMed] [Google Scholar]
- 51.Shaw P.J., Forrest V., Ince P.G., Richardson J.P., Wastell H.J. CSF and plasma amino acid levels in motor neuron disease: elevation of CSF glutamate in a subset of patients. Neurodegeneration. 1995;4(2):209–216. doi: 10.1006/neur.1995.0026. [DOI] [PubMed] [Google Scholar]
- 52.Hirsch E.C., Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8(4):382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- 53.Amor S., Puentes F., Baker D., van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129(2):154–169. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bartels A.L., Leenders K.L. Cyclooxygenase and neuroinflammation in Parkinson’s disease neurodegeneration. Curr. Neuropharmacol. 2010;8(1):62–68. doi: 10.2174/157015910790909485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bovolenta R., Zucchini S., Paradiso B., Rodi D., Merigo F., Navarro Mora G., Osculati F., Berto E., Marconi P., Marzola A., Fabene P.F., Simonato M. Hippocampal FGF-2 and BDNF overexpression attenuates epileptogenesis-associated neuroinflammation and reduces spontaneous recurrent seizures. J. Neuroinflammation. 2010;7:81. doi: 10.1186/1742-2094-7-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu S-Y., Wang T-F., Yu L., Jen C.J., Chuang J-I., Wu F-S., Wu C-W., Kuo Y-M. Running exercise protects the substantia nigra dopaminergic neurons against inflammation-induced degeneration via the activation of BDNF signaling pathway. Brain Behav. Immun. 2011;25(1):135–146. doi: 10.1016/j.bbi.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 57.Xapelli S., Bernardino L., Ferreira R., Grade S., Silva A.P., Salgado J.R., Cavadas C., Grouzmann E., Poulsen F.R., Jakobsen B., Oliveira C.R., Zimmer J., Malva J.O. Interaction between neuropeptide Y (NPY) and brain-derived neurotrophic factor in NPY-mediated neuroprotection against excitotoxicity: a role for microglia. Eur. J. Neurosci. 2008;27(8):2089–2102. doi: 10.1111/j.1460-9568.2008.06172.x. [DOI] [PubMed] [Google Scholar]
- 58.Maeda K., Yasuda M., Kaneda H., Maeda S., Yamadori A. Cerebrospinal fluid (CSF) neuropeptide Y- and somatostatin-like immunoreactivities in man. Neuropeptides. 1994;27(6):323–332. doi: 10.1016/0143-4179(94)90058-2. [DOI] [PubMed] [Google Scholar]
- 59.Bedoui S., Miyake S., Lin Y., Miyamoto K., Oki S., Kawamura N., Beck-Sickinger A., von Hörsten S., Yamamura T., Neuropeptide Y., Neuropeptide Y. NPY) suppresses experimental autoimmune encephalomyelitis: NPY1 receptor-specific inhibition of autoreactive Th1 responses in vivo. J. Immunol. 2003;171(7):3451–3458. doi: 10.4049/jimmunol.171.7.3451. [DOI] [PubMed] [Google Scholar]
- 60.Kowiański P., Lietzau G., Czuba E., Waśkow M., Steliga A., Moryś J. BDNF: A key factor with multipotent impact on brain signaling and synaptic plasticity. Cell. Mol. Neurobiol. 2018;38(3):579–593. doi: 10.1007/s10571-017-0510-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eyileten C., Sharif L., Wicik Z., Jakubik D., Jarosz-Popek J., Soplinska A., Postula M., Czlonkowska A., Kaplon-Cieslicka A., Mirowska-Guzel D. The relation of the brain-derived neurotrophic factor with microRNAs in neurodegenerative diseases and ischemic stroke. Mol. Neurobiol. 2021;58(1):329–347. doi: 10.1007/s12035-020-02101-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reibel S., Larmet Y., Lê B.T., Carnahan J., Marescaux C., Depaulis A. Brain-derived neurotrophic factor delays hippocampal kindling in the rat. Neuroscience. 2000;100(4):777–788. doi: 10.1016/S0306-4522(00)00351-1. [DOI] [PubMed] [Google Scholar]
- 63.Koyama R., Ikegaya Y. To BDNF or not to BDNF: that is the epileptic hippocampus. Neuroscientist. 2005;11(4):282–287. doi: 10.1177/1073858405278266. [DOI] [PubMed] [Google Scholar]
- 64.Takahashi M., Hayashi S., Kakita A., Wakabayashi K., Fukuda M., Kameyama S., Tanaka R., Takahashi H., Nawa H. Patients with temporal lobe epilepsy show an increase in brain-derived neurotrophic factor protein and its correlation with neuropeptide Y. Brain Res. 1999;818(2):579–582. doi: 10.1016/S0006-8993(98)01355-9. [DOI] [PubMed] [Google Scholar]
- 65.Croce N., Gelfo F., Ciotti M.T., Federici G., Caltagirone C., Bernardini S., Angelucci F. NPY modulates miR-30a-5p and BDNF in opposite direction in an in vitro model of Alzheimer disease: a possible role in neuroprotection? Mol. Cell. Biochem. 2013;376(1-2):189–195. doi: 10.1007/s11010-013-1567-0. [DOI] [PubMed] [Google Scholar]
- 66.McKhann G., Drachman D., Folstein M., Katzman R., Price D., Stadlan E.M. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–944. doi: 10.1212/WNL.34.7.939. [DOI] [PubMed] [Google Scholar]
- 67.Jellinger K.A. Pathology of Parkinson’s disease. Changes other than the nigrostriatal pathway. Mol. Chem. Neuropathol. 1991;14(3):153–197. doi: 10.1007/BF03159935. [DOI] [PubMed] [Google Scholar]
- 68.DiFiglia M., Sapp E., Chase K.O., Davies S.W., Bates G.P., Vonsattel J.P., Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277(5334):1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 69.Paulson H.L., Perez M.K., Trottier Y., Trojanowski J.Q., Subramony S.H., Das S.S., Vig P., Mandel J.L., Fischbeck K.H., Pittman R.N. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19(2):333–344. doi: 10.1016/S0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 70.Schmidt T., Landwehrmeyer G.B., Schmitt I., Trottier Y., Auburger G., Laccone F., Klockgether T., Völpel M., Epplen J.T., Schöls L., Riess O. An isoform of ataxin-3 accumulates in the nucleus of neuronal cells in affected brain regions of SCA3 patients. Brain Pathol. 1998;8(4):669–679. doi: 10.1111/j.1750-3639.1998.tb00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nixon R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013;19(8):983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 72.Aveleira C.A., Botelho M., Cavadas C. NPY/neuropeptide Y enhances autophagy in the hypothalamus: a mechanism to delay aging? Autophagy. 2015;11(8):1431–1433. doi: 10.1080/15548627.2015.1062202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aveleira C.A., Botelho M., Carmo-Silva S., Pascoal J.F., Ferreira-Marques M., Nóbrega C., Cortes L., Valero J., Sousa-Ferreira L., Álvaro A.R., Santana M., Kügler S., Pereira de Almeida L., Cavadas C. Neuropeptide Y stimulates autophagy in hypothalamic neurons. Proc. Natl. Acad. Sci. USA. 2015;112(13):E1642–E1651. doi: 10.1073/pnas.1416609112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mattson M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000;1(2):120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 75.Lee D.Y., Hong S.H., Kim B., Lee D-S., Yu K., Lee K-S. Neuropeptide Y mitigates ER stress-induced neuronal cell death by activating the PI3K-XBP1 pathway. Eur. J. Cell Biol. 2018;97(5):339–348. doi: 10.1016/j.ejcb.2018.04.003. [DOI] [PubMed] [Google Scholar]
- 76.Yarosh H.L., Angulo J.A. Modulation of methamphetamine-induced nitric oxide production by neuropeptide Y in the murine striatum. Brain Res. 2012;1483:31–38. doi: 10.1016/j.brainres.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kir H.M., Sahin D., Oztaş B., Musul M., Kuskay S. Effects of single-dose neuropeptide Y on levels of hippocampal BDNF, MDA, GSH, and NO in a rat model of pentylenetetrazole-induced epileptic seizure. Bosn. J. Basic Med. Sci. 2013;13(4):242–247. doi: 10.17305/bjbms.2013.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Drechsel D.A., Estévez A.G., Barbeito L., Beckman J.S. Nitric oxide-mediated oxidative damage and the progressive demise of motor neurons in ALS. Neurotox. Res. 2012;22(4):251–264. doi: 10.1007/s12640-012-9322-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Calabrese E.J., Baldwin L.A. Defining hormesis. Hum. Exp. Toxicol. 2002;21(2):91–97. doi: 10.1191/0960327102ht217oa. [DOI] [PubMed] [Google Scholar]
- 80.Calabrese E.J., Bachmann K.A., Bailer A.J., Bolger P.M., Borak J., Cai L., Cedergreen N., Cherian M.G., Chiueh C.C., Clarkson T.W., Cook R.R., Diamond D.M., Doolittle D.J., Dorato M.A., Duke S.O., Feinendegen L., Gardner D.E., Hart R.W., Hastings K.L., Hayes A.W., Hoffmann G.R., Ives J.A., Jaworowski Z., Johnson T.E., Jonas W.B., Kaminski N.E., Keller J.G., Klaunig J.E., Knudsen T.B., Kozumbo W.J., Lettieri T., Liu S.Z., Maisseu A., Maynard K.I., Masoro E.J., McClellan R.O., Mehendale H.M., Mothersill C., Newlin D.B., Nigg H.N., Oehme F.W., Phalen R.F., Philbert M.A., Rattan S.I., Riviere J.E., Rodricks J., Sapolsky R.M., Scott B.R., Seymour C., Sinclair D.A., Smith-Sonneborn J., Snow E.T., Spear L., Stevenson D.E., Thomas Y., Tubiana M., Williams G.M., Mattson M.P. Biological stress response terminology: Integrating the concepts of adaptive response and preconditioning stress within a hormetic dose-response framework. Toxicol. Appl. Pharmacol. 2007;222(1):122–128. doi: 10.1016/j.taap.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 81.Calabrese V., Cornelius C., Dinkova-Kostova A.T., Calabrese E.J., Mattson M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal. 2010;13(11):1763–1811. doi: 10.1089/ars.2009.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Govindan S., Amirthalingam M., Duraisamy K., Govindhan T., Sundararaj N., Palanisamy S. Phytochemicals-induced hormesis protects Caenorhabditis elegans against α-synuclein protein aggregation and stress through modulating HSF-1 and SKN-1/Nrf2 signaling pathways. Biomed. Pharmacother. 2018;102:812–822. doi: 10.1016/j.biopha.2018.03.128. [DOI] [PubMed] [Google Scholar]
- 83.Brunetti G., Di Rosa G., Scuto M., Leri M., Stefani M., Schmitz-Linneweber C., Calabrese V., Saul N. Healthspan maintenance and prevention of Parkinson’s-like phenotypes with hydroxytyrosol and oleuropein aglycone in C. elegans. Int. J. Mol. Sci. 2020;21(7):E2588. doi: 10.3390/ijms21072588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Siracusa R., Scuto M., Fusco R., Trovato A., Ontario M.L., Crea R., Di Paola R., Cuzzocrea S., Calabrese V. Anti-inflammatory and anti-oxidant activity of hidrox® in rotenone-induced Parkinson’s disease in Mice. Antioxidants. 2020;9(9):E824. doi: 10.3390/antiox9090824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Marini A.M., Jiang H., Pan H., Wu X., Lipsky R.H. Hormesis: a promising strategy to sustain endogenous neuronal survival pathways against neurodegenerative disorders. Ageing Res. Rev. 2008;7(1):21–33. doi: 10.1016/j.arr.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 86.Yulyaningsih E., Zhang L., Herzog H., Sainsbury A. NPY receptors as potential targets for anti-obesity drug development. Br. J. Pharmacol. 2011;163(6):1170–1202. doi: 10.1111/j.1476-5381.2011.01363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lochhead J.J., Thorne R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012;64(7):614–628. doi: 10.1016/j.addr.2011.11.002. [DOI] [PubMed] [Google Scholar]