

Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disorder worldwide. Specifically, typical late-onset AD is a sporadic form with a complex etiology that affects over 90% of patients. The current gold standard for AD diagnosis is based on the determination of amyloid status by analyzing cerebrospinal fluid samples or brain positron emission tomography. These procedures can be used widely as they have several disadvantages (expensive, invasive). As an alternative, blood metabolites have recently emerged as promising AD biomarkers. Small molecules that cross the compromised AD blood-brain barrier could be determined in plasma to improve clinical AD diagnosis at early stages through minimally invasive techniques. Specifically, lipids could play an important role in AD since the brain has a high lipid content, and they are present ubiquitously inside amyloid plaques. Therefore, a systematic review was performed with the aim of identifying blood lipid metabolites as potential early AD biomarkers. In conclusion, some lipid families (fatty acids, glycerolipids, glycerophospholipids, sphingolipids, lipid peroxidation compounds) have shown impaired levels at early AD stages. Ceramide levels were significantly higher in AD subjects, and polyunsaturated fatty acids levels were significantly lower in AD. Also, high arachidonic acid levels were found in AD patients in contrast to low sphingomyelin levels. Consequently, these lipid biomarkers could be used for minimally invasive and early AD clinical diagnosis.
Keywords: Alzheimer’s disease, blood, lipid, biomarker, diagnosis, PET
1. INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the most common type of dementia [1]. The late-onset AD characterized by a complex etiology is the most common form of AD, affecting over 90% of patients [2]. Aging clearly contributes to AD [3] since specific regions of the brain are vulnerable to aging. The major AD neuropathological hallmarks are well-known. First, extracellular accumulation of amyloid protein forming senile plaques could be due to the over-production or impaired clearance of β-amyloid [Aβ) peptide [4]. Second, tau protein intracellular accumulation forms neurofibrillary tangles (NFTs) [5], involving or reflecting synaptic loss and neurodegeneration, which could trigger cognitive dysfunction, progressive memory loss, and behavioral changes. Also, alterations in molecular mechanisms, like inflammation and oxidative stress, play crucial roles in AD development, as shown in Fig. (1).
Fig. (1).
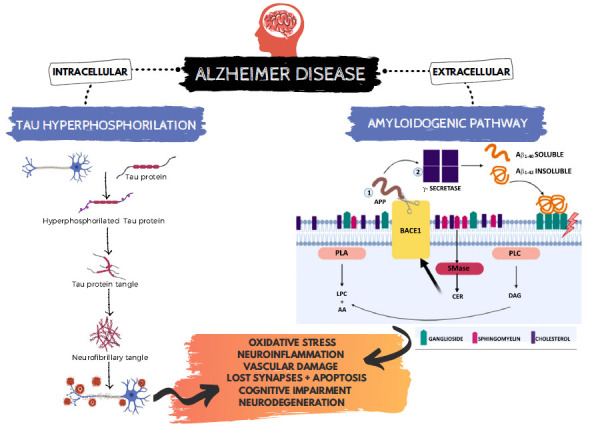
Neuropathological hallmarks of Alzheimer's Disease. AA: arachidonic acid, APP: amyloid precursor protein, BACE1: β-secretase 1, CER: ceramides, DAG: diacylglycerol, LPC: lysophosphatidylcholine, SMase: sphingomyelinase, PLA: phospholipase A, PLC: phospholipase C.
The current “gold standard” for AD diagnosis includes cerebrospinal fluid (CSF) biomarkers, or brain positron emission tomography (PET), and magnetic resonance imaging (MRI) [6]. These techniques have some disadvantages since they are invasive, time-consuming, prone to patient’s discomfort, and expensive [7]. Thus, there is a need to identify accessible biomarkers and minimally invasive samples, as well as to develop an early diagnosis model.
Most of the AD biofluid biomarkers studies are based on CSF samples [8]. However, CSF analysis constitutes a restricted assessment of inflammatory and degenerative diseases because not all brain areas are reflected in CSF biomarkers levels [9]. In fact, two barriers separate central nervous system components from systemic circulation. They are the blood-CSF barrier (BCB) and blood-brain barrier (BBB), which are not completely impermeable [9]. Functional BBB allows specific, small, and lipophilic molecules transport [10], depending on the oil-water partition coefficient, so lipophilic molecules are more likely to cross the BBB [9]. Over the years, damaged lipids accumulate in the membrane, requiring increased lipid clearance [11]. In addition, amyloid deposits in the brain vasculature of AD patients could alter BBB function, causing its physical breakdown [10] and possibly cerebral amyloid angiopathy. Also, tau-induced activation of glial cells could favor the transport of leukocytes across the BBB and increase endothelium adhesion molecules expression [12]. Post-mortem studies have shown BBB damage in AD, including pericytes degeneration and blood-derived proteins accumulation in the hippocampus and cortex. Hence, changes in the regulation of cerebral blood flow caused by reduced microvascular density, loss of tight junctions, increased endothelial pinocytosis, reduced mitochondrial content, and BBB breakdown have been demonstrated. Also, BBB dysfunction can induce tau hyperphosphorylation and vice-versa. Moreover, neuroinflammation and oxidative stress contribute to BBB damage and NFTs formation [12]. Consequently, small molecules can be found in the blood after crossing the permeable BBB [13-18]. In this sense, blood metabolites have recently emerged as promising biomarkers in early and minimally invasive AD diagnoses [19]. Specifically, plasma Aβ, p-tau, and tau are determined by ultrasensitive assay methods, showing some correlation with brain amyloid and tau physiopathology in AD [20, 21]. However, there is a lack of clinical validation studies to corroborate these preliminary results. These plasma metabolites could be useful as screening tools and used in longitudinal evaluations [22]. Hence, blood-based biomarkers constitute a promising approach in early AD detection.
Among blood biomarkers for AD, lipids could play an important role because the brain is one of the richest organs in lipid content, comprising around 50-60% of its dry weight. These lipid components are involved in energy storage, neurogenesis, membrane and myelin sheath formation, cellular transport, or signaling pathways [11]. Possibly, lipid levels in the human brain decrease after the age of 50 [23]. Also, the brains of AD patients show a higher number of lipoid granules in glia, suggesting aberrant lipid metabolism [5]. Consequently, recent research has focused on lipids as potential early AD biomarkers. Peña-Bautista et al. [24, 25] proposed an early diagnostic model based on plasma levels of some lipid peroxidation products. Similarly, other studies evaluating some lipid peroxidation compounds, such as 4-hydroxynonenal (4-HNE) or malondialdehyde (MDA), were also found in the literature [26, 27]. In fact, it has been found that brain microvessels from AD patients release increased levels of multiple inflammatory mediators compared to the age-matched controls [10]. Indeed, lipoxidative stress is significantly higher in AD patients, increasing with advanced AD stages [23].
Lipids are found ubiquitously inside amyloid plaques, so their functional roles in Aβ generation, clearance, and deposition have been intensively investigated [28]. In fact, lipids could modulate the Aβ aggregation and tau hyperphosphorylation. Lipid bilayer has a strong impact on amyloid precursor protein (APP) processing because APP, BACE-1, and γ-secretase are found together in membrane microdomains enriched in cholesterol, gangliosides, and other sphingolipids (lipid rafts), where the Aβ generation takes place [4]. These biochemical microstructures are characterized by their PUFAs content [23]. Moreover, Aβ peptides interact with lipid raft components (e.g., gangliosides, phospholipids, cholesterol) [29], as well as with apolipoprotein E (ApoE) and tau protein [30]. Therefore, this would promote Aβ aggregation on the cell membrane and tau hyperphosphorylation, disrupting membrane integrity, neuroinflammation, and increased oxidative damage [5]. Subsequently, they could affect intracellular calcium homeostasis, triggering neurotoxic cascades in AD [28]. In addition, an important risk factor for late-onset AD is the ApoE genotype E4, which is implicated in the lipid transport to various organs, including the delivery of cholesterol to the brain [2]. As a result, cholesterol metabolism is related to the pathogenesis of AD, constituting a risk factor in its development [4]. APOE is one of the major apolipoproteins in brain lipid metabolism. Moreover, APOE4 is the least efficient isoform in regulating the cholesterol efflux from cells, promoting the strongest inflammatory effects in microglia and astrocytes [31]. Furthermore, some studies have shown a correlation between plasma lipid levels and CSF AD biomarkers (Aβ, tau) [32, 33], corroborating the role of lipid metabolism in AD.
On the other hand, there is a lack of studies related to evaluating potential plasma lipid biomarkers in asymptomatic individuals with familial AD and unimpaired individuals with Down syndrome. Therefore, further studies are required to identify potential AD biomarkers in these specific AD variants.
In general, lipid compounds can be classified into eight categories proposed by the international consortium LIPID MAPS [34, 35]. They are fatty acids (FA), glycerolipids (GL), glycerophospholipids (GP), sphingolipids (SPs), saccharolipids (SL), polyketides (derived from the condensation of ketoacyl subunits), sterol lipids, and prenol lipids (derived from the condensation of isoprene subunits). Depending on their nature and function, lipids could be differentially affected at different stages of disease progression [2, 36]. So, the most interesting lipid classes involved in early AD are summarized in Table 1.
Table 1.
Lipid classes in early AD diagnosis.
| Lipid Class | Description | AD Relationship |
|---|---|---|
| Fatty acids(AA, DHA) | Linked to cognitive impairment | Impairment at early AD stage |
| Glycerolipids (DAG, TG) | Related to memory loss | Higher levels in plasma and frontal cortex at early AD stages |
| Glycerophopholipids (PE, PC) | Found in brain cell membranes | Lower levels in frontal and temporal AD cortex (neurodegeneration) Correlated with AD severity |
| Sphingolipids (SMs, CER) | SM: Lipid rafts (amyloid hotspot) CER: BACE1 |
Correlation with amyloidogenic pathway |
| Lipid peroxidation (isoprostanoids, 4-HNE, MDA) | Under oxidative stress conditions | Impairment levels in plasma from AD patients |
Abbreviations: 4-HNE: 4-hydroxynonenal, AA: arachidonic acid, BACE1: β-secretase 1, CER: ceramides, DAG: diacylglycerol, DHA: docosahexaenoic acid, MDA: malondialdehyde PC: phosphatidylcholine, PE: phosphatidylethanolamine, SMs sphingomyelins, TG: triglyceride.
Regarding treatments, FDA has approved a new drug (aducanumab) for AD treatment, which may be potentially disease-modifying. Until now, available treatments have only achieved symptomatic relief, and they seem to exhibit more efficacy and utility at early AD stages. In general, more studies are required to advance the knowledge of physiological mechanisms involved in AD development, identify more potential biomarkers to carry out the follow-up of the disease progression, evaluate response to treatment [37], facilitate recruitment into clinical trials, and identify potential new therapeutic targets [38]. Evidencing the multifactorial nature of AD, a panel of biomarkers may offer suitable diagnostic indices (sensitivity, specificity, positive or negative predictive values) [37] and reliable prognosis.
The aim of this review is to identify potential lipids which could be promising biomarkers for early and minimally invasive AD diagnosis.
2. METHODS
The present review was carried out following the PRISMA method. Data searching was completed on 20th June, 2020, using PubMed, Scopus, and Science Direct databases and supplemented through manual searching. It was limited to the last 11 years, using the combination of search terms “Alzheimer Disease” AND “Lipid biomarkers” AND “blood” OR “Early diagnosis.” Therefore, studies in the English language, since 2009, using minimally invasive human samples from AD patients and determining lipid compounds were included. Finally, 155 articles were screened to check if they met the scope of this review (Fig. 2).
Fig. (2).
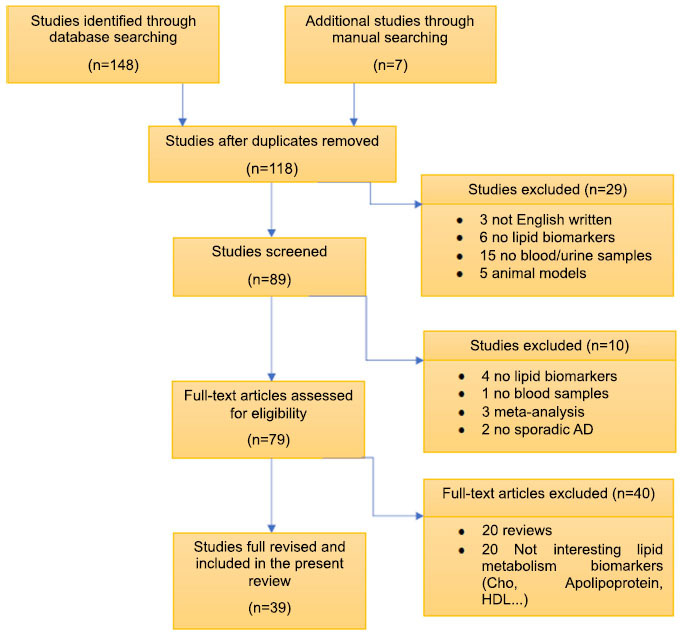
Flow diagram summarizing the systematic search, screening, and study selection for this review.
3. RESULTS AND DISCUSSION
The present review is focused on the crucial relationship between aberrant brain lipid metabolism, lipid peroxidation, and neurodegeneration. In fact, several works focusing on early AD diagnosis and determining different lipid classes in minimally invasive samples as potential biomarkers were found in the literature. These studies can be classified according to different characteristics, such as sample type (blood, plasma, serum), study subjects, lipid compounds, and analytical techniques. Although CSF biomarkers greatly improve AD-specific diagnosis, only a few studies have used them in biologically defined cohorts. As can be seen, the results are summarized in Table 2.
Table 2.
AD studies evaluating lipid compounds and derivatives as early and minimally invasive samples.
| 1. FATTY ACIDS | |||||
|---|---|---|---|---|---|
| Reference | Sample | Participants* | Lipid Biomarkers |
Analytical
Technique |
Results |
| Wang et al. [43] | Serum | Centre of Harbin Elderly Care Service, Heilongjiang (North of China), 58–92 yrs AD (n=46) HC (n=39) |
4 SFA (C14:0, C16:0, C18:0, C24:0), 3 MFA (C16:1n-7, C18:1n-9, C24:1n-9) and 8 PUFA (C18:2n-6, C18:3n-6, C18:3n-3, C20:2n-6, C20:4n-6, C20:5n-3, C22:5n-6, and C22:6n-3). | GC-MS | 2 SFA= C14:0 (p<0.001) + C16:0 (p<0.05) + 3 PUFA (C18:1 (p< 0.05) + C18:3p<0.05), +C22:6 (p<0.001)): ↓AD< HC. Total n-6 FFAs: ↓AD< HC (p < 0.05) |
| Abdullah et al. [46] | Serum | Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT) MCI (n=15) AD (n=8) NCI (n=172) |
PL (AA, DHA) | HPLC-MS | AA, DHA, Aβ42/Aβ40 ratios and ApoE-ε4 genotype →AUC=0.910, whereas PL species alone →AUC=0.880 |
| Lin et al. [50] | Serum | Volunteers from Taoyuan City, Taiwan 55-90 yrs. NCI (n=15) MCI (n=10) AD (n=15) |
40 ACs 15 SMs 40 GPs |
LC-MS/MS | ACs (C3 and C5): ↓AD<HC<↑MCI (p<0.05) ACs +PC (C38:2) + age→ discriminate NCI (AUC=0.77), AD (AUC=0.72), MCI (AUC=0.59) |
| Olazaran et al. [44] | Plasma | 60-85 yrs From 6 Spanish university hospitals + 1 Spanish research institution HC (n=93), aMCI (n=58) AD (n=100) |
210 metabolites; ACs, NEFAs, LPC, LPE, LPI 256 metabolites; DAG, TG, SM, CER, CMH, PC, PE, and PI |
UPLC-MS | PI (40:6), PC (36:5), PC (37:6). PC (38:5), PC (38:6), PC (40:5) + PC (40:6): ↓AD+ ↓aMCI<<HC (p<0.05) Exceptions: PE (36:4), PE (38:5), PE (18:0/0:0) + PE (18:1/0:0): ↑aMCI+ ↑AD patients>HC (p<0.05) NEFAs (16:0, 18:1n-9, ω-3 FA (EPA, DPA, DHA)): ↓aMCI+↓ AD <HC (p<0.05) SM(39:1)+SM(41:1)+SM(42:1)+ CER(39:1)+CER(40:1)+ CER(41:1)+ CER(42:1)+ CER(43:1). +TGs: ↓↓aMCI+ ↓↓AD<<HC (p<0.05) |
| Whiley et al. [47] | Plasma | AddNeuroMed cohort + King’s College London Dementia Case Register (UK). Age- and sex-matched Screen phase: AD (n=10), MCI (n=10), control (n=15) Validation phase:(1) AD (n=42), MCI (n=50), control (n=49) |
(1) PCs (2) ω-fatty acids (AA, DHA, EPA) (3) choline and phosphocholine, glycerophosphocholine |
Screen phase: LC-MS (1, 2), NMR Validation phase: LC-MS (1,2) |
Screen phase: 3 PCs (16:0/20:5 (p <0.001) + 16:0/22:6 (p <0.05) + 18:0/22:6 (p<0.005): ↓↓AD<↓MCI< HC. Validation phase: 3 PCs (16:0/20:5 (p <0.001) + 16:0/22:6 (p <0.05) + 18:0/22:6 (p<0.005): ↓AD< HC. EPA: HC<↑AD (p=0.023) |
| Wang et al. [48] | Plasma | Department of Neurology, Rui Jin Hospital (Shanghai, China) AD (n= 57) aMCI (n=58) HC (n=57) |
6 metabolites (AA) 5 metabolites (AA, α-AAA, 2-aminoadipic acid) |
UPLC-QTOF-MS GC-TOF-MS |
14 FA: ↓AD<HC. 40 metabolites: aMCI ≠ HC. Panel 6 metabolites; ROC curves=1.00. Panel 5 metabolites; AUC = 0.998, 95% CI = (0.993,1.000) between aMCI and HC |
| Xicota et al. [49] | Plasma | INSIGHT-preAD cohort Amyloid positive (n=47) Amyloid negative (n=47) |
3 MCFA; octanoic, undecanoic and hydroxyl-nonanoic acids | LC-MS | 3 MCFA: ↑amyloid positive group> amyloid negative group. And correlated over time in a subset of 22 subjects (p<0.05). |
| Goozee et al. [45] | Blood: erythrocyte | McCusker Kerr Anglican Retirement Village Initiative in Ageing Health (KARVIAH) study cohort, 65–90 yrs High NAL(n=35) Low NAL (n=65) |
SFA (myristic acid, palmitic acid, stearic acid, arachidic acid, lignoceric acid), MUFAs, n-6 PUFA, n-3 PUFA | GC | SFA, MUFA. Not different n-6 PUFA+ AA: ↑high NAL >low NAL (p <0.05) DPA (C22:5n-3, p<0.05): high NAL<↑low NAL Positive trend= AA-NAL (β = 0.197, p = 0.050) and inverse trend= linoleic acid-NAL (β = −0.172, p = 0.088) |
| 2. GLYCEROLIPIDS AND GLYCEROPHOSPHOLIPIDS | |||||
| Reference |
Sample
Type |
Participants* | Lipid Biomarkers |
Analytical
Technique |
Results |
| Al-khateeb et al. [51] | Serum | Jordan University Hospital AD (mild-moderate) (n=41) HC (n=40) |
TG | Enzymatic colorimetric method | No difference in lipid profile |
| Anand et al. [54] | Serum | Knight Alzheimer’s Disease Research Center (Knight ADRC) 1)MCI (n=10), mild AD (n=10), moderate AD (n= 9) HC (n=32) 2) MCI (n=9), mild AD (n=9), moderate AD(n=9) HC (n=30) |
87 lipids | ESI-TOF/MS | 35 markers (SMs, PCs, PEs, LPCs, DAGs, TGs...) can differentiate AD vs. HC (p<0.05), sensitivity=93%, specificity=80%. |
| Varma et al. [56] | Serum | Alzheimer's Disease Neuroimaging Initiative (ADNI) database (prodromal AD); HC (n=216), MCI (n=366), AD (n=185) mean age = 75.19, %female = 42.63 Baltimore Longitudinal Study of Aging (BLSA) database (preclinical AD); Converters (n=92), Non-converters (n=115) mean age =78.68, %female = 42.63 |
26 metabolite panel (ACs, SMs, PCs) | FIA-MS/MS | BLSA database: ↑↑SM C16:0+ ↑↑SM C16:1+↑↑SM (OH) C14:1+ ↑↑SM C18:1→↑↑risk of AD. ↓/↑ PC (38:4), PC (C34:2) =↑ risk conversion to AD. ↑SLs =↑declines in cognition. ADNI database: ↓PC (C40:6) =AD-like pattern brain atrophy. ↑SM C18:1= ↑ risk AD among individuals MCI and HC. ↑PC (38:4) = ↑ risk of conversion to AD. |
| Arnold et al. [59] | Serum | *ADNI cohort (n=1517) HC (n=362) MCI(n=490) AD (n=302) |
139 metabolites (PCs, SM..) | UPLC-MS/MS | 108 metabolites significantly gender associated. SMs+ PCs: ↑women >men ACs: ↑men>women (p<0.05). No differences by MCI/AD status. |
| González-Domínguez et al. [61] | Serum | Over 65 yrs Neurologic Service of Hospital Juan Ramón Jiménez (Huelva, Spain) AD (n=19) HC (n=17) |
LPC, LPE, PC, PE | UPLC-ESI-QTOF-MS | LPC, LPE: ↓AD<HC (p<0.05). All PEs + total plasmalogens: ↓AD< HC (p<0.05) |
| Proitsi et al. [38] | Plasma | Dementia Case Register at King’s College London + AddNeuroMed study ≥60 yrs AD (n=142) HC (n=135) |
2 PCs (40:4 y 36:3), 85 TG | UPLC-Q-ToF MS | 54 TG: ↓AD <HC (p<0.05) 2 PCs (40:4 y 36:3) ↑AD >HC (p<0.05) PC 36:3 is associated with faster ROD |
| Olazaran et al. [44] | Plasma | 60-85 yrs From 6 Spanish university hospitals + 1 Spanish research institution HC (n=93), aMCI (n=58) AD (n=100) |
210 metabolites; ACs, NEFAs, LPC, LPE, LPI 256 metabolites; DAG, TG, SM, CER, CMH, PC, PE, and PI |
UPLC-MS | PI (40:6), PC (36:5), PC (37:6). PC (38:5), PC (38:6), PC (40:5) + PC (40:6):↓↓AD+ ↓↓aMCI<<HC (p<0.05) Exceptions: PE (36:4), PE (38:5), PE (18:0/0:0) + PE (18:1/0:0): ↑aMCI+ ↑AD patients>HC (p<0.05) NEFAs (16:0, 18:1n-9, ω-3 FA (EPA, DPA, DHA)): ↓aMCI+ ↓AD <HC (p<0.05) SM(39:1)+SM(41:1)+SM(42:1)+ CER(39:1)+CER(40:1)+ CER(41:1)+ CER(42:1)+ CER(43:1). +TGs: ↓↓aMCI+ ↓↓AD<<HC (p<0.05) |
| Leeuw et al. [52] | Plasma | *Amsterdam Dementia Cohort AD (n=127) HC (n=121) |
26 metabolites (14 TG) | UPLC-MS/MS | 14 TG: ↓AD<HC, SM d18:1/20:1: ↑AD>HC (p<0.05) Combining 8,12-iPF-2a IV+ nitro-fatty acid NO2-aLA (C18:3) predictive AD vs. HC |
| Kim et al. [53] | Plasma | Memory clinic of University-affiliated general hospital (Seoul, Korea) Female, 65-80 yrs NCI(n=13) MCI (n=23) AD (n=14) |
14 lipid classes (PE, PA, CER, PI, TG, DAG…) | UHPLC-ESI-MS/MS | TG 50:1, DG 18:1_18:1+ PE 36:2 MCI group + MMSE (→AUC =0.833, 0.847, and 0.917, respectively) |
| Kim et al. [55] | Plasma | Dementia Case Register at King’s College London + AddNeuroMed study AD (n=205) Mean age= 77.35 Male/female= 81/123 NCI (n=207) Mean age= 74.88 Male/female= 77/130 |
CER, PCs | MS | CER 16:0 (p<0.01), CER 18:0 (p<0.01) + CER 24:1 (p<0.05) ↑AD>NCI CER 20:0, CER 22:0, CER 24:0= not different. PC36:5 +PC38:6: ↓AD<NCI (p<0.05). PC40:6 (n.s) PC36:5-+hippocampal volume positively associated (p<0.01) |
| Costa et al. [57] | Plasma | Institute of Psychiatry, Faculty of Medicine, University of Sao Paulo, Brazil AD (n=34) Male/female=10/24 Mean age=75 yrs aMCI (n=20) Male/female=3/17 Mean age=73.9 yrs HC (n=25) Male/female=7/18 Mean age=74.4 yrs |
7 PCs, 1 LPC, 2 ACs | MS/MS | PC (C36:6), PC (C40:6), C16:1-OH: ↓MCI<HC (p<0.05) PC (C40:2): MCI<↑AD PC (C40:2), PC (C40:6): ↓MCI-AD<HC. (p≤0.05) |
| Peña-Bautista et al. [60] | Plasma | *Neurology Unit of the Hospital La Fe, Valencia (Spain) 50-80 yrs Early AD (n=29) HC (n=29) |
GPs, LPC, CER, LPE, PC, cardiolipins | UPLC-Q-ToF MS | LPC (18:1) early AD> HC (p<0.05) |
| Oberacher et al. [58] | Blood Lysate human platelets |
Hall/Tirol State Hospital, Austria. Training set (n=54): HC(n=18) MCI(n=15) AD(n=21) Validation set (n=26): HC(n=11), MCI(n=6), AD(n=9) Blinded follow up conversion study (9 months) (n=10) HC (n=3) AD(n=7) |
163 endogenous metabolites, including 40 ACs, hydroxyACs and dicarboxylACs and 15 SM and hydroxySMs, 77 PCs and 15 LPCs | FIA-MS/MS | Total PCs, LPCs+ SMs different between groups (p<0.05). Ratio of total PCs: LPCs = 2-fold ↑MCI >AD (p<0.001). SFA: MCI-AD ≠HC. (p<0.001) Training set + C4.5 decision tree algorithm→ accuracy 85.2%. Most discriminative variables→PC ae C32:2, PC ae C34:1, PC aa C36:5, lysoPC a C18:1, lysoPC a C16:0, (SM(OH) C14:1) Follow up study: PC ae C40:4→verify the clinical diagnosis (19/20 cases, cut-off of <0.30 μM) |
| 3. SPHINGOLIPIDS | |||||
| Reference |
Sample
Type |
Participants | Lipid Biomarkers |
Analytical
Technique |
Results |
| Mielke et al. [68] | Serum | Women’s Health and Aging Study (WHAS) II 70-79 yrs Females only Probable cognitive impairment (n=100) HC (n=123) |
SM, CER | ESI/MS/MS | ↑↑ total SMs+ CER 16:0, CER 18:0, CER 22:0, CER 24:1, CER 24:0, stearoyl + sulfatide significantly ↑↑increased risk of incident impairment on HVLT-delayed. No association with SMs/ CER-risk of TMT-B impairment. ↑↑SM/total cholesterol= triple risk of HVLT-delayed impairment (p=0.074). |
| Olazaran et al. [44] | Plasma | 60-85 yrs From 6 Spanish university hospitals + 1 Spanish research institution HC (n=93), aMCI (n=58) AD (n=100) |
210 metabolites; ACs, NEFAs, LPC, LPE, LPI 256 metabolites; DAG, TG, SM, CER, CMH, PC, PE, and PI |
UPLC-MS | PI (40:6), PC (36:5), PC (37:6). PC (38:5), PC (38:6), PC (40:5) + PC (40:6):↓↓AD+ ↓↓aMCI<<HC (p<0.05) Exceptions: PE (36:4), PE (38:5), PE (18:0/0:0) + PE (18:1/0:0): ↑aMCI+ ↑AD patients>HC (p<0.05) NEFAs (16:0, 18:1n-9, ω-3 FA (EPA, DPA, DHA)): ↓aMCI+ ↓AD <HC (p<0.05) SM(39:1)+SM(41:1)+SM(42:1)+ CER(39:1)+CER(40:1)+ CER(41:1)+ CER(42:1)+ CER(43:1). +TGs: ↓↓aMCI+ ↓↓AD<<HC (p<0.05) |
| Kim et al. [55] | Plasma | Dementia Case Register at King’s College London + AddNeuroMed study AD (n=205) Mean age= 77.35 Male/female= 81/123 NCI (n=207) Mean age= 74.88 Male/female= 77/130 |
CER, PCs | MS | CER 16:0 (p<0.01), CER 18:0 (p<0.01) + CER 24:1 (p<0.05) ↑AD>NCI CER 20:0, CER 22:0, CER 24:0=not different. PC36:5 +PC38:6: ↓AD<NCI (p<0.05). PC40:6 (n.s) PC36:5-+hippocampal volume positively associated (p<0.01) |
| Mielke et al. [64] | Plasma | BLSA cohort Over 55 yrs NCI (n=992; 119/626 men developed AD; 73/366 women developed AD) |
CER, SMs | LC-ESI/MS/MS | Men: Most CER= ↑AD risk (→C16:0 (2-fold) +SMs: C18:0, C 18:1, C20:1 + C22:1(p≤0.001) Women: No associations CER-AD risk All SMs (exception C24:1) =↓ AD risk (p<0.001) |
| Mielke et al. [65] | Plasma | Clinical Core of the Johns Hopkins Alzheimer's Disease Research Center Over 55 yrs HC (n=25) aMCI (n=17) Early probable AD (n=21) |
CER: C24:0, C24:1, C22:0, C16:0, C18:0, C20:0, C26:0, C26:1. | HPLC-ESI-MS/MS | CER: C22:0, C24:0+ C26:0: ↓MCI<HC+ ↑AD group. (p<0.05) CER: C16:0, C18:0+ C22:0: MCI < ↑AD (p<0.05) CER= C24:1, C26:1 (not different) |
| Han et al. [67] | Plasma | Joseph and Kathleen Bryan Alzheimer’s Disease Research Center (Bryan ADRC) + Department of Psychiatry, (Duke University). AD (n=26) Mean age=77.2 % Male= 46 yrs NCI (n=26) Mean age=73 yrs % Male=42 |
9 lipid classes; 65 PC, 86 PE, 25 PI, 33 SM, 14 LPC, 29 CER, TG>500 | MDMS-SL | 8 SMs: ↓AD<NCI (p<0.05) 6 different levels SMs (p<0.1). 2 CER=N16:0+ N21:0 ↑AD>NCI (p<0.05) + 5 CER: ↑AD> NCI (p<0.1) |
| Mielke et al. [69] | Plasma | Alzheimer’s Disease and Memory Disorders Center (ADMDC) at Baylor College of Medicine Probable AD (n=120) Mean age=71.8 yrs 60.8% female |
CER, DHCER, SM, DHSM | LC-ESI/MS/MS | ↓ DHCER + ↑ SM and ↑DHSM related to slower decline cognitive measures. DHSM/DHCER + SM/Ceramide ratios are strongly related to cognitive decline. TGs are not associated with progression. |
| 4. LIPID PEROXIDATION COMPOUNDS | |||||
| Reference |
Sample
Type |
Participants | Lipid Biomarkers |
Analytical
Technique |
Results |
| Padurariu et al. [26] | Serum | Psychiatry University Hospital, Iasi (Romania) HC (n=15) Females/males=7/8 Mean age=62.5 yrs MCI(n=15) Female/male= 5/10 Mean age: 63.2 yrs AD (n=15) Female/male=6/9 Mean age=65.8 yrs |
MDA | TBARS assay | MDA: ↑MCI + ↑AD >HC (p<0.0005) and between MCI and AD groups (p<0.0005). |
| Ademowo et al.[27] | Serum | Carotenoid and AGE-Related Dementia Study (CARDS) at the Nutrition Research Centre Ireland (NCRI). AD mild-moderate (n=21) Mean age=79 yrs, %female=43 HC (n=16) Mean age=75 yrs 5female=50% |
oxPLs (POVPC + IsoP) | LC-MS ELISA |
POVC: 2-fold ↑AD>HC (p <0.05) IsoP not difference |
| Peña-Bautista et al. [24] | Plasma | * Neurology Unit of the University and Polytechnic Hospital La Fe, Valencia (Spain) 50-80 yrs HC (n=26) MCI-AD(n=68) |
18 lipid peroxidation compounds; IsoPs, prostaglandins, NeuroPs, dihomo-IsoPs, dihomo-IsoFs | UPLC-MS/MS | 15(R)-15-F2t-IsoP+15-keto-15-E2t-IsoP+ 15-keto-15-F2t-IsoP+ 15-E2t-IsoP+ 4(RS)-F4t-NeuroP + ent-7(RS)-7-F2t-dihomo-IsoP:↑AD>HC (p<0.001) PGF2α+5-F2t-IsoP+ 7(RS)-ST-Δ8-11-dihomo-IsoF:↓MCI-AD<HC (p<0.001) |
| Leeuw et al. [52] | Plasma | *Amsterdam Dementia Cohort AD (n=127) HC (n=121) |
26 metabolites (14 TG) | UPLC-MS/MS | 14 TG: ↓AD<HC, SM d18:1/20:1: ↑AD>HC (p<0.05) Combining 8,12-iPF-2a IV+ nitro-fatty acid NO2-aLA (C18:3) predictive AD vs. HC |
| Peña-Bautista et al. [74] | Plasma | * Neurology Unit of the University and Polytechnic Hospital La Fe, Valencia (Spain) 50-80 yrs Early stages AD (n=80) HC (n=32) |
NeuroPs, IsoPs, neurofurans, isofurans, 17-epi-17-F2-dihomo-IsoP, PGF2α | UPLC-MS/MS | 8-iso-15(R)-PGF2α, 4(RS)-F4-NeuroP, neurofurans, IsoPs 17-epi-17-F2t-dihomo-IsoP=different levels between groups (p<0.05). PGF2α: HC>↓AD(p<0.05) Sensitivity=72.5%, specificity=100%. |
| Yoshida et al. [75] | Plasma | Aoisoranosato Geriatric Health Services Facility + Arimakogen Hospital AD patients (n=39) Mean age= 76.2 yrs Male/females=15/24 VD (n=25) Mean age=80.4 yrs Male/female=8/17 HC (n=24) Mean age=68.4yrs Male/female=8/16 |
tHODE, t8-iso-PGF2α | HPLC-ESI-MS/MS | tHODE: ↑AD> HC. t8-iso-PGF2α: ↑AD > HC→ sensitivity 82%, specificity 71%. |
| Peña-Bautista et al. [73] | Plasma Urine |
* Neurology Unit of the University and Polytechnic Hospital La Fe, Valencia (Spain) MCI-AD (n=70) Mean age=70 yrs % female=58.6 HC (n=26) Mean age=66 yrs % female=34.6 |
22 analytes of lipid peroxidation | UPLC-MS/MS | Plasma: 15(R)-15-F2t-IsoP+ 4(RS)-4-F4t-NeuroP+NeuroPs, IsoPs+ ent-7(RS)-7-F2t-dihomo IsoP +neurofurans +isofurans: ↑MCI-AD >HC as well as higher age +female proportion (p<0.05) ANN model→ sensitivity=88.2%, specificity=76.9%. Urine: 15(R)-15-F2t-IsoP+ 2,3-dinor-15-epi-15-F2t-IsoP+ 4(RS)-4-F4t-NeuroP+ ent-7(RS)-7-F2t-dihomo-IsoP+ 17-epi-17-F2t-dihomo-IsoP+ 10-epi-10-F4t-NeuroP+ 17-F2t-dihomo-IsoP+ neurofurans: ↑MCI-AD>HC as well as higher age+ female proportion(p<0.05). ANN model→sensitivity=80,9%, specificity=76.9%. |
| Mufson et al. [71] | Plasma Urine |
Religious Orders Study NCI (n=134) Mean age= 79.6 yrs MCI (n=32) Mean age=83.5 yrs AD (n=14) Mean age= 86.3 yrs |
F2-IsoPs | GC/MS | Urine; F2-IsoPs: ↑men>women. Plasma + urine F2-IsoPs not correlated (p<0.0001) No differences among clinical groups. |
| Sundelof et al. [72] | Urine | Uppsala Longitudinal Study of Adult Men (ULSAM) 70-77 yrs Males only n=679 (n=40 developed AD and n=86 developed all-cause dementia) |
F2-IsoPs (8-iso-PGF2α) | Radioinmuno-assay | F2-IsoPs are not associated with a higher incidence of AD or dementia. |
| 5. METABOLITES COMBINED PANEL | |||||
| Reference |
Sample
Type |
Participants | Lipid Biomarkers |
Analytical
Technique |
Results |
| Casanova et al. [76] | Serum | BLSA cohort: Converters AD (n=93) baseline age 77.9 yrs Non-converters AD (n=99) baseline age 76.6 yrs AGES-RS cohort: Converters (n=100) baseline age 78.18 yrs Non-converters (n=100) baseline age 78.23 yrs |
10 metabolite panel (by Mapstone) 187 targeted metabolites |
FIA-MS/MS | BLSA→10 metabolite panel: AUC=0.64, sensitivity/specificity=51.6%/65.7% converters vs. non-converters. AGES-RS→ 10 metabolite panel: AUC=0.394, sensitivity/specificity=47%/36% converters vs. non-converters. For symptoms onset; BLSA (AUC= 0.58, sensitivity/specificity= 53.8%/62.6%) AGES-RS (AUC=0.481, sensitivity/specificity=52%/48%) AD vs. non-converters. sensitivity/specificity=67.7%/66.7% |
| Oresic et al. [77] | Serum | *PredictAD project, University of Kuopio HC (n=46) Baseline age=71 yrs Male/female= 21/25 MCI (n=143) Baseline age=71.5 yrs Male/female=47/96 AD (n=37) Baseline age=75 yrs Male/female=17/20 |
139 lipids in co-regulated clusters; PCs, LPCs, SMs… | UPLC-MS | Metabolic signature = 2 PC (18:0/18:2) from cluster 1 and PC (16:0/20:4) from cluster 5, lactic acid and ketovaline predicted AD→AUC=0.77 |
| Toledo et al. [79] | Serum | *ADNI-1 cohort (n=702) HC (n=199) 75.3yrs, 49.7% male MCI (n=358) 75.1 yrs, 35.4% male AD (n=175) 75.6 yrs, 48.6% male ERF study (N = 905, CN) + Rotterdam Study (N = 2480, CN) Indiana Memory and Aging Study (IMAS)(n=34) CN (n=17) 68.4 yrs, 76.5% women MCI(n=10) 72.1 yrs, 60%women AD (n=7) 72.4 yrs, 71.4% women |
138 metabolites (PC, SM) | UPLC-MS/MS | 13 metabolites→ cognitive scores, CSF, and MRI measures (p<0.05). 6 metabolites-CSF Aβ1–42 positivity; PC (C36:2) + PC (C40) +, PC (C42:4) + PC (C44:4) + SM (OH)(C14:1)+ SM (C16:0), 4 associated with t-tau/Aβ1–42 ratio (C18+ PC (C36:2), SM (C16:0)+ SM (C20:2), 5 associated with ADAS-Cog13 scores (C14:1+ C16:1, SM C20:2 + α-aminoadipic acid [α-AAA], and valine), 6 associated with SPARE-AD scores (C12+ C16:1+ PC (C42:4)+PC (C44:4) + α-AAA,+ valine). |
| Barupal et al. [33] | Serum | *ADNI database cohort (n=806) | 349 lipids (AC=9, FA=29, 8 CE, LPC=22, LPE=4, PC=53, PE=11, PI=11, CER=19, SM=34, DAG=13, TG=84…) |
LC-MS/MS | 168 lipids associated with at least 1 AD phenotype (p<0.05). 28 coregulated lipids sets (LM1 to LM28) established. Significant associations of ω-3+ ω-6 lipids with AD diagnosis + cognitive functions (p<0.05). PUFA +TG: ↓AD <HC (p<0.05) |
| Mapstone et al. [7] | Plasma | Rochester/Orange County Aging Study(≥70yrs) (5 yrs follow-up) Discovery phase: aMCI/AD+ Converters (n=53) HC (n=53) Matched based on sex, age, and education level Blinded cross-validation: aMCI/AD+ HC (n=20) aMCI/AD (n=20) Converters (n=10) Discovery and validation groups did not differ on clinical measures (F(4,170) = 1.376, P = 0.244) or on any composite z-score (F(5,169) = 2.118, P = 0.066) |
Set 10 metabolites; PC diacyl (aa) C36:6, PC aa C38:0, PC aa C38:6, PC aa C40:1, PC aa C40:2, PC aa C40:6, PC acyl-alkyl (ae) C40:6, LPC a C18:2, ACs (C3, C16:1-OH) | SID-MRM-MS UPLC-ESI-QTOF-MS |
Discovery phase: Untargeted metabolomic analysis→PI ↓Converters<HC LASSO analysis→PC, AC ↓Converters < HC Targeted analysis→Set 10 metabolites in discovery phase + blinded cross-validation: ↓Converters+ ↓aMCI/AD < HC (→Accuracy=90%)(p<0.05) |
| Fiandaca et al. [78] | Plasma | Rochester/Orange County Aging Study (≥70yrs) (5 yrs follow-up) matched for age, gender, and education and featured similar APOE allele status Discovery cohort. NCI (n=53), Phenoconverters (aMCI/AD) (n=18) 3/18 +ApoE ε4 alelle Internal Validation Cohort. NCI (n=20), Phenoconverters (aMCI/AD) (n=10) 2/10 +ApoE ε4 alelle |
24 Metabolites (13 PCs, 9 ACs…) | MRM-SID-MS | Discovery cohort: 174 metabolites significantly expressed differently (p<0.05) Panel 24/174 metabolites→ Discovery + internal validation cohort: 3/24 metabolites (ACs) ↑Phenoconverters > NCI (p<0.05) 21/24 metabolites ↓Phenoconverters < NCI (p<0.05) Panel 24 metabolites→AUC=1.00 (discovery group) and 0.995 (internal validation) |
*Studies with biologically defined cohorts (CSF biomarkers Aβ1-42, p-tau) to identify specifically AD groups.
Regarding lipid compounds, some of them are brain constituents, and they showed some changes at a peripheral level under AD conditions. The reviewed studies used blood samples, mainly plasma or serum. Nevertheless, they showed some controversy in their results about the relationship between some lipid classes and AD (Table 2).
3.1. Fatty Acids
Fatty acids are considered the main structural elements of complex lipids. Fatty acids can be monounsaturated (MUFAs), polyunsaturated (PUFAs), and saturated fatty acids (SFAs). PUFAs have double bonds; therefore, they are extremely sensitive to oxidative attack by reactive oxygen species compared to SFA or MUFA chains. Martín et al. [30] revealed that lipid rafts from AD brains displayed abnormally low levels of PUFAs, mainly DHA and oleic acid, at the earliest stages of AD. Whereas SFAs play an important role in activating early inflammatory pathways [39]. Kao et al. [5] found decreased SFAs and PUFAs levels in lipid rafts composition. Also, PUFAs are important in maintaining brain function (e.g., membrane fluidity, signal transduction, etc.), its structure (neural membranes), and its vascular integrity [40,41]. Some of the most important PUFAs are arachidonic acid (AA) and docosahexaenoic acid (DHA), which are precursors for the biosynthesis of the lipid mediators. AA could be metabolized by the 5-lipoxygenase (5-LO) enzyme to leukotrienes or by the cyclooxygenase (COX) enzyme to prostaglandins or thromboxane, enhancing the pro-inflam-matory pathway. DHA is considered the most abundant PUFA in all brain regions (nearly 30% of the lipid in the human brain). It is mainly obtained from the diet, although astrocytes can synthesize DHA from α-linoleic acid (ALA) [42]. It is proposed that neurons can actively regulate DHA content through an active uptake of DHA from different sources to ensure an optimal DHA pool for neuronal needs [23]. DHA is known to have anti-inflammatory, neuroprotective, and antioxidant effects. In addition, DHA reduces Aβ deposition and tau phosphorylation, thereby enhancing APP canonical cleavage by α-secretase [5]. Moreover, DHA synthesis requires oxidative conditions, corroborating its target role in oxidative stress [23].
According to the articles screened in this review, two SFAs (myristic and palmitic acid (C14:0, C16:0)) and three unsaturated fatty acids (oleic acid (C18:1), ALA (C18:3), and DHA (C22:6)) showed statistically significant lower serum levels in AD patients than in healthy controls (HC). Also, total PUFAs levels were found to be statistically lower in samples from AD patients than in HC (p<0.05) [43]. Supporting this, Olazarán et al. [44] found lower plasma levels of several free fatty acids (FFA) and some ω-3 PUFAs in mild cognitive impairment (MCI) and AD patients versus HC, while AA showed higher levels in AD groups than in HC [45, 46]. In addition, docosapentaenoic acid (DPA) showed lower levels of lysates in erythrocytes obtained from AD patients by Goozee et al. [45]. Therefore, they proposed that the AA: DPA ratio, which showed a positive trend between AA levels and neocortical amyloid load (NAL), was linked to NAL. Conversely, non-significant AA and DHA variations and a moderate decrease in eicosapentaenoic acid (EPA) were observed between HC and AD subjects (p =0.023) by Whiley et al. [47]. Interestingly, Abdullah et al. [46] demonstrated an excellent accuracy (AUC 0.91) of a panel of serum combined biomarkers, including AA, DHA, and other late-onset AD biomarkers (e.g., Aβ42/Aβ40 ratios, Apoε4 genotype) in detecting MCI caused by AD. Also, another model combining five plasma metabolites, including AA or α-aminoadipic acid (α-AAA), yielded a satisfactory discrimination capacity (AUC = 0.998, 95% CI 0.993-1.000) between amnestic MCI (aMCI) and control groups [48]. In the case of medium-chain fatty acids (MCFAs) (6-12 carbon atoms), the positive amyloid group (Aβ+) showed higher plasma levels than the negative amyloid group (Aβ-) (p<0.05) [49].
Regarding acylcarnitines (ACs), which transport fatty acids inside mitochondria for energy production, Lin et al. [50] reported lower serum concentrations in AD patients (p<0.05) than in HC, while higher levels were obtained for MCI patients (p<0.05) than HC.
In general, most fatty acids showed lower levels in MCI and AD patients than HC, mainly for ω-3 PUFAs. Specifically, DHA is the most abundant FA in the brain under normal conditions, showing a neuroprotective role. It could be a gene-diet interaction since APOE4 impairs DHA transport into the brain, thereby increasing AD risk [42]. However, AA is found to be increased in AD, while DPA is diminished. It could be explained by the fact that AA is being metabolized to lipid pro-inflammatory mediators. In addition, PUFAs have double bonds, which are susceptible to being damaged by peroxidation. Also, increased levels of MCFAs in AD patients, as well as ACs in MCI, could be attributed to the compensatory effect in response to amyloid deposition, preventing mitochondrial dysfunction. Finally, the developed models combining some of these biomarkers serve as important tools in AD patients’ detection.
3.2. Glycerolipids and Glycerophospholipids
Glycerolipids (GLs) are membrane lipids that constitute the storage form of fatty acids. They play an important role in metabolism as energy sources. Glycerol linked to two or three fatty acids forms diglycerides (DG) and triglycerides (TG), respectively.
In reviewed studies, Al-Khateeb et al. [51] did not observe differences in TG profile between AD and HC groups, so they were not considered biomarkers for early AD diagnosis. However, Leeuw et al. [52] observed an underexpression of 14 TGs in AD. Also, 54 features, including TGs, were associated with the clinical diagnosis and brain atrophy [38]. Moreover, Kim et al. [53] showed that TG 50:1 and DG 18:1_18:1 are strongly correlated with brain atrophy. In fact, by combining this data with Mini-Mental State Examination (MMSE) scores, the predicting AUC values increased to 0.833 for TG 50:1 and 0.847 for DG 18:1_18:1, suggesting these compounds as peripheral candidates to clinical diagnose AD. In addition, a combined approach identified and confirmed 35 lipid biomarkers (PCs, PEs, LPCs, TG, DAGs, SMs) that were able to distinguish any AD stage from the HC group. For this, two independent studies were carried out, obtaining suitable diagnosis indices (sensitivity 93%, specificity 80%) [54].
Regarding glycerophospholipids (GPs), which are formed by glycerolipids and a phosphate group, there are different types depending on the linked alcohol, such as phosphatidylethanolamines (PE) and the phosphatidylcholines (PC) lipid class. Some GPs showed dramatically reduced plasma levels in AD and aMCI patients in comparison to HC. Among these metabolites, it is important to highlight phosphatidylinositol (PI) (40:6) and PC (36:5), PC (37:6), PC (38:5), PC (38:6), PC (40:5) and PC (40:6) [44]. Furthermore, Kim et al. [55] observed that PC (36:5) and PC (38:6) were significantly reduced in the AD group compared to the HC group (p<0.05), while PC (40:6) did not show a significant change between groups. Thus, PC (36:5) was positively associated with hippocampal volume (p<0.01) [55], and lower blood concentration of PC (40:6) was associated with an AD-like pattern of brain atrophy [56]. Another study has reported lower levels for PC (36:6) or PC (40:6) in MCI than in HC patients. In addition, lower PC (40:2) levels were found in MCI than in AD patients (p<0.05). Interestingly, the converters MCI-AD showed lower levels of PC (40:2) and PC (40:6) at baseline than controls (p≤0.05) [57]. Also, a higher blood concentration of PC (38:4) was associated with a significantly greater risk of conversion to incident AD [56]. In addition, data provided suggested that PC (40:4) was useful to verify the clinical diagnosis with a cut-off of 0.30 μM [58]. Moreover, Arnold et al. [59] reported that serum levels for all SMs and most PCs were higher in women.
In a follow-up study, the ratio of total PCs versus lysophosphatidylcholines (LPCs) levels was two-fold higher in MCI than in AD patients, but no statistically significant differences were found between AD patients and controls. The most discriminative variables identified were three PCs (PC (32:2), PC (34:1), PC (36:5)), two LPCs (LPC (18:1), LPC (16:0)), and a sphingomyelin (SM) (SM(OH) (14:1)) [58]. Peña-Bautista et al. [60] reported a significant increase in LPC (18:1) plasma levels in early AD compared to the HC group (p<0.05).
Otherwise, reduced lysophospholipid levels were found in the serum of AD patients, and all PEs were found to be decreased in AD samples [61]. However, several PE, such as PE (36:4), PE (38:5), PE (18:0/0:0), and PE (18:1/0:0), were detected at higher levels in aMCI and AD patients than in HC [44]. In fact, a combination of PE (36:2) with the MMSE scores increased AUC values from 0.800 to 0.917 [53].
The inner part of the lipid bilayer is enriched in antioxidant lipids (PE), and at some AD stages, it could generate a response to oxidative stress damage by increasing the levels of these lipids. Although some controversial findings are found in literature, compounds such as LPC (18:1), several PE, and some PCs (PC (40:2), PC (40:6), PC (38:4), PC (36:5), PC (38:6)) could be proposed as promising biomarkers. Nevertheless, more studies are needed to support these results.
3.3. Sphingolipids
Sphingolipids (SPs) are major components of lipid rafts. Cell signaling pathways and mitochondrial function are tightly regulated by sphingolipids such as sphingomyelin, ceramide, and sphingosine-1-phosphate [3]. In fact, they can modulate the APP processing and Aβ peptides aggregation. SPs are grouped into different classes, such as ceramides (CER), gangliosides, or sphingomyelins (SMs). CER are potential products of sulfatides (synthesized by oligodendrocytes, predominantly in myelin sheath), and they are considered lipid second messenger. It could be synthesized by both neurons and glia. Accumulation of CER causes up-regulation of inflammatory cytokines, reactive oxygen species generation, mitochondrial damage, and apoptosis [62].
In general, AD patients showed high CER levels in the brain that can enhance Aβ peptides production and can mediate target-specific mitochondria degradation by autophagy [63]. Also, Aβ peptides stimulate CER synthesis, establishing a feed-forward cycle between CER-Aβ peptides. Conversely, cerebral sphingosine-1-phosphate (neuroprotective) levels are shown to decline in AD patients and negatively correlate with Aβ peptides load and NFTs number. Therefore, lower sphingosine-1-phosphate levels are associated with higher AD severity [4] (Fig. 3).
Fig. (3).
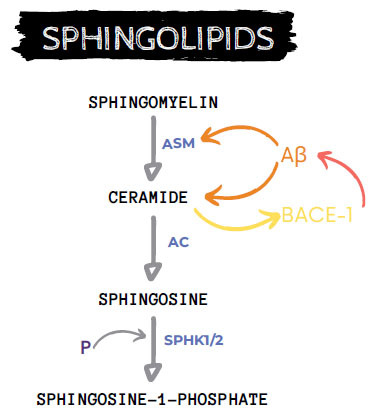
Sphingolipids metabolism cascade and AD interaction. AC: acid ceramidase, ASM: acid sphingomyelinase, Aβ: β-amyloid peptides, P: phosphate, SPHK1/2: Sphingosine kinase type 1 or 2.
Mielke et al. [64] statistically analyzed SPs levels. As a result, the highest tertiles of most CER were associated with an increased risk of AD, showing the strongest association for C16:0 (two-fold increased risk of developing AD) in men. Among women, no interesting associations were found between CER and risk of AD. In this sense, the middle tertile of SM (C18:0) and the highest tertile of some SMs (C18:1, C20:1, C22:1) were associated with an increased risk of AD in men, whereas all SMs in the highest tertile (except C24:1) showed a reduced risk of AD in women. In another study, higher blood concentrations of four SMs (SM C16:0, SM C16:1, SM (OH) C14:1, SM C18:1) were associated with a significantly greater risk of future conversion to incident AD in cognitively normal older individuals, especially SM C18:1 [56]. According to this, Leeuw et al. [52] showed that the SM (d18:1/20:1) was overexpressed in AD, while some CER (C22:0, C24:0, and C26:0) were significantly lower in the MCI group than HC and AD groups. However, no significant differences were found for CER C20:0, C22:0, and C24:0 plasma levels by Kim et al. [55]. Also, in previous studies, mean levels of CER (C16:0, C18:0) were lower in the MCI group than in the AD group, and no differences were observed for unsaturated CER (C24:1, C26:1) [65]. Recent studies have shown that plasma levels for CER (C16:0) (p<0.01), CER (C18:0) (p<0.01), and CER (C24:1) (p<0.05) were elevated in AD patients [55]. Moreover, the other seven CERs (N16:0, N21:0 at p<0.05 level and N26:0, OH-N24:1, OH-N24:2, N28:2, N23:0 at p<0.1 level) showed statistically higher plasma levels in AD than in the control group. On contrast, aMCI and AD patients showed marked decreased levels of SM (C39:1), SM (C41:1), SM(C42:1), CER (C39:1), CER (C40:1), CER (C41:1), CER (C42:1), and CER (C43:1) [44].
Therefore, several authors found an increase in CER and DHCer [66] associated with rapidly progressive dementia. High ceramide levels could be responsible for the increased susceptibility of neurons and oligodendrocytes to cell death.
Consequently, the ratios of CER: SMs appeared to discriminate AD versus controls more robustly than the differences between individual species [67]. Lower serum levels of total SMs and CER were associated with memory impairment, specifically delayed memory, while higher levels could predict future impairment [68]. The ratios of dihydrosphingomyelin/dihydroceramide (DHSM/DHCER) and SM/ CER were strongly related to cognitive decline, and they were the most sensitive predictors of progression compared to any individual molecular species of SMs or CER [69].
In general, SPs could play an important role in AD pathogenesis. Hence, different SPs ratios and some specific CER and SMs would be promising AD biomarkers.
3.4. Lipid Peroxidation Compounds
It is well-known that both inflammation and oxidative stress play a crucial role in AD development. The oxidative stress can damage double bonds of unsaturated molecules and promote the peroxidation of lipid compounds. An excess of reactive oxygen species can lead to increased lipid peroxidation within the brain, altering membrane permeability and activity of membrane receptors. For example, 4-HNE can indirectly increase NFTs in AD hippocampal tissue, modifying tau protein [70].
Reactive oxygen species generated by oxidative cell metabolism are more soluble in the fluid lipid bilayer than in the aqueous solution. Consequently, neural cell membrane lipids become primary targets of lipid peroxidation, which generates isoprostanes (IsoPs) and neuroprostanes (NeuroPs) [23].
Moreover, an oxidized phospholipid (oxPL) as 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphatidylcholine (POVC) showed two-fold higher serum levels in AD than in controls, showing an inverse correlation with cognitive function in AD. However, no differences were found in serum isoprostane concentrations in AD versus HC patients [27]. Moreover, plasma and urine F2-isoprostane levels did not correlate (p<0.0001), and no differences were observed between evaluated clinical groups [71]. Therefore, it could be stated that F2-IsoPs levels were neither associated with a higher incidence of AD nor with dementia [72]. In contrast, Peña-Bautista et al. [24] conducted a study on early AD plasma samples showing that some IsoPs and NeuroPs levels (15(R)-15-F2t-IsoP, 15-keto-15-E2t-IsoP, 15-keto-15-F2t-IsoP, 15-E2t-IsoP, 4(RS)-F4t-NeuroP, ent-7(RS)-7-F2t-dihomo- IsoP) were higher in AD patients than in HC, whereas other isoprostanoids (PGF2α, 5-F2t-IsoP, 7(RS)-ST-Δ8-11-dihomo-IsoF) showed lower values in the case group versus the control group. From these results, an MCI-AD diagnosis model was developed, considering some isoprostanoids plasma levels, as well as age and gender. Thus, the obtained sensitivity and specificity were 88.2% and 76.9%, respectively. Similarly, another MCI-AD diagnosis model was developed considering isoprostanoids levels in urine, age, and gender, with satisfactory sensitivity (80.9%) and specificity (76.9%) [73]. Hence, Peña-Bautista et al. [74] observed a significant increase (p<0.05) in some plasma compounds (8-iso-15(R)-PGF2α, 4(RS)-F4t- NeuroP, NeuroPs, IsoPs, 17-epi-17-F2t-dihomo-IsoP) in early stages of AD (subjects biologically defined by CSF biomarkers), while PGF2α showed higher levels in the control group. As a result, a negative correlation was observed between PGF2α and isoprostanes/isofurans. Finally, they reached satisfactory discrimination at the early stages of AD, improving the clinical diagnosis with a sensitivity of 72.5% and a specificity of 100%.
In addition, Leeuw et al. [52] suggested three markers of oxidative stress with strong predictive power in demarcating AD patients from HC. These markers were isoprostane-pathway derivatives (8,12-iPF-2a IV, PGD2) and the nitro-fatty acid NO2-aLA (C18:3)). Nevertheless, Padurariu et al. [26] found a statistically significant increase in serum MDA level of MCI and AD patients compared to the HC group, as well as between MCI and AD groups (p<0.0005). Furthermore, the values of hydroxyoctadecadienoic acid (tHODE) in AD patients were significantly higher than in HC, although there was a considerable overlap. Also, the plasma level of t8-iso-PGF2α in AD patients was higher than in HC, as observed from Yoshida et al. [75], yielding a sensitivity of 82% and a specificity of 71%. In general, these studies demonstrated that lipid peroxidation was higher in AD patients than in the HC group.
Therefore, promising lipid peroxidation compounds could serve as AD biomarkers since they proportionate satisfactory discrimination between groups.
3.5. Multi-metabolites Analysis
Finally, some panels combining metabolites were found in the literature. For set metabolites, including PC diacyl (aa) C36:6, PC aa C38:0, PC aa C38:6, PC aa C40:1, PC aa C40:2, PC aa C40:6, PC acyl-alkyl (ae) C40:6, LPC aa C18:2, and ACs (C3, C16:1-OH), Mapstone et al. [7] found diminished plasma levels in the AD participants, but not in HC. Nevertheless, levels were found to be similar in the aMCI and AD groups. Even so, this panel robustly identified cognitively normal individuals who would become aMCI or AD patients within two to three years with an accuracy above 90%. Casanova et al. [76] repeated the panel of Mapstone et al. [7]; however, worse results have been obtained, with an AUC of 0.64 and sensitivity/specificity of 51.6%/65.7% for discriminating baseline converter samples from non-converters in the Baltimore Longitudinal Study of Aging (BLSA) cohort, and poor values (AUC of 0.394 and sensitivity/specificity of 47/36%) in the Age, Gene/Environment Susceptibility- Reykjavic Study (AGES-RS) cohort. Also, unsatisfactory data were obtained for discriminate symptoms onset in converters versus non-converters. In the case of BLSA samples, they provided an AUC of 0.58 and sensitivity/specificity of 53.8/62.6%, whereas AGES-RS samples showed an AUC of 0.481 and sensitivity/specificity of 52/48%. In addition, phospholipids with long fatty acid chains (summed to C30 to C44 carbon-carbon bonds) appeared to be important for discriminating between pre- and post-conversion samples relative to non-converters. In the study conducted by Oresic et al. [77], the best model derived from logistic regression analysis was obtained by combining four metabolites (PC (18:0/18:2), PC (16:0/20:4), lactic acid, ketovaline), which predicted AD with an AUC of 0.77. On the other hand, Fiandaca et al. [78] designed a 24-metabolite panel, including PCs and ACs, with an AUC of 1.00 and 0.995 for the discovery and internal validation groups, respectively. It provided an improved risk assessment for subjects who developed aMCI or AD. Moreover, Toledo et al. [79] showed significant associations (p<0.05) between 13 metabolites with cognitive scores, CSF and MRI biomarker measures; specifically, six metabolites (PC ae C36:2, PC ae C40:3, PC ae C42:4, PC ae C44:4, SM (OH) C14:1, SM C16:0) were associated with CSF Aβ1–42 positivity and four compounds were associated with t-tau/Aβ1–42 ratio (C18, PC ae C36:2, SM C16:0, SM C20:2). In addition, five compounds were associated with ADAS-Cog13 scores (C14:1, C16:1, SM C20:2, α-aminoadipic acid [α-AAA], valine), and six compounds were associated with spatial pattern of abnormality for recognition of early Alzheimer’s disease (SPARE-AD) scores (C12, C16:1, PC ae C42:4, PC ae C44:4, α-AAA, valine). In all these analyses, higher ACs, PCs, and SMs values were associated with worse clinical features and biomarker measures, whereas the opposite associations were observed for valine and α–AAA values. Barupal et al. [33] proposed a total of 168 lipids from 349 that were associated with at least one AD phenotype. This study categorized all these lipids in 28 co-regulated lipid sets with heterogeneous compositions. Finally, data showed significant associations of ω-3 and ω-6 containing complex lipids with AD diagnosis and cognitive functions. Consequently, it could be interesting to use different lipid biomarkers in combined panels to improve early AD diagnosis.
Some limitations should be considered in the present review. Firstly, some studies were based on only women [53] [68] or men [72] samples. However, Arnold et al. [59] studied sex-associated differences in blood lipid levels among participants with probable AD, MCI, and HC in ADNI cohorts, revealing that sexual dimorphism of peripheral lipid levels was not significantly affected by MCI and probable AD.
Secondly, the use of CSF biomarkers improved participants’ classification, mainly for AD patients’ identification. In fact, Arnold et al. [59] identified eight significant homogeneous metabolite-phenotype associations with Aβ1-42 pathology. Moreover, Barupal et al. [33] suggested that CSF Aβ1-42 was significantly associated with 4 of their lipid sets, and CSF total tau correlated with 12 lipid sets. However, a few reviewed studies have used these diagnostic biomarkers to carry out an accurate biological classification of participants [25, 52, 59, 77, 79], ameliorating the quality of their cohorts. Most of the articles reviewed used a neurocognitive battery test to diagnose participants with either probable AD or MCI. Participants were classified according to criteria from the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) [80]. Also, internationally recognized cognition tests were completed using the mini-mental state examination (MMSE) [81] (Controls had an MMSE score of >26) or Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog) [82], while severity levels were determined by Clinical Dementia Rating (CDR) [83]. Moreover, neuroimaging studies (Magnetic Resonance Imaging (MRI) [84] or PET [85]) were performed to confirm the severity of AD-related brain atrophy.
Thirdly, a lot of articles reviewed their results for multiple medication classes or nutritional intake [52, 72, 75, 79]. For example, Al-Khateeb et al. [51] divided the cohort into 3 placebo-controlled prevention trial subgroups (43% placebo, 28% celecoxib, 29% naproxen), and no effect was observed on MCI/AD diagnosis. Also, samples included in the study conducted by Ademowo et al. [27] were recruited from a randomized, double-blind clinical trial on the effect of macular carotenoid supplementation on macular pigment, vision, and cognitive function. However, they suggest that pharmacological treatment did not have any effect on serum POVPC levels. In the case of cohort included in the study conducted by Wang et al. [43], the participants followed an isocaloric diet (45% energy from carbohydrate, 30% fat, and 25% protein for 3 days with n-6/n-3 ratio ~ 5.4) before the study and AD patients took some acetylcholinesterase inhibitors (donepezil, rivastigmine, and galantamine). In general, several co-morbidities are considered exclusion criteria of participants’ eligibility in these studies. In fact, major medical illness, such as hypercholesterolemia, hypertension, renal disease, diabetes, severe hepatic disease, severe kidney disease, disseminated cancer, alcohol or drug abuse, Down syndrome, moderate or severe cranial trauma, vascular diseases, metabolic or endocrine disturbances, and inflammation and psychiatric or neurological diseases could be considered exclusion criteria.
To summarize, the lack of studies with high-quality cohorts could difficult the identification of lipid compounds as specific AD biomarkers. Moreover, only a few studies have evaluated the potential of AD lipids biomarkers for detecting comorbidities, constituting an explanation for the lack of overall strength in these studies [86, 87].
CONCLUSION
Nowadays, an early and simple AD diagnosis and disease follow-up are crucial points in improving the prognosis and advancing the research on new effective treatments, focusing on personalized medicine. Also, aging is associated with metabolic changes, including impaired brain lipid metabolism, making the diagnosis of AD difficult. From this review, it was observed that some lipid metabolites and lipid peroxidation compounds could be a promising approach to early and minimally invasive AD diagnosis. It seems that ceramide levels are significantly higher in MCI and AD subjects than in HC, while PUFAs levels, specifically DHA, are found to be lower. Also, high AA levels and low SMs levels are found in AD patients. Nevertheless, there is still some discrepancy about which are the most interesting lipid classes and about the specific lipid species that are relevant at the different disease stages. The reviewed studies generally select well-defined cohorts and reliable analytical techniques, but there is a lack of clinically validated biomarkers. In this sense, more studies focusing on the determination of specific lipid compounds in blood samples from well-defined participants involving large samples sizes are required to develop an early and minimally invasive AD diagnosis technique. To conclude, lipids in the blood are considered a promising research field because of their potential to serve as early and minimally invasive AD biomarkers. In fact, they provided information on events, like oxidative stress, neurodegeneration, or neuroinflammation, that occur in early AD stages. Therefore, aberrant lipid metabolism pathways could play a crucial role in AD pathogenesis.
ACKNOWLEDGEMENTS
CC-P would like to acknowledge a postdoctoral “Miguel Servet” grant CP16/00082 and a FIS PI19/00570 grant from the Health Institute Carlos III (Spanish Ministry of Economy, Industry, and Innovation).
LIST OF ABBREVIATIONS
- AA
Arachidonic Acid
- ACs
Acylcarnitines
- AD
Alzheimer’s Disease
- aMCI
Amnestic Mild Cognitive Impairment
- APP
Amyloid Precursor Protein
- AUC
Area Under the Curve
- Aβ
β-amyloid Peptides
- CER
Ceramides
- CSF
Cerebrospinal Fluid
- DAG
Diacylglycerol
- DHA
Docosahexaenoic Acid
- DPA
Docosapentaenoic Acid
- HC
Healthy Control
- IsoP
Isoprostane
- MCI
Mild Cognitive Impairment
- NCI
No Cognitive Impairment
- PCs
Phosphatidylcholines
- PE
Phosphatidylethanolamine
- PUFAs
Polyunsaturated Fatty Acids
- SMs
Sphingomyelins
- SP
Sphingolipids
- TG
Triglycerides.
CONSENT FOR PUBLICATION
Not applicable.
STANDARDS OF REPORTING
PRISMA guidelines and methodologies were followed for this study.
FUNDING
This study was supported by the FIS PI19/00570 grant from the Health Institute Carlos III (Spanish Ministry of Economy, Industry, and Innovation).
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
SUPPLEMENTARY MATERIAL
PRISMA checklist is available on the publisher’s website along with the published article.
REFERENCES
- 1.Martin P.A., Wimo A., Guerchet M., Gemma-Claire Ali M., Wu Y-T., Prina M. World Alzheimer Report 2015. The global impact of dementia an analysIs of prevalence, incidence, cost and trends Alzheimer’s Disease International. London, UK: ADI; 2015. [Google Scholar]
- 2.El F., Jing P., Xia J., Cai D. Alzheimer ’ s disease risk genes and lipid regulators. Alzheimers Dis. 2016;53:15–29. doi: 10.3233/JAD-160169. [DOI] [PubMed] [Google Scholar]
- 3.Wang G., Bieberich E. Sphingolipids in neurodegeneration (with focus on ceramide and S1P). Adv. Biol. Regul. 2018;70:51–64. doi: 10.1016/j.jbior.2018.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Penke B. Bogár, F.; Fülöp, L. β-Amyloid and the Pathomechanisms of Alzheimer’s Disease: A Comprehensive View. Molecules. 2017;22(10):1692. doi: 10.3390/molecules22101692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kao Y.C., Ho P.C., Tu Y.K., Jou I.M., Tsai K.J. Lipids and Alzheimer’s disease. Int. J. Mol. Sci. 2020:21. doi: 10.3390/ijms21041505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jack C.R., Bennett D.A., Blennow K., Carrillo M.C., Dunn B., Haeberlein S.B. NIA-AA research framework: Toward a biological defi-nition of Alzheimer’s disease. Alzheimer’s and Dementia. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mapstone M., Cheema A.K., Fiandaca M.S., Zhong X., Mhyre T.R., MacArthur L.H., Hall W.J., Fisher S.G., Peterson D.R., Haley J.M., Nazar M.D., Rich S.A., Berlau D.J., Peltz C.B., Tan M.T., Kawas C.H., Federoff H.J. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014;20(4):415–418. doi: 10.1038/nm.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grasso G. Mass spectrometry is a multifaceted weapon to be used in the battle against Alzheimer’s disease: Amyloid beta peptides and beyond. Mass Spectrom. Rev. 2019;38(1):34–48. doi: 10.1002/mas.21566. [DOI] [PubMed] [Google Scholar]
- 9.Bachhuber F., Tumani H. The cerebrospinal fluid and barriers – anatomic and physiologic considerations. Handb. Clin. Neurol. 2018:146. doi: 10.1016/B978-0-12-804279-3.00002-2. [DOI] [PubMed] [Google Scholar]
- 10.Zlokovic B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011;12(12):723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hussain G., Anwar H., Rasul A., Imran A., Qasim M., Zafar S. Lipids as biomarkers of brain disorders. Crit. Rev. Food Sci. Nutr. 2019:83–98. doi: 10.1080/10408398.2018.1529653. [DOI] [PubMed] [Google Scholar]
- 12.Michalicova A., Majerova P., Kovac A., Alonso A. Tau protein and its role in blood – brain barrier dysfunction. Front. Mol. Neurosci. 2020;13(September):570045. doi: 10.3389/fnmol.2020.570045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y.C., Lauwers E., Verstreken P. Presynaptic protein homeostasis and neuronal function. Curr. Opin. Genet. Dev. 2017;44:38–46. doi: 10.1016/j.gde.2017.01.015. [DOI] [PubMed] [Google Scholar]
- 14.Rosenberg G.A. Blood-brain barrier permeability in aging and Alzheimer’s disease. J. Prev. Alzheimers Dis. 2014;1(3):138–139. doi: 10.14283/jpad.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jannic B., Karl F., Nicole L., Richard R. The neurobiology of aging and Alzheimer’s disease: walking down the same road? Eur. J. Neurosci. 2013;37(12):1885–1886. doi: 10.1111/ejn.12261. [DOI] [PubMed] [Google Scholar]
- 16.Montagne A., Barnes S.R., Sweeney M.D., Halliday M.R., Sagare A.P., Zhao Z., Toga A.W., Jacobs R.E., Liu C.Y., Amezcua L., Harrington M.G., Chui H.C., Law M., Zlokovic B.V. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85(2):296–302. doi: 10.1016/j.neuron.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sweeney M.D., Sagare A.P., Zlokovic B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018;14:133–150. doi: 10.1038/nrneurol.2017.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de la Torre J.C. Vascular risk factor detection and control may prevent Alzheimer’s disease. Ageing Res. Rev. 2010;9(3):218–225. doi: 10.1016/j.arr.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Hampel H., O’Bryant S.E., Molinuevo J.L., Zetterberg H., Masters C.L., Lista S. Blood-based biomarkers for Alzheimer disease: map-ping the road to the clinic. Nat. Rev. Neurol. 2018;14:639–652. doi: 10.1038/s41582-018-0079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding X., Zhang S., Jiang L., Wang L., Li T., Lei P. Ultrasensitive assays for detection of plasma tau and phosphorylated tau 181 in Alzheimer’s disease: a systematic review and meta-analysis. Transl. Neurodegener. 2021;10(1):10. doi: 10.1186/s40035-021-00234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thijssen E.H., Verberk I.M.W., Vanbrabant J., Koelewijn A., Heijst H., Scheltens P., van der Flier W., Vanderstichele H., Stoops E., Teunissen C.E. Highly specific and ultrasensitive plasma test detects Abeta(1-42) and Abeta(1-40) in Alzheimer’s disease. Sci. Rep. 2021;11(1):9736. doi: 10.1038/s41598-021-89004-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blennow K., Zetterberg H. Biomarkers for Alzheimer’s disease: current status and prospects for the future. J. Intern. Med. 2018;284:643–663. doi: 10.1111/joim.12816. [DOI] [PubMed] [Google Scholar]
- 23.Naudí A., Cabré R., Jové M., Ayala V., Gonzalo H., Portero-Otín M. Lipidomics of Human Brain Aging and Alzheimer’s Disease Pa-thology. Int. Rev. Neurobiol. 2015:133–189. doi: 10.1016/bs.irn.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 24.Peña-bautista C., Vigor C., Galano J., Oger C., Durand T., Ferrer I. Free radical biology and medicine plasma lipid peroxidation bi-omarkers for early and non-invasive Alzheimer’s disease detection. Free Radic. Biol. Med. 2018;124:388–394. doi: 10.1016/j.freeradbiomed.2018.06.038. [DOI] [PubMed] [Google Scholar]
- 25.Peña-Bautista C., Roca M., Hervás D., Cuevas A., López-Cuevas R., Vento M., Baquero M., García-Blanco A., Cháfer-Pericás C. Plasma metabolomics in early Alzheimer’s disease patients diagnosed with amyloid biomarker. J. Proteomics. 2019;200(January):144–152. doi: 10.1016/j.jprot.2019.04.008. [DOI] [PubMed] [Google Scholar]
- 26.Padurariu M., Ciobica A., Hritcu L., Stoica B., Bild W., Stefanescu C. Changes of some oxidative stress markers in the serum of pa-tients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 2010;469(1):6–10. doi: 10.1016/j.neulet.2009.11.033. [DOI] [PubMed] [Google Scholar]
- 27.Ademowo O.S., Dias H.K.I., Milic I., Devitt A., Moran R., Mulcahy R. Phospholipid oxidation and carotenoid supplementation in Alz-heimer’s disease patients. Free Radic. Biol. Med. 2017;108:77–85. doi: 10.1016/j.freeradbiomed.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morgado I., Garvey M. Lipids in Amyloid-β processing, aggregation, and toxicity. Adv. Exp. Med. Biol. 2015;855:67–94. doi: 10.1007/978-3-319-17344-3_3. [DOI] [PubMed] [Google Scholar]
- 29.Mattson M.P., Cutler R.G., Jo D.G. Alzheimer peptides perturb lipid-regulating enzymes. Nat. Cell Biol. 2005;7(11):1045–1047. doi: 10.1038/ncb1105-1045. [DOI] [PubMed] [Google Scholar]
- 30.Martín V., Fabelo N., Santpere G., Puig B., Marín R., Ferrer I., Díaz M. Lipid alterations in lipid rafts from Alzheimer’s disease hu-man brain cortex. J. Alzheimers Dis. 2010;19(2):489–502. doi: 10.3233/JAD-2010-1242. [DOI] [PubMed] [Google Scholar]
- 31.Rebeck G.W. The role of APOE on lipid homeostasis and inflammation in normal brains. J. Lipid Res. 2017;58(8):1493–1499. doi: 10.1194/jlr.R075408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernath M.M., Bhattacharyya S., Nho K., Barupal D.K., Fiehn O., Baillie R., Risacher S.L., Arnold M., Jacobson T., Trojanowski J.Q., Shaw L.M., Weiner M.W., Doraiswamy P.M., Kaddurah-Daouk R., Saykin A.J. Serum triglycerides in Alzheimer disease: Relation to neuroimaging and CSF biomarkers. Neurology. 2020;94(20):e2088–e2098. doi: 10.1212/WNL.0000000000009436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barupal D.K., Baillie R., Fan S., Saykin A.J., Meikle P.J., Arnold M. Sets of coregulated serum lipids are associated with Alzheimer’s disease pathophysiology. Alzheimer’s Dement. Diagnosis, Assess. Dis. Mon. 2019;11:619–627. doi: 10.1016/j.dadm.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fahy E., Subramaniam S., Brown H.A., Glass C.K., Merrill A.H., Jr, Murphy R.C., Raetz C.R., Russell D.W., Seyama Y., Shaw W., Shimizu T., Spener F., van Meer G., VanNieuwenhze M.S., White S.H., Witztum J.L., Dennis E.A. A comprehensive classification system for lipids. J. Lipid Res. 2005;46(5):839–861. doi: 10.1194/jlr.E400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 35.Fahy E., Subramaniam S., Murphy R.C., Nishijima M., Raetz C.R.H., Shimizu T. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009;50:59. doi: 10.1194/jlr.R800095-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Touboul D., Gaudin M. Lipidomics of Alzheimer’s disease. Bioanalysis. 2014;6(4):541–561. doi: 10.4155/bio.13.346. [DOI] [PubMed] [Google Scholar]
- 37.Craig-Schapiro R., Fagan A.M., Holtzman D.M. Biomarkers of Alzheimer’s disease. Neurobiol. Dis. 2009;35(2):128–140. doi: 10.1016/j.nbd.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Proitsi P., Kim M., Whiley L., Simmons A., Sattlecker M., Velayudhan L., Lupton M.K., Soininen H., Kloszewska I., Mecocci P., Tsolaki M., Vellas B., Lovestone S., Powell J.F., Dobson R.J., Legido-Quigley C. Association of blood lipids with Alzheimer’s dis-ease: A comprehensive lipidomics analysis. Alzheimers Dement. 2017;13(2):140–151. doi: 10.1016/j.jalz.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 39.Barrier B.B., Van Dyken P., Lacoste B., Talbot S., Menard C. Impact of metabolic syndrome on neuroinflammation. Front. Neurosci. 2018;12:1–19. doi: 10.3389/fnins.2018.00930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bazinet R.P., Layé S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014;15:771–785. doi: 10.1038/nrn3820. [DOI] [PubMed] [Google Scholar]
- 41.Salem N., Vandal M., Calon F. The benefit of docosahexaenoic acid for the adult brain in aging and dementia. Prostaglandins Leukot. Essent. Fatty Acids. 2015;92:15–22. doi: 10.1016/j.plefa.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 42.Patrick R.P. Role of phosphatidylcholine-DHA in preventing APOE4-associated Alzheimer’s disease. FASEB J. 2019;33(2):1554–1564. doi: 10.1096/fj.201801412R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang D.C., Sun C.H., Liu L.Y., Sun X.H., Jin X.W., Song W.L., Liu X.Q., Wan X.L. Serum fatty acid profiles using GC-MS and multivariate statistical analysis: potential biomarkers of Alzheimer’s disease. Neurobiol. Aging. 2012;33(6):1057–1066. doi: 10.1016/j.neurobiolaging.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 44.Olazarán J., Gil-de-Gómez L., Rodríguez-Martín A., Valentí-Soler M., Frades-Payo B., Marín-Muñoz J., Antúnez C., Frank-García A., Acedo-Jiménez C., Morlán-Gracia L., Petidier-Torregrossa R., Guisasola M.C., Bermejo-Pareja F., Sánchez-Ferro Á., Pérez-Martínez D.A., Manzano-Palomo S., Farquhar R., Rábano A., Calero M. A blood-based, 7-metabolite signature for the early diagnosis of Alz-heimer’s disease. J. Alzheimers Dis. 2015;45(4):1157–1173. doi: 10.3233/JAD-142925. [DOI] [PubMed] [Google Scholar]
- 45.Goozee K., Chatterjee P., James I., Shen K., Sohrabi H.R., Asih P.R., Dave P., Ball B., ManYan C., Taddei K., Chung R., Garg M.L., Martins R.N. Alterations in erythrocyte fatty acid composition in preclinical Alzheimer’s disease. Sci. Rep. 2017;7(1):676. doi: 10.1038/s41598-017-00751-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abdullah L., Evans J.E., Emmerich T., Crynen G., Shackleton B., Keegan A.P. APOE ε4 specific imbalance of arachidonic acid and docosahexaenoic acid in serum phospholipids identifies individuals with preclinical mild cognitive impairment/Alzheimer’s disease. Aging (Albany NY) 2017;9(3):964–985. doi: 10.18632/aging.101203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whiley L., Sen A., Heaton J., Proitsi P., García-Gómez D., Leung R., Smith N., Thambisetty M., Kloszewska I., Mecocci P., Soin-inen H., Tsolaki M., Vellas B., Lovestone S., Legido-Quigley C. Evidence of altered phosphatidylcholine metabolism in Alzheimer’s disease. Neurobiol. Aging. 2014;35(2):271–278. doi: 10.1016/j.neurobiolaging.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang G., Zhou Y., Huang F.J., Tang H.D., Xu X.H., Liu J.J., Wang Y., Deng Y.L., Ren R.J., Xu W., Ma J.F., Zhang Y.N., Zhao A.H., Chen S.D., Jia W. Plasma metabolite profiles of Alzheimer’s disease and mild cognitive impairment. J. Proteome Res. 2014;13(5):2649–2658. doi: 10.1021/pr5000895. [DOI] [PubMed] [Google Scholar]
- 49.Xicota L., Ichou F., Lejeune F.X., Colsch B., Tenenhaus A., Leroy I., Fontaine G., Lhomme M., Bertin H., Habert M.O., Epelbaum S., Dubois B., Mochel F., Potier M.C. Multi-omics signature of brain amyloid deposition in asymptomatic individuals at-risk for Alz-heimer’s disease: The INSIGHT-preAD study. EBioMedicine. 2019;47:518–528. doi: 10.1016/j.ebiom.2019.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin C.N., Huang C.C., Huang K.L., Lin K.J., Yen T.C., Kuo H.C. A metabolomic approach to identifying biomarkers in blood of Alz-heimer’s disease. Ann. Clin. Transl. Neurol. 2019;6(3):537–545. doi: 10.1002/acn3.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Al-khateeb E., Althaher A., Al-khateeb M., Al-Musawi H., Azzouqah O., Al-Shweiki S., Shafagoj Y. Relation between uric acid and Alzheimer’s disease in elderly Jordanians. J. Alzheimers Dis. 2015;44(3):859–865. doi: 10.3233/JAD-142037. [DOI] [PubMed] [Google Scholar]
- 52.de Leeuw F.A., Peeters C.F.W., Kester M.I., Harms A.C., Struys E.A., Hankemeier T., van Vlijmen H.W.T., van der Lee S.J., van Duijn C.M., Scheltens P., Demirkan A., van de Wiel M.A., van der Flier W.M., Teunissen C.E. Blood-based metabolic signatures in Alzheimer’s disease. Alzheimers Dement. (Amst.) 2017;8:196–207. doi: 10.1016/j.dadm.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim S.H., Yang J.S., Lee J.C., Lee J.Y., Lee J.Y., Kim E., Moon M.H. Lipidomic alterations in lipoproteins of patients with mild cog-nitive impairment and Alzheimer’s disease by asymmetrical flow field-flow fractionation and nanoflow ultrahigh performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A. 2018;1568:91–100. doi: 10.1016/j.chroma.2018.07.018. [DOI] [PubMed] [Google Scholar]
- 54.Anand S., Barnes J.M., Young S.A., Garcia D.M., Tolley H.D., Kauwe J.S.K., Graves S.W. Discovery and confirmation of diagnostic serum lipid biomarkers for Alzheimer’s disease using direct infusion mass spectrometry. J. Alzheimers Dis. 2017;59(1):277–290. doi: 10.3233/JAD-170035. [DOI] [PubMed] [Google Scholar]
- 55.Kim M., Nevado-Holgado A., Whiley L., Snowden S.G., Soininen H., Kloszewska I., Mecocci P., Tsolaki M., Vellas B., Thambi-setty M., Dobson R.J.B., Powell J.F., Lupton M.K., Simmons A., Velayudhan L., Lovestone S., Proitsi P., Legido-Quigley C. Associ-ation between plasma ceramides and phosphatidylcholines and hippocampal brain volume in late onset Alzheimer’s disease. J. Alzheimers Dis. 2017;60(3):809–817. doi: 10.3233/JAD-160645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Varma V.R., Oommen A.M., Varma S., Casanova R., An Y., Andrews R.M. Brain and blood metabolite signatures of pathology and progression in Alzheimer disease: A targeted metabolomics study. PLoS Med. 2018;15(1):e1002482. doi: 10.1371/journal.pmed.1002482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Costa A.C., Joaquim H.P.G., Forlenza O., Talib L.L., Gattaz W.F. Plasma lipids metabolism in mild cognitive impairment and Alz-heimer’s disease. World J. Biol. Psychiatry. 2019;20(3):190–196. doi: 10.1080/15622975.2017.1369566. [DOI] [PubMed] [Google Scholar]
- 58.Oberacher H., Arnhard K., Linhart C., Diwo A., Marksteiner J., Humpel C. Targeted metabolomic analysis of soluble lysates from platelets of patients with mild cognitive impairment and Alzheimer’s Disease compared to healthy controls: Is PC aeC40:4 a promising di-agnostic tool? J. Alzheimers Dis. 2017;57(2):493–504. doi: 10.3233/JAD-160172. [DOI] [PubMed] [Google Scholar]
- 59.Arnold M., Nho K., Kueider-Paisley A., Massaro T., Huynh K., Brauner B. Sex and APOE ε4 genotype modify the Alzheimer’s dis-ease serum metabolome. Nat. Commun. 2020;11(1):148. doi: 10.1038/s41467-020-14959-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peña-Bautista C., Roca M., López-Cuevas R., Baquero M., Vento M., Cháfer-Pericás C. Metabolomics study to identify plasma bi-omarkers in Alzheimer disease: ApoE genotype effect. J. Pharm. Biomed. Anal. 2020;180:113088. doi: 10.1016/j.jpba.2019.113088. [DOI] [PubMed] [Google Scholar]
- 61.González-Domínguez R., García-Barrera T., Gómez-Ariza J.L. Combination of metabolomic and phospholipid-profiling approaches for the study of Alzheimer’s disease. J. Proteomics. 2014;104:37–47. doi: 10.1016/j.jprot.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 62.García-Ruiz C., Colell A., Marí M., Morales A., Fernández-Checa J.C. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J. Biol. Chem. 1997;272(17):11369–11377. doi: 10.1074/jbc.272.17.11369. [DOI] [PubMed] [Google Scholar]
- 63.Hernandez-Diaz S., Soukup S.F. The role of lipids in autophagy and its implication in neurodegeneration. Cell Stress. 2020;4(7):167–186. doi: 10.15698/cst2020.07.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mielke M.M., Haughey N.J., Han D., An Y., Bandaru V.V.R., Lyketsos C.G., Ferrucci L., Resnick S.M. The association between plasma ceramides and sphingomyelins and risk of Alzheimer’s disease differs by sex and APOE in the baltimore longitudinal study of ag-ing. J. Alzheimers Dis. 2017;60(3):819–828. doi: 10.3233/JAD-160925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mielke M.M., Haughey N.J., Bandaru V.V., Schech S., Carrick R., Carlson M.C., Mori S., Miller M.I., Ceritoglu C., Brown T., Al-bert M., Lyketsos C.G. Plasma ceramides are altered in mild cognitive impairment and predict cognitive decline and hippocampal volume loss. Alzheimers Dement. 2010;6(5):378–385. doi: 10.1016/j.jalz.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alessenko A.V., Albi E. Exploring Sphingolipid Implications in Neurodegeneration. Front. Neurol. 2020;11:437. doi: 10.3389/fneur.2020.00437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Han X., Rozen S., Boyle S.H., Hellegers C., Cheng H., Burke J.R. Metabolomics in early Alzheimer ’ s disease: Identification of altered plasma sphingolipidome using shotgun lipidomics. PLoS One. 2011;6(7):e21643. doi: 10.1371/journal.pone.0021643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mielke M.M., Bandaru V.V.R., Haughey N.J., Rabins P.V., Lyketsos C.G., Carlson M.C. Serum sphingomyelins and ceramides are early predictors of memory impairment. Neurobiol. Aging. 2010;31(1):17–24. doi: 10.1016/j.neurobiolaging.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mielke M.M., Haughey N.J., Bandaru V.V.R., Weinberg D.D., Darby E., Zaidi N., Pavlik V., Doody R.S., Lyketsos C.G. Plasma sphingomyelins are associated with cognitive progression in Alzheimer’s disease. J. Alzheimers Dis. 2011;27(2):259–269. doi: 10.3233/JAD-2011-110405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chew H., Solomon V.A., Fonteh A.N., Martins I.J. Involvement of lipids in Alzheimer ’ s Disease pathology and potential therapies the importance of cellular lipid membranes. Front. Physiol. 2020;11(June):1–28. doi: 10.3389/fphys.2020.00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mufson E.J., Leurgans S. Inability of plasma and urine F2A-isoprostane levels to differentiate mild cognitive impairment from Alz-heimer’s disease. Neurodegen. Dis. 2010:139–142. doi: 10.1159/000289224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sundelöf J., Kilander L., Helmersson J., Larsson A., Rönnemaa E., Degerman-Gunnarsson M., Sjögren P., Basun H., Lannfelt L., Basu S. Systemic tocopherols and F2-isoprostanes and the risk of Alzheimer’s disease and dementia: a prospective population-based study. J. Alzheimers Dis. 2009;18(1):71–78. doi: 10.3233/JAD-2009-1125. [DOI] [PubMed] [Google Scholar]
- 73.Peña-bautista C., Durand T., Oger C., Baquero M., Vento M., Cháfer-pericás C. Assessment of lipid peroxidation and artificial neural network models in early Alzheimer disease diagnosis. Clin. Biochem. 2019;72:64–70. doi: 10.1016/j.clinbiochem.2019.07.008. [DOI] [PubMed] [Google Scholar]
- 74.Peña-Bautista C., López-Cuevas R., Cuevas A., Baquero M., Cháfer-Pericás C. Lipid peroxidation biomarkers correlation with medial temporal atrophy in early Alzheimer Disease. Neurochem. Int. 2019;129:104519. doi: 10.1016/j.neuint.2019.104519. [DOI] [PubMed] [Google Scholar]
- 75.Yoshida Y., Yoshikawa A., Kinumi T., Ogawa Y., Saito Y., Ohara K., Yamamoto H., Imai Y., Niki E. Hydroxyoctadecadienoic acid and oxidatively modified peroxiredoxins in the blood of Alzheimer’s disease patients and their potential as biomarkers. Neurobiol. Aging. 2009;30(2):174–185. doi: 10.1016/j.neurobiolaging.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 76.Casanova R., Varma S., Simpson B., Kim M., An Y., Saldana S., Riveros C., Moscato P., Griswold M., Sonntag D., Wahrheit J., Klavins K., Jonsson P.V., Eiriksdottir G., Aspelund T., Launer L.J., Gudnason V., Legido Quigley C., Thambisetty M. Blood me-tabolite markers of preclinical Alzheimer’s disease in two longitudinally followed cohorts of older individuals. Alzheimers Dement. 2016;12(7):815–822. doi: 10.1016/j.jalz.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Orešič, M.; Hyötyläinen, T.; Herukka, S.K.; Sysi-Aho, M.; Mattila, I.; Seppänan-Laakso, T.; Julkunen, V.; Gopalacharyulu, P.V.; Hal-likainen, M.; Koikkalainen, J.; Kivipelto, M.; Helisalmi, S.; Lötjönen, J.; Soininen, H. Metabolome in progression to Alzheimer’s disease. Transl. Psychiatry. 2011;1(12):e57–e57. doi: 10.1038/tp.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fiandaca M.S., Zhong X., Cheema A.K., Orquiza M.H., Chidambaram S., Tan M.T. Plasma 24-metabolite panel predicts preclinical transition to clinical stages of Alzheimer’s disease. Front. Neurol. 2015;6:12. doi: 10.3389/fneur.2015.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Toledo J.B., Arnold M., Kastenmüller G., Chang R., Baillie R.A., Han X., Thambisetty M., Tenenbaum J.D., Suhre K., Thompson J.W., John-Williams L.S., Mahmoudian D.S., Rotroff D.M., Jack J.R., Motsinger-Reif A., Risacher S.L., Blach C., Lucas J.E., Massa-ro T., Louie G., Zhu H., Dallmann G., Klavins K., Koal T., Kim S., Nho K., Shen L., Casanova R., Varma S., Legido-Quigley C., Moseley M.A., Zhu K., Henrion M.Y.R., van der Lee S.J., Harms A.C., Demirkan A., Hankemeier T., van Duijn C.M., Trojanowski J.Q., Shaw L.M., Saykin A.J., Weiner M.W., Doraiswamy P.M., Kaddurah-Daouk R. Metabolic network failures in Alzheimer’s dis-ease: A biochemical road map. Alzheimers Dement. 2017;13(9):965–984. doi: 10.1016/j.jalz.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McKhann G., Drachman D., Folstein M., Katzman R., Price D., Stadlan E.M. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of department of health and human services task force on Alzheimer’s disease. Neurology. 1984;34(7):939–944. doi: 10.1212/WNL.34.7.939. [DOI] [PubMed] [Google Scholar]
- 81.Folstein M.F., Folstein S.E., McHugh P.R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clini-cian. J. Psychiatr. Res. 1975;12(3):189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 82.Mohs R.C., Knopman D., Petersen R.C., Ferris S.H., Ernesto C., Grundman M., Sano M., Bieliauskas L., Geldmacher D., Clark C., Thal L.J. Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer’s Disease As-sessment Scale that broaden its scope. Alzheimer Dis. Assoc. Disord. 1997;11(Suppl. 2):13–21. [PubMed] [Google Scholar]
- 83.Hughes C.P., Berg L., Danziger W.L., Coben L.A., Martin R.L. A new clinical scale for the staging of dementia. Br. J. Psychiatry. 1982;140(6):566–572. doi: 10.1192/bjp.140.6.566. [DOI] [PubMed] [Google Scholar]
- 84.Frisoni G.B., Fox N.C., Jack C.R., Jr, Scheltens P., Thompson P.M. The clinical use of structural MRI in Alzheimer disease. Nat. Rev. Neurol. 2010;6(2):67–77. doi: 10.1038/nrneurol.2009.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nordberg A., Rinne J.O., Kadir A., Långström B. The use of PET in Alzheimer disease. Nat. Rev. Neurol. 2010;6(2):78–87. doi: 10.1038/nrneurol.2009.217. [DOI] [PubMed] [Google Scholar]
- 86.Anoop A., Singh P.K., Jacob R.S., Maji S.K. CSF biomarkers for Alzheimer’s disease diagnosis. Int. J. Alzheimers Dis. 2010;2010 doi: 10.4061/2010/606802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Blennow K., Dubois B., Fagan A.M., Lewczuk P., de Leon M.J., Hampel H. Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer’s disease. Alzheimers Dement. 2015;11(1):58–69. doi: 10.1016/j.jalz.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PRISMA checklist is available on the publisher’s website along with the published article.