


Abstract
Splicing is one key mechanism determining the state of any eukaryotic cell. Apart from linear splice variants, circular splice variants (circRNAs) can arise via non-canonical splicing involving a back-splice junction (BSJ). Most existing methods only identify circRNAs via the corresponding BSJ, but do not aim to estimate their full sequence identity or to identify different, alternatively spliced circular isoforms arising from the same BSJ. We here present CYCLeR, the first computational method for identifying the full sequence identity of new and alternatively spliced circRNAs and their abundances while simultaneously co-estimating the abundances of known linear splicing isoforms. We show that CYCLeR significantly outperforms existing methods in terms of F score and quantification of transcripts in simulated data. In a in a comparative study with long-read data, we also show the advantages of CYCLeR compared to existing methods. When analysing Drosophila melanogaster data, CYCLeR uncovers biological patterns of circRNA expression that other methods fail to observe.
INTRODUCTION
One major source of complexity in eukaryotes is splicing, whereby one gene can give rise to a number of splicing products depending on the cell’s state (tissue, developmental stage, disease state etc.). Splicing not only gives rise to linear splicing isoforms, but—interestingly—has also been recently shown to yield so-called circular splice variants or isoforms (circRNAs). These arise via a so-called back-splice junction, when a downstream 5’ splice site is covalently linked to an upstream 3’ splice site (1). The corresponding junction is called a back-splice junction (BSJ).
For decades, scientists have focused almost exclusively on linear splice variants and their functional roles. This focus, however, has recently shifted towards circRNAs (2-6). While the molecular functions of select circRNAs have already been identified (3–7), we are only beginning to understand the mechanisms by which circRNAs arise and the functional roles they play in vivo.
Since circRNAs constitute a form of alternative splicing of the nascent RNA transcript (3), the raw RNA-seq reads deriving from linear and circular splicing isoforms of one and the same gene cannot be easily used to reliably infer any new circRNAs and their abundance, see Figure 1. Only RNA-seq reads spanning a back-splice junction provide direct evidence for circRNAs. These reads, however, provide only limited information on the full sequence identity of the underlying circRNAs, thus making the proper identification and quantification of new circRNAs impossible.
Figure 1.

Challenge of identifying circRNAs from RNA-seq data. Typical, raw transcriptome data from linear and circular splicing isoforms (top left and right) comprises a multitude of pair-end reads covering the exons of these isoforms (E1 etc., colouring of pair-end reads according to the exon from which they derive). In order to infer the original splicing products from these raw transcriptome reads, they are typically first mapped to the genome (bottom). Most of the mapped reads will not cover splice sites (exon-intron boundaries) and could either derive from a linear and circular splicing isoform. One challenge is that only reads spanning a back-splice junction provide direct evidence for circRNAs (marked in light green). As is also clear from this picture, the correct identification and quantification of circRNAs cannot be achieved without the simultaneous identification and quantification of the linear splicing isoforms. Thus, if the linear splicing isoforms of a gene are known up-front, their correct quantification needs to be estimated in conjunction with the identification and correct quantification of unknown circRNAs.
The ultimate goal in transcriptome assembly is to identify the full sequence identity of all linear and circular splice variants and to estimate their relative abundances. The relative expression levels are key requirements for differential expression analyses and co-expression correlation analyses. Most existing methods for transcriptome assembly, however, ignore circRNAs and only focus on linear splicing isoforms. Commonly used transcriptome assembly programs (8,9) are based on creating a directed acyclic splice graph whose nodes and edges are determined by the presence of forward junction spanning reads in the raw transcriptome data. These programs, however, cannot handle the cyclic splice graphs that the circRNAs represent.
One added challenge is that circRNAs typically constitute only a small fraction of the transcriptome (1–3% of linear poly-A transcripts in common cell lines (10)), thus making the identification of circRNAs via an increased RNA-seq library depth no viable option. In addition, the expression of linear splicing isoforms from the same gene locus can significantly skew the circular transcript assembly. For efficient circRNA detection, there is thus the need for an enrichment of circRNAs in a sample. Due to the lack of free 5’- and 3’-ends, circRNAs are resistant to exonuclease enzymes (3,11). By using an exonuclease treatment, transcriptome libraries can thus be enriched in circRNAs. Alternatively, circRNA-enriched libraries can also be produced by poly-A depletion (12). These circRNA-enriched libraries help circRNA-identification, however, they cannot be used to improve circRNA-quantification as the depletion steps are known to effect different isoforms differently (13). A combination of treatments can lead to an almost exclusive circular RNA-seq library (14). In all of this manuscript, we refer to the RNA-seq library generated by any method for circular enrichment as circRNA enriched library. To allow for the simultaneous quantification of linear and circular transcripts and to facilitate genomic feature selection, our tool also requires a standard library of ribo-depleted total RNA-seq.
In order to enable a fair assessment of CYCLeR compared to existing tools for circRNA identification and quantification, it is key to first classify the existing methods based on their goals.
We define class I to comprise well-known circRNA tools that aim to identify and quantify circRNAs solely on reads spanning BSJs. Unlike CYCLeR, these tools do not aim to identify the full-sequence identity of new circRNAs, let alone multiple, alternatively splicing circRNAs overlapping the same BSJ (10).
Class II contains circRNA characterisation tools that take as input the predictions generated by methods from class I in order to produce a set of potential splicing events for each sequence interval defined by a BSJ. Since the alternative splicing of linear RNAs may obstruct the detection of circular alternative splicing (AS) events, class II tools require circRNA enriched libraries as input information in order to function properly. Two approaches to detect the alternative splicing of circRNAs from transcriptome RNA-seq data have emerged. The first approach is conceptionally based on exon and alternative splicing detection, using mate-pair information of pair-end reads spanning a BSJ (15,16). This approach, however, can only detect AS events within the range of the insert size used for making the RNA-seq library. The second approach compares circle enriched and total ribo-depleted libraries, similarly to CYCLeR. This strategy has the advantage of removing the dependency on the library insert size and is employed by the CIRCexplorer2 (12) pipeline. Reads spanning a BSJ typically represent only around 0.1% of a library, thereby making quantification based solely on those reads unreliable.
The challenges in circRNA quantification prompted the release of special quantification tools (defined as class III) that take as input the output of class II or class I tools and produce as output estimated circRNA levels. The tools of class III can be divided into two sub-classes. One sub-class (class IIIa) comprises tools that provide BSJ quantification as well as expression level ratios of the BSJ and FSJ in the locus of the same circRNA (13,17). While those values are reportedly in agreement with qPCR results (13,17), they do not allow to derive the relative expression levels of all alternatively-spliced linear and/or circular isoforms that happen to overlap the same BSJ and FSJ, respectively, which is the goal of our method CYCLeR. An interesting case is sailfish-cir (18), the sole member of the sub-class IIIb. sailfish-cir takes as an input a list of BSJs and known linear transcripts and makes a pseudo-linear reference of potential circRNA transcripts. sailfish-cir later uses a combination of the pseudo-linear reference and known linear transcripts to quantify linear and circular transcripts simultaneously. To conclude, all class III tools provide a circRNA abundance estimation that is only based on a BSJ, but ignore any additional, alternatively spliced circRNA isoforms that may be sharing the same BSJ.
The tools (19,20) which constitute class IV aim to recover the full-sequence identity of circRNAs. These tools also require as an input the output of tools from class I or class II. The need for BSJ identification is essential, as the experimental circRNA enrichment procedures are known to be imperfect and as some linear RNA transcripts will always remain. A BSJ set as input allows to focus only on the circRNAs in the data. The coordinates of the BSJ resolve a typical issue during linear transcript assembly - the identification of start and end exons of a transcript. One example is CIRI-full which employs an extension of the mate-pair approach of the class II tools in order to derive quantitative information on full-length circRNA isoforms. The recommended input to CIRI-full are 2 × 250 nt paired-end libraries, which allows for the full sequence-identity recovery of some circRNAs. This insert size used for making the RNA-seq library thus limits the scope of the algorithm’s output, thereby allowing only for the identification of circular isoforms of up to 600 nt length (19). The assembly strategy applied in CircAST is less ambitious, as the tool does not utilise quantitative information for the reconstruction (20). Class IV tools provide relative abundances of circRNA isoforms as counts based on fractions of the BSJ spanning reads. All class IV tools require circRNA enriched libraries as input.
To conclude, the existing methods for circRNA investigation constitute a wide set of diverse tools with distinct goals and features, see classes I to IV defined above as well as Table 1 (13,15–23). Right now, however, there exists no method for identifying the full-sequence identity of circRNAs including their potential alternatively spliced variants and for simultaneously estimating the expression levels of known linear splicing isoforms. This is the challenge that our method CYCLeR (Co-estimate Your Circular and Linear RNAs) aims to address.
Table 1.
Classification of existing methods for circRNA identification and quantification according to their goals and the input they require. Tools in column CE (circle enriched) with an entry ‘yes’ require circRNA enriched libraries as input for optimal performance
| Class | Common reference name | Practical purpose | CE* | Representatives |
|---|---|---|---|---|
| Class I | CircRNA identification tools | BSJ Identification and quantification | no | CIRI2, CIRCexplorer KNIFE, etc. |
| Class II | CircRNA characterisation tools | CircRNA AS event identification | yes | CIRCexplorer2, CIRI-AS, fuchs |
| Class IIIa | CircRNA quantification tools | Improved CircRNA quantification, based on BSJ to FSJ ratios | yes | clear, CIRIquant |
| Class IIIb | CircRNA quantification tools | Improved CircRNA quantification by using model-based framework | no | sailfish-cir |
| Class IV | Tools for full-length assembly of CircRNAs | Full-length assembly of CircRNAs and relative CircRNA isoform abundances | yes | CIRI-full, CircAST |
| Class V | - | Full-length assembly of CircRNAs and simultaneous linear and circular RNA abundance estimation | yes | CYCLeR |
| Class VI | - | Full-length assembly of circRNAs based on specifically generated Nanopore library | yes | CIRI-long, isoCirc |
By comparing total ribo-depleted (control) and circle-enriched RNA-seq libraries as input, the algorithm underlying CYCLeR first captures circle-specific features and then reconstructs full-length circRNA isoforms via a flow-based algorithm. The circRNA transcripts are then converted into a pseudo-linear isoform profile in order to estimate the abundance of linear and circular transcripts via expectation-maximisation (EM).
As CYCLeR has several unique features, we benchmark different aspects of it by comparing it to the most adequate state-of-the-art methods that share the same feature. We thus assess the assembly of circRNAs in CYCLeR separately from isoform quantification. For the assembly benchmarking, we compare CYCLeR against class IV tools as well as CIRCexplorer2 from class II. This is because CIRCexplorer2’s isoform reconstruction module deduces all potential isoforms involving the identified alternative splicing events (and thus does not account for correlated pairs of alternative splicing events such as the skipping of two neighbouring exons, or mutually exclusive exons). For benchmarking the isoform quantification of CYCLeR, only class IV tools are considered. Note that class III tools cannot be utilised for this purpose, as they have no ability to distinguish multiple circular isoforms sharing the same BSJ. For an overview of relevant existing methods, please refer to Table 2.
Table 2.
Overview of relevant circRNA transcript reconstruction and quantification tools
| Software | CircRNA feature selection | De novo feature identification | Transcript reconstruction | Transcript quantification | Flexibility* |
|---|---|---|---|---|---|
| sailfish-cir | exons within circRNA boundaries selected based on known linear annotation | — | available linear annotation is used to infer AS | EM quantification based created pseudo-linear reference | yes |
| CIRIquant | — | FSJ within circRNA boundaries selected based on hisat (36) mapping and StringTie (9) assembly of circRNA enriched libraries | — | fitting circRNA levels to a gaussian mixture model, combining circRNA enriched and total RNA-seq data | yes |
| CircAST | — | exon boundaries detected based on splice junctions derived from Tophat2 (37) mapping | minimum set of paths between BSJs that include all splice junctions | EM algorithm based on adjusted fragment length distribution | yes |
| CIRC-explorer suite | comparison between total and circle enriched RNA-seq libraries | RABT assembly with Cufflinks (8) (StringTie (9) in latter versions), based on TopHat2 (hisat (36)) mapping | statistical test to determine AS events and reconstruct all potential isoforms | clear (17) add-on quantification of circRNA as a ratio based on the levels of the most predominant equivalent linear transcript | no |
| ciri suite | exon selection based of pair-end reads | internal Perl script detecting junctions and retained introns from bwa (38) mapping | AS events are detected with statistical test based on difference of coverage between exons | transcript quantification though iterative optimisation of exon abundances within a pre-constructed splice graph (32) | no |
| CYCLeR | comparison between total and circle enriched RNA-seq libraries with DEXSeq (39) package | feature detection through SGSeq (40) package based on star (26) mapping | transcript reconstruction using a greedy algorithm on splice graph | EM quantification based created pseudo-linear reference | yes |
Abbreviations used: AS, alternative splicing; BSJ, back-splice junction; EM, expectation maximisation.* indicates that the tool is fully compatible with various BSJ identification tools.
Alternatively, circRNA isoforms can be discovered by nanopore sequencing linearized molecules that were generated via a rolling circle amplification of the corresponding circRNA (24,25). The tools that utilize this type of experimental data as input are marked as class VI. This procedure first requires linear transcript depletion ahead of rolling circle amplification of circRNA. This strategy for circRNA full-isoform identification, however, has issues with reproducibility and requires a high number of replicates to identify all circRNA isoforms (24). These methods also have known inherent biases. For example, not all known BSJs yield corresponding nanopore reads. While most of the BSJs with high splicing rates can be recovered, there is a lack of evidence for some isoforms (24,25). To summarize, conclusions drawn from this strategy are not reliable enough to serve as a reference annotation. Even with these issues fixed, this strategy would still not be able to simultaneously quantify both linear and circular transcripts. The corresponding tools that employ this strategy, however, can still serve as partial verification of the results in a comparative study between short-read circRNA assembly tools.
We make our new method CYCLeR available at https://github.com/stiv1n/CYCLeR. The repository contains information about a trial run of the core script, as well as all required command line tools which we conveniently provide as a Docker package.
MATERIALS AND METHODS
In this section, we introduce the steps and algorithms employed by CYCLeR. CYCLeR takes as an input a set of BSJs, total ribo-depleted and circRNA enriched libraries and—optionally—annotation information on linear transcripts. CYCLeR first prepares gene specific splice graphs and iteratively reconstructs potential isoforms. After CYCLeR assembles the isoforms from every sample, it prepares a complete combined set of isoforms to serve as a reference for quantification.
In the following, we explain the reasoning behind the parameters of the different benchmarking strategies. We test the assembly efficacy as well as the quantification accuracy, as is common, with a simulated dataset. We also analyse real transcriptome data to illustrate the difference of the output produced by class III tools and by CYCLeR and to highlight the advantage that full-sequence information brings to an analysis. A qPCR benchmark is used to show the advantage of CYCLeR with respect to class IIIa tools. We also conduct an exploratory transcriptome analysis to highlight the advantages of CYCLeR over class IIIb tools.
Selection of a reliable BSJ set
The detection of BSJs relies on the capability of a mapper to detect chimeric reads. Different mappers detect different sets of chimeric reads per sample. To be certain that a reliable set of BSJ sites is identified, we use CIRI2 and CIRCexplorer2 as input. They employ BWA-MEM and star for chimeric detection, respectively. Optionally, CYCLeR allows the user to manually add a set of BSJs via an additional TSV input file. Since our method requires circRNA enriched libraries as input, we can readily adjust for false positives by comparing the BSJ set identified from total RNA-seq to the set identified from circRNA enriched RNA-seq. The possibility of combining BSJ sets from various tools gives an added advantage to CYCLeR, as all comparable existing methods are limited to a single type of BSJ input, see Table 2.
Creation of circular splice graphs
CYCLeR utilises the two-pass mapping mode of star (26) to recover all split-mapped reads. These mapped reads are then utilized to identify both annotated and new genomic features such as exons, retained introns and splice junctions. For each gene, CYCLeR subsequently creates a corresponding splice graph that also contains information on feature abundances (needed for the reconstruction algorithm). Then, only the genomic features that fall between a BSJ-start and a corresponding BSJ-end coordinate (i.e. a circle generating loci) get extracted and a circle-specific splice graph is constructed. The corresponding features (exons and splice-site junctions) within this graph are then re-quantified, normalised and adjusted for GC-content and length biases. Features that are depleted in circRNA-enriched samples are detected either through a direct quantity comparison or a negative binomial test (based on replicates) and excluded from the final splice graph in order to subsequently minimise the number of false reconstructions, see Supplementary Figure S3. CIRCexplorer2 employs a similar strategy, but the statistics is performed exclusively on the junction spanning reads in comparison of a single pair of ribo-depleted and circRNA enriched libraries. CIRCexplorer2 therefore suffers from a reduced sensitivity of feature detection. CIRI-full does not require such a genomic feature filter, but its feature detection is limited by the insert size of the RNA-seq library.
Reconstruction of circRNA transcripts
One common approach towards transcriptome assembly is to use a so-called flow-based algorithm to disentangle the splice graph (9).
In CYCLeR, we employ a greedy algorithm for the iterative reconstruction of transcripts to ensure a low number of false-positive-assembled transcripts. To this end, we use the comprehensive splice graph created in the previous step and start by selecting the exon with the lowest abundance. We then identify the maximum flow though this exon inside the splice graph and reconstruct the corresponding circular transcript. The corresponding exon abundances then need to be subtracted from the respective features of the original graph and any fully depleted features be removed. These steps are repeated until no more transcripts can be reconstructed, see Figure 2 for an example. Of the relevant existing tools, only CIRI-full possesses an algorithm for the optimization of exon abundances across circRNA transcripts. Its underlying algorithm is designed to take into account a rolling circle cDNA product. We avoid these bias-inducing experimental complications in our simulated dataset, please refer to section ‘Simulated dataset’.
Figure 2.

Circle transcript reconstruction within CYCLeR for the example of the 5-HT2A gene. Starting with the full splice graph for the entire gene locus and its respective exon abundances (see top line, left, in grey for the graph and the FPKM-plot at the top right), CYCLeR extracts the circle-specific sub-graph corresponding to splice-site junction J4 which falls between a BSJ-start and a corresponding BSJ-end coordinate (see second line from top left, in blue for the graph). This blue sub-graph correponds to a single circular splicing isoform. This blue sub-graph and the corresponding exon abundances are subsequently subtracted from the original, full splice graph (see graph at the top left and middle FPKM-plot) to yield the remaining splice graph (third line from top, in grey). Similarly to before, CYCLeR then extracts the next circle-specific sub-graph, this time corresponding to a back-splice junction-spanning splice-site junction between exon E6 and E4 (fourth line from top, in red). This sub-graph provides evidence for a circular splicing isoform comprising three exons E4, E5 and E6 (note the different exon abundances). The quantities corresponding to this circular isoform are subsequently deleted from the remaining grey splice graph, resulting in a sub-graph (second line from bottom, left, grey) that corresponds to another circle-specific graph, this time comprising only exons E4 and E6, but not E5 (bottom line, in green). This sub-graph and its abundances provide evidence for a single circular isoform (bottom FPKM-plot).
Combining circRNA transcript sets across samples
Since different samples and replicates produce different sets of predicted transcripts, assembly tools often have modules for merging transcripts (8,9). In CYCLeR, transcripts from different samples are merged into a single reference, while simultaneously using the differences between sets to fix discrepancies caused by mapping artefacts. The final set of transcripts can then be exported in terms of an annotation file (in GTF format or as flat file) and a sequence file in FASTA format.
CircRNA transcript quantification
CYCLeR performs transcript quantification similarly to sailfish-cir (18). For each circRNA transcript, a corresponding pseudo-linear reference is created, for details see Supplementary Figure S5 and supplementary section ‘CircRNA transcript quantification’. This strategy allows us to account for the fact that circRNAs can produce more diverse fragments than a linear RNA with the same sequence, stemming from the sequence around the BSJ. Afterwards, the set of pseudo-linear transcripts is combined with annotated linear transcripts and used to create an index for quantification with Kallisto (27). The EM abundance estimation assigns read counts to transcripts that serve as a marker for relative quantification. False transcript reconstructions are inevitable and a portion of them are due to mapping artifacts. Conveniently, the transcripts associated with mapping artifacts have a very low number of reads assigned to them by the EM algorithm. Therefore, a hard threshold based on the number of assigned reads can be used as a secondary filter for erroneous transcript assemblies. The optimal threshold value, however, will naturally depend on the library depth, the type of library, the organism and the read length. Keeping the quantification process separate from the assembly gives CYCLeR thereby an advantage over tools that rely indiscriminately on transcript assembly.
Benchmarking with simulated data
The benchmarking of tools for transcriptome assembly and quantification is commonly based on dedicated, simulated data. Since CYCLeR aims to identify full-length circular isoforms and alternative splice variants and their abundance, we can only benchmark against the tools from class IV. In principle, CIRCexplorer 2 from class II can also be included in the benchmarking as it outputs potential transcripts derived from on all potential combinations of AS events. The parameters used for the programs are summarised in Supplementary Figure S18. Sensitivity, precision and F score are defined as usual:
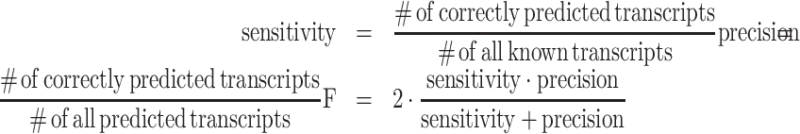 |
The quantification correlations are based on the estimated values of correctly assembled transcripts. The Pearson product correlation between estimated and simulated abundances is calculated differently with regard to the difference in the output. For CYCLeR, it is done as follows:
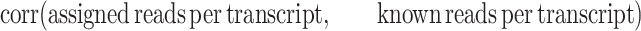 |
and CIRI-full as:
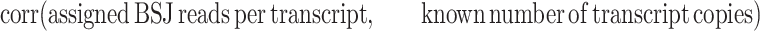 |
Simulated dataset
The transcript reconstruction in CYCLeR requires as input a total ribo-depleted and a circRNA enriched library, preferably as a pair of replicates. CIRCexplorer 2 has similar requirements. The Class IV methods require long library inserts with preferably 250 bases sequenced on both sides. With CYCLeR, we focus solely on library preparation involving RNA fragmentation in order to avoid a rolling circle amplification which would introduce unknown biases. For the benchmarking of CYCLeR and existing methods, we thus simulate two types of RNA-seq libraries: one library with a median fragment length of a 280 bp and 75 bp sequencing and one library with a median fragment length of 500 and 250 bp sequencing. From both, we simulate a pair of total ribo-depleted and circle-enriched libraries with two replicates each, for details see supplementary section ‘Simulation of RNA-seq data’ as well as Supplementary Tables S1–S3 and Supplementary Figures S1 and S2.
Benchmarking with real data
We perform a series of comparative studies on real data to illustrate the merits of CYCLeR, please refer to Supplementary Table S4 for the reference sequence and the annotation versions used.
Benchmarking with qPCR
A quantitative benchmarking involving qPCR with primers converging on one BSJ only makes sense for tools of class I and class IIIa which have no ability to detect alternative splicing isoform mapping to the same BSJ. Those tools output the relative abundance of a circRNA based on BSJ-spanning reads and do not account for AS events that may occur within the locus of the same circle. Since CYCLeR has the ability to quantify multiple, alternatively spliced transcripts per BSJ, it is important to realise that the abundance values of our method are not directly comparable to qPCR results. We thus run CYCLeR on dataset GSE75733 and PA1 total ribominus libraries from GSE73325 (12,17,28) and use the qPCR values reported in (17) for BSJs associated with a unique circular isoform. For this, we average the estimated CYCLeR abundance for two PA1 replicates and correlate it to the average qPCR value per BSJ. This comparison is made with the values reported in (17). Note that CIRIquant is not included in this benchmarking, as it cannot handle single-end RNA-seq input data.
Analysis of D. melanogaster data
As stated previously, every tool for circular transcript reconstruction relies on circRNA enriched libraries as input to function well. The lack of large, circRNA enriched data sets, however, is a major problem in the field. We chose D. melanogaster as a model organism for this study due the availability of RNase R treated samples from mature fly head, S2 cell line and early embryo (GSE69212, GSE55872) (3,29). After creating an index based on the aforementioned samples, CYCLeR quantification is performed on a data set containing 103 D. melanogaster samples by the Lai lab (30). We compare the predictions of CYCLeR to those predicted by representative methods of class I and class III. As representative for class I, we chose the BSJ identification module of CIRCexplorer 2, since this has been shown to outperform other tools in class I (13). The representatives of class IIIa produce optimal output when provided with circRNA enriched libraries for each sample. Since this dataset does not fit this requirement and as sailfish-cir is the only class III tool that outputs full transcriptome information and simultaneously quantifies linear and circular RNAs, we use sailfish-cir as representative for class III. The observed difference in performance between CYCLeR and sailfish-cir can be attributed to the lack of de novo assembly in sailfish-cir. Benchmarking against this tool allows us to highlight the advantage of circRNA transcriptome assembly and its influence on the full-isoform quantification. Note that class IV methods do not work efficiently without circRNA enriched data as indicated by simulations.
We normalise BSJ counts from CIRCexplorer 2 as counts-per-billion (CPB) and convert abundance of CYCLeR and sailfish-cir to RPKM. We variance-stabilise the corresponding values using the DESeq2 package and use them to calculate sample adjacencies (similarity) as Spearman’s rank correlation coefficient. Based on the adjacencies, we calculated sample distances with the topological overlap matrix calculation from the wgcna package (31).
Comparative study with long-read data
As previously stated, long-read circRNA sequencing protocols provide an alternative way for detecting full-length circRNA isoforms. However, as those protocols are known to have biases, the resulting data cannot serve as comprehensive benchmark. Nonetheless, the results of long-read sequencing can serve as a partial verification of the short-read results.
With this in mind, we selected the data used in the CIRI-long publication (25). This dataset contains long-read sequencing data, as well as ribo-depleted and circRNA enriched short-read data, generated from the same pool of RNAs. This test compares the results of CYCLeR CIRI-full and CIRCexplorer2 in the context of the CIRI-long results.
The different tools have varying default thresholds and parameters for assembly, which are not optimized for the high sequencing depth of this study. To put the tools on an equivalent base level of assembly parameters, we used a threshold that only circRNAs with at least five BSJ-spanning reads will be considered.
RESULTS
In this section, we show that CYCLeR outperforms all competing, existing methods based on simulated data. We also analyse D. melanogaster transcriptome data to highlight the merits of correct isoform information. Finally, we showcase the difference of output between BSJ-centric tools and CYCLeR and discuss the limitations of qPCR benchmarks.
Reconstruction of circRNA transcripts from simulated data
We had to omit CircAST from our resulting plots, because the script failed to run due to virtual memory issues, even on 400 GB RAM compute nodes of our high-performance computer cluster. We combined the transcripts reconstructed from simulated replicates into one set per tool. For CIRI-full, only the transcripts reconstructed from circRNA enriched libraries are considered.
We separate the simulated transcripts into two reference sets, a low complexity one (that serves as our reference set) and a high complexity one, see Supplementary Figure S2 and Supplementary Table S2. We show sensitivity and precision plots based on the reference set in Figure 3. Please refer to Supplementary Figure S4 for the sensitivity and precision plot of the corresponding results for the high complexity reference set. For both data sets, CYCLeR clearly outperforms the existing tools both in terms of sensitivity and precision.
Figure 3.

Comparative benchmarking of CYCLeR and comparable tools. Bar plot of sensitivity precision and F score of CYCLeR and different existing tools based on the simulated reference dataset. The superior F score for CYCLeR shows a good balance between sensitivity and precision. CYCLeR outperforms CIRI-full on all metrics. CIRCexplorer2 matches the sensitivity of CYCLeR, but the number of false positive assemblies shown by the precision measure makes CIRCexplorer2 an unreliable choice. Please also note that CYCLeR output is only marginally affected by the library read length.
As can be seen from the benchmarking, CIRI-full achieves high precision, but has only limited sensitivity. This is due to the fact that the algorithm outputs the full sequence of circRNAs only when there is no break in the coverage of its putative exons. This strategy essentially prevents the detection of any circle larger than the fragment length of the library. For the 2 × 75 bp dataset, CIRI-full covers mostly cases with single circRNA isoform. For the 2 × 250 bp dataset, the sensitivity increases, but the precision drops, due to the increased complexity of the alternative splicing landscape. CIRCexplorer2 was designed as a tool for detecting alternative splicing events in circRNAs and reports as output transcripts corresponding to all potential combinations of splice events, hence the resulting low precision.
Any analysis steps such as quantification, rely on prior knowledge of the sequence identity of the transcripts. Sensitivity and precision of the assembly are equally important for generating a useful set of splicing isoforms. We therefore also report the F score, i.e. the harmonic mean of the sensitivity and precision. As Figure 3 shows, CYCLeR significantly outperforms both CIRI-full and CIRCexplorer2 in terms of F score.
One advantage of CYCLeR is that it does not have any implicit or explicit limitations in terms of the insert sizes or the read lengths of the RNA-seq input libraries. In addition, the quantification of genomic features within CYCLeR does also not rely as strongly on high sequencing depths compared to other tools as our method solely relies on the quantification by junction reads. CYCLeR thus utilises the entire RNA-seq library for transcript assembly, not only a fraction of around 20% of the reads that happen to span splice sites. This unique feature of CYCLeR, requires a dedicated scaling and normalisation of read counts for exons which we apply. We optimise the scaling for reads spanning no more than two exons. In the 2 × 250 bp dataset, there are naturally more reads spanning multiple exons, thus leading to a decrease in performance of CYCLeR compared to the 2 × 75 bp set. With CYCLeR, we observe a minor difference between the results for the 2 × 75 bp and the 2 × 250 bp data sets. CIRCexplorer2 is less affected by the read length than CIRI-full. Nevertheless, the output decreases for the shorter length sequencing mode. This gives CYCLeR a slight advantage in sensitivity in 2 × 75 mode, while CIRCexplorer2 slightly outperforms in 2 × 250 mode. In the benchmark with the high complexity 2 × 250 bp reference set, CIRCexplorer 2 manages to outperform CYCLeR in terms of sensitivity, see Supplementary Figure S4, yet CYCLeR simultaneously significantly outperforms in terms of precision, thereby outperforming CIRCexplorer2 overall. Compared to all existing tools, CYCLeR’s strongest advantage is the graph algorithm that is tailor-made for circRNA transcript assembly. A second significant advantage is CYCLeR’s ability to be able to utilize a combination of BSJ identification tools. In its current version, we deliberately chose to exercise extreme caution in reconstruction from low abundance loci. This yields a high precision, but also filters out several low abundance features, i.e. implies a slightly reduced sensitivity. The primary advantage of CYCLeR is the assembly algorithm, yet attributing the success exclusively to the algorithm would be too simplistic. The difference in performance can be largely attributed to CYCLeR’s better genomic feature selection, which facilitates a lower number of erroneous reconstructions. In contrast to this, CIRI-full is limited to the assembly of only short circRNAs. CIRCexplorer2 performs poorly in assembly as it employs a tool for de novo linear assembly—Cufflinks/Stringtie. As these tools do not handle circRNA cases well, they induce errors that then propagate within the pipeline. CIRCexplorer2 has the additional disadvantage of being overly reliant on the annotation for detecting AS events. As additional evidence for CYCLeR’s merits, we provide the results in Supplementary Figure S13. The example shows the advantage of CYCLeR’s feature selection and how it yields a better assembly. We opted to show a case from the 2 × 250 sequencing mode, since this is optimal for CIRI-full assembly. For every tool, it is quite possible to select specific examples in the simulated data where any particular algorithm has an advantage. Nevertheless, it is clear that CYCLeR consistently outperforms in all cases with unannotated genomic features. It is important to note that the secure feature selection of CYCLeR is a statistical test that requires replicates based on the DEXSeq package. CIRCexplorer2—which uses a single pair of libraries for comparison—significantly underperforms when eliminating linear-isoform-specific features.
CircRNA transcript quantification from simulated data
Table 3 presents the quantification of circRNAs from total ribo-depleted RNA-seq and circRNA enriched RNA-seq simulated data. We designed the simulated dataset in order for the linear transcripts to provide maximum disruption for the correct processing of the circular transcripts. For this benchmark, we considered only class IV tools as only these tools are able to quantify full isoforms, not only BSJ-spanning reads. Note that CIRI-vis (32) is the tool in the final step of the CIRI-full pipeline.
Table 3.
Correlation of predicted versus simulated circRNA transcript counts. Correlation of predicted transcript abundances versus simulated. Correlations are based only on the values of correctly identified transcripts by both tools. The values are based on correlations for the transcripts of the high complexity set
| Ribo-depleted | CircRNA enriched | ||||
|---|---|---|---|---|---|
| Tool | Replicate1 | Replicate2 | Replicate1 | Replicate2 | Type |
| CYCLeR | 0.57 | 0.64 | 0.66 | 0.66 | 2 × 75 |
| CIRI-vis | 0.54 | 0.64 | 0.66 | 0.67 | 2 × 75 |
| CYCLeR | 0.84 | 0.85 | 0.87 | 0.88 | 2 × 250 |
| CIRI-vis | 0.67 | 0.66 | 0.76 | 0.74 | 2 × 250 |
The quantification benchmark is based on the high complexity dataset. The results in Table 3 are based only on the estimated quantities of the correctly identified transcripts by both tools. In this way, we judge the output of these programs as well as the influence of the precision of the assembly on the transcript quantification.
It is important to note that CYCLeR is the only existing method to simultaneously quantify both known linear and newly assembled circular transcripts.
Consistency of the assembly
To evaluate the consistency of isoform assembly between samples, we compared the sets of transcripts reconstructed from different samples. The summary is shown in Table 5. The overlap between biological replicates from the same strain diverges by about 10%. When we compare head RNA-seq libraries between strains, the difference substantially increases. When comparing the reconstructed transcripts between different stages of development or cell lines, the overlap is minimal, see Supplementary Figure S9.
Table 5.
Summary of transcript assembly between different transcriptome samples
| Sample A & Sample B | A\B | A∩B | B\A |
|---|---|---|---|
| D. melanogaster(Head) RNase R WT19: Rep1 & Rep2 | 311 | 3017 | 298 |
| D. melanogaster(Head) RNase R WT28: Rep1 & Rep2 | 199 | 2343 | 196 |
| D. melanogaster(Head) RNase R: WT19 & WT28 | 1889 | 1737 | 1001 |
| PA1 cell line: PolyA(–) & PolyA(–)/RNase R | 1003 | 6075 | 761 |
Pair-wise set difference and set overlaps between samples. Column 1 provides information on the two samples in the pair-wise comparison of reconstructed transcripts, columns 2 and 4 specify information about the number of different transcripts between samples and column 3 contains the number of overlapping transcripts.
We also compare the reconstruction from different treatments for circRNA enrichment. The PA1 cell line has available treatments, only polyA-depletion and a combination of polyA-depletion and RNase R treatment. Naturally, the sample with two types of depletion substantially increases the number of detected BSJs compared to a single type of depletion: ∼8000 versus ∼34 500. The set of reconstructed transcripts is dependent on the starting set of BSJs. We resolve the conflict by selecting a BSJ set that is derived from the total RNA-seq of the PA1 cell line (∼2500), since those are the BSJs that belong to the circRNAs quantified later. Based on that set of BSJs, we compare the sets of assembled transcripts. The difference is comparable to the different between biological replicates, please see Table 5. This leads to the conclusion that—for the generation of a general purpose set of circRNA transcripts—a single type of depletion is sufficient.
Benchmark with respect to qPCR values
The benchmark from (17) focuses on 13 BSJs. The BSJ locus of CAMSAP1 (Chr9:135881632–135883078), however, has been experimental evidence for two alternative isoforms sharing the same BSJ site, see Supplementary Figure S11 A. Based on the output from CYCLeR, we can also conclude that the BSJ locus CORO1C (Chr12:108652271–108654410) yields at least two alternative isoforms, see Supplementary Figure S11 B.
clear and CYCLeR are in very good agreement, as shown by a correlation of 0.95, see Supplementary Figure S12 A and B. The difference derives from the fact that clear uses the BSJ levels to estimate abundances, while CYCLeR uses a pseudoalignment strategy. The experimental biases that affect clear output are introduced by experimental biases in the short region around the BSJ. Therefore, the most likely sources of discrepancy are the differences in the length of the sequence and the GC-content between the full transcript sequence versus the BSJ region. Supplementary Figure S12 A and B show that the difference in GC-content can explain most of the discrepancy between clear and CYCLeR. Compared to clear, CYCLeR has a lower correlation with qPCR results (0.67 vs 0.75). As the region that is evaluated by clear mostly overlaps the region covered by the qPCR primers, the bias in GC-difference is minimized. Naturally, there are many experimental biases that can cause a difference between estimated qPCR and RNA-seq abundances. Thus, neither of the tools manages to match the qPCR results, but the poor performance of CYCLeR can be attributed to the fact that the quantification by CYCLeR is more affected by the difference in GC-content. The results of the abundance estimations can be seen in Supplementary Figure S10. Note that CIRIquant is not included in this benchmark as it cannot handle single-end RNA-seq data.
Information on the length and exon number of PA1 transcripts assembled by CYCLeR can be seen in Supplementary Figure S15A and B.
Analysis of D. melanogaster data
In addition to investigating the merits of CYCLeR with simulated data, where the isoforms and their respective abundances are known, we also explore the merits of CYCLeR and other tools on real transcriptome data. For this, we investigate RNA-seq data from D. melanogaster, see the section above for more details.
For CIRCexplorer 2, we normalise the counts as CPM, whereas we normalise those from sailfish-cir and CYCLeR as RPKM. All counts are then variance stabilised using vst from the DESeq2 package (33). It is important to note the number of BSJs that are included in this analysis, see Table 4. CIRCexplorer 2 includes all the BSJs identified in the analysis, whereas sailfish-cir filters out the BSJs that are not part of the linear annotation. CYCLeR uses the BSJs that correspond to the RNase R treated dataset.
Table 4.
D. melanogaster data set: total number of identified transcripts
| BSJs | Transcripts | |
|---|---|---|
| CIRCexplorer2 | 12 554 | – |
| sailfish-cir | 11 117 | 11 515 |
| CYCLeR | 4371 | 5659 |
Summary of the full number of BSJs and transcripts that have been identified by the corresponding tools.
Figure 4A and B shows the UMAP dimensional scaling of all 103 samples from the dataset of the Lai lab (34). There is no indication of overall loss of information due to the decrease of the BSJ set. The procedure is also repeated for sailfish-cir and shown in Supplementary Figure S7. Within the dataset, the subset with most time points corresponds to different stages during embryo development. The embryo development samples were extracted and the UMAP scaling was repeated, see also Figure 5. It is obvious that the data is heavily affected by multiple batch effects. We thus assign batches based on SRA accession numbers.
Figure 4.

Comparison of CIRCexplorer2 and CYCLeR for the D. melanogaster transcriptome sets. The comparison is made based exclusively on circRNA abundance estimations. (A) and (B) show the UMAP dimensional scaling of the abundances inferred by CIRCexplorer2’s BSJ detection module and CYCLeR for all 103 samples of the Lai dataset. (C) (CIRCexplorer2) and (D) (CYCLeR) show a dendrogram of the subset of data corresponding to embryo stages which are based on between sample distance calculations. The scale of the dendrograms represents the samples’ distances.
Figure 5.

Batch effect in Lai 2014 dataset. (A) The BSJ-spanning reads count per samples in counts per billion (CPB). (B–D) The UMAP dimensional scaling of the abundances estimated by CIRCexplorer2 BSJ detection module, sailfish-cir and CYCLeR respectively. We annotated the experimental batches based on SRA accession numbers and colour-coded them. Only the CYCLeR results reflect the underlying biological trend in the distribution of the sample points indicated by the dotted curve. The trend is not perfect, due to the influence of strong experimental biases, but sufficient to reliably identify outliers (marked with straight arrow) and to improve downstream analyses.
There are two noteworthy differences to observe. Using CYCLeR for quantification, it is possible to identify a gradient in the data that reproduces the known developmental stages. In addition, the quantification by CYCLeR makes the outliers in the data easily distinguishable. The reason for this advantage of CYCLeR derives from its variance stability between replicates, see Supplementary Figure S6. This difference can also be clearly seen within the dendrogram representing the sample distances based on circRNA transcript counts, see Figure 4C and D. The clustering by CYCLeR in Figures 4 and 5 is in complete agreement with the results from (3) depicting two segregated stages of fly embryo development, namely pre-mbl expression and post-mbl expression. The corresponding MBL protein is known to affect circRNA expression by binding intronic sequences. The most notable difference in the circRNA profile is circ-mbl (exon 2) becoming the most expressed circRNA. Overall, the distances between replicate samples inferred by CYCLeR are a significantly better reflection of their true biological relationships, emphasizing that the correct assembly of full-sequence isoforms is key for the correct clustering of biological samples.
The improved separation of samples from the embryo stage shown in Figure 5 reflects the true similarity between the D. melanogaster samples well. An example is shown in Supplementary Figure S8, where we observe that the embryo-derived cell lines have a similar circRNA pattern to early stage (0-14h) embryo samples, whereas the samples from later embryo stages (16-24h) adopt a pattern that is already similar to subsequent stages in D. melanogaster development.
Information on the length and exon number of D. melanogaster circRNA transcripts assembled by CYCLeR can be seen in Supplementary Figure S15C and D.
Comparative study with long-read data
The CIRI-long protocol has multiple steps for circRNA enrichment. This allows for the detection of circRNA isoforms beyond the capabilities of Illumina sequencing (25). There is, however, still a bias in the CIRI-long strategy. It is known that ∼50% of the BSJs recovered from Illumina data are missing in CIRI-long data. It is therefore safe to assume that this bias does not only affect the detection of BSJs, but also the AS detection within circRNA loci. Most likely, the majority of the bias is due to the 1000 nt fragment selection in the protocol, creating a bias against longer circRNAs. The CIRI-long data therefore cannot be used to determine true positives and false positives. The high recall of CIRI-long for low abundance isoforms makes it a poor benchmark for false negatives. The only reasonable statistic that can be performed is an evaluation of the overlaps of predictions between the Illumina based tools (CYCLeR , CIRI-full, CIRCexplorer2) and CIRI-long. The predictions that are supported by both—Illumina and Nanopore data—are thus considered to reflect the ground truth. What is important to track is the number of verified isoforms that are predicted by multiple Illumina-based tools (‘shared’), as well as the ones that are verified and predicted by a single tool (‘unique’). An additional significant statistic is the number of ‘unverified’ isoforms per Illumina-based tool, i.e. those isoforms that are not verified by the CIRI-long data. To compare CIRI-full on an equivalent level as the other tools, we adjust all results to account for the length limitation of CIRI-full. We thus limit all results to 2000 nt length. The full results are provided in Supplementary Figure S17. This length matches the CIRI-long limit well, as there is minor difference in the CIRI-long output in Figure 6 and Supplementary Figure S17. In Figure 6A and B, we observe the aforementioned comparison. We find that every tool manages to identify a unique set of circRNA isoforms that are verified by CIRI-long. We also observe that CIRCexplorer2 outputs very high number of isoforms compared to the other Illumina based tools. CIRCexplorer2 does provide the highest number of verified isoforms. However, the number of unverified isoforms from CIRCexplorer2 is disproportionately high.
Figure 6.

Comparative study with CIRI-long data. (A, B) The results of the comparison between Illumina-based methods and a Nanopore-based method. (A) shows a Venn diagram of the length adjusted (<2000) set of assembled transcripts for each tool. (B) A bar graph representation of the same data, but with emphasis on overlapping regions from the Venn diagram. In (B), the assembled transcripts for each Illumina-based tool are divided into verified (by CIRI-long) or unverified. The latter are further subdivided into unique—the transcripts that are shared only by one Illumina-based tool and CIRI-long, and shared—the transcripts that are shared by two or more Illumina-based tools and CIRI-long. CIRI-full has the lowest transcript count in every category. This is due to the length limit of its underlying assembly based on the library insert size. When comparing CIRCexplorer2 and CYCLeR, we notice that CIRCexplorer2 has only ∼100 more verified transcripts, while simultaneously having ∼3000 more unverified transcripts. Based on the information provided by the simulated benchmark, it is a safe assumption that the extra isoforms produces by CIRCexplorer2 are primarily erroneous assemblies.
To shed more light on the assembly results, we also provide Supplementary Figure S16 which specifies the unique BSJs that correspond to the assembled transcripts. CYCLeR has a higher number of verified isoforms than CIRI-full as well as higher number of unverified isoforms. We observe that CIRCexplorer2 has the lowest number of unique BSJs in the output. Thus, the exceptionally high number of isoforms cannot be explained by the BSJ input alone. The number of unverified isoforms produced by CIRCexplorer2 goes beyond the expectations due to biases in the experimental procedures. The logical conclusion is that CIRCexplorer2 has a high number of false positives, due to the disadvantages of the algorithm. We observe that CYCLeR has more verified results than CIRI-full. Both tools, however, have a similar ratio of ∼60% unverified isoforms. This indicates that the precision of CYCLeR and CIRI-full is comparable when adjusting for the length of the sequence. There is an increase in the number of isoforms reported by CYCLeR while the number of unique BSJs is similar to CIRI-full. This can be explained by the fact that longer circRNAs are more likely to have alternative isoforms. Thus, we conclude that the increased number of isoforms predicted by CYCLeR as compared to CIRI-full is due to the ability of CYCLeR to correctly assemble even long splicing isoforms. The advantage of CYCLeR over CIRCexplorer2 and CIRI-full can be summarized by Supplementary Figure S14. In conclusion, both CYCLeR and CIRCexplorer2 outperform CIRI-full in terms of verified transcripts. However, the number of false positives in CIRCexplorer2 makes the tool unsuitable for isoform assembly. CIRI-full is limited by the isoform length and—even with reasonable precision statistics—the tool overlooks a significant number of verified cases. To conclude, even when analysing long-read data, CYCLeR is once again the only tool with a good balance between precision and recall.
DISCUSSION
We here present CYCLeR, the first computational method for identifying the full sequence identify of new circRNAs and their abundances while simultaneously co-estimating the abundances of known linear splicing isoforms. These linear isoforms can be specified as optional input by the user.
CYCLeR outperforms existing tools for circRNA identification and quantification on all accounts on simulated data. Our reference set provides a benchmark similar to existing circRNA studies and allows us to gain a better understanding of tool-specific biases caused by the challenging mapping of chimeric fragments. Our high complexity data set was devised to represent the full complexity of real data, where the low abundances of transcripts and the overlap between multiple circular and linear isoforms can render the identification of the full isoform profile a near impossible task. When dealing with complex data, the true value of the tool becomes less centered around the recall and more focused on precision. The full set of circRNA isoforms is impossible to reconstruct due to natural limitation imposed by the depth of the RNA-seq library. Our results from the analysis of simulated data are complemented by conclusions drawn from analysing long-read data generated by Nanopore sequencing. The algorithm of CYCLeR ensures that the full sequence identity of the most abundant isoforms is correctly reconstructed. This advantage of the assembly allows for reliable quantification of the predominant isoforms. The quantification of circRNAs in CYCLeR is enhanced by the fact its assembly and abundance estimations are performed in two separate steps. This makes CYCLeR very robust, as erroneously assembled transcripts are estimated with significantly lower abundance than the correctly assembled ones. Existing tools that depend on long library insert size typically fail to detect circRNAs that fall below the detection limit of the libraries (19). In contrast to this, CYCLeR is independent of the library insert size, thereby allowing for short insert sizes and retaining the ability to identify both short and long circRNAs.
Last, but not least, the experimental workload required for transcriptome analyses performed with CYCLeR is substantially lower than that for existing methods.
A natural question that arises is whether the need for the generation of an additional set of libraries is justified by the improvement in performance. As our as well as previously published results clearly show, circRNA enriched libraries are strictly required for proper circRNA isoform assembly. Moreover, a total RNA-seq library is also strictly necessary for correct isoform quantification. An optional, additional polyA-selected library used as control could further improve the circRNA genomic feature selection. Since the total RNA-seq library can serve as control for the feature selection, however, the additional cost for an additional polyA-selected library will typically not be justified.
We show in a study of D. melanogaster transcriptomes that samples with RNase R treatment for a few key time points are sufficient to produce results that reveal biologically important relationships. We also showcase the merits of proper circRNA isoform detection for correct circRNA quantification. Finally, we find that CYCLeR improves sample clustering and facilitates outlier sample detection. This is an important feature that will play a key role in the technological transition towards single cell experiments.
The common Bioinformatics approach towards identifying circRNAs is primarily based on tracing the levels of BSJ-spanning reads across different samples. Some existing tools supplement this BSJ information by ratios of BSJ/FSJ spanning reads (13,17). This forces the user to implement a circRNA-specific analysis pipeline (13). Even these analysis pipelines, however, are still not able to distinguish between several alternatively spliced circRNAs mapping to the same BSJ. While the BSJ/FSJ ratio is very consistent with qPCR results for circRNAs comprising only two exons (17), we currently completely lack methods that are able to identify the full sequence identity of all circRNAs and their alternative splice variants in order to be able to apply standard differential expression analyses or clustering pipelines. This is the gap that our method CYCLeR fills. CIRI-full provides full isoform sequence identification, but its underlying algorithm cannot correctly reconstruct and quantify the isoforms from non-enriched data. This forces the user to perform statistical analyses of the circRNAs based primarily on the BSJ-spanning reads, even when full isoform data is available. A further disadvantage is that circular and linear transcripts need to be quantified separately and can only subsequently be combined in a co-expression network (35) which yields a biased clustering for both types of isoforms. In contrast to this, CYCLeR simultaneously quantifies linear and circular transcripts from the same sample in an integrated manner. In addition to the thus resulting superior prediction performance of CYCLeR, this feature gives the user the added advantage of being able to proceed with standard downstream pipelines for quantitative analyses for both types of isoforms. This is essential for enabling correct co-expression and simultaneous analyses of both linear and circular RNAs.
DATA AVAILABILITY
No new data were generated or analysed in support of this research.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank two out of our three reviewers for their helpful and constructive comments. I.M.M. would also like to thank Katjes Fassin GmbH + Co. KG, Germany, for their liquorice products which provided moments of joy and pleasure when these would have otherwise been lacking.
Contributor Information
Stefan R Stefanov, Berlin Institute for Medical Systems Biology, Max Delbrück Center for Molecular Medicine in the Helmholtz Association, Hannoversche Str. 28, 10115 Berlin, Germany; Freie Universität Berlin, Department of Biology, Chemistry and Pharmacy, Institute of Chemistry and Biochemistry, Thielallee 63, 14195 Berlin, Germany.
Irmtraud M Meyer, Berlin Institute for Medical Systems Biology, Max Delbrück Center for Molecular Medicine in the Helmholtz Association, Hannoversche Str. 28, 10115 Berlin, Germany; Freie Universität Berlin, Department of Biology, Chemistry and Pharmacy, Institute of Chemistry and Biochemistry, Thielallee 63, 14195 Berlin, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Helmholtz Association, Germany (to I.M.M.). Funding for open access charge: Helmholtz Association, Germany (to I.M.M.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Cape B., Swain A., Nicolis S., Hacker A., Walter M., Koopman P.. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell. 1993; 73:1019–1030. [DOI] [PubMed] [Google Scholar]
- 2. Salzman J., Gawad C., Wang P.L., Lacayo N., Brown P.O.. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012; 7:e30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ashwal-fluss R., Meyer M., Pamudurti N.R., Ivanov A., Bartok O., Hanan M., Evantal N., Memczak S., Rajewsky N., Kadener S.. Article circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell. 2014; 56:55–66. [DOI] [PubMed] [Google Scholar]
- 4. Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M.et al.. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013; 495:333–338. [DOI] [PubMed] [Google Scholar]
- 5. Li Z., Huang C., Bao C., Chen L., Lin M., Wang X., Zhong G., Yu B., Hu W., Dai L.et al.. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2016; 22:256–264. [DOI] [PubMed] [Google Scholar]
- 6. Pamudurti N.R., Bartok O., Jens M., Ashwal-Fluss R., Stottmeister C., Ruhe L., Hanan M., Wyler E., Perez-Hernandez D., Ramberger E.et al.. Translation of circRNAs. Mol. Cell. 2017; 66:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang Y., Fan X., Mao M., Song X., Wu P., Zhang Y., Jin Y.. Extensive translation of circular RNAs driven by N6 -methyladenosine. Nat. Publ. Gr. 2017; 27:626–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L.. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010; 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L.. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015; 33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Szabo L., Salzman J.. Detecting circular RNAs: bioinformatic and experimental challenges. Nat. Rev. Genet. 2016; 17:679–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jeck W.R., Sorrentino J.A., Wang K.A.I., Slevin M.K., Burd C.E., Liu J., Marzluff W.F., Sharpless N.E.. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013; 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang X.o., Dong R., Zhang Y., Zhang J.l., Luo Z., Zhang J., Chen L.l., Yang L.. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016; 26:1277–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang J., Chen S., Yang J., Zhao F.. Accurate quantification of circular RNAs identifies extensive circular isoform switching events. Nat. Commun. 2020; 11:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pandey P.R., Rout P.K., Das A., Gorospe M., Panda A.C.. RPAD (RNase R treatment, polyadenylation, and poly(A)+ RNA depletion) method to isolate highly pure circular RNA. Methods. 2019; 155:41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gao Y., Wang J., Zheng Y., Zhang J., Chen S., Zhao F.. Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat. Commun. 2016; 7:12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Metge F., Czaja-hasse L.F., Reinhardt R., Dieterich C.. FUCHS—towards full circular RNA characterization using RNAseq. PeerJ. 2017; 5:e2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ma X.K., Wang M.R., Liu C.X., Dong R., Carmichael G.G., Chen L.L., Yang L.. CIRCexplorer3: A CLEAR pipeline for direct comparison of circular and linear RNA expression. Genom. Prot. Bioinforma. 2019; 17:511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li M., Xie X., Zhou J., Sheng M., Yin X., Ko E.a., Zhou T., Gu W.. Quantifying circular RNA expression from RNA-seq data using model-based framework. Bioinformatics. 2017; 33:2131–2139. [DOI] [PubMed] [Google Scholar]
- 19. Zheng Y., Ji P., Chen S., Hou L., Zhao F.. Reconstruction of full-length circular RNAs enables isoform-level quantification. Genome Med. 2019; 11:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu J., Li Y., Wang C., Cui Y., Xu T., Wang C., Wang X., Sha J., Jiang B., Wang K.et al.. CircAST: full-length assembly and quantification of alternatively spliced isoforms in circular RNAs. Genom. Prot. Bioinform. 2019; 17:522–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang X., Wang H.b., Zhang Y., Lu X., Chen L.l., Yang L.. Complementary sequence-mediated exon circularization. Cell. 2014; 159:134–147. [DOI] [PubMed] [Google Scholar]
- 22. Szabo L., Morey R., Palpant N.J., Wang P.L., Afari N., Jiang C., Parast M., Murry C.E., Laurent L.C., Salzman J.. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015; 16:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao Y., Zhang J., Zhao F.. Circular RNA identification based on multiple seed matching. Brief. Bioinform. 2018; 19:803–810. [DOI] [PubMed] [Google Scholar]
- 24. Xin R., Gao Y., Gao Y., Wang R., Kadash-Edmondson K.E., Liu B., Wang Y., Lin L., Xing Y.. isoCirc catalogs full-length circular RNA isoforms in human transcriptomes. Nat. Commun. 2021; 12:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang J., Hou L., Zuo Z., Ji P., Zhang X., Xue Y., Zhao F.. Comprehensive profiling of circular RNAs with nanopore sequencing and CIRI-long. Nat. Biotechnol. 2021; 39:836–845. [DOI] [PubMed] [Google Scholar]
- 26. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bray N.L., Pimentel H., Melsted P., Pachter L.. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016; 34:525–527. [DOI] [PubMed] [Google Scholar]
- 28. Yang Z., Xue W., Li X., Chen L, Zhang Y., Fang H., Zhang J., Yang L., Chen L.. The biogenesis of nascent circular RNAs. Cell Rep. 2016; 15:611–624. [DOI] [PubMed] [Google Scholar]
- 29. Pek J.W., Osman I., Tay M.L., Zheng R.T.. Stable intronic sequence RNAs have possible regulatory roles in Drosophila melanogaster. J. Cell Biol. 2015; 211:243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Westholm J.O., Miura P., Graveley B.R., Lai E.C., Westholm J.O., Miura P., Olson S., Shenker S., Joseph B., Sanfilippo P.. Genome-wide analysis of Drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Reports. 2014; 9:1966–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Langfelder P., Horvath S.. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008; 9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zheng Y., Zhao F., Zhao F.. Visualization of circular RNAs and their internal splicing events from transcriptomic data. Bioinformatics. 2020; 36:2934–2935. [DOI] [PubMed] [Google Scholar]
- 33. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McInnes L., Healy J., Melville J.. UMAP: uniform manifold approximation and projection for dimension reduction. 2018; arXiv doi:18 September 2020, preprint: not peer reviewedhttps://arxiv.org/abs/1802.03426.
- 35. Ji P., Wu W., Chen S., Zheng Y., Zhou L., Zhang J., Cheng H., Yan J., Zhang S., Yang P.et al.. Expanded expression landscape and prioritization of circular RNAs in mammals. Cell Rep. 2019; 26:3444–3460. [DOI] [PubMed] [Google Scholar]
- 36. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L.. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013; arXiv doi:26 May 2013, preprint: not peer reviewedhttps://arxiv.org/abs/1303.3997.
- 39. Anders S., Reyes a., Huber W.. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012; 22:2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goldstein L.D., Cao Y., Pau G., Lawrence M., Wu T.D., Seshagiri S., Gentleman R.. Prediction and quantification of splice events from RNA-seq data. PLoS One. 2016; 11:e0156132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new data were generated or analysed in support of this research.