

Abstract
Dementia with Lewy bodies (DLB) is clinically defined by the presence of visual hallucinations, fluctuations, REM sleep behavioral disorder, and parkinsonism. Neuropathologically, it is characterized by the presence of Lewy pathology. However, neuropathological studies have demonstrated the high prevalence of coexistent Alzheimer’s disease, TDP-43, and cerebrovascular pathologic cases. Due to their high prevalence and clinical impact on DLB individuals, clinical trials should account for these co-pathologies in their design and selection and the interpretation of biomarkers values and outcomes. Here, we discuss the frequency of the different co-pathologies in DLB and their cross-sectional and longitudinal clinical impact. We then evaluate the utility and possible applications of disease-specific and disease non-specific biomarkers and how co-pathologies can impact these biomarkers. We propose a framework for integrating multi-modal biomarker fingerprints and step-wise selection and assessment of DLB individuals for clinical trials, monitoring target engagement, and interpreting outcomes in the setting of co-pathologies.
Keywords: Dementia with Lewy bodies, Lewy body dementia, α-synuclein, dementia, parkinsonism, PET, MRI, cerebrospinal fluid, neurodegeneration, clinical trials
Introduction
The principle of parsimony guides clinical diagnosis. In neurodegenerative disorders, the clinical signs and symptoms have previously been correlated with neurodegeneration in discrete and selectively vulnerable nervous system regions. A prior prevailing view has been that a combination of distinctive clinical and brain changes is characteristic of singular neurodegenerative processes [1]. However, neuropathological studies have challenged this concept, showing that cognitively impaired individuals almost invariably have multiple pathologies [2, 3]. Dementia with Lewy bodies (DLB), the second most common neurodegenerative dementia, exemplifies this, with various co-pathologies being the norm rather than the exception [4–16]. However, the clinical impact of co-pathologies in DLB has been incompletely studied, as most studies are based on autopsy retrospective clinic-pathological correlation [9, 13, 14, 17–21]. In addition, some studies have not distinguished DLB and Parkinson’s disease dementia participants, which, whilst representing different points along a Lewy body disease continuum, have differing clinical presentations and patho-aetiological mechanisms [7, 22].
DLB is clinically defined by the presence of dementia together with its core clinical features [5]: fluctuating cognition, well-formed recurrent visual hallucinations, REM behavioral sleep disorders, and parkinsonism. Research criteria now include the DLB prodromal stages for individuals with mild cognitive impairment, delirium-onset, and psychiatric-onset presentations [23]. These DLB clinical diagnostic and prodromal DLB research criteria also include indicative biomarkers tuned predominantly to impacts of α-synuclein-led neurodegeneration [5, 23]. Co-pathologies in DLB can impact clinical phenotype, disease progression, and structural and functional biomarker findings. Neurodegenerative co-pathologies are defined by the deposition of specific misfolded proteins [4, 24–33]. There are several biomarkers available to quantify Aβ and tau brain deposits in-vivo, thanks to the development of sensitive and specific biofluid and imaging biomarkers. In addition, several tissue and CSF assays are now available to detect α-synuclein presence [34–43]. Cerebrovascular pathology is the most prevalent non-neurodegenerative co-pathology with multiple manifestations that can be evaluated using MRI [2, 44, 45].
Evaluation of these co-pathologies may offer the opportunity to better predict clinical progression, enable individualized approaches to treatment, and improve the clinical trial design. Here, we evaluate the clinical features and presentation of DLB in the context of underlying co-pathologies and emerging biomarkers to their potential to quantify neuropathological change. We then consider the implications of these findings for future clinical trials.
Neuropathology and Prevalence of Co-Pathologies
DLB’s hallmark is Lewy pathology, which encompasses Lewy bodies and Lewy neurites, defined by the presence of misfolded α-synuclein. Lewy pathology is also the hallmark lesion of Parkinson’s disease (PD), and together both conditions are collectively classified as Lewy body disease. The DLB neuropathological criteria enable the evaluation of the likelihood of Lewy pathology leading to a DLB presentation [5]. However, the amygdala-predominant and brainstem stages do not lead to a DLB clinical presentation. DLB prevalence increases with age, but aging is also associated with an increased prevalence of multiple neurodegenerative and cerebrovascular diseases that contribute to cognitive impairment [2, 46]. Therefore, it is not surprising to observe multiple brain pathologies in aging individuals with cognitive and motor disorders [2, 3, 47]. In addition, neurodegenerative and cerebrovascular pathology have different distribution patterns and injury mechanisms, further adding to the heterogeneity of brain changes [44, 48–53]. The prevalence of these pathologies is outlined below and summarized in Table 1.
Table 1.
Co-pathology in dementia with Lewy bodies
| Type of Neurodegenerative Condition | Type of Pathology | Prevalence in DLB |
|---|---|---|
| Alzheimer’s disease | Amyloid-beta (Aβ) plaques Tau neurofibrillary tangles |
48–88% of individuals with DLB have intermediate- or high-level AD pathology versus 17–62% in PDD and 7–10% in those with PD who are dementia free [7–10]. AD pathology is associated with a greater Lewy pathology burden [51]. |
| TDP-43 and limbic-predominant age-related TDP-43 encephalopathy (LATE) | TDP-43 | 13–60% of individuals with DLB [11–15] More prevalent in the advanced neocortical Lewy pathology (LP) stages, in the presence of AD co-pathology in DLB vs. individuals with PD who are dementia-free [11, 13, 15] |
| Frontotemporal lobar degenerations Tau (FTLD) | Tau | Less prevalent compared to other co-pathologies |
| Other Tau-related neurodegenerative conditions, including aging-related Tau astrogliopathy (ARTAG) | Tau | Not studied in DLB alone In Lewy body disease overall (PD + DLB): 31% of participants without AD co-pathology [22] 72% in the presence of AD co-pathology [22] |
| Cerebrovascular Pathology | Microinfarcts Gross infarcts |
Neuropathological samples [16] Microinfarcts 26.7% Gross infarcts 6.7%16 MRI-based samples] [57, 62 18.8–27% |
| Cerebral amyloid angiopathy (CAA) |
Neuropathological samples CAA 66.7% none to intermediate AD neuropathological changes versus 94.7% CAA prevalence in individuals with high AD neuropathological changes [13] CAA is highest in DLB (82%−91%), followed by PDD (50%), and lowest in cognitively unimpaired PD (21.7%) [13, 17] |
|
| Cerebral microbleeds (usually associated with CAA) | Are also more frequent in DLB (30%−45.2%) than in PDD (26.1%), PD (11.5%), and control participants (17.1%) [58–60]. However, one study that compared participants with DLB with and without microbleeds did not find differences in florbetapir binding and found that high systolic blood pressure was the factor associated with microbleeds in DLB [61] | |
| White matter hyperintensities (WMH) burden | It may be ↑ in DLB vs. healthy controls [59, 62, 66–68], although reports are inconsistent [69–71] |
Alzheimer’s Disease Pathology
Alzheimer’s disease (AD) co-pathology, defined by the presence of amyloid-beta (Aβ) plaques and tau neurofibrillary tangles, is present in more than 50% of DLB individuals, with a higher prevalence compared to both Parkinson’s disease (PD) and PD dementia (PDD) [6–10]. AD has been linked to a greater Lewy pathology burden [22, 51, 54] and is the only co-pathology considered when interpreting neuropathological findings in DLB that is recognized in the 2005 and 2017 guidelines [5, 55].
TDP-43 Pathology
Previous studies reported a wide prevalence range of TDP-43 pathology in DLB. Study differences are likely the result of differences in sampled areas and the definition of additional TDP-43 co-pathology. TDP-43 co-pathology in DLB follows an anatomical distribution consistent with limbic-predominant age-related TDP-43 encephalopathy (LATE) [12, 13, 27]. TDP-43 pathology burden is associated with greater Lewy pathology burden and the presence of AD co-pathology, exemplifying the complex interrelations between the different neuropathologies.
Frontotemporal Lobar Degeneration Tau and other Neurodegenerative Conditions
Frontotemporal lobar degeneration (FTLD) Tau pathology is less prevalent [22], and likely plays a minor role as a DLB co-pathology. Aging-related Tau astrogliopathy (ARTAG) is prevalent in individuals with dementia [56], including Lewy body disease (PD and DLB combined) [22].
Cerebrovascular Pathology
Cerebrovascular pathology inversely correlates with Lewy pathology [16] and DLB clinical features [57], which indicate that a lower threshold of neurodegenerative pathology may be necessary for clinical dementia expression in the presence of cerebrovascular co-pathology. Autopsy studies have reported a high prevalence of cerebral amyloid angiopathy (CAA) in up to two-thirds of individuals with DLB [13]. Increased CAA is more prevalent in DLB individuals with AD co-pathology. Within the Lewy body disease group, CAA is highest in DLB, followed by PDD, and lowest in cognitively unimpaired PD [13, 17]. Similarly, microbleeds are more frequent in DLB than in PDD, PD, and control participants [58–60]. In one neuroimaging study, microbleeds were associated with the systolic blood pressure but not with amyloid-beta (Aβ) PET values [61]. Compared to AD, DLB showed a similar burden of microbleeds [58]. There are inconsistent findings regarding the prevalence of infarcts in DLB compared to controls [16, 62], which might result from differences in the inclusion criteria of studies. A recent review showed no relationship between large cortical or small subcortical infarcts or intracerebral hemorrhage and the presence of Lewy body dementia [45]. Nevertheless, the same review showed increased MRI-assessed white matter hyperintensity (WMH) burden in individuals with DLB compared to cognitively unremarkable participants, which was consistently supported by neuropathological data. An ongoing challenge is that whilst WMH are considered a marker of cerebrovascular disease, other etiologies, including AD-associated pathology and axonal loss, may associate with WMH [63–65].
Clinical Impact of Co-pathologies in DLB
Co-pathologies are not only prevalent, but they will also likely affect the clinical presentation and disease progression by compounding brain dysfunction. Therefore, the impact of each co-pathology needs to be evaluated to predict clinical progression and understand the outcomes of treatments.
Cross-Sectional Clinical Associations of Co-Pathologies in DLB
From a cognitive standpoint, DLB individuals with an AD biomarker profile show poorer performance in memory and orientation tests than those without AD [72]. AD is the co-pathology with the greatest clinical impact, and pathological Aβ and tau levels have been related to worse global cognition [73–77]. Conversely, there are conflicting results regarding the effects of AD biomarkers values on the clinical presentation and core features of DLB. Several studies have reported that a higher burden of AD co-pathology or abnormally phosphorylated tau (p-tau) cerebrospinal fluid (CSF) levels decrease the odds of presenting core DLB features [9, 73, 78]. [79]. However, Aβ and tau PET studies have not found associations between AD biomarker positivity and DLB core features [74, 80, 81]. One study identified a higher frequency of visual hallucinations in DLB individuals with an AD CSF profile [82].
Limited data exist on the implications of TDP-43 co-pathology on clinical phenotype in DLB. One recent study found that individuals with TDP-43 co-pathology had a lower likelihood of presenting visual hallucinations and parkinsonism and were, therefore, less frequently diagnosed as probable DLB during life [15]. Although the presence of TDP-43 is also associated with older age and a higher likelihood of concomitant tau deposition, the lower likelihood of a clinical DLB diagnosis in individuals with Lewy pathology and TDP-43 co-pathology persisted even when considering these factors [15].
As noted above, cerebrovascular lesions in DLB correlate negatively with the severity of Lewy pathology [16, 83]. This association is consistent with cerebrovascular lesions lowering the threshold for dementia in individuals with AD and Lewy pathology [2]. However, the evaluation of WMH associations has led to conflicting results. WMH burden has shown inconsistent associations with visual hallucinations [57, 62], which may suggest that the location of the WMH makes a contribution [66, 84]. Several studies found no overall association of WMH with cognition [66, 69, 70, 84]. However, some studies point to the importance of WMH, which affect cholinergic white matter pathways and a modulating effect of apolipoprotein E (APOE) ε4 [57, 85, 86]. Parkinsonism and cognitive fluctuations are not associated with WMH burden [57]. The etiology of WMH in MR has also been debated and may reflect axonal degeneration due to cortical neuronal loss rather than ischemia/small vessel disease per se [65].
Limited information is available on the impact of ethnicity on DLB. One study reported a higher prevalence of Lewy pathology in African Americans than whites, although the study did not specifically evaluate DLB [87]. No results are available specifically considering co-pathologies. In a study assessing sex differences, AD co-pathology led to a lower frequency of RBD and parkinsonism in men and women, with men also presenting with a lower frequency of cognitive fluctuations and visual hallucinations in the presence of AD co-pathology [88]. How the presence of co-pathologies impacts caregivers is unknown.
Longitudinal Associations of Co-Pathologies in DLB
DLB individuals with an AD CSF profile have a faster cognitive decline [89]. MMSE decline correlated with Aβ plaques, neurofibrillary tangles, and Lewy pathology [90]. The age of onset of dementia is lower in patients with high tau and amyloid beta [90, 91]. Overall, shorter survival appears to be linked with increased Aβ pathology [8, 92, 93], with a lower impact of tau pathology [8, 93]. The severity and distribution of Lewy pathology also have an effect; DLB patients with diffuse neocortical and occipital Lewy pathology showed a more rapid disease course than those with brainstem and limbic Lewy pathology [51, 93].
Implications of Genetic Findings
DLB shares its genetic risk factors with AD (APOE) [94–97] and PD (α-synuclein -SNCA- and β-glucocerebrosidase -GBA-) [96, 97]. However, different regions within SNCA have been associated with PD and DLB [96, 98], and APOE remains significantly associated with DLB in individuals with no or low burden of AD pathology [95]. Most of DLB’s heritability is based on genetic risk variants associated with a small increase in DLB risk, with recent studies showing rare monogenic pathogenic mutations [99]. Clinically, APOE ε4 is associated with a faster disease progression and shorter survival in DLB [100, 101]. Future studies will need to evaluate the pathological changes that mediate this progression. Conversely, AD co-pathology is less prevalent in DLB individuals with GBA mutations [7]. These results indicate that genetic risk factors play an important role in the frequency of co-pathologies and clinical outcomes in DLB.
Biomarkers in the Setting of DLB Co-pathologies
An inherent limitation of the neuropathological studies is the cross-sectional evaluation of pathology at the time of death, which does not inform when these pathologies appear and how they interact during the disease’s course. The recent explosion of available biomarkers has made it more possible to assess co-pathologies in vivo, enabling their detection even at prodromal disease stages. Biomarkers can be classified based on their modality or the pathological feature they quantify. From a modality perspective, biomarkers can be subdivided into neuroimaging, biofluid, neurophysiological, tissue-based (biopsy), and technology-based objective measures. Biomarkers can also be broadly divided into disease-specific and disease-nonspecific regarding the measured pathological feature. Disease-specific biomarkers include biomarkers that quantify specific changes to an underlying pathology, like Aβ biomarkers. Conversely, disease non-specific biomarkers measure changes that are not specific to a pathology, like brain atrophy evaluated using structural MRI sequences. We will discuss the utility of the biomarkers based on the pathological feature they measure because this aligns better with their role within a diagnostic and outcomes framework. These biomarkers are described below and summarized in Table 2.
Table 2:
Biomarkers in the Setting of DLB Co-Pathologies
| Pathological change | Biomarker modality | Implications in dementia with Lewy bodies |
|---|---|---|
| Disease-Specific Biomarkers | ||
| Aβ | CSF and PET biomarkers Plasma assay [109] |
Both modalities accurately predict the presence of Aβ plaques in the brain [102, 133] and are in good agreement with each other [103]. Conversion to pathological values of CSF Aβ tends to precede PET abnormalities [134]. |
| Tau | CSF and PET biomarkers Plasma assay [110] |
Correlates with tau brain deposition [102, 135]; typically detect neurofibrillary tangle pathology present in AD rather than FTLD-Tau pathology [136, 137]. New tracers are being developed to quantify FTLD-Tau pathology [138]. |
| α-synuclein | CSF real-time quaking-induced conversion (RT-QuIC) [39, 42] and protein misfolding cyclic amplification (PMCA) [43] Skin [34–36, 39, 41] Olfactory Mucosa [37–39] Submandibular gland [39, 40] Colon [39] |
Binary detection of the presence/absence of α-synuclein only. Less useful to quantify disease progression. Further studies are needed in DLB. |
| Disease Nonspecific Biomarkers | ||
| Synaptic loss | CSF and blood biomarkers [113] | Structural and functional imaging alterations are strongly correlated with fluid-based biomarkers of synaptic and neurodegenerative tissue change [139]. |
| Brain atrophy | Imaging techniques using either MRI | Correlates with the degree of cognitive impairment and is known to play a significant role in disease presentation and progression [114]. Present in brain aging and can follow a different pattern in cognitively impaired individuals [140, 141]. |
| Neuronal dysfunction and damage | CSF and blood biomarkers [128, 142] | Structural and functional imaging alterations strongly correlate with fluid-based biomarkers of synaptic and neurodegenerative tissue change [139] and can track change in-vivo [113]. |
| Glial activation | CSF and blood biomarkers [130] | |
| Structural and Functional Imaging Biomarkers | ||
| Relative preservation of medial temporal lobe structures | MRI CT |
Included in the diagnostic criteria for DLB [5]. |
| Reduced basal ganglia dopamine transporter uptake | SPECT | |
| Increased focal and diffuse abnormalities | EEG | |
| Reduced occipital metabolism/cingulate island sign | FDG-PET | |
| Insular thinning and gray matter atrophy | MRI | Reported as requiring further investigation in the research criteria for prodromal DLB. |
| Medial temporal lobe structures atrophy | Visual rating scales | Greater atrophy in multiple brain regions than in controls and relative preservation of medial temporal lobe structures compared to AD [143–145]. AD co-pathology is associated with greater medial temporal lobe atrophy in DLB, demonstrating that AD co-pathology (and possibly TDP-43) modifies imaging patterns in people with DLB [143, 146, 147]. |
| Abnormalities in the cholinergic system | Volumetry and cortical thickness quantifications | Atrophy of nucleus basalis of Meynert in prodromal MCI stages [148] Associations between nucleus basalis of Meyner atrophy, Aβ, and cognitive changes differ in AD and DLB [149]. |
| Limbic TDP-43 co-pathology | RT-QuIC [42] | Studies are needed in DLB. |
| Higher WMH burden | MRI | Associated with more neurodegeneration in DLB [57, 67, 150], particularly in brain areas that receive dense cholinergic input [57], which may have implications for the cholinergic treatment of DLB. |
| Structural connectivity changes | Diffusion tensor imaging (DTI) technique and higher tensor modeling techniques such as fixel-based analysis | Seen in at-risk groups for PDD [118]. Studies are needed in DLB. |
| Quantitative susceptibility mapping Quantitative multiparameter maps |
Applied in PD [120], revealing a correlation with clinical change. Studies are needed in DLB. |
|
| Temporoparietal and occipital hypometabolism | 18-Fluorine 18F fluorodeoxyglucose (FDG) and SPECT perfusion imaging | Further studies are needed in DLB with co-pathologies. |
| “Cingulate island sign” | FDG-PET | May indicate a lower Braak neurofibrillary tangle stage at autopsy [123]. |
| Abnormalities in serotonergic systems | Volumetric measures and PET radiotracers | Further studies are needed in DLB with co-pathologies. |
| CSF and blood markers of neurodegeneration, synaptic dysfunction, and glial activation | ||
| Neuro-axonal damage | Neurofilament Light (NfL) | Elevated already in prodromal DLB stages, higher levels in the presence of AD co-pathology [151]. |
| Glial-related change | Glial fibrillary acidic protein (GFAP), the soluble triggering receptor expressed on myeloid cells 2 (sTREM2, mainly expressed by microglia), and S100 calcium-binding protein B (S100B, mainly expressed by astrocytes). | Elevated in DLB compared to controls [130]. |
| Synaptic dysfunction | Fluid-based biomarkers: synaptosomal-associated protein 25 (SNAP-25), Synaptogamin-1, neurogranin, and β-synuclein. | Further studies are needed in DLB with co-pathologies. |
Disease-Specific Biomarkers
Disease-specific biomarkers quantifying Aβ, tau, TDP-43, and α-synuclein are valuable biomarkers with the potential to indicate the presence of each co-pathology. Traditionally these biomarkers included CSF assays and PET scans [102–104], although, despite recent advances, there is still a lack of a sensitive and specific ligand for α-synuclein. Several studies show that PD individuals present lower CSF tau levels than controls [105–107]. It is unclear what is the reason for this phenomenon or whether this occurs in DLB. Further studies with autopsy validation will need to evaluate if different CSF tau biomarker cutpoints are needed in Lewy body disease, including DLB. Initial flortaucipir PET studies in DLB individuals indicate higher binding than healthy controls and cognitively unremarkable PD individuals, with a wide range of binding and correlate with MMSE scores [80, 108]. The newly developed plasma Aβ and tau assays will offer less invasive, easily deployable biomarkers [109–111]. Plasma tau levels are elevated in DLB individuals with pathological CSF Aβ values, and higher levels also predict worse baseline cognition and faster cognitive decline in the same individuals [110]. CSF real-time quaking-induced conversion (RT-QuIC) and protein misfolding cyclic amplification (PMCAand α-synuclein [42]assays can detect α-synuclein [43]. One manuscript presented a CSF TDP-43 RT-QuIC assay [112]. Initial studies in PD participants have shown high sensitivity and specificity of α-synuclein seeding assays, which have been tested in multiple tissues and biofluids with variable sensitivities [34–40], in addition to CSF. Some of these studies have also included DLB participants showing high accuracy in CSF and skin samples [36, 39]. In addition, skin α-synuclein immunohistochemical evaluation can accurately classify PD individuals [41].
Disease-Nonspecific Biomarkers
Disease-nonspecific biomarkers can measure synaptic loss, brain atrophy, neuronal dysfunction, and glial activation across different neurodegenerative diseases. These changes correlate with the degree of cognitive impairment in dementia and are known to play a significant role in disease presentation and progression [4, 113]. However, these biomarkers reflect changes secondary to a range of pathologies either in singular or in combination and must be interpreted accordingly in clinical practice and trials. These characteristics make these biomarkers suitable for investigating the underlying pathology’s impact on DLB [4, 114].
Structural and Functional Imaging Biomarkers
Multiple structural and functional approaches have been evaluated in DLB [114], including MRI, CT, SPECT, EEG, and FDG-PET. As noted previously, biomarkers are now included in the diagnostic criteria for DLB [5] and, more recently, the research criteria for prodromal DLB criteria [23]. These neuroimaging modalities capture downstream, cumulative changes resulting from the combined pathologies present in DLB individuals (or any other dementia). The processed images can be scored using visual rating scales and easily performed in the routine clinical setting. Conversely, more quantitative measures like volumetry and cortical thickness quantifications are more sensitive techniques [115] and are desirable as outcome measures in clinical trials. They have been used to evaluate differences in brain atrophy patterns in people with DLB with and without underlying AD co-pathology [116, 117]. Neuroimaging implications of limbic TDP-43 co-pathology in DLB remain to be studied.
Changes in structural connectivity are evaluated using the diffusion tensor imaging (DTI) technique and higher tensor modeling techniques such as fixel-based analysis. These techniques have been applied in at-risk groups for PDD [118]; whether this approach is sensitive to tau and other co-pathologies within the DLB spectrum is still a topic of debate [119]. Emerging techniques sensitive to tissue changes, such as quantitative susceptibility mapping or quantitative multiparameter maps, have not yet been directly evaluated in DLB alongside pathological data but show greater promise based on correlations with clinical measures in PD [120]. Similarly, MRI substantia nigra free water values are increased in PD and atypical parkinsonism and could serve as diagnostic and imaging outcome measures [121, 122], but further work is needed in DLB.
Beyond structural changes, functional MRI and metabolic/perfusion nuclear medicine imaging techniques can track dysfunction in specific brain regions, including temporoparietal and occipital hypometabolism in DLB seen on 18F fluorodeoxyglucose (FDG) and SPECT perfusion imaging. The “cingulate island sign” (occipital hypometabolism with relative sparing of the posterior cingulate cortex) appears to distinguish those with DLB from other dementias [81]. It could indicate either nonsignificant or a low burden of AD co-pathology [123]. Different metabolic patterns have already been used to detect different underlying pathologies and progression risks in PD [124, 125]. However, their use is still limited in the DLB and needs further evaluation.
CSF and blood markers of neurodegeneration, synaptic dysfunction, and glial activation
Several non-specific neurodegenerative biomarkers have been developed to evaluate axonal damage, glial involvement, and synaptic dysfunction in CSF and blood samples.
Axonal damage can be quantified by measuring the neurofilament light chain (NfL), a structural component of the neural cytoskeleton. In axonal injury, NfL is released into the extracellular space leading to its increase in CSF and plasma [126, 127]. Higher CSF and blood NfL levels are already present in the prodromal DLB phase and correlate with short-term outcomes [127, 128]. The available evidence suggests that co-pathologies influence NfL because its values increase across multiple types of brain injuries, including neurodegenerative, traumatic, inflammatory, and vascular conditions [127, 129]. Glial-related markers are elevated in DLB compared to controls [130]. Proposed biofluid biomarkers of synaptic dysfunction have not yet been assessed in DLB [113].
Integration of Multiple Biomarkers
Traditionally, a single diagnosis has been assigned to patients with motor or cognitive disorders. Following this approach, biomarker studies aim to distinguish one pathology from the other, assuming they are mutually exclusive (Figure 1A). However, neuropathological studies have demonstrated that cognitively impaired individuals have multiple pathologies leading to their cognitive changes [46, 47]. The AT(N) framework accommodates the integration of different processes (Aβ, tau, and neurodegeneration) to classify individuals within the AD spectrum [131, 132]. The latest version of the AT(N) framework also discusses the possibility of categorizing biomarker values into a three-range approach and evaluating biomarkers targeting other pathologies to assess their potential contribution in AD. We propose a new biomarker quantitative approach (Figure 1B) in DLB that integrates pathology disease burden (Aβ, tau -differentiating different tau species-, TDP-43, and vascular changes) and processes such as inflammation and neurodegeneration into different quantitative axes leading to a specific biomarker fingerprint for each individual. It also includes normal and pathological range definitions. The integration of quantitative values in the fingerprint allows for potential changes in biomarker cutpoints (red and green shading boundary) on an individual basis based on established factors that impact biomarker performance (adjusting plasma biomarker cutpoint values based on renal clearance, or gray matter volume/thickness cutpoints based on age).
Figure 1. Application of diagnostic biomarkers.
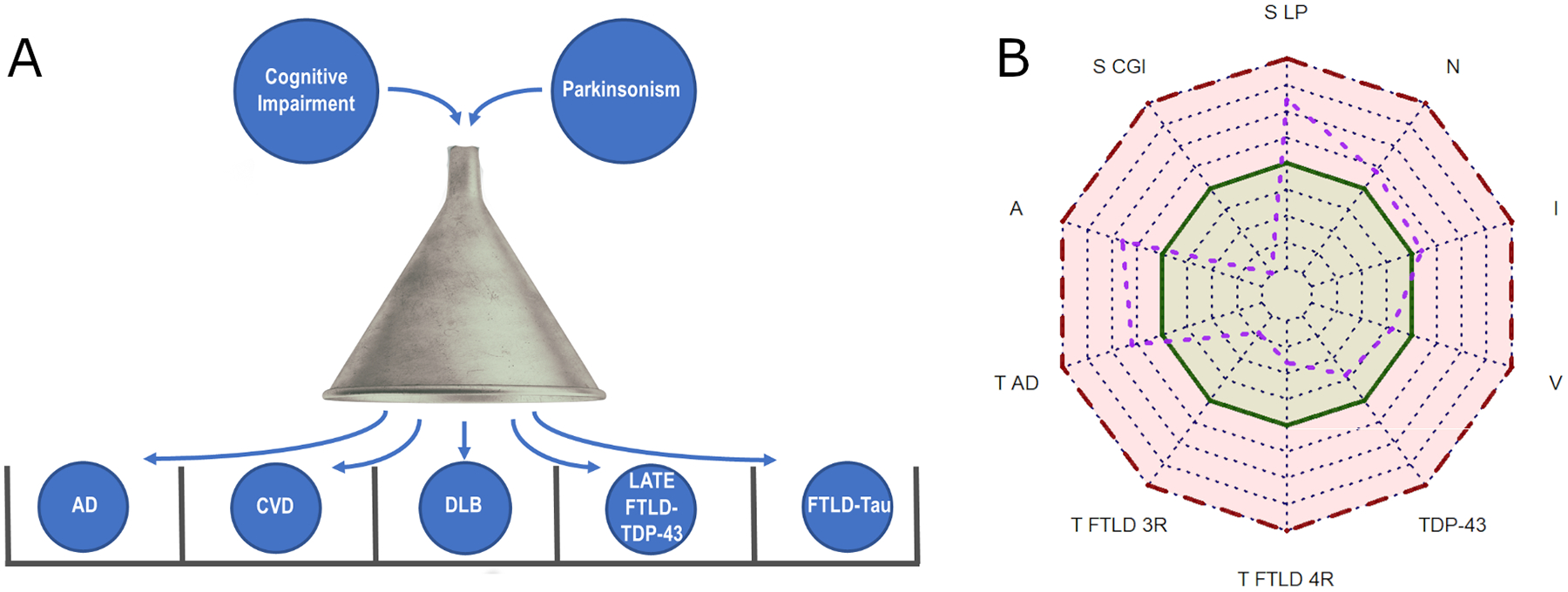
A) Biomarker applied to identify a single pathology/diagnosis and exclude other pathologies/diagnoses (differential exclusion diagnosis). B) Application of biomarkers to independently identify the different underlying processes leading to cognitive and movement disorders. The radar chart approach accommodates the inclusion of quantitative biomarker data along each axis, including a threshold into normal (green) and pathological (red) biomarker value ranges and offering a specific biomarker fingerprint for each individual. The purple line and dots represent a hypothetical patient. A: Amyloid β; AD: Alzheimer’s disease; CGI: Cytoplasmatic glia inclusion; CVD: Cerebrovascular disease; DLB: Dementia with Lewy bodies; FTLD: Frontotemporal lobar degeneration; I: Inflammation; LATE: Limbic-predominant age-related TDP-43 encephalopathy; LP: Lewy pathology; N: Neurodegeneration; S: α-Synuclein; T: Tau; S: α-synuclein; V: Cerebrovascular pathology; 3R: 3-repeat tauopathy; 4R: 4-repeat tauopathy.
Implications for Clinical Trial Design
The new diagnostic criteria and the increasing number of Lewy pathology biomarkers will facilitate the recruitment into clinical trials by including participants at potential earlier stages and greater access to biomarkers to increase the diagnostic sensitivity and exclude participants without Lewy pathology.
Most clinical trials have not evaluated the impact of co-pathologies on treatment response. Evaluating co-pathologies in future clinical trials will be crucial because of their impact on clinical presentation, cognitive performance, disease progression, and biomarkers, as summarized above [6, 7, 73–77, 82, 89, 152, 153]. One study suggested that treatment with acetylcholinesterase inhibitors (AChEI) was associated with a slower rate of cognitive decline in DLB patients with concomitant AD (although, in this study, DLB without AD were not evaluated) [154]. Conversely, DLB individuals with negative amyloid PET scans experience a more significant response to AChEI treatment than those with positive Aβ PET scans [155].
There are two overall consequences of co-pathologies for DLB clinical trial design. First, co-pathologies in DLB will need to be treated, for example, the administration of AD disease-modifying therapies (and symptomatic therapies) in DLB participants with AD co-pathology. Second, clinical trials evaluating DLB-specific treatments should stratify DLB participants and account for the potential effect of co-pathologies. This approach will be relevant for symptomatic therapies or treatments that do not specifically target Lewy pathology. However, in clinical trials targeting Lewy pathology (or another disease-defining protein deposit), participants could be selected based on their biomarker-defined presence instead of the clinical presentation.
In theory, participants with DLB with coincident AD pathology or vascular risk factors may benefit from disease-modifying therapies that successfully treat these conditions. This approach could also be extended to symptomatic therapies. To test this hypothesis, successful disease-modifying therapies and symptomatic treatments for co-pathologies should be evaluated in randomized clinical trials recruiting DLB individuals with co-pathologies confirmed by appropriate disease-specific biomarkers.
Current neurodegenerative disease clinical trials confirm the presence of the targeted pathology using biomarkers, but these trials usually do not account for the presence of co-pathologies. Therefore, clinical trials design calls for a combination of biomarkers to confirm the pathology of interest and to identify relevant co-pathologies. This information would have multiple applications: 1) influencing the inclusion and exclusion criteria for study entry, 2) trial stratification and evaluation of outcomes during the clinical trial, which includes integration of a multi-modal biomarker approach that is able to model the impact of each co-pathology. Biomarker use could decrease participant heterogeneity to increase effect sizes and decrease sample sizes, but current evidence in DLB is insufficient to evaluate the potential benefits.
These disease-specific biomarkers identify the misfolded proteins that define the different neurodegenerative conditions and therefore detect the different co-pathologies. However, these same biomarkers might not be adequate clinical trial outcome measures, at least based on recent AD clinical trials targeting Aβ [156, 157]. Therefore, disease non-specific biomarkers that define downstream changes closer to the cognitive outcomes could represent better outcome measures [131, 156, 157]. Future prospective cohort studies need to investigate how quantifying these co-pathologies informs longitudinal clinical changes and neurodegeneration biomarkers that could serve as outcomes in clinical trials [158].
There is insufficient data from clinical cohorts characterizing biomarkers performance, genetic heterogeneity, and co-pathology prevalence in DLB individuals with multiple comorbidities or belonging to different minority and underserved populations leading to health disparity. This lack of diversity and access difficulty in research studies and clinical trials is not unique to DLB [159]. In addition, stringent clinical criteria and lack of diverse recruitment in clinical trials also can limit the generalizability of their results and an equitable access to them as highlighted in other medical fields [160]. Studies including AD participants have shown differences in biomarker values in these groups [158, 161]. Ongoing and upcoming DLB clinical studies and trials need to consider the multiple aspects laid out by the National Institute on Aging Health Disparities Research Framework [162]. Strategies to increase enrollment and access to research studies and clinical trials are vital to achieving these goals [163]. In the future, plasma biomarkers could offer a less invasive and more cost-effective diagnosis. Disease-specific plasma biomarkers are at a more advanced stage in AD; however, they are currently not ready to be used as a stand-alone diagnostic marker o in primary care [164].
Figure 2 summarizes a framework to evaluate and account for co-pathologies in DLB clinical trials. It aims to integrate multi-modal biomarker approaches in DLB to develop personalized treatment selection and outcome evaluation approaches. The first stage includes the selection of the disease stage and phenotype based on clinical criteria. Earlier disease stages, including prodromal DLB [23] and rapid eye movement behavioral disorder (RBD) [165], offer earlier treatment windows. The second step includes DLB biomarkers to confirm the underlying Lewy pathology. The third step would consist of biomarkers that assess the presence of co-pathologies and quantify neurodegeneration. All this information is integrated during the fourth step to identify subgroups and different progression rates for endophenotyping and stratification. This leads to personalized treatment selection in step 5. Step 6 evaluates target engagement biomarkers that track changes that correlate with future clinical outcomes.
Figure 2. DLB clinical trial framework accounting for the presence of co-pathologies.
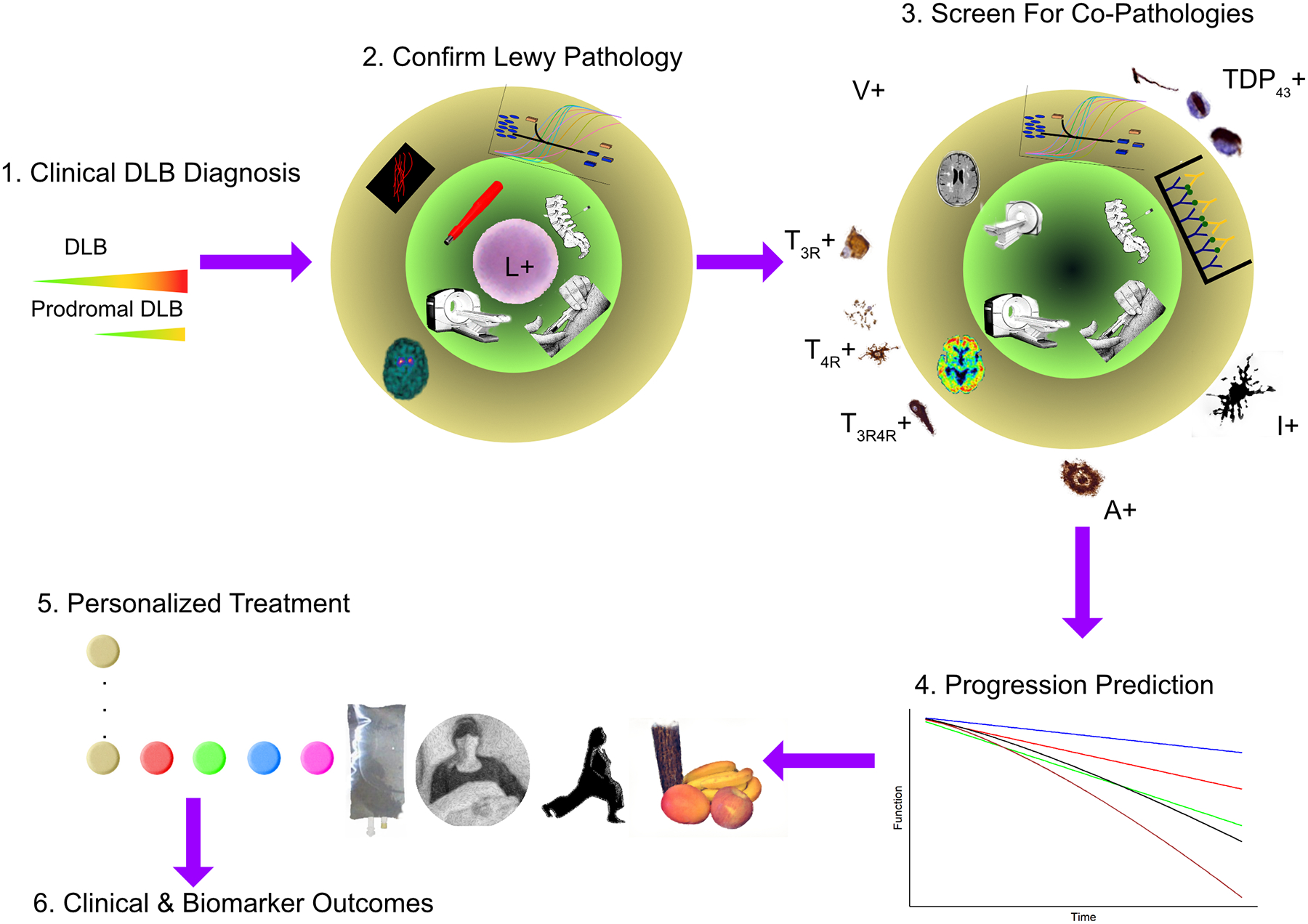
After the initial selection of participants based on clinical disease severity (step 1), Lewy pathology needs to be confirmed based on α-synuclein disease-specific biomarkers (step 2). Co-pathologies are then screened using additional biomarkers (step 3). The combined biomarker information could be part of exclusion or inclusion criteria or be considered during the clinical trial for stratification (step 4) or as co-variates. The biomarker fingerprint (See figure 1B) will estimate the predicted disease progression rate and identify the combination of treatments for each individual (step 5). Finally, biomarkers can be used to verify target engagement and as clinical trial outcome measures (step 6). In steps 2 and 3, the inner green circle represents the biomarker modalities like skin biopsies, neuroimaging (PET, SPECT, and MRI), blood, and cerebrospinal fluid). The outer circle represents the biomarker measurement, including radiotracer binding, quantification of vascular pathology through MRI, immuno-assays, real-time quaking-induced conversion, or protein misfolding cyclic amplification performed on biofluid samples.
This framework can also accommodate innovative clinical trial designs such as adaptive methodologies and master protocols [166]. Adaptive methodologies are pre-specified modifications to a clinical trial protocol during the data collection period [167], which include: changes to eligibility criteria, endpoints, dosage or patient allocation, sample size re-estimation, and addition or termination of treatment arms [168]. These modifications can be implemented in master protocols, a type of clinical trial that uses a single protocol to test a single drug in different diseases (basket trial), multiple drugs in a single disease (umbrella trials), or multiple therapies (separately or in combination) in multiple diseases in parallel (platform trials) [169]. These novel designs improve operational efficacy, include broader patient populations, share a single common control group (thus requiring fewer participants), cycle between therapies, simplify comparison across sub-studies, and increase the number of treatments tested [169]. These new methodologies could improve our ability to account for co-pathologies.
Conclusion
Individuals with DLB frequently present concurrent co-pathologies that impact clinical presentation and progression. Therefore, it is likely that combined disease-specific disease-modifying therapies will be required to affect all the pathologies contributing to the clinical signs and symptoms. However, our current understanding of co-pathologies’ impact is limited because current evidence is mainly derived from retrospective autopsy studies. Another challenge is identifying ideal disease non-specific biomarkers that closely correlate with clinical outcomes and serve as reliable clinical outcomes. An additional caveat is that these disease non-specific biomarkers pathologies might reflect changes from multiple pathologies.
Future approaches will require integrated multi-modal biomarkers with different functions. To inform clinical trials and care, we need cohort studies that evaluate disease-specific biomarkers to characterize the prevalence, impact, and progression of co-pathologies in prodromal and early dementia stages. Clinical trials will also need disease-nonspecific biomarkers that closely correlate with meaningful clinical outcomes and serve as outcomes in clinical trials. The design of these cohort studies should further evaluate the studied biomarkers’ current evidence to advance them towards clinical application and provide initial evidence of the emerging biomarkers [170]. The cohorts should recruit DLB individuals representing diverse socioeconomic, races, ethnicities, and risk factors.
Here, we proposed a multi-axial biomarker integration approach and a multi-step process for selection, stratification, evaluation of target engagement, and interpretation of clinical trial results in DLB. This multi-layered biomarker approach will provide a personalized assessment of pathologies guiding recruitment into clinical trials and interpreting its outcomes. Once disease-modifying therapies are available, this process will help predict different response rates to treatments and guide potential treatment approaches that combine various drugs targeting each pathology present in the brain.
Acknowledgments
This manuscript was facilitated by the Alzheimer’s Association International Society to Advance Alzheimer’s Research and Treatment (ISTAART), through the Lewy body dementias professional interest area (PIA). The views and opinions expressed by authors in this publication represent those of the authors and do not necessarily reflect those of the greater PIA membership, ISTAART or the Alzheimer’s Association.
The authors are particularly grateful for the support and diligence of Jodi Titiner and Chris Weber in supporting the LBD PIA and the production of this manuscript.
JBT has been supported by the Edmond J. Safra and is the Harrison Endowed Scientific Director of the Nantz National Alzheimer Center.
MJG is supported by the “Miguel Servet” program [CP19/00031] and a research grant [PI20/00613] of the Instituto de Salud Carlos III-Fondo Europeo de Desarrollo Regional (ISCIII-FEDER).
DF is supported by the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and Karolinska Institutet; Center for Innovative Medicine (CIMED); the Swedish Brain Foundation; Demensfonden; Gun och Bertil Stohnes Stiftelse; Gamla Tjänarinnor; Karolinska Institutet Research Funding and Karolinska Institutet Funding for Geriatric Diseases.
RW is supported by a Wellcome Clinical Research Career Development Fellowship (205167/Z/16/Z).
DA is a Royal Society Wolfson Research Merit Award Holder and would like to thank the Wolfson Foundation and the Royal Society for their support. Professor Aarsland is supported by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
IMcK and JPT are supported by the NIHR Newcastle Biomedical Research Centre.
SWS was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Neurological Disorders and Stroke (program #: 1ZIANS003154).
BB is supported in part by NIH (grants P30 AG62677, U01 NS100620, U19 AG 071754, U54 NS110435), the Lewy Body Dementia Association, the Mayo Clinic Dorothy and Harry T. Mangurian Jr. Lewy Body Dementia Program, the Little Family Foundation, and Ted Turner and Family Foundation.
JBL was supported through National Institutes of Health (National Institute of Neurological Disorders and Stroke, U01NA100610 and National Institute on Aging, P30AG072959) and the Lewy Body Dementia Association (Reseach Center of Excellence program)
SL is partly supported through NHMRC Investigator Grant #1195830.
JK is partially supported by the Alzheimer’s Research UK (Pump priming grant).
CA, JK, KWC have no funding sources to acknowledge.
Conflicts of interest
DF, MJG, JK, IMcK, JPT, JBT and KWC report no conflicts of interest.
RW has received speaker fees from GE Healthcare and honoraria from Britannia.
SWS serves on the scientific advisory council of the Lewy Body Dementia Association and received grants/contracts from Cerevel Therapeutics.
DA has received research support and/or honoraria from Astra-Zeneca, H. Lundbeck, Novartis Pharmaceuticals and GE Health, and serves as paid consultant for H. Lundbeck, Eisai, and Axovant.
BB has received research support for clinical trials from Alector, Biogen and Transposon. He receives royalties from the publication of a book entitled Behavioral Neurology of Dementia (Cambridge Medicine). He serves on the Scientific Advisory Board of the Tau Consortium which is funded by the Rainwater Charitable Foundation.
JBL serves on the Lewy Body Dementia Association Scientific Advisory Council, the ISTAART Lewy Body Dementia Professional Interest Area (Vice Chair), receives Grant support from GE Healthcare, and served on an Advisory Board for Vaxxinity.
SL has received research support and/or honoraria from Acceler8, Pharmaxis.
CA has received research support and/or honoraria from F. Hoffmann-La Roche Ltd, Zambonm, Nutricia, and Schwabe Farma Ibérica S.A.U, and meeting/travel support from Nutricia and Biogen.
References
- [1].Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain : a journal of neurology. 2013;136:2697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain : a journal of neurology. 2018;141:2181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Attems J, Toledo JB, Walker L, Gelpi E, Gentleman S, Halliday G, et al. Neuropathological consensus criteria for the evaluation of Lewy pathology in post-mortem brains: a multi-centre study. Acta neuropathologica. 2021;141:159–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology. 2017;89:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ruffmann C, Calboli FC, Bravi I, Gveric D, Curry LK, de Smith A, et al. Cortical Lewy bodies and Abeta burden are associated with prevalence and timing of dementia in Lewy body diseases. Neuropathology and applied neurobiology. 2016;42:436–50. [DOI] [PubMed] [Google Scholar]
- [7].Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX, et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet neurology. 2017;16:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hepp DH, Vergoossen DL, Huisman E, Lemstra AW, Netherlands Brain B, Berendse HW, et al. Distribution and Load of Amyloid-beta Pathology in Parkinson Disease and Dementia with Lewy Bodies. Journal of neuropathology and experimental neurology. 2016;75:936–45. [DOI] [PubMed] [Google Scholar]
- [9].Merdes AR, Hansen LA, Jeste DV, Galasko D, Hofstetter CR, Ho GJ, et al. Influence of Alzheimer pathology on clinical diagnostic accuracy in dementia with Lewy bodies. Neurology. 2003;60:1586–90. [DOI] [PubMed] [Google Scholar]
- [10].Fujishiro H, Iseki E, Higashi S, Kasanuki K, Murayama N, Togo T, et al. Distribution of cerebral amyloid deposition and its relevance to clinical phenotype in Lewy body dementia. Neuroscience letters. 2010;486:19–23. [DOI] [PubMed] [Google Scholar]
- [11].Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE, et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta neuropathologica. 2007;114:221–9. [DOI] [PubMed] [Google Scholar]
- [12].Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K, et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain research. 2007;1184:284–94. [DOI] [PubMed] [Google Scholar]
- [13].McAleese KE, Walker L, Erskine D, Thomas AJ, McKeith IG, Attems J. TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain Pathol. 2017;27:472–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Arai T, Mackenzie IR, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K, et al. Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta neuropathologica. 2009;117:125–36. [DOI] [PubMed] [Google Scholar]
- [15].Buciuc M, Whitwell JL, Boeve BF, Ferman TJ, Graff-Radford J, Savica R, et al. TDP-43 is associated with a reduced likelihood of rendering a clinical diagnosis of dementia with Lewy bodies in autopsy-confirmed cases of transitional/diffuse Lewy body disease. Journal of neurology. 2020;267:1444–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ghebremedhin E, Rosenberger A, Rub U, Vuksic M, Berhe T, Bickeboller H, et al. Inverse relationship between cerebrovascular lesions and severity of lewy body pathology in patients with lewy body diseases. Journal of neuropathology and experimental neurology. 2010;69:442–8. [DOI] [PubMed] [Google Scholar]
- [17].Jellinger KA. Significance of cerebral amyloid angiopathy and other co-morbidities in Lewy body diseases. J Neural Transm (Vienna). 2021;128:687–99. [DOI] [PubMed] [Google Scholar]
- [18].Lopez OL, Litvan I, Catt KE, Stowe R, Klunk W, Kaufer DI, et al. Accuracy of four clinical diagnostic criteria for the diagnosis of neurodegenerative dementias. Neurology. 1999;53:1292–9. [DOI] [PubMed] [Google Scholar]
- [19].Peavy GM, Salmon DP, Edland SD, Tam S, Hansen LA, Masliah E, et al. Neuropsychiatric features of frontal lobe dysfunction in autopsy-confirmed patients with lewy bodies and “pure” Alzheimer disease. The American journal of geriatric psychiatry : official journal of the American Association for Geriatric Psychiatry. 2013;21:509–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jellinger KA. Are there morphological differences between Parkinson’s disease-dementia and dementia with Lewy bodies? Parkinsonism Relat Disord. 2022;100:24–32. [DOI] [PubMed] [Google Scholar]
- [21].Jellinger KA. Dementia with Lewy bodies and Parkinson’s disease-dementia: current concepts and controversies. J Neural Transm (Vienna). 2018;125:615–50. [DOI] [PubMed] [Google Scholar]
- [22].Coughlin D, Xie SX, Liang M, Williams A, Peterson C, Weintraub D, et al. Cognitive and Pathological Influences of Tau Pathology in Lewy Body Disorders. Annals of neurology. 2019;85:259–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].McKeith IG, Ferman TJ, Thomas AJ, Blanc F, Boeve BF, Fujishiro H, et al. Research criteria for the diagnosis of prodromal dementia with Lewy bodies. Neurology. 2020;94:743–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta neuropathologica. 2014;128:755–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H, et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta neuropathologica. 2016;131:87–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta neuropathologica. 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain : a journal of neurology. 2019;142:1503–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y, et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta neuropathologica. 2009;117:137–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain : a journal of neurology. 2009;132:2922–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, et al. A harmonized classification system for FTLD-TDP pathology. Acta neuropathologica. 2011;122:111–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Litvan I, Hauw JJ, Bartko JJ, Lantos PL, Daniel SE, Horoupian DS, et al. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. Journal of neuropathology and experimental neurology. 1996;55:97–105. [DOI] [PubMed] [Google Scholar]
- [32].Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, et al. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. Journal of neuropathology and experimental neurology. 2002;61:935–46. [DOI] [PubMed] [Google Scholar]
- [33].Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta neuropathologica. 2010;119:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Manne S, Kondru N, Jin H, Serrano GE, Anantharam V, Kanthasamy A, et al. Blinded RT-QuIC Analysis of alpha-Synuclein Biomarker in Skin Tissue From Parkinson’s Disease Patients. Movement disorders : official journal of the Movement Disorder Society. 2020;35:2230–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang Z, Becker K, Donadio V, Siedlak S, Yuan J, Rezaee M, et al. Skin alpha-Synuclein Aggregation Seeding Activity as a Novel Biomarker for Parkinson Disease. JAMA neurology. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mammana A, Baiardi S, Quadalti C, Rossi M, Donadio V, Capellari S, et al. RT-QuIC Detection of Pathological alpha-Synuclein in Skin Punches of Patients with Lewy Body Disease. Movement disorders : official journal of the Movement Disorder Society. 2021;36:2173–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Stefani A, Iranzo A, Holzknecht E, Perra D, Bongianni M, Gaig C, et al. Alpha-synuclein seeds in olfactory mucosa of patients with isolated REM sleep behaviour disorder. Brain : a journal of neurology. 2021;144:1118–26. [DOI] [PubMed] [Google Scholar]
- [38].De Luca CMG, Elia AE, Portaleone SM, Cazzaniga FA, Rossi M, Bistaffa E, et al. Efficient RT-QuIC seeding activity for alpha-synuclein in olfactory mucosa samples of patients with Parkinson’s disease and multiple system atrophy. Transl Neurodegener. 2019;8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bargar C, Wang W, Gunzler SA, LeFevre A, Wang Z, Lerner AJ, et al. Streamlined alpha-synuclein RT-QuIC assay for various biospecimens in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol Commun. 2021;9:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Manne S, Kondru N, Jin H, Anantharam V, Huang X, Kanthasamy A, et al. alpha-Synuclein real-time quaking-induced conversion in the submandibular glands of Parkinson’s disease patients. Movement disorders : official journal of the Movement Disorder Society. 2020;35:268–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kim JY, Illigens BM, McCormick MP, Wang N, Gibbons CH. Alpha-Synuclein in Skin Nerve Fibers as a Biomarker for Alpha-Synucleinopathies. J Clin Neurol. 2019;15:135–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Groveman BR, Orru CD, Hughson AG, Raymond LD, Zanusso G, Ghetti B, et al. Rapid and ultra-sensitive quantitation of disease-associated alpha-synuclein seeds in brain and cerebrospinal fluid by alphaSyn RT-QuIC. Acta Neuropathol Commun. 2018;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Shahnawaz M, Mukherjee A, Pritzkow S, Mendez N, Rabadia P, Liu X, et al. Discriminating alpha-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature. 2020;578:273–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Skrobot OA, Attems J, Esiri M, Hortobagyi T, Ironside JW, Kalaria RN, et al. Vascular cognitive impairment neuropathology guidelines (VCING): the contribution of cerebrovascular pathology to cognitive impairment. Brain : a journal of neurology. 2016;139:2957–69. [DOI] [PubMed] [Google Scholar]
- [45].Hijazi Z, Yassi N, O’Brien JT, Watson R. The influence of cerebrovascular disease in dementia with Lewy bodies and Parkinson’s disease dementia. European journal of neurology : the official journal of the European Federation of Neurological Societies. 2022;29:1254–65. [DOI] [PubMed] [Google Scholar]
- [46].Boyle PA, Yu L, Leurgans SE, Wilson RS, Brookmeyer R, Schneider JA, et al. Attributable risk of Alzheimer’s dementia attributed to age-related neuropathologies. Annals of neurology. 2019;85:114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person-specific contribution of neuropathologies to cognitive loss in old age. Annals of neurology. 2018;83:74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Vogel JW, Young AL, Oxtoby NP, Smith R, Ossenkoppele R, Strandberg OT, et al. Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nature medicine. 2021;27:871–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Brettschneider J, Del Tredici K, Irwin DJ, Grossman M, Robinson JL, Toledo JB, et al. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta neuropathologica. 2014;127:423–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Annals of neurology. 2013;74:20–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Toledo JB, Gopal P, Raible K, Irwin DJ, Brettschneider J, Sedor S, et al. Pathological alpha-synuclein distribution in subjects with coincident Alzheimer’s and Lewy body pathology. Acta neuropathologica. 2016;131:393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, et al. Updated TDP-43 in Alzheimer’s disease staging scheme. Acta neuropathologica. 2016;131:571–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Deramecourt V, Slade JY, Oakley AE, Perry RH, Ince PG, Maurage CA, et al. Staging and natural history of cerebrovascular pathology in dementia. Neurology. 2012;78:1043–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Robinson JL, Richardson H, Xie SX, Suh E, Van Deerlin VM, Alfaro B, et al. The development and convergence of co-pathologies in Alzheimer’s disease. Brain : a journal of neurology. 2021;144:953–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–72. [DOI] [PubMed] [Google Scholar]
- [56].Kovacs GG, Xie SX, Robinson JL, Lee EB, Smith DH, Schuck T, et al. Sequential stages and distribution patterns of aging-related tau astrogliopathy (ARTAG) in the human brain. Acta Neuropathol Commun. 2018;6:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ferreira D, Nedelska Z, Graff-Radford J, Przybelski SA, Lesnick TG, Schwarz CG, et al. Cerebrovascular disease, neurodegeneration, and clinical phenotype in dementia with Lewy bodies. Neurobiology of aging. 2021;105:252–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Gungor I, Sarro L, Graff-Radford J, Zuk SM, Tosakulwong N, Przybelski SA, et al. Frequency and topography of cerebral microbleeds in dementia with Lewy bodies compared to Alzheimer’s disease. Parkinsonism Relat Disord. 2015;21:1101–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kim SW, Chung SJ, Oh YS, Yoon JH, Sunwoo MK, Hong JY, et al. Cerebral Microbleeds in Patients with Dementia with Lewy Bodies and Parkinson Disease Dementia. AJNR American journal of neuroradiology. 2015;36:1642–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Takemoto M, Yamashita T, Ohta Y, Tadokoro K, Omote Y, Morihara R, et al. Cerebral Microbleeds in Patients with Parkinson’s Disease and Dementia with Lewy Bodies: Comparison Using Magnetic Resonance Imaging and 99 mTc-ECD SPECT Subtraction Imaging. Journal of Alzheimer’s disease : JAD. 2021;80:331–5. [DOI] [PubMed] [Google Scholar]
- [61].Donaghy PC, Firbank M, Mitra D, Petrides G, Lloyd J, Barnett N, et al. Microbleeds in dementia with Lewy bodies. Journal of neurology. 2020;267:1491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sarro L, Tosakulwong N, Schwarz CG, Graff-Radford J, Przybelski SA, Lesnick TG, et al. An investigation of cerebrovascular lesions in dementia with Lewy bodies compared to Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2017;13:257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].van Veluw SJ, Arfanakis K, Schneider JA. Neuropathology of Vascular Brain Health: Insights From Ex Vivo Magnetic Resonance Imaging-Histopathology Studies in Cerebral Small Vessel Disease. Stroke; a journal of cerebral circulation. 2022;53:404–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].McAleese KE, Walker L, Graham S, Moya ELJ, Johnson M, Erskine D, et al. Parietal white matter lesions in Alzheimer’s disease are associated with cortical neurodegenerative pathology, but not with small vessel disease. Acta neuropathologica. 2017;134:459–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].McAleese KE, Miah M, Graham S, Hadfield GM, Walker L, Johnson M, et al. Frontal white matter lesions in Alzheimer’s disease are associated with both small vessel disease and AD-associated cortical pathology. Acta neuropathologica. 2021;142:937–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Barber R, Scheltens P, Gholkar A, Ballard C, McKeith I, Ince P, et al. White matter lesions on magnetic resonance imaging in dementia with Lewy bodies, Alzheimer’s disease, vascular dementia, and normal aging. Journal of neurology, neurosurgery, and psychiatry. 1999;67:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Joki H, Higashiyama Y, Nakae Y, Kugimoto C, Doi H, Kimura K, et al. White matter hyperintensities on MRI in dementia with Lewy bodies, Parkinson’s disease with dementia, and Alzheimer’s disease. Journal of the neurological sciences. 2018;385:99–104. [DOI] [PubMed] [Google Scholar]
- [68].Koikkalainen J, Rhodius-Meester H, Tolonen A, Barkhof F, Tijms B, Lemstra AW, et al. Differential diagnosis of neurodegenerative diseases using structural MRI data. NeuroImage Clinical. 2016;11:435–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Burton EJ, McKeith IG, Burn DJ, Firbank MJ, O’Brien JT. Progression of white matter hyperintensities in Alzheimer disease, dementia with lewy bodies, and Parkinson disease dementia: a comparison with normal aging. The American journal of geriatric psychiatry : official journal of the American Association for Geriatric Psychiatry. 2006;14:842–9. [DOI] [PubMed] [Google Scholar]
- [70].Oppedal K, Aarsland D, Firbank MJ, Sonnesyn H, Tysnes OB, O’Brien JT, et al. White matter hyperintensities in mild lewy body dementia. Dement Geriatr Cogn Dis Extra. 2012;2:481–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].De Reuck J, Auger F, Durieux N, Cordonnier C, Deramecourt V, Pasquier F, et al. Topographic distribution of white matter changes and lacunar infarcts in neurodegenerative and vascular dementia syndromes: A post-mortem 7.0-tesla magnetic resonance imaging study. Eur Stroke J. 2016;1:122–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Andersson M, Zetterberg H, Minthon L, Blennow K, Londos E. The cognitive profile and CSF biomarkers in dementia with Lewy bodies and Parkinson’s disease dementia. International journal of geriatric psychiatry. 2011;26:100–5. [DOI] [PubMed] [Google Scholar]
- [73].Ferreira D, Przybelski SA, Lesnick TG, Lemstra AW, Londos E, Blanc F, et al. beta-Amyloid and tau biomarkers and clinical phenotype in dementia with Lewy bodies. Neurology. 2020;95:e3257–e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Gomperts SN, Locascio JJ, Marquie M, Santarlasci AL, Rentz DM, Maye J, et al. Brain amyloid and cognition in Lewy body diseases. Movement disorders : official journal of the Movement Disorder Society. 2012;27:965–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].van Steenoven I, van der Flier WM, Scheltens P, Teunissen CE, Lemstra AW. Amyloid-beta peptides in cerebrospinal fluid of patients with dementia with Lewy bodies. Alzheimer’s research & therapy. 2019;11:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Maetzler W, Liepelt I, Reimold M, Reischl G, Solbach C, Becker C, et al. Cortical PIB binding in Lewy body disease is associated with Alzheimer-like characteristics. Neurobiol Dis. 2009;34:107–12. [DOI] [PubMed] [Google Scholar]
- [77].Mak E, Nicastro N, Malpetti M, Savulich G, Surendranathan A, Holland N, et al. Imaging tau burden in dementia with Lewy bodies using [(18)F]-AV1451 positron emission tomography. Neurobiology of aging. 2021;101:172–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Di Censo R, Abdelnour C, Blanc F, Bousiges O, Lemstra AW, van Steenoven I, et al. CSF tau proteins correlate with an atypical clinical presentation in dementia with Lewy bodies. Journal of neurology, neurosurgery, and psychiatry. 2020;91:109–10. [DOI] [PubMed] [Google Scholar]
- [79].Del Ser T, Hachinski V, Merskey H, Munoz DG. Clinical and pathologic features of two groups of patients with dementia with Lewy bodies: effect of coexisting Alzheimer-type lesion load. Alzheimer disease and associated disorders. 2001;15:31–44. [DOI] [PubMed] [Google Scholar]
- [80].Kantarci K, Lowe VJ, Boeve BF, Senjem ML, Tosakulwong N, Lesnick TG, et al. AV-1451 tau and beta-amyloid positron emission tomography imaging in dementia with Lewy bodies. Annals of neurology. 2017;81:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Graff-Radford J, Murray ME, Lowe VJ, Boeve BF, Ferman TJ, Przybelski SA, et al. Dementia with Lewy bodies: basis of cingulate island sign. Neurology. 2014;83:801–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Lemstra AW, de Beer MH, Teunissen CE, Schreuder C, Scheltens P, van der Flier WM, et al. Concomitant AD pathology affects clinical manifestation and survival in dementia with Lewy bodies. Journal of neurology, neurosurgery, and psychiatry. 2017;88:113–8. [DOI] [PubMed] [Google Scholar]
- [83].Jellinger KA, Attems J. Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta neuropathologica. 2008;115:427–36. [DOI] [PubMed] [Google Scholar]
- [84].Fukui T, Oowan Y, Yamazaki T, Kinno R. Prevalence and clinical implication of microbleeds in dementia with lewy bodies in comparison with microbleeds in Alzheimer’s disease. Dement Geriatr Cogn Dis Extra. 2013;3:148–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Mirza SS, Saeed U, Knight J, Ramirez J, Stuss DT, Keith J, et al. APOE epsilon4, white matter hyperintensities, and cognition in Alzheimer and Lewy body dementia. Neurology. 2019;93:e1807–e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Park HE, Park IS, Oh YS, Yang DW, Lee KS, Choi HS, et al. Subcortical whiter matter hyperintensities within the cholinergic pathways of patients with dementia and parkinsonism. Journal of the neurological sciences. 2015;353:44–8. [DOI] [PubMed] [Google Scholar]
- [87].Graff-Radford NR, Besser LM, Crook JE, Kukull WA, Dickson DW. Neuropathologic differences by race from the National Alzheimer’s Coordinating Center. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2016;12:669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Bayram E, Coughlin DG, Litvan I. Sex Differences for Clinical Correlates of Alzheimer’s Pathology in People with Lewy Body Pathology. Movement disorders : official journal of the Movement Disorder Society. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Abdelnour C, van Steenoven I, Londos E, Blanc F, Auestad B, Kramberger MG, et al. Alzheimer’s disease cerebrospinal fluid biomarkers predict cognitive decline in lewy body dementia. Movement disorders : official journal of the Movement Disorder Society. 2016;31:1203–8. [DOI] [PubMed] [Google Scholar]
- [90].Howlett DR, Whitfield D, Johnson M, Attems J, O’Brien JT, Aarsland D, et al. Regional Multiple Pathology Scores Are Associated with Cognitive Decline in Lewy Body Dementias. Brain Pathol. 2015;25:401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Compta Y, Parkkinen L, Kempster P, Selikhova M, Lashley T, Holton JL, et al. The significance of alpha-synuclein, amyloid-beta and tau pathologies in Parkinson’s disease progression and related dementia. Neuro-degenerative diseases. 2014;13:154–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Wakisaka Y, Furuta A, Tanizaki Y, Kiyohara Y, Iida M, Iwaki T. Age-associated prevalence and risk factors of Lewy body pathology in a general population: the Hisayama study. Acta neuropathologica. 2003;106:374–82. [DOI] [PubMed] [Google Scholar]
- [93].Ferman TJ, Aoki N, Crook JE, Murray ME, Graff-Radford NR, van Gerpen JA, et al. The limbic and neocortical contribution of alpha-synuclein, tau, and amyloid beta to disease duration in dementia with Lewy bodies. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2018;14:330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Geiger JT, Ding J, Crain B, Pletnikova O, Letson C, Dawson TM, et al. Next-generation sequencing reveals substantial genetic contribution to dementia with Lewy bodies. Neurobiol Dis. 2016;94:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, et al. APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA neurology. 2013;70:223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Guerreiro R, Ross OA, Kun-Rodrigues C, Hernandez DG, Orme T, Eicher JD, et al. Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet neurology. 2018;17:64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Rongve A, Witoelar A, Ruiz A, Athanasiu L, Abdelnour C, Clarimon J, et al. GBA and APOE epsilon4 associate with sporadic dementia with Lewy bodies in European genome wide association study. Scientific reports. 2019;9:7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Bras J, Guerreiro R, Darwent L, Parkkinen L, Ansorge O, Escott-Price V, et al. Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Human molecular genetics. 2014;23:6139–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Orme T, Hernandez D, Ross OA, Kun-Rodrigues C, Darwent L, Shepherd CE, et al. Analysis of neurodegenerative disease-causing genes in dementia with Lewy bodies. Acta Neuropathol Commun. 2020;8:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Larsson V, Torisson G, Londos E. Relative survival in patients with dementia with Lewy bodies and Parkinson’s disease dementia. PloS one. 2018;13:e0202044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Ballard C, O’Brien J, Morris CM, Barber R, Swann A, Neill D, et al. The progression of cognitive impairment in dementia with Lewy bodies, vascular dementia and Alzheimer’s disease. International journal of geriatric psychiatry. 2001;16:499–503. [DOI] [PubMed] [Google Scholar]
- [102].Toledo JB, Brettschneider J, Grossman M, Arnold SE, Hu WT, Xie SX, et al. CSF biomarkers cutoffs: the importance of coincident neuropathological diseases. Acta neuropathologica. 2012;124:23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Toledo JB, Bjerke M, Da X, Landau SM, Foster NL, Jagust W, et al. Nonlinear Association Between Cerebrospinal Fluid and Florbetapir F-18 beta-Amyloid Measures Across the Spectrum of Alzheimer Disease. JAMA neurology. 2015;72:571–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Ossenkoppele R, Smith R, Mattsson-Carlgren N, Groot C, Leuzy A, Strandberg O, et al. Accuracy of Tau Positron Emission Tomography as a Prognostic Marker in Preclinical and Prodromal Alzheimer Disease: A Head-to-Head Comparison Against Amyloid Positron Emission Tomography and Magnetic Resonance Imaging. JAMA neurology. 2021;78:961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Kang JH, Irwin DJ, Chen-Plotkin AS, Siderowf A, Caspell C, Coffey CS, et al. Association of cerebrospinal fluid beta-amyloid 1–42, T-tau, P-tau181, and alpha-synuclein levels with clinical features of drug-naive patients with early Parkinson disease. JAMA neurology. 2013;70:1277–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Hatcher-Martin JM, McKay JL, Pybus AF, Sommerfeld B, Howell JC, Goldstein FC, et al. Cerebrospinal fluid biomarkers in Parkinson’s disease with freezing of gait: an exploratory analysis. NPJ Parkinsons Dis. 2021;7:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Hall S, Surova Y, Ohrfelt A, Zetterberg H, Lindqvist D, Hansson O. CSF biomarkers and clinical progression of Parkinson disease. Neurology. 2015;84:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Gomperts SN, Locascio JJ, Makaretz SJ, Schultz A, Caso C, Vasdev N, et al. Tau Positron Emission Tomographic Imaging in the Lewy Body Diseases. JAMA neurology. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Janelidze S, Teunissen CE, Zetterberg H, Allue JA, Sarasa L, Eichenlaub U, et al. Head-to-Head Comparison of 8 Plasma Amyloid-beta 42/40 Assays in Alzheimer Disease. JAMA neurology. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Gonzalez MC, Ashton NJ, Gomes BF, Tovar-Rios DA, Blanc F, Karikari TK, et al. Association of Plasma p-tau181 and p-tau231 Concentrations With Cognitive Decline in Patients With Probable Dementia With Lewy Bodies. JAMA neurology. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Karikari TK, Ashton NJ, Brinkmalm G, Brum WS, Benedet AL, Montoliu-Gaya L, et al. Blood phospho-tau in Alzheimer disease: analysis, interpretation, and clinical utility. Nature reviews Neurology. 2022;18:400–18. [DOI] [PubMed] [Google Scholar]
- [112].Scialo C, Tran TH, Salzano G, Novi G, Caponnetto C, Chio A, et al. TDP-43 real-time quaking induced conversion reaction optimization and detection of seeding activity in CSF of amyotrophic lateral sclerosis and frontotemporal dementia patients. Brain Commun. 2020;2:fcaa142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Nilsson J, Gobom J, Sjodin S, Brinkmalm G, Ashton NJ, Svensson J, et al. Cerebrospinal fluid biomarker panel for synaptic dysfunction in Alzheimer’s disease. Alzheimers Dement (Amst). 2021;13:e12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Scamarcia PG, Agosta F, Caso F, Filippi M. Update on neuroimaging in non-Alzheimer’s disease dementia: a focus on the Lewy body disease spectrum. Curr Opin Neurol. 2021;34:532–8. [DOI] [PubMed] [Google Scholar]
- [115].Velickaite V, Ferreira D, Lind L, Ahlstrom H, Kilander L, Westman E, et al. Visual rating versus volumetry of regional brain atrophy and longitudinal changes over a 5-year period in an elderly population. Brain and behavior. 2020;10:e01662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Spotorno N, Coughlin DG, Olm CA, Wolk D, Vaishnavi SN, Shaw LM, et al. Tau pathology associates with in vivo cortical thinning in Lewy body disorders. Ann Clin Transl Neurol. 2020;7:2342–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Abdelnour C, Ferreira D, Oppedal K, Cavallin L, Bousiges O, Wahlund LO, et al. The combined effect of amyloid-beta and tau biomarkers on brain atrophy in dementia with Lewy bodies. NeuroImage Clinical. 2020;27:102333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Zarkali A, McColgan P, Leyland LA, Lees AJ, Weil RS. Visual Dysfunction Predicts Cognitive Impairment and White Matter Degeneration in Parkinson’s Disease. Movement disorders : official journal of the Movement Disorder Society. 2021;36:1191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Donaghy PC, Firbank M, Petrides G, Lloyd J, Barnett N, Olsen K, et al. Diffusion imaging in dementia with Lewy bodies: Associations with amyloid burden, atrophy, vascular factors and clinical features. Parkinsonism Relat Disord. 2020;78:109–15. [DOI] [PubMed] [Google Scholar]
- [120].Thomas GEC, Leyland LA, Schrag AE, Lees AJ, Acosta-Cabronero J, Weil RS. Brain iron deposition is linked with cognitive severity in Parkinson’s disease. Journal of neurology, neurosurgery, and psychiatry. 2020;91:418–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Ofori E, Krismer F, Burciu RG, Pasternak O, McCracken JL, Lewis MM, et al. Free water improves detection of changes in the substantia nigra in parkinsonism: A multisite study. Movement disorders : official journal of the Movement Disorder Society. 2017;32:1457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Burciu RG, Ofori E, Shukla P, Pasternak O, Chung JW, McFarland NR, et al. Free-water and BOLD imaging changes in Parkinson’s disease patients chronically treated with a MAO-B inhibitor. Human brain mapping. 2016;37:2894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Graff-Radford J, Lesnick TG, Savica R, Chen Q, Ferman TJ, Przybelski SA, et al. (18)F-fluorodeoxyglucose positron emission tomography in dementia with Lewy bodies. Brain Commun. 2020;2:fcaa040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Kantarci K, Boeve BF, Przybelski SA, Lesnick TG, Chen Q, Fields J, et al. FDG PET metabolic signatures distinguishing prodromal DLB and prodromal AD. NeuroImage Clinical. 2021;31:102754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Pillai JA, Wu G, Tousi B, Larvie M, Leger GC, Leverenz JB. Amygdala sign, a FDG-PET signature of dementia with Lewy Bodies. Parkinsonism Relat Disord. 2019;64:300–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Bacioglu M, Maia LF, Preische O, Schelle J, Apel A, Kaeser SA, et al. Neurofilament Light Chain in Blood and CSF as Marker of Disease Progression in Mouse Models and in Neurodegenerative Diseases. Neuron. 2016;91:494–6. [DOI] [PubMed] [Google Scholar]
- [127].Ashton NJ, Janelidze S, Al Khleifat A, Leuzy A, van der Ende EL, Karikari TK, et al. A multicentre validation study of the diagnostic value of plasma neurofilament light. Nature communications. 2021;12:3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Pilotto A, Imarisio A, Carrarini C, Russo M, Masciocchi S, Gipponi S, et al. Plasma Neurofilament Light Chain Predicts Cognitive Progression in Prodromal and Clinical Dementia with Lewy Bodies. Journal of Alzheimer’s disease : JAD. 2021;82:913–9. [DOI] [PubMed] [Google Scholar]
- [129].Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T, et al. Neurofilaments as biomarkers in neurological disorders. Nature reviews Neurology. 2018;14:577–89. [DOI] [PubMed] [Google Scholar]
- [130].Schulz I, Kruse N, Gera RG, Kremer T, Cedarbaum J, Barbour R, et al. Systematic Assessment of 10 Biomarker Candidates Focusing on alpha-Synuclein-Related Disorders. Movement disorders : official journal of the Movement Disorder Society. 2021. [DOI] [PubMed] [Google Scholar]
- [131].Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2018;14:535–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].van de Beek M, Ooms FAH, Ebenau JL, Barkhof F, Scheltens P, Teunissen CE, et al. Association of the ATN Research Framework With Clinical Profile, Cognitive Decline, and Mortality in Patients With Dementia With Lewy Bodies. Neurology. 2022;98:e1262–e72. [DOI] [PubMed] [Google Scholar]
- [133].Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA : the journal of the American Medical Association. 2011;305:275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Palmqvist S, Mattsson N, Hansson O, Alzheimer’s Disease Neuroimaging I. Cerebrospinal fluid analysis detects cerebral amyloid-beta accumulation earlier than positron emission tomography. Brain : a journal of neurology. 2016;139:1226–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Lowe VJ, Lundt ES, Albertson SM, Min HK, Fang P, Przybelski SA, et al. Tau-positron emission tomography correlates with neuropathology findings. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2020;16:561–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Yap SY, Frias B, Wren MC, Scholl M, Fox NC, Arstad E, et al. Discriminatory ability of next-generation tau PET tracers for Alzheimer’s disease. Brain : a journal of neurology. 2021;144:2284–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Soleimani-Meigooni DN, Iaccarino L, La Joie R, Baker S, Bourakova V, Boxer AL, et al. 18F-flortaucipir PET to autopsy comparisons in Alzheimer’s disease and other neurodegenerative diseases. Brain : a journal of neurology. 2020;143:3477–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Levy JP, Bezgin G, Savard M, Pascoal TA, Finger E, Laforce R, et al. 18F-MK-6240 tau-PET in genetic frontotemporal dementia. Brain : a journal of neurology. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Mielke MM, Przybelski SA, Lesnick TG, Kern S, Zetterberg H, Blennow K, et al. Comparison of CSF neurofilament light chain, neurogranin, and tau to MRI markers. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2021;17:801–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Habes M, Grothe MJ, Tunc B, McMillan C, Wolk DA, Davatzikos C. Disentangling Heterogeneity in Alzheimer’s Disease and Related Dementias Using Data-Driven Methods. Biological psychiatry. 2020;88:70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Habes M, Pomponio R, Shou H, Doshi J, Mamourian E, Erus G, et al. The Brain Chart of Aging: Machine-learning analytics reveals links between brain aging, white matter disease, amyloid burden, and cognition in the iSTAGING consortium of 10,216 harmonized MR scans. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2021;17:89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Olsson B, Portelius E, Cullen NC, Sandelius A, Zetterberg H, Andreasson U, et al. Association of Cerebrospinal Fluid Neurofilament Light Protein Levels With Cognition in Patients With Dementia, Motor Neuron Disease, and Movement Disorders. JAMA neurology. 2019;76:318–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].van der Zande JJ, Steenwijk MD, Ten Kate M, Wattjes MP, Scheltens P, Lemstra AW. Gray matter atrophy in dementia with Lewy bodies with and without concomitant Alzheimer’s disease pathology. Neurobiology of aging. 2018;71:171–8. [DOI] [PubMed] [Google Scholar]
- [144].Harper L, Fumagalli GG, Barkhof F, Scheltens P, O’Brien JT, Bouwman F, et al. MRI visual rating scales in the diagnosis of dementia: evaluation in 184 post-mortem confirmed cases. Brain : a journal of neurology. 2016;139:1211–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Oppedal K, Ferreira D, Cavallin L, Lemstra AW, Ten Kate M, Padovani A, et al. A signature pattern of cortical atrophy in dementia with Lewy bodies: A study on 333 patients from the European DLB consortium. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2019;15:400–9. [DOI] [PubMed] [Google Scholar]
- [146].Josephs KA, Dickson DW, Tosakulwong N, Weigand SD, Murray ME, Petrucelli L, et al. Rates of hippocampal atrophy and presence of post-mortem TDP-43 in patients with Alzheimer’s disease: a longitudinal retrospective study. Lancet neurology. 2017;16:917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Josephs KA, Whitwell JL, Knopman DS, Hu WT, Stroh DA, Baker M, et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology. 2008;70:1850–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Schumacher J, Taylor JP, Hamilton CA, Firbank M, Cromarty RA, Donaghy PC, et al. In vivo nucleus basalis of Meynert degeneration in mild cognitive impairment with Lewy bodies. NeuroImage Clinical. 2021;30:102604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Yoo HS, Jeon S, Cavedo E, Ko M, Yun M, Lee PH, et al. Association of beta-Amyloid and Basal Forebrain With Cortical Thickness and Cognition in Alzheimer and Lewy Body Disease Spectra. Neurology. 2022;98:e947–e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Barber R, Gholkar A, Scheltens P, Ballard C, McKeith IG, O’Brien JT. MRI volumetric correlates of white matter lesions in dementia with Lewy bodies and Alzheimer’s disease. International journal of geriatric psychiatry. 2000;15:911–6. [DOI] [PubMed] [Google Scholar]
- [151].Delaby C, Alcolea D, Carmona-Iragui M, Illan-Gala I, Morenas-Rodriguez E, Barroeta I, et al. Differential levels of Neurofilament Light protein in cerebrospinal fluid in patients with a wide range of neurodegenerative disorders. Scientific reports. 2020;10:9161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Jellinger KA, Wenning GK, Seppi K. Predictors of survival in dementia with lewy bodies and Parkinson dementia. Neuro-degenerative diseases. 2007;4:428–30. [DOI] [PubMed] [Google Scholar]
- [153].Bostrom F, Hansson O, Blennow K, Gerhardsson L, Lundh T, Minthon L, et al. Cerebrospinal fluid total tau is associated with shorter survival in dementia with Lewy bodies. Dementia and geriatric cognitive disorders. 2009;28:314–9. [DOI] [PubMed] [Google Scholar]
- [154].Nelson PT, Kryscio RJ, Abner EL, Schmitt FA, Jicha GA, Mendiondo MS, et al. Acetylcholinesterase inhibitor treatment is associated with relatively slow cognitive decline in patients with Alzheimer’s disease and AD + DLB. Journal of Alzheimer’s disease : JAD. 2009;16:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Graff-Radford J, Boeve BF, Pedraza O, Ferman TJ, Przybelski S, Lesnick TG, et al. Imaging and acetylcholinesterase inhibitor response in dementia with Lewy bodies. Brain : a journal of neurology. 2012;135:2470–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Khachaturian ZS. The ‘Aducanumab Story’: Will the Last Chapter Spell the End of the ‘Amyloid Hypothesis’ or Mark a New Beginning? The Journal Of Prevention of Alzheimer’s Disease. 2022. [DOI] [PubMed] [Google Scholar]
- [157].Alzheimer’s Disease Interventions: Implications of therapeutic promises amidst questions and doubts about clinically meaningful outcomes. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2021;17:1591–4. [DOI] [PubMed] [Google Scholar]
- [158].Mielke MM, Dage JL, Frank RD, Algeciras-Schimnich A, Knopman DS, Lowe VJ, et al. Performance of plasma phosphorylated tau 181 and 217 in the community. Nature medicine. 2022;28:1398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Sharma A, Palaniappan L. Improving diversity in medical research. Nat Rev Dis Primers. 2021;7:74. [DOI] [PubMed] [Google Scholar]
- [160].Karim S, Xu Y, Kong S, Abdel-Rahman O, Quan ML, Cheung WY. Generalisability of Common Oncology Clinical Trial Eligibility Criteria in the Real World. Clin Oncol (R Coll Radiol). 2019;31:e160–e6. [DOI] [PubMed] [Google Scholar]
- [161].Gleason CE, Zuelsdorff M, Gooding DC, Kind AJH, Johnson AL, James TT, et al. Alzheimer’s disease biomarkers in Black and non-Hispanic White cohorts: A contextualized review of the evidence. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2022;18:1545–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Hill CV, Perez-Stable EJ, Anderson NA, Bernard MA. The National Institute on Aging Health Disparities Research Framework. Ethn Dis. 2015;25:245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Brewster P, Barnes L, Haan M, Johnson JK, Manly JJ, Napoles AM, et al. Progress and future challenges in aging and diversity research in the United States. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2019;15:995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Hansson O, Edelmayer RM, Boxer AL, Carrillo MC, Mielke MM, Rabinovici GD, et al. The Alzheimer’s Association appropriate use recommendations for blood biomarkers in Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Videnovic A, Ju YS, Arnulf I, Cochen-De Cock V, Hogl B, Kunz D, et al. Clinical trials in REM sleep behavioural disorder: challenges and opportunities. Journal of neurology, neurosurgery, and psychiatry. 2020;91:740–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Zeissler ML, Li V, Parmar MKB, Carroll CB. Is It Possible to Conduct a Multi-Arm Multi-Stage Platform Trial in Parkinson’s Disease: Lessons Learned from Other Neurodegenerative Disorders and Cancer. Journal of Parkinson’s disease. 2020;10:413–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Thorlund K, Haggstrom J, Park JJ, Mills EJ. Key design considerations for adaptive clinical trials: a primer for clinicians. BMJ. 2018;360:k698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Administration USDoHaHSFaD. Adaptive Designs for Clinical Trials of Drugs and Biologics. Guidance for industry. 2019. [Google Scholar]
- [169].Lu CC, Li XN, Broglio K, Bycott P, Jiang Q, Li X, et al. Practical Considerations and Recommendations for Master Protocol Framework: Basket, Umbrella and Platform Trials. Ther Innov Regul Sci. 2021;55:1145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Pepe MS, Etzioni R, Feng Z, Potter JD, Thompson ML, Thornquist M, et al. Phases of biomarker development for early detection of cancer. Journal of the National Cancer Institute. 2001;93:1054–61. [DOI] [PubMed] [Google Scholar]