

Abstract
Objectives
The complement system is an important component of innate immunity. The alternative pathway (AP) amplification loop is considered an essential feed forward mechanism for complement activation. However, the role of the AP in classical pathway (CP) activation has only been studied in ELISA settings. Here, we investigated its contribution on physiologically relevant surfaces of human cells and bacterial pathogens and in antibody‐mediated complement activation, including in autoimmune haemolytic anaemia (AIHA) setting with autoantibodies against red blood cells (RBCs).
Methods
We evaluated the contribution of the AP to complement responses initiated through the CP on human RBCs by serum of AIHA patients and recombinant antibodies. Moreover, we studied complement activation on Neisseria meningitidis and Escherichia coli. The effect of the AP was examined using either AP‐depleted sera or antibodies against factor B and factor D.
Results
We show that the amplification loop is redundant when efficient CP activation takes place. This is independent of the presence of membrane‐bound complement regulators. The role of the AP may become significant when insufficient CP complement activation occurs, but this depends on antibody levels and (sub)class. Our data indicate that therapeutic intervention in the amplification loop will most likely not be effective to treat antibody‐mediated diseases.
Conclusion
The AP can be bypassed through efficient CP activation. The AP amplification loop has a role in complement activation during conditions of modest activation via the CP, when it can allow for efficient complement‐mediated killing.
Keywords: alternative pathway, amplification loop, antibodies, autoimmune haemolytic anaemia, classical pathway, complement activation
The alternative pathway (AP) amplification loop is considered an essential feed forward mechanism for activation of the complement system. On physiologically relevant surfaces of red blood cells and bacteria, we show that the AP can be bypassed if classical pathway (CP) activation is strong, due to high antibody levels or antibody class. The AP has a role in complement activation during low activation via the CP, when it can allow for efficient complement‐mediated killing.

Introduction
The complement system is part of innate immunity and consists of approximately 50 circulating and membrane‐bound proteins. The system is essential for clearance of pathogens, immune complexes and cellular debris from the human body. Moreover, the complement system is important for homeostasis and has intracellular functions. 1 , 2 , 3 Despite its importance in the innate immune system, it is well‐recognised that the potent inflammatory effects of complement activation can be destructive for host cells. To prevent this, the system is tightly regulated by complement regulatory proteins. 4 , 5 The importance of these regulators is shown by the fact that mutations in complement regulators are linked to a large variety of complement‐mediated diseases. 6 , 7 , 8 , 9 Furthermore, hyperactivation of the complement system by autoantibodies may result in autoimmune diseases such as autoimmune haemolytic anaemia (AIHA), which is characterised by autoantibodies directed against red blood cells (RBCs) that activate the complement system, causing clearance of RBCs through extravascular or intravascular haemolysis. 10 , 11
The complement system can be activated via three pathways: the classical (CP), the lectin (LP) or the alternative pathway (AP) dependent on the nature of the activating ligand. The CP is activated by antibody–antigen complexes, the LP by pathogen‐specific carbohydrates and the AP is either activated spontaneously or by C3b as downstream amplifier of the CP or LP. During activation of the CP and the LP, complement proteins C4 and C2 are cleaved, forming the C3 convertase C4b2b on surfaces. The spontaneous activation of the AP is activated by the natural hydrolysis of C3 into C3(H2O), allowing for the binding of factor B (FB), which in turn is cleaved by the serine protease factor D (FD) resulting in the C3 convertase C3(H2O)Bb. Furthermore, the AP can be activated by C3b formed by the CP or LP, also known as the amplification loop. 1 , 12 After the formation of C3 convertases, C3 cleavage induces C5 convertase formation and terminal complement activation. 1
Complement activation drives several diseases, and although the main complement route at play is often clear, the role of additional pathways is not always fully understood. Some pathologies, such as age‐related macular degeneration, atypical haemolytic uremic syndrome and C3 glomerulopathy, are thought to depend on AP activation. 7 , 13 , 14 , 15 This is demonstrated by the fact that these diseases are linked to mutations in (regulators of) the AP. 16 , 17 , 18 , 19 For other, mainly antibody‐driven diseases such as AIHA and rheumatoid arthritis (RA), CP activation is key, but the role of the LP and AP in disease development and progression is often unclear. 11 , 20 , 21 This knowledge is highly relevant when developing disease‐specific complement‐targeting drugs. 6 The two complement therapeutics currently available in the clinic block C3 and C5 22 , 23 and thus do not target a specific activation pathway. However, complement therapeutics targeting specific complement activation pathways are emerging, such as the recently approved anti‐C1s sutimlimab, 24 for which a better understanding of the role of the AP in different diseases and contexts will be important. 6 , 11 , 25 , 26
Based on experiments using artificial surfaces devoid of complement regulators, it has been postulated that the AP has a large contribution to total complement activation (up to 80%) through amplification of the response after initial CP or LP activation. 27 , 28 The role of the AP‐mediated amplification during complement activation on physiologically relevant cell surfaces is poorly described, but previous evidence points towards a far smaller contribution during antibody‐mediated complement‐dependent cytotoxicity on cancer cells. 29 As several novel therapeutics are in development that aim to block the AP, 11 , 30 , 31 , 32 it is crucial to generate fundamental understanding of the role of AP amplification after CP and LP activation on physiological relevant surfaces. This may help to determine whether blocking the AP in certain diseases is beneficial and to which extent complement activation via the other routes remains intact upon AP inhibition. Here, we aimed at getting a further understanding of the conditions that influence the role of the amplification loop after CP activation. We evaluate its contribution to complement activation on human cells upon induction by autoantibodies from AIHA serum or recombinant antibodies, as well as to complement‐mediated bacterial killing. We show that the role of the AP‐mediated amplification after CP‐mediated complement activation depends on the strength of the initial CP activation, since the effect of AP amplification is mainly evident at low antibody concentrations. The amplification via the AP can become redundant at efficient CP activation by high antibody concentrations, leaving the CP‐mediated opsonisation and lysis intact upon AP inhibition. Thus, inhibition of the AP could be beneficial in diseases where the AP is shown to be involved, but our data indicate that AP inhibition may not be beneficial in antibody‐mediated disease.
Results
Complement deposition on human cells in AIHA is solely CP‐mediated
Previous studies, using artificial surfaces devoid of complement regulators, suggested that the AP contributes majorly to total complement activation after initial CP or LP activation. 27 , 28 Here, we set out to determine the effect of the AP during CP activation on human cells. To this end, we used a previously developed assay 33 in which we mimic the antibody‐mediated disease AIHA by incubating healthy donor RBCs with heat‐inactivated AIHA patient serum as an antibody source, supplemented with normal human serum (NHS) as an exogenous complement source. Complement activation was determined by detecting C4 and C3 deposition on the RBC surface 33 , 34 (see Supplementary figure 1a for the gating strategy). C4 and C3 deposition was completely abrogated using a blocking monoclonal anti‐C1q antibody, confirming CP initiated complement activation (Figure 1a). To investigate the contribution of the amplification loop to the total amount of complement activation, FB‐ or FD‐depleted serum was used as complement source, with or without the addition of purified FB or FD, respectively. To verify that FB‐ and FD‐depleted serum were indeed AP inactive, we incubated these sera (with or without the addition of purified FB or FD, respectively) in a Mg‐containing buffer on LPS‐coated 96‐wells plates and measured the amount of C3 deposition via ELISA. No C3 deposition was detected in the depleted sera, while C3 deposition was completely restored to the level of NHS when the depleted sera were supplied with purified FB or FD, respectively (Supplementary figure 2a), confirming that the depleted sera were complement active but devoid of AP activity. C4 (Figure 1a and b) and C3 (Figure 1a and c) deposition levels varied between patients, with highest levels for Patient 1. This was the only patient with IgM autoantibodies, which are known to result in strong complement activation. 11 , 35 Strikingly, the levels of both C4 and C3 deposition did not change between serum containing FB or FD or not, suggesting that AP does not play a detectable role in this set‐up. The addition of anti‐C1q resulted in C4 and C3 deposition levels comparable to the negative control, heat‐inactivated NHS (HI‐NHS) (Figure 1a–c), indicating that complement activation in these sera was induced by the CP. Next to C4 and C3 deposition, we determined the haemolysis caused by end‐stage complement activation on RBCs. Incubation of RBCs with AIHA sera mixed with various sera as complement source resulted in comparable levels of complement‐mediated lysis for all three patients, irrespective of whether the additional serum contained FB or FD (Figure 1d). Anti‐C1q inhibited the lysis of RBCs to background levels (HI‐NHS) in all patients (Figure 1d). We observed consistently that lack of the AP did not decrease complement activation, whereas inhibiting the CP reduced it to background level (Figure 1a–d). However, statistical significance for the latter observation was not reached due to high patient heterogeneity and the limited patient numbers.
Figure 1.
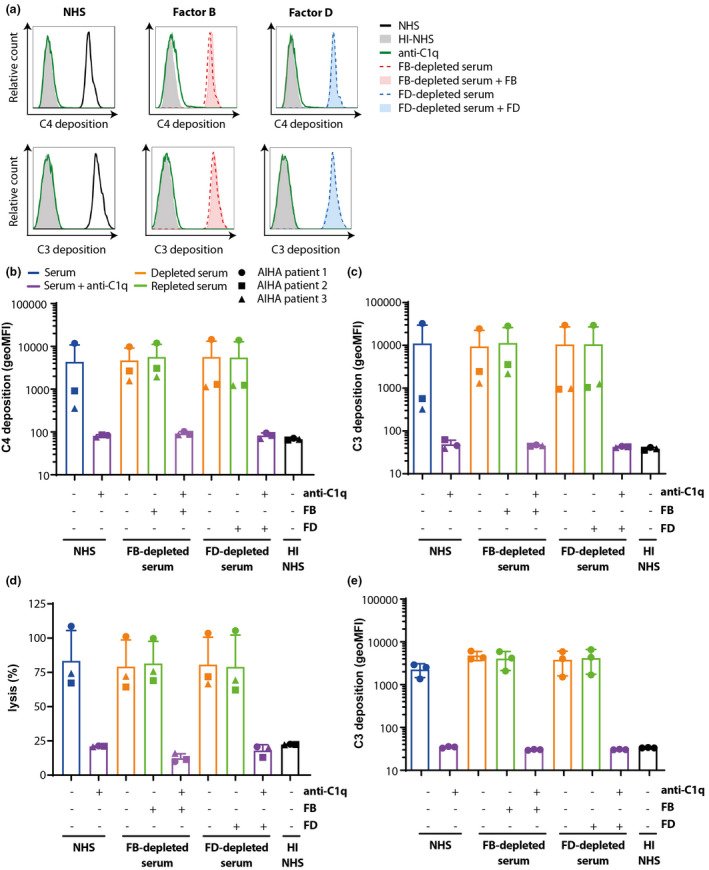
Complement activation in AIHA is fully CP‐mediated. (a) Sera of AIHA patients and NHS as complement source (25%, v/v) were incubated with healthy donor RBCs in the presence or absence of CP inhibitor anti‐C1q. C4 and C3 deposition was determined (black line) showing that incubation with anti‐C1q (green line) completely inhibited complement activation to background levels (HI‐NHS solid grey). (b, c) Sera of three AIHA patients (with ●, ▲ and ■ depicting individual patients) and FB‐depleted serum (red line) or FD‐depleted serum (blue line) supplemented with or without FB and FD respectively (solid blue/red versus dotted line) as complement source (25%, v/v) were incubated with healthy donor RBCs in the presence or absence of CP inhibitor anti‐C1q (green line) and C4 (a, b) or C3 (a–c) deposition was determined. C4 and C3 deposition in the absence or presence of FB or FD was equal in all situations. Anti‐C1q completely inhibited complement activation under all conditions. (d) Haemolysis of RBCs induced by AIHA patient antibodies, relative to 100% lysis by incubation with milliQ. (e) C3 deposition was measured on HAP1 CD46/CD55 KO cells, showing that in the absence of these complement regulators, the presence of FB or FD did not affect complement activation, while anti‐C1q could completely block complement activation. (a) Histograms for AIHA patient 1. (b–d) Mean with SD of three different AIHA patient sera. (e) Mean with SD of n = 3 independent experiments. Statistical significance was tested using the Friedman test with Dunn's multiple comparisons test for b, c, e and using repeated measures ANOVA with Dunnett's multiple comparisons test for d.
As these results were not in agreement with previous research in plate set‐ups in which a large role for the AP was found, 27 , 28 we hypothesised that this difference might be explained by the presence of membrane‐bound complement regulatory proteins on human cells, which do not exist in an ELISA set‐up. 4 Therefore, we next investigated the contribution of the amplification loop to complement deposition on human HAP1 cells that lack complement regulators CD46, CD55 and complement receptor 1, which inhibit surface C3 convertases 36 (see Supplementary figure 1b for the gating strategy). Incubation of these human complement regulator‐deficient cells with NHS resulted in C3 and C4 depositions (Figure 1e and Supplementary figure 3a–d). Similar to our results on RBCs, no difference in C3 and C4 deposition was observed after incubation with FB‐ or FD‐depleted sera with or without purified FB or FD supplementation, and complete inhibition of complement deposition was observed with anti‐C1q (Figure 1e and Supplementary figure 3b, c). This suggested that membrane‐bound complement regulators did not affect the contribution of the AP to total complement activation. When repeating these experiments with factor H (FH)‐depleted serum reconstituted with either FH or FH62V, a less potent regulator that allows higher AP activity, 19 we found no difference between the two variants. Control serum and AP inhibited serum resulted in high levels of C4 and C3 deposition for both FH variants, while inhibiting the CP strongly reduced complement deposition (Supplementary figure 3e, f). Thus, even with higher AP activity, no role for AP amplification following CP activation was observed. Altogether, these data demonstrated that in vitro complement activation on human RBCs by antibodies from AIHA serum, but also on HAP1 cells lacking membrane‐bound complement regulators, was predominantly mediated by the CP with no detectable contribution of the AP.
The contribution of the AP in CP‐mediated complement activation is determined by antibody density and class
As our first results on cells contradicted earlier studies using ELISA set‐ups, we set out to replicate the previous data. These ELISAs were also utilised to further investigate which conditions influence AP contribution and could be followed up upon in cellular assays. As CP activation depends on antibody density, 37 we varied the antibody concentrations to induce CP activation. To this end, ELISA plates were coated with 10‐fold difference concentrations of heat‐aggregated antibody (AHG), after which 25% human serum was added in the presence or absence of anti‐FB, anti‐FD or anti‐C1q blocking antibodies. The effectiveness of these anti‐FB and anti‐FD antibodies to block the AP was confirmed using an AP‐based ELISA set‐up detecting C3 deposition on coated LPS (Supplementary figure 2b). We observed that with high‐density AHG coat, C3 deposition was not inhibited by anti‐FB and anti‐FD antibodies (Figure 2). Only with low‐density AHG coat, and thus conditions for low CP activation, we observed inhibition at the level of C3b deposition on the ELISA plates when the AP was blocked. Anti‐C1q inhibited complement activation significantly in all conditions. This indicated that the AP contributes to C3 deposition following conditions of low CP activation.
Figure 2.
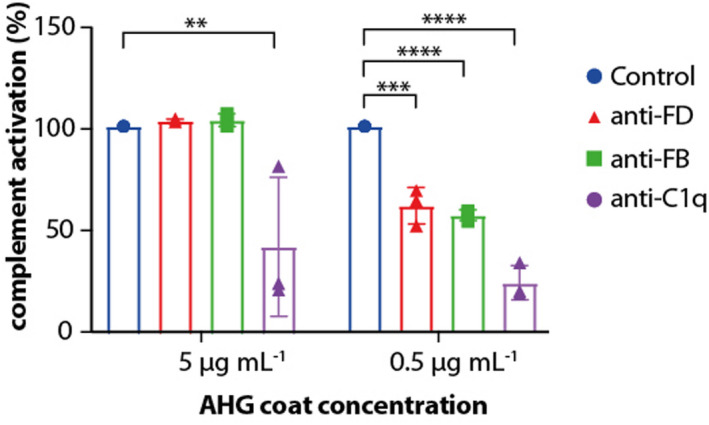
Contribution of the AP to complement activation depends on antibody concentration. In ELISA setting, C3b deposition was measured on varying densities of heat agglutinated IgG (AHG) coat, with 25% (v/v) serum and antibodies to block the alternative or classical pathway, respectively. At high AHG densities, inhibiting the AP with anti‐FB or anti‐FD had no effect on C3b deposition. Bars show mean + SD of n = 3 independent experiments, statistical significance was tested using one‐way ANOVA with Dunnett's multiple comparisons test. **P < 0.01, ***P < 0.001, ****P < 0.0001.
To determine the AP contribution to CP‐initiated complement activation on RBCs, we utilised biotinylated RBCs in combination with monoclonal antibiotin antibodies of different (sub)class. 38 , 39 As we used O erythrocytes and AB serum, this system allowed us to control the specific amount and (sub)class of antibody present. We investigated the role of the AP in CP‐mediated haemolysis using human antibiotin IgG1, IgG2, IgG3, IgG4 and IgM. IgG2 and IgG4, which have been described as less potent CP activators in the literature, 40 did not induce any haemolysis in our system, so the role of the AP could not be determined (data not shown). CP inhibition almost completely reduced haemolysis in all conditions, regardless of antibody (sub)class (Figure 3a–c). Using IgG1 as a complement activation trigger (Figure 3a), the results for control and anti‐FB were similar and the antibiotin EC50 was not affected by AP inhibition. With IgG3, AP inhibition resulted in a curve that is no longer parallel to the control and significantly increased the EC50 of antibiotin (Figure 3b). For IgM, there was no change in parallelism between the anti‐FB and control curve, and the EC50 was unaffected (Figure 3c). IgM is a highly potent CP activator that does not require oligomerisation before C1q binding, unlike IgGs, 35 , 37 which could explain the lack of AP contribution at all tested IgM concentrations.
Figure 3.
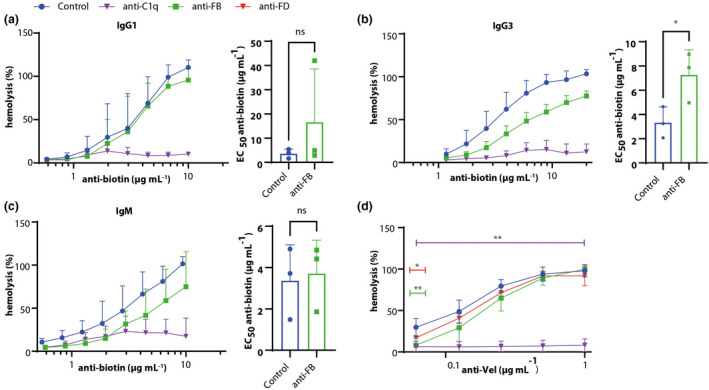
AP contributes to haemolysis upon IgG3‐induced complement activity. Biotinylated RBCs were incubated with IgG1 (a), IgG3 (b) or IgM (c) antibiotin antibodies, with or without anti‐FB or anti‐C1q to induce haemolysis. The addition of anti‐C1q was able to inhibit haemolysis as induced by IgG1, IgG3 and IgM almost completely. EC50 was calculated to compare control to anti‐FB. With IgG1‐induced activation (a), there was no inhibition by anti‐FB. In IgG3‐induced activation (b), anti‐FB reduced haemolysis at most concentrations and statistically significant increased the EC50. In IgM‐induced activation (c), anti‐FB did not significantly reduce haemolysis or increase the EC50. (d) Haemolysis as initiated by an anti‐Vel IgM antibody in a more sensitive system, adding a FH‐inhibiting antibody and bromelain‐treated RBCs. At high anti‐Vel concentrations, there was no effect of inhibiting the AP, but at lowest antibody concentrations, AP inhibition had a statistical significant effect. Blocking the CP inhibited haemolysis completely. (a–c) Haemolysis as induced by milliQ was set to 100%. (a–c) Mean + SD from n = 3, statistical significance tested for EC50 using the paired t‐test; (d) mean + range from n = 2, statistical significance tested using one‐way ANOVA with Dunnett's multiple comparisons test, compared with the serum only condition. * P < 0.05, ** P < 0.01.
In order to further understand whether the AP can play a role upon IgM‐triggered CP activation, we used an additionally sensitised haemolytic assay, utilising bromelain‐treated RBCs and a FH‐inhibiting antibody combined with anti‐Vel IgM antibodies to induce complement activation. Anti‐C1q inhibited the anti‐Vel‐mediated complement activation completely showing a significant reduction in all anti‐Vel concentrations, clearly indicating that lysis of the RBCs in this system was predominantly CP‐mediated. AP inhibition only decreased the complement‐mediated lysis of RBCs at low anti‐Vel concentrations (Figure 3d), with a significant reduction only at 0.06125 μg mL−1 anti‐Vel. In agreement with our ELISA data and the antibiotin IgM data, this indicated that the AP is an important contributor to the CP when the initial activation, due to lower antibody titers, is less robust, even when the AP is dysregulated by FH inhibition.
Overall, using ELISA and cell‐based assays, we show that the contribution of the AP on CP activation is only observed when limited antibody levels, or less potent activators, spark the initial CP‐mediated complement activation.
Classical pathway complement activation on bacterial surfaces bypasses the amplification loop in the presence of antibodies
The complement system is vital to ward off invading pathogens, such as bacteria. To take a broader look at the contribution of the AP to CP complement activation on biological surfaces, we investigated the role of the amplification loop during opsonisation and lysis of the bacterial pathogen Neisseria meningitidis. To this end, we used serum of a genetically FD‐deficient patient before and after vaccination against N. meningitidis. 41 , 42 A previous study reported antibodies reactive with N. meningitidis in this patient prevaccination, and increased antibody titre after vaccination. 42 C3 deposition (Figure 4a), C5b‐9 deposition (Figure 4b) in FACS (see Supplementary figure 1c for gating strategy) and killing (Figure 4c) of N. meningitidis by FD‐deficient serum was low and could be significantly increased by addition of FD in prevaccination serum. This difference by FD addition was lost in serum of the same patient after vaccination against N. meningitidis. Anti‐C1q completely blocked complement activation and bacterial killing in all conditions (Figure 4a–c). This indicates that the AP only plays a measurable role during complement opsonisation and killing of N. meningitidis in the absence of a robust antibody response, similar as observed for RBCs and HAP1 cells (Figure 2d and e and Figure 3).
Figure 4.
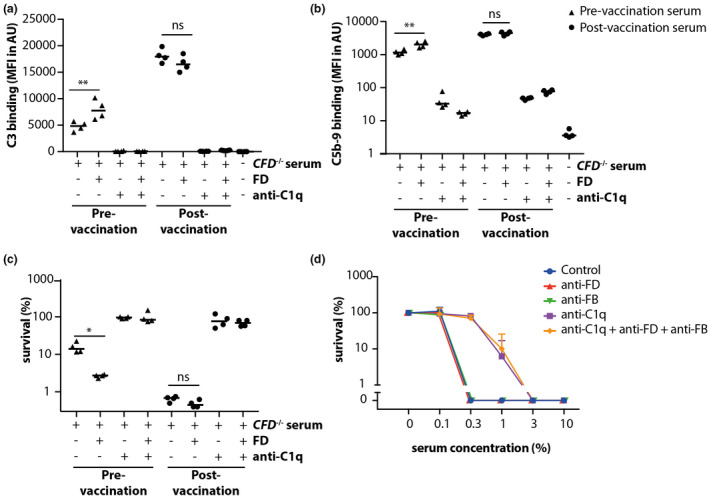
Complement activation on Neisseria meningitidis bypasses the amplification loop in post‐vaccination serum. Neisseria meningitidis was incubated with 10% FD‐deficient patient serum before (▲) and after (●) MenB‐4C vaccination. (a, b) show C3 and C5b‐9 binding, respectively, as determined by flow cytometry on whole bacteria. Both are statistically significant increased by the addition of FD to the deficient serum before vaccination, but this effect was lost postvaccination. The addition of anti‐C1q resulted in complete inhibition of C3 deposition and strong reduction of C5b‐9 deposition in all circumstances. (c) Bacterial survival upon serum incubation as determined by CFU, demonstrating that bacterially killing was increased by addition of FD in the prevaccination serum, but not the postvaccination serum. When anti‐C1q was added, bacterial survival was around 100%, indicating that CP activation was required for bacterial killing. (d) Complement‐mediated E. coli killing was inhibited by addition of anti‐C1q alone to a similar extent as addition of anti‐C1q, anti‐FB and anti‐FD combined. Anti‐FB and anti‐FD alone were unable to prevent complement‐mediated bacterial killing. (a–c) Mean and individual data points from four independent experiments, (d) mean + SD from n = 3 independent experiments. Statistical significance in a–c was tested using the paired t‐test, * P < 0.05, ** P < 0.01.
As N. meningitidis has specific complement evasion mechanisms, such as FH binding to inhibit AP activation, results observed with N. meningitidis might not be applicable to other bacteria. To explore whether these findings are consistent with other bacteria, we also studied the role of the AP using Escherichia coli as a model organism. Bacterial killing did not occur upon incubation with HI‐NHS (data not shown) or NHS with eculizumab (Supplementary figure 4), indicating that bacterial killing was terminal complement‐mediated. Upon the addition of anti‐C1q to serum, we observed strong inhibition of bacterial killing. No additional effect was observed when combining anti‐C1q, anti‐FB and anti‐FD (Figure 4d). Individually, the addition of anti‐FB or anti‐FD to NHS showed no inhibition of complement‐mediated killing.
In conclusion, the role of the AP on bacterial surfaces in vitro was similar to our observations with human RBCs and seems restricted to situations in which limiting amounts of antibodies are present.
Discussion
Here, we studied the role of the AP during CP‐initiated complement activation on physiologically relevant human cell surfaces and bacteria. In contrast to what was reported previously in ELISA‐based assays, 28 the role of the AP on cell surfaces is not as prominent and is determined by the robustness of the initial antibody trigger of the CP.
Understanding the role of the AP in disease is important as a large variety of specific AP blocking therapeutics are currently in development. 11 , 30 , 31 , 32 To date, the role of the AP in CP‐ and LP‐mediated complement activation is considered to be extensive based on ELISA data detecting terminal complement activation. 27 , 28 However, ELISA plates are substantially different from human cells, as they are devoid of membrane‐bound complement regulators which are known to be of great importance during several complement‐mediated diseases. 5 , 6 We show that the AP plays a significant role in ELISA at low antibody levels, where the residual complement activation upon anti‐C1q addition could be explained by spontaneous AP activation on the plastic plate or by LP activation. We then assessed the role of the AP in CP‐mediated complement activation on human and bacterial cell surfaces. We found that the role of the AP in CP‐mediated complement activation on surfaces is limited and restricted to situations where only modest levels of antibodies are present, resulting in less robust complement activation. AP contribution was not affected by membrane‐bound complement regulators. This even holds when the AP has become dysregulated by the presence of less potent FH. Thus, the AP‐mediated amplification seems mainly relevant when complement activation by the CP is modest. This means that the AP becomes redundant when antibodies can activate the CP strongly because of a high titre, or because of strong C1q binding as is seen for IgM. 43 This observation is in agreement with earlier studies where the role of the AP was nearly undetectable when efficient CP activation was induced by HexaBodies 29 and in vitro blocking of the AP did not affect complement activation on Streptococcus pneumoniae and N. meningitidis in vaccinated serum. 44 , 45
Previous research has mainly used murine models to assess the contribution of the AP in CP‐mediated diseases. Models for RA, antiphospholipid syndrome and myasthenia gravis indicated involvement of AP activation in disease outcome. 46 , 47 , 48 , 49 This seems contradictory to data presented in this paper. However, several details should be taken into account to put our data in perspective with previous animal research. First, the murine models for complement‐mediated diseases cannot directly be translated to human conditions as complement activation varies between mouse strains and even the gender of the mice seems to have influence on complement activity. 50 Mice also have different complement regulatory proteins than humans, which makes it difficult to extrapolate results from murine models to the human situation. 50 , 51 For example, mice only have three instead of the human five FH related‐proteins (FHRs), 52 a different expression pattern of most membrane‐bound complement regulators and mice express Crry instead of CR1. 51 Second, our results indicate differences between antibody (sub)classes and titres, especially between IgG and IgM, which other studies did not compare (sub)classes. Lastly, when looking into RA specifically, inflammation and complement activation occurs in the joints and on cartilage, as well as systemically. Although it is generally accepted that the CP is strongly involved in RA via both IgG and IgM antibodies in the joints, 53 previous clinical research showed systemic involvement of both the AP and the CP. 54 It is suggested that immune complexes on the articular surface initiate an inflammatory cascade via the AP in mice. 46 , 55 The mice model in these papers mainly depends on IgG1 antibodies, which are known to be a poor activator of the murine CP. 46 This could explain the observation of the strong involvement of the AP here: the amplification loop of the AP kicks into play when CP activation is low as we have shown in Figures 3 and 4.
Relevance of the AP in the killing of bacteria has been shown in vivo in some studies, particularly mouse models. Many pathogenic bacteria use FH binding as complement evasion strategy, 56 , 57 indicating the importance of the AP in vivo. One patient with FB deficiency was reported to suffer from pneumococcal infections and meningitis caused by N. meningitidis, 58 which seized upon vaccination and antibiotic prophylaxis. Furthermore, genetic FD deficiency has been described to be associated with meningococcal infections in humans. 41 , 59 , 60 These studies do not report on initial antibody titers against meningococci, and most cases describe young, unvaccinated infants. With regard to animal studies, C3 opsonisation of S. pneumoniae was lower when serum of FD‐deficient mice was used, than serum of FD‐sufficient mice. 61 In line with these results, C3 opsonisation of S. pneumoniae in FB‐deficient mice was reduced compared to wild‐type mice, in addition to lower phagocytic capacity. 62 Discrepancies between these previously published papers and our experiments on bacterial surfaces regarding the role of the AP might be explained by the fact that laboratory mice most likely do not have specific antibodies, and therefore have minimal CP activation, against the described bacterial pathogens. This is in agreement with our current data (Figure 4a–c) and previous publications that show that the role of the AP diminishes upon vaccination. 44 , 45
Taken together, lacking a functional AP by missing FD or FB seems to result in higher susceptibility for certain bacteria in both mice and humans. Based on our current results, we conclude that the AP is important for the clearance of pathogens by C3b opsonisation and membrane attack complex formation upon first encounter with the pathogen, when there is no efficient antibody response yet. When antibodies have been formed through exposure or vaccination, the CP may be activated via these antibodies in which case the AP amplification loop is not required, as underlined by our current data. Future research could investigate how the combination of different antibody (sub)classes within a polyclonal antibody response would influence the contribution of the different activation pathways. One interesting follow‐up question would then be to investigate whether the presence of IgG2 or IgG4 antibodies, which do not (strongly) activate complement, would lead to competition with complement‐activating (sub)classes for epitope binding, or reduce hexamerisation efficiency, and thus result in less optimal CP activation.
In conclusion, we show that the importance of the AP amplification in CP‐mediated complement activation depends on the robustness of the initial activation trigger. Although we have not studied this for complement activation via the LP, we hypothesise, based on the similarities between the CP and LP complement activation, 1 , 63 that this will also apply to the contribution of the AP amplification in LP dependent complement activation. Therefore, intervention in the amplification loop may not be an effective therapy for human CP‐mediated diseases such as AIHA, in which complement activation via antibodies is the sole activation route. Treatment of CP‐initiated autoimmune diseases may be most effective when using complement inhibitory drugs targeting the very initiation of the CP, for example by C1‐esterase inhibitor or sutimlimab, 24 both inhibiting the CP protease C1s. 64 , 65 , 66 , 67 Targeting the AP remains relevant in AP‐mediated disease. We have shown that CP activation is not abrogated by AP inhibition if antibody titres are high. This would indicate that the AP can probably be safely inhibited in patients without diminishing CP activity with the additional advice of vaccination against pathogens such as N. meningitidis.
Methods
Serum material
From a previous study, 68 three anonymised serum samples of AIHA patients were used as antibody source in which complement was inactivated by incubation for 30 min at 56°C. Patient 1 was diagnosed with IgM class autoantibodies, Patients 2 and 3 with IgG autoantibodies. A NHS pool was created from eight different healthy volunteers after informed consent as used in the C3 and C4 deposition assays on RBCs and HAP1 cells, except for the HAP1 assay using FH62V where we used a pool of 148 healthy volunteers after informed consent. An NHS pool consisting of 30 different healthy volunteers after informed consent was used for the ELISAs and E. coli experiments. AB serum was obtained from individual healthy donors after informed consent and used for the haemolytic assays. As a negative control for complement activation, NHS was heat inactivated for 30 min at 56°C (HI‐NHS). FB‐, FD‐ and FH‐depleted serum were obtained from CompTech (Tyler, TX, USA). Serum of a patient with homozygous CFD mutation before and after meningococcal ACYW and 4CMenB vaccination was obtained as described by Sprong et al. 41 and van den Broek et al. 42
Antibodies and proteins
To detect complement deposition in‐house monoclonal antibody (mAb), anti‐C3‐19 (which binds C3 fragments C3b, iC3b, C3d and C3dg) and anti‐C4‐10 (which binds C4b and C4d) were used. 33 , 69 In‐house inhibitory mAb anti‐C1q‐85 was used to block the CP 70 at three times excess of C1q serum concentration, in‐house inhibitory mAb anti‐FB‐1 and anti‐FD (Lampalizumab; Genentech, San Francisco, CA, USA) were used to inhibit the AP at 2.5 times excess of the serum concentration of their respective target. Inhibitory mAb anti‐C5 (Eculizumab; Alexion Pharmaceutical, Cheshire, CT) was obtained from remnants of used Soliris® injection bottles and used to block terminal complement activation to prevent lysis during the experiments. Antibiotin and anti‐Vel antibodies for the haemolytic assays were produced in‐house, using sequences described by Bagçi et al., 39 Kohen et al., 38 van der Rijst et al. 71 and Oskam et al. 72 and methods described by Falkenburg et al. 73 In‐house inhibiting mAb anti‐FH.09 was used in the haemolytic assay with anti‐Vel at 2.5 times excess of FH serum concentration. 74 Purified FB, FD and FH were obtained from CompTech. In all experiments, 75 μg mL−1 purified FB and 0.5 μg mL−1 purified FD were added to substitute 25% (v/v) FB‐ or FD‐depleted serum, respectively, to approach normal levels of these proteins. FH62V was affinity purified from EDTA plasma. 19 300 μg mL−1 FH or FH62V was used to reconstitute FH‐depleted serum.
Alternative pathway and CP C3 deposition ELISA
ELISAs were performed as described by Dekkers et al. 75 For the alternative pathway ELISA, Nunc Polysorp flat‐bottom plates (Thermo Scientific, Rockland, IL, USA) were coated overnight with 40 μg mL−1 lipopolysaccharide from Salmonella typhosa (LPS, Sigma‐Aldrich, St. Louis, MI, USA) in phosphate‐buffered saline (PBS) at room temperature (RT). Plates were washed and incubated for 1 h at 37°C with 20% (v/v) NHS or FB‐or FD‐depleted serum, with or without purified FB (162.5 μg mL−1) or FD (1.05 μg mL−1) supplementation in Veronal Buffer (1.8 mm sodium barbital and 3.1 mm barbituric acid, pH 7.3–7.4; VB) with 0.1% (w/v) Tween‐20, 0.3% (w/v) BSA, 5 mm MgCl2 and 10 mm EDTA. For the CP ELISA, Nunc Maxisorp flat‐bottom plates (ThermoFisher Scientific, Whaltam, MA, USA) were coated overnight with 5 or 0.5 μg mL−1 agglutinated human IgG (AHG, Sanquin Reagents) in PBS at RT. Plates were washed and incubated for 1 h at RT with 25% (v/v) NHS with or without anti‐FB (202 μg mL−1), anti‐FD (1.3 μg mL−1) or anti‐C1q (31.75 μg mL−1) in VB with 0.1% (w/v) Tween‐20, 0.3% (w/v) BSA, 1 mm CaCl2 and 0.5 mm MgCl2. ELISA was performed as described above. Biotinylated anti‐C3.19 was used as conjugate and ELISA was developed as described by Dekkers et al., 75 with the exception of the usage of streptavidin‐HRP for the classical pathway ELISA.
Complement activation on RBCs and human cells deficient for CD46/CD55
C3 and C4 deposition on RBCs was assessed as described previously. 33 In short, 0.08% (v/v) bromelain‐treated O‐typed RBCs were incubated with 3% (v/v) AIHA patient serum as antibody source, 25% (v/v) serum as complement source (NHS, FB‐ or FD‐depleted serum) and in the presence of at least equimolar inhibitory mAb anti‐C5 in VB with 0.1% (w/v) gelatin (VBG) supplemented with 10 mm CaCl2 and 2 mm MgCl2 (VBG++) for 1.5 h at 37°C. As described previously, 36 human CD46/55‐deficient HAP1 cells (1.0 × 105 cells) were incubated with 25% (v/v) complement source (NHS, FB‐, FD‐ or FH‐depleted human serum) and at least equimolar inhibitory mAb anti‐C5 in VB++ for 1 h at 37°C.DAPI was used at 1 μm to detect dead cells. C3 and C4 deposition was detected by using 1.0 μg mL−1 DyLight 488‐conjugated anti‐C3‐19 and 1.0 μg mL−1 DyLight 647‐conjugated anti‐C4‐10. LSR Canto II flow cytometer (BD Biosciences, San Jose, CA, USA) was used for measuring and data analysis was performed using the FlowJo software v1.0 (BD Biosciences, San Jose, CA, USA).
Haemolytic assay induced by AIHA serum
0.4% (v/v) O‐typed RBCs were treated with bromelain (Sanquin Reagents, Amsterdam, NL) opsonised with 10% (v/v) AIHA patient sera were incubated for 1.5 h at 37° C with 25% (v/v) serum as complement source (NHS, FB‐ or FD‐depleted human serum) with or without purified FB or FD supplementation. Lysis was measured as absorbance of the supernatant at 412 nm and corrected for the absorbance at 690 nm with a Synergy 2 plate reader (BioTek Instruments, Winooski, VT, USA) and expressed as percentage of a 100% lysis control (RBCs incubated in MilliQ).
Haemolytic assay induced by anti‐Vel and antibiotin
To determine the effect of antibody concentration and (sub)class on AP contribution on cells, we used an antibiotin haemolytic assay. O‐typed RBCs were biotinylated in 0.11 mm Pierce Premium Grade Sulfo‐NHS‐LC‐Biotin (Thermo Scientific). Cells were diluted to 1% (v/v) in VBG with 2 mm CaCl2 and 1 mm MgCl2 and incubated with antibiotin antibodies and 25% (v/v) healthy AB serum, with or without anti‐FB (162.5 μg mL−1) and anti‐C1q (75 μg mL−1) to inhibit the AP and CP, respectively, at 2–3 times excess. Antibiotin antibodies were added at a maximum concentration of 20 μg mL−1 for IgG1 and IgM and at 40 μg mL −1 for IgG2, −3 and − 4 and diluted 1:1.5. Cells were incubated at 37°C for 1 h while shaking. Lysis was measured as absorbance of the supernatant at 412 nm and corrected for the absorbance at 690 nm with a Synergy 2 plate reader (BioTek Instruments) and expressed as percentage of a 100% lysis control (RBCs incubated in MilliQ).
To further assess the role of the AP on RBC surfaces, we used our previously set‐up haemolytic assay that was optimised to detect complement‐activating antibodies by the addition of a FH inhibiting antibody 74 and using bromelain‐treated RBCs. 25% (v/v) healthy AB serum was pre‐incubated with inhibiting anti‐FH.09 (59.5 μg mL−1) and with or without anti‐FD (1.38 μg mL−1), anti‐FB (150 μg mL−1) and anti‐C1q (75 μg mL−1) in VBG with 1 mm CaCl2 and 0.5 mm MgCl2. Anti‐Vel IgM was added at 1 μg mL−1 and titrated 1:2. 0.5% (v/v) bromelain‐treated O‐typed RBCs were added and incubated with serum and antibodies at 37 °C for 1 h while shaking. Lysis was measured as absorbance of the supernatant at 412 nm and corrected for the absorbance at 690 nm with a Synergy 2 plate reader (BioTek Instruments, Winooski, VT, USA) and expressed as percentage of a 100% lysis control (RBCs incubated in MilliQ).
C3 and C5b‐9 binding to Neisseria meningitidis
Bacteria were grown in Tryptic Soy Broth to OD620 ~ 0.23, washed once with HBSS + Ca2+/Mg2+ + 0.1% (w/v) gelatin (HBSS3+) and diluted to an OD620 of 0.2 with HBSS3+. Twenty‐five microlitre bacteria was mixed with 25 μL 20% (v/v) patient serum diluted in HBSS3+ with or without reconstitution of 0.2 μg mL−1 FD (CompTech) and/or 30 μg mL−1 anti‐C1q and incubated 30 min at 37°C. Bacteria were pelleted by centrifugation at 3200× g and fixed for 20 min in 2% (w/v) paraformaldehyde in PBS at RT. Surface‐bound complement C3 and complement complex C5b‐9 were detected with 1:500‐diluted FITC‐labelled polyclonal goat anti‐human C3 (MP biomedicals) and 1:100‐diluted monoclonal mouse antihuman C5b‐9 (Clone aE11, Santa Cruz Biotechnology, Santa Cruz, CA, USA) followed by 1:500‐diluted Alexa647‐labelled donkey antimouse IgG (Invitrogen). Surface binding of C3 and C5b‐9 was determined by flow cytometry using a FACS LSR II instrument (BD Biosciences, San Jose, CA, USA) and expressed in mean fluorescence intensity (MFI) in arbitrary units (AU). Data were analysed by using FlowJo version 10.4.1 (BD Biosciences).
Neisseria meningitidis killing assay
Neisseria meningitidis was grown fresh in GC broth at 37°C and 5% CO2 to an OD620 of 0.23, washed once with HBSS3+ and diluted 800‐fold in HBSS3+ to obtain a concentration of ~200 000 colony forming unit (CFU) mL−1. Twenty microlitre bacteria was mixed with 20 μL 20% (v/v) patient serum or HI‐NHS diluted in HBSS3+ with or without reconstitution of 0.2 μg mL−1 FD (CompTech) and/or 30 μg mL−1 anti‐C1q blocking antibody and incubated 30 min at 37°C. Samples were diluted 10‐, 100‐, 1000‐fold with PBS and 10 μL droplets were plated on GC agar plates and grown overnight at 37°C and 5% CO2. Survival was determined by dividing the CFU counts in patient serum by the CFU count in HI‐NHS after a 30‐min incubation and presented as percentage.
Escherichia coli killing assay
An overnight E. coli (MG1655) culture was diluted (1:100) and then grown to an OD620 of 0.5 at 37°C. Bacteria were diluted to 10 000 CFU mL−1 in sterile RPMI/HAS, and 50 μL of bacteria was incubated with a 1:3 serial dilution of NHS and HI‐NHS, starting at 10% (v/v), with or without anti‐C1q (17 μg mL−1), anti‐FD (1.5 μg mL−1) and anti‐FB (65 μg mL−1) and incubated for 1 h while shaking at 37°C. Bacteria were plated at four serial dilutions of 1:10 in PBS on agar plates and incubated overnight at 37°C. CFU were counted the next day.
Statistics
Analysis and statistical tests were performed using GraphPad Prism (version 9.1.1; GraphPad Software, San Diego, CA, USA).
Author contributions
Esther CW de Boer: Conceptualization; data curation; formal analysis; investigation; methodology; visualization; writing – original draft; writing – review and editing. Astrid JF Thielen: Conceptualization; data curation; investigation; methodology; writing – original draft; writing – review and editing. Jeroen D Langereis: Data curation; formal analysis; investigation; writing – review and editing. Angela Kamp: Investigation. Mieke C Brouwer: Investigation. Nienke Oskam: Methodology; writing – review and editing. Marlieke LM Jongsma: Methodology. April J Baral: Methodology; writing – review and editing. Robbert M Spaapen: Methodology; writing – review and editing. Sacha Zeerleder: Conceptualization; methodology. Gestur Vidarsson: Methodology; writing – review and editing. Theo Rispens: Methodology; writing – review and editing. Diana Wouters: Conceptualization; methodology; writing – review and editing. Richard B Pouw: Methodology; supervision; writing – review and editing. Ilse Jongerius: Conceptualization; funding acquisition; methodology; supervision; writing – original draft; writing – review and editing.
Conflict of interest
MCB, TR, DW, RBP and IJ are co‐inventors of (multiple) patents and/or patent applications describing the therapeutic use of anti‐FH antibodies. All other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflict of interest.
Supporting information
Supplementary figures 1–4
Acknowledgments
This work was supported by internal grants of Sanquin (PPOC‐12‐038‐PRG and PPOC‐19‐25/L2476) and part of the research programme Aspasia with project number 015.014.069, which is (partially) financed by the Netherlands Organisation for Scientific Research (NWO) to IJ. RMS was supported by an NWO‐VENI personal grant (016.131.047). We kindly thank prof. Claire Harris for providing us with the FH62V.
References
- 1. Merle NS, Church SE, Fremeaux‐Bacchi V, Roumenina LT. Complement system part I ‐ molecular mechanisms of activation and regulation. Front Immunol 2015; 6: 1778–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hess C, Kemper C. Regulation of metabolism and basic cellular processes. Immunity 2016; 45: 240–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reichhardt MP, Meri S. Intracellular complement activation—an alarm raising mechanism? Semin Immunol 2018; 38: 54–62. [DOI] [PubMed] [Google Scholar]
- 4. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol 2009; 9: 729–740. [DOI] [PubMed] [Google Scholar]
- 5. Defendi F, Thielens NM, Clavarino G, Cesbron JY, Dumestre‐Pérard C. The immunopathology of complement proteins and innate immunity in autoimmune disease. Clin Rev Allergy Immunol 2020; 58: 229–251. [DOI] [PubMed] [Google Scholar]
- 6. de Boer ECW, van Mourik AG, Jongerius I. Therapeutic lessons to be learned from the role of complement regulators as double‐edged sword in health and disease. Front Immunol 2020; 11: e578069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang T, Lu J, Liang S et al. Comprehensive analysis of complement genes in patients with atypical hemolytic uremic syndrome. Am J Nephrol 2016; 43: 160–169. [DOI] [PubMed] [Google Scholar]
- 8. Brodsky R. Paroxysmal nocturnal hemoglobinuria. Blood 2014; 124: 2804–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lorés‐Motta L, van Beek AE, Willems E et al. Common haplotypes at the CFH locus and low‐frequency variants in CFHR2 and CFHR5 associate with systemic FHR concentrations and age‐related macular degeneration. Am J Hum Genet 2021; 108: 1367–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am 2017; 101: 351–359. [DOI] [PubMed] [Google Scholar]
- 11. Jalink M, de Boer ECW, Evers D et al. Halting targeted and collateral damage to red blood cells by the complement system. Semin Immunopathol 2021; 43: 799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol 2016; 12: 383–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haydock L, Garneau AP, Tremblay L, Yun H, Hanlin Y, Raphaël G. Genetic abnormalities in biopsy ‐ proven, adult ‐ onset hemolytic uremic syndrome and C3 glomerulopathy. J Mol Med 2022; 100: 269–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raina R, Krishnappa V, Blaha T et al. Atypical hemolytic‐uremic syndrome: an update on pathophysiology, diagnosis, and treatment. Ther Apher Dial 2019; 23: 4–21. [DOI] [PubMed] [Google Scholar]
- 15. Armento A, Ueffing M, Clark SJ. The complement system in age ‐ related macular degeneration. Cell Mol Life Sci 2021; 78: 4487–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nester CM, Barbour T, de Cordoba SR et al. Atypical aHUS: state of the art. Mol Immunol 2015; 67: 31–42. [DOI] [PubMed] [Google Scholar]
- 17. Arjona E, Huerta A, Goicoechea De Jorge E, Rodríguez de Córdoba S. The familial risk of developing atypical hemolytic uremic syndrome. Blood 2020; 136: 1558–1561. [DOI] [PubMed] [Google Scholar]
- 18. Moore I, Strain L, Pappworth I et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood 2010; 115: 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heurich M, Martínez‐Barricarte R, Francis NJ et al. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc Natl Acad Sci USA 2011; 108: 8761–8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dijkstra DJ, Joeloemsingh JV, Bajema IM, Trouw LA. Seminars in immunology complement activation and regulation in rheumatic disease. Semin Immunol 2019; 45: 1–9. [DOI] [PubMed] [Google Scholar]
- 21. Berentsen S, Sundic T. Red blood cell destruction in autoimmune hemolytic anemia: role of complement and potential new targets for therapy. Biomed Res Int 2015; 2015: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hillmen P, Szer J, Weitz I et al. Pegcetacoplan versus eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med 2021; 384: 1028–1037. [DOI] [PubMed] [Google Scholar]
- 23. Lee JW, de Fontbrune FS, Lee LWL et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood 2019; 133: 530–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Röth A, Berentsen S, Barcellini W et al. Sutimlimab in patients with cold agglutinin disease: results of the randomized placebo‐controlled phase 3 CADENZA trial. Blood 2022; 140: 980–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harris CL, Pouw RB, Kavanagh D, Sun R, Ricklin D. Developments in anti‐complement therapy; from disease to clinical trial. Mol Immunol 2018; 102: 89–119. [DOI] [PubMed] [Google Scholar]
- 26. Mastellos DC, Ricklin D, Lambris JD. Clinical promise of next‐generation complement therapeutics. Nat Rev Drug Discov 2019; 18: 707–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harboe M, Garred P, Karlstrøm E, Lindstad JK, Stahl GL, Mollnes TE. The down‐stream effects of mannan‐induced lectin complement pathway activation depend quantitatively on alternative pathway amplification. Mol Immunol 2009; 47: 373–380. [DOI] [PubMed] [Google Scholar]
- 28. Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol 2004; 138: 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cook EM, Lindorfer MA, van der Horst H et al. Antibodies that efficiently form hexamers upon antigen binding can induce complement‐dependent cytotoxicity under complement‐limiting conditions. J Immunol 2016; 197: 1762–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Holz FG, Sadda SR, Busbee B et al. Efficacy and safety of lampalizumab for geographic atrophy due to age‐related macular degeneration: chroma and spectri phase 3 randomized clinical trials. JAMA Ophthalmol 2018; 136: 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mainolfi N, Ehara T, Karki RG et al. Discovery of 4‐((2 S,4 S)‐4‐Ethoxy‐1‐((5‐methoxy‐7‐methyl‐1 H‐indol‐4‐yl)methyl)piperidin‐2‐yl)benzoic acid (LNP023), a factor B inhibitor specifically designed to be applicable to treating a diverse array of complement mediated diseases. J Med Chem 2020; 63: 5697–5722. [DOI] [PubMed] [Google Scholar]
- 32. Risitano AM, Kulasekararaj AG, Lee JW et al. Danicopan: an oral complement factor D inhibitor for paroxysmal nocturnal hemoglobinuria. Haematologica 2020; 106: 3188–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meulenbroek EM, Wouters D, Zeerleder S. Methods for quantitative detection of antibody‐induced complement activation on red blood cells. J Vis Exp 2014; 83: e51161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baas I, Delvasto‐Nuñez L, Ligthart PB et al. Complement C3 inhibition by compstatin Cp40 prevents intra‐ and extravascular hemolysis of red blood cells. Haemotologica 2020; 105: 471–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brüggeman M, Williams GT, Bindon CI et al. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med 1987; 166: 1351–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thielen AJF, van Baarsen IM, Jongsma ML, Zeerleder S, Spaapen RM, Wouters D. CRISPR/Cas9 generated human CD46, CD55 and CD59 knockout cell lines as a tool for complement research. J Immunol Methods 2018; 456: 15–22. [DOI] [PubMed] [Google Scholar]
- 37. Diebolder CA, Beurskens FJ, De Jong RN et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014; 343: 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kohen F, Bagci H, Barnard G et al. Preparation and properties of anti‐biotin antibodies. Methods Enzymol 1997; 279: 451–463. [DOI] [PubMed] [Google Scholar]
- 39. Bagçi H, Kohen F, Kusçuoglu U, Bayer EA, Wilchek M. Monoclonal anti‐biotin antibodies simulate avidin in the recognition of biotin. FEBS Lett 1993; 322: 47–50. [DOI] [PubMed] [Google Scholar]
- 40. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol 2014; 5: e520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sprong T, Roos D, Weemaes C et al. Deficient alternative complement pathway activation due to factor D deficiency by 2 novel mutations in the complement factor D gene in a family with meningococcal infections. Blood 2006; 107: 4865–4870. [DOI] [PubMed] [Google Scholar]
- 42. van den Broek B, van Els CACM, Kuipers B et al. Multi‐component meningococcal serogroup B (MenB)‐4C vaccine induces effective opsonophagocytic killing in children with a complement deficiency. Clin Exp Immunol 2019; 198: 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sharp TH, Boyle AL, Diebolder CA, Kros A, Koster AJ, Gros P. Insights into IgM‐mediated complement activation based on in situ structures of IgM‐C1‐C4b. Proc Natl Acad Sci USA 2019; 116: 11900–11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Muri L, Ispasanie E, Schubart A et al. Alternative complement pathway inhibition abrogates pneumococcal Opsonophagocytosis in vaccine‐Naïve, but not in vaccinated individuals. Front Immunol 2021; 12: e732146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ispasanie E, Muri L, Schubart A et al. Alternative complement pathway inhibition does not abrogate meningococcal killing by serum of vaccinated individuals. Front Immunol 2021; 12: e747594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ji H, Ohmura K, Mahmood U et al. Arthritis critically dependent on innate immune system players. Immunity 2002; 16: 157–168. [DOI] [PubMed] [Google Scholar]
- 47. Thurman JM, Kraus DM, Girardi G et al. A novel inhibitor of the alternative complement pathway prevents antiphospholipid antibody‐induced pregnancy loss in mice. Mol Immunol 2005; 42: 87–97. [DOI] [PubMed] [Google Scholar]
- 48. Subías M, Tortajada A, Gastoldi S et al. A novel antibody against human factor B that blocks formation of the C3bB proconvertase and inhibits complement activation in disease models. J Immunol 2014; 193: 5567–5575. [DOI] [PubMed] [Google Scholar]
- 49. Banda NK, Thurman JM, Kraus D et al. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen‐induced arthritis. J Immunol 2006; 177: 1904–1912. [DOI] [PubMed] [Google Scholar]
- 50. Kotimaa J, Klar‐Mohammad N, Gueler F et al. Sex matters: systemic complement activity of female C57BL/6J and BALB/cJ mice is limited by serum terminal pathway components. Mol Immunol 2016; 76: 13–21. [DOI] [PubMed] [Google Scholar]
- 51. Miwa T, Song WC. Membrane complement regulatory proteins: insight from animal studies and relevance to human diseases. Int Immunopharmacol 2001; 1: 445–459. [DOI] [PubMed] [Google Scholar]
- 52. Pouw RB, Vredevoogd DW, Kuijpers TW, Wouters D. Of mice and men: the factor H protein family and complement regulation. Mol Immunol 2015; 67: 12–20. [DOI] [PubMed] [Google Scholar]
- 53. Nakagawa K, Sakiyama H, Tsuchida T et al. Complement C1s activation in degenerating articular cartilage of rheumatoid arthritis patients: immunohistochemical studies with an active form specific antibody. Ann Rheum Dis 1999; 58: 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bemis EA, Norris JM, Seifert J et al. Complement and its environmental determinants in the progression of human rheumatoid arthritis. Mol Immunol 2019; 112: 256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Matsumoto I, Maccioni M, Lee DM et al. How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint‐specific autoimmune disease. Nat Immunol 2002; 3: 360–365. [DOI] [PubMed] [Google Scholar]
- 56. Hovingh ES, van den Broek B, Jongerius I. Hijacking complement regulatory proteins for bacterial immune evasion. Front Microbiol 2016; 7: e2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Moore SR, Menon SS, Cortes C, Ferreira VP. Hijacking factor H for complement immune evasion. Front Immunol 2021; 12: e602277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Slade C, Bosco J, Unglic G, Bleasel K, Nagel M, Winship I. Deficiency in complement factor B. N Engl J Med 2013; 369: 1664–1667. [DOI] [PubMed] [Google Scholar]
- 59. Hiemstra PS, Langeler E, Compier B et al. Complete and partial deficiencies of complement factor D in a Dutch family. J Clin Invest 1989; 84: 1957–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Biesma DH, Hannema AJ, Van Velzen‐Blad H et al. A family with complement factor D deficiency. J Clin Invest 2001; 108: 233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xu Y, Ma M, Ippolito GC, Schroeder HW, Carroli MC, Volanakis JE. Complement activation in factor D‐deficient mice. Proc Natl Acad Sci USA 2001; 98: 14577–14582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li Q, Li YX, Douthitt K, Stahl GL, Thurman JM, Tong HH. Role of the alternative and classical complement activation pathway in complement mediated killing against Streptococcus pneumoniae colony opacity variants during acute pneumococcal otitis media in mice. Microbes Infect 2012; 14: 1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Matsushita M, Endo Y, Fujita T. Structural and functional overview of the lectin complement pathway: its molecular basis and physiological implication. Arch Immunol Ther Exp (Warsz) 2013; 61: 273–283. [DOI] [PubMed] [Google Scholar]
- 64. Wouters D, Stephan F, Strengers P et al. C1‐esterase inhibitor concentrate rescues erythrocytes from complement‐mediated destruction in autoimmune hemolytic anemia. Blood 2013; 121: 1242–1244. [DOI] [PubMed] [Google Scholar]
- 65. DeZern AE, Uknis M, Yuan X et al. Complement blockade with a C1 esterase inhibitor in paroxysmal nocturnal hemoglobinuria. Exp Hematol 2014; 42: 857–861.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shi J, Rose EL, Singh A et al. TNT003, an inhibitor of the serine protease C1s, prevents complement activation induced by cold agglutinins. Blood 2014; 123: 4015–4022. [DOI] [PubMed] [Google Scholar]
- 67. Wouters D, Zeerleder S. Complement inhibitors to treat igM‐mediated autoimmune hemolysis. Haematologica 2015; 100: 1388–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Meulenbroek EM, de Haas M, Brouwer C, Folman C, Zeerleder SS, Wouters D. Complement deposition in autoimmune hemolytic anemia is a footprint for difficult‐to‐detect IgM autoantibodies. Haematologica 2015; 100: 1407–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hack CE, Paardekooper J, Smeenk RJ, Abbink J, Eerenberg J, Nuijens JH. Disruption of the internal thioester bond in the third component of complement (C3) results in the exposure of neodeterminants also present on activation products of C3. An analysis with monoclonal antibodies. J Immunol 1988; 141: 1602–1609. [PubMed] [Google Scholar]
- 70. Hoekzema R, Martens M, Brouwer MC, Hack CE. The distortive mechanism for the activation of complement component C1 supported by studies with a monoclonal antibody against the ‘arms’ of C1q. Mol Immunol 1988; 25: 485–494. [DOI] [PubMed] [Google Scholar]
- 71. van der Rijst MVE, Lissenberg‐Thunnissen SN, Ligthart PC et al. Development of a recombinant anti‐Vel immunoglobulin M to identify Vel‐negative donors. Transfusion 2019; 59: 1359–1366. [DOI] [PubMed] [Google Scholar]
- 72. Oskam N, Ooijevaar‐de Heer P, Derksen NIL et al. At critically low antigen densities, IgM hexamers outcompete both IgM Pentamers and IgG1 for human complement deposition and complement‐dependent cytotoxicity. J Immunol 2022; 209: 16–25. [DOI] [PubMed] [Google Scholar]
- 73. Falkenburg WJJ, Oskam N, Koers J et al. Identification of clinically and Pathophysiologically relevant rheumatoid factor epitopes by engineered IgG targets. Arthritis Rheumatol 2020; 72: 2005–2016. [DOI] [PubMed] [Google Scholar]
- 74. Pouw RB, Brouwer MC, de Gast M et al. Potentiation of complement regulator factor H protects human endothelial cells from complement attack in aHUS sera. Blood Adv 2019; 3: 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dekkers G , Brouwer MC , Jeremiasse J et al. Unravelling the effect of a potentiating anti‐factor H antibody on atypical hemolytic uremic syndrome associated factor H variants. J Immunol 2020; 205: 1778–1786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures 1–4