

Abstract
Clear cell carcinoma (CCC) of the cervix (cCCC) is a rare and aggressive type of human papillomavirus (HPV)-negative cervical cancer with limited effective treatment options for recurrent or metastatic disease. Historically, CCCs of the lower genital tract were associated with in utero diethylstilbestrol (DES) exposure, however the genetic landscape of sporadic cCCCs remains unknown. Here, we sought to define the molecular underpinning of cCCCs. Using a combination of whole-exome, targeted capture and RNA-sequencing, we identified pathogenic genetic alterations in the Hippo signaling pathway in 50% (10/20) of cCCCs, including recurrent WWTR1 S89W somatic mutations in 40% (4/10) of the cases harboring mutations in the Hippo pathway. Irrespective of the presence or absence of Hippo pathway genetic alterations, however, all primary cCCCs analyzed in this study (n=20) harbored features of Hippo pathway deregulation at the transcriptomic and protein levels. In vitro functional analysis revealed that expression of the WWTR1 S89W mutation leads to reduced binding of TAZ to 14–3-3, promoting constitutive nuclear translocation of TAZ and Hippo pathway repression. WWTR1 S89W expression was found to lead to acquisition of oncogenic behavior, including increased proliferation, migration and colony formation in vitro as well as increased tumorigenesis in vivo, which could be reversed by targeted inhibition of the TAZ/YAP1 complex with verteporfin. Finally, xenografts expressing WWTR1 S89W displayed a shift in tumor phenotype, becoming more infiltrative as well as less differentiated, and were found to be composed of cells with conspicuous cytoplasmic clearing as compared to controls. Our results demonstrate that Hippo pathway alterations are likely drivers of cCCCs and likely contribute to the clear cell phenotype. Therapies targeting this pathway may constitute a new class of treatment for these rare, aggressive tumors.
Keywords: Clear cell, cervix, Hippo pathway, massively parallel sequencing, WWTR1
INTRODUCTION
Clear cell carcinoma (CCC) of the cervix (cCCC) is a rare subtype of endocervical adenocarcinoma accounting for 3% of all cervical adenocarcinomas [1]. In contrast to the most prevalent type of cervical cancer, squamous cell carcinoma (SCC), cCCC is not related to human papillomavirus (HPV) infection [1,2]. Historically, CCCs of the lower genital tract were associated with in utero exposure to diethylstilbestrol (DES)[3,4], however today most CCCs are sporadic without a known etiology [2]. Although early-stage cCCCs have a good overall prognosis following radical surgical resection, cCCCs have a greater propensity to recur and metastatize, and advanced-stage disease is often incurable and associated with high mortality [5,6]. Current treatment options for recurrent or metastatic disease are limited, and novel therapeutic approaches for cCCCs are needed.
The etiology and genomic features of non-HPV related cervical cancers, including cCCCs, remain active areas of investigation. Boyd et al [7] performed a molecular analysis of 24 vaginal and cervical CCCs and found microsatellite instability in 50% and 100% of non-DES and DES-exposed cases, respectively. As part of The Cancer Genome Atlas (TCGA) project, common-type HPV-associated SCCs and adenocarcinomas were found to display an enrichment in APOBEC signatures, recurrent PIK3CA mutations (26%), 3q copy number gains (66%) and amplification of the Hippo signaling pathway component YAP1 (16%)[8]. Based on our previous genomic studies of rare epithelial malignancies that have resulted in the identification of novel genotypic-phenotypic correlations and convergent phenotypes [9–13], we posited that if cCCCs were driven by a highly recurrent if not pathognomonic genetic alteration these could potentially serve as therapeutic targets. Here, we sought to define the molecular underpinning of cCCCs using a combination of targeted, whole-exome and RNA-sequencing. Our analyses revealed recurrent alterations affecting the Hippo pathway in cCCCs, including WWTR1 S89W mutations. Functional in vitro and in vivo studies further demonstrate that WWTR1 S89W mutations are not only oncogenic and associated with phenotypic changes in these tumors but may also constitute the basis for new targeted therapeutics for patients with this disease.
MATERIALS AND METHODS
Samples
Following approval by the institutional review boards (IRBs) of the authors’ institutions, unstained tissue sections from formalin-fixed paraffin-embedded (FFPE) cCCCs were retrieved from Weill Cornell Medical Center (New York, NY) and Memorial Sloan Kettering Cancer Center (MSK; New York, NY). Patient consents were obtained following the respective IRB protocols approved by the authors’ institutions, and samples were anonymized before analysis. All tumors were centrally reviewed by two pathologists (K.J.P., C.M.), and a total of 20 tumors were classified as cCCCs and included in this study. The histopathologic criteria are detailed in Supplementary materials and methods.
Whole-exome, targeted capture and RNA-sequencing
Microdissected tumor and normal DNA from five cCCCs were subjected to whole-exome sequencing (WES) and from fifteen cCCCs to MSK-IMPACT sequencing, a massively parallel sequencing assay targeting all exons and selected introns of 468 cancer-related genes [14](see Supplementary materials and methods for details). Somatic mutations and copy number alterations (CNAs) were identified using validated bioinformatics methods [10,15,16]. TERT promoter mutations in the WES data were manually inspected using Integrative Genomics Viewer (IGV)[17]. CNAs and loss of heterozygosity (LOH) were defined using FACETS [18], as described [10,15]. ABSOLUTE (v1.0.6)[19] was employed to determine the cancer cell fraction (CCF) of each mutation [10,15]. DAVID pathway analysis was performed utilizing the results obtained from all cCCCs (n=20) based on genes affected by non-synonymous somatic mutations, amplifications or homozygous deletions [20]. Seven cCCCs were subjected to paired-end RNA-sequencing using validated protocols, as previously described [11,15],(see Supplementary materials and methods and [13,15,21–28]).
Cell lines
Parental immortalized human keratinocyte HaCaT (CVCL_0038) and HEK-293 (CVCL_0045) cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cell line identification was confirmed by short tandem repeat (STR) profiling. Cells were tested for mycoplasma infection using the Universal Mycoplasma Detection kit (ATCC) and cultured according to the vendor’s instructions.
WWTR1 mutagenesis, vector construction and generation of stable cell lines
Introduction of the S89W mutation in pDONR223-WWTR1-WT (Addgene, Watertown, MA, USA, #82253) sequence changing the 89 codon of WWTR1 from TCG (Serine (Ser/S)) to TGG (Tryptophan (Trp/W)) was performed using the Q5 Mutagenesis Kit (New England) following the manufacturer’s protocol and transformed in NEB 5-alpha competent E. coli. The mutagenesis primers were F 5-CCGCTCGCACtggTCGCCCGCGT-3 and 5-ACATGCTGGGCACCCCCAGC-3, and the introduced mutation was confirmed by Sanger sequencing. Vector construction and generation of stable cell lines are described in Supplementary materials and methods.
RT-qPCR
Total RNA was reverse-transcribed transcription into cDNA using SuperScript VILO Master Mix (ThermoFisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions. Quantitative TaqMan RT-PCR for WWTR1 (Hs00210007), AXL (Hs01064444), ITGB2 (Hs00164957), LMNB2 (Hs00383326) and CYR61 (Hs00155479) was performed using QuantStudio3 (Applied Biosystems; ThermoFisher Scientific). Expression data were normalized to GAPDH (Hs02786624), as described [10].
Western blotting, immunohistochemistry and immunofluorescence
Protein extraction, standard western blotting, immunohistochemical and immunofluorescence analyses were performed as described [11,29,30], see also Supplementary materials and methods.
Immunoprecipitation
For Pan-14–3-3 immunoprecipitation, 8 µg of TAZ (E5P2N) antibody (Cell Signaling Technology, Danvers, MA, USA, #71192) was crosslinked to 25 µl of Protein G Dynabeads using a Pierce Crosslink Magnetic IP/Co-IP Kit (ThermoFisher Scientific) and following manufacturer’s instructions. Protein from HEK-293 cells was extracted with IP buffer (ThermoFisher Scientific), and 500 µg protein lysate was incubated with TAZ beads in 500 µl IP buffer for 1 h at room temperature. Supernatant containing NuPAGE LDS buffer was subjected to western blotting (TAZ and tubulin antibodies, 1:1000) as described [11].
Luciferase experiments
HEK-293 cells expressing WWTR1 wild-type (WT), WWTR1 S89W mutation and empty vector as control were transfected with 90 ng of Hippo pathway reporter (8xGTIIC-luciferase Addgene #34615) and 10 ng of transfection efficiency control Renilla-SV40 (Promega) using lipofectamine 3000 in 96-well plates (25,000 cells per well), as described [10].
Proliferation, scratch wound healing and colony formation assay
Cells were seeded, then proliferation, scratch wound healing and colony formation assays were performed as previously described [10–12], see also Supplementary materials and methods.
Xenograft studies
NOD Scid Gamma (NSG) mice (Jackson Laboratory, Bar Harbor, ME, USA) were used for in vivo studies and were cared for in accordance with guidelines approved by MSK’s Institutional Animal Care and Use Committee and Research Animal Resource Center. Six eight-week-old female mice were injected subcutaneously with 7.5 million HaCaT cells with Matrigel (Corning, Oneonta, NY, USA) in a 1:1 ratio. Once tumors reached an average volume of 100 mm3, mice were randomized to receive either verteporfin 100 mg/kg or vehicle control intraperitoneally every other day for one month. Mice were observed daily throughout the treatment period for signs of morbidity/mortality. Tumors were measured twice weekly using calipers, and volume was calculated (length x width2 × 0.52). Body weight was assessed twice weekly. After 4 weeks of treatment, tumor samples were collected for histological and immunohistochemical analysis. All tumors were reviewed by two pathologists (J.S.R.-F. and K.J.P.).
Inhibitors
The TEAD/YAP/TAZ association inhibitor verteporfin (Visudyne; #S1786, Selleckchem, Radnor, PA, USA) was resuspended in DMSO and used in proliferation, clonogenic, and scratch wound healing assays at 2 μM. The protein synthesis inhibitor cycloheximide (#HY-12320, MedChemExpress, Monmouth Junction, NJ, USA), resuspended in DMSO, was used for western blotting assay at 50 μM.
Statistical analyses
Statistical analysis was performed using Prism 7 (GraphPad Software, San Diego, CA, USA). Student’s two-tailed t-test was employed for comparison of means in parametric data. Heteroscedasticity was assessed for each comparison, and homoscedastic or heteroscedastic t-tests were employed as appropriate, as described [10,11]. A p-value<0.05 was considered significant.
RESULTS
Clinical and histologic features of cCCCs
Following central pathology review, twenty cCCCs were included in this study (supplementary material, Table S1). Histologically, these tumors displayed the characteristic histologic features of cCCCs, consisting of tubulocystic, papillary and solid structures composed of cells with clear or eosinophilic cytoplasm and ‘hobnail’ cells with moderate-to-marked atypia and infrequent mitotic figures (Figure 1A). Patient median age at diagnosis was 50.5 years (range 8–79). At the time of diagnosis, all stages, following the 2018 FIGO staging system for cervical cancer [31], were represented, with IB1 (8/20), IB2, IB3, and IIA (all 3/20) being the most common. None of the patients in our cohort was documented to have had a history of DES exposure. Eight patients recurred (40%) with a median disease-free survival of 21 (range 5 – 69) months (supplementary material, Table S1).
Figure 1. Recurrent alterations affecting genes in the canonical Hippo signaling pathway in clear cell carcinomas of the cervix.
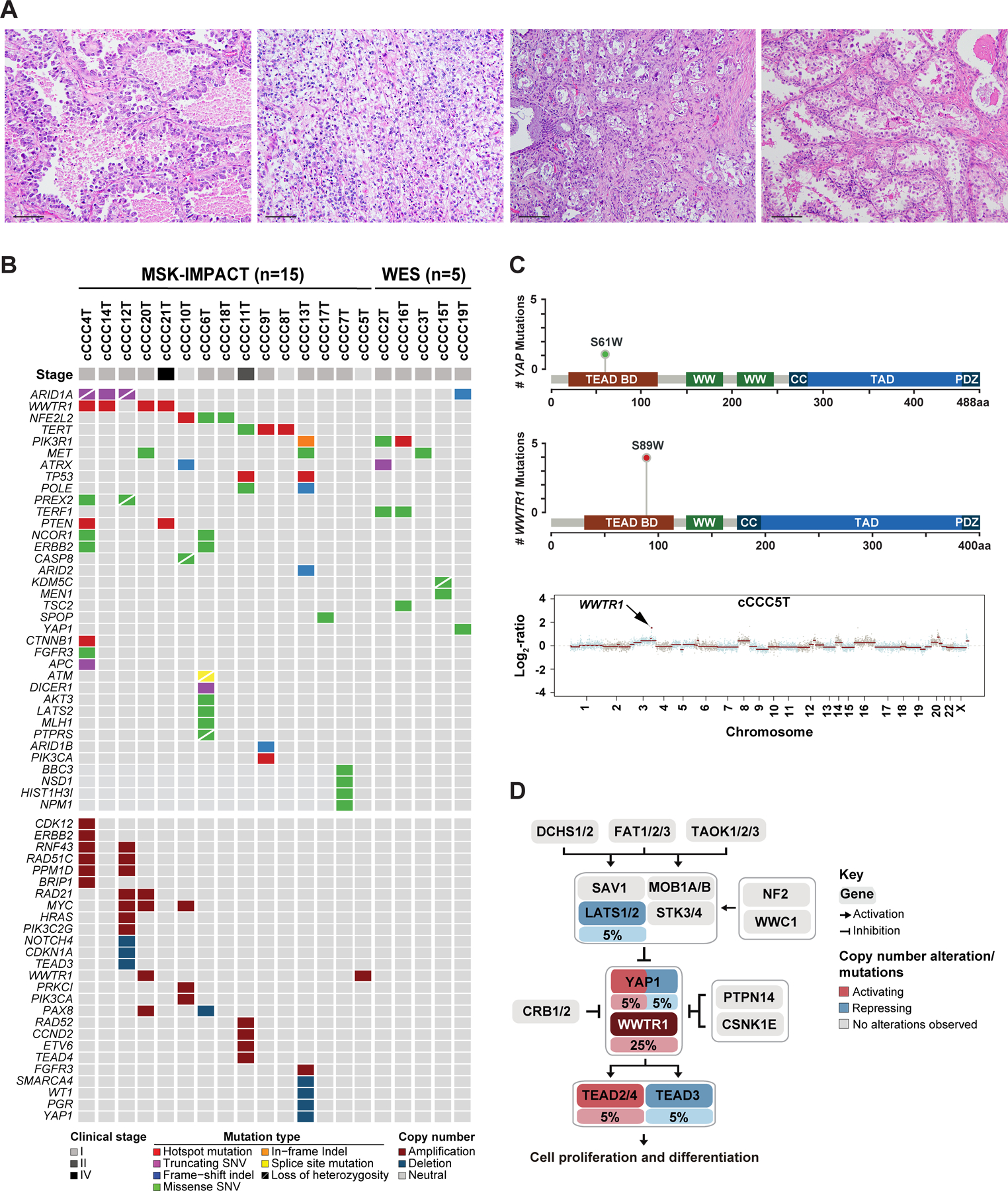
(A) Micrographs of representative hematoxylin and eosin-stained (H&E) sections of clear cell carcinoma of the cervix (scale bar 50 µm). (B) Heatmap depicting non-synonymous somatic mutations, amplifications and homozygous deletions identified in cCCCs subjected to MSK-IMPACT (n=15) or whole-exome sequencing (WES; n=5). Cases are shown in columns and genes in rows. Genetic alterations are color-coded according to the legend. Clinical stage is depicted in the phenotype bar (top). (C) Lollipop plots depicting the frequency of YAP1 S61W (top) and WWTR1 S89W (middle) mutations in cCCCs, including the protein domains of each gene. TEAD BD, TEAD binding domain; CC, coiled-coil region. Copy number plot of the cCCC5T harboring a WWTR1 amplification (bottom). The Log2-ratios are plotted on the y-axis according to genomic positions (x-axis). Chromosomes are depicted by alternating blue and red bands. (D) Frequency of activating (red) or repressing (blue) somatic genetic alterations affecting genes in the canonical Hippo signaling pathway. The percentage of cCCCs harboring a somatic mutation or gene copy number alteration is depicted under the gene name. Pathway reported in Sanchez-Vega et al [33].
cCCCs harbor recurrent genetic alterations in the Hippo pathway
To define their genomic landscape, twenty tumor-normal cCCC pairs were subjected to WES (n=5) or targeted MSK-IMPACT sequencing due to limited quantity/quality of available DNA (n=15). cCCCs were found to have a relatively low mutation burden with a median of 0.94 somatic mutations/Mb (range 0.43–1.47) on WES, and of 2.63 somatic mutations/Mb (range 0–9.5) by MSK-IMPACT (supplementary material, Table S2). In addition to ARID1A mutations (4/20, 20%), a known genomic feature of other Mullerian clear cell carcinomas [32], this analysis revealed the presence of recurrent TERT (3/20, 15%), NFE2L2 (3/20, 15%) and WWTR1 mutations (4/20, 20%; Figure 1B). The WWTR1 mutations affected the S89W residue, which maps to the TEAD binding domain of TAZ, an effector of the Hippo signaling cascade (Figure 1C).
A pathway analysis based on the presence of pathogenic somatic mutations, amplifications and homozygous deletions further revealed a significant enrichment for alterations in curated Hippo pathway-related genes in these tumors (p<0.01, EASE Score, Fisher’s exact test; Figure 1D)[33]. Of the twenty cCCCs analyzed, ten (50%) were found to harbor pathogenic alterations affecting components of the Hippo signaling pathway, including WWTR1 (5/10, 50%), YAP1 (2/10, 20%), TEAD3 (1/10, 10%), TEAD4 (1/10, 10%), and LATS2 (1/10, 10%), which occurred in a mutually exclusive pattern. Specifically, we found four hotspot mutations and one amplification in WWTR1, one likely pathogenic S61W missense mutation [34,35] and one deletion in YAP1, one homozygous deletion in TEAD3, one amplification in TEAD4 and one likely pathogenic K784N missense mutation in LATS2 (Figure 1B and supplementary material, Table S2). Both the WWTR1 S89W and the YAP1 S61W mutations were found to be clonal (supplementary material, Figure S1) and to affect residues in the TEAD binding domains of their respective proteins, TAZ and YAP1, which contain the 14–3-3 binding motifs [36](Figure 1C). We subjected five cCCCs lacking mutations, amplifications or homozygous deletions affecting Hippo pathway-related genes to RNA-sequencing, but no fusion transcripts affecting Hippo pathway genes were identified (supplementary material, Table S3). Importantly, irrespective of the presence or absence of genetic alterations in the Hippo pathway, RT-qPCR analysis provided transcriptomic evidence of deregulation of the Hippo pathway in all cCCCs analyzed (supplementary material, Figure S2), suggesting the presence of epigenetic or genetic alterations not detected by the targeted sequencing approaches employed. Furthermore, using RNA-sequencing, cCCCs displayed a significant increase in expression of WWTR1 and TAZ/YAP transcriptional targets (15/26; p<0.05; supplementary material, Figure S2) [36], providing further evidence of alteration of the Hippo pathway in cCCCs.
WWTR1 S89W expression results in the acquisition of oncogenic properties
There is burgeoning evidence to demonstrate that the Hippo pathway plays important roles in the pathogenesis of different types of cancer [37], with recurrent amplification of YAP1 and WWTR1 being reported in SCCs of the head and neck, esophagus, lung, cervix [33] and ovarian cancers [38]. Somatic hotspot mutations have not been previously described as a recurrent mechanism of Hippo pathway alteration in cancer. Based on a study that experimentally assessed the effect of 19 WWTR1 non-synonymous mutations in MCF10A non-malignant breast epithelial cells, which identified the S89W mutation as functionally activating [36], we hypothesized that expression of WWTR1 S89W would be associated with oncogenic properties. To test this, we performed functional in vitro studies by expressing WWTR1 wild-type (WT), WWTR1 S89W mutation and empty vector as control (supplementary material, Figure S3). Given the scarcity of commercially available HPV-negative non-malignant cervical cell lines, we selected primary non-tumorigenic differentiation-competent HaCaT keratinocytes as model system, which have been employed by others for the study of cervical tumorigenesis [39–41]. As a second model, and to confirm the results obtained in HaCaT cells, we employed HEK-293 cells, which are widely used in cell biology/cancer research studies given their reliable growth and propensity for transfection [42]. In both HaCaT and HEK-293 cells, WWTR1 S89W expression resulted in a significant increase in proliferation and colony formation as compared to cells expressing empty vector control (control) or WWTR1 WT (Figure 2A,B). A scratch assay analysis revealed increased migration of HaCaT and HEK-293 cells upon stable expression of WWTR1 S89W when compared to cells expressing WWTR1 WT or control (Figure 2C). As expected, based on observations of WWTR1 amplification/overexpression [37], WWTR1 WT expressing cells showed increased proliferation, colony formation and migration as compared to controls; however, cells harboring WWTR1 S89W consistently exhibited a significantly stronger oncogenic phenotype (Figure 2A–C).
Figure 2. WWTR1 S89W expression results in the acquisition of oncogenic properties in vitro.
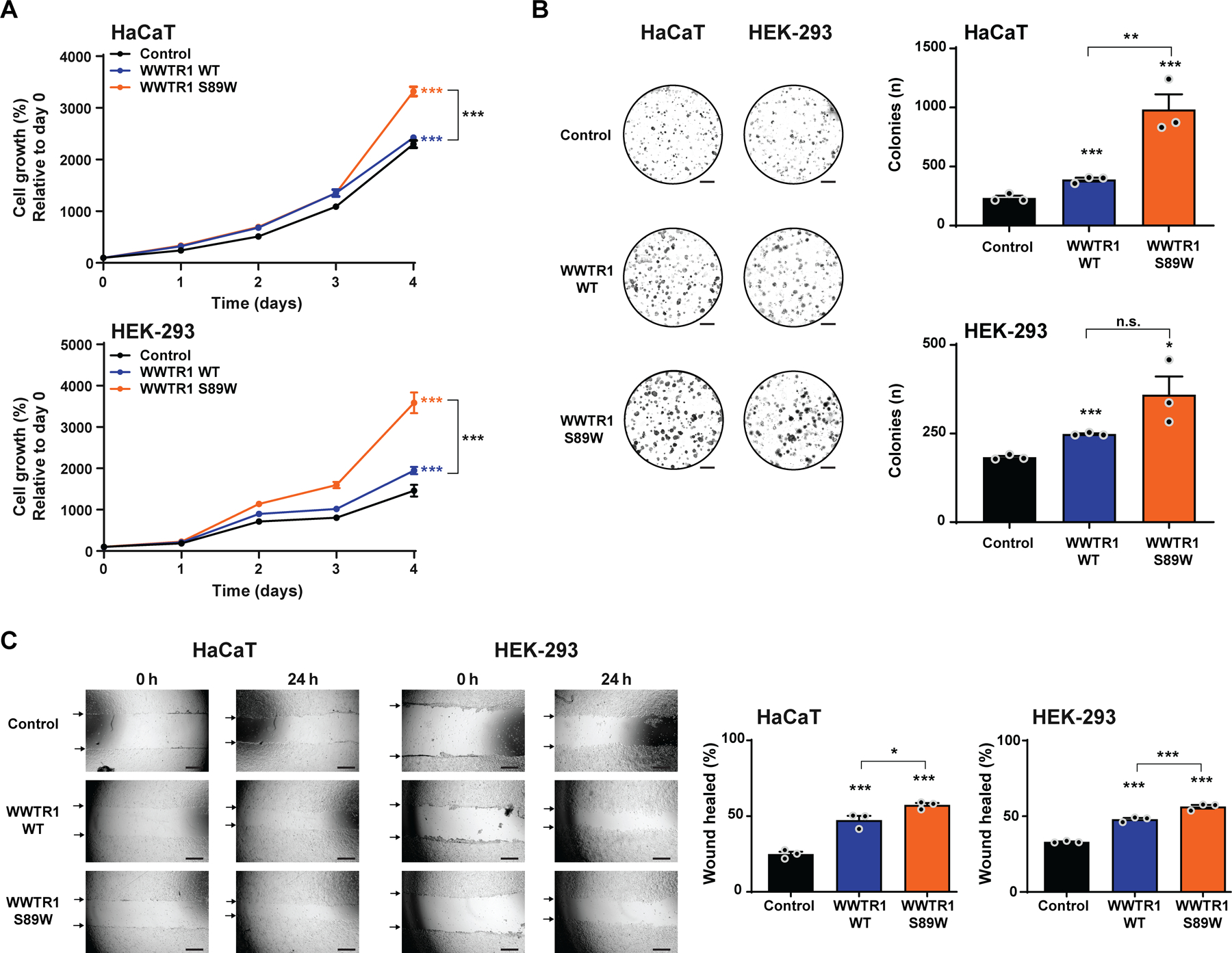
(A) CellTiter-blue proliferation assay of immortalized human keratinocyte HaCaT and HEK-293 cells stably expressing empty vector (Control), WWTR1 wild-type (WWTR1 WT) and WWTR1 S89W. Quantification of cell growth (%) relative to day 0. (B) Representative images of colony formation assay of HaCaT and HEK-293 cells stably expressing Control, WWTR1 WT and WWTR1 S89W (scale bars, 5 mm; bottom). Quantification of the number of colonies/ well (right). (C) Wound healing assay of HaCaT and HEK-293 cells stably expressing Control, WWTR1 WT and WWTR1 S89W. The migratory effects/ wound area was assessed at 0 and 24 h (Scale bar, 500 μm; bottom) and quantified (right). All experiments were performed in technical triplicate in at least 3 independent replicates, and statistical analysis were performed comparing each condition to Control. Mean ± SD. n.s., not significant; *P<0.05, **P<0.01, ***P<0.001; two-tailed unpaired t-test.
Expression of WWTR1 S89W results in constitutive deregulation of the Hippo signaling pathway
TAZ has been reported to play pivotal roles in regulation of the Hippo signaling pathway [43,44]. Upon nuclear translocation, along with YAP, TAZ acts as a co-transcriptional activator of the pathway, thereby promoting tissue growth and cell viability [37]. We investigated whether WWTR1 S89W expression would result in alteration of this pathway in our cell models. We found that both HaCaT and HEK-293 cells expressing WWTR1 S89W displayed increased expression levels of key Hippo pathway target genes, including AXL, ITGB2, LMNB2 and CYR61 [44,45], when compared to the respective control and WWTR1 WT cells (Figure 3A). This finding was further supported by quantitative western blot analyses, which revealed increased levels of AXL, ITGB2, LMNB2 and CYR61 proteins upon WWTR1 S89W expression in both cell models (Figure 3B). Again, the expression of WWTR1 WT was also associated with increased AXL, ITGB2, LMNB2 and CYR61 gene expression, however, at significantly lower levels than in those expressing the WWTR1 S89W mutation (Figure 3A). Our in vitro findings suggest that the oncogenic behavior of cCCCs harboring the WWTR1 S89W mutation can, at least in part, be explained by the deregulation of the Hippo pathway, corroborating the observation that this tumor suppressor pathway is consistently repressed in human cCCCs (supplementary material, Figure S2).
Figure 3. WWTR1 S89W expression results in the overexpression of Hippo pathway targets in vitro and in deregulation of the Hippo pathway.
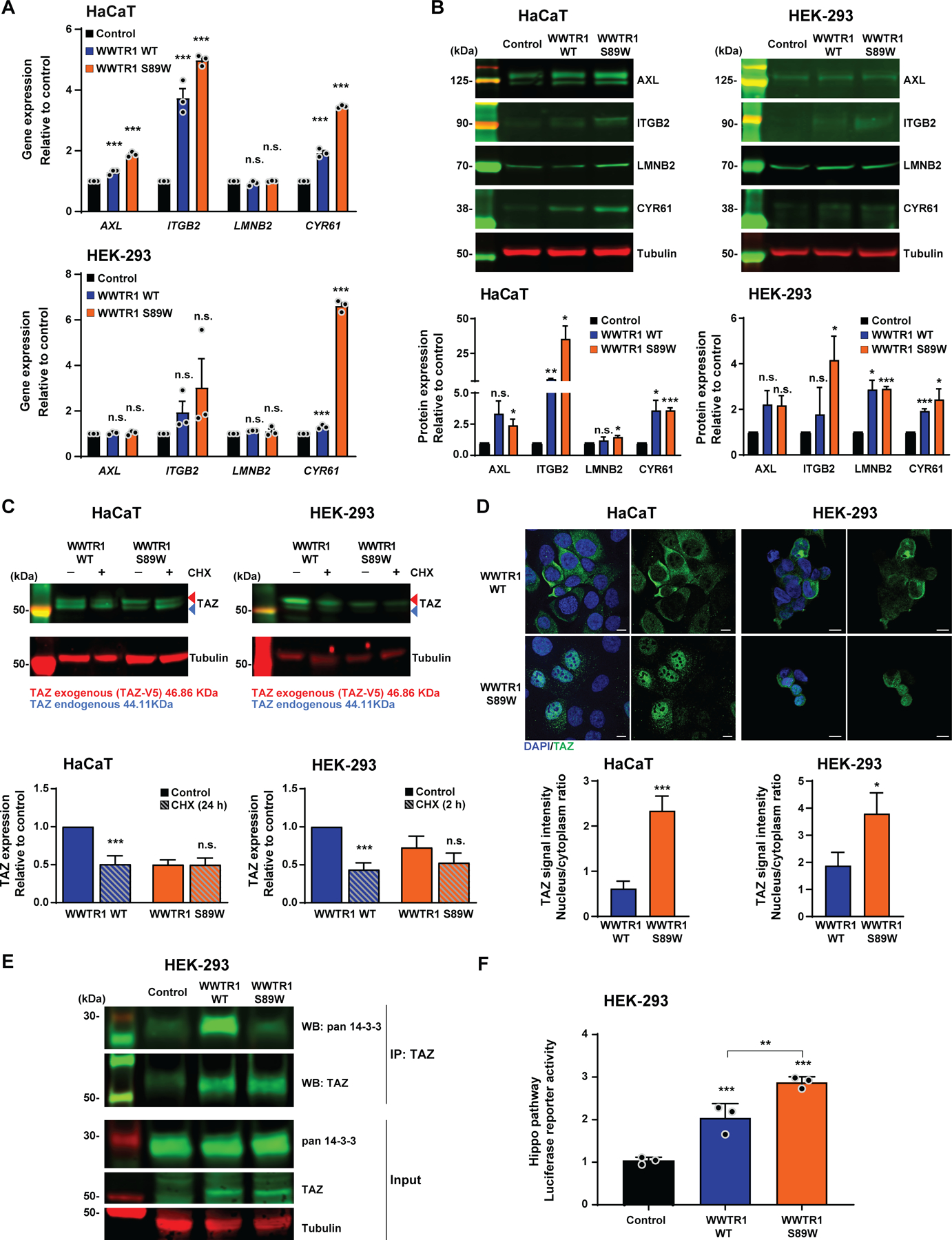
(A) Quantitative assessment of Hippo pathway targets AXL, ITGB2, LMNB2 and CYR61 transcripts in immortalized human keratinocyte HaCaT and HEK-293 cells stably expressing empty vector (Control), WWTR1 wild-type (WWTR1 WT) and WWTR1 S89W. Expression levels were normalized to GAPDH expression, and comparisons of mRNA expression levels were performed relative to Control. (B) Representative western blot analysis of Hippo pathway targets AXL, ITGB2, LMNB2 and CYR61 protein levels in HaCaT and HEK-293 cells stably expressing Control, WWTR1 WT and WWTR1 S89W. Tubulin was used as protein loading control. Quantification (bottom) of protein levels as compared to Control. (C) Representative western blot analysis of TAZ protein levels in immortalized human keratinocyte HaCaT and HEK-293 cells stably expressing WWTR1 WT and WWTR1 S89W treated with 50 μM Cycloheximide (CHX). Tubulin was used as protein loading control. Quantification (bottom) of protein levels as compared to untreated WWTR1 WT cells. (D) Representative confocal micrographs of immunofluorescence analysis of TAZ (green) and 4–6-diamidino-2-phenylindole (DAPI, blue) in HaCaT and HEK-293 cells stably expressing Control, WWTR1 WT and WWTR1 S89W (scale bars, 50 μm). Quantification (bottom) of TAZ intensity/ cell in a nucleus/cytoplasm ratio. (E) Immunoprecipitation assay with TAZ antibody of HEK-293 cells stably expressing Control, WWTR1 WT or WWTR1 S89W. Western blot was performed using anti-TAZ and anti-pan 14–3-3 antibodies, and tubulin as loading control. (F) Hippo pathway luciferase reporter assay of HEK-293 cells stably expressing Control, WWTR1 WT and WWTR1 S89W. SV40-Renilla was used to normalize transfection efficiency. All experiments were performed in technical triplicate in at least 3 independent replicates and statistical analysis was performed comparing each condition to Control (A,B,F), or to WWTR1 WT (C,D). Mean ± SD; n.s., not significant; *P<0.05, **P<0.01, ***P<0.001; two-tailed unpaired t-test.
WWTR1 S89W mutation stabilizes TAZ protein deregulating the Hippo pathway
We investigated the molecular mechanisms underlying the deregulation of the Hippo pathway in cells expressing the WWTR1 S89W mutation. TAZ activity is regulated through phosphorylation by LATS2, promoting proteolytic degradation or retention in the cytoplasm due to sequestering by the 14–3-3 protein [46,47]. To define whether TAZ stability is affected by the presence of the S89W mutation, we treated HaCaT and HEK-293 cells with the protein synthesis inhibitor cycloheximide (CHX) and observed that in cells expressing WWTR1 S89W, TAZ had a longer half-life than in those cells expressing WWTR1 WT in both HaCaT and HEK-293 cell lines (Figure 3C). Additionally, we hypothesized that WWTR1 S89W cells would exhibit constitutive Hippo pathway repression, and thereby be functionally activating, with expression of Hippo pathway targets due to higher rates of TAZ nuclear translocation. Immunofluorescence analyses revealed increased TAZ signal intensity in the nucleus of HaCaT and HEK-293 cells expressing WWTR1 S89W than in cells expressing WWTR1 WT, in which TAZ localization was limited to the cytoplasm (Figure 3D). Moreover, immunoprecipitation of TAZ in HEK-293 cells indicated that in WWTR1 S89W cells, the TAZ protein did not bind efficiently to 14–3-3, demonstrating that the S89W mutation reduces 14–3-3 cytoplasmic sequestration of TAZ, leading to TAZ nuclear translocation [46](Figure 3E). Finally, we performed a luciferase reporter assay with a specific Hippo/TEAD reporter containing eight TEAD binding sites [48] in HEK-293 cells stably expressing control, WWTR1 WT and WWTR1 S89W. Increased activation of the Hippo/TEAD reporter was observed in cells expressing WWTR1 WT and WWTR1 S89W, however, Hippo/TEAD activation was significantly enhanced in WWTR1 S89W expressing cells (Figure 3F). Taken together, these findings demonstrate that the WWTR1 S89W mutation increases TAZ stability and promotes nuclear localization, thereby resulting in downstream constitutive repression of the Hippo signaling pathway.
Oncogenic and phenotypic properties can be reversed by inhibition of the TAZ/YAP/TEAD transcriptional complex
Verteporfin (Vysudine) targets the Hippo pathway by repressing the interaction between TAZ, YAP1 and TEAD, and is FDA approved for photodynamic treatment of vascular disorders of the eye [49]. Currently, verteporfin is being studied in clinical trials for the treatment of different tumor types, including cutaneous metastasis of breast and pancreatic cancer [50]. Given the deregulation of the Hippo pathway upon WWTR1 S89W expression in our in vitro models, we assessed whether inhibition of the TAZ/YAP1/TEAD transcriptional complex would result in reversal of the observed oncogenic phenotype induced by WWTR1 S89W expression. Upon treatment of HaCaT and HEK-293 cells with verteporfin, we observed significant inhibition of proliferation and colony formation in cells expressing WWTR1 S89W (Figure 4A,B and supplementary material, Figure S4). Verteporfin treatment also led to a significant decrease in migration rates of HaCaT and HEK-293 cells expressing WWTR1 S89W, which was not observed in controls or cells expressing WWTR1 WT (Figure 4C).
Figure 4. Inhibition of the Hippo pathway transcriptional complex by verteporfin results in the reversal of oncogenic properties in WWTR1 S89W expressing cells in vitro.
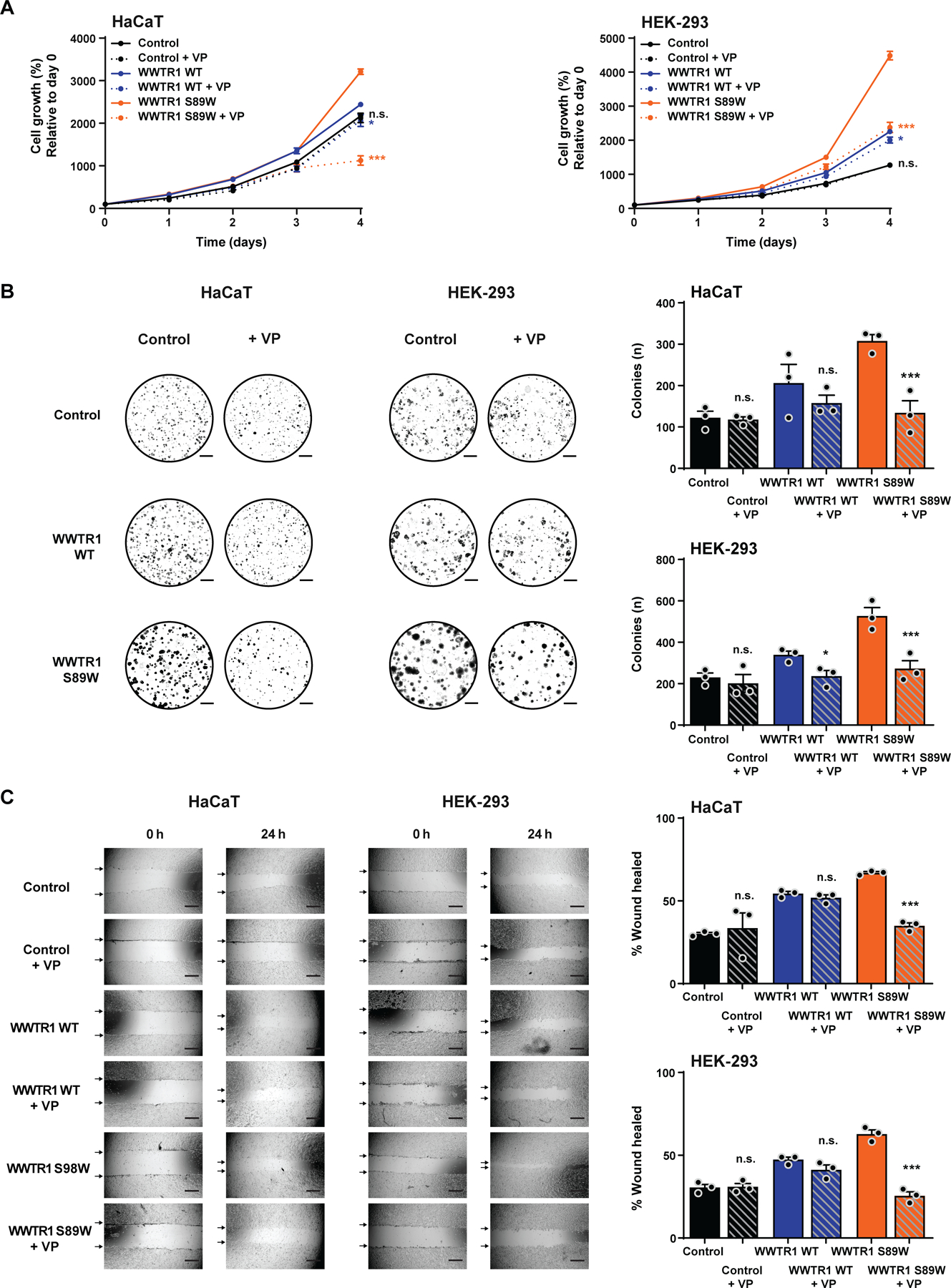
(A) CellTiter-blue proliferation assay of immortalized human keratinocyte HaCaT and HEK-293 cells stably expressing empty vector (Control), WWTR1 wild-type (WWTR1 WT) and WWTR1 S89W treated with 2 µM verteporfin (VP) or untreated. Quantification of cell growth (%) relative to day 0. (B) Representative images of colony formation assay of HaCaT and HEK-293 cells stably expressing Control, WWTR1 WT and WWTR1 S89W treated with 2 µM VP or untreated (scale bars, 5 mm; bottom). Quantification of the number of colonies/ well (right). (C) Wound healing assay of HaCaT and HEK-293 cells stably expressing Control, WWTR1 WT and WWTR1 S89W treated with 2 µM VP or untreated. The migratory effects/ wound area was assessed at 0 and 24 h (Scale bar, 500 μm; bottom) and quantified (right). VP, Verteporfin. All experiments were performed in technical triplicate in at least 3 independent replicates and statistical analysis was performed comparing each condition to its corresponding untreated pair. Mean ±SD; n.s., not significant; *P<0.05, **P<0.01, ***P<0.001; two-tailed unpaired t-test.
Impact of the WWTR1 S89W mutation on tumorigenesis
We next sought to define the impact of WWTR1 S89W on tumorigenicity. HaCat cells expressing control, WWTR1 WT and WWTR1 S89W were injected subcutaneously into NSG mice. Consistent with our observations made in vitro, HaCaT cells expressing WWTR1 S89W grew faster and formed significantly larger tumors than cells expressing WWTR1 WT or control (Figure 5A). In addition, treatment of mice with verteporfin for 33 days inhibited the acquired oncogenic potential of WWTR1 S89W expressing cells to control levels (Figure 5A), indicating that Hippo pathway deregulation is responsible for the increased tumor growth. Consistent with the in vitro observations and the notion that WWTR1 WT or WWTR1 S89W expressing xenografts are dependent on the Hippo pathway, verteporfin treatment resulted in necrosis only in the xenografts expressing WWTR1 WT or WWTR1 S89W (supplementary material, Figure S5). Importantly, mice treated with verteporfin did not show detectable clinical side effects or significant loss of body weight (supplementary material, Figure S5), indicating that verteporfin treatment is likely safe [51,52].
Figure 5. Tumorigenicity and phenoptypic assessment in xenograft assays.
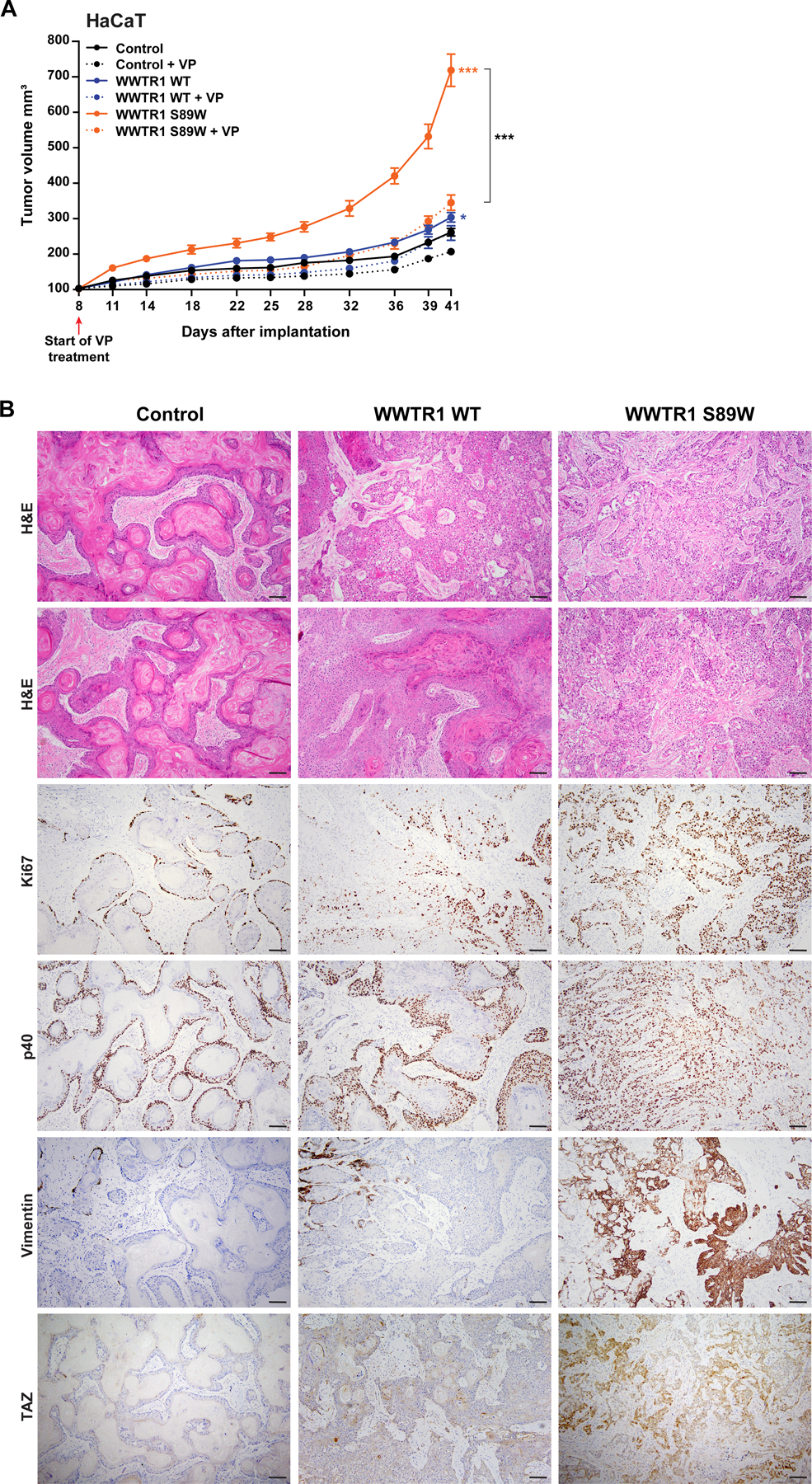
(A) Tumor growth of xenografts derived from immortalized human keratinocyte HaCaT cells expressing empty vector (Control), WWTR1 wild-type (WWTR1 WT) and WWTR1 S89W treated with 2 µM Verteporfin (VP) or DMSO as control. VP, Verteporfin. Experiments were performed in 5 mice per condition. Mean ±SEM; n.s., not significant; *P < .05, **P < .01, ***P < .001; two‐tailed unpaired t-test. (B) Representative micrographs of sections of xenograft tumors derived from HaCaT cells expressing Control, WWTR1 WT and WWTR1 S89W subjected to hematoxylin‐and‐eosin (H&E) staining, Ki67, p40, Vimentin and TAZ immunohistochemical analysis (scale bar, 100 µm).
Histologic review of the xenografts revealed that whilst control HaCaT cells resulted in pure SCCs with squamous pearls and overt keratinization (Figure 5B), xenografts expressing WWTR1 WT and, in particular, WWTR1 S89W, displayed a phenotype shift, with tumors becoming more infiltrative and less differentiated, being composed of infiltrating ragged gland-like structures, nests and cords of tumor cells, with marked atypia. A systematic histologic analysis of the xenografts revealed that the degree of differentiation decreased in a stepwise manner according to the expression of EV, WWRT1 WT to WWTR1 S89W, whereas immunohistochemical analysis of the Ki67 labeling indices demonstrated that the proliferation levels increased in a stepwise manner from EV, WWRT1 WT to WWTR1 S89W (Ki67 labeling index 33, 60 and 89, respectively; Figure 5B). Consistent with these observations, the expression and distribution of p40, a protein expressed in cells of the squamous lineage apart from terminally differentiated squamous cells, was higher in WWTR1 WT and WWTR1 S89W xenografts than in models expressing EV (p40-positive cells: EV 58%, WWTR1 WT 77%, WWTR1 S89W 92%). These findings are further supported by immunohistochemical analysis of vimentin expression (which is detected in poorly differentiated squamous cell carcinomas and in clear cell carcinomas (Figure 6), but either absent or only focally present in well differentiated squamous cell carcinomas), with an antibody that reacts with the human but not mouse vimentin, revealed a higher level of vimentin expression in xenografts expressing WT WWTR1 and WWTR1 S89W (Figure 5B).
Figure 6. TAZ and vimentin expression in human primary clear cell cervical cancers.
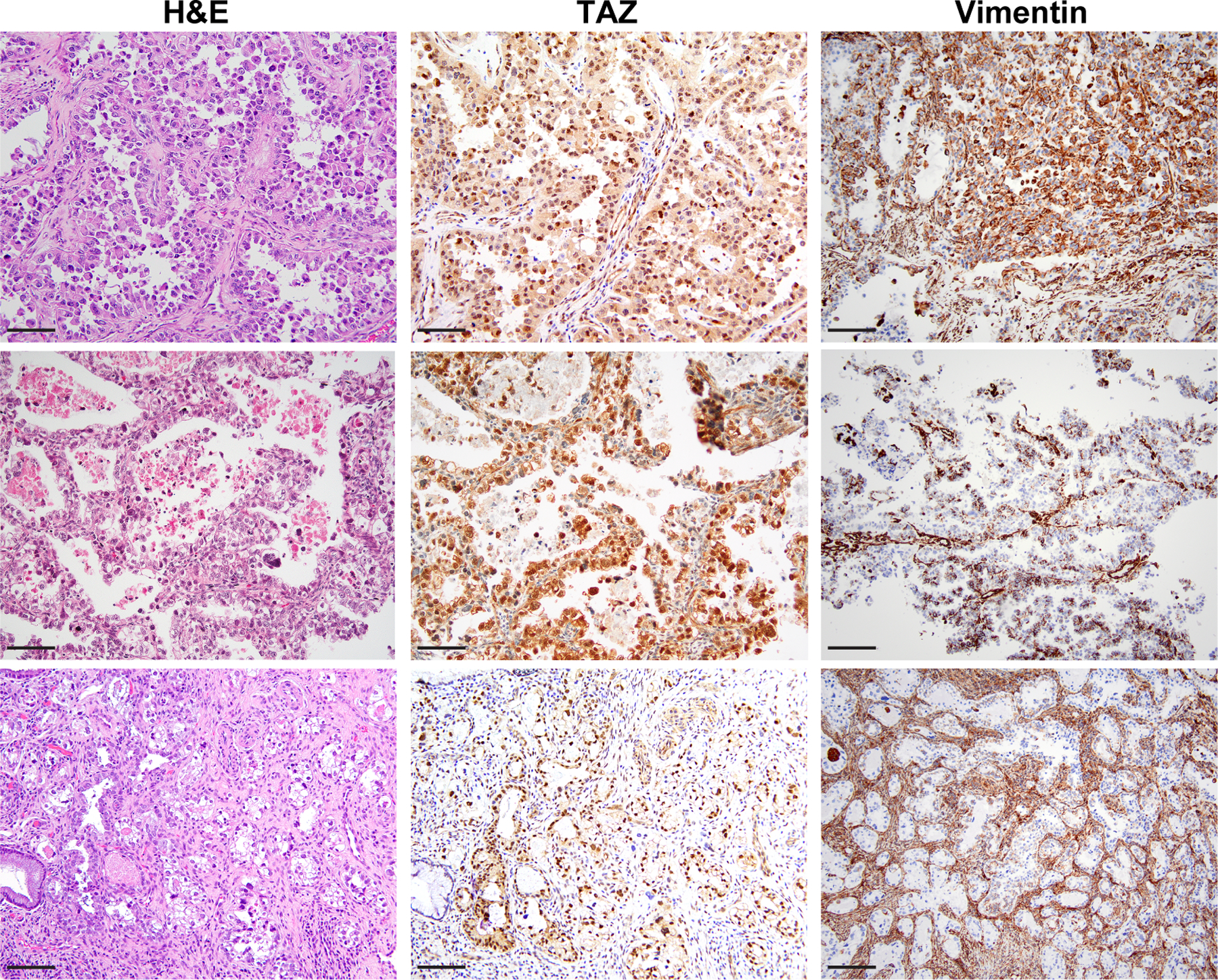
Micrographs of representative sections of primary clear cell carcinomas of the cervix (cCCCs) subjected to H&E staining,TAZ and vimentin immunohistochemical analysis (scale bar, 50 µm).
Immunohistochemical analysis further revealed that while TAZ expression was either absent or primarily cytoplasmic in the control and WWTR1 WT expressing xenografts, WWTR1 S89W tumors showed increased nuclear TAZ expression (Figure 5B). Given that we observed WWTR1 S89W expression to lead to nuclear TAZ expression in both the in vitro and in vivo settings, we performed TAZ immunohistochemical analysis of primary cCCCs (n=13). Consistent with our in vitro and in vivo findings, strong nuclear TAZ expression was present in the human primary cCCCs (Figure 6).
Taken together, our results not only implicate the Hippo pathway in the pathogenesis of human cCCCs and tumor phenotype changes, but also support the contention that pharmacologic inhibition of the Hippo pathway may constitute a potential novel targeted therapeutic approach for cCCCs.
DISCUSSION
cCCCs, while curable in the early stage, are associated with poor prognosis in the advanced setting [6]. Treatment options for metastatic or recurrent cCCC remain limited in part due to a lack of understanding of its genetic basis and pathogenesis. Here we demonstrate that cCCCs harbor recurrent alterations in the Hippo signaling pathway, with WWTR1 being the most commonly affected gene. At variance with other cancer types, including renal clear cell carcinomas, in which WWTR1 amplification has been documented [36], in cCCCs, we detected the presence of a recurrent WWTR1 S89W somatic mutation. In cases lacking this mutation, other somatic genetic alterations affecting the Hippo pathway, including those affecting YAP1, TEAD3/4 and LATS2, and/or activation of downstream targets were identified, consistent with the notion that CCCs may constitute a convergent phenotype, which is dependent on deregulation of this signaling pathway.
The Hippo pathway, critically important in the regulation of organ size and tissue homeostasis, is recurrently altered in certain cancers including mesothelioma, glioma, SCCs of the head and neck, and cervix [33,36]. Upregulation and/or overexpression of YAP1 and TAZ, transcriptional co-activators of the pathway, contribute to tumor progression in multiple cancer types, including breast, lung, liver, colon and ovarian cancer [52,53]. The canonical Hippo pathway comprises a serine/threonine kinase cascade, in which MST1/2 interacts with Salvador homolog 1 (SAV1) and phosphorylates LATS1/2, thereby activating them. Activated LATS1/2 in turn phosphorylates conserved serine residues on YAP1/TAZ, leading to cytoplasmic retention of YAP1/TAZ via 14–3-3 binding and degradation of YAP1/TAZ [47]. Recent studies have highlighted the oncogenic effects of mutating the LATS2 phosphorylation site of TAZ, S89, resulting in constitutive activation of TAZ (TAZ-S89A), leading to increased cellular proliferation in multiple cell models [54–56]. Here, we demonstrate that the S89W missense mutation in WWTR1 increases TAZ stability and nuclear localization, resulting in constitutive repression, and thereby functional activation, of the Hippo pathway. As expected, we observed increased proliferation, colony formation and migration in our cell models in cells expressing WWTR1 S89W. Furthermore, we observed increased tumorigenesis in xenograft models injected with WWTR1 S89W cells, which could be reversed by verteporfin, a potent Hippo pathway transcriptional complex inhibitor, both in vitro and in vivo. Expression of WWTR1 S89W was not only found to be oncogenic but may also result phenotypic changes including the clearing of the cytoplasm and increased expression of markers of dedifferentiation, corroborating the notion that constitutive repression of the Hippo pathway plays a role in the pathogenesis of this aggressive form of HPV-negative cervical cancer.
In addition to the recurrent S89W mutation in WWTR1, we report on the S61W mutation in YAP1. The downstream effects of the S61W YAP1 mutation, akin to those of the WWTR1 S89W mutation described here, have been reported to include reduced phosphorylation, and subsequent protein stabilization and increased nuclear localization of YAP1, thereby resulting in an increase in TAZ/YAP1/TEAD transcriptional activity [34,35]. Interestingly, one cCCC studied here contained a YAP1 deletion, which would presumably have the opposite effect, however there are reports that YAP inversely regulates TAZ protein abundance [57,58].
Whilst previous studies have reported cervical cancers to have alterations in Hippo pathway genes, specifically YAP1 and WWTR1 amplification, the majority of these findings were reported irrespective of histologic subtype or in the more common type of HPV-driven SCCs [8,59,60]. The AACR Project GENIE reported the WWTR1 S89W mutation to be present in 0.02% of all cancers, with the majority being cancers of gynecologic origin, including two cCCCs [61]. In the TCGA pan-cancer study [62], the WWTR1 S89W mutation was only detected in a single case (1/10967 samples, 0.009%), namely a FIGO grade 1 endometrioid endometrial cancer. Given that expression of WWTR1 S89W in HaCaT cells resulted in conspicuous cytoplasmic clearing in vivo, we sought to define whether this mutation would constitute a feature of not only cCCCs but also of other Mullerian carcinomas with clear cell phenotype. Re-analysis of the MSK-IMPACT sequencing data (v6/7) of ovarian and endometrial cancers revealed the presence of a WWTR1 S89W mutation in 1/1790 (0.06%) ovarian cancer, which was of clear cell histology, and WWTR1 S89W or S89L mutations in 5/2,093 (0.24%) endometrial carcinomas, two of which were CCCs (supplementary material, Figure S6). None of the remaining CCCs of the ovary (n=141) or endometrium (n=78) harbored WWTR1 S89 mutations. These data suggest that mutations affecting codon S89 of WWTR1 may be causally linked to a clear cell phenotype not only in cCCCs, but also in a subset of non-cervical Mullerian cancers. Of note, a subset of ovarian CCCs is known to express high levels of YAP at the protein level, correlating with poor prognosis [63,64]. Aside from a single case with a YAP1 missense mutation, we did not observe YAP1 amplification in the 141 ovarian CCCs analyzed here.
This study has several limitations. Given the scarcity of non-malignant HPV-negative cervical cancer cell lines, the functional studies were performed in immortalized human keratinocytes (HaCaT) and in HEK-293 cells, which exact cell type is still a matter of contention [42]. HaCaT WWTR1 S89W xenografts were found to display phenotypic features distinct from the control cells, including clear cytoplasm, supporting the notion that these cells may be an adequate disease model. Although a likely pathogenic genetic alteration affecting the Hippo pathway was detected in only 50% (10/20) of the cases, features of Hippo pathway deregulation, including Hippo downstream target and/or nuclear TAZ protein expression, were found in all cCCCs analyzed here, supporting the importance of this pathway in the pathogenesis of these aggressive cancers. Further studies are warranted to determine alternative mechanisms that lead to increased TAZ/YAP1 complex activity. Given that this study was based on WES, MSK-IMPACT and RNA-sequencing, we cannot rule out genetic alterations affecting non-coding regions of Hippo pathway genes or translocations resulting in the deregulation of this pathway in cases not subjected to RNA-sequencing or epigenetic regulation of this pathway. Whole-genome sequencing and epigenetic profiling of cCCCs lacking the canonical Hippo pathway alterations reported here are warranted. Finally, given the small number of patients in our cohort, and the multi-institutional nature of the study, correlations with mutation status and patient outcomes could not be performed. Larger studies are required to elucidate the impact of Hippo pathway alterations on clinical outcomes of cCCC patients.
Despite these limitations, here we report on the observation that Hippo pathway alterations, particularly the somatic WWTR1 S89W mutation, likely constitute drivers of cCCCs, and may also be causative of the clear cell phenotype in a subset of cCCCs and, potentially, of Mullerian cancers. Our findings also offer a new potential therapeutic target for patients with these aggressive forms of HPV-negative cervical cancers, with FDA-approved inhibitors of the Hippo pathway available and currently under investigation in clinical trials for patients with other cancer types.
Supplementary Material
Supplementary materials and methods
Figure S1. Cancer cell fractions of somatic mutations and copy number alterations of clear cell carcinomas of the cervix.
Figure S2. Overexpression of Hippo pathway targets in human clear cell carcinomas of the cervix and in xenograft tumors
Figure S3. TAZ protein and gene expression validation
Figure S4. IC50 of verteporfin in HEK-293 and HaCaT cells
Figure S5. Quantification of cytoplasmic clearing, necrosis and mice body weight in xenograft models
Figure S6. WWTR1 S89 mutations in ovarian and endometrial cancers
Table S1. Clinicopathologic characteristics of patients with clear cell carcinoma of the cervix included in this study
Table S2. Non-synonymous somatic mutations identified in clear cell carcinomas of the cervix subjected to whole-exome or targeted massively parallel sequencing
Table S3. Fusion candidate genes and/or readthroughs identified by RNA-sequencing analysis of clear cell carcinomas of the cervix
ACKNOWLEDGEMENTS
We thank the Integrated Genomics Operating, Gene Editing and Screening, Molecular Cytology, Pathology Core Facilities and Institutional Animal Care and Use Committee and Research Animal Resource Center at MSK.
FINANCIAL SUPPORT
B. Weigelt is funded in part by Cycle for Survival, Breast Cancer Research Foundation and Stand Up to Cancer grants, J.S. Reis-Filho in part by the Breast Cancer Research Foundation, F. Pareja in part by an NIH K12 CA184746 grant, D. Zamarin by the Ovarian Cancer Research Foundation Liz Tilberis Award and the Department of Defense Ovarian Cancer Research Academy (OC150111). F. Pareja, B. Weigelt and J.S. Reis-Filho are funded in part by the NIH/NCI P50 CA247749 01 grant. J.R. Lapa e Silva is supported by a senior investigator (1A) grant from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brazil. Research reported in this publication was funded in part by the NIH/ NCI Cancer Center Core Grant No. P30-CA008748.
Footnotes
CONFLICT OF INTEREST STATEMENT
D. Zamarin reports consulting fees from Merck, Synlogic Therapeutics, Tizona Therapeutics, and Tesaro, outside the submitted work. D. Zamarin has a patent for use of Newcastle Disease Virus for cancer therapy, outside the submitted work. N.R. Abu-Rustum reports institutional grants from GRAIL, outside the submitted work. J.S. Reis-Filho reports receiving personal/consultancy fees from Goldman Sachs, REPARE Therapeutics, Paige.AI and Eli Lilly, membership of the scientific advisory boards of VolitionRx, REPARE Therapeutics, Paige.AI and Personalis, membership of the Board of Directors of Grupo Oncoclinicas, and ad hoc membership of the scientific advisory boards of Roche Tissue Diagnostics, Ventana Medical Systems, Novartis, Genentech and InVicro, outside the scope of this study. J.S. Reis-Filho is an Associate Editor of The Journal of Pathology. B. Weigelt reports ad hoc membership of the scientific advisory board of REPARE Therapeutics.No other conflicts of interest were declared
Data availability statement
Sequencing data that support the findings of this study will be available at cBioPortal (www.cbioportal.org) upon publication of the manuscript.
REFERENCES
- 1.Park KJ. Cervical adenocarcinoma: integration of HPV status, pattern of invasion, morphology and molecular markers into classification. Histopathology 2020; 76: 112–127. [DOI] [PubMed] [Google Scholar]
- 2.Stolnicu S, Hoang L, Soslow RA. Recent advances in invasive adenocarcinoma of the cervix. Virchows Arch 2019; 475: 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med 1971; 284: 878–881. [DOI] [PubMed] [Google Scholar]
- 4.Selected item from the FDA drug bulletin-november 1971: diethylstilbestrol contraindicated in pregnancy. Calif Med 1972; 116: 85–86. [PMC free article] [PubMed] [Google Scholar]
- 5.Huo D, Anderson D, Herbst AL. Follow-up of Patients with Clear-Cell Adenocarcinoma of the Vagina and Cervix. N Engl J Med 2018; 378: 1746–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stolnicu S, Karpathiou G, Guerra E, et al. Clear Cell Carcinoma (CCC) of the Cervix Is a Human Papillomavirus (HPV)-independent Tumor Associated With Poor Outcome: A Comprehensive Analysis of 58 Cases. Am J Surg Pathol 2022. doi: 10.1097/PAS.0000000000001863. Epub ahead of print. [DOI] [PMC free article] [PubMed]
- 7.Boyd J, Takahashi H, Waggoner SE, et al. Molecular genetic analysis of clear cell adenocarcinomas of the vagina and cervix associated and unassociated with diethylstilbestrol exposure in utero. Cancer 1996; 77: 507–513. [DOI] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research Network, et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017; 543: 378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashworth A, Lord CJ, Reis-Filho JS. Genetic interactions in cancer progression and treatment. Cell 2011; 145: 30–38. [DOI] [PubMed] [Google Scholar]
- 10.Geyer FC, Li A, Papanastasiou AD, et al. Recurrent hotspot mutations in HRAS Q61 and PI3K-AKT pathway genes as drivers of breast adenomyoepitheliomas. Nat Commun 2018; 9: 1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim SH, Da Cruz Paula A, Basili T, et al. Identification of recurrent FHL2-GLI2 oncogenic fusion in sclerosing stromal tumors of the ovary. Nat Commun 2020; 11: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinreb I, Piscuoglio S, Martelotto LG, et al. Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat Genet 2014; 46: 1166–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pareja F, Brandes AH, Basili T, et al. Loss-of-function mutations in ATP6AP1 and ATP6AP2 in granular cell tumors. Nat Commun 2018; 9: 3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 2015; 17: 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pareja F, Lee JY, Brown DN, et al. The Genomic Landscape of Mucinous Breast Cancer. J Natl Cancer Inst 2019; 111: 737–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang MT, Bhattarai TS, Schram AM, et al. Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer Discov 2018; 8: 174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013; 14: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 2016; 44: e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 2012; 30: 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 21.Basturk O, Weigelt B, Adsay V, et al. Sclerosing epithelioid mesenchymal neoplasm of the pancreas - a proposed new entity. Mod Pathol 2020; 33: 456–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ciriello G, Gatza ML, Beck AH, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015; 163: 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shugay M, Ortiz de Mendibil I, Vizmanos JL, et al. Oncofuse: a computational framework for the prediction of the oncogenic potential of gene fusions. Bioinformatics 2013; 29: 2539–2546. [DOI] [PubMed] [Google Scholar]
- 24.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lawrence M, Huber W, Pages H, et al. Software for computing and annotating genomic ranges. PLoS Comput Biol 2013; 9: e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016; 32: 2847–2849. [DOI] [PubMed] [Google Scholar]
- 29.Weigelt B, Warne PH, Downward J. PIK3CA mutation, but not PTEN loss of function, determines the sensitivity of breast cancer cells to mTOR inhibitory drugs. Oncogene 2011; 30: 3222–3233. [DOI] [PubMed] [Google Scholar]
- 30.Li Z, Razavi P, Li Q, et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018; 34: 893–905 e898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corrigendum to “Revised FIGO staging for carcinoma of the cervix uteri” [Int J Gynecol Obstet 145(2019) 129–135]. Int J Gynaecol Obstet 2019; 147: 279–280. [DOI] [PubMed] [Google Scholar]
- 32.Wiegand KC, Shah SP, Al-Agha OM, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 2010; 363: 1532–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanchez-Vega F, Mina M, Armenia J, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018; 173: 321–337 e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang J, Wu S, Barrera J, et al. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005; 122: 421–434. [DOI] [PubMed] [Google Scholar]
- 35.Zhao B, Wei X, Li W, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 2007; 21: 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Xu X, Maglic D, et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep 2018; 25: 1304–1317 e1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015; 163: 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berger AC, Korkut A, Kanchi RS, et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018; 33: 690–705 e699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seiki T, Nagasaka K, Kranjec C, et al. HPV-16 impairs the subcellular distribution and levels of expression of protein phosphatase 1gamma in cervical malignancy. BMC Cancer 2015; 15: 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panayiotou T, Michael S, Zaravinos A, et al. Human papillomavirus E7 binds Oct4 and regulates its activity in HPV-associated cervical cancers. PLoS Pathog 2020; 16: e1008468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campos-Viguri GE, Peralta-Zaragoza O, Jimenez-Wences H, et al. MiR-23b-3p reduces the proliferation, migration and invasion of cervical cancer cell lines via the reduction of c-Met expression. Sci Rep 2020; 10: 3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stepanenko AA, Dmitrenko VV. HEK293 in cell biology and cancer research: phenotype, karyotype, tumorigenicity, and stress-induced genome-phenotype evolution. Gene 2015; 569: 182–190. [DOI] [PubMed] [Google Scholar]
- 43.Saucedo LJ, Edgar BA. Filling out the Hippo pathway. Nat Rev Mol Cell Biol 2007; 8: 613–621. [DOI] [PubMed] [Google Scholar]
- 44.Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol 2011; 13: 877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim MK, Jang JW, Bae SC. DNA binding partners of YAP/TAZ. BMB Rep 2018; 51: 126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boopathy GTK, Hong W. Role of Hippo Pathway-YAP/TAZ Signaling in Angiogenesis. Front Cell Dev Biol 2019; 7: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rausch V, Hansen CG. The Hippo Pathway, YAP/TAZ, and the Plasma Membrane. Trends Cell Biol 2020; 30: 32–48. [DOI] [PubMed] [Google Scholar]
- 48.Dupont S, Morsut L, Aragona M, et al. Role of YAP/TAZ in mechanotransduction. Nature 2011; 474: 179–183. [DOI] [PubMed] [Google Scholar]
- 49.Paszko E, Ehrhardt C, Senge MO, et al. Nanodrug applications in photodynamic therapy. Photodiagnosis Photodyn Ther 2011; 8: 14–29. [DOI] [PubMed] [Google Scholar]
- 50.Huggett MT, Jermyn M, Gillams A, et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br J Cancer 2014; 110: 1698–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wei H, Wang F, Wang Y, et al. Verteporfin suppresses cell survival, angiogenesis and vasculogenic mimicry of pancreatic ductal adenocarcinoma via disrupting the YAP-TEAD complex. Cancer Sci 2017; 108: 478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Q, Zhang N, Xie R, et al. Homeostatic control of Hippo signaling activity revealed by an endogenous activating mutation in YAP. Genes Dev 2015; 29: 1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steinhardt AA, Gayyed MF, Klein AP, et al. Expression of Yes-associated protein in common solid tumors. Hum Pathol 2008; 39: 1582–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lei QY, Zhang H, Zhao B, et al. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol 2008; 28: 2426–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mohamed A, Sun C, De Mello V, et al. The Hippo effector TAZ (WWTR1) transforms myoblasts and TAZ abundance is associated with reduced survival in embryonal rhabdomyosarcoma. J Pathol 2016; 240: 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu J, Alharbi A, Shan H, et al. TAZ induces lung cancer stem cell properties and tumorigenesis by up-regulating ALDH1A1. Oncotarget 2017; 8: 38426–38443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finch-Edmondson ML, Strauss RP, Passman AM, et al. TAZ Protein Accumulation Is Negatively Regulated by YAP Abundance in Mammalian Cells. J Biol Chem 2015; 290: 27928–27938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.LeBlanc L, Ramirez N, Kim J. Context-dependent roles of YAP/TAZ in stem cell fates and cancer. Cell Mol Life Sci 2021; 78: 4201–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu T, Liu Y, Gao H, et al. Clinical significance of yes-associated protein overexpression in cervical carcinoma: the differential effects based on histotypes. Int J Gynecol Cancer 2013; 23: 735–742. [DOI] [PubMed] [Google Scholar]
- 60.Deng J, Zhang W, Liu S, et al. LATS1 suppresses proliferation and invasion of cervical cancer. Mol Med Rep 2017; 15: 1654–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 2017; 7: 818–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoadley KA, Yau C, Hinoue T, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018; 173: 291–304 e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang X, George J, Deb S, et al. The Hippo pathway transcriptional co-activator, YAP, is an ovarian cancer oncogene. Oncogene 2011; 30: 2810–2822. [DOI] [PubMed] [Google Scholar]
- 64.Azar WJ, Christie EL, Mitchell C, et al. Noncanonical IL6 Signaling-Mediated Activation of YAP Regulates Cell Migration and Invasion in Ovarian Clear Cell Cancer. Cancer Res 2020; 80: 4960–4971. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Figure S1. Cancer cell fractions of somatic mutations and copy number alterations of clear cell carcinomas of the cervix.
Figure S2. Overexpression of Hippo pathway targets in human clear cell carcinomas of the cervix and in xenograft tumors
Figure S3. TAZ protein and gene expression validation
Figure S4. IC50 of verteporfin in HEK-293 and HaCaT cells
Figure S5. Quantification of cytoplasmic clearing, necrosis and mice body weight in xenograft models
Figure S6. WWTR1 S89 mutations in ovarian and endometrial cancers
Table S1. Clinicopathologic characteristics of patients with clear cell carcinoma of the cervix included in this study
Table S2. Non-synonymous somatic mutations identified in clear cell carcinomas of the cervix subjected to whole-exome or targeted massively parallel sequencing
Table S3. Fusion candidate genes and/or readthroughs identified by RNA-sequencing analysis of clear cell carcinomas of the cervix
Data Availability Statement
Sequencing data that support the findings of this study will be available at cBioPortal (www.cbioportal.org) upon publication of the manuscript.