

Abstract
Chiral amines are among the most important organic compounds and have widespread applications. Enantioselective construction of chiral amines is a major aim in organic synthesis. Among synthetic methods, direct functionalization of omnipresent C–H bonds with common organic nitrogen compounds represents one of the most attractive strategies. However, C–H amination strategies are largely limited to constructing a specific type of N-heterocycles or amine derivatives. To maximize the synthetic potential of asymmetric C–H amination, we report here an approach that unites the complementary reactivities of radical and ionic chemistry for streamlined synthesis of functionalized chiral amines. This synthesis merges the development of an enantioselective radical process for 1,5-C(sp3)–H amination of alkoxysulfonyl azides via Co(II)-based metalloradical catalysis with an enantiospecific ionic process for ring-opening of the resulting five-membered chiral sulfamidates by nucleophiles. Given that alkoxysulfonyl azides are derived from the corresponding alcohols, this approach offers a powerful synthetic tool for enantioselective β-C–H amination of common alcohols while converting the hydroxy group to other functionalities through formal nucleophilic substitution.
Radical chemistry has been increasingly explored for the development of alternative synthetic tools that are complementary to traditional ionic reactions for construction of molecular structures1–4. Despite notable advancements, there remain formidable challenges in the synthetic applications of radical reactions, such as the control of enantioselectivity and reactivity. Among recent developments5–9, metalloradical catalysis (MRC) explores how to utilize metal-centred radical species as open-shell catalysts for catalytically generating metal-stabilized organic radicals and for controlling their subsequent one-electron homolytic reactions10–15. In particular, Co(II) complexes of porphyrins as a family of stable 15e-metalloradicals have been demonstrated as effective catalysts for homolytic activation of organic azides to generate α-Co(III)-aminyl radicals as key intermediates for MRC16. Supported by proper D2-symmetric chiral amidoporphyrins with cavity-like environments, the Co(II)-based catalysts can effectively govern the desired course of MRC towards enantioselective aziridination17 and C(sp3)–H amination18. Given the complementary reactivities of radical and ionic chemistry, we were intrigued by how effective, practical and general a synthetic methodology could become when merging a catalytic radical C–H functionalization process with traditional ionic functional group interconversions. To this end, 1,2-difunctionalization of alcohols into chiral amines was proposed (Fig. 1a). The synthetic pathway begins with a facile nucleophilic azide transfer to common alcohols and entails an envisioned enantioselective radical 1,5-C–H amination of these alcohol-derived organic azides to generate five-membered cyclic sulfamidates. Nucleophilic ring-opening reactions can then produce a wide range of highly enantioenriched β-functionalized chiral amines. While the nucleophilic ring-opening of five-membered cyclic sulfamidates would be expected to be stereospecific19,20, it was unclear if the unprecedented radical 1,5-C–H amination with alkoxysulfonyl azides could proceed efficiently with effective control of enantioselectivity in view of their unknown reactivity and higher flexibility, which significantly disfavours the efficiency of hydrogen atom abstraction and radical substitution compared with the previously explored sulfamoyl azides18. In addition to the prerequisite for metalloradical activation of alkoxysulfonyl azides, how successful this methodology will be is exclusively contingent on the identification of a suitable D2-symmetric chiral amidoporphyrin as the supporting ligand that will not only facilitate both the 1,5-H-atom abstraction of the corresponding α-Co(III)-aminyl radical intermediate I and subsequent radical substitution of the resulting ε-Co(III)-alkyl radical II (Fig. 1b) for a broad type of C–H substrates, but also control the enantioselectivity of the C–N bond formation. If this feat could be achieved, the combination of enantioselective radical 1,5-C–H amination of alkoxysulfonyl azides with stereospecific ring-opening of the resulting optically active cyclic sulfamidates by different nucleophiles would create a streamlined approach for divergent synthesis of chiral amines bearing diverse β-functionalities from simple alcohols (Fig. 1a). Such amines are key structural motifs in many biologically important molecules (Supplementary Fig. 1)21–23.
Fig. 1 |. Combined radical and ionic approach for enantioselective synthesis of chiral amines from alcohols.
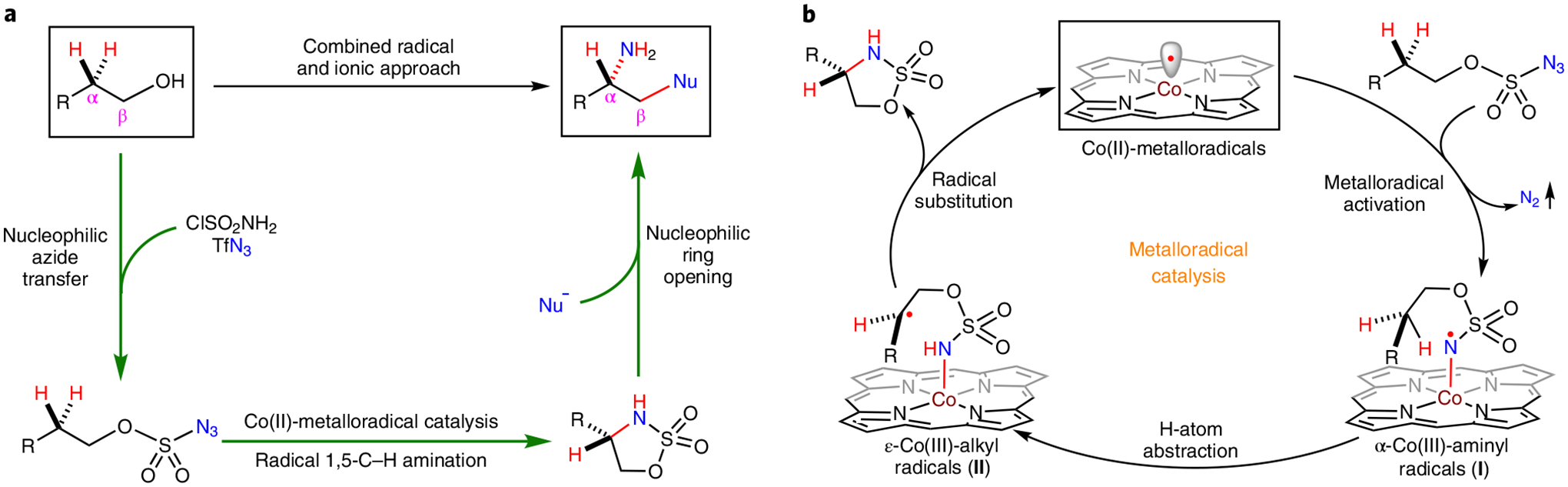
a, 1,2-Difunctionalization of alcohols for asymmetric synthesis of β-functionalized chiral amines by merging enantioselective radical process for 1,5-C–H amination of alkoxysulfonyl azides with enantiospecific ionic process for ring-opening of the resulting five-membered chiral sulfamidates by nucleophiles. b, Proposed mechanism for enantioselective radical 1,5-C–H amination of alkoxysulfonyl azides via Co(II)-based metalloradical catalysis to synthesize optically active cyclic sulfamidates. Tf, trifluoromethanesulfonyl; Nu, nucleophile.
Metal-catalysed asymmetric 1,5-C–H amination represents an attractive strategy for construction of synthetically valuable five-membered chiral cyclic sulfamidates24. While advancements have been recently made in the development of catalytic systems for 1,6-C(sp3)–H amination of sulfamate esters25–30, there has been only limited success in synthesizing five-membered N-heterocycles through catalytic 1,5-C(sp3)–H amination31. This is perhaps not a surprise given that most of the existing catalytic systems proceed through a concerted C–H insertion pathway involving metallonitrene intermediates, which would inevitably encounter the difficulty associated with the highly strained [3.1.0]-bicyclic transition state for the 1,5-C–H amination process32. Because of the electrophilic nature of metallonitrene intermediates, amination of allylic and propargylic C–H bonds poses an additional issue of chemoselectivity over competitive addition reactions to C=C and C≡C bonds33,34. Apart from the reactivity issues, control of enantioselectivity in catalytic 1,5-C–H amination has remained largely unsolved, with only a few examples for benzylic C–H bonds25,35. As an application of Co(II)-MRC for addressing these synthetic challenges, we report here a catalytic radical process for enantioselective 1,5-C(sp3)–H amination of alkoxysulfonyl azides that enables stereoselective synthesis of five-membered cyclic sulfamidates. We unveil a remarkable ligand effect on the Co(II)-based catalytic system, leading to the identification of a bridged D2-symmetric chiral amidoporphyrin as the optimal supporting ligand36. The optimized catalytic system is capable of activating various alkoxysulfonyl azides at room temperature for 1,5-amination of different types of C–H bonds, including those at benzylic, allylic, propargylic and even unactivated sites, delivering five-membered chiral sulfamidates in high yields with excellent diastereoselectivities and enantioselectivities. We also present mechanistic studies that shed light on the underlying stepwise radical mechanism for this catalytic system. We then demonstrate the versatility of subsequent ring-opening reactions of the resulting enantioenriched five-membered cyclic sulfamidates by various types of nucleophiles, giving rise to a wide range of chiral amines bearing diverse β-functionalities with retention of the original enantiopurity.
Results and discussion
Reaction optimization.
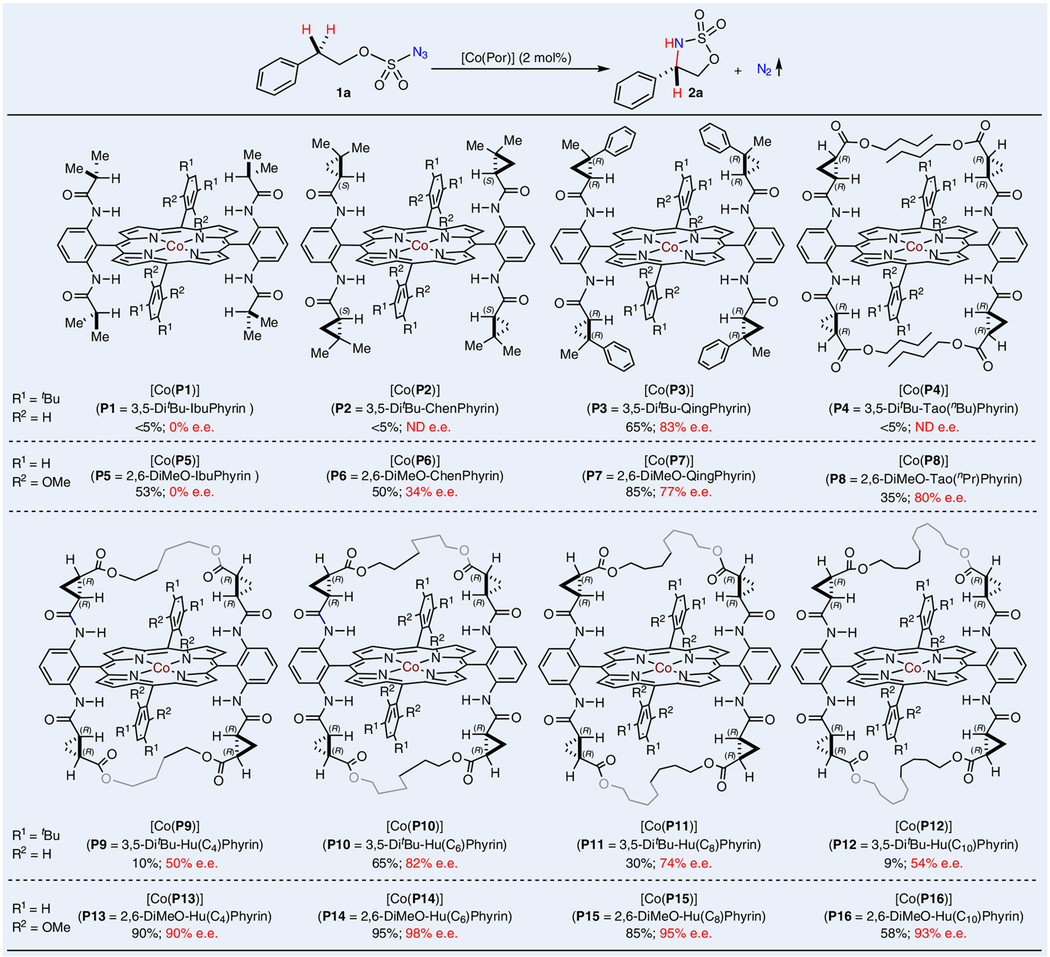
At the outset of this project, 2-phenylethoxysulfonyl azide (1a) was selected as the model alkoxysulfonyl azide to explore 1,5-C–H amination via Co(II)-based metalloradical catalysis (Table 1). After some initial evaluation of reaction conditions, we decided to systematically investigate the ligand effect of amidoporphyrins on the Co(II)-catalysed intramolecular C–H amination in benzene at room temperature. It was found that both achiral metalloradical catalyst [Co(P1)] (P1 = 3,5-DitBu-IbuPhyrin)37 and first-generation chiral metalloradical catalyst [Co(P2)] (P2 = 3,5-DitBu-ChenPhyrin)38 were ineffective for 1,5-C–H amination reaction of azide 1a, producing only a trace amount of the desired 5-membered chiral cyclic sulfamidate 2a. When the second-generation metalloradical catalyst [Co(P3)] (P3 = 3,5-DitBu-QingPhyrin)39 was employed, a substantial improvement in the yield of product 2a was observed while inducing a high level of asymmetry. However, switching to another second-generation catalyst [Co(P4)] (P4 = 3,5-DitBu-Tao(nBu) Phyrin) resulted in almost no formation of 2a. These initial results appeared to indicate the importance of rigidifying the pocketlike environment of the amidoporphyrin ligand. Accordingly, analogous metalloradical catalysts [Co(P5)] (P5 = 2,6-DiMeOIbuPhyrin), [Co(P6)] (P6 = 2,6-DiMeO-ChenPhyrin), [Co(P7)] (P7 = 2,6-DiMeO-QingPhyrin) and [Co(P8)] (P8 = 2,6-DiMeOTao(nPr)Phyrin), where the porphyrins bear the same corresponding amide units but contain 2,6-dimethoxyphenyl instead of 3,5-di-tert-butylphenyl groups as 5,15-aryl substituents, were used to catalyse the C–H amination reaction. As expected, significantly improved results were generally observed for catalytic reactions by [Co(P5)]–[Co(P8)] in comparison with [Co(P1)]–[Co(P4)]. Notably, changing from [Co(P4)] to analogous [Co(P8)] led to formation of 2a in significantly improved yield with a high level of enantioselectivity. This promising result prompted us to explore the direction to further rigidify D2-symmetric chiral amidoporphyrins by employing new-generation catalysts [Co(HuPhyrins)] with cavity-like environments, which are created from [Co(P4)] and [Co(P8)] by linking across two chiral amide units on both sides of the porphyrin plane with alkyl bridges36. Although C4-bridged catalyst [Co(P9)] (P9 = 3,5-DitBu-Hu(C4)Phyrin) was inferior to [Co(P8)], further improvements in both yield and enantioselectivity were obtained with the use of C6-bridged catalyst [Co(P10)] (P10 = 3,5-DitBu-Hu(C6)Phyrin). Interestingly, when C8-bridged catalyst [Co(P11)] (P11 = 3,5-DitBu-Hu(C8)Phyrin) was used, the improvements disappeared. The downward trend in both reactivity and enantioselectivity continued with the use of C10-bridged catalyst [Co(P12)] (P12 = 3,5-DitBu-Hu(C10)Phyrin), indicating the pronounced influence of the bridge length. To combine the bridging effect with the influence of 5,15-aryl substituents, analogous bridged catalysts [Co(P13)] (P13 = 2,6-DiMeO-Hu(C4)Phyrin), [Co(P14)] (P14 = 2,6-DiMeO-Hu(C6)Phyrin), [Co(P15)] (P15 = 2,6-DiMeOHu(C8)Phyrin) and [Co(P16)] (P16 = 2,6-DiMeO-Hu(C10)Phyrin) were then employed to catalyse the C–H amination reaction. Excitingly, further improvements in both yield and enantioselectivity were observed for the reactions catalysed by [Co(P13)]–[Co(P16)]. Among them, C6-bridged [Co(P14)] emerged as the optimal catalyst, affording the desired five-membered chiral cyclic sulfamidate 2a in 95% yield with 98% e.e.
Table 1 |.
Ligand effect on Co(ii)-catalysed enantioselective radical 1,5-C(sp3)–H amination of alkoxysulfonyl azide
|
Performed in benzene at room temperature for 24 h using [Co(Por)] (2 mol%) under N2 in the presence of 4 Å molecularsieves; [azide 1a] = 0.1 M; isolated yields and e.e. values of 2a are shown for each of the ligands examined. The e.e. was determined by chiral HPLC. Differential scanning calorimetry (DSC) measurement (see Supplementary Section 2.2 for details) showed that azide 1a decomposed at an onset temperature of 134.7 °C with a peak temperature of 135.8 °C. ND, not determined.
Substrate scope.
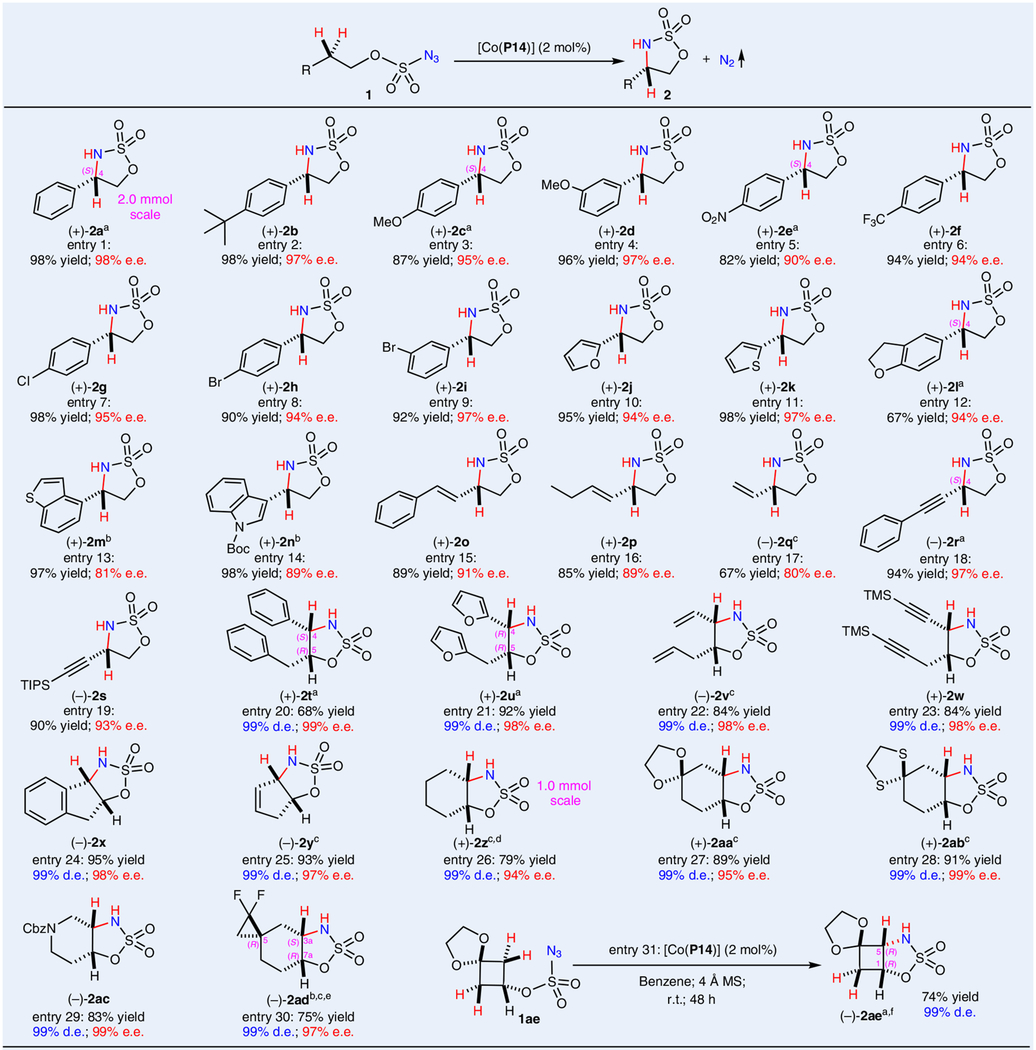
Under the optimized conditions, metalloradical catalyst [Co(P14)] was found to be generally effective in catalysing asymmetric intramolecular amination of different types of C(sp3)–H bonds in various alkoxysulfonyl azides (Table 2). Like azide 1a, 2-arylethoxysulfonyl azides containing aryl substituents with varied electronic and steric properties, including 4-tert-butyl (1b), 4-methoxy (1c), 3-methoxy (1d), 4-nitro (1e), 4-trifluoromethyl (1f), 4-chloro (1g), 4-bromo (1h) and 3-bromo (1i) groups, were all suitable substrates for [Co(P14)]-catalysed 1,5-C–H amination, forming the corresponding five-membered cyclic sulfamidates (+)-2a–(+)-2i in high yields with excellent enantioselectivities (entries 1–9). It is worth mentioning that the catalytic amination process could be readily scaled up under the standard conditions without affecting the high yield and excellent enantioselectivity, as demonstrated by the efficient synthesis of five-membered chiral cyclic sulfamidate (+)-2a on a 2.0 mmol scale in 98% yield with 98% e.e. (entry 1). The absolute configurations of (+)-2a, (+)-2c and (+)-2e were all established as (4S) by X-ray crystallography. The Co(II)-based metalloradical system was shown to be compatible with alkoxysulfonyl azides containing heteroaryl groups, such as furan (1j), thiophene (1k), dihydrobenzofuran (1l), benzothiophene (1m) and indole (1n), leading to high-yielding formation of the corresponding heteroaryl-substituted sulfamidates (+)-2j–(+)-2n with excellent enantioselectivities (entries 10–14). The absolute configuration of (+)-2l was also established as (4S) by X-ray crystallography. Furthermore, the catalytic system could chemoselectively aminate allylic and propargylic C–H bonds without affecting the otherwise more reactive C=C and C≡C π bonds, as shown by the productive preparation of the alkenyl- and alkynylsubstituted cyclic sulfamidate (−)-2o–(−)-2s in highly enantioenriched forms (entries 15–19). The absolute configuration of (−)-2r was also established as (4S) by X-ray crystallography. In addition to the neutral and non-oxidative conditions, the demonstrated compatibility and chemoselectivity are attributed to the underlying stepwise radical mechanism of the Co(II)-based amination process.
Table 2 |.
Enantioselective intramolecular radical 1,5-C(sp3)–H amination of alkoxysulfonyl azides by [Co(P14)]
|
Performed in benzene at room temperature for 24 h using [Co(P14)] (2 mol%) under nitrogen in the presence of 4 Å molecular sieves; [azide 1] = 0.1 M; isolated yields; e.e. determined by chiral HPLC. Diastereomeric excess (d.e.) was determined by 1H NMR analysis of crude reaction mixture. aAbsolute configuration determined by X-ray crystallography. b[Co(P14)] (4 mol%). cThe e.e. was determined through derivatization. dSimilar yield with identical e.e. obtained for reaction run on 0.10 mmol scale. eAbsolute configuration determined by X-ray crystallography of the benzylation derivative. fNo determination of e.e. due to unavailability of racemic sample.
To further broaden the synthetic utility of the asymmetric catalytic system, we then explored the application of the 1,5-C–H amination by [Co(P14)] to a desymmetrization process that would stereoselectively create two contiguous stereogenic centres in one catalytic operation. To this end, 1,3-diphenyl-2-propanol-derived alkoxysulfonyl azide 1t was prepared as a substrate for the catalytic system. To our delight, the two benzylic sites in azide 1t could be desymmetrized by [Co(P14)] for 1,5-C–H amination, affording 4,5-disubstituted cyclic sulfamidate (+)-2t in good yield with almost perfect control of both diastereoselectivity and enantioselectivity (entry 20). The absolute configurations of (+)- were established as (4S,5R) by X-ray crystallography, confirming the unexpected formation of cis-diastereomer. In addition to benzylic C–H bonds, [Co(P14)] could effectively catalyse asymmetric desymmetrization amination of α-heteroaryl C–H bonds and of allylic and propargylic C–H bonds as manifested by the high-yielding formation of (+)-2u–(+)-2w as cis-diastereomers with nearly complete control of the two newly generated contiguous stereogenic centres from the reactions of azides 1u–1w, with full tolerance of the heteroaryl, alkenyl and alkynyl functionalities (entries 21–23). The absolute configurations of (+)-2u were established as (4R,5R) by X-ray crystallography, again affirming the formation of the less common cis-diastereomer. Prompted by these exciting results, we further exploited the use of alkoxysulfonyl azides derived from cyclic secondary alcohols as potential substrates for Co(II)-catalysed desymmetrization amination, which would lead to asymmetric formation of bicyclic sulfamidates with highly strained fused structures. Accordingly, 2-indanol-based alkoxysulfonyl azide 1x was prepared as a substrate for the catalytic asymmetric desymmetrization amination. Remarkably, the two cyclic benzylic sites could be effectively desymmetrized by [Co(P14)] for 1,5-C–H amination, constructing cis-fused tricyclic sulfamidate (−)-2x in excellent yield with close to complete control of stereoselectivities for the two newly generated contiguous stereogenic centres (entry 24). Similarly, the C–H bonds at the two allylic sites in alkoxysulfonyl azide 1y derived from 3-cyclopentenol could chemoselectively undergo asymmetric desymmetrization amination, affording cis-bicyclic sulfamidate (−)-2y in high yield with excellent stereoselectivities (entry 25). Such desymmetrization protocol could be even applied to asymmetric amination of unactivated C–H bonds in saturated cyclic substrates (bond dissociation energy, ~100 kcal mol−1)40 as demonstrated by the productive transformation of cyclohexanol-based alkoxysulfonyl azide 1z to cis-fused bicyclic sulfamidate (+)-2z with excellent asymmetric control on either small (0.1 mmol) or large (1.0 mmol) scale (entry 26). Furthermore, ketal and thioketal functionalities in related cyclohexanol-based alkoxysulfonyl azides 1aa and 1ab were well tolerated in the Co(II)-catalysed asymmetric desymmetrization amination, generating the corresponding cis-fused spirotricyclic sulfamidate (+)-2aa and (+)-2ab in excellent yields with excellent control of stereoselectivities (entries 27 and 28). Moreover, alkoxysulfonyl azides based on heterocyclic alcohols were shown to be suitable substrates as exemplified by the asymmetric desymmetrization of 1-Cbz-4-piperidinol-derived alkoxysulfonyl azide 1ac for 1,5-C–H amination to generate cyclic sulfamidate-fused piperidine (−)-2ac in high yield as cis-isomer with perfect control of stereoselectivities (entry 29). Building on these remarkable results, we carried on further exploration of the potentiality of catalyst [Co(P14)] for concurrent construction of three stereogenic centres from achiral substrates through asymmetric desymmetrization amination. Hence, alkoxysulfonyl azide 1ad prepared from the difluorocyclopropane/cyclohexane spirobicylic alcohol was tested as the substrate for this purpose. Excitingly, the two unactivated 2° C–H sites on the cyclohexane ring of azide 1ad could be effectively desymmetrized by smooth 1,5-C–H amination, leading to productive formation of cis-fused spirotricyclic sulfamidate (−)-2ad with simultaneous creation of three stereogenic centres, of which both the diastereoselectivity and enantioselectivity could be excellently controlled. The absolute configurations of (−)-2ad were established as (3aS,5R,7aR) by X-ray crystallography from its N-benzyl derivative (+)-N-Bn-2ad (entry 30). As another direction of our further exploration, we decided to scrutinize the catalytic system for potential construction of fused bicyclic five-membered sulfamidates with even higher ring strain. Consequently, alkoxysulfonyl azide 1ae derived from ketal-containing cyclobutanol was prepared as the substrate for the asymmetric desymmetrization amination system. To our delight, it was found that catalyst [Co(P14)] could even enable asymmetric 1,5-amination of the secondary C–H bonds on the four-membered cyclobutane ring (bond dissociation energy, >100 kcal mol−1)40 of azide 1ae with effective desymmetrization of the two reaction sites, giving rise to highly-strained [3.2.0]-bicyclic sulfamidate (−)-2ae without affecting the dioxolane/cyclobutane spirobicyclic functional unit (entry 31). The absolute configurations of (−)-2ae were established as (1R,5R) by X-ray crystallography. It should be mentioned that the first- and second-generation non-bridged catalysts [Co(P1)]–[Co(P8)] were completely ineffective for the amination reaction of azide 1ae, demonstrating the remarkable effect of ligand engineering by distal bridging (see Supplementary Fig. 3 for additional examples)36.
Mechanistic investigation.
Several mechanistic experiments were performed to shed light on the proposed stepwise radical pathway of the Co(II)-catalysed 1,5-C(sp3)–H amination of alkoxysulfonyl azides. To observe the radical intermediates, isotropic electron paramagnetic resonance (EPR) spectroscopy was carried out at room temperature for the reaction mixture of azide 1a by [Co(P14)] in benzene (Fig. 2a). The obtained EPR spectrum displays diagnostic signals at a g value of ~ 2.00 that are indicative of organic radicals (Fig. 2a). Among other possibilities, the observed broad signals could be simulated as a mixture of α-Co(III)-aminyl radical and ε-Co(III)-alkyl radical on the basis of hyperfine couplings by 59Co (I = 7/2), 14N (I = 1) and 1H (I = 1/2): 10% of N-centred radical at α-position I[Co(P14)]/1a (g = 1.99599; A(Co) = 8.1 MHz; A(N) = 48.7 MHz; A(H) = 0 MHz) and 90% of C-centred radical at ε-position II[Co(P14)]/1a (g = 2.00458; A(Co) = 0 MHz; A(N) = 0 MHz; A(H) = 13.4 MHz). Although it is not unique, this simulation is consistent with the proposed stepwise radical mechanism involving the key step of 1,5-H-atom abstraction. Furthermore, these intermediates could be detected by high-resolution mass spectrometry (HRMS) with ESI ionization (Fig. 2a). The obtained spectrum evidently exhibited a signal corresponding to [I [Co(P14)]/la]+ and/or [II[Co(P14)]/1a]+ (m/z = 1,662.4981), which resulted from the corresponding neutral radicals by the loss of one electron. Both the exact mass and the isotope distribution pattern measured experimentally match well with those calculated from the formula of [(P14)Co(C8H9NO3S]+ (m/z = 1,662.5009) (see Supplementary Fig. 5 for details). To gain insight on the origin of asymmetric induction in the process, isotopomeric alkoxysulfonyl azides (S)-1aD and (R)-1aD were prepared in optically pure forms as substrates for Co(II)-catalysed asymmetric radical 1,5-C–H amination to study the kinetic isotope effect (KIE) (Fig. 2b). With the use of achiral catalyst [Co(P5)] (P5 = 2,6-DiMeOIbuPhyrin), the intramolecular KIEs for reactions of isotopomers (S)-1aD and (R)-1aD were determined to have nearly identical values of 11.3 and 11.4, respectively (Fig. 2b, entries 1 and 2). These high values of primary KIEs are in accordance with the proposed step of C–H bond cleavage through 1,5-H-atom abstraction by the initially formed α-Co(III)-aminyl radical I (Fig. 1b). If a chiral catalyst is employed, it is anticipated to result in deviation from the intrinsic KIE (higher in the chirality-matched case while lower in the chirality-mismatched case) if the step of H-atom abstraction is enantioselective. Experimentally, it was found that the use of chiral catalyst [Co(P14)] raised the KIE to 44.0 for (R)-1aD (Fig. 2b, entry 4) while lowering it to 0.2 for (S)-1aD (Fig. 2b, entry 3), suggesting highly preferential abstraction of pro-S over pro-R hydrogen atom by [Co(P14)]. The measured KIE values could be translated to the ratios of the initially established two prochiral faces (Re)-II1a to (Si)-II1a (Re:Si of II1a). If no racemization of the facial chirality occurs in the subsequent step of radical substitution, the Re:Si of II1a ratios could then be used to calculate the predicted enantiomeric excesses (e.e.%cal) of the amination product 2a. The fact that the observed enantiomeric excesses (e.e.%exp) were much lower than the predicted values for the reactions by achiral catalyst [Co(P5)] indicated significant racemization of the initial facial chirality via α-C–C bond rotation before the radical substitution step (Fig. 2b, entries 1 and 2). When chiral catalyst [Co(P14)] was used, however, it was found that e.e.%exp was significantly higher than e.e.%cal for reactions of both (S)-1aD (Fig. 2b, entry 3: 92% versus 68%) and (R)-1aD (Fig. 2b, entry 4: 99% versus 96%). These results revealed that the steps of both H-atom abstraction and radical substitution cooperatively controlled the enantioselectivity of the Co(II)-catalysed 1,5-C–H amination. Moreover, the results in Table 1 seem to suggest that the catalysts with the higher yields generally exhibit the better enantioselectivities. To study the relative kinetics among the catalysts, we performed NMR studies on the 1,5-C–H amination reaction of azide 1a under standard conditions by the optimized catalyst [Co(P14)] in comparison with the less-effective catalyst [Co(P9)] as they differ dramatically in both yield (95% versus 10% yield) and enantioselectivity (98% versus 50% e.e.). The observed rate constants (kobs) for the catalytic reaction by [Co(P14)] and [Co(P9)] were determined to be 7.5 × 10−2 h−1 and 4.5 × 10−3 h−1, respectively, showing that the reaction by [Co(P14)] proceeded approximately 16 times faster than that by [Co(P9)] (see Supplementary Fig. 6 for details). Collectively, these results indicate that the catalysts with the higher yields and better enantioselectivities also catalysed the reactions at faster rates.
Fig. 2 |. Mechanistic studies on Co(ii)-catalysed radical 1,5-C–H amination of alkoxysulfonyl azides.
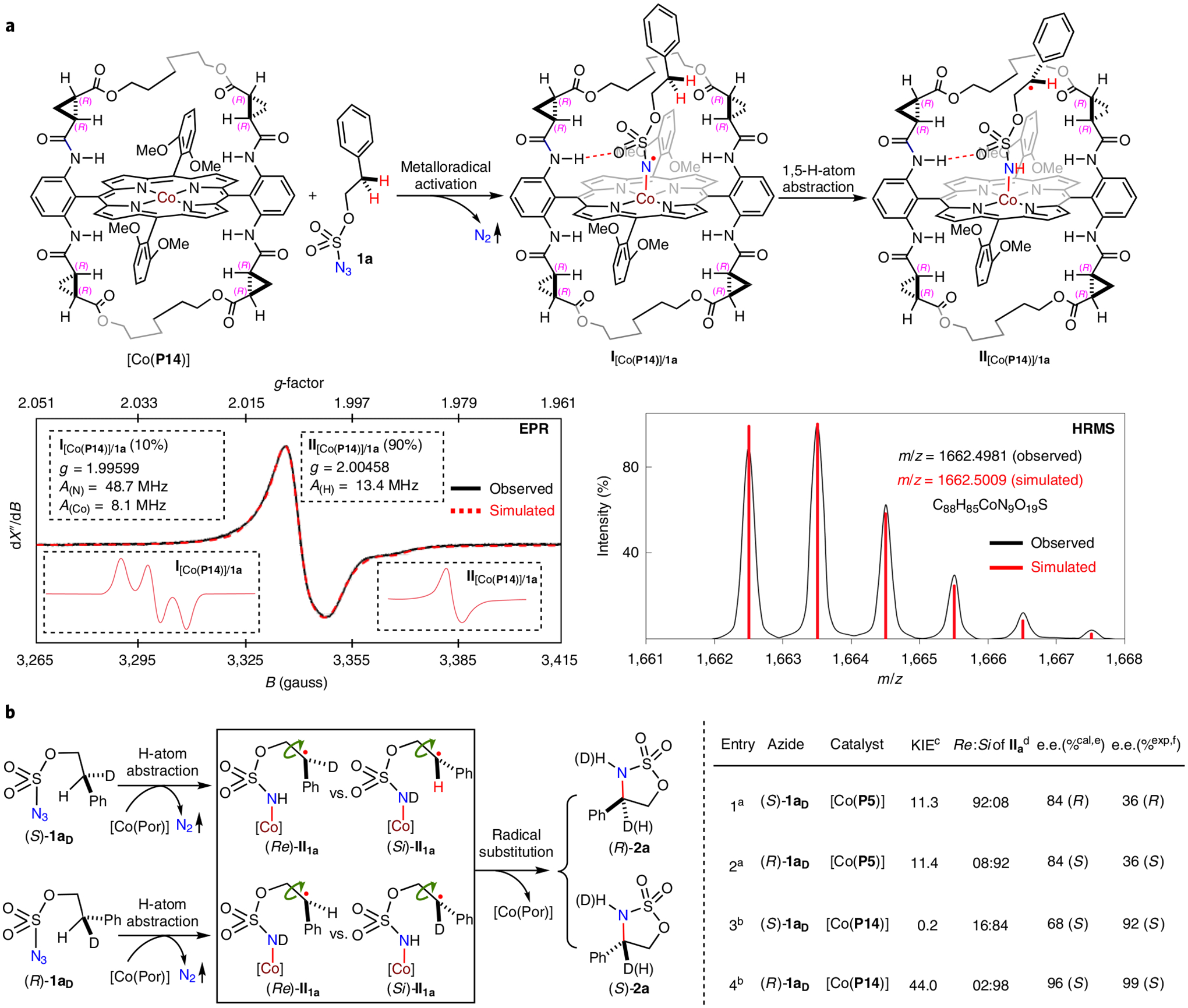
a, Detection of α-Co(III)-aminyl radical and ε-Co(III)-alkyl radical intermediates by electron paramagnetic resonance (EPR; dX”/dB: the first derivative of the imaginary part of molecular magnetic susceptibility (X”) with respect to the external static magnetic field (B) in arbitrary units) and high-resolution mass spectrometry (HRMS). b, Assessment of asymmetric induction mode through KIE studies on 1,5-C–H amination of enantiopure isotopomeric azides.aPerformed in benzene at 40 °C for 24 h using [Co(Por)] (2 mol%) on 0.10 mmol scale under nitrogen in the presence of 4 Å molecular sieves; [azide 1aD] = 0.1 M; see Supplementary Table 1 for yields. bCarried out at room temperature for 24 h. cRatios of H:D determined by 1H NMR spectroscopy, suggesting highly preferential abstraction of pro-S over pro-R hydrogen atoms with [Co(P14)]. dCalculated ratios of the initially established two prochiral faces (Re)-ii1a to (Si)-ii1a based on the ratio of H:D. eCalculated enantiomeric excesses of 2a on the basis of stereoretentive radical substitution. fObserved enantiomeric excesses of 2a determined by chiral HPLC analysis, which offered no separation of (R)-2aH from (R)-2aD and (S)-2aH from (S)-2aD, indicating significant racemization of the initial facial chirality via α-C–C bond rotation before the step of radical substitution with [Co(P5)] while further enantioenrichment of the initial facial chirality during the step of C–N bond formation with [Co(P14)].
Synthetic transformations.
Given that five-membered cyclic sulfamidates have a high propensity to be ring-opened by nucleophiles with site-selectivity at the carbon next to the oxygen19, this newly developed enantioselective radical process for 1,5-C(sp3)–H amination of alkoxysulfonyl azides could be potentially merged with the ionic ring-opening process for asymmetric synthesis of β-functionalized chiral amines20,41. To this end, we systematically investigated the ring-opening reactions of the resulting enantioenriched five-membered cyclic sulfamidates 2 with various classes of nucleophiles (Fig. 3). Different carbon nucleophiles were employed to evaluate the opening of five-membered ring structures for stereospecific synthesis of chiral amine derivatives (Fig. 3a). As demonstrated with the highly enantioenriched N-Boc-sulfamidate N-Boc-2a (98% e.e.) and N-Bn-sulfamidate N-Bn-2a (98% e.e.) as the model substrates, a series of sp-, sp2- and sp3-hybridized carbon nucleophiles could effectively open the cyclic structures, leading to stereospecific production of β-functionalized chiral amines (+)-3a–(+)-3d in high yields with full retention of the original enantiopurities. Potassium cyanide could similarly act as a carbon nucleophile for stereospecific ring-opening reaction of N-Boc-2a to form β-amino nitrile (+)-3e in excellent yield. In addition to being readily converted to Boc-l-β-phenylalanine through hydrolysis42, (−)-3e was utilized as a key intermediate for the synthesis of (+)-(S)-dihydroperiphylline43. It was further shown that indole could function as carbon nucleophile at the C3 position (rather than as nitrogen nucleophile) to open the cyclic sulfamidate N-Boc-2a stereospecifically in the presence of MeMgCl and CuCl (ref.44), generating β-phenyl chiral tryptamine (+)-3f in good yield without affecting the original high enantiopurity. Besides carbon nucleophiles, nitrogen nucleophiles were examined for the ring-opening process to synthesize vicinal 1,2-diamine derivatives (Fig. 3b)45. With sodium azide and pyrazole as nucleophiles, β-azido-α-chiral amine (+)-3g and β-pyrazole α-chiral amine (+)-3h were formed in excellent yields with preservation of the original stereochemistry. Under similar conditions, the amide moiety in ethyl tosylglycinate could open the ring structure of N-Bn-2a smoothly to generate the corresponding diamine product, which could in situ undergo effective lactamization upon heating to afford the piperazinone derivative (−)-3i, an important class of recurring moieties in drug research and development46. The absolute configuration of (−)-3i was established as (6S) by X-ray crystallography, confirming the retention of the original stereochemistry in 2a. Additionally, primary and secondary amines, and the sterically hindered natural product cytisine, could all act as effective nucleophiles for productive formation of 1,2-diamines (+)-3j, (+)-3k, and the interesting cytisine-derived47 diamine (−)-3l in excellent yields without affecting the original enantiopurity. Other heteroatom-based nucleophiles could also be employed for this stereospecific opening process (Fig. 3c). For instance, even sodium acetate served as an effective oxygen nucleophile to react with N-Boc-2a, providing enantioenriched β-amino alcohol derivative (+)-3m in excellent yield. The absolute configuration of (+)-3m was established as (2S) by X-ray crystallography. When 17α-ethynylestradiol, a derivative of the natural product estradiol that contains three potential nucleophilic sites (phenol, tertiary alcohol and terminal alkyne), was reacted with N-Boc-2a under basic conditions, it afforded (+)-3n as the sole product in excellent yield. Furthermore, even fluoride could function as a competent nucleophile, generating β-fluoro-α-chiral amine (+)-3o in high yield with excellent stereospecificity48. Similarly, the use of phosphine nucleophiles such as potassium diphenylphosphanide allowed the synthesis of (+)-3p, which may function as chiral N,P-chelating ligand to support metal-based asymmetric catalysis. 1-Thio-β-d-glucose as its tetraacetate form was shown to be a highly effective sulfur nucleophile in reacting with N-Boc-2a to form sugar-derived β-amino thiol (−)-3q in excellent yield.
Fig. 3 |. regioselective ring-opening of enantioenriched sulfamidates for stereospecific synthesis of β-functionalized chiral amines.
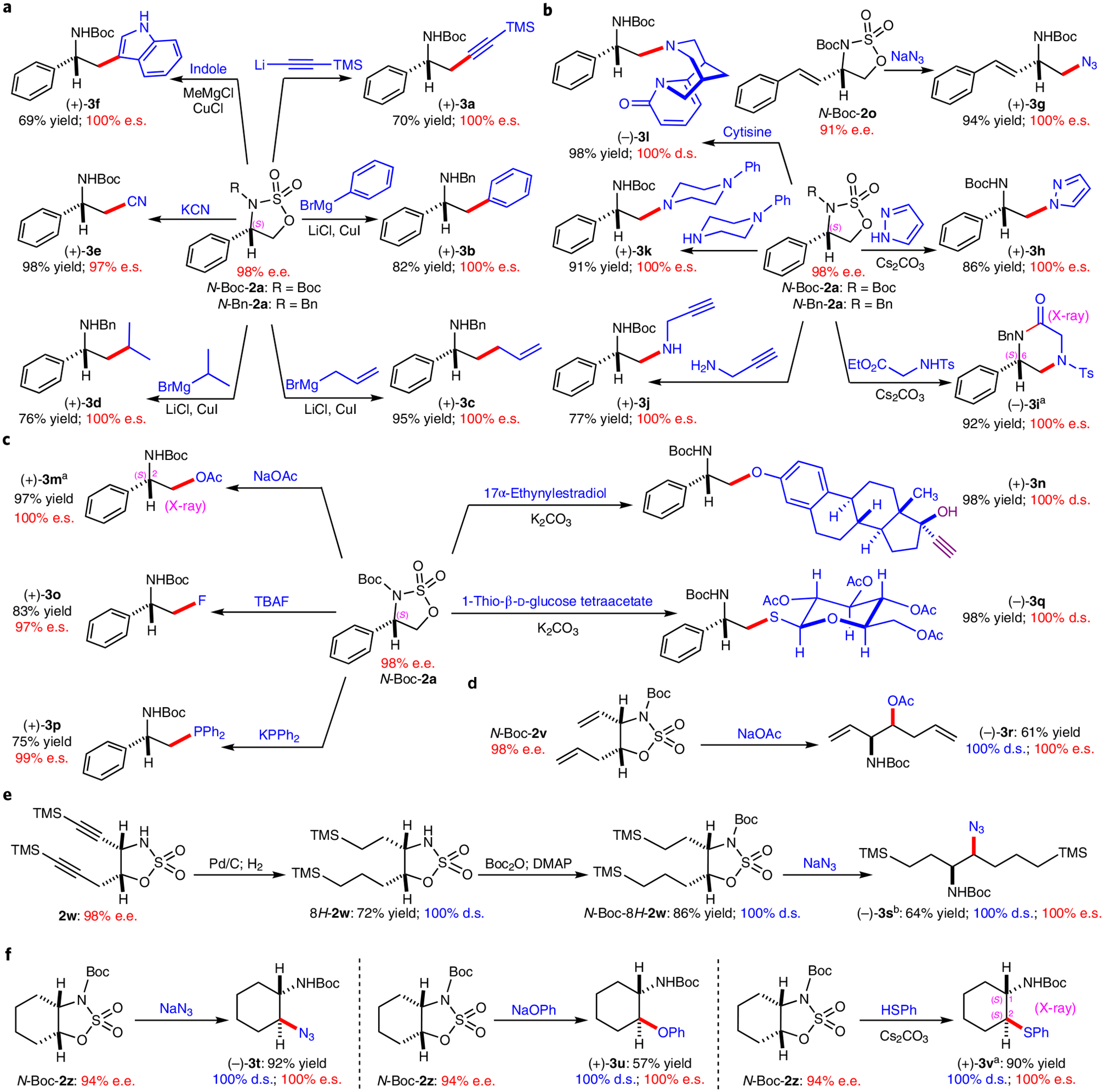
a, Ring-opening by carbon nucleophiles. b, Ring-opening by nitrogen nucleophiles. c, Ring-opening by oxygen, fluorine, phosphine and sulfur nucleophiles. d, Ring-opening of 4,5-disubstituted cyclic sulfamidates. e, Synthesis of 1,2-diamine derivatives with two similar vicinal stereocentres via ring-opening of 4,5-disubstituted cyclic sulfamidates. f, Ring-opening of fused bicyclic sulfamidates. Isolated yields; e.e. determined by chiral HPLC; d.e. determined by 1H NMR analysis of crude reaction mixture; enantiospecificity (e.s.) calculated as ((% e.e. of product)/(% e.e. of starting material)) × 100; diastereospecificity (d.s.) calculated as ((% d.e. of product)/(% d.e. of starting material)) × 100. aAbsolute configuration determined by X-ray crystallography. be.e. determined by 1H NMR analysis of the samples with chiral shift reagent. TBAF, tetrabutylammonium fluoride; Boc, tert-butyloxycarbonyl; Bn, benzyl; Ts, tosyl; DMAP, 4-dimethylaminopyridine; DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene; TMS, trimethylsilyl.
We next evaluated the ring-opening reactions that occur directly at the existing chiral carbon centre next to the oxygen atom by using 4,5-disubstituted cyclic sulfamidates such as 2t–2ad generated through the Co(II)-catalysed desymmetrization amination. It was shown that 4,5-disubstituted cyclic sulfamidate N-Boc-2v could be site-selectively ring-opened at the C5-stereogenic centre with sodium acetate as a nucleophile, generating the syn-vicinal amino alcohol derivative (−)-3r in good yield with full retention of the original enantiopurity (Fig. 3d). It is noteworthy that the vicinal stereocentres in the highly enantioenriched (−)-3r contain terminal vinyl and allyl functionalities, respectively, a stereochemical pattern that is attractive for synthesis yet difficult to achieve. The olefin moieties in (−)-3r may be selectively oxidized towards the synthesis of polyols and can be converted into threo-β-hydroxy-d-glutamic acid49. As another synthetic application of 4,5-disubstituted cyclic sulfamidates, both C≡C triple bonds in 4-alkynyl-5-propargyl cyclic sulfamidate (+)-2w were fully reduced to form 4,5-dialkyl cyclic sulfamidate 8H-2w, followed by Boc protection to give N-Boc-8H-2w, and then underwent site-selective ring-opening by sodium azide, leading to stereospecific formation of the synthetically valuable β-azido α-chiral amine (−)-3s in good yield with preservation of the original diastereopurity and enantiopurity (Fig. 3e). The structural motif in (−)-3s is notable as the two vicinal stereocentres bear similar linear alkyl chains that differ only by one methylene unit. This distinctive pattern of structures would be highly challenging to construct through either aminohydroxylation50 or diamination51 of the corresponding linear alkenes. Considering the synthetic utilities of 1,2-disubstituted carbocycles bearing two contiguous stereogenic centres, ring-opening of cis-fused bicyclic sulfamidate N-Boc-2z was explored with representative nucleophiles, such as sodium azide, sodium phenoxide and benzenethiol, in the presence of Cs2CO3. Trans-β-substituted-α-chiral cyclohexylamine derivatives (−)-3t, (+)-3u and (+)-3v were produced as single diastereomers in high yields with complete retention of the original optical activity (Fig. 3f). Among these 1,2-disubstituted carbocycles, β-azido-cyclohexylamine (−)-3t, which was previously prepared in nine steps, served as a key intermediate for the synthesis of unique peptidomimetic foldamers52. The absolute configurations of β-phenylthiocyclohexylamine (+)-3v were established as (1S,2S) by X-ray crystallography, indicating inversion of the original stereochemistry at the reacting carbon centre in (+)-2z.
In addition to the nucleophilic ring-opening reactions, these cyclic sulfamidates can also undergo site-selective elimination20 to generate the valuable chiral allyl amines as demonstrated for the high-yielding formation of the optically active 2-cyclohexenylamine (−)-3w from the cyclohexane-fused sulfamidate N-Boc-2z (Fig. 4). To further demonstrate synthetic utilities, cyclohexane-fused sulfamidate N-Boc-2z was shown to serve as a useful intermediate for the stereoselective construction of 1,2,3-trisubstituted carbocycles bearing three contiguous stereogenic centres. After exchanging the protective group from N-Boc to N-Bn through benzylation of (−)-3w and Boc deprotection of bis-protected intermediate 4w, the resulting secondary amine 5w was further transformed to the sulfamoyl azide 6w in high yield. Under the catalysis of achiral metalloradical [Co(P1)], 6w underwent intramolecular aziridination through diastereoselective radical bicyclization53, affording the sulfamide-fused tricyclic heterocycle 7w in 77% yield. In view of the high strain in the tricyclic structure, the aziridine unit in 7w could be selectively opened at the exo-position by various nucleophiles as demonstrated with the regioselective ring-opening reactions with benzenethiol in the presence of NaH, benzylamine and TMSN3 in the presence of TBAF, producing 1,2,3-trifuncionalized cyclohexanes (+)-8w, (+)-9w and (+)-10w, respectively, in good to excellent yields with full retention of the original optical purity. The sulfamide-fused cyclohexanes such as (+)-8w, (+)-9w and (+)-10w which bear three contiguous stereogenic centres may serve as valuable intermediates for target synthesis relevant to pharmaceutical research and development (Supplementary Fig. 1e)54.
Fig. 4 |. Synthesis of 1,2,3-trifunctionalized cyclohexane derivatives via ring-opening of cyclohexane-fused sulfamidates.
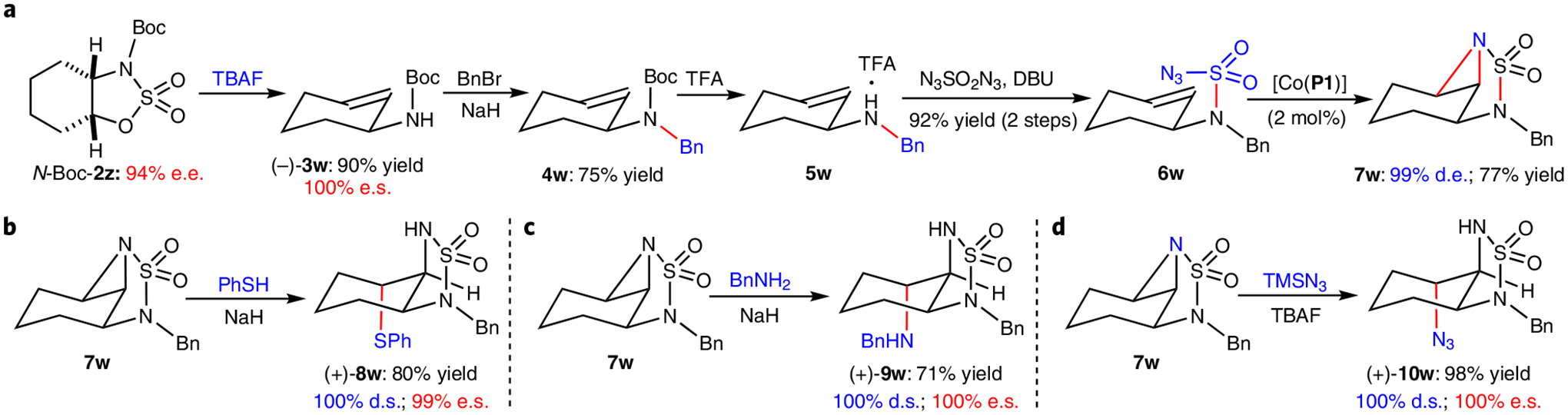
a, Synthesis of sulfamide-fused tricyclic heterocycle 7w through site-selective elimination of 2z and intramolecular aziridination of 6w. b, Regioselective ring-opening of 7w by benzenethiol. c, Regioselective ring-opening of 7w by benzylamine. d, Regioselective ring-opening of 7w by TMSN3. Isolated yields; e.e. determined by chiral HPLC; d.e. determined by 1H NMR analysis of crude reaction mixture. TFA, trifluoroacetic acid.
Conclusions
We have demonstrated a general approach for stereoselective synthesis of chiral amines bearing various β-functionalities from readily available alcohols through the union of complementary radical and ionic chemistry. This streamlined synthesis of chiral amines, which is realized through a formal 1,2-difuntionalization of alcohols, consists of enantioselective 1,5-C–H radical amination of alkoxysulfonyl azides and stereospecific ring-opening of the resulting five-membered cyclic sulfamidates by nucleophiles. In addition to demonstrating the site-selectivity, stereospecificity and nucleophile diversity of the ionic ring-opening process, we have developed an efficient catalytic radical process for enantioselective 1,5-C(sp3)–H amination of alkoxysulfonyl azides via Co(II)-based MRC. The key to the successful development of the Co(II)-based C–H amination system is the unveiling of the remarkable ligand effect that leads to the identification of 2,6-DiMeO-Hu(C6)Phyrin as the optimal supporting ligand. The optimized catalytic system enables the effective activation of various alkoxysulfonyl azides for 1,5-amination of different types of C(sp3)–H bonds at room temperature, affording five-membered chiral sulfamidates in high yields with excellent diastereoselectivities and enantioselectivities. We have presented mechanistic studies that shed light on the understanding of the underlying stepwise radical mechanism and asymmetric induction of the Co(II)-based metalloradical system. In addition to the efficient preparation of highly enantioenriched chiral amines bearing diverse β-functionalities, this combined radical and ionic approach allows for stereoselective construction of both linear and cyclic molecular structures bearing multiple contiguous stereocentres that would be difficult or impossible to construct through existing methods. Considering the ubiquity of chiral amines as structural motifs in biologically important molecules, this streamlined synthetic technology will find wide applications.
Methods
Diazotransfer reagents reported in the literature.
Trifluoromethanesulfonyl azide (TfN3)55 and fluorosulfuryl azide (FSO2N3)56 have previously been developed as diazotransfer reagents. In comparison with TfN3, FSO2N3 is safer to use and easier to handle. If the situation permits, FSO2N3 is recommended as a preferred reagent for diazotransfer reactions.
Notes on safety.
Some azides can be explosive and should be handled carefully. When common sense is employed, the azides can be prepared, stored and used without risk in the standard organic chemistry laboratory56. Although no issues were encountered for alkoxysulfonyl azides throughout our research for this project, face shields, leather gloves and protective leather clothing are highly recommended. The concentration of solvents via rotary evaporation during the synthesis of alkoxysulfonyl azides was done at 0 °C behind a blast shield.
General procedure for synthesis of alkoxysulfonyl azides.
At 0 °C, formic acid (2.0 equiv.) was added dropwise to a solution of chlorosulfonyl isocyanate (2.0 equiv.) in dichloromethane (DCM) (2.5 M). The reaction mixture was then slowly warmed up to room temperature and stirred overnight. To this freshly prepared sulfamoyl chloride, a solution of starting alcohol (1.0 equiv.) and triethylamine (2.2 equiv.) in DCM (2.5 M) was slowly added at 0 °C. The reaction was stirred at room temperature for another hour. Upon complete consumption of the starting alcohol as monitored by thin-layer chromatography, the reaction was quenched with H2O. The aqueous layer was then extracted with DCM (20 ml × 3) and the organic layers were combined, dried and concentrated. The residue was purified by flash chromatography on silica gel (eluent, DCM:EtOAc = 15:1) to give the desired sulfamate ester, which was used directly for the next step.
Diazotransfer reaction using method A.
To a round-bottom flask was added the above-synthesized sulfamate ester (1.0 equiv.) in DCM solution (0.25 M) and DBU (1.5 equiv.). The reaction mixture was cooled to −78 °C. A solution of freshly made TfN3 in hexanes (0.5 M; 1.5 equiv.) was then added slowly and the reaction was monitored by thin-layer chromatography to completion (typically in 5 min). The reaction mixture was then directly loaded into the silica gel column and purified by flash column chromatography. The fractions containing product were collected and concentrated at 0 °C to afford the desired alkoxysulfonyl azides, which were stored immediately in a −20 °C freezer. See Supplementary Section 2.0 on how to handle excess TfN3 waste.
Diazotransfer reaction using method B.
To a round-bottom flask was added sequentially the above-synthesized sulfamate ester (1.0 equiv.) and FSO2N3 in methyl tert-butyl ether (0.2 M; 1.0 equiv.), diluted with an equal volume of DMF and then followed by the addition of aqueous KHCO3 solution (3.0 M; 4.0 equiv.). The reaction mixture was stirred for 1 h at room temperature. After completion, EtOAc (40 ml) was added and the mixture was washed sequentially with brine (60 ml × 6), water (60 ml × 2) and brine (60 ml). After drying over Na2SO4, the organic layer was concentrated by rotary evaporation. The residue was then loaded into the silica gel column and purified by flash column chromatography. The fractions containing product were collected and concentrated at 0 °C to afford the desired alkoxysulfonyl azides, which were stored immediately in a −20 °C freezer.
General procedure for intramolecular C–H amination.
An oven-dried Schlenk tube that was precharged with Co(II)-metalloradical catalyst (0.002 mmol) and 4 Å molecular sieves (20 mg) was evacuated and backfilled with nitrogen gas. After replacing the Teflon screw cap of the Schlenk tube with a rubber septum, 0.5 ml of anhydrous benzene was added before the addition of alkoxysulfonyl azide (0.1 mmol), followed by adding the remaining solvent (a total of 1.0 ml). The Schlenk tube was then purged with nitrogen for 2 min before the Teflon screw cap was placed back to replace the rubber septum. The reaction mixture was then stirred at the desired temperature for the required time. Following completion of the reaction, the reaction mixture was purified via flash chromatography on silica gel. The fractions containing product were collected and concentrated by rotary evaporation to afford the purified compound.
Supplementary Material
Acknowledgements
We are grateful for financial support by the NIH (R01-GM132471) (X.P.Z.) and in part by the NSF (CHE-1900375) (X.P.Z.).
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s44160-022-00107-3.
Data availability
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2097080 (2l), 2097081 (2t), 2097082 (2ae), 2097083 (2e), 2097084 (2a), 2097085 (3m), 2097086 (3v), 2097087 (3i), 2097088 (N-Bn-2ad), 2097089 (2u), 2097090 (2r) and 2097091 (2c). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data that support the findings of this study, which include experimental procedures and compound characterization, are available within the paper and its Supplementary Information.
References
- 1.Renaud P & Sibi MP Radicals in Organic Synthesis (Wiley-VCH, 2001). [Google Scholar]
- 2.Zard SZ Radical Reactions in Organic Synthesis (Oxford Univ. Press, 2003). [Google Scholar]
- 3.Curran DP, Porter NA & Giese B Stereochemistry of Radical Reactions: Concepts, Guidelines, and Synthetic Applications (John Wiley & Sons, 2008). [Google Scholar]
- 4.Sibi MP, Manyem S & Zimmerman J Enantioselective radical processes. Chem. Rev 103, 3263–3296 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Kern N, Plesniak MP, McDouall JJW & Procter DJ Enantioselective cyclizations and cyclization cascades of samarium ketyl radicals. Nat. Chem 9, 1198–1204 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Li J et al. Site-specific allylic C–H bond functionalization with a copper-bound N-centred radical. Nature 574, 516–521 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Wang F, Chen P & Liu G Copper-catalyzed radical relay for asymmetric radical transformations. Acc. Chem. Res 51, 2036–2046 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Dong X-Y et al. A general asymmetric copper-catalysed Sonogashira C(sp3)–C(sp) coupling. Nat. Chem 11, 1158–1166 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Gu Q-S, Li Z-L & Liu X-Y Copper(I)-catalyzed asymmetric reactions involving radicals. Acc. Chem. Res 53, 170–181 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Demarteau J, Debuigne A & Detrembleur C Organocobalt complexes as sources of carbon-centered radicals for organic and polymer chemistries. Chem. Rev 119, 6906–6955 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Nugent WA & RajanBabu TV Transition-metal-centered radicals in organic synthesis. Titanium(III)-induced cyclization of epoxy olefins. J. Am. Chem. Soc 110, 8561–8562 (1988). [Google Scholar]
- 12.Gansäuer A et al. Catalysis via homolytic substitutions with C−O and Ti−O bonds: oxidative additions and reductive eliminations in single electron steps. J. Am. Chem. Soc 131, 16989–16999 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Yao C, Dahmen T, Gansäuer A & Norton J Anti-Markovnikov alcohols via epoxide hydrogenation through cooperative catalysis. Science 364, 764–767 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Ye K-Y, McCallum T & Lin S Bimetallic radical redox-relay catalysis for the isomerization of epoxides to allylic alcohols. J. Am. Chem. Soc 141, 9548–9554 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Das SK, Roy S, Khatua H & Chattopadhyay B Iron-catalyzed amination of strong aliphatic C(sp3)–H bonds. J. Am. Chem. Soc 142, 16211–16217 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Goswami M et al. Characterization of porphyrin-Co(III)-’nitrene radical’ species relevant in catalytic nitrene transfer reactions. J. Am. Chem. Soc 137, 5468–5479 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang H, Lang K, Lu H, Wojtas L & Zhang XP Asymmetric radical bicyclization of allyl azidoformates via cobalt(II)-based metalloradical catalysis. J. Am. Chem. Soc 139, 9164–9167 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li CQ et al. Catalytic radical process for enantioselective amination of C(sp3)–H bonds. Angew. Chem. Int. Ed 57, 16837–16841 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bower JF, Rujirawanich J & Gallagher T N-Heterocycle construction via cyclic sulfamidates. Applications in synthesis. Org. Biomol. Chem 8, 1505–1519 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Meléndez RE & Lubell WD Synthesis and reactivity of cyclic sulfamidites and sulfamidates. Tetrahedron 59, 2581–2616 (2003). [Google Scholar]
- 21.Yin Q, Shi Y, Wang J & Zhang X Direct catalytic asymmetric synthesis of α-chiral primary amines. Chem. Soc. Rev 49, 6141–6153 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Roughley SD & Jordan AM The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem 54, 3451–3479 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Blakemore DC et al. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem 10, 383–394 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Park Y, Kim Y & Chang S Transition metal-catalyzed C–H amination: scope, mechanism, and applications. Chem. Rev 117, 9247–9301 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Milczek E, Boudet N & Blakey S Enantioselective C–H amination using cationic ruthenium(II)–pybox catalysts. Angew. Chem. Int. Ed 47, 6825–6828 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Zalatan DN & Du Bois J A chiral rhodium carboxamidate catalyst for enantioselective C−H amination. J. Am. Chem. Soc 130, 9220–9221 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams CS, Boralsky LA, Guzei IA & Schomaker JM Modular functionalization of allenes to aminated stereotriads. J. Am. Chem. Soc 134, 10807–10810 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alderson JM, Phelps AM, Scamp RJ, Dolan NS & Schomaker JM Ligand-controlled, tunable silver-catalyzed C–H amination. J. Am. Chem. Soc 136, 16720–16723 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams CS, Grigg RD & Schomaker JM Complete stereodivergence in the synthesis of 2-amino-1,3-diols from allenes. Chem. Sci 5, 3046–3056 (2014). [Google Scholar]
- 30.Miyazawa T et al. Chiral paddle-wheel diruthenium complexes for asymmetric catalysis. Nat. Catal 3, 851–858 (2020). [Google Scholar]
- 31.Nakafuku KM et al. Enantioselective radical C–H amination for the synthesis of β-amino alcohols. Nat. Chem 12, 697–704 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roizen JL, Harvey ME & Du Bois J Metal-catalyzed nitrogen-atom transfer methods for the oxidation of aliphatic C–H bonds. Acc. Chem. Res 45, 911–922 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thornton AR, Martin VI & Blakey SB π-Nucleophile traps for metallonitrene/alkyne cascade reactions: a versatile process for the synthesis of α-aminocyclopropanes and β-aminostyrenes. J. Am. Chem. Soc 131, 2434–2435 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Burke EG & Schomaker JM Oxidative allene amination for the synthesis of azetidin-3-ones. Angew. Chem. Int. Ed 54, 12097–12101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang J-L, Yuan S-X, Huang J-S, Yu W-Y & Che C-M Highly diastereo- and enantioselective intramolecular amidation of saturated C–H bonds catalyzed by ruthenium porphyrins. Angew. Chem. Int. Ed 41, 3465–3468 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Hu Y et al. Next-generation D2-symmetric chiral porphyrins for cobalt(II)-based metalloradical catalysis: catalyst engineering by distal bridging. Angew. Chem. Int. Ed 58, 2670–2674 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruppel JV et al. A highly effective cobalt catalyst for olefin aziridination with azides: hydrogen bonding guided catalyst design. Org. Lett 10, 1995–1998 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Chen Y, Fields KB & Zhang XP Bromoporphyrins as versatile synthons for modular construction of chiral porphyrins: cobalt-catalyzed highly enantioselective and diastereoselective cyclopropanation. J. Am. Chem. Soc 126, 14718–14719 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Xu X et al. Highly asymmetric intramolecular cyclopropanation of acceptor-substituted diazoacetates by Co(II)-based metalloradical catalysis: iterative approach for development of new-generation catalysts. J. Am. Chem. Soc 133, 15292–15295 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Tian Z, Fattahi A, Lis L & Kass SR Cycloalkane and cycloalkene C–H bond dissociation energies. J. Am. Chem. Soc 128, 17087–17092 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Rousseau J-F, Chekroun I, Ferey V & Labrosse JR Concise preparation of a stable cyclic sulfamidate intermediate in the synthesis of a enantiopure chiral active diamine derivative. Org. Process Res. Dev 19, 506–513 (2015). [Google Scholar]
- 42.Diosdado S, López R & Palomo C Ureidopeptide-based Brønsted bases: design, synthesis and application to the catalytic enantioselective synthesis of β-amino nitriles from (arylsulfonyl)acetonitriles. Chem. Eur. J 20, 6526–6531 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Kaseda T, Kikuchi T & Kibayashi C Enantioselective total synthesis of (+)-(S)-dihydroperiphylline. Tetrahedron Lett 30, 4539–4542 (1989). [Google Scholar]
- 44.Wolfard J, Xu J, Zhang H & Chung CK Synthesis of chiral tryptamines via a regioselective indole alkylation. Org. Lett 20, 5431–5434 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Lucet D, Le Gall T & Mioskowski C The chemistry of vicinal diamines. Angew. Chem. Int. Ed 37, 2580–2627 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Rathi AK, Syed R, Shin H-S & Patel RV Piperazine derivatives for therapeutic use: a patent review (2010–present). Expert Opin. Ther. Pat 26, 777–797 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Tasso B et al. Synthesis, binding, and modeling studies of new cytisine derivatives, as ligands for neuronal nicotinic acetylcholine receptor subtypes. J. Med. Chem 52, 4345–4357 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Vara BA & Johnston JN Enantioselective synthesis of β-fluoro amines via β-amino α-fluoro nitroalkanes and a traceless activating group strategy. J. Am. Chem. Soc 138, 13794–13797 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishizuka T, Ishibuchi S & Kunieda T Chiral synthons for 2-amino alcohols. Facile preparation of optically active amino hydroxy acids of biological interest. Tetrahedron 49, 1841–1852 (1993). [Google Scholar]
- 50.Muñiz K Imido-osmium(VIII) compounds in organic synthesis: aminohydroxylation and diamination reactions. Chem. Soc. Rev 33, 166–174 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Cardona F & Goti A Metal-catalysed 1,2-diamination reactions. Nat. Chem 1, 269–275 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Pendem N et al. Helix-forming propensity of aliphatic urea oligomers incorporating noncanonical residue substitution patterns. J. Am. Chem. Soc 135, 4884–4892 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Jiang H, Lang K, Lu H, Wojtas L & Zhang XP Intramolecular radical aziridination of allylic sulfamoyl azides by cobalt(II)-based metalloradical catalysis: effective construction of strained heterobicyclic structures. Angew. Chem. Int. Ed 55, 11604–11608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reitz AB, Smith GR & Parker MH The role of sulfamide derivatives in medicinal chemistry: a patent review (2006–2008). Expert Opin. Ther. Pat 19, 1449–1453 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Cavender CJ & Shiner VJ Trifluoromethanesulfonyl azide. Its reaction with alkyl amines to form alkyl azides. J. Org. Chem 37, 3567–3569 (1972). [Google Scholar]
- 56.Meng G et al. Modular click chemistry libraries for functional screens using a diazotizing reagent. Nature 574, 86–89 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2097080 (2l), 2097081 (2t), 2097082 (2ae), 2097083 (2e), 2097084 (2a), 2097085 (3m), 2097086 (3v), 2097087 (3i), 2097088 (N-Bn-2ad), 2097089 (2u), 2097090 (2r) and 2097091 (2c). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data that support the findings of this study, which include experimental procedures and compound characterization, are available within the paper and its Supplementary Information.