

Abstract
Repeat expansions in the C9orf72 gene are the most common genetic cause of amyotrophic lateral sclerosis and familial frontotemporal dementia (ALS/FTD). To identify molecular defects that take place in the dorsolateral frontal cortex of patients with C9orf72 ALS/FTD, we compared healthy controls with C9orf72 ALS/FTD donor samples staged based on the levels of cortical phosphorylated TAR DNA binding protein (pTDP-43), a neuropathological hallmark of disease progression. We identified distinct molecular changes in different cell types that take place during disease progression. These alterations include downregulation of nuclear and mitochondrial ribosomal protein genes in early disease stages that become upregulated as the disease progresses. High ratios of premature oligodendrocytes expressing low levels of genes encoding major myelin protein components are characteristic of late disease stages and may represent a unique signature of C9orf72 ALS/FTD. Microglia with increased reactivity and astrocyte specific transcriptome changes in genes involved in glucose/glycogen metabolism are also associated with disease progression. Late stages of C9orf72 ALS/FTD correlate with sequential changes in the regulatory landscape of several genes in glial cells, namely MBP/MAG/MOG in oligodendrocytes, CD83/IRF8 in microglia, and GLUT1/GYS2/AGL in astrocytes. Only layer 2-3 cortical projection neurons with high expression of CUX2/LAMP5 are significantly reduced in C9orf72 ALS/FTD patients with respect to controls. Our findings reveal previously unknown progressive functional changes in cortical cells of C9orf72 ALS/FTD patients that shed light on the mechanisms underlying the pathology of this disease.
Keywords: Neurodegeneration, Frontotemporal dementia, Amyotrophic lateral sclerosis, TDP-43, snATAC-seq, snRNA-seq, Chromatin, Epigenetics, Transcription
Introduction
Amyotrophic lateral sclerosis (ALS) and Frontotemporal Dementia (FTD) are progressive neurodegenerative disorders characterized by the loss of neuronal cell populations in the central nervous system (CNS). In ALS, upper motor neurons in the primary motor cortex, and lower motor neurons in the spinal cord degenerate, leading to paralysis and respiratory failure, typically within 2-5 years of disease onset1. FTD is a heterogenous disorder characterized pathologically by the degeneration of the frontal and temporal cortex, leading to progressive cognitive impairments2. Despite being symptomatically distinct, ALS and FTD have considerable clinical, genetic, and neuropathological overlap, supporting the notion that these two disorders lie on a disease continuum3.
The coexistence of ALS and FTD in the same patient or members of the same family has long been observed and reported in several case studies4. Indeed, cross-sectional studies suggest that approximately 50% of ALS patients develop cognitive impairments and ~30% of patients diagnosed with FTD present with motor neuron symptoms5. Interestingly, a multi-center, retrospective study found that the order of symptom onset affected survival in ALS-FTD, with ALS onset resulting in shorter survival. As a major breakthrough in our understanding of ALS and FTD, a G4C2 hexanucleotide repeat expansion in the gene C9orf72 was identified as the most common cause of both ALS and FTD6. Individuals harboring the repeat expansion can present clinically with ALS, FTD, or both7. This variable clinical presentation is also associated with varying disease durations; C9orf72 patients that present with ALS or ALS-FTD have a median survival of 2.8 and 3 years, respectively, compared to 9 years in patients presenting with FTD only8.
A neuropathological hallmark of ALS and FTD is the mislocalization, phosphorylation, and aggregation of TAR DNA binding protein 43 (TDP-43)9. TDP-43 is a ubiquitously expressed, nuclear RNA/DNA-binding protein that performs important functions associated with RNA metabolism, including alternative splicing and mRNA stability. The neuropathological confirmation of FTD is referred to as Frontotemporal Lobar Degeneration (FLTD) and positive phosphorylated TDP-43 (pTDP-43) immunoreactivity distinguishes FTLD-TDP from other FTLD pathologies. Specifically, the FTLD-TDP Type B pathology, defined by cytoplasmic pTDP-43 inclusions in cortical layers II-V neurons as well as oligodendroglial inclusions in white matter9–11, is most often observed in C9orf72 cases that develop clinical features of FTD and ALS. Present in approximately 95% of ALS cases and ~50% of FTD cases, pTDP-43 burden has been shown to correlate with degeneration of affected cell populations in both ALS and FTD 12–14. In C9orf72 carriers specifically, semi-quantitative analyses suggest that the extent of TDP-43 pathology in an affected CNS region correlates with clinical phenotypes 15. However, the molecular changes associated with quantitative measurements of relative pTDP-43 abundance in a disease-relevant brain region for all frontal cortical cell types have not previously been explored. Given the variability in symptom onset (ALS vs FTD) and discordant timing of clinical progression between ALS and FTD despite a shared genetic etiology, postmortem samples from C9orf72 donors with quantitative measurements of pTDP-43 abundance provide a unique opportunity to identify molecular cascades that promote and/or result from TDP-43 dysfunction.
Here we present a multiomic single-nucleus analysis of postmortem human brain cortex tissue from 14 C9orf72 ALS/FTD patients and cognitively healthy age-matched controls to provide a more complete picture of the cellular and molecular events altered in different cell types and stages of disease progression based on pTDP-43 abundance. We find that the expression of a large number of ribosomal protein genes is downregulated in multiple cell types in patients with low levels of pTDP-43, whereas these genes become upregulated in cases with increased pTDP-43 accumulation. Donors with high levels of pTDP-43 exhibit several distinct features in non-neuronal cells compared to donors in early stages. The frontotemporal cortex of patients with high TDP-43 burden include a specific cluster of premature oligodendrocytes with downregulated genes encoding for myelin components, increased microglia reactivity characterized by upregulation of IRF8, and astrocyte specific changes in transcription of genes involved in glucose/glycogen metabolism. Collectively, we propose a sequential mechanism of alterations in the regulatory landscape of C9orf72 ALS/FTD, highlighting the contribution of pTDP-43 accumulation during the progression of neurodegeneration in cell types affected in FTD.
Results
Single nucleus multiome analysis of the human dorsolateral prefrontal cortex from C9orf72 ALS/FTD donors
To address cell type and pTDP-43 based stage specific dysregulation in C9orf72 ALS/FTD, we utilized a multiome approach to simultaneously analyze the transcriptome and epigenome of the dorsolateral prefrontal cortex of postmortem human brain tissue (Brodmann area 9) from C9orf72 ALS/FTD donors (n=9) and age-matched, healthy controls with normal cognitive and motor function (n=5) (Fig. 1A and Table S1). The average ages are 69 and 68 for control and C9orf72 ALS/FTD donors, respectively. Repeat primed PCR was performed to confirm the presence of expanded repeats in the C9orf72 locus. Immunohistochemistry and quantitative immunoassay measurements for dipeptide repeat proteins were also performed on all cases (Table S1) as an alternative method to confirm the C9orf72 repeat expansion. C9orf72 ALS/FTD donors were grouped into terciles based on quantitative measurements of phosphorylated TDP-43 (pTDP-43) abundance in lysates from the same cortical tissue used for multiome analysis (TDPneg, TDPmed, TDPhigh) (Fig. 1B; fig. S1A and table S1). The presence of pTDP-43 cytoplasmic aggregates in dorsolateral prefrontal cortex is the defining neuropathological hallmark of FTD and it has been reported to associate with more rapid cognitive decline and often found in patients with dementia but not in patients with mild cognitive impairment16. All C9orf72 ALS/FTD donors have a clinical diagnosis of ALS and/or FTD, with neuromuscular abnormalities and different degrees of cognitive impairment. However, donors in the TDPmed and TDPhigh donor group have late Braak stages suggestive of a more extensive burden of neurofibrillary tangles (Table S1).
Fig. 1. Multiomic single-nucleus analyses identify diverse cortical cell types in the dorsolateral prefrontal cortex of controls and C9orf72 ALS/FTD patients with different levels of pTDP-43.
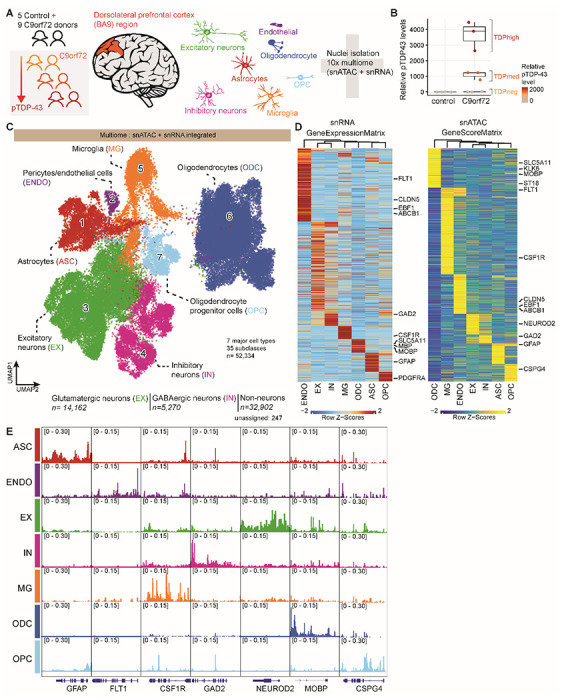
(A) Schematic representation of single-nucleus multiome profiling (snATAC-seq and snRNA-seq in the same nuclei) of dorsolateral prefrontal cortex samples from 5 control and 9 c9orf72 ALS/FTD donors analyzed in this study. (B) pTDP-43 levels in control and C9orf72 ALS/FTD patient cortical tissues. (C) snATAC-seq and snRNA-seq integrated UMAP visualization of major cortical cell types where each dot corresponds to each of the 52,334 nuclei profiled simultaneously for transcriptome and chromatin accessibility using the 10x multiome platform. (D) Row-normalized single-nucleus gene expression (left) or gene score (right) heatmaps of cell-type marker genes. (E) Pseudo-bulk chromatin accessibility profiles for each cell type at cell-type marker genes.
We obtained a total of 52,334 single-nucleus multiomes from the 14 frontal cortex samples (snRNA-seq and snATAC-seq) after quality control filtration using the ArchR multiome pipeline17 (see Materials and Methods; Fig. 1C; fig. S1 and S2A-C; table S2). Dimensionality reduction was performed for each snRNA-seq and snATAC-seq dataset using the ArchR optimized iterative LSI method17 and batch effect correction was performed using Harmony 18. Uniform manifold approximation and projection (UMAP) and unsupervised clustering with Seurat19 were applied to the combined snATAC-seq and snRNA-seq, resulting in a total of 35 distinct cell clusters (Fig. 1C; fig. S1B-C). One possible pitfall of droplet based single cell RNA sequencing techniques is the potential inclusion of cell-free RNA, which is commonly referred to as ambient RNA contamination. To ensure that the results of downstream analyses are not due to ambient RNA contamination, we used SoupX 20 and obtained the same results with and without this correction (see, for example, Fig. S3; table S5). Gene activity scores derived from snATAC-seq chromatin accessibility at proximal promoter regions were used to identify marker genes in each cell cluster (Fig. 1C–E; Fig. S1A; table S3). A total of seven major cortical cell types were identified, including excitatory neurons (EX; 14,162 nuclei; 9 clusters), inhibitory neurons (IN; 5,270 nuclei; 7 clusters), astrocytes (ASC; 4,437 nuclei; 5 clusters), microglia (MG; 5,019 nuclei; 4 clusters), oligodendrocytes (ODC; 20,520 nuclei; 6 clusters), oligodendrocyte progenitor cells (OPC; 2,394 nuclei; 3 clusters), and endothelial cells (ENDO; 532 nuclei; 1 cluster) (Fig. 1C). The cell type identification was verified by module score composed of known cell-type specific marker genes (Fig. S2D).
Pan-cortical dysregulation of ribosomal protein gene expression in C9orf72 ALS/FTD
To address whether the transcriptome and chromatin accessibility are altered progressively and correlate with pTDP-43 levels in each cell type of C9orf72 ALS/FTD cortex tissue, we performed a large-scale pair-wise comparison between controls and different groups of C9orf72 ALS/FTD donors in each of the 35 cell clusters separately, for gene expression and chromatin accessibility (Fig. 2; table S4). Differentially expressed genes are found in all C9orf72 ALS/FTD donor groups and all cortical cell types, whereas the largest number of differentially expressed genes are found in excitatory neurons independent of the levels of pTDP-43 (Fig. 2D, table S4). Interestingly, many differentially expressed genes are altered in both neurons and non-neuronal cells, suggesting a possible common dysregulated network that may be universally disrupted in C9orf72 ALS/FTD (Fig. 2F). Genes encoding for structural constituents of ribosomes were among the most significantly dysregulated genes in both neurons and non-neurons and in all C9orf72 ALS/FTD donors (Fig. 2E). These genes are highly expressed in all cells, and the levels of ribosomal proteins are carefully controlled through a strict feedback mechanism, both at the transcriptional and translational level21. We found distinct patterns of expression of ribosomal protein genes in different cell types of C9orf72 ALS/FTD donors depending on the levels of pTDP-43. Ribosomal protein genes are down-regulated in all cell types of TDPneg donors (Fig. 2G, left), whereas TDPmed donors show down-regulation of these genes in most non-neuronal cells and up-regulation in neurons (Fig. 2G, middle). Furthermore, a subset of ribosomal protein genes is upregulated in the TDPhigh donor group. For example, MRPS6, is upregulated in most cell types of TDPhigh donors (Fig. 2G, right). Interestingly, cells of the EX-C20 neuronal cluster, with gene features of deep cortical layers 4-6, display the most distinct changes when comparing C9orf72 ALS/FTD donors with different pTDP-43 levels (Fig. 2 G, H). Indeed, pTDP-43 neuronal cytoplasmic inclusions in deep cortical layers distinguishes TDP-43 Type B pathology commonly associated with C9orf72 mutations from Type A pathology associated with GRN mutations, which is predominately observed in only layer 211. Thus, these progressive gene expression changes observed in deep cortical layer neurons based on distinct pTDP-43 abundance are consistent with the observed pTDP-43 pathology in C9orf72. In TDPhigh donors, ribosomal protein genes are upregulated in the EX-C20 cluster, while these genes are downregulated in the same EX-C20 cluster in TDPmed and TDPneg donors (Fig. 2G). These results suggest a correlation between ribosomal protein gene dysregulation and disease progression based on pTDP-43 levels. In donors lacking pTDP-43, changes in gene expression might be a direct result of the presence of repeat expansions in C9orf72, either because of effects of DPRs encoded by the repeats, or effects of the repeats on C9orf72 protein levels. Interestingly, the gene encoding fibroblast growth factor 1 (FGF1) is down-regulated in expression and chromatin accessibility in the EX-C20 cluster of TDPneg donors when compared to control (Fig. 2C; table S4). Using published FGFR1 ChIP-seq data in MCF-7 cells22, we found that FGFR1 binds to the promoters of TDPneg down-regulated nuclear (48/74, 65%) and mitochondrial (14/33, 42%) ribosomal protein genes. This provides a plausible mechanism to explain the extensive downregulation of ribosomal protein genes at early disease stages. Mislocalization of pTDP-43 in the cytoplasm of cells from TDPhigh donors, and to a lesser extend TDPmed donors, may explain upregulation of ribosomal protein genes at late disease stages. TDP-43 is a nuclear RNA-binding protein that has been reported to shuttle between the nucleus and the cytoplasm and transport mRNAs encoding ribosomal proteins23. Therefore, it is possible that the observed changes in nuclear ribosomal protein RNAs in TDPmed and TDPhigh donors might be due to a defect in TDP-43 shuttling leading to their accumulation in the nucleus. This pan-cortical neuronal and non-neuronal dysregulation in the frontal cortex of C9orf72 ALS/FTD patients accompanied by pTDP-43 level specific changes has not been reported in single cell or bulk studies of ALS/FTD.
Fig. 2. Cell-type specific dysregulation of gene expression and chromatin accessibility in C9orf72 ALS/FTD donors.
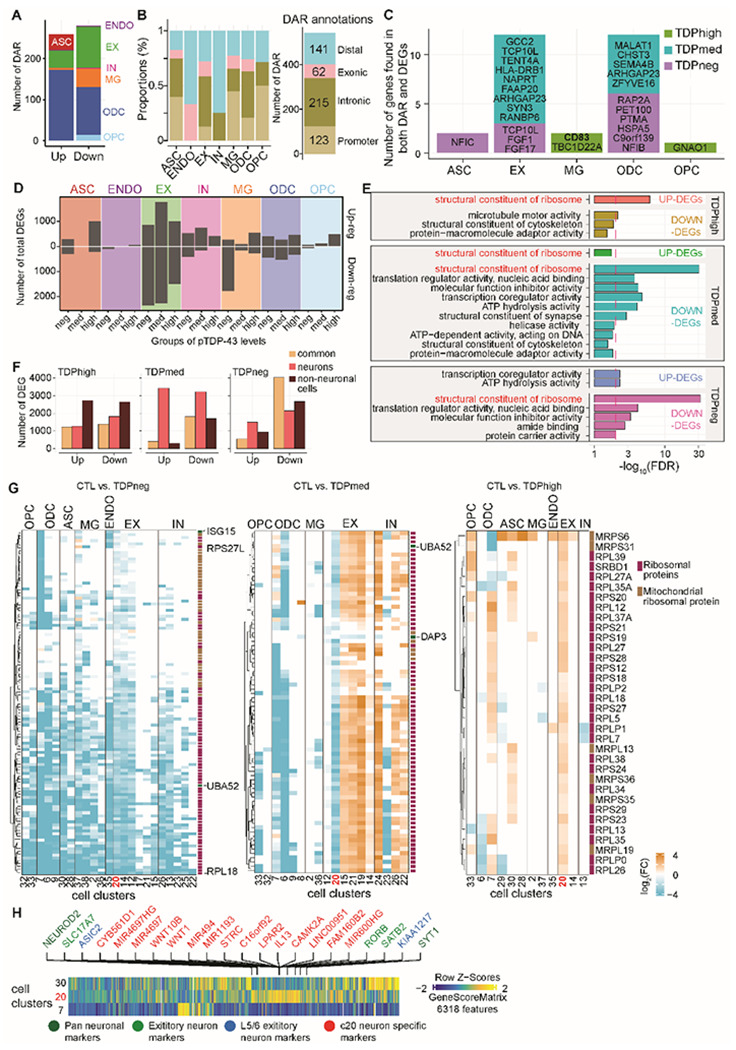
(A) Total number of differential chromatin accessible regions (DARs) for each cell type (left) and for each pTDP-43 donor group (right). (B) Distribution of DARs related to functional annotations in each cell type (left); summary of counts for each annotation group (right). (C) DEGs located closest to DARs in each major cell type and pTDP-43 group. (D) Differential gene analysis comparing C9orf72 ALS/FTD with control donors. Number of total differentially expressed genes (DEGs) grouped by each major cell type (two-sided Wilcoxon rank-sum test, FDR <10−4, Log2(C9orf72 donor / control) > 1). (E) Number of differentially expressed genes for each pTDP-43 donor group that are common among neuronal and non-neuronal cells. (F) Gene Ontology enrichment analysis of differentially expressed genes common to neuronal and non-neuronal cells (FDR <0.05; Fisher’s exact test and adjusted by Benjamini–Hochberg correction). (G) Differentially expressed genes encoding for structural constituents of the ribosome for each cluster. Left: fold change in TDPhigh donors vs control; Middle: fold change in the TPDmed donor group vs control; Right: fold change in TPDneg donors vs control. (H) Marker genes for EX-C20 are shown and labeled in green, red or blue. Red indicates genes specific to the EX-C20 cluster, green indicates SATB2 neuron clusters, and blue marks L5/6 neurons.
C9orf72 ALS/FTD is associated with impaired oligodendrocyte maturation in late disease stages
Nine distinct cell populations were identified within the oligodendrocyte lineage (n=22,914 nuclei), encompassing the largest cell population in the multiome dataset (Fig. 3A; fig. S2D), including oligodendrocyte precursor cells (OPCs) and differentiated oligodendrocytes (ODCs). Oligodendrocytes function in the central nervous system by establishing the myelin layer and providing metabolic support to neurons. Importantly, grey matter demyelination has been observed in the motor cortex and the spinal cord of ALS patients24. Furthermore, pTDP-43 inclusions in oligodendrocytes are a characteristic neuropathological finding in brains of C9orf72 ALS/FTD patients9,10. These observations suggest that oligodendrocyte dysfunction plays an important role in C9orf72 ALS/FTD. Clusters OPC-C32, OPC-C33, and OPC-C34 contain oligodendrocyte precursor cells (OPCs) with high expression of PDGFRA and CSPG4 (Fig. 3D–E; fig. S2D). The remaining six oligodendrocyte clusters are differentiated oligodendrocytes with higher levels of OPALIN and PLP1. We noticed that there are proportionally more cells in ODC clusters ODC-C4, ODC-C6, and ODC-C7 in TDPhigh compared to other donor groups and controls (Fig. 3B). Specifically, 40% of all oligodendrocytes in TDPhigh donors are present in the ODC-C6 cluster (Fig. 3C), while only 2% of oligodendrocytes are present in this cluster in TDPmed and TDPneg donors, suggesting that ODC-C6 is unique to the late disease stage. Based on gene score and hierarchical clustering of marker genes, ODC-C6 cells are transcriptionally distinct from other ODC clusters (Fig. 3D), with lower expression of MOG, MOBP, and MBP, which encode for the major protein components of myelin (Fig. 3E). ODC-C6 cells also have higher expression levels of TCF7L2 and ITPR2 (Fig. 3E). Expression of these two genes in ODCs is a marker of newly differentiated oligodendrocytes, suggesting that ODC-C6 cells represent newly formed promyelinating oligodendrocytes, a unique cell type found in TDPhigh patients at late disease stages. In contrast to ODC-C6, the rest of ODC clusters are composed of mature myelinating ODCs with strong expression of genes involved in myelinating processes25,26 (Fig. 3E). These normal mature myelinating ODCs are found mainly in TDPmed and TDPneg donor groups in earlier disease stages (Fig. 3B). Interestingly, compared with oligodendrocytes from control donors, oligodendrocytes from the TDPhigh donor group exhibited downregulation of myelin-associated genes (MBP, MOG, MAG, PLLP, and PMP22; Fig. 3F, left and table S4). Many of these genes encode for major protein components of myelin, including MBP, MOG, and MAG, which are down-regulated in ODC-C6 premyelinating oligodendrocytes of TDPhigh donors (Fig. 3F, right). These findings further strengthen our suggestion that a large portion of oligodendrocytes in the dorsolateral prefrontal cortex in late FTD disease stages are defective in myelination. mRNAs encoded by these genes are bound by pTDP-43 in the mouse brain27,28 and the development of oligodendrocytes has been shown to be regulated by TDP-4329. Given prior evidence of pTDP-43 inclusions in oligodendrocytes, we speculate that pTDP-43 aggregates may play a direct role in dysregulating mRNAs encoding myelin components, thus affecting the maturation of oligodendrocytes and their ability to myelinate neurons, a unique feature to C9orf72 ALS/FTD patients with significant pTDP-43 burden.
Fig. 3. Premature and premyelinating oligodendrocytes are unique to high pTDP-43 donors in late disease stages, specific to severe TDP-43 proteinopathies.
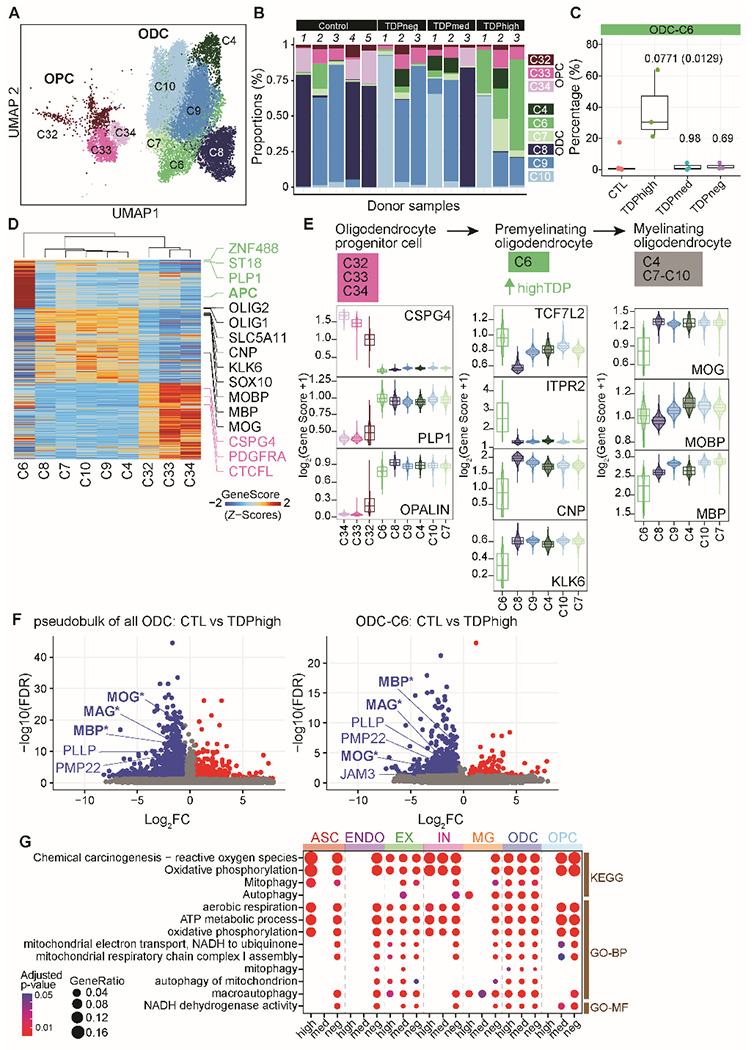
(A) UMAP plot of oligodendrocyte lineage cells. (B) Proportion of oligodendrocyte precursor cells (OPC) and oligodendrocytes (ODC) clusters in each sample, including donors with different levels of pTDP-43 and cognitively normal controls. (C) Proportion of ODC-C6 in different pTDP-43 donor groups (Kruskal–Wallis test with Benjamini–Hochberg correction (p-adj=0.0771) and without correction (p-value=0.0129). (D) Plot of snATAC-seq gene scores ordered by hierarchical clustering with marker genes distinguishing each ODC cell cluster. (E) Illustration of developmental stages of the oligodendrocyte lineage cells. Developmental stage specific genes and their gene scores are shown for each cluster (bottom), highlighting the unique characteristics of ODC-C6 with high expression of promyelinating oligodendrocyte genes. (F) Differentially expressed genes in all oligodendrocytes (left) and ODC-C6 (right) in control versus TDPhigh donors. Myelin related DEGs are labeled in blue (GO cellular component, FDR, 2.85 x 10−4). Genes whose mRNA are bound by pTDP-43 based on CLIP28 experiments are marked by an asterisk. (G) Down-regulated genes specifically enriched for mitochondria and respiration ontology terms (FDR <0.05; Fisher’s exact test and adjusted by Benjamini–Hochberg correction).
Microglia reactivity increases in C9orf72 ALS/FTD with high pTDP-43 burden
Microglia typically account for 5% of all brain cells30 and have the highest expression of C9orf72 compared to other cortical cell types31 (Fig. S2E). As the resident immune cells, microglia are thought to contribute to the increased inflammation reported in the ALS/FTD disease spectrum32,33. In our study, 4 out of a total of 35 frontal cortex cell clusters with a total of 5,019 nuclei have microglia identity with known microglia markers, including AIF1, RUNX1, PTPRC (CD45), CX3CR1, P2RY12, TMEM119, and ITGAM (CD11b)34 (Fig. 4A; fig. S2D). Recent snRNA-seq studies of human cortical tissues found that microglia form one diffuse cluster, suggesting that instead of distinct cell types, human microglia populations vary gradually in their transcriptome states34,35. Each of the four microglia clusters in our results exhibits a distinct set of expressed genes, snATAC-seq peaks, and transcription factor binding motifs (Fig. 4B and C), suggesting they correspond to distinct sets of microglia cells. It is possible that the ability to discover these four distinct microglia populations is based on using snRNA-seq and snATAC-seq from the same nuclei, instead of snRNA-seq only, thus achieving a more defined cell cluster identify. Although the four microglia clusters are observed in both control and ALS/FTD donors (Fig. 4D), we observe a large number of differentially expressed genes when comparing diseased individuals with healthy controls (Fig. 2F). To understand these unique microglia clusters, we analyzed each one for differences in the transcriptome, chromatin accessibility and the inferred TF motif enrichment. MG-C2 is the largest microglia cluster (n=2,900 nuclei) and appears to be in a combination of homeostatic and activating states based on the presence of marker genes in the multiome data. This cluster exhibits the highest expression of microglia homeostatic marker genes7, including CX3CR1, TMEM119 and CSF1R (Fig. 4A). Cells in this cluster also express genes characteristic of the activating state, including inflammatory genes involved in antigen presentation (CD86, CD80; MHC II – C1QA, C1QB, C1QC), reactive chemokines (CCL2, CCL3), and interleukin (IL-1a, IL18) (Fig. 4B). TF binding motifs for SPI1 (also known as PU.1) (Fig. 4C), a TF that is essential for microglia activation36,37, are specifically enriched in the MG-C2 cluster at chromatin accessible regions. IRF8, another critical TF that transforms microglia into a reactive phenotype36, is uniquely highly expressed in the MG-C2 cluster. Interestingly, the expression of IRF8 and IL18 proinflammatory cytokines and GPNMB that is associated with neuroinflammation, are specifically upregulated in TDPhigh patients compared to TDPmed or TDPneg donor groups and control (Fig. 4B and E, table S4). The homeostatic marker gene TMEM119 is specifically downregulated in TDPhigh patients (Table S4). These results suggest that cells in the main microglia cluster are more reactive in C9orf72 ALS/FTD patients with high pTDP-43 levels, while the majority of microglia remains in the homeostatic state in the TDPmed and TDPneg donor groups at earlier disease stages.
Fig. 4. Dysregulation of microglia function in C9orf72 ALS/FTD donors with low or high pTDP-43.
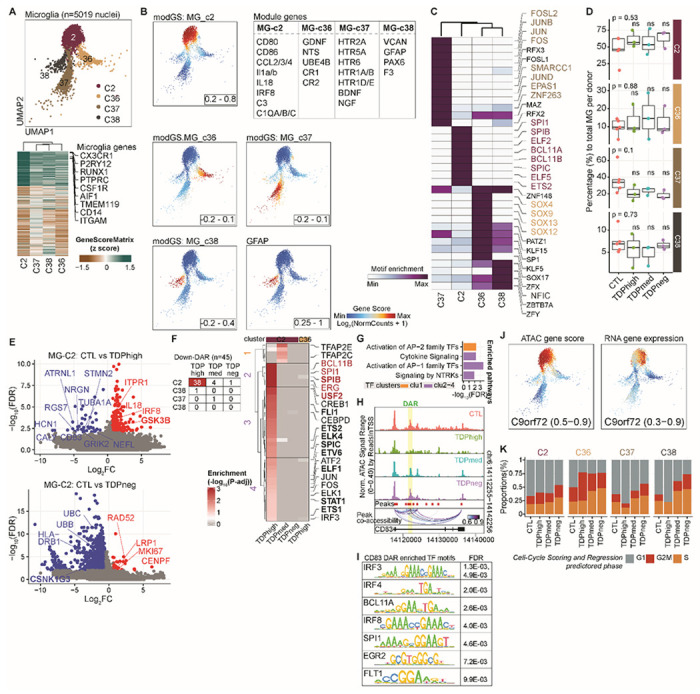
(A) UMAP plots of the four microglia clusters (top). Heatmap showing the row-normalized pseudo-bulk gene score in each snATAC-seq cluster split by nuclei from each of the four MG clusters; rows are organized based on hierarchical clustering and the key genes that define the microglia lineages are marked (bottom). (B) Same UMAP as in (A) colored by expression of cluster specific genes, rendered with imputation of ATAC-seq module gene scores and the module genes are listed in the table in the same panel. (C) Heatmap of motif enrichment within differential marker peaks of each microglia cluster. Color indicates the motif enrichment (−log10(P value)) based on the hypergeometric test. Specifically enriched TFs for each MG cluster are highlighted using the same cell cluster specific colors as in (A). (D) Fraction of each MG cluster in control and pTDP-43 donor groups (Kruskal−Wallis test with Benjamini–Hochberg correction; p>0.05, n.s.). (E) Volcano plot of genes differentially expressed between control and C9orf72 ALS/FTD donors with different levels of pTDP-43 in MG-C2 proinflammatory microglia. Selected DEGs enriched in gene ontology analysis are indicated (FC>1.5; FDR < 0.01). (F) Differential chromatin accessible regions in microglia clusters. Left: Numbers of DARs for each MG cluster and in each pTDP-43 group (FDR < 0.05; log2(fold change of TDP donors / control); Right: TF motifs enriched at DARs in MG-C2 cells comparing control with different pTDP-43 donor groups. TFs associated with immune cells based on Suo et al38 are marked in bold. The most significantly enriched TF motifs in cluster MG-C2 are highlighted in red. (G) Top GO enrichment of cluster specific TFs shown in F (FDR < 0.01). (H) Genome browser view of the CD83 gene, which is down-regulated in expression specifically in high pTDP-43 donors. Also shown is the location of an intronic enhancer with decreased chromatin accessibility in TDPhigh donors. (I) The CD83 intronic enhancer contains motifs for TFs enriched in cells of the immune system. (J) Imputed gene activity score (left) and gene expression (right), highlighting high gene activity scores and expression of the C9orf72 gene in the MG-C2 cluster. (K) Proportion of estimated cells in each cell cycle phase in each MG cluster of C9orf72 ALS/FTD donors. Prediction of cell cycle phase of all cells in each microglia cluster was estimated based on expression of G2/M and S phase markers.
Chromatin accessibility is also most dysregulated in MG-C2 when comparing other donor groups to control (Fig. 4F). Specifically, binding motifs for 45 different TFs are enriched at down-regulated genes that are differentially expressed between C9orf72 ALS/FTD patients and healthy controls. Of the 45 TFs, 10 are involved in defining immune cell fate38 (Fig. 4F, right). Based on GO analysis, these TFs are involved in different immune pathways, including cytokine signaling, activation of AP-1, and signaling by receptor tyrosine kinases (NTRKs) (Fig. 4G). One of the sequences with decreased accessibility is located in the second intron of CD83, whose expression is also down-regulated in the TDPhigh donor group (Fig. 4H). This region has been shown to be an enhancer that regulates the expression of CD83 in dendritic cells39, where it is bound by interferon regulatory factors (IRFs) and NFkB. Motif analysis at the summits of the ATAC-seq peak suggests that this intronic enhancer can be bound by IRF3,4,8 as well as BCL11A, SPI1, EGR2, and FLT1 (Fig. 4I). Additionally, CD83 has been shown to be dysregulated in monocytes of ALS patients40. As a member of the immunoglobulin (Ig)G superfamily, CD83 protein exists in two isoforms, a membrane bound and a soluble form. Although the function of CD83 is largely unknown in microglia of the CNS, reduced T cell stimulation is associated with inhibition of CD83 expression41 and the soluble form of CD83 can inhibit dendritic and T cell proliferation42. Perhaps CD83 represents another layer of immune balance specific to the TDPhigh donor group. Given that proinflammatory responsive genes are upregulated in cells of the MG-C2 cluster in TDPhigh donors, the results suggest a more reactive state when cortical microglia exhibit high levels of pTDP-43 in advanced disease stages. Additionally, MG-C2 cells distinctly exhibit high gene activity scores and expression of the C9orf72 gene (Fig. 4J; Table S 3), in agreement with previous reports indicating that C9orf72 gene expression is particularly high in dendritic immune cells and microglia43. C9orf72 mRNA expression is reduced in the frontal cortex of C9orf72 ALS/FTD subjects compared to healthy donors tissues6 and C9orf72 is required for proper microglial function in mice31. However, only TDPneg patients show significant down-regulation of C9orf72 gene expression in the MG-C2 cluster (Table S4) while other C9orf72 ALS/FTD donor groups show normal C9orf72 expression, suggesting that downregulation of C9orf72 is unique to the initial disease stages before the onset of pTDP-43 accumulation and microglia activation. Our data suggest that the more reactive MG-C2 cells found in the TDPhigh donor group may not be to the result of changes of C9orf72 gene expression and that the downregulation of C9orf72 may not lead to an increase in microglia reactivity. These results suggest that microglia may represent a dysregulated cell type that distinguishes the stage of disease progression in different ALS/FTD donors as a result of changes in transcription regulation in response to increased cytoplasmic pTDP-43 accumulation. Casein Kinases 1 and 244,45 and glycogen synthase kinase (GSK3)46 have been shown to phosphorylate TDP-43. Changes in the expression of casein kinases are not statistically significant when comparing control to C9orf72 ALS/FTD samples. However, the expression of GSK3B is specifically upregulated in the MG-C2 cluster in the TDPhigh donor group (Fig. 4E, table S4), suggesting that high levels of GSK3B might be responsible for the phosphorylation of TDP-43 in the TDPhigh donor group. In contrast, Casein Kinase 1 Gamma 3 (CSNK1G3) is specifically downregulated in the MG-C2 cluster in the TDPneg donor group (Fig. 4E, table S4). Casein Kinase 1 has been shown to predominantly phosphorylate Ser409 and Ser41044 of TDP-43 found in the cytoplasmic inclusions of ALS/FTD patient tissues47. Downregulation of CSNK1G3 may thus explain the lack of pTDP-43 aggregates in TDPneg patients. These observations could represent an alternative explanation to the increase in microglia reactivity and increase in inflammatory responses associated with high level of pTDP-43.
Distinct from MG-C2, the other three MG clusters exhibit lower expression of genes involved in microglia homeostasis (Fig. 4A), suggesting that the shift away from the homeostatic state may be due to downregulation of these genes. Clusters MG-C36 and MG-C37 exhibit moderate expression of markers for the alternative M2-like microglia state48, which has been proposed to be an anti-inflammatory state that plays a protective role in the brain, in contrast to reactive microglia. Specifically, MG-C36 and MG-C37 are defined by different sets of neurotrophic factors (MG-C36: BDNF/GDNF/NTS; MG-C37: BDNF/GDNF/NGF; Fig. 4B) to support neuron survival and modulate the formation of long-term memories49,50. In addition, MG-C36 is marked by genes involved in cell-adhesion, pro-proliferation (UBE4B), interferon type I interferon receptor binding, and the Complement receptor genes CR1 and CR2 (Fig. 4B). In contrast, MG-C37 cells are marked by genes encoding serotonin receptors and genes involved in G-protein-coupled receptor signaling. The expression of UBE4B prompted us to analyze genes involved in cell cycle regulation for each MG cluster in each C9orf72 ALS/FTD donor group and healthy controls. We found that the cell cycle of MG-C36 cells is disrupted in all C9orf72 ALS/FTD donors when compared to controls (Fig. 4K). Among the differentially expressed genes found in MG-C36 and MG-C37, BDNF and C9orf72 (log2(FC) = −2.17 and −2.36, respectively) are downregulated specifically in the TDPneg group (Table S4). Since microglia and astrocytes are the main producers of neurotrophic factors, the downregulation of these genes may contribute to the dysregulation of neurotrophic-based neuronal support, especially in earlier disease stages before pTDP-43 accumulation.
MG-C38 is a distinct cluster that expresses markers of microglia and astrocytes, such as GFAP and VCAN (Fig. 4B), suggesting this cluster might correspond to a specific subset of microglia cells that is phenotypically transitioning into astrocyte-like cells. This type of cells has been shown to be present in an inherited model of ALS51. The expression of PAX6 in MG-C38 further confirms the glial cell origin of cells in this cluster (Fig. 4B, table S3). These cells also express high levels of F3 (CD142 protein) RNA, another distinctive marker of this cluster in comparison to other microglia cell clusters. Only TDPneg donors exhibit differential gene expression in MG-C38 cells (Table S4), and the most downregulated gene is APOE (log2(FC) = −8.014), which may explain aspects of clinical pathology in TDPneg donors lacking the typical cortical pTDP-43 aggregates. Given the different combinations of changes in the transcriptome and epigenome found in microglia from all groups of C9orf72 ALS/FTD donors with different pTDP-43 levels, it is possible that microglia may be one of the initial drivers of ALS/FTD pathology due to the high C9orf72 gene expression found in microglia compared to other cell types in the frontal cortex. Only TDPneg donors exhibit downregulation of C9orf72 expression in microglia, suggesting a unique feature of initial disease stages before the onset of pTDP-43 pathology. This initial event in microglia may then trigger proinflammatory responses and dampen neurotrophic factor expression, ultimately affecting neuronal survival.
Astrocyte pseudotime trajectory recapitulates astrocyte reactiveness in C9orf72 ALS/FTD
Astrocytes (ASCs) represent another cortical cell type known to respond and become reactive to disease state, particularly via dysregulation of metabolic pathways in neurodegenerative diseases. Cell clusters ASC-C27 to ASC-C31 with a total of 4,437 nuclei can be identified as having astrocyte identity based on high expression of GFAP, AQP4 and SLC1A2 (Fig. 5A; fig. S2D). Each astrocyte subpopulation exhibits a distinct set of expressed genes (Table S3). By comparing changes in the transcriptome between control and C9orf72 ALS/FTD donor groups with different levels of pTDP-43, we found that differentially expressed genes are cluster and pTDP-43 level specific (Fig. 5B). Genes involved in glucose/glycogen metabolism are specifically enriched in astrocytes from TDPhigh donors, and many of these genes upregulated in TDPhigh donors with respect to control are found in cluster ASC-C30 (Fig. 5B–D). In comparison to other ASC clusters, ASC-C30 has lower levels of GFAP, which encodes the main astrocyte intermediate filament protein and it is a signature of reactive astrocytes52. Cells in ASC-30 also have higher levels of MT2A, which encodes a metallothionein protein associated with neuronal injury53 (Fig. 5D–E), suggesting that ASC-C30 contains astrocytes with less reactivity. TDPhigh donors exhibit significant up-regulation of the glucose transporter GLUT1 in ASC-C30, which may result in higher glucose uptake. Indeed, glucose hypermetabolism in the frontal cortex is unique to C9orf72 ALS/FTD patients54. Also, specific to TDPhigh patients, significant up-regulation of glycogen synthase (GYS2) and glycogen debranching enzyme (AGL) may alter the balance of glucose to glycogen metabolism (Fig. 5D). These data suggest that glucose uptake is increased in astrocytes with lower reactivity in later disease stages, and the additional glucose may be converted to glycogen instead of lactate, which is typically made available to neurons to sustain their energy demands. In contrast, TDPneg patients who lack pTDP-43 pathology exhibit downregulation of genes encoding for enzymes involved in fermentation of pyruvate to lactate, leading to less lactate production and affecting astrocyte-neuron lactate shuttle (Fig. 5D). This TDPneg-specific finding agrees with published data suggesting that C9orf72 ALS/FTD derived astrocytes are more vulnerable to glucose starvation induced cell stress55.
Fig. 5. pTDP-43 level specific dysregulation of astrocytes correlates with the pseudotime trajectory.
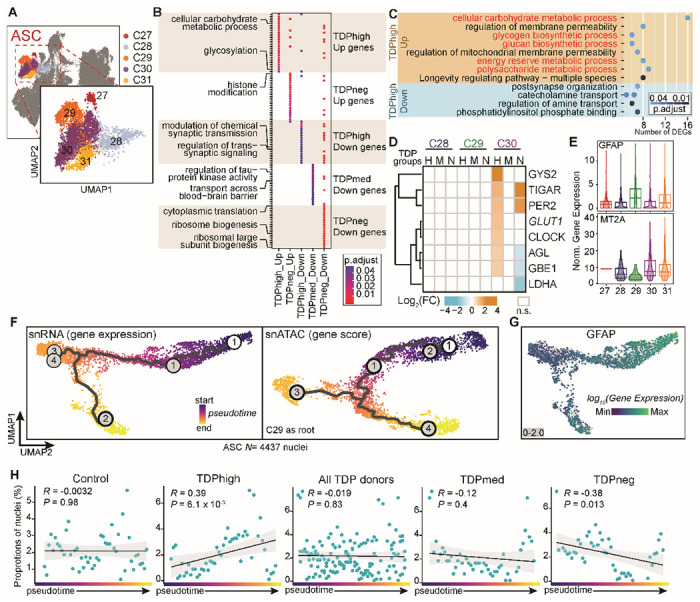
(A) UMAP plots of astrocyte (ASC) clusters. (B) DEG clusters enriched for specific pTDP-43 level groups in ASC subpopulations; distinct GO terms are marked for each DEG cluster. (C) Enriched ontology terms for DEGs in the TDPhigh donor group include glucose/glycogen metabolism and are highlighted in red. (D) Genes involved in glucose/glycogen metabolism and their fold change in different ASC subpopulations and pTDP-43 level groups. (E) Cluster ASC-C30 is distinct from other ASC subpopulations and it is defined by higher MT2A and lower GFAP. (F) Trajectory analysis: UMAP dimensionality reduction of astrocytes using snRNA-seq gene expression (left) and snATAC-seq gene score matrices (right). Each cell is colored by pseudotime trajectory assignment using ASC-C29 as the root node. (G) GFAP expression of the same trajectory UMAP as in F. (H) Scatterplot showing the proportion of ASC nuclei from C9orf72 ALS/FTD and control samples at 50 evenly sized bins across the trajectory. The black line shows a linear regression, and the gray outline represents the 95% confidence interval. Pearson correlation coefficient and P value from two-sided test are shown. Each panel represents the analysis of different C9orf72 donor groups.
To further understand the heterogeneity of astrocytes in C9orf72 ALS/FTD, we performed pseudotime trajectory analysis using Monocle 356 on the snRNA-seq and snATAC-seq data in astrocyte cell clusters (Fig. 5F). We found that snRNA-seq and snATAC-seq have the same pseudotime trajectory and the trajectory is ordered based on GFAP expression, where the lowest GFAP levels are found at the end of the trajectory (Fig. 5G). This suggests that the astrocyte pseudotime trajectory recapitulates astrocyte reactiveness that correlates with the levels of GFAP expression, where the reactivity of astrocyte decreases along the pseudotime trajectory. The proportion of nuclei from TDPhigh donors appears to increase along the trajectory (Fig. 5H; Pearson correlation R=0.39, p-value = 6.1 × 103), and the proportion of nuclei from the TDPneg donors appears to decrease along the trajectory (Pearson correlation R= −0.38, p-value = 0.013). This trend is not found in control nor TDPmed donor groups (Fig. 5H). Therefore, a larger proportion of astrocytes express higher levels of GFAP in the earlier disease stages and the levels of astrocyte GFAP decrease in more advanced disease stages, suggesting that GFAP expression inversely correlates with progression of C9orf72 ALS/FTD.
Heterogeneity of pTDP-43 accumulation in inhibitory and excitatory neurons
Neurons are the most diverse cell type in the single nucleus multiome dataset. A total of 19,432 nuclei can be annotated as excitatory or inhibitory neurons using gene activity scores for key linage genes, and each category consists of 10 and 6 clusters, respectively (Fig. 6A and B). Multiple neuronal subtypes can be annotated based on known marker genes57. Excitatory neurons can be categorized by their cortical layer position (layer 2-6) and their axonal projections (Fig. 6C); while inhibitory interneurons can be grouped by their developmental origin from the medial, lateral/caudal ganglionic eminences and classified based on their subtypes (Fig. 6D). Although cytoplasmic aggregates of pTDP-43 are observed in nearly all ALS/FTD clinical cases, it is not known which neuronal cell types are more vulnerable to pTDP-43 levels. NeuN+ cortical neurons from control and ALS/FTD neocortex have been previously fractionated based on levels of nuclear TDP-43, allowing the characterization of nuclear TDP-43 positive and negative sp6ecific transcriptomes using bulk RNA sequencing58. However, this study was not able to identify the neuronal cell types contributing to the ensemble of TDP-43 positive and negative RNA-seq profiles. To estimate the contribution of neuronal subtypes present in this TDP-43 sorted neuronal population, we employed the cell composition deconvolution algorithm CIBERSORTx 59 and compared our single nucleus datasets with the published TDP-43 sorted transcriptomes. We were thus able to quantify the contribution of each individual neuronal subtype identified in our multiome dataset in the published TDP43-negative and TDP43-positive transcriptomes. NeuN positive neurons are composed of 70% excitatory neurons and 30% inhibitory neurons57. Therefore, expected excitatory neuron clusters, specifically, EX-C14, EX-C15, EX-C17, and EX-C20, have the higher contribution to the bulk neuronal transcriptome compared to inhibitory neurons, regardless of pTDP-43 levels (Fig. 6E). Among all neuronal clusters, the cortical (EX-C15, EX-C21) and corticothalamic (EX-C11, EX-C12, EX-C14) projection neurons, appear to have significantly higher contribution to the transcriptome of nuclear TDP43-negative cells (Fig. 6E), suggesting that these neuronal subtypes may be more sensitive to nuclear TDP-43 loss. In contrast, inhibitory neuron clusters IN-C13, IN-C22, and IN-C25 appear to have a higher contribution to the TDP43-positive transcriptome (Fig. 6E). These clusters share a similar identity based on lower gene activity of PVALB (Parvalbumin), suggesting that Parvalbumin neurons are less vulnerable to nuclear TDP-43 loss. This finding is consistent with a recent study60 in the motor cortex demonstrating that TDP-43 pathology was more prevalent in excitatory neurons and absent from Parvalbumin interneurons. Interestingly, Parvalbumin overexpression in transgenic mice rescues the motor neuron loss phenotype of mutant SOD1 mice, an animal model of familial ALS61, suggesting that Parvalbumin expressing inhibitory neurons may have the same protective effect in C9orf72 ALS/FTD.
Fig. 6. Neuronal cell types in the prefrontal cortex of control and C9orf72 ALS/FTD donors.
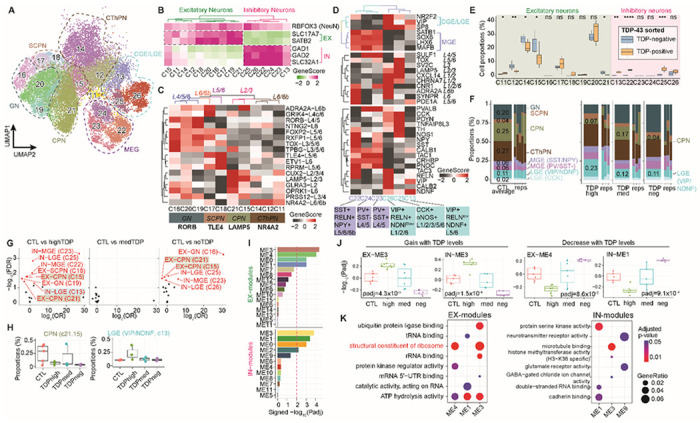
(A) UMAP plots of neuronal clusters. (B) Gene activity scores for marker genes of excitatory and inhibitory neurons. (C) Heatmap of gene activity scores of cortical layer specific marker genes for excitatory neurons and axonal projection subclassification is indicated below. CPN, cortical projection neuron; GN, granule neuron; SCPN, subcortical projection neuron; CThPN, corticothalamic projection neuron. (D) Heatmap of gene activity scores of marker genes associated with inhibitory neurons of subpallial origin (top), cortical layers (middle) and subclassification (bottom). CGE, caudal ganglionic eminence; MGE, medial caudal ganglionic eminence; LGE, lateral ganglionic eminence; SST, somatostatin; RELN, reelin; NPY, neuropeptide Y; PV, parvalbumin; VIP, vasoactive intestinal peptide; NDNF, neuron-derived neurotrophic factor; CCK, cholecystokinin; nNOS, neuronal nitric oxide synthase. (E) Cell proportion deconvolution with pTDP-43 positive and negative nuclei (n.s. not statistically significant; P≤0.05 is considered statistically significant: * P≤0.05, ** P≤0.01, *** P≤0.001, **** P≤0.0001). (F) Proportion of neuronal subtypes defined by cortical projection or developmental origins in all sample groups. (G) Volcano plot showing odd ratio (OR) and FDR computed by MASC101 for all 16 neuronal clusters. Red labeled clusters are significantly increased or depleted in association with specific C9orf72 ALS/FTD donor groups (FDR-adjusted P < 0.05; absolute OR >0). (H) Proportion of nuclei in the significantly increased or depleted clusters found in panel G. (I) Significance of WGCNA modules with different levels of pTDP-43. (J) Pearson correlation of WGCNA module eigengenes and pTDP-43 levels that correspond to disease progression (dashed line, p-adj ≤0.01, statistically significant). Modules positively correlated (left) and negatively correlated (right) with disease progression. (K) Gene ontology analysis of the hub genes in each disease progression correlated module (same modules as shown in panels I and J).
Cortical projection excitatory neurons are significantly reduced in the frontal cortex of C9orf72 ALS/FTD donors
ALS/FTD with pTDP-43 inclusions is typically accompanied by frontal cortex atrophy and neuronal loss9. We therefore grouped neurons based on their excitatory projection classification and developmental origin for interneurons to avoid cell clusters with few nuclei. We found that cortical projection neurons (EX-C21 and EX-C15) are more than threefold lower in proportion in TDPhigh and TDPneg patient groups compared to control (Fig. 6F), suggesting that these neurons are especially susceptible to C9orf72 ALS/FTD frontal cortex degeneration. We systematically assessed the differential abundance between C9orf72 ALS/FTD donor and control groups for all neuronal clusters. Among the 16 neuronal clusters, a total of 8 and 6 clusters showed significant proportional changes in TDPhigh and TDPneg donor groups, respectively (FDR-adjusted P < 0.05, absolute log2(odds ratio (OR)) > 0; Methods; Fig. 6G). One proportionally decreased population in both TDPhigh and TDPneg compared to control was the layer 2-3 cortical projection neurons with high expression of CUX2/LAMP5 (EX-C21 and EX-C15; Fig. 6C and 6G-H), which further confirms our previous conclusion on the vulnerability of cortical projection neurons in C9orf72 ASL/FTD degeneration, despite the differences in pTDP-43 accumulation. In addition to cortical projection neurons, IN-C13 VIP+/NDNF+ inhibitory neurons originating from the lateral ganglionic eminence are significantly increased in proportion in the TDPhigh donor group compared to control (Fig. 6H). Based on our cell composition deconvolution analysis against TDP-43 positive and negative specific transcriptomes, IN-C13 cells have a higher contribution to the TDP43-positive compared to the TDP43-negative transcriptome (Fig. 6E). Our data suggest that these IN-C13 VIP+/NDNF+ neurons might be resistant to neurodegeneration, possibly because they are less vulnerable to nuclear TDP-43 loss. Interestingly, the TDPmed donor group does not have significant changes in the relative proportion of neural clusters, perhaps because this group represents a transitional state in disease progression that reflects the mixture of cortical neuronal populations.
Since neurons exhibit a large number of differentially expressed genes in all C9orf72 ALS/FTD donor groups (Fig. 2A), we set out to identify putative gene regulatory networks that correlate with pTDP-43 accumulation. We used the weighted gene co-expression network analysis (WGCNA) to cluster co-expressed and differentially expressed genes into modules and to identify highly correlated genes using unsupervised clustering. Using pseudobulk snRNA-seq data for each neuronal cluster, we found 16 modules for excitatory and 12 modules for inhibitory neurons (Fig. 6I; table S7). Among these, 4 excitatory neuron modules and 4 inhibitory neuron modules significantly correlate with pTDP-43 levels. EX-ME3 and IN-ME3 positively correlate while EX-ME4 and IN-ME1 negatively correlate with the amount of pTDP-43 (Fig. 6J). Using the hub genes in these pTDP-43 level correlated modules, we found that the correlated excitatory neuron modules are enriched for gene ontology terms related to structural constituent of ribosomes and ATP hydrolysis activity, while the hub genes of the inhibitory neuron modules do not share common enriched terms (Fig. 6K; table S6). The WGCNA analysis further supports our earlier conclusions that dysregulation of ribosomal protein genes may play a significant role in C9orf72 ALS/FTD and track pTDP-43 and disease progression.
Mitochondria may play a central role in C9orf72 ALS/FTD
Oligodendrocytes are essential not only for action potential propagation in neurons through myelin, but also to provide metabolic support to neurons62. Given the oligodendrocyte maturation defects found in late stage TDPhigh donors, the crucial role of these cells in neuron metabolism, and the glucose metabolic dysregulation found in astrocytes, we propose that the metabolic dysfunction rooted in glial cells may contribute to the defective neuronal phenotypes. Indeed, when we examined down-regulated genes, mitochondria and respiration gene ontology terms were enriched in oligodendrocytes and excitatory neurons independent of the levels of pTDP-43 (Fig. 3G). Specifically, 44 genes encoding mitochondrial respiratory chain complex I assembly are downregulated in all C9orf72 ALS/FTD patients in excitatory neurons and oligodendrocytes (Fig. 3G; table S6). Mutations in one particular downregulated gene, CHCHD10, which encodes a mitochondria protein located in the intermembrane space, have been identified to be associated with the disease in multiple ALS cohorts63,64. The shared dysregulation of genes encoding mitochondrial components in oligodendrocytes and excitatory neurons may not be a coincidence, since mitochondria are located in the cytoplasmic compartment of the myelin sheath65. A recent study has provided evidence that the C9orf72 protein is localized to the inner mitochondrial membrane and is essential for mitochondria complex I assembly, and thus crucial for OXPHOS function and energy metabolism66. This suggests that C9orf72 function in mitochondria plays an important role in excitatory neurons and oligodendrocyte phenotypes, independent of nuclear loss of TDP-43. Based on the nature of the genes differentially expressed in microglia clusters, the TDPhigh group is not significant for mitochondria terms in microglia, suggesting that once the onset of pTDP-43 accumulation starts, the mitochondria defects in microglia lead to the transition to the highly reactive proinflammatory state observed in the TDPhigh donor group, which is not observed in TDPneg donors.
Discussion
A G4C2 repeat expansion in the first intron of the C9orf72 gene is the most common genetic cause of both FTD and ALS. ALS neuromuscular abnormalities are caused by neurodegeneration of motor neurons in the motor cortex of ALS/FTD patients67. However, the contribution of other cell types present in the frontotemporal cortex to neural degeneration and cognitive decline during disease progression and the accompanying molecular changes remain largely unexplored. In this study, we utilized a unique C9orf72 ALS/FTD staging paradigm by selecting cases based on the abundance of pTDP-43. Cortical cytoplasmic accumulation of pTDP-43 has been found to correlate with neuropathological burden and severity of FTD clinical symptoms, and the progression of pTDP-43 distribution in the CNS has been proposed to stage patients in different phases of the disease68. Using a multiome approach to simultaneously analyze changes in chromatin accessibility and gene expression in the same cell, we identified several systematic changes that have not been reported before, including a correlation between ribosomal protein gene dysregulation and pTDP-43 levels across all cortical cell types, abnormalities in oligodendrocytes, microglia and astrocytes specifically associated with high pTDP-43 levels, and the selective vulnerability of cortical projection neurons to C9orf72 ALS/FTD progression.
Pan-cortical dysregulation of ribosomal protein genes, as previously observed in ALS/FTD and other age-related neurodegenerative disorders69, suggests a broad role of protein translation in FTD pathology. We observe a striking dichotomy in the expression of ribosomal protein genes, which are downregulated during early FTD disease stages but become upregulated when levels of pTDP-43 are high. While the role of TDP-43 in RNA metabolism is well established, several studies have provided evidence that TDP-43 interacts with the translational machinery70 and cytoplasmic TDP-43 inclusions can result in global translational inhibition70–72. However, the finding of extensive downregulation of nuclear and mitochondrial ribosomal protein genes in multiple cell types that lack TDP-43 pathology suggests alternative mechanisms to explain the decreased ribosomal proteins gene expression in these cells. G4C2repeats present in C9orf72 RNA form RNA G-quadruplex inclusions that sequester RNA-binding proteins such as hnRNPs, thus disrupting splicing73. Furthermore, C9orf72 dipeptide repeat proteins (DPRs) made from these repeats associate with U2 snRNP and prevent spliceosome assembly and splicing74, including that of ribosomal protein genes. In addition, our findings suggest that these genes could be downregulated at early stages of FTD at the level of transcription initiation, since the amount of FGFR1, which regulates the expression of many ribosomal protein genes, decreases dramatically. Since the expression of these genes is regulated through multiple feedback mechanisms, in addition to affecting protein translation by disrupting the assembly of mature ribosomes, their dysregulation could also have indirect effects on splicing. For example, it has been shown that some ribosomal proteins bind near splice sites and inhibit U2 snRNP binding75, thus interfering with splicing. Our finding of broad downregulation of ribosomal protein gene expression in early FTD stages is supported by previous work indicating that several ribosomal protein genes were downregulated in spinal cord and frontal cortex of C9orf72 ALS/FTD patients and in patient derived fibroblast and iPSCs76. Furthermore, downregulation of the RPS15A, RPL30, and RPL23A ribosomal protein genes in oligodendrocytes significantly correlates with AD diagnosis77. snRNA-seq of C9orf72 ALS/FTD patients indicates that ribosomal protein genes are upregulated in excitatory neurons of the motor cortex67, supporting our findings in dorsolateral prefrontal cortex in donors with significant pTDP-43 burden. Our results suggest the possibility that alteration of ribosomal gene expression may be a common event in both the motor and the dorsolateral prefrontal cortices, which are responsible for distinct functions on this continuous disease spectrum.
Microglia activation is observed in various neurological diseases78, and our results suggest that this proinflammatory state is not an early event during C9orf72 ALS/FTD disease progression, since upregulation of IRF8 and IL18 in microglia is only observed in patients with high levels of pTDP-43 at late disease stages. It has been suggested that microglia activation leads to an increase in the number of mitochondria79 while undergoing a metabolic switch from oxidative phosphorylation to aerobic glycolysis80. Mitochondrial dysfunction has been implicated in the activation of microglia in several neurodegenerative diseases, raising the possibility that misregulation of mitochondrial genes observed in our results is responsible for microglia activation. Genes encoding for proteins of the mitochondrial respiratory chain complex I are initially downregulated in all cell types of TDPneg donors. With increasing pTDP-43 burden, downregulation of these genes becomes restricted to excitatory neurons and oligodendrocytes from C9orf72 ALS/FTD donors with high accumulation of pTDP-43. A recent study has demonstrated that the C9orf72 protein is imported to the mitochondria intermembrane space, and C9orf72 haploinsufficiency impairs mitochondria function66. However, the fact that genes encoding for mitochondrial respiratory chain complex I components are not downregulated in microglia of TDPhigh donors suggests that the highly reactive proinflammatory microglia state is not a consequence of mitochondria defects. Therefore, the proinflammatory state of microglia that is unique to the TDPhigh donor group might be a consequence of the cytoplasmic accumulation of pTDP-43, and GSK-3β might be the protein kinase involved in TDP-43 phosphorylation in MG-C2 cells of the TDPhigh donor group.
Many other observed alterations in gene expression are specific to donors with high levels of pTDP-43. As is the case with microglia activation, dysregulation of these processes may be a direct consequence of the formation of pTDP-43 cytoplasmic inclusions. Indeed, pTDP-43 inclusions in oligodendrocytes are a hallmark of C9orf72 ALS/FTD. Our data demonstrate that cases with high pTDP-43 burden contain a high proportion of newly differentiated/pre-mature oligodendrocytes (ODC-C6). The relatively low expression of genes encoding myelin protein components in this cell cluster may be due to high cytoplasmic pTDP-43 accumulation, since this protein is involved in the posttranscriptional regulation of myelin proteins27,29. We do not observe a significant change in oligodendrocyte progenitor cells, suggesting that the cluster of premature oligodendrocytes is likely a result of its inability to become mature due to the downregulation of myelin components. Several lines of evidence converge on oligodendrocyte dysfunction as an important contributor to ALS/FTD pathogenesis24,81–83. Tissues from sporadic ALS patients show significant regions of demyelination and decreased expression of myelin related proteins24. Genetic studies have also provided insights into the role of oligodendrocytes in ALS/FTD. Recent GWAS studies have implicated single nucleotide polymorphisms (SNPs) in the MOBP gene, which encodes for myelin-associated oligodendrocyte basic protein, as a risk factor for ALS84,85. SNPs in MOBP are also associated with shorter disease duration and more severe white matter degeneration in FTD86. It is thus possible that the cause of cortical projection neuron loss in TDPhigh donors is the lack of sufficient mature oligodendrocytes, which are essential for neuron myelination and metabolic support. The impairment of myelination is not limited to C9orf72 FTD-TDP pathology as downregulation of myelin-associated genes is also observed in the prefrontal cortex of patients with AD pathology87,88. In AD donors carrying two copies of the APOE4 variant, cholesterol homeostasis is responsible for the downregulation of myelin-associated genes in oligodendrocytes88. However, the downregulation of myelin-associated genes in C9orf72 TDPhigh oligodendrocytes is not accompanied by changes in cholesterol homeostasis, suggesting that cortical myelination defects are common in patients with cognitive impairments with different genetic mutations. Further dissection of oligodendrocyte-neuron interactions may give additional insights into the mechanisms underlying the progression of FTD. Ultimately, the systematic identification of cell type specific defects in pathways common to all C9orf72 ALS/FTD donors as well as disease stage specific alterations will inform targets and timing of therapeutic interventions.
Methods
Human tissue samples.
Post-mortem brain samples from the dorsolateral prefrontal cortex (DLPFC) of C9orf72 ALS/FTD patients and controls were obtained from the Goizueta Emory Alzheimer’s Disease Center brain bank with approval from the Emory Institutional Review Board. All C9orf72 patients had a clinical diagnosis of ALS and/or FTD. Controls consisted of normal individuals with no clinical history of neurological disease. Patient information is provided in Table S1. All brains underwent thorough neuropathologic evaluation, including hematoxylin and eosin stains, silver stains, and immunohistochemistry for β-amyloid, tau, α-synuclein, and phosphorylated TDP-43.
Quantification of cortical phosphorylated TDP-43 levels.
Sequential biochemical fractionation was performed first and followed by Meso-Scale Discovery (MSD) immunoassay. Tissue lysates were fractionated according to previously published protocols9. In brief, ~200 mg of DLPFC tissue were homogenized in low salt buffer (10 mM Tris, 5 mM EDTA, 1 mM DTT, 10% sucrose, 1x HALT protease/phosphatase inhibitors). Lysates were pelleted at 25,000 g for 30 min. Supernatants were collected as the “low salt” fraction. The resulting pellet was solubilized in Triton-X buffer (1% triton X-100 and 0.5 M NaCl in low salt buffer). Lysates were subsequently pelleted at 180,000 g for 30 min. Supernatants were collected as the “Triton-X” fraction. The resulting pellet was solubilized in Triton-X buffer with 30% sucrose. Lysates were subsequently pelleted at 180,000 g for 30 min. The resulting pellet was solubilized in Sarkosyl buffer (1% sarkosyl and 0.5 M NaCl in low salt buffer). Lysates were subsequently pelleted at 180,000 g for 30 min. Supernatants from these fractions were not used for analysis. The resulting insoluble pellet was resolubilized in 8 M urea and used for MSD immunoassay to measure blinded the abundance of phosphorylated TDP-43 in the detergent insoluble and urea soluble fractions using a previously described sandwich immunoassay that utilizes MSD electrochemiluminescence detection technology89. The capture antibody was a mouse monoclonal antibody that detects TDP-43 phosphorylated at serines 409/410 (1:500, no. CAC-TIP-PTD-M01, Cosmo Bio USA), and the detection antibody was a sulfo-tagged rabbit polyclonal C-terminal TDP-43 antibody (2 μg/mL, 12892-1-AP, Proteintech). Lysates were diluted in 8 M urea such that all samples of a given type were made up to the same concentration and an equal amount of protein for samples was tested in duplicate wells. Response values corresponding to the intensity of emitted light upon electrochemical stimulation of the assay plate using the MSD QUICKPLEX SQ120 were acquired. These response values were background corrected by subtracting the average response values from corresponding negative controls e.g., lysates from tissues or cells lacking a repeat expansion.
Isolation of nuclei from frozen brain tissue.
Tissue sections were snap frozen and stored at −80°C and the nuclei were isolated as previously described90,91. Briefly, 20 mg frozen tissues were thawed in 1 mL cold homogenization buffer (260 mM sucrose, 30 mM KCl, 10 mM NaCl, 20 mM Tricine-KOH pH 7.8, 1 mM DTT, 0.5 mM Spermidine, 0.2 mM Spermine, 0.3% NP40, complete Protease inhibitor (Roche), and Ribolock) and homogenized in a pre-chilled Dounce. Cell lysates were passed through a 70 μm Flowmi cell strainer before separation using a discontinuous iodixanol gradient and centrifugation at 1480 g at 4°C for 20 min in a swinging bucket centrifuge with the brake off. The nuclei band located at the interface between 30% and 40% iodixanol was collected and washed in RSB-T wash buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20)
Single-nucleus multiome library preparation and sequencing.
Libraries were generated using the 10x Genomics Chromium Single Cell Multiome ATAC + Gene Expression kit following the manufacturer’s instructions, with the following modifications. Per sample, 16,100 nuclei were resuspended in 1x diluted nuclei buffer (10x Genomics) with 2% BSA (Sigma) with a capture target of 10,000 nuclei. ATAC-seq libraries were sequenced to target of 25,000 read-pairs per nucleus and RNA libraries were sequenced to 20,000 read-pairs per nucleus on an Illumina NovaSeq 6000 instrument at the Florida State University Translational Science Laboratory. The matched RNA-seq and ATAC-seq libraries were processed using the 10x Genomics Cell Ranger ARC (cellranger-arc-2.0.0) pipeline with default parameters and aligned to the hg38 human genome assembly (refdata-cellranger-arc-GRCh38-2020-A-2.0.0).
Processing and analyses of single nucleus multiome data.
Reads mapping to the mitochondrial genome, chromosome Y, and common blacklisted regions were excluded from downstream analysis. ArchR (v1.0.1)17 was used for processing the fragment data, quality control, dimensionality reduction, and clustering with default parameters. Harmony batch correction was performed prior to unsupervised dimensionality reduction and clustering 18. Nuclei with a TSS score <4, less than 1000 unique nuclear fragments, and without matched RNA reads were excluded in the downstream analysis. Additionally, for snRNA-seq quality control, we also removed nuclei with more than 1% levels of mitochondrial RNAs A total of 2,399 doublets were removed using the ArchR doublet detection tool and default parameters. As a result of the stringent quality control, a total of 52,581 nuclei were used for downstream analysis with median TSS score of 9.963 and median fragments per nucleus of 9548. The quality control matrix is provided in Table S2.
Identification of cluster and cell type assignments.
Cell type identification was performed based on gene activity scores calculated using ArchR with default parameters; the gene activity scores are correlated with gene expression and calculated based on chromatin accessibility at the gene body, promoter and distal regulatory regions17,92. Marker genes for each cluster were identified using ArchR getMarkerFeatures() function (filtering threshold: FDR ≤ 0.01 & log2(Fold change) ≥ 0.5; Table S3) and manually compared to known marker genes of cortical cell types. Clusters with less than 150 nuclei were excluded from further analysis resulting in a total of 35 distinct cell clusters that can be assigned to known cortical cell types. The cell classification was further verified by gene modules computed using ArchR addModuleScore() function with geneScoreMatrix with the following genes for each cell type (Fig. S2): Neurons: SNAP25 and SYT1; excitatory neurons: SLC17A7, SATB2, RORB, NEUROD2; inhibitory neurons: GAD1, GAD2, NXPH1; astrocytes: GFAP, AQP4, SLC1A2; microglia: CSF1R, CD74, P2RY12, PTPRC,TMEM119; oligodendrocytes: MOBP, MBP, ST18, KLK6, SLC5A11; oligodendrocyte precursor cells: PDGFRA, CSPG4; and endothelial cells: FLT1, CLDN5, ABCB1, EBF1.
snATAC-seq peak calling.
ArchR was used to call peaks with default parameters. Briefly, a pseudo-bulk dataset was created for each of the 35 cell clusters. The reproducible peak sets were analyzed using addReproduciblePeakSet() with MACS293 with a fixed-width peak size of 501 bp and iterative overlap peak merging. The resulting PeakMatrix, with a total of 362,205 peaks, was used for downstream analysis.
Comparison of snRNA-seq differential gene expression and snATAC-seq differential chromatin accessibility analysis.
For each C9orf72 ALS/FTD sample, pseudo-bulk data from each 35 distinct clusters were compared to that from the healthy control samples using ArchR getMarkerFeatures() with default parameters. The Wilcoxon rank sum test was used since this is most widely acceptable single-cell differential comparison method. The three groups of patients, grouped based on pTDP-43 levels, were compared with control samples. GeneExpressionMatrix and PeakMatrix generated by ArchR were used for differential genes and differential accessibility analysis, respectively. Genes and chromatin accessibility regions were considered significant differentially if they have an FDR-corrected p-value < 0.05 and an absolute log2(fold change) > 0.5 relative to the control group. To ensure a high confidence of differential gene expression comparison, we performed at least 5 permutation tests by randomizing the sample tag per nucleus while keeping the same proportion of each cell type per groups of patients. None of the permutation tests identified any significant differently expressed genes (FDR-corrected p-value < 0.05), ensuring high confidence in the differential gene expression comparison.
Enrichment of TF motifs in differentially accessible regions.
The regions identified as differentially accessible were tested for motif enrichment using ArchR peakAnnoEnrichment() after motif annotation analysis with addMotifAnnotations() using the cisbp motifs database94. The TF motifs were considered significantly enriched if FDR ≤ 0.1 & Log2FC ≥ 1.
Pseudotime analysis of astrocyte cell clusters.
Monocle 395 was used for pseudotime analysis with default parameters. The gene expression and gene activity score matrices were extracted from ArchR’s ArchRProject and the identities of the 35 cell clusters were retained when using Monocle. Cells in astrocyte cluster ASC-C29 were set as the root cells.
Ambient RNA analysis.
SoupX20 was used to estimate the levels of ambient RNA in each snRNA-seq dataset. The automated method was used for estimating the ambient RNA contaminated fraction and adjustCounts() was used to compute the final adjustment of RNA expression count matrix based on the estimated RNA contamination profile.
Weighted correlation network analysis on neuronal cell clusters.
WGCNA96 (weighted gene co-expression network analysis) was used to identify gene coexpression networks of neuronal clusters. This method identifies highly correlated gene clusters (termed modules) via unsupervised clustering. Pseudo-bulk expression for each excitatory and inhibitory neuronal cell cluster (n=10 and n=6, respectively) was analyzed separately with WGCNA using default parameters. Only differentially expressed genes in neuronal clusters were used for WGCNA analysis and a soft threshold power of 9 was used when constructing the network using blockwiseModules(). Hub genes were identified using signedKME() for each module. Correlation analysis between WGCNA modules and disease progression by grouping levels of pTDP-43 as described above was done using linear models on each module with Limma97 with multiple testing corrections, and the correlation was considered significant if p-adj ≤0.01. Gene ontology enrichment for each module was performed using clusterProfiler98 using the protein-coding hub genes with kME value > 0.8.
Analysis of cell-cycle scoring and regression.
The analysis was performed following the default vignette in Seurat99 with the list of cell cycle markers100. The gene expression matrix was extracted from ArchR’s ArchRProject to create the Seurat object before using Seurat’s CellCycleScoring().
Differential cell abundance analysis.
To identify differences in cell composition across the donor groups with different levels of pTDP-43 in each cell cluster in each major cell type, we calculated the relative percentage of each cluster in each major cell type for each sample. The differential cell proportions were estimated using Kruskal-Wallis test with Benjamini-Hochberg correction comparing the control with different pTDP-43 level groups. P-values > 0.05 were considered not significant. For the neuronal clusters, the significance of differential abundance was further analyzed using MASC101, which considers the mixed-effect model with a binomial distribution accounting for technical confounders and biological variation. We included the following fixed covariates in the model: sex, sample status (control and C9orf72 ALS/FTD cases), and level of pTDP-43. Cell clusters were considered significant at FDR-adjusted P < 0.05 and absolute odds ratio >0. The results of MASC analysis are shown in Fig. 6G.
Reverse deconvolution with pTDP-43 sorted bulk RNA-seq datasets.
Published RNA-seq datasets of FACS sorted pTDP-43 NeuN+ neurons were downloaded from the Gene Expression Omnibus (GEO) database under accession GSE12654358. Pseudo-bulk snRNA-seq data for each cell clusters were analyzed using CIBERSORTx 59 with default parameters.
Supplementary Material
Acknowledgements
We would like to thank Dr. Cynthia Vied at the Translational Science Laboratory of Florida State University for help with Illumina sequencing; Dr. Steven Sloan from the Department of Human Genetics at Emory University for assistance with the initial stage of astrocyte lineage analysis; Dr. Ryan Corces at the Gladstone Institute of Neurological Disease for assistance in the initial analysis of snATAC-seq data. This work was supported by U.S. Public Health Service Awards R01 ES027859 and P30ES019776 (VGC); R35 NS111602, R01 HG008935, U01 MH116441 (PJ) from the National Institutes of Health. H-LW was supported by NIH F32 ES031827. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Hardiman O. et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers 3, 17071 (2017). 10.1038/nrdp.2017.71 [DOI] [PubMed] [Google Scholar]
- 2.Warren J. D., Rohrer J. D. & Rossor M. N. Clinical review. Frontotemporal dementia. BMJ 347, f4827 (2013). 10.1136/bmj.f4827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferrari R., Kapogiannis D., Huey E. D. & Momeni P. FTD and ALS: a tale of two diseases. Curr Alzheimer Res 8, 273–294 (2011). 10.2174/156720511795563700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hudson A. J. Amyotrophic lateral sclerosis and its association with dementia, parkinsonism and other neurological disorders: a review. Brain 104, 217–247 (1981). 10.1093/brain/104.2.217 [DOI] [PubMed] [Google Scholar]
- 5.Chio A. et al. Cognitive impairment across ALS clinical stages in a population-based cohort. Neurology 93, e984–e994 (2019). 10.1212/WNL.0000000000008063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeJesus-Hernandez M. et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Majounie E. et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 11, 323–330 (2012). 10.1016/S1474-4422(12)70043-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glasmacher S. A., Wong C., Pearson I. E. & Pal S. Survival and Prognostic Factors in C9orf72 Repeat Expansion Carriers: A Systematic Review and Meta-analysis. JAMA Neurol 77, 367–376 (2020). 10.1001/jamaneurol.2019.3924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neumann M. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006). 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- 10.Murray M. E. et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol 122, 673–690 (2011). 10.1007/s00401-011-0907-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackenzie I. R. et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122, 111–113 (2011). 10.1007/s00401-011-0845-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brettschneider J. et al. TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol 128, 423–437 (2014). 10.1007/s00401-014-1299-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geser F. et al. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol 66, 180–189 (2009). 10.1001/archneurol.2008.558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawakami I., Arai T. & Hasegawa M. The basis of clinicopathological heterogeneity in TDP-43 proteinopathy. Acta Neuropathol 138, 751–770 (2019). 10.1007/s00401-019-02077-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mackenzie I. R. et al. Dipeptide repeat protein pathology in C90RF72 mutation cases: clinicopathological correlations. Acta Neuropathol 126, 859–879 (2013). 10.1007/s00401-013-1181-y [DOI] [PubMed] [Google Scholar]
- 16.Wilson R. S. et al. TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurol 70, 1418–1424 (2013). 10.1001/jamaneurol.2013.3961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Granja J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat Genet 53, 403–411 (2021). 10.1038/s41588-021-00790-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korsunsky I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296 (2019). 10.1038/s41592-019-0619-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stuart T. et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821 (2019). 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young M. D. & Behjati S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience 9 (2020). 10.1093/gigascience/giaa151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petibon C., Malik Ghulam M., Catala M. & Abou Elela S. Regulation of ribosomal protein genes: An ordered anarchy. Wiley Interdiscip Rev RNA 12, e1632 (2021). 10.1002/wrna.1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Servetto A. et al. Nuclear FGFR1 Regulates Gene Transcription and Promotes Antiestrogen Resistance in ER(+) Breast Cancer. Clin Cancer Res 27, 4379–4396 (2021). 10.1158/1078-0432.CCR-20-3905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagano S. et al. TDP-43 transports ribosomal protein mRNA to regulate axonal local translation in neuronal axons. Acta Neuropathol 140, 695–713 (2020). 10.1007/s00401-020-02205-y [DOI] [PubMed] [Google Scholar]
- 24.Kang S. H. et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci 16, 571–579 (2013). 10.1038/nn.3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marques S. et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 352, 1326–1329 (2016). 10.1126/science.aaf6463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Bruggen D., Agirre E. & Castelo-Branco G. Single-cell transcriptomic analysis of oligodendrocyte lineage cells. Curr Opin Neurobiol 47, 168–175 (2017). 10.1016/j.conb.2017.10.005 [DOI] [PubMed] [Google Scholar]
- 27.Wang J. et al. Cell-autonomous requirement of TDP-43, an ALS/FTD signature protein, for oligodendrocyte survival and myelination. Proc Natl Acad Sci USA 115, E10941–E10950 (2018). 10.1073/pnas.1809821115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polymenidou M. et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14, 459–468 (2011). 10.1038/nn.2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heo D. et al. Stage-specific control of oligodendrocyte survival and morphogenesis by TDP-43. Elife 11 (2022). 10.7554/eLife.75230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Q. & Barres B. A. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 18, 225–242 (2018). 10.1038/nri.2017.125 [DOI] [PubMed] [Google Scholar]
- 31.O’Rourke J. G. et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329 (2016). 10.1126/science.aaf1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Appel S. H., Beers D. R. & Zhao W. Amyotrophic lateral sclerosis is a systemic disease: peripheral contributions to inflammation-mediated neurodegeneration. Curr Opin Neurol 34, 765–772 (2021). 10.1097/WCO.0000000000000983 [DOI] [PubMed] [Google Scholar]
- 33.Umoh M. E. et al. A proteomic network approach across the ALS-FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain. Embo Mol Med, 48–62 (2018). 10.15252/emmm.201708202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sankowski R. et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat Neurosci 22, 2098–2110 (2019). 10.1038/s41593-019-0532-y [DOI] [PubMed] [Google Scholar]
- 35.Olah M. et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun 11, 6129 (2020). 10.1038/s41467-020-19737-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masuda T. et al. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Rep 1, 334–340 (2012). 10.1016/j.celrep.2012.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou N., Liu K., Sun Y., Cao Y. & Yang J. Transcriptional mechanism of IRF8 and PU.1 governs microglial activation in neurodegenerative condition. Protein Cell 10, 87–103 (2019). 10.1007/s13238-018-0599-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suo S. et al. Revealing the Critical Regulators of Cell Identity in the Mouse Cell Atlas. Cell Rep 25, 1436–1445 e1433 (2018). 10.1016/j.celrep.2018.10.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stein M. F. et al. Multiple interferon regulatory factor and NF-kappaB sites cooperate in mediating cell-type-and maturation-specific activation of the human CD83 promoter in dendritic cells. Mol Cell Biol 33, 1331–1344 (2013). 10.1128/MCB.01051-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao W. et al. Characterization of Gene Expression Phenotype in Amyotrophic Lateral Sclerosis Monocytes. JAMA Neurol 74, 677–685 (2017). 10.1001/jamaneurol.2017.0357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kruse M. et al. Inhibition of CD83 cell surface expression during dendritic cell maturation by interference with nuclear export of CD83 mRNA. J Exp Med 191, 1581–1590 (2000). 10.1084/jem.191.9.1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lechmann M., Berchtold S., Hauber J. & Steinkasserer A. CD83 on dendritic cells: more than just a marker for maturation. Trends Immunol 23, 273–275 (2002). 10.1016/s1471-4906(02)02214-7 [DOI] [PubMed] [Google Scholar]
- 43.Zhang Y. et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947 (2014). 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kametani F. et al. Identification of casein kinase-1 phosphorylation sites on TDP-43. Biochem Biophys Res Commun 382, 405–409 (2009). 10.1016/j.bbrc.2009.03.038 [DOI] [PubMed] [Google Scholar]
- 45.Hasegawa M. et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64, 60–70 (2008). 10.1002/ana.21425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sreedharan J., Neukomm L. J., Brown R. H. Jr. & Freeman M. R. Age-Dependent TDP-43-Mediated Motor Neuron Degeneration Requires GSK3, hat-trick, and xmas-2. Curr Biol 25, 2130–2136 (2015). 10.1016/j.cub.2015.06.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neumann M. et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathologica 117, 137–149 (2009). 10.1007/s00401-008-0477-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jurga A. M., Paleczna M. & Kuter K. Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front Cell Neurosci 14, 198 (2020). 10.3389/fncel.2020.00198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Batchelor P. E., Liberatore G. T., Porritt M. J., Donnan G. A. & Howells D. W. Inhibition of brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor expression reduces dopaminergic sprouting in the injured striatum. Eur J Neurosci 12, 3462–3468 (2000). 10.1046/j.1460-9568.2000.00239.x [DOI] [PubMed] [Google Scholar]
- 50.Fujita R., Ma Y. & Ueda H. Lysophosphatidic acid-induced membrane ruffling and brain-derived neurotrophic factor gene expression are mediated by ATP release in primary microglia. J Neurochem 107, 152–160 (2008). 10.1111/j.1471-4159.2008.05599.x [DOI] [PubMed] [Google Scholar]
- 51.Trias E. et al. Phenotypic transition of microglia into astrocyte-like cells associated with disease onset in a model of inherited ALS. Front Cell Neurosci 7, 274 (2013). 10.3389/fncel.2013.00274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Orre M. et al. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol Aging 35, 2746–2760 (2014). 10.1016/j.neurobiolaging.2014.06.004 [DOI] [PubMed] [Google Scholar]
- 53.Chung R. S. et al. Neuron-glia communication: metallothionein expression is specifically up-regulated by astrocytes in response to neuronal injury. J Neurochem 88, 454–461 (2004). 10.1046/j.1471-4159.2003.02193.x [DOI] [PubMed] [Google Scholar]
- 54.Cistaro A. et al. The metabolic signature of C9ORF72-related ALS: FDG PET comparison with nonmutated patients. Eur J Nucl Med Mol Imaging 41, 844–852 (2014). 10.1007/s00259-013-2667-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Allen S. P. et al. C9orf72 expansion within astrocytes reduces metabolic flexibility in amyotrophic lateral sclerosis. Brain 142, 3771–3790 (2019). 10.1093/brain/awz302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trapnell C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386 (2014). 10.1038/nbt.2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lake B. B. et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590 (2016). 10.1126/science.aaf1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu E. Y. et al. Loss of Nuclear TDP-43 Is Associated with Decondensation of LINE Retrotransposons. Cell Rep 27, 1409–1421 e1406 (2019). 10.1016/j.celrep.2019.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Newman A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 37, 773–782 (2019). 10.1038/s41587-019-0114-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nolan M. et al. Quantitative patterns of motor cortex proteinopathy across ALS genotypes. Acta Neuropathol Commun 8, 98 (2020). 10.1186/s40478-020-00961-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beers D. R. et al. Parvalbumin overexpression alters immune-mediated increases in intracellular calcium, and delays disease onset in a transgenic model of familial amyotrophic lateral sclerosis. J Neurochem 79, 499–509 (2001). 10.1046/j.1471-4159.2001.00582.x [DOI] [PubMed] [Google Scholar]
- 62.Griffiths I. et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 280, 1610–1613 (1998). 10.1126/science.280.5369.1610 [DOI] [PubMed] [Google Scholar]
- 63.Bannwarth S. et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137, 2329–2345 (2014). 10.1093/brain/awu138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cozzolino M., Rossi S., Mirra A. & Carri M. T. Mitochondrial dynamism and the pathogenesis of Amyotrophic Lateral Sclerosis. Front Cell Neurosci 9, 31 (2015). 10.3389/fncel.2015.00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rinholm J. E. et al. Movement and structure of mitochondria in oligodendrocytes and their myelin sheaths. Glia 64, 810–825 (2016). 10.1002/glia.22965 [DOI] [PubMed] [Google Scholar]
- 66.Wang T. et al. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab 33, 531–546 e539 (2021). 10.1016/j.cmet.2021.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pineda S. S. et al. Single-cell profiling of the human primary motor cortex in ALS and FTLD. biorxiv (2021). 10.1101/2021.07.07.451374 [DOI] [Google Scholar]
- 68.Brettschneider J. et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 74, 20–38 (2013). 10.1002/ana.23937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lehmkuhl E. M. & Zarnescu D. C. Lost in Translation: Evidence for Protein Synthesis Deficits in ALS/FTD and Related Neurodegenerative Diseases. Adv Neurobiol 20, 283–301 (2018). 10.1007/978-3-319-89689-211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Freibaum B. D., Chitta R. K., High A. A. & Taylor J. P. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res 9, 1104–1120 (2010). 10.1021/pr901076y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Charif S. E., Luchelli L., Vila A., Blaustein M. & Igaz L. M. Cytoplasmic Expression of the ALS/FTD-Related Protein TDP-43 Decreases Global Translation Both in vitro and in vivo. Front Cell Neurosci 14, 594561 (2020). 10.3389/fncel.2020.594561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Russo A. et al. Increased cytoplasmic TDP-43 reduces global protein synthesis by interacting with RACK1 on polyribosomes. Hum Mol Genet 26, 1407–1418 (2017). 10.1093/hmg/ddx035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Conlon E. G. et al. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife 5 (2016). 10.7554/eLife.17820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yin S. et al. Evidence that C9ORF72 Dipeptide Repeat Proteins Associate with U2 snRNP to Cause Mis-splicing in ALS/FTD Patients. Cell Rep 19, 2244–2256 (2017). 10.1016/j.celrep.2017.05.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Macias S., Bragulat M., Tardiff D. F. & Vilardell J. L30 binds the nascent RPL30 transcript to repress U2 snRNP recruitment. Mol Cell 30, 732–742 (2008). 10.1016/j.molcel.2008.05.002 [DOI] [PubMed] [Google Scholar]
- 76.Tank E. M. et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nature Communications 9, 2845 (2018). 10.1038/s41467-018-05049-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morabito S. et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat Genet 53, 1143–1155 (2021). 10.1038/s41588-021-00894-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leng F. & Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol 17, 157–172 (2021). 10.1038/s41582-020-00435-y [DOI] [PubMed] [Google Scholar]
- 79.Banati R. B. et al. Mitochondria in activated microglia in vitro. J Neurocytol 33, 535–541 (2004). 10.1007/s11068-004-0515-7 [DOI] [PubMed] [Google Scholar]
- 80.Orihuela R., McPherson C. A. & Harry G. J. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol 173, 649–665 (2016). 10.1111/bph.13139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee Y. et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448 (2012). 10.1038/nature11314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Philips T. et al. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 136, 471–482 (2013). 10.1093/brain/aws339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Philips T. et al. MCT1 Deletion in Oligodendrocyte Lineage Cells Causes Late-Onset Hypomyelination and Axonal Degeneration. Cell Rep 34, 108610 (2021). 10.1016/j.celrep.2020.108610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van Rheenen W. et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet 48, 1043–1048 (2016). 10.1038/ng.3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Saez-Atienzar S. et al. Genetic analysis of amyotrophic lateral sclerosis identifies contributing pathways and cell types. Sci Adv 7 (2021). 10.1126/sciadv.abd9036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Irwin D. J. et al. Myelin oligodendrocyte basic protein and prognosis in behavioral-variant frontotemporal dementia. Neurology 83, 502–509 (2014). 10.1212/WNL.0000000000000668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mathys H. et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019). 10.1038/s41586-019-1195-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blanchard J. W. et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature (2022). 10.1038/s41586-022-05439-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Prudencio M. et al. Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum Mol Genet 26, 3421–3431 (2017). 10.1093/hmg/ddx233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Corces R., J. Greenleaf W. & Y. Chang H. Isolation of nuclei from frozen tissue for ATAC-seq and other epigenomic assays, protocols.io (2019). 10.17504/protocols.io.6t8herw [DOI] [Google Scholar]
- 91.Corces M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nature Methods 14, 959-962-962 (2017). 10.1038/nmeth.4396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Granja J. M. et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat Biotechnol 37, 1458–1465 (2019). 10.1038/s41587-019-0332-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu T. Use model-based Analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein-DNA interactions in embryonic stem cells. Methods in molecular biology (Clifton, N.J.) 1150, 81 95 (2014). 10.1007/978-1-4939-0512-64 [DOI] [PubMed] [Google Scholar]
- 94.Weirauch M. T. et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443 (2014). 10.1016/j.cell.2014.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cao J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496 502 (2019). 10.1038/s41586-019-0969-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Langfelder P. & Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ritchie M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wu T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2, 100141 (2021). 10.1016/j.xinn.2021.100141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hao Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529 (2021). 10.1016/j.cell.2021.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tirosh I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). 10.1126/science.aad0501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fonseka C. Y. et al. Mixed-effects association of single cells identifies an expanded effector CD4(+) T cell subset in rheumatoid arthritis. Sci Transl Med 10 (2018). 10.1126/scitranslmed.aag0305 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.