

SUMMARY
Precision oncology relies on the accurate molecular characterization of individual patients with cancer at the time of treatment initiation. However, tumor molecular profiles are not static, and cancers continually evolve because of ongoing mutagenesis and clonal selection. Here, we performed genomic analyses of primary tumors, metastases, and plasma collected from individual patients to define the concordance of actionable genomic alterations and to identify drivers of metastatic disease progression. We observed a high degree of discordance of actionable genomic alterations, with 23% discordant between primary and metastatic disease sites. Among chromatin-modifying genes, ARID1A mutations, when discordant, were exclusive to the metastatic tumor samples. Our findings indicate that the high degree of lesion-to-lesion genomic heterogeneity may be a barrier to precision oncology approaches for bladder cancer and that circulating tumor DNA profiling may be preferred to tumor sequencing for a subset of patients.
In brief
Clinton et al. define the concordance of genomic alterations in urothelial carcinoma from a localized to metastatic state to identify drivers of progression. Within individual patients, there is significant discordance between primary and metastatic sites. Additionally, plasma cell-free DNA (cfDNA) can identify alterations not detected by tumor sequencing.
Graphical Abstract
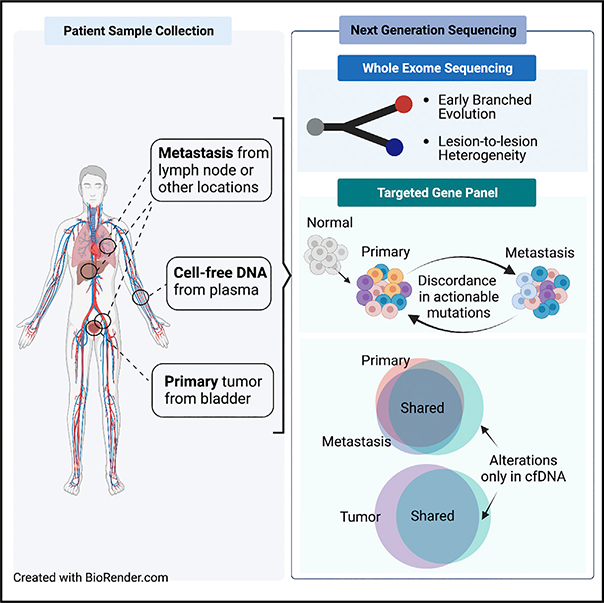
INTRODUCTION
Therapy selection is increasingly guided by prospective molecular analyses designed to identify clinically actionable molecular alterations in individual patients with cancer. In molecularly selected patients with cancer, targeted therapies that inhibit mutated oncoproteins can induce dramatic and durable responses.1 In addition to serving as predictive biomarkers of drug response, genomic alterations can also influence the likelihood of disease recurrence and patterns of metastatic spread.2 However, a major hurdle to the broader adoption of precision oncology paradigms is the evolution of tumor genomes as cancers progress from a localized to metastatic disease state. New somatic mutations and structural alterations arise in tumors over time because of impaired DNA replication, ongoing exposure to mutagens, or in response to the selective pressure of systemic therapy.3–6 This ongoing mutagenesis and clonal selection results in intratumoral and lesion-to-lesion genomic heterogeneity that can influence treatment response and clinical outcomes.
To date, most large-scale tumor profiling studies of patients with bladder cancers have focused on the analysis of primary tumor samples.7 Only recently have metastatic samples been profiled, and the number of patients with matched primary and metastatic samples analyzed was limited, thus the concordance of actionable genomic alterations between primary and metastatic disease sites remains poorly characterized.8–10 With the recent FDA approval of erdafitinib, a selective fibroblast growth factor receptor (FGFR) inhibitor for the treatment of FGFR2/3-mutated metastatic urothelial cancers, tumor genomic profiling is now recommended for all patients with advanced urothelial cancers as a guide to treatment selection.11 There is thus an urgent clinical need to define the mutational concordance of primary and metastatic bladder cancers for FGFR3 and other potentially actionable genomic alterations. Advances in sequencing and bioinformatic methodology have also made feasible the detection and mutational profiling of tumor-derived DNA circulating in plasma (cell-free DNA [cfDNA]) as an alternative to tumor sequencing.12 cfDNA provides the opportunity for serial sampling and may be preferable to analysis of archival primary tumor samples in patients with metastatic disease who have received extensive prior therapy.
Here, we leveraged a prospective institution-wide tumor sequencing initiative to define the concordance of oncogenic alterations in primary and metastatic disease sites. Through paired analysis of primary and metastatic tumors and cfDNA isolated from plasma collected from the same individual, we also sought to identify genomic alterations that contribute to metastatic disease progression, the primary mediator of morbidity and cancer-specific death in bladder cancer and most other cancer types.
RESULTS
Patient and tumor characteristics
To identify differences in the genomic landscape of primary and metastatic urothelial cancers, we collected detailed demographic, clinical, and treatment data from patients with urothelial cancer who underwent prospective tumor DNA profiling as a guide to treatment selection. Between 2014 and 2021, 1,313 patients with bladder urothelial carcinoma were prospectively analyzed using the MSK-IMPACT next-generation sequencing assay (Figures 1A and 1B; Table 1). As a primary objective of the MSK-IMPACT initiative was to identify mutations that could serve as predictive biomarkers of systemic therapy response, this cohort was enriched for patients who had metastatic disease at diagnosis or developed recurrent disease during their disease course (57% versus 42% for The Cancer Genome Atlas [TCGA] bladder cancer cohort).
Figure 1. Clinical characteristics of prospectively sequenced urothelial carcinomas.
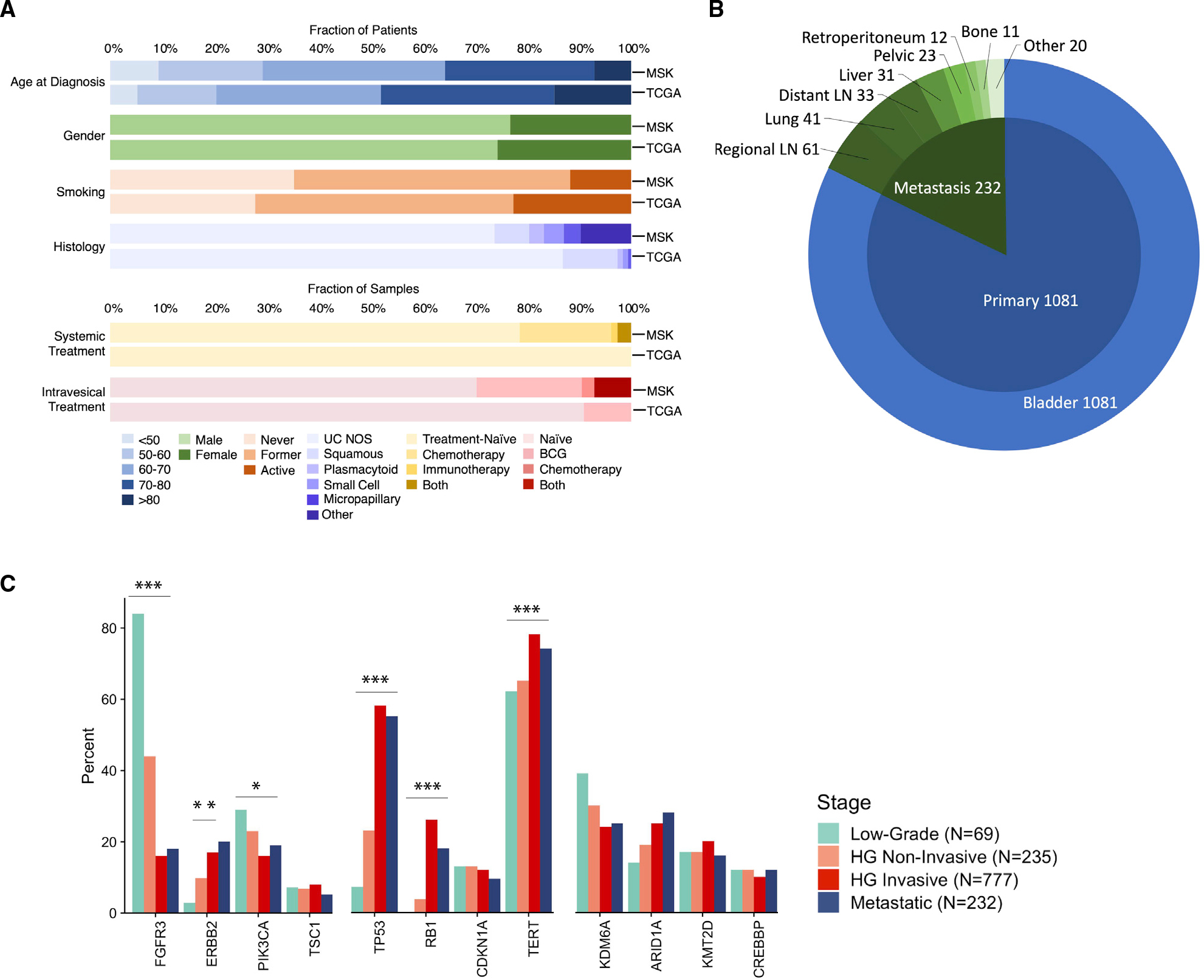
(A) Clinical and tumor features of the prospective Memorial Sloan Kettering Cancer Center (MSK) and retrospective The Cancer Genome Atlas (TCGA) bladder urothelial carcinoma cohorts.
(B) Distribution of the biopsied metastatic disease sites in the MSK urothelial carcinoma study cohort.
(C) Frequency of alterations in frequently mutated oncogenes in the MSK urothelial cancer cohort stratified by disease state (low-grade primary tumors, non-invasive and invasive high-grade [HG] primary tumors, and metastatic sites). Significant values are labeled as adjusted p value (q value): *q < 0.05, **q < 0.01, ***q < 0.001. Adjustment for multiple comparisons using the false discovery method demonstrated no loss in significance of the highlighted genes but a loss of significance for KDM6A and ARID1A (p = 0.03, q = 0.06).
Table 1.
Characteristics of patients profiled by MSK-IMPACT compared with TCGA
| MSK (n = 1,313) |
TCGA (n = 407) |
p | |
|---|---|---|---|
|
| |||
| Age | 66 (58,73) | 69 (60,76) | <0.001 |
| Gender (%) | 0.32 | ||
| Male | 1,009 (77%) | 303 (74%) | |
| Female | 304 (23%) | 104 (26%) | |
| Smoking (%) | <0.001 | ||
| Never | 444 (35%) | 110(28%) | |
| Former | 669 (53%) | 197 (50%) | |
| Active | 146(12%) | 89 (22%) | |
| Unknown | 54 | 11 | |
| Intravesical treatment (%) | <0.001 | ||
| Naive | 902 (70%) | 370 (91%) | |
| BCG | 258 (20%) | 37 (9%) | |
| Chemo | 30 (2%) | 0 (0%) | |
| Both | 91 (7%) | 0 (0%) | |
| Unknown | 32 | 0 | |
| Systemic treatment (%) | <0.001 | ||
| Naive | 1,000 (79%) | 407 (100%) | |
| Chemotherapy | 221 (17%) | 0 (0%) | |
| Immunotherapy | 16(1%) | 0 (0%) | |
| Both | 30 (2%) | 0 (0%) | |
| Unknown | 46 | 0 | |
We compared the frequency of commonly mutated genes in tumors collected from primary and metastatic disease sites to understand genomic alterations associated with an increased risk for metastatic progression. For tumors collected from the primary tumor site, we stratified patients into clinically relevant groups based on tumor grade and stage: low-grade non-muscle-invasive tumors (pTa/pT1, n = 69); high-grade non-invasive tumors (pTis/pTa, n = 235); high-grade invasive tumors (≥pT1, n = 777); and metastatic tumors (n = 232). The frequency of oncogenic mutations in targetable kinases, chromatin-modifying genes, and TP53 pathway genes were then compared across the different tumor stage groups (Figure 1C). The frequency of all genes with ≥5% mutational frequency stratified by tumor grade and stage are summarized in Table S1. Further subgroup analyses of mutational frequencies stratified by low-grade, carcinoma in situ, Ta/T1 high-grade non-muscle-invasive papillary, muscle-invasive/locally advanced, and metastatic sites are summarized in Figure S1.
The frequency of mutations in TP53 (q < 0.001) and RB1 (q < 0.001) and alterations in ERBB2 (q = 0.001) were more frequent in higher-grade and -stage tumors as reported in previous studies.7,13 Conversely, mutations in FGFR3 (q < 0.001), and PIK3CA (q = 0.04) were more frequently altered in lower-grade and -stage patients. Notably, ARID1A was the only chromatin-modifying gene more commonly mutated in high-grade invasive and metastatic samples (25% and 28%, respectively) compared with low- and high-grade non-invasive tumors (14% and 19% respectively; 4-group comparison q = 0.06). Mutations in chromatin-modifying genes are common in bladder cancer, and recent studies have shown that these mutations are often present in the surrounding benign-appearing urothelium of patients with urothelial cancer, suggesting that mutations in genes that regulate chromatin state may be an early initiating event in at least a subset of bladder cancers.14,15
Matched pairs of primary bladder and upper tract urothelial carcinomas and metastases
To determine whether differences in the mutation rate of individual genes in primary versus metastatic tumors were reflective of the timing at which mutations in these genes arose during tumorigenesis, or rather the impact of the mutation on the likelihood of metastatic spread, we performed whole-exome sequencing (WES) of primary-metastasis urothelial cancer pairs from the same patient. Only the 22 primary-metastasis pairs with an estimated tumor purity of 25% or greater based on FACETS were included in the subsequent concordance analysis to avoid the confounding effects of tumor purity on sensitivity of mutation detection (Figure 2A). While estimates of tumor mutational burden (TMB) were not significantly different between the primary and metastatic tumor sites (one-sided Wilcoxon signed rank, p = 0.1), the average mutational concordance rate was low at 42% for all mutations (ranged between 6% and 84%; Figure 2B). Furthermore, although TMB was often similar between the primary tumors and paired metastasis, phylogenetic analysis revealed that this was often due to a large, but similar, number of private mutations being present in both tumors, a pattern consistent with an early branched evolution (Figures 2C and 2D). As mutational concordance between primary and metastatic tumors is likely influenced by exogenous pressures, we correlated the percentage of shared mutations between primary and metastatic tumors and the length of time between specimen collection but did not observe a significant trend (goodness of fit, R2 = 0.17, p = 0.053). Furthermore, exclusion of patients with intervening treatment did not change the directionality or significance of this trend.
Figure 2. Whole-exome sequencing of 22 paired primary and metastatic urothelial cancers.
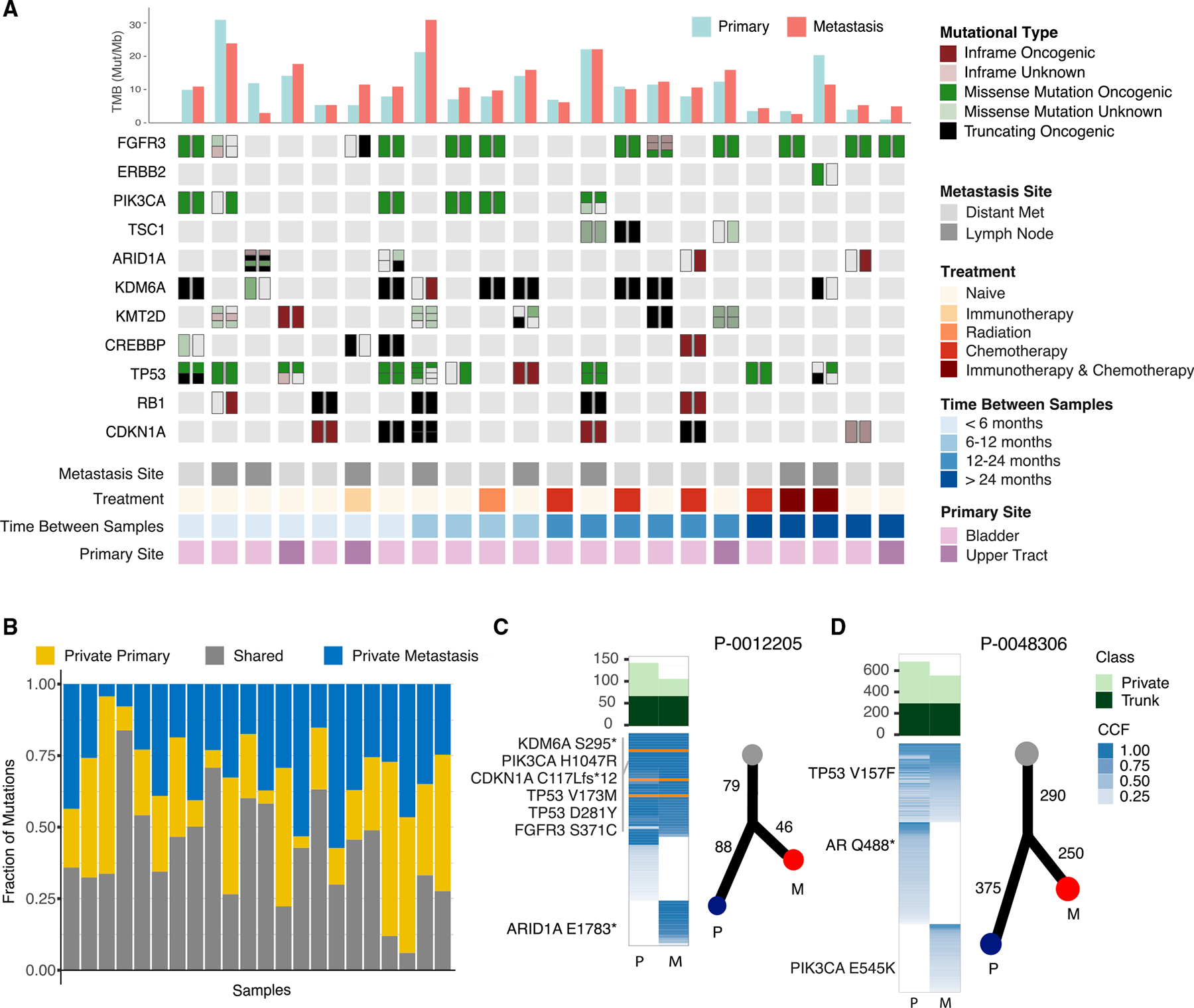
(A) OncoPrint of whole-exome sequencing data from 22 primary-metastasis urothelial cancer pairs. Tumor mutational burden (mutations/megabase [MB]) and select recurrently mutated genes are shown. Each column represents an individual patient with the mutational status of the primary tumor on the left and the metastatic specimen on the right.
(B) Mutational concordance and discordance between primary and metastatic tumor samples shown as the fraction of mutations that were shared, exclusive to the primary tumor or to the metastasis.
(C and D) Phylogenic analysis of the primary and metastatic tumors from two representative patients. Shown are the mutation matrix colored as trunk (dark green) or exclusive (light green) for the respective phylogeny, fraction of tumor cells mutated (cancer cell fractions, shades of blue) and inferred evolutionary relationship.
Numbers indicate shared or private mutation counts.
See also Figure S2.
Given the high degree of genomic discordance between primary and metastatic pairs, we next performed mutation signature deconvolution to determine whether the predominant mutational signatures differed. While the mutational processes in the primary and metastatic lesions were similar (Figure S2A), an analysis of concordance of known or likely oncogenic alterations revealed several notable findings (Figures S2B–S2D). Most notably, ARID1A mutations were present in the metastases of three patients but absent in their corresponding primary tumors (Figure 2A). As a representative example, WES of the primary tumor from patient P-0012205 collected at the time of radical cystectomy and a lung metastasis that developed 6 months later revealed shared mutations in TERT, KDM6A, FGFR3, PIK3CA, TP53, CDKN1A, and CREBBP but an ARID1A mutation (E1783*) exclusive to the metastatic sample (Figure 2C). Discordance of potentially actionable kinase mutations was also observed in multiple primary/metastatic pairs analyzed by WES. For example, patient P-0048306 presented with carcinoma in situ (CIS), which was treated with 3 courses of intravesical BCG, but later developed a lymph node metastasis. WES of the primary tumor and lymph node metastasis revealed a shared TP53 mutation (V157F) but an actionable PIK3CA (E545K) mutation exclusive to the lymph node metastasis (Figure 2D).
While WES has the potential to identify alterations not covered by the MSK-IMPACT panel design, MSK-IMPACT can detect some targetable alterations including gene fusions often missed by WES. As an example, one patient was found to have an FGFR3-TACC3 fusion detected by MSK-IMPACT that was exclusive to the metastatic samples. Therefore, to determine whether the higher depth and uniformity of sequencing coverage afforded by targeted sequencing would identify evidence of additional discordance in known or likely oncogenic mutations and to increase our statistical power to detect mutational discordance in known and presumed cancer-associated genes, we expanded the dataset by performing MSK-IMPACT targeted sequencing on the original 22 paired samples, as well as on an additional 124 urothelial carcinoma primary-metastasis pairs. Among these 148 matched primary-metastatic pairs, two were excluded from the mutational concordance analysis as they were hypermutated, microsatellite instability-high (MSI-H) tumors. There were also three patients with primary urothelial carcinomas and presumed lung metastases that, upon review of the MSK-IMPACT results, were deemed to be separate primary tumors. Four additional primary-metastasis pairs were excluded and evaluated separately as the metastatic sites were collected prior to the primary site with intervening systemic treatment. Most metastatic specimens were collected from distant metastases (74%), with the remainder from sites of regional lymph node metastasis. The median time between tumor collection was 10.5 months (interquartile range [IQR] 2.6–25.5), with 41% of patients receiving systemic treatment between collection of the primary and metastatic tumors. Intervening systemic treatment was chemotherapy in 38 (32%) patients, immunotherapy in 5 (4%), and both in 6 (5%) (Figure 3A).
Figure 3. Frequent discordance of actionable genomic alterations in primary and metastatic urothelial carcinomas.
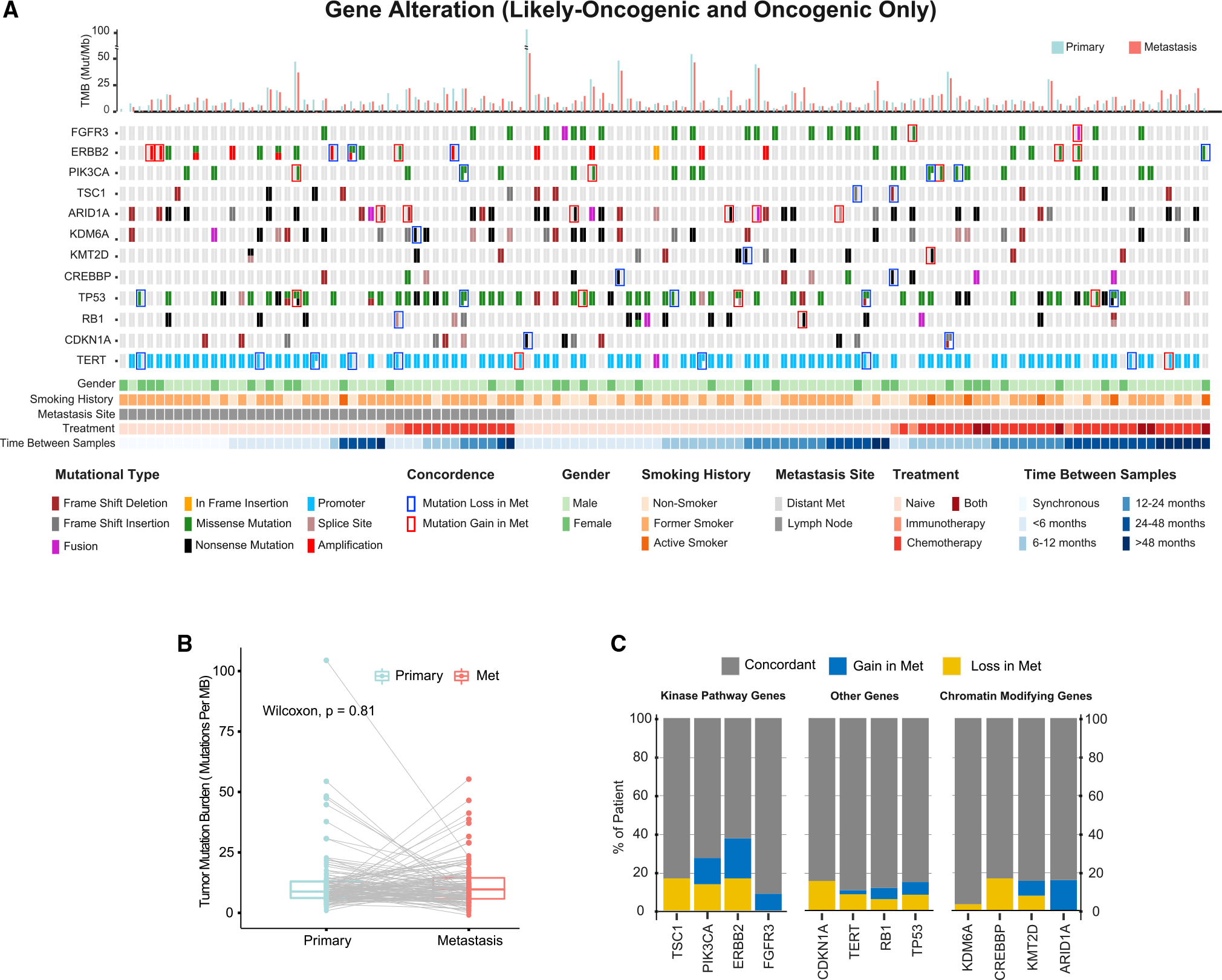
(A) Targeted sequencing of 119 paired primary and metastatic urothelial cancer samples. Each column represents an individual patient with the mutational status of the primary tumor on the left and the metastatic specimen on the right. Only oncogenic and likely oncogenic mutations, fusions, and ERBB2 amplifications were included in the OncoPrint.
(B) Comparison of tumor mutational burden (TMB) in primary and metastatic tumor sites.
(C) Mutational concordance of select frequently mutated genes including targetable kinases and chromatin-modifying genes. Percentages reflect only patients with a mutation in the designated gene in either the primary or metastasis or both.
See also Figure S3.
Consistent with the WES data above, the mean TMB as inferred from MSK-IMPACT sequencing of the primary and metastatic cohorts was similar (11.7 and 11.2, respectively; Figure 3B). Overall, in the expanded 119 patient primary-metastasis tumor pair cohort, the mutational concordance for known and likely oncogenic alterations was 85% (Figure 3A). Of relevance to ongoing efforts to develop targeted therapies for patients with urothelial carcinoma, 23% of potentially actionable gene mutations (FGFR3, PIK3CA, TSC1, ERBB2) were discordant in the primary-metastasis pairs (Figure 3C). For example, in patients with oncogenic and targetable FGFR3 alterations in at least one tumor, the FGFR3 alteration was exclusive to the metastatic sample in 9% of patients, with both patients with discordant FGFR3 mutational status having received intervening systemic therapy. These results suggest that archival tumor tissue may not always be adequate for FGFR3 genotyping in patients with urothelial carcinoma being considered for erdafitinib therapy. Mutational discordance was also common for ERBB2, PIK3CA, and TSC1, with 38%, 27%, and 17% of patients with mutations in these genes exhibiting discordance between the primary and metastatic tumor samples, respectively.
We also assessed the concordance of the chromatin-modifying genes commonly mutated in bladder cancer as several of these epigenetic regulators are potentially targetable with EZH2 inhibitors. As observed in the WES analysis, mutations in ARID1A were never present only in the primary tumor but were exclusive to the metastatic samples of 16% of patients in which an ARID1A mutation was detected in either. These results indicate that ARID1A mutations often arise later in tumor pathogenesis, consistent with the higher rates of ARID1A mutation noted in high-grade, muscle-invasive, and metastatic urothelial carcinoma samples (Figure 1C). To assess whether intratumoral heterogeneity within the primary sample accounted for the ARID1A discordance among select primary-metastatic pairs, we resampled and sequenced multiple spatially distinct regions of 3 of the ARID1A discordant pairs. As shown in Figure S3, we did not identify the oncogenic ARID1A mutation in the resampled regions of the primary tumors of any of the discordant cases. Additionally, for patients with discordant ARID1A mutations by MSK-IMPACT analysis, a manual review of the BAM files was performed to ensure that the primary tumor did not have evidence of the ARID1A mutation at a variant allele frequency below the threshold for mutation calling by our pipeline. Notably, this manual review of the BAM files did not detect any ARID1A mutant reads in the primary tumors of discordant cases.
Mutational concordance of plasma-derived cfDNA and tumor in patients with metastatic urothelial cancer
As analysis of a single metastatic tumor site cannot assess for genomic discordance between metastatic tumors, we performed ultra-high depth sequencing (~20,000× raw coverage, 1,000× collapsed duplex coverage) of 129 cancer-associated genes using plasma cfDNA collected from 45 patients for whom we had sequenced a primary and metastatic tumor pair. Among these 45 patients, 20% of targetable mutations as defined by the OncoKB knowledgebase16 were identified only by the plasma cfDNA analysis (Figures 4A and 4B). Given this high degree of discordance between tumor and plasma cfDNA, we expanded the analysis to include 123 patients with at least one bladder cancer tumor sample (either a primary or metastatic disease site) and cfDNA (Figure S4). While 60% of targetable alterations were concordant between tumor and plasma cfDNA, 17% were exclusive to cfDNA only, and 23% were exclusive to tumor samples (Figure 4C). These results suggest that the analysis of primary-metastatic tumor pairs underestimated the discordance of targetable alterations in patients with bladder cancer. One notable patient had an FGFR3 wild-type primary tumor but four oncogenic FGFR3 alterations (three oncogenic hotspot FGFR3 mutations and an FGFR3-TACC3 fusion) detected in cfDNA, only one of which was detected in the metastatic tumor sample, suggesting convergent evolution (Figure 4D). Additional FGFR3 mutations were identified by cfDNA analysis in blood samples collected during erdafitinib therapy, including a N540S mutation, which is paralogous to N546 of FGFR1, and K650E, which is paralogous to K660 of the FGFR2 IIIb isoform (Figure S5), mutations that have been shown previously to be associated with resistance to FGFR kinase inhibitors.17,18 cfDNA analysis also identified a V553M FGFR3 mutation, a previously unreported FGFR3 mutation that is near the putative gatekeeper and therefore a likely contributor to acquired erdafitinib resistance in this patient. In sum, the results suggest that cfDNA analysis is complementary to analyses of archival primary tumor tissue or biopsy of a single metastatic site for guiding treatment selection in patients with metastatic urothelial cancer and that analysis of both tissue and cfDNA may be required for some patients.
Figure 4. Concordance of oncogenic mutations between tumor and cell-free DNA (cfDNA) in patients with metastatic urothelial cancer.
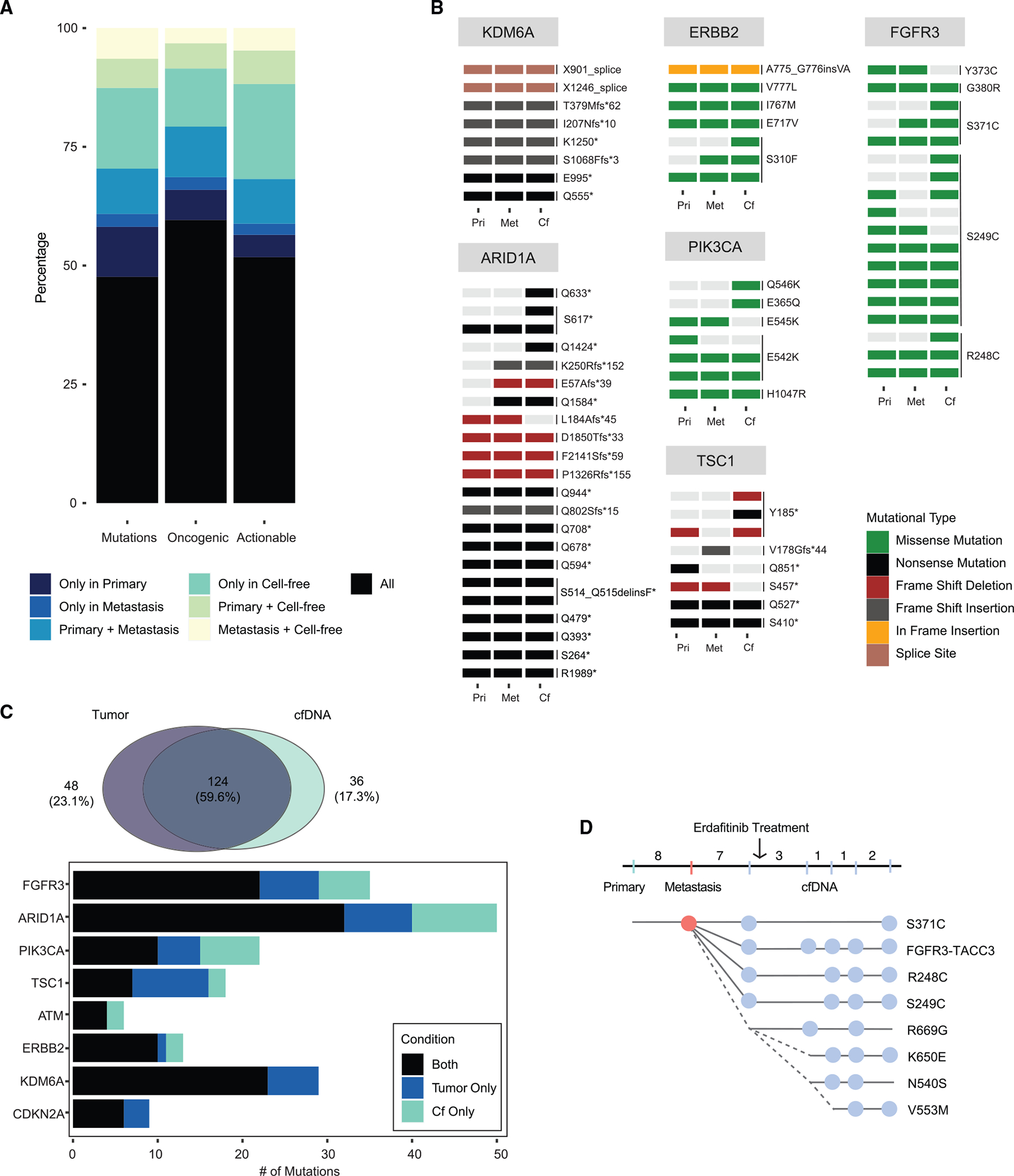
(A) Concordance between primary and metastatic tumor sites and plasma-derived cfDNA in 45 patients with metastatic urothelial cancer stratified by all mutations, oncogenic/likely oncogenic mutations only, and actionable mutations only as defined by OncoKB levels 1–4.
(B) OncoPrint of select actionable/oncogenic genes in patient-matched primary, metastatic, and cfDNA samples.
(C) Paired comparison of tumor and cfDNA samples from 123 patients with metastatic urothelial cancer.
(D) Patient P-0033799 presented with localized bladder cancer and was treated with neoadjuvant chemotherapy followed by radical cystectomy. The patient later developed multiple lung metastases and was treated with pembrolizumab. Upon further progression, a biopsy of a lung metastasis identified an actionable FGFR3 mutation (S371C) not present in the primary tumor sample. Plasma collected for cfDNA analysis prior to initiation of erdafitinib identified two additional actionable FGFR3 mutations (R248C and S249C) and a FGFR3-TACC3 fusion. While on therapy, cfDNA analyses identified 4 additional FGFR3 mutations, a subset of which have been previously been shown to confer resistance to FGFR-directed therapy. Numbers correspond to the interval in months between each specimen collection.
See also Figures S4 and S5.
DISCUSSION
Precision oncology, or the tailoring of cancer therapy to individual patients, requires knowledge of the timing at which oncogenic and potentially actionable genomic alterations arise during tumor initiation and metastatic progression and the influence of genomic heterogeneity on treatment response. In the current study, we sought to explore the degree to which tumor heterogeneity may be a barrier to precision oncology strategies in bladder cancer by examining the concordance of potentially actionable genomic alterations in primary and metastatic disease sites and circulating tumor DNA in plasma. While TMB was not significantly different between primary and patient-matched metastatic tumors, less than half of mutations, on average, were present at both disease sites. The results are consistent with earlier smaller series of paired primary and metastasis samples that suggested that bladder cancers are characterized by early branched evolution, which gives rise to significant intratumoral and lesion-to-lesion genomic heterogeneity, including discordance of FGFR3 mutations between primary and metastatic disease sites.9,10,19 However, the larger size of the current cohort provided sufficient power to detect a high degree of discordance of potentially actionable genomic alterations including mutations in FGFR3, ERBB2, TSC1, and PIK3CA. Finally, comparisons of cfDNA with tumor sequencing suggested that analysis of circulating tumor-derived DNA in plasma could identify some targetable genomic alterations, such as FGFR3 mutation, not present in archival tissue samples.12
Intrinsic and acquired drug resistance remain major barriers to the broader adoption of precision oncology paradigms. Second-site mutations that impair drug binding and co-alterations that reduce dependence on the mutated oncoprotein (oncogenic bypass) are well-described mechanisms of targeted therapy resistance.20–23 Whether mutational subclonality and discordance among disease sites of targetable mutated oncogenes is a common mechanism of treatment resistance remains largely unknown. This is because the vast majority of clinically validated actionable mutated oncoproteins such as EGFR, KRAS (G12C), ALK, ROS1, and RET in lung cancer and BRAF V600E in melanoma are drivers of tumor initiation that arise early during disease pathogenesis and are almost always clonal and concordant between primary and metastatic disease sites in the cancer subtypes in which drugs targeting these oncoproteins have proven effective.22,24–27 Studies of mutational clonality have also been hindered by technical limitations of older polymerase chain reaction- and mass spectrometry-based companion diagnostics that were incapable of quantitating the clonality of individual genetic alterations. With the adoption of high-depth-of-coverage next-generation sequencing clinical assays, including methods that use cfDNA from plasma, oncologists will increasingly be confronted with patients who have theoretically actionable but sub-clonally mutated oncogenes. Our results suggest that a major contributing factor to the lower success to date with targeted therapies in patients with bladder cancer compared with lung cancer may be the higher likelihood that potentially actionable mutations identified by clinical tumor profiling arise later in disease pathogenesis and are subclonal or discordant among disease sites.10,28
An additional noteworthy finding of our study was that ARID1A mutations, when discordant, were exclusive to the metastatic samples of 16% of patients with an ARID1A mutation detected at either tumor site. ARID1A is an essential regulatory component of SWI/SNF chromatin remodeling complexes and a tissue-specific regulator of chromatin structure and assembly, with prior studies suggesting that ARID1A function is highly cell-type and tissue specific.29–31 Loss of ARID1A has been shown to promote tumor cell invasion and metastatic progression in animal models of hepatocellular and endometrial cancers, and our data suggest it may play a similar role in patients with urothelial cancer. Our finding of frequent discordance in ARID1A mutational status in primary and metastatic samples may have therapeutic implications, as ARID1A has emerged as a potential biomarker of immune checkpoint inhibitor sensitivity and may also be predictive of EZH2 inhibitor sensitivity.32,33
Current guidelines recommend routine clinical testing for FGFR alterations in patients with locally advanced or metastatic urothelial cancer as a guide to erdafitinib therapy.11,34 However, these guidelines do not specify whether primary or metastatic tumor specimens should be utilized. Our analysis of pairs of patient-matched primary and metastatic urothelial carcinomas revealed a high rate of discordance of oncogenic mutations including targets for FDA-approved and investigational therapies. The results suggest that analysis of archival tumor tissue collected from the primary bladder tumor site will fail to detect clinically actionable mutations in as many as 35% of patients with metastatic bladder cancer and that both tissue and cfDNA analyses may be required for some patients to maximize the likelihood of detecting an actionable genomic alteration when present.
Limitations of the study
There was variability in the time between collection of the primary and metastatic sites among patients as well as the details of intervening treatment. However, this heterogeneity reflects real-world practice patterns and, in fact, allowed for hypothesis-generating subset analyses to evaluate the influence of these factors. Additionally, the current analysis focused exclusively on alterations detectable through analysis of DNA. Given the frequency and co-occurrence of mutations in genes that regulate chromatin state in bladder tumors, there are likely significant epigenetically mediated differences in gene regulation and tumor microenvironmental differences between primary and metastatic disease sites that contribute to disease progression and influence treatment response. Finally, there were differences among patients in the interval between the metastatic biopsy and collection of cfDNA, and thus mutational discordance in some cases may have been the result of ongoing mutagenesis or clonal selection driven by intervening treatment. Nevertheless, the current study represents the largest series to date of sequenced metastatic bladder cancer specimens, providing insights that may lead to clinically useful prognostic markers of disease progression and targets for future drug development.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, David B. Solit (solitd@mskcc.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The raw sequencing data for the MSK-IMPACT cohort are protected and are not broadly available due to privacy laws. The clinical and processed genomic data for the MSK-IMPACT cohort are publicly available through the cBioPortal for Cancer Genomics.35 WES data has been deposited in dbGAP. Link and accession number are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Biological samples | ||
|
| ||
| Human tumor and matched normal samples (blood) | This paper | N/A |
| Human cell-free DNA | This paper | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| xGen Exome Research Panel | IDT | v1.0 |
| HiSeq 3000/4000 SBS Kit | illumina | Cat# FC-410-1001 |
| HiSeq Rapid SBS Kit v2 | illumina | Cat# FC-402-4022 |
| NovaSeq 6000 v1 S2 Reagent Kit | illumina | Cat# 20012862 |
|
| ||
| Deposited data | ||
|
| ||
| Somatic mutations and clinical data | This paper | https://cbioportal.mskcc.org/study/summary?id=paired_bladder_2022 |
| Whole Genome Sequencing | This paper | dbGAP:phs001783 |
| Human reference genome NCBI build 37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
|
| ||
| Software and algorithms | ||
|
| ||
| cBioPortal | Cerami et al., 201235 | https://www.cbioportal.org/ |
| OncoKB | Chakravarty et al., 201716 | https://github.com/oncokb/oncokb |
| TEMPO | Center for Molecular Oncology, MSKCC | https://github.com/mskcc/tempo |
| MSK-ACCESS pipeline | Rose Brannon et al., 202136 | https://github.com/mskcc/ACCESS-Pipeline |
| Burrows-Wheeler Aligner | Li and Durbin, 200937 | https://sourceforge.net/projects/bio-bwa/files/ |
| Genome Analysis Toolkit | DePristo et al., 201138 | https://gatk.broadinstitute.org/hc/en-us |
| MSIsensor | Middha, et al., 201739 | https://github.com/ding-lab/msisensor |
| Strelka2 | Kim et al., 201740 | https://github.com/Illumina/strelka |
| Manta | Chen et al., 201641 | https://github.com/Illumina/manta |
| Delly | Rausch et al., 201242 | https://github.com/dellytools/delly |
| Samtools | Li et al., 200943 | http://samtools.sourceforge.net/ |
| R version 3.6.2 | R CRAN | https://www.r-project.org/ |
| Gtsummary | Sjoberg et al., 202144 | https://cran.r-project.org/package=gtsummary |
| Clinfun | CRAN Repository | https://cran.r-project.org/package=clinfun |
| Survminer | CRAN Repository | https://cran.r-project.org/package=survminer |
| Ape | Paradis and Schliep, 201945 | https://cran.r-project.org/package=ape |
| Mutation Signature | Alexandrov et al., 201346 | https://github.com/mskcc/mutation-signatures |
| FACETS | Shen and Seshan, 201647 | https://github.com/mskcc/facets |
| Jalview | Waterhouse et al., 200948 | https://www.jalview.org/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient eligibility
Following institutional review board approval (ClinicalTrials.gov, NCT01775072), demographic, pathologic, genomic, and treatment data were collected on patients with localized or metastatic bladder urothelial carcinoma evaluated at Memorial Sloan Kettering Cancer Center (MSK) from 1999 to 2021. Blood was collected as a source of germline DNA. All tumor specimens were examined by a board-certified genitourinary pathologist (H.A-A.) to confirm the histologic diagnosis, to assess for evidence of divergent differentiation and macro-dissected to enrich tumor content. Tumors with pure variant histology were excluded. However, tumors with a mixture of urothelial and divergent differentiation were included. Clinical information including patient demographics, smoking, treatment history and survival outcomes were extracted from electronic medical records and are summarized in Table 1.
Matched primary-metastasis pairs
Patients enrolled in the MSK-IMPACT sequencing cohort who had undergone biopsy or resection and tumor genomic analysis of both primary and metastatic tumor samples were identified. Additional patients were included when corresponding primary-metastasis paired samples (either primary or metastatic) sufficient for genomic analysis could be obtained from the MSK institutional tumor bank or from an outside institution. Metastases included both synchronous and metachronous lesions. Locoregional recurrences deemed to be secondary to positive surgical margins or tumor spillage were excluded.
METHOD DETAILS
Next-generation sequencing
Targeted deep sequencing was performed using the Memorial Sloan Kettering Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) assay on DNA derived from patient-matched tumor and blood (as a source of germline DNA). MSK-IMPACT is a hybridization, capture-based next-generation sequencing platform that detects somatic mutations, copy number alterations, and structural variants in the coding regions and select noncoding regions of up to 505 cancer-associated genes, depending on the assay version.49,50 Oncogenic and likely oncogenic alterations were identified using the OncoKB knowledgebase.16 Specimens that were processed but lacked any somatic alterations and had an estimated tumor purity <20% were excluded. Mutational concordance rate of primary-metastatic pairs was calculated based on altered pairs only.
Whole Exome Sequencing (WES) was performed from newly extracted DNA or through re-capture of existing MSK-IMPACT sequencing libraries using the xGen Exome Research Panel v1.0 (IDT). For WES re-capture, PCR amplification for 8 cycles was performed on post-capture MSK-IMPACT libraries. Samples were analyzed on either a HiSeq 4000 or HiSeq 2500 in rapid mode in a 100bp or 125bp paired-end run using the HiSeq 3000/4000 SBS Kit or HiSeq Rapid SBS Kit v2 (Illumina) or on a NovaSeq 6000 in a 100bp paired-end run using the NovaSeq 6000 SBS v1 Kit and an S2 flow cell (Illumina).38 The median WES target coverage for tumor and normal samples was 151X and 86X, respectively.
WES sequencing data was analyzed using the TEMPO (Time-Efficient Mutational Profiling in Oncology; https://github.com/mskcc/tempo) pipeline. Briefly, demultiplexed FASTQ files were converted to BAM files and aligned to the b37 assembly of the human reference genome.37,43 Somatic mutations (point mutations and small insertions and deletions) were identified using MuTect251 and Strelka2.40 Structural variants were identified using Manta41 and Delly.42 Tumor mutation burden (TMB) was calculated as the number of non-synonymous exonic mutations per megabase of the target capture. MSI was assessed genomically using MSIsensor39 with MSI status defined based on MSIsensor scores: <3, microsatellite stable (MSS); ≥3 and <10, microsatellite indeterminate (MSI-I); and ≥10, MSI-high (MSI-H). Mutational signatures were inferred from single-nucleotide mutations for all sequenced samples with five or more such mutations. The fraction of mutations attributable to each of 30 known mutational signatures46 was determined using a basin-hopping algorithm (https://github.com/mskcc/mutation-signatures), which assigns a weight to each of the 30 signatures based on the distribution of six types of single-nucleotide substitutions (C to A, G, or T; T to A, C, or G) and their trinucleotide context in a sample. Signatures with a known common source of somatic hypermutation were considered together e.g. signatures 6, 14, 15, 20, 21 and 26 as mismatch-repair deficiency/MSI-associated.
Copy number analysis was performed using FACETS (https://github.com/mskcc/facets) and processed using facets-suite (https://github.com/mskcc/facets-suite). 47 In cases where FACETS failed to estimate tumor purity, mutation-based purity was calculated externally as the median variant allele frequency of the mutations located in diploid regions. Clonality was estimated in each affected tumor specimen (fraction of tumor cells harboring the indicated mutation) as described previously.52,53 Briefly, we inferred the cancer cell fraction (CCF) for all mutations using the mutant allele fraction, locus-specific read coverage, and an analytical estimate of tumor purity using a binomial distribution and maximum likelihood estimation to generate posterior probabilities. For clonality analysis, a somatic mutation was considered clonal if the CCF was greater than 0.8, or greater than 0.7 while the upper bound of the 95% CI of its CCF was greater than 0.9. Additionally, if the clonal fraction of the segment harboring the mutation loci was less than 0.6 times purity then we considered the clonality call to be indeterminable. Mutation phylogeny between primary and metastatic tumors were inferred using the union of somatic mutations called in any of the paired samples and performed with R package ape.45
Cell Free DNA (cfDNA) analysis was performed using the Memorial Sloan Kettering Analysis of Circulating cfDNA to Evaluate Somatic Status (MSK-ACCESS) assay, an ultrasensitive, capture-based liquid biopsy test for the detection of somatic alterations in select coding and noncoding regions of 129 key cancer-associated genes.36,54 Concordance of mutations between primary, metastasis and cfDNA sample pairs was performed on the shared regions covered by the MSK-IMPACT and MSK-ACCESS assays. cfDNA samples without any mutations were excluded from analysis. In those patients with multiple samples, the samples closest in time were selected. After assessing concordance between primary, metastasis and cfDNA sample pairs, the cohort was expanded to include any patient with a primary/metastatic bladder cancer tumor sequenced and a cfDNA sample. Paralogy analysis of FGFR1/2/3 was performed using Jalview.48 Alignment conservation annotation was calculated to characterize the physico-chemical properties seen at each position in a multiple protein sequence alignment.55
QUALIFICATION AND STATISTICAL ANALYSIS
Chi-square test was used to define statistical significance of difference between clinical and demographic categorical variables and Wilcoxon rank-sum test for continuous variable comparisons. In a comparison of cancer-related genes by stage, a Chi-square test was utilized, the results were adjusted for the false discovery rate with q-value <0.05 considered as statistically significant. Analyses were conducted using R version 3.6.2 (http://www.R-project.org/). R packages used included gtsummary44 (https://CRAN.R-project.org/package=gtsummary), clinfun (https://CRAN.R-project.org/package=clinfun) and survminer (https://CRAN.R-project.org/package=survminer).
Supplementary Material
Highlights.
Mutations of chromatin-modifying genes vary between grade/stage in bladder cancer
Characterized by early branched evolution and lesion-to-lesion genomic heterogeneity
Primary and metastatic sites have 23% discordance in actionable genomic alterations
Plasma cfDNA identifies targetable genes not detected in tumor specimens
ACKNOWLEDGMENTS
The authors thank members of the Memorial Sloan Kettering Kravis Center for Molecular Oncology, Integrated Genomics Organization and Diagnostic Molecular Pathology for their work establishing and maintaining the MSK-IMPACT and whole-exome recapture datasets. This work was supported by the Mark Foundation; the Sidney Kimmel Center for Prostate and Urologic Cancers; the Michael and Zena Wiener for Therapeutics Program in Bladder Cancer; Pin Down Bladder Cancer; the Kaufthal Fund; Cycle for Survival; the Marie-Josee and Henry R. Kravis Center for Molecular Oncology; NIH/NCATS grant number UL1-TR002384; NIH grants R01-CA233899 and P01-CA221757; and SPORE in Bladder Cancer P50-CA221745 and P30-CA008748.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
D.B.S. has served as a consultant for/received honorarium from Pfizer, Loxo/Lilly Oncology, Vividion Therapeutics, Scorpion Therapeutics, Fore Therapeutics, FOG Pharma, Rain Therapeutics, and BridgeBio. H.A.A.-A. provided consultation to AstraZeneca, Janssen Biotech, Bristol-Myers-Squibb, and Paige.ai. J.E.R. has served as a consultant for Astellas, Seagen, Merck, Roche, Genentech, AstraZeneca, Janssen Biotech, Gilead, Pfizer, EMD-Serono, Mirati, Boehringer Ingelheim, Pharmacyclis, GSK, Infinity, Tyra BioSciences, Bayer, and QED Therapeutics and received honoraria from EMD-Serono. M.B. has served as a consultant for Eli Lilly and PetDx and has a patent pending on “Systems and Methods for Detecting Cancer Via cfDNA Screening.” S.D. has served as a consultant for Roche.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111859.
REFERENCES
- 1.Taniguchi H, Sen T, and Rudin CM (2020). Targeted therapies and biomarkers in small cell lung cancer. Front. Oncol. 10, 741. 10.3389/fonc.2020.00741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilson TR, Udyavar AR, Chang CW, Spoerke JM, Aimi J, Savage HM, Daemen A, O’Shaughnessy JA, Bourgon R, and Lackner MR (2019). Genomic alterations associated with recurrence and TNBC subtype in high-risk early breast cancers. Mol. Cancer Res. 17, 97–108. 10.1158/1541-7786.MCR-18-0619. [DOI] [PubMed] [Google Scholar]
- 3.Cairns J (1975). Mutation selection and the natural history of cancer. Nature 255, 197–200. 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]
- 4.Nowell PC (1976). The clonal evolution of tumor cell populations. Science 194, 23–28. 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 5.Greaves M, and Maley CC (2012). Clonal evolution in cancer. Nature 481, 306–313. 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yates LR, and Campbell PJ (2012). Evolution of the cancer genome. Nat. Rev. Genet. 13, 795–806. 10.1038/nrg3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, Hinoue T, Laird PW, Hoadley KA, Akbani R, et al. (2017). Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 171, 540–556.e25. 10.1016/j.cell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakauma-González JA, Rijnders M, van Riet J, van der Heijden MS, Voortman J, Cuppen E, Mehra N, van Wilpe S, Oosting SF, Rijstenberg LL, et al. (2022). Comprehensive molecular characterization reveals genomic and transcriptomic subtypes of metastatic urothelial carcinoma. Eur. Urol. 81, 331–336. 10.1016/j.eururo.2022.01.026. [DOI] [PubMed] [Google Scholar]
- 9.Winters BR, De Sarkar N, Arora S, Bolouri H, Jana S, Vakar-Lopez F, Cheng HH, Schweizer MT, Yu EY, Grivas P, et al. (2019). Genomic distinctions between metastatic lower and upper tract urothelial carcinoma revealed through rapid autopsy. JCI Insight 5, e128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faltas BM, Prandi D, Tagawa ST, Molina AM, Nanus DM, sternberg C, Rosenberg J, Mosquera JM, Robinson B, Elemento O, et al. (2016). Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nat. Genet. 48, 1490–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loriot Y, Necchi A, Siefker-Radtke AO, Garcia-Donas J, Huddart R, Burgess E, Fleming M, Rezazadeh A, Mellado B, Varlamov S, et al. (2019). Erdafitinib in locally advanced or metastatic urothelial carcinoma. N. Engl. J. Med. 381, 1594–1595. [DOI] [PubMed] [Google Scholar]
- 12.Vandekerkhove G, Lavoie JM, Annala M, Murtha AJ, Sundahl N, Walz S, Sano T, Taavitsainen S, Ritch E, Fazli L, et al. (2021). Plasma ctDNA is a tumor tissue surrogate and enables clinical-genomic stratification of metastatic bladder cancer. Nat. Commun. 12, 184. 10.1038/s41467-020-20493-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang MT, Penson A, Desai NB, Socci ND, Shen R, Seshan VE, Kundra R, Abeshouse A, Viale A, Cha EK, et al. (2018). Small-cell carcinomas of the bladder and lung are characterized by a convergent but distinct pathogenesis. Clin. Cancer Res. 24, 1965–1973. 10.1158/1078-0432.CCR-17-2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li R, Du Y, Chen Z, Xu D, Lin T, Jin S, Wang G, Liu Z, Lu M, Chen X, et al. (2020). Macroscopic somatic clonal expansion in morphologically normal human urothelium. Science 370, 82–89. [DOI] [PubMed] [Google Scholar]
- 15.Lawson ARJ, Abascal F, Coorens THH, Hooks Y, O’Neill L, Latimer C, Raine K, Sanders MA, Warren AY, Mahbubani KTA, et al. (2020). Extensive Heterogeneity in somatic mutation and selection in the human bladder. Science 370, 75–82. [DOI] [PubMed] [Google Scholar]
- 16.Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, Nissan MH, et al. (2017). On-cokb: a precision oncology knowledge base. JCO Precis. Oncol. 2017. 10.1200/PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoza K, Himeno R, Amano S, Kobashigawa Y, Amemiya S, Fukuda N, Kumeta H, Morioka H, and Inagaki F (2016). Biophysical characterization of drug-resistant mutants of fibroblast growth factor receptor 1. Gene Cell. 21, 1049–1058. 10.1111/gtc.12405. [DOI] [PubMed] [Google Scholar]
- 18.Goyal L, Saha SK, Liu LY, Siravegna G, Leshchiner I, Ahronian LG, Lennerz JK, Vu P, Deshpande V, Kambadakone A, et al. (2017). Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 7, 252–263. 10.1158/2159-8290.CD-16-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pouessel D, Neuzillet Y, Mertens LS, van der Heijden MS, de Jong J, Sanders J, Peters D, Leroy K, Manceau A, Maille P, et al. (2016). Tumor heterogeneity of fibroblast growth factor receptor 3 (FGFR3) mutations in invasive bladder cancer: implications for perioperative anti-FGFR3 treatment. Ann. Oncol. 27, 1311–1316. 10.1093/annonc/mdw170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo J, Solimini NL, and Elledge SJ (2009). Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823–837. 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, and Sawyers CL (2001). Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293, 876–880. 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 22.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. (2011). RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480, 387–390. 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al. (2014). Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 4, 80–93. 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al. (2007). Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131, 1190–1203. 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 25.Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, Italiano A, Schuler M, Borghaei H, Barlesi F, et al. (2021). Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med. 384, 2371–2381. 10.1056/NEJMoa2103695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, Asaka R, Hamanaka W, Ninomiya H, Uehara H, et al. (2012). RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 18, 378–381. 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 27.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, and Chapman PB (2010). Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 363, 809–819. 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaufler D, Ast DF, Tumbrink HL, Abedpour N, Maas L, Schwäbe AE, Spille I, Lennartz S, Fassunke J, Aldea M, et al. (2021). Clonal dynamics of BRAF-driven drug resistance in EGFR-mutant lung cancer. NPJ Precis. Oncol. 5, 102. 10.1038/s41698-021-00241-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mashtalir N, D’Avino AR, Michel BC, Luo J, Pan J, Otto JE, Zullow HJ, McKenzie ZM, Kubiak RL, St. Pierre R, et al. (2018). Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell 175, 1272–1288.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun X, Wang SC, Wei Y, Luo X, Jia Y, Li L, Gopal P, Zhu M, Nassour I, Chuang JC, et al. (2017). Arid1a has context-dependent oncogenic and tumor suppressor functions in liver cancer. Cancer Cell 32, 574–589.e6. 10.1016/j.ccell.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson MR, Reske JJ, Holladay J, Wilber GE, Rhodes M, Koeman J, Adams M, Johnson B, Su R-W, Joshi NR, et al. (2019). ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat. Commun. 10, 3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okamura R, Kato S, Lee S, Jimenez RE, Sicklick JK, and Kurzrock R (2020). ARID1A alterations function as a biomarker for longer progression-free survival after anti-PD-1/PD-L1 immunotherapy. J. Immunother. Cancer 8, e000438. 10.1136/jitc-2019-000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rehman H, Chandrashekar DS, Balabhadrapatruni C, Nepal S, Balasubramanya SAH, Shelton AK, Skinner KR, Ma AH, Rao T, Agarwal S, et al. (2022). ARID1A-deficient bladder cancer is dependent on PI3K signaling and sensitive to EZH2 and PI3K inhibitors. JCI Insight 7, e155899. 10.1172/jci.insight.155899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Network NCC (2022). NCCN Clinical Practice Guidelines in Oncology: Bladder Cancer Version 2.2022 (National Comprehensive Cancer Network; ). 2.2022. [DOI] [PubMed] [Google Scholar]
- 35.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rose Brannon A, Jayakumaran G, Diosdado M, Patel J, Razumova A, Hu Y, Meng F, Haque M, Sadowska J, Murphy BJ, et al. (2021). Enhanced specificity of clinical high-sensitivity tumor mutation profiling in cell-free DNA via paired normal sequencing using MSK-ACCESS. Nat. Commun. 12, 3770. 10.1038/s41467-021-24109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Middha S, Zhang L, Nafa K, Jayakumaran G, Wong D, Kim HR, Sadowska J, Berger MF, Delair DF, Shia J, et al. (2017). Reliable pan-cancer microsatellite instability assessment by using targeted next-generation sequencing data. JCO Precis. Oncol. 2017, 1–17. 10.1200/po.17.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, Chen X, Beyter D, Krusche P, and Saunders CT (2017). Strelka2: fast and accurate variant calling for clinical sequencing applications. Preprint at bioRxiv. 10.1101/192872. [DOI] [PubMed] [Google Scholar]
- 41.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, and Saunders CT (2016). Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 42.Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, and Korbel JO (2012). DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339. 10.1093/bioinformatics/bats378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sjoberg D, Whiting K, Curry M, Lavery J, and Larmarange J (2021). Reproducible summary tables with the gtsummary package. R J. 13, 570–580. 10.32614/RJ-2021-053. [DOI] [Google Scholar]
- 45.Paradis E, and Schliep K (2019). Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. 10.1093/bioinformatics/bty633. [DOI] [PubMed] [Google Scholar]
- 46.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen R, and Seshan VE (2016). FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Waterhouse AM, Procter JB, Martin DMA, Clamp M, and Barton GJ (2009). Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, et al. (2015). Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT). J. Mol. Diagn. 17, 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, et al. (2017). Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10, 000 patients. Nat. Med. 23, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benjamin D, Sato T, Cibulskis K, Getz G, Stewart C, and Lichtenstein L (2019). Calling somatic SNVs and indels with Mutect2. Preprint at bioRxiv. 10.1101/861054. [DOI] [Google Scholar]
- 52.Bielski CM, Zehir A, Penson AV, Donoghue MTA, Chatila W, Armenia J, Chang MT, Schram AM, Jonsson P, Bandlamudi C, et al. (2018). Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 50, 1189–1195. 10.1038/s41588-018-0165-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, and Swanton C (2015). Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 7, 283ra54. 10.1126/scitranslmed.aaa1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsui DWY, Cheng ML, Shady M, Yang JL, Stephens D, Won H, Srinivasan P, Huberman K, Meng F, Jing X, et al. (2021). Tumor fraction-guided cell-free DNA profiling in metastatic solid tumor patients. Genome Med. 13, 96. 10.1186/s13073-021-00898-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Livingstone CD, and Barton GJ (1993). Protein sequence alignments: a strategy for the hierarchical analysis of residue conservation. Comput. Appl. Biosci. 9, 745–756. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequencing data for the MSK-IMPACT cohort are protected and are not broadly available due to privacy laws. The clinical and processed genomic data for the MSK-IMPACT cohort are publicly available through the cBioPortal for Cancer Genomics.35 WES data has been deposited in dbGAP. Link and accession number are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Biological samples | ||
|
| ||
| Human tumor and matched normal samples (blood) | This paper | N/A |
| Human cell-free DNA | This paper | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| xGen Exome Research Panel | IDT | v1.0 |
| HiSeq 3000/4000 SBS Kit | illumina | Cat# FC-410-1001 |
| HiSeq Rapid SBS Kit v2 | illumina | Cat# FC-402-4022 |
| NovaSeq 6000 v1 S2 Reagent Kit | illumina | Cat# 20012862 |
|
| ||
| Deposited data | ||
|
| ||
| Somatic mutations and clinical data | This paper | https://cbioportal.mskcc.org/study/summary?id=paired_bladder_2022 |
| Whole Genome Sequencing | This paper | dbGAP:phs001783 |
| Human reference genome NCBI build 37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
|
| ||
| Software and algorithms | ||
|
| ||
| cBioPortal | Cerami et al., 201235 | https://www.cbioportal.org/ |
| OncoKB | Chakravarty et al., 201716 | https://github.com/oncokb/oncokb |
| TEMPO | Center for Molecular Oncology, MSKCC | https://github.com/mskcc/tempo |
| MSK-ACCESS pipeline | Rose Brannon et al., 202136 | https://github.com/mskcc/ACCESS-Pipeline |
| Burrows-Wheeler Aligner | Li and Durbin, 200937 | https://sourceforge.net/projects/bio-bwa/files/ |
| Genome Analysis Toolkit | DePristo et al., 201138 | https://gatk.broadinstitute.org/hc/en-us |
| MSIsensor | Middha, et al., 201739 | https://github.com/ding-lab/msisensor |
| Strelka2 | Kim et al., 201740 | https://github.com/Illumina/strelka |
| Manta | Chen et al., 201641 | https://github.com/Illumina/manta |
| Delly | Rausch et al., 201242 | https://github.com/dellytools/delly |
| Samtools | Li et al., 200943 | http://samtools.sourceforge.net/ |
| R version 3.6.2 | R CRAN | https://www.r-project.org/ |
| Gtsummary | Sjoberg et al., 202144 | https://cran.r-project.org/package=gtsummary |
| Clinfun | CRAN Repository | https://cran.r-project.org/package=clinfun |
| Survminer | CRAN Repository | https://cran.r-project.org/package=survminer |
| Ape | Paradis and Schliep, 201945 | https://cran.r-project.org/package=ape |
| Mutation Signature | Alexandrov et al., 201346 | https://github.com/mskcc/mutation-signatures |
| FACETS | Shen and Seshan, 201647 | https://github.com/mskcc/facets |
| Jalview | Waterhouse et al., 200948 | https://www.jalview.org/ |