

Abstract
Prostate cancer (PCa), the second leading cause of death in American men, includes distinct genetic subtypes with distinct therapeutic vulnerabilities. The DACH1 gene encodes a winged helix/Forkhead DNA-binding protein that competes for binding to FOXM1 sites. Herein, DACH1 gene deletion within the 13q21.31-q21.33 region occurs in up to 18% of human PCa and was associated with increased AR activity and poor prognosis. In prostate OncoMice, prostate-specific deletion of the Dach1 gene enhanced prostatic intraepithelial neoplasia (PIN), and was associated with increased TGFβ activity and DNA damage. Reduced Dach1 increased DNA damage in response to genotoxic stresses. DACH1 was recruited to sites of DNA damage, augmenting recruitment of Ku70/Ku80. Reduced Dach1 expression was associated with increased homology directed repair and resistance to PARP inhibitors and TGFβ kinase inhibitors. Reduced Dach1 expression may define a subclass of PCa that warrants specific therapies.
Keywords: DACH1, prostate cancer, DNA repair
INTRODUCTION
Prostate cancer (PCa), the second leading cause of death in American men, is a genetically heterogeneous disease, likely reflecting distinct genetic drivers [1]. While substratification of PCa into genetic subtypes forms the basis of rational therapy for PCa, current diagnostic tools fail to reliably distinguish aggressive tumors from non-aggressive ones in order to predict therapeutic response [2], and the lack of markers to stratify PCa cases into low- and high-risk groups results in overtreatment of 20–42% of patients [3]. New biomarkers are urgently needed for therapeutic stratification. A better molecular understanding of the disease is necessary to develop novel targeted therapies for metastatic PCa.
Defects in DNA damage repair (DDR) pathways are a hallmark of human cancer, with somatic events present in up to 20% of primary PCa [1], including BRCA2 [4], which participates in homology-directed DNA repair (HR). Defective HR due to defects in BRCA1 or BRCA2 has led to the use of poly(adenosine diphosphate(ADP)-ribose) polymerase (PARP) inhibitors in prostate cancer therapy [5]. Target-region sequencing, array-based gene expression, copy number variation (CNV) analysis, and whole-genome sequencing of tumors have reported several PCa-related genomic alterations, including copy number gains of 8q, and copy number losses of 3p, 8p, 10q, 13q, and 17p [6–8]. In PCa, known genetic drivers for tumor initiation include PTEN and NKX3.1 deletions, rearrangements/fusions of multiple genes (including TMPRSS2 and the oncogenic ETS transcription factor, ERG) [8], and predisposing genetic factors (including germline DNA-repair gene mutations) [9], (reviewed in [1]). Loss of heterozygosity or deletion also occurs within the 13q21 region in PCa, may include BRCA2, and is associated with high-grade prostate cancer [10–12].
In addition to genetic drivers of PCa, hyperactivity of the androgen receptor, inflammation [13], TGFβ activity, and DNA damage contribute to tumor progression [14]. Transforming growth factor β (TGFβ) has tumor-inhibitory activity in the early stages of prostate tumorigenesis but promotes migration, epithelial-mesenchymal transition (EMT), invasion, and metastasis in late-stage disease [15, 16]. DNA-dependent protein kinase (DNA-PK) is a serine/threonine kinase that, with Ku70, Ku80, XRCC4, ligase IV, and Artemis, drives non-homologous end joining (NHEJ) repair [17]. The heterodimer of Ku70 and Ku80 binds to double-strand breaks (DSBs) and recruits and activates the catalytic subunit DNA-PKC, which in turn recruits the XRCC4/ligase IV heterodimer that is responsible for rejoining the break. Inactivation of the Ku70 or Ku80 genes in mice leads to hypersensitivity to radiation, malignant transformation [18, 19], and an associated increase in HR [20, 21], as binding to both ends of a two-ended DSB stabilizes contacts between Ku heterodimers, tethering the DNA ends and preventing access by the HDR machinery[21].
The Drosophila Dac gene was initially cloned as a dominant inhibitor of the hyperactive EGFR, Ellipse [22]. The human Dachshund1 (DACH1) gene encodes a DNA-binding protein similar to the winged helix/Forkhead subgroup of the helix-turn-helix family. Cyclic amplification and selection of target (CAST), together with ChIP, identified DACH1 DNA binding sequences that resemble Forkhead binding sites [23]. Furthermore, expression of the DACH1 gene is reported to be reduced in PCa, and DACH1 overexpression inhibited PCa cell line growth [24].
Given the importance of identifying molecular genetic events governing PCa onset and progression, in the work reported here we investigated the role of the Dach1 gene in PCa progression in transgenic mice. We identify a novel role for DACH1 in maintaining genomic stability through the regulation of DNA repair. A reduced abundance of DACH1 in human prostate cancer, associated with either gene deletion or promoter DNA methylation, correlated with poor outcomes. In prostate OncoMice, prostate-specific deletion of the Dach1 gene enhanced prostatic intraepithelial neoplasia (PIN), and was associated with increased TGFβ activity and DNA damage. Mechanistically, we show that DACH1 is recruited to sites of DNA damage, augmenting recruitment of Ku70/Ku80, restraining homologous end joining (HR). Reduced Dach1 expression was associated with resistance to PARP inhibitors and TGFβ kinase inhibitors. Our results suggest reduced Dach1 expression may define a subclass of PCa that warrants specific therapies.
RESULTS
The DACH1 gene is frequently deleted in human prostate cancer (PCa).
In order to determine whether the DACH1 gene is deleted in PCa we interrogated the genomic sequencing analysis from four cBioPortal cohorts (Mich/MCTP, N=59; Broad/Cornell, N=57; SU2C/PCF (2015), N=150; FHCRC, N=54). Genomic deletion at 13q21 (~18 Mb) has been associated with aggressive prostate cancer [11, 12], although specific genetic drivers were not identified. In recent studies of PCa in a Chinese population, PCDH9 was noted to be one of the most frequently deleted genes in 13q21.31-q21.33 (~11 Mb) [25]. In TCGA data for 492 primary tumors (firebrowse.org), we identified DACH1 as a frequently deleted gene within this region (Fig. 1A, B). GISTIC2 analysis identified 51 (10%) ‘deep’ (i.e., homozygous) deletions of DACH1. In six distinct cohorts, homozygous deletions of DACH1 were identified in between 3 and 18% of prostate cancers (Fig. 1C, Fig. S1). Median z-scores for DACH1 RNA-seq were progressively larger for DACH1 homozygously deleted, heterozygously deleted, and diploid, indicating that somatic copy number can influence DACH1 expression (Fig. S3A). In two cohorts for which CNV data was available for primary and metastatic sites, we identified the relative prevalence of deep and shallow (heterozygous) genomic deletions in both sites (Fig. 1C), finding DACH1 homozygous deletions more frequent in the metastatic site than in the primary tumors (Mich: 9.1% vs. 20%, N=59; FHCRC: 2.3% vs. 9.3%, N= 54). The prevalence of DACH1 heterozygous deletions was higher in the metastatic lesions than in primary tumors within a given cohort for three of six cohorts (Mich: 27.3% vs. 36%, N=59; FHCRC: 8.5% vs. 48.8%, N=54, respectively) (Fig. 1C).
Figure 1. The DACH1 gene is deleted in human prostate cancer.

(A). Landscape of somatic copy number alteration (SCNA) [26] in the TCGA PCa cohort (firebrowse.org) shown as profiles of GISTIC2 G-scores [76] for GRCh37/hg19 SNP6-based data for n=492 primary tumors. (B) as from (A), for chromosome 13, vertical lines indicate positions of candidate tumor suppressor genes (TSGs) PCDH9-DACH1-KLF5. The grey rectangle at the left indicates that TCGA/firebrowse.org reported no GISTIC2 data for the 13p arm. (C). Analysis of DACH1 gene status in human prostate cancer (PCa) from two cBioPortal cohorts (for which copy number data was available for primary and the metstatic sites) shows DACH1 homozygous deletions (dark blue) in 2.3% to 20% of patients and a higher frequency of heterozygous deletions (light blue). (D). DACH1 copy number and overall survival data was determined by combining three cBioPortal cohorts (TCGA PanCancer Atlas 2018, SU2C 2019, and MCTP) (N=667 tumor samples). Kaplan-Meier plot for overall survival is shown using a copy number threshold of −2 to segregate the data into samples with altered vs. unaltered DACH1 [26]. Patients with homozygous DACH1 deletions (“Altered” in the figure caption) showed reduced overall survival (log-rank P<9.3 ×10−3) (“Altered” 50/17 vs. “Unaltered” 617/114). The numbers (50/17, 617/114) indicate the number of samples in the group (e.g., Altered = 50, Unaltered = 617) and the number of events, i.e., for overall survival, deaths (e.g., Altered = 17, Unaltered = 114). (E). The range of DNA methylation beta values for probe cg13726218 (reported for DACH1 at cBioPortal) and RNA-Seq by Expectation Maximization (RSEM) DACH1 normalized expression (n=333 cohort) [26]. The (negative) Spearman correlation between beta and RSEM DACH1 normalized expression was rho=−0.41, FDR=7.6×10−14. (F). Kaplan-Meier plot data from Gerhausen et al.[27] showing low DACH1 gene expression (expressed as a z-score with a z-score threshold of −1.25 ) is significantly correlated with earlier biochemical recurrence (BCR). (log rank p value = 4.7×10−4 (n=79).
To assess the relationship between DACH1 homozygous deletions and outcomes, we queried copy number and overall survival data for three cBioPortal cohorts (TCGA PanCancer Atlas 2018, SU2C 2019, and MCTP) (n=667 tumor samples). Using a somatic copy number threshold of −2 to segregate the data into samples with altered vs. unaltered DACH1, we generated a Kaplan-Meier plot for overall survival (Fig. 1D) using R’s survival v3.2–7 package, and calculated median times for altered vs. unaltered DACH1 with a custom R script. The patients with homozygous DACH1 deletions (referred to as “Altered” in the figure caption) had reduced overall survival (medians of 84 vs. 120 months, N=667, log-rank test P=9.3×10−3) (Altered 50/17, Unaltered 617/114) (Fig. 1D).
PTEN-deletion PCa showed infrequent concurrent DACH1 deletions in 5 of the 6 cohorts (not all datasets are independent), with a strong trend toward mutual exclusivity (Fig. S1). In 5 cohorts, the majority of heterozygous DACH1 deletions were associated with RB1 heterozygous deletions (Fig. S2). However, both DACH1 homozygous and heterozygous deletions occur in the absence of RB1 deletions (79/150, 25/444, 13/492), and discordance between RB1 and DACH1 genetic deletions was also observed (Fig. S2C, D).
In addition to gene deletion, the abundance of DACH1 mRNA may be affected by DNA methylation. We assessed DACH1 DNA methylation and RNAseq expression data from firebrowse.org, for the n=333 primary tumor samples and n=43 adjacent tissue normal samples in the cBioPortal TCGA 2015 cohort [26]. Of 29 Illumina 450K probes associated with DACH1, 26 had substantially complete data, and 15 of these probes were within ~3kb of the DACH1 transcriptional start site (TSS) (Fig. S3B). Beta values (β) estimate DNA methylation level using the ratio of intensities between methylated and unmethylated alleles, and are continuous variables between 0 and 1, with 0 being unmethylated and one fully methylated. Probe cg13726218, which cBioPortal associates with DACH1, had both a wide range of beta values (Fig. S3B, probe indicated by a red asterisk) and a strongly-negative Spearman coefficient with DACH1 RNA-Seq expression (rho=−0.41, FDR=7.6×10−14) (Fig. 1E). The additional fourteen of the 15 probes in proximity to the TSS had narrow ranges of low beta values in primary tumor samples (Fig. S3B). Median beta values were similar in primary tumors and adjacent tissue normals for 25 of the 26 probes (Fig. S3C).
The relationship between cg13726218 DNA methylation and gene expression could be expressed as a linear negative correlation (Fig. S3; n=308 DACH1-diploid samples, Pearson correlation cor = −0.40, p = 2.9×10−13, alternative hypothesis: true correlation ≠ 0). As a nonlinear alternative, a fit Generalized Additive Model (GAM) had a minimized generalized cross-validation (GCV) score of 0.83 and an adjusted R2 of 0.16 (Fig. S3E). Collectively, these results indicate that increasing promoter DNA methylation contributed to a reduction in DACH1 mRNA.
As both gene deletion and increasing DNA methylation reduced DACH1 mRNA abundance, we next investigated the potential role of DACH1 gene expression in outcome using SCNA, interrogating data from Gerhausen et al., [27]. Low DACH1 gene expression (expressed as a z-score < −1.25 ) was significantly correlated with earlier biochemical recurrence (BCR, log-rank p-value = 4.7×10−4, n=79, Fig. 1F). We further assessed RSEM expression for DACH1 (TCGA cohort [26], n=290 of 333 samples). Defining “altered DACH1” samples as those in which DACH1’s RSEM z-score was below −2.0, and using PanCancer outcomes [28], we found a Kaplan-Meier log-rank P value of 0.028 for Progression Free Interval (PFI) outcomes (Fig. S3F).
Immunostaining for DACH1 protein was conducted in tissue microarrays (TMAs) of 68 cases in triplicate 1 mm biopsy cores of primary prostate cancer with matched normal/benign tissue. DACH1 was positive in all normal/benign prostate epithelia but was negative in 9 of the 68 cases (13.2%). 89% (8/9) of DACH1-negative cases were of high grade (Gleason 4+3=7 or higher) whereas of DACH1-positive cases only 54% (32/59) were high grade (likelihood-ratio test p=0.034). Representative images of DACH1-positive and -negative cases of prostate cancer are shown in Fig. S4.
DACH1-deletion tumors constitute a prostate cancer subtype.
Using DACH1 copy number data for samples from The Cancer Genome Atlas (TCGA) [26], we identified a group of 29 of 333 tumors with DACH1 homozygous deletion (DACH1 subtype, Fig. 2A). In a prior study, androgen receptor (AR) activity varied widely and in a subtype-specific manner, with SPOP and FOXA1 mutant tumors having the highest levels of AR-induced transcripts [26]. Comparison of AR activity levels (using an AR score derived from the expression of AR target genes [26]) showed a significant increase in the DACH1-deletion group as compared to normal samples (P=2×10−5 by t-test) and ERG mutation groups (P=0.003 by t-test) (Fig. 2A). AR activity expressed as a z score for each prostate cancer subtype also showed an increase in the DACH1 deletion group as compared to normal samples (Fig. S5A). However, the levels of AR mRNA and protein were not statistically significantly different (p>0.05) between DACH1 genotypes (Fig. 2B). Immunohistochemical (IHC) staining of human PCa samples for AR, categorized by DACH1 IHC status, also showed no significant difference based on the DACH1 tumor status (Fig. S5B,C, N=64). DACH1 homozygous deletions were enriched for iCluster 2 and 3 [29], mRNA cluster 2 (P=0.0003 by Fisher exact test), SCNA (“more” somatic copy-number alteration, P=0.0004 by Fisher exact test), but not for DNA methylation (Fig. 2C,D).
Figure 2. DACH1 deletion PCa enhances AR signaling.

(A). Interrogation of human PCa gene expression data [26], showing candidate genetic drivers ERG, ETV1/ETV4/FLI1, SPOP, FOXA1, and unknown. Samples with DACH1 homozygous (deep) genetic deletions (29/333) are shown as an additional subtype. The AR score (the average of the AR target gene expression) refers to a group of AR-responsive genes[26], and together with the expression Z-score of the AR target genes, are shown as colorimetric scales. The AR score-based gene names are shown. The androgen receptor (AR) activity, inferred by the induction of AR target genes, was increased in DACH1 homozygous (‘deep’) deletion PCa compared with normal (P=2×10−5 by t-test) and ERG mutation groups (P=0.003 by t-test). (B). AR mRNA and AR protein levels, shown for each DACH1 deletion sample, were not significantly different. (C). The iCluster[29], mRNA cluster, and SCNA (somatic copy-number alteration), and DNA methylation status are shown for the PCa classified by the corresponding gene deletion subtypes. (D). DACH1 homozygous deletions were enriched for iCluster 2 and 3[29], mRNA cluster 2 (P=0.0003 by Fisher exact test, SCNA (“more” somatic copy-number alteration, P=0.0004 by Fisher exact test), but not for DNA methylation.
The mutation frequencies of SPOP and FOXA1 differed (P < 0.05) between DACH1 deleted and diploid before correcting P values for multiple testing (Fig. S6A). After Bonferroni correcting, only SPOP’s mutation frequency differed (Bonferroni adjusted p-value =0.012, Fig. S6A). In an oncoprint that included mutations and copy number, DACH1 alterations co-occurred with those for SPOP (q < 0.001), and ERG’s alterations were mutually exclusive with SPOP’s (q = 0.006) (Fig. S6B-C). BRCA2 deletions were identified in 8% of cases, co-occurring significantly with DACH1 deletions (~1/3 of cases, P value <0.001, Fig. S6D).
Given the association of DACH1 promoter methylation and mRNA abundance in human PCa (Fig. 1E), we determined whether DNA methylation restrained DACH1 expression in cultured PCa cells (Fig. S7). The DNA methylase inhibitor 5-Aza-dC (10 μM) induced DACH1 abundance in LNCaP and C4–2 cells (Fig. S7, lanes 1 vs. 4, and 7 vs. 10). Although 5-Aza-dC slightly increased p53, the 26S proteasome inhibitors MG132 (20 μM) or N-acetyl-L-leucyl-L-leucyl-L-nor leucinal (LLNL) induced a robust increase in p53 abundance (Fig. S7).
Dach1 deletion in the prostate promotes prostatic intraepithelial neoplasia (PIN).
Given that DACH1 and RB1 deletion may co-occur in prostate cancer (Fig. 1B), we sought to determine the functional significance of DACH1 gene deletion, independently of RB1, in the onset and progression of PCa, using a murine model. Dach1 homozygous null mice die at birth. Given this, in order to determine the role of endogenous Dach1 in mediating prostate transformation, multigenic mice were generated by crossing conditional Dach1 gene deletion mice [30] with prostate-specific Cre transgenics, using Probasin-Cre4 (Pb-Cre4) transgenic mice, which express Cre in both the basal and luminal prostatic epithelia, then further intercrossing with the TRAMP model of prostate cancer (Fig. 3A). The TRAMP model has been extensively characterized, and TRAMP mice develop PIN after 12 weeks. To follow efficient temporal and spatial regulation of Cre recombination in vivo, we intercrossed these bi-transgenic mice with a double-fluorescent Cre reporter mouse that, prior to Cre-mediated excision, expresses membrane-targeted tandem dimer Tomato (mT) and, after excision, expresses membrane-targeted green fluorescent protein (mG). The mice thus cointegrate four transgenes (Fig. 3A). 3T3 cells derived from Dach1−/− mouse showed deletion of the Dach1 gene did not reduce the abundance of pRB (Fig. S8). Rather the abundance of pRBSer807/811 was increased by Dach1 gene deletion when normalized to the protein loading control lamin B1 (Fig. S8). Genomic analysis of tail DNA by PCR confirmed the presence of the transgenes in these mice (Fig. S9A). Cre-induced GFP was expressed in the prostate epithelium (Fig. S9B), whereas no GFP was observed in the absence of Cre recombinase (Fig. S9B).
Figure 3. Prostate-specific Dach1 gene deletion promotes prostate hyperplasia and dysplasia in OncoMice (15 weeks).

(A). Schematic representation of transgenes integrated into mice. (B). Representative immunohistochemistry for Dach1, with data quantitated as mean ± standard error of the mean (SEM) for N=20 (4 separate mice, with 5 views per mouse, in each group). (C). Blinded quantitative histology grading of prostate of multigenic mice at 15 weeks. Data are shown as mean ± SEM for N=15 (5 separate mice, with 3 prostate areas [anterior, ventral, lateral] per mouse) in each group). H&E staining demonstrates the presence of a focal atypical intraductal proliferation in Dach1−/− prostate, compatible with prostatic intraepithelial neoplasia (PIN). Representative immunohistochemistry with results shown as mean ± SEM for Ki-67 (n=20, 4 separate mice for each genotype, 5 views per mouse) (D), Beclin 1 (n=9, 3 separate mice for each genotype, 3 views per mouse) (E); and AR (n=15 for Dach1wt/wt mice, 3 separate mice, 5 views per mouse) (n=12 for Dach1fl/fl mice, 3 separate mice, 2 views for one mouse and 5 views for other two mice) (F). Scale bars, 50 μm. A Student’s t-test was performed for all comparisons.
Dach1 abundance was identified by immunohistochemistry in the prostate epithelial cells, primarily in the luminal compartment, with additional staining of basal cells in the Dach1wt/wt with Probasin-Cre-ROSA26mTmGfl-TRAMP mice (shown Dach1+/+) (Fig. 3B). Prostate epithelial cell Dach1 abundance was abrogated in the Probasin-Cre-Dach1fl/fl ROSA26mT/mG-TRAMP line (shown as Dach1−/−)(Fig. 3B). In order to determine the role of Dach1 in the development of murine PIN, Dach1+/+ and Dach1−/− mice were examined at 15 weeks. Based on H&E staining, the prostates of Dach1−/− mice showed morphological changes characteristic of PIN, including a larger stromal layer with increased cellularity and nuclear atypia (Fig. 3C). The Dach1−/− prostate showed increased prostatic hyperplasia grade (1.8 ± 0.21 vs. 2.5 ± 0.22, P=0.03, t-test, n=15, 5 separate mice with 3 areas [ventral, anterior, lateral prostate] per mouse of each genotype) and increased dysplasia degree (0.86 ± 0.13 vs. 1.26 ± 0.17, P=0.08, t-test, n=15, 5 separate mice with 3 prostate areas, [vental, anterior, lateral] per mouse in each group) (Fig. 3C). Ki-67 expression is a marker of cellular proliferation and is highly correlated with PIN. The Ki-67 staining intensity was increased in the Dach1−/− (Fig. 3D) (2.14×104 ± 0.32×104 vs. 3.92×104 ± 0.27×104, P=0.0001, t-test). Beclin 1, an autophagy marker, was increased by >60% (Fig. 3E, P<0.05, t-test). AR staining, which was primarily nuclear in the epithelial cells and stroma, was increased in the Dach1−/− mouse prostate (Fig. 3F, P=0.004, t-test).
Dach1 restrains a TGFβ gene expression signaling node in the prostate and in PCa cells.
Enrichment analysis of mRNA from ventral prostates from 15-week Dach1+/+ versus Dach1−/− mice identified regulators that were either inhibited or induced by endogenous Dach1 (Fig. 4A–B). TGFβ1 was identified as the gene node with the largest number of target genes repressed by Dach1. Dach1 expression correlated with the inhibition of several additional known pro-proliferative nodes, including c-Myc, ESR1, KRAS, INSR and EGFR (Fig. 4A–B). In addition, phospho-SMAD2 was increased in the prostate epithelium of Dach1−/− PIN (Fig. 4C,D; P<0.008), consistent with a role for endogenous Dach1 in reducing TGFβ activity.
Figure 4. Prostate-specific Dach1 gene deletion in TRAMP mice induces PIN lesions with increased TGFβ activity.

(A-B). Genome-wide expression analysis of TRAMP Dach1+/+ vs. Dach1−/− PIN lesions was analyzed for enrichment of known targets of upstream regulators using Ingenuity Pathway Analysis (IPA) and represented as (A) barplot with calculated by IPA activation Z-score labeled and as (B) bubble plot with size of the bubbles proportional to –log10 p values. (C). IHC was conducted for SMAD activation using SMAD2P, quantitated and shown as (D) mean ± SEM (n=15 for Dach1wt/wt mice, 3 separate mice, 5 views per mouse) (n=10 for Dach1fl/fl mice, 2 separate mice, 5 views per mouse). (E, F). Western blot of either PCa cell lines for the presence of DACH1 or (G) TGFβ-treated (10 ng/ml for 24 hrs) PC3 cells illustrating induction of nuclear vimentin and cytoplasmic cyclin D1. Protein loading controls are β-tubulin (a marker of cytoplasmic proteins) and Lamin B1 (a marker for nuclear protein enrichment). (H). Microarray-based gene expression analysis of PC3 cells stably expressing DACH1, showing restraint of genes mediating TGFβ signaling (shown with blue arrows), including reduction of TGFB2 and TGFBR2 [30].
Induction of an epithelial-mesenchymal transition (EMT) plays a role in both PCa metastatic progression and resistance to treatment [31]. DACH1 was detectable in human PCa cell lines (Fig. 4E,F), including PC3 cells, in which TGFβ induced EMT, as evidenced by induction of the mesenchymal marker vimentin (Fig. 4G), and an increase in the proportion of cyclin D1 located in the cytoplasm. In addition, PC3 cell lines stably expressing DACH1 showed inhibition of TGFβ target gene expression (Fig. 4H, blue arrows)[30].
DACH1 governs the DNA damage response to genotoxic stress in PCa cells.
γH2AX staining, a marker of the DNA damage response (DDR), was increased by 50% in the Dach1−/− mice ventral prostate (Fig. 5A,B) (P=0.00002, Student’s t-test). The basal level γH2AX abundance was increased 2-fold in Dach1−/− 3T3 cells (Fig. 5C) with the number of γH2AX bodies increased >15-fold in Dach1−/− compared with Dach1+/+ 3T3 cells (Fig. 5D,E). Arsenic trioxide (ATO) induces DNA damage through induction of oxidative stress, which mediates cell-cycle arrest and repair of damaged DNA via an ATR/Chk2/Chk1 pathway. ATO increased the number of γH2AX bodies to ~5 in Dach1+/+ and ~20 in Dach1−/− 3T3 cells (Fig. 5D,E). shDACH1 increased the number of γH2AX bodies in LNCaP cells and enhanced ATO-induced γH2AX bodies from approximately 5 to almost 30 per cell (Fig. 5F,G). The induction of γH2AX in ATO-treated LNCaP cells was further enhanced approximately 2-fold by DACH1 shRNA (Fig. 5H,I). ATO-induced γH2AX abundance remained elevated in DACH1 shRNA transduced cells until 48 hrs (Fig. 5I). Re-expression of DACH1 with a doxycycline-inducible expression vector in LNCaP cells reduced γH2AX (Fig. 5J).
Figure 5. Prostatic Dach1 governs the DNA damage response in TRAMP mice.

(A). Immunohistochemical staining for markers of DNA damage (γH2AX) in TRAMP mice prostate, with (B). Quantitation data shown as mean ± SEM for percentage of γH2AX positive cells (P=2.07×10−7 by Student’s t-test) (n=19 for Dach1wt/wt mice, 3 separate mice, 6 views for two mice, 7 views for one mouse) (n=23 for Dach1fl/fl mice, 4 separate mice, 7 views for two mice, 6 views for one mouse, 3 views for one mouse) (left panel). Data shown as mean ± SEM for relative intensity of γH2AX (n=16 for Dach1wt/wt mice, 3 separate mice, 5 views for two mice, 6 views for one mouse) (n=20 for Dach1fl/fl mice, 4 separate mice, 6 views for two mice, 5 views for one mouse, 3 views for one mouse) (P=2.2×10−5 by Student’s t-test) (right panel). (C). Western blot of Dach1+/+ or Dach1−/− 3T3 cells. (D). γH2AX immunofluorescent staining of Dach1+/+ or Dach1−/− 3T3 cells, with (E). quantitation shown as mean ± SEM, (n=20 separate cells). (F). LNCaP cells transduced with shDACH1, treated with arsenic trioxide (ATO 1 hrs) (10 μM) and (G). quantitation shown as mean ± SEM (n=20 cells). (H). LNCaP cells stably transduced with control vector or shDACH1 were treated with ATO (1 μM) for the time points indicated. Western blotting was conducted for γH2AX, with quantitation of a representative experiment shown in (I). (J). LNCaP cell line stably expressing doxycycline-inducible DACH1 were analyzed for the abundance of γH2AX and other proteins as indicated. GAPDH was used as a protein loading control. S.E., short exposure; L.E., long exposure.
DACH1 facilitates the recruitment of, and co-accumulates with, Ku70/Ku80 proteins, at sites of DNA damage.
The Ku70/80 heterodimer is the DNA-binding component of DNA-dependent protein kinase and serves as an early upstream event in NHEJ. In order to determine the functional significance of DACH1 in the recruitment of Ku70/Ku80, we conducted laser micro irradiation of Dach1−/− 3T3 cells. In Dach1+/+ 3T3 cells, Ku70 and Ku80 accumulated rapidly at sites of DNA damage after 1 min (Fig. 6A). In Dach1−/− 3T3 cells, EGFP-DACH1 protein accumulated in ~1 min at sites of DNA damage after laser micro irradiation, and co-localized with RFP-Ku70 or RFP-Ku80 (Fig. 6B–C). Quantitation of protein accumulation at the sites of the laser injury was conducted, and the fold increase in foci intensity is shown as mean ± SEM for N=5 separate cells (Fig. S11A-D). Dach1−/− 3T3 cells transduced with EGFP-DACH1 co-accumulated RFP-Ku70 and RFP-Ku80 at DSB sites (Fig. 6B–C, Fig. S11A-D). All three proteins responded to micro irradiation within 1 min, with DACH1 continuing to accumulate at 10 min (Fig. S11A-D). In contrast, RFP-Ku70 and RFP-Ku80 failed to localize at the sites of laser irradiation in Dach1−/− 3T3 cells transduced with the control empty vector construct (Fig. 6B–C, Fig. S11A-D).
Figure 6. DACH1 facilitates the recruitment of, and co-accumulates with, Ku70/Ku80 proteins at sites of DNA damage.

(A). Co-accumulation of Ku-70/Ku-80 at laser micro irradiation-induced DSBs sites in Dach1+/+ 3T3 cells. (B,C). 24 h after transfection, the accumulation of DACH1 and Ku70/Ku80 in Dach1−/− 3T3 cells transfected with EGFP or EGFP-tagged DACH1 and red fluorescent protein (RFP)-tagged Ku70 or RFP-tagged Ku80 expression vectors were treated with laser micro-irradiation (403 nm) to induce DSBs. Time is shown after micro-irradiation. Accumulation of the transfected proteins was indicated by EGFP (green) or RFP (red) fluorescence at laser-irradiated sites. Co-accumulation was visualized in yellow merged images. Time is shown in minutes and -fold increase in foci intensity is shown as mean ± SEM for N=5 separate cells.
In order to examine further the interaction between DACH1 and Ku70/Ku80 a mass spectrometry analysis was conducted. DACH1 protein complexes were prepared from HEK 293T cells transfected with a FLAG-DACH1 expression vector (Fig. S12A). DACH1-associated proteins were resolved on a 4–10% Tris-HCl gel and silver-stained. The proteins recovered from the gel were subjected to in-gel tryptic digestion and sequential MS/MS. Two excised bands, corresponding to 70 and 80 kDa were identified as ATP-dependent DNA helicase 2 subunit Ku70 and ATP-dependent DNA helicase 2 subunit 2 (Ku80). 293T cells were transfected with expression vectors for FLAG-tagged DACH1 wild type, FLAG-DACH1 DS domain deletion (DDS), FLAG-DACH1 C term (Fig. S12B). Immune precipitation of FLAG-tagged DACH1 co-precipitated Ku70 and Ku80. Therefore, we considered the possibility that DACH1 may interface with Ku70 via a conserved N-BOX (the dac and ski/sno DS domain) which consists of ~100 amino acids conserved with various Sno/Ski family members, predicted to form a highly organized structure of α-helices and β-strands referred to as the DS domain. Expression of the DACH mutant deleted of the DS domain (DDS) (Fig. S12B) abolished binding to Ku70 and Ku80 (Fig. S12C).
DACH1 enhances DNA repair and homologous recombination.
Nuclear bodies marked by the DNA damage response protein p53 binding protein 1 (53BP1) [32] participate in the cellular response to DNA damage. 53BP1 relocates to nuclear foci within minutes after exposure of cells to ionizing radiation (IR). Consistent with the finding that endogenous DACH1 governs the DDR, shDACH1 increased basal and ATO-induced 53BP1 nuclear foci formation in LNCaP cells (Fig. 7A).
Figure 7. DACH1 enhances DNA repair.
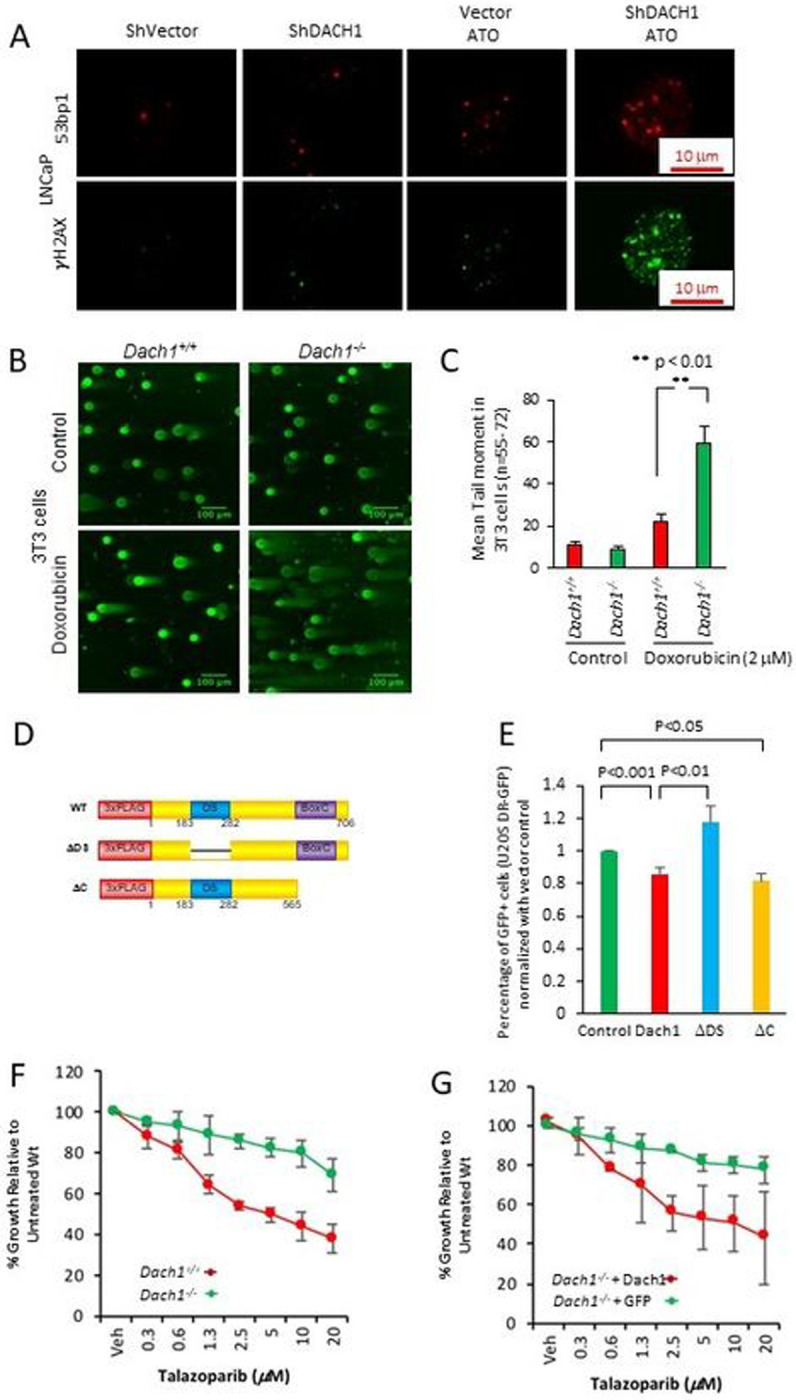
(A). LNCaP cells stably transduced with control vector or shDACH1 were treated with ATO (1μM), and immunofluorescence for 53BP1 or γH2AX was conducted. (B). The neutral pH comet assay, which mainly detects DNA double-strand breaks (DSBs), was conducted as a single-cell DNA damage assay. Dach1+/+ and Dach1−/− 3T3 cells were treated with 2 μM doxorubicin for 18 hrs. Scale bar, 100 μm with (C). data shown as mean ± SEM. (D). Schematic representation of DACH1 expression vectors, which were introduced into (E) U2OS cells expressing I-SceI based reporter assays for homologous repair (DR-GFP). Cells were analyzed after 48 hrs with data shown as mean ± SEM for N=12 (DACH1), N=7 (DACH1DDS), and N=5 (DACH1DC). (F). Dach1+/+ and Dach1−/− 3T3 cells or (G). Dach1−/− 3T3 transduced with a MSCV/DACH1-IRES-GFP expression vector or GFP vector control, were treated for 3 days with increasing doses of Talazoparib, a PARP inhibitor. Data are shown as mean ± SEM for N=3 separate experiments in triplicate.
The comet assay is a surrogate assay for measuring double-stranded DNA breaks in the cell. When cells are electrophoresed in neutral pH, the image looks like a comet with a distinct head composing intact DNA and a tail consisting of damaged DNA, primarily double-strand breaks (DSBs). In doxorubicin-treated 3T3 cells (2 μM, 18 hrs) the mean comet tail moment was 22 in Dach1+/+ cells, but 59 in Dach1−/− cells (Fig. 7B,C), indicating that endogenous Dach1 increases repair of damaged DNA.
To determine the type of DNA repair decreased by Dach1 we deployed I-SceI-based reporter assays for the homology-directed repair (DR-GFP) pathway [33, 34] (Fig. 7D,E). DACH1 expression restrained DR-GFP activity ~15% in U2OS DR-GFP cells (Fig. 7E, P<0.001, N=12). Expression of a DACH1 mutant, deleted of the DS domain, abolished the restraint of DR-GFP activity, whereas expression of a DACH1 mutant deleted of the carboxyl terminus maintained repression of DR-GFP activity ~20% ((Fig. 7D,E, P<0.05, N=5). Cell killing with PARP inhibitors (PARPi) was enhanced in cells with reduced homologous repair (HR) [5]. As DACH1 restrained homologous repair based on DR-GFP activity, we investigated the functional significance by assessing the impact of the PARPi Talazoparib. The growth of Dach1+/+ cells was inhibited in a dose-dependent manner (Fig. 7F, T50 ~4.5 μM); however, the Dach1−/− cells were relatively resistant to PARPi (Fig. 7F, T50~1098 μM (projected)). The reintroduction of DACH1 with retroviral-expressing MSCV-DACH1-IRES-GFP into the Dach1−/− cells restored sensitivity to Talazoparib (Fig. 7G, T50 ~10.2 μM) when compared to the Dach1−/− cells+ GFP vector (T50 ~7258 μM (projected)).
DACH1 determines TGFβ response in cell proliferation and DNA damage.
Our analysis of the Dach1−/− TRAMP mice showed Dach1-restrained TGFβ signaling in vivo. In order to determine the role of Dach1 in TGFβ-mediated DNA repair, the TGF-β receptor type I (TGF-βRI) kinase inhibitors (LY2157299 and LY364947) were deployed. Dach1−/− cells showed enhanced sensitivity to doxorubicin-induced cell-killing that was reversed by the reintroduction of DACH1 (Fig. S13A,B). Consistent with hyperactivated TGFβ signaling upon Dach1 deletion, Dach1−/− cells were correspondingly resistant to the antiproliferative effect of LY2157299 and LY364947 in the presence of a DNA-damaging agent (Fig. S13A,B).
DNA damage repair was assessed by the mean tail movement in comet assays. To avoid an artifact of different cellular proliferation rates we deployed a concentration of LY2157299 and LY364947 in which the antiproliferative effect of the TGFβRki (LY2157299 and LY364947) was similar between Dach1+/+ vs. Dach1−/− 3T3 cells (LY2157299 and LY364947). Pretreatment with LY2157299 and LY364947 for 3 days decreased doxorubicin-induced comet tail formation in Dach1+/+ 3T3 cells (Doxorubicin vs. Doxorubicin+LY2157299 or Doxorubicin vs. Doxorubicin + LY364947) (Figure S14A-C). Consistent with the resistance of Dach1−/− to cell killing by TGFBki in the presence of doxorubicin, the TGFβRki did not reduce the DNA damage induced by doxorubicin in Dach1−/− cells (Fig. S14A-C).
In the presence of 4 Gy X-ray irradiation, reduced endogenous DACH1 by shRNA in LNCaP cells, reduced colony formation (survival fraction representing colony number) and colony size (representing cell numbers per colony) in the presence of the DNA-PK inhibitor NU7026 (Fig. S15). Endogenous DACH1, therefore, conveyed resistance to NU7026 (Fig. S15).
DISCUSSION
Gene deletion within the 13q21 region has been associated with high-grade prostate cancer. In an independent cohort enriched for early-onset prostate cancer [27] respectively. In the current studies, we identified homozygous deletions of DACH1 in between 3 and 18% of prostate cancers in six distinct cohorts. DACH1 is located within the 13q21.31-q21.33 region, which is deleted in poor-prognosis prostate cancer [10–12]. Herein, homozygous DACH1 deletions correlated with reduced overall survival (medians of 84 vs. 120 months, N=667). DACH1 deletion co-occurred with deletion of SPOP and BRCA2, consistent with TCGA analysis demonstrating multiple pathways may be disrupted in a given tumor type [35]. The abundance of DACH1 was also reduced by DNA methylation, and low DACH1 gene expression was significantly correlated with earlier biochemical recurrence.
DACH1 is located on chromosome 13q. In a subset of DACH1-deleted prostate cancers, we showed that DACH1 was co-deleted with RB (in 5 databases, co-homozygous-deletion of RB with DACH1 occurred in 15%, 5.3%, 0%, 1.4%, and 9%, Fig. S2). The current studies were therefore designed to test the importance of the additional DACH1 deletion in the presence of inactivated RB. Such an approach is necessary to define the additional impact of DACH1 in the onset and progression of PCa. Our studies were therefore designed to distinguish whether DACH1 deletion was an “innocent bystander”, given the genomic proximity of DACH1 to RB. The TRAMP mouse[36–39], one of the most widely accepted in PCa research[40], inactivates pRB via the SV40 Large T antigen, and recapitulates with ~100% penetrance multiple aspects of the human disease, including prostatic intraepithelial neoplasia (PIN) lesions, and multifocal invasive carcinoma; and emulates histological and molecular events of human PCa[41]. With time, beyond which our studies were conducted, progression to castration-resistant prostate cancer[42–44], including castrate-resistant PCa (CRPC)[45] and the aggressive therapy-induced neuroendocrine prostate cancer tNEPC[42–44], which occurs with metastasis to distant organs[40] including the skeleton[46]. Analysis of the prostates of transgenic mice at 15 weeks demonstrated that genetic deletion of Dach1 in TRAMP mice correlated with increased Ki-67, histological features of PIN progression, increased TGFβ activity, and increased evidence of DNA damage. An increased PIN in the DACH1 prostate-specific deletion samples (PbCre:Dach1fl/fl) was demonstrated by increased cellular proliferation, loss of cellular polarity (black arrow) (Fig. S10), nuclear enlargement (white arrow) (Fig. S10), and the presence of nucleoli (gray arrow) (Fig. S10). Dach1 deletion in the prostate resulted in PIN but not tumorigenesis in the time frame assessed. Thus, deregulation of Dach1 alone, like CHD[47], ERG1[48], or ETV1[49], is insufficient to drive prostate cancer in the mouse prostate. The genetic disruption of Dach1 did not disrupt RB1 gene expression and was associated with enhanced pRBSer807/811P. In PCa cell lines, the reintroduction of DACH1 expression reduced RB abundance (Fig. S8B). These findings suggest that the effect of Dach1 deletion in the prostate is not mediated by coincident loss of RB1. These results are the first to show that endogenous Dach1 restrains features of tumorigenesis in vivo, and are consistent with prior correlative studies showing reduced DACH1 abundance in tumors of the brain, ovary, lung, uterus, non-small cell lung cancer, hepatocellular carcinoma, breast, and prostate cancer [24, 50].
In prior tissue culture-based analysis, DACH1 retrained TGFβ signaling by association with NCoR/SMAD4 [51, 52]. The current studies extend these findings by showing that endogenous DACH1 restrains TGFβ signaling in the murine prostate in vivo. TGFβ signaling was the most upregulated pathway by gene expression analysis in the prostate of prostate-specific Dach1-deletion prostate cancer oncomice. TGFβ activity was induced in the Dach1 knockout prostate tissue in vivo and assessed by the induction of SMAD2Ser465/467P. Conversely, increased expression of DACH1 in PC3 cells reduced TGFβ activity and expression of TGFβ target genes. In proliferation assays, DACH1 deletion-mediated TGFβ hyperactivation led to relative resistance to the growth-inhibitory effects of the TGFβKRi in the presence of doxorubicin. In the current studies, the inhibition of TGFβ with the TGFβRki (LY2157299) reduced comet tail formation, consistent with recent studies in which Tgfβ reduced DNA repair and increased comet tail formation in a lung cancer cell line [53]. The reduction in mean tail movement by the TGFbRki (LY2157299) was defective in the Dach1−/− cells, consistent with increased TGFβ signaling in the Dach1−/− cells. Increased TGFβ signaling has been strongly linked to PCa[54–56] promoting therapeutic resistance, cell invasiveness and tumor metastasis [57]. The role of TGFβ hyperactivation in DACH1 deletion prostate cancer warrants further analysis.
In the work reported here, DACH1 was shown to promote the repair of damaged DNA. Prostate-specific deletion of the Dach1 gene in TRAMP mice correlated with increased γH2AXSer139 and 53BP1 staining in prostate tissues and in cultured cells (LNCaP and 3T3). Comet assays of Dach1−/− cells showed increased tail formation, consistent with defective DNA repair. Analysis of the mechanisms by which DACH1 participates in DNA damage repair identified a chaperone function in which DACH1 augmented the recruitment of Ku70/Ku80 to sites of laser-induced double-stranded DNA breaks. Dach1 gene deletion reduced recruitment of Ku70/Ku80 to sites of DSB induced by microirradiation, and reintroduction of DACH1 restored Ku70/Ku80 recruitment to DSB. Proteomic analysis of DACH1-binding proteins identified Ku70 and Ku80 by direct sequence analysis and immune precipitation (IP)-Western blotting showed that DACH1 co-precipitated Ku70 and Ku80. DACH1 overexpressed cells had decreased HR, like Ku70 or Ku80 deletion cells, had increased HR assessed using DR-GFP reporter assays [58]. Herein the restraint of homologous repair by DACH1 required the DS domain. Consistent with the increased HR, Dach1−/− cells, showed resistance to PARP inhibition. As with Dach1−/− cells, defective Ku70/Ku80 function is associated with PARP resistance and restoration of HR[59, 60]. Mechanistically, Ku proteins bind to both ends of a two-ended DSB, stabilizing contacts between Ku heterodimers and tethering the DNA ends, thereby preventing access to the HDR machinery [21]. The mechanism by which DACH1 binding to Ku70/Ku80 induces PARP resistance remains to be further determined; however, Dach1−/− cells appear to be functionally defective in Ku protein recruitment. HR is increased in Ku70 or Ku80 deletion cells [20, 21]. Ku and PARP-1 have been found to compete for binding to DNA ends[61], and PARP1 binds to Ku70/Ku80[62], blocking its activity[63].
Inactivation of the Ku70 or Ku80 genes in mice leads to hypersensitivity to radiation and malignant transformation [18, 19]. Herein, DACH1 shRNA enhanced the radiation sensitivity to DNA-PK inhibition in colony assays. Collectively, these studies show that a loss of Dach1 abundance is associated with resistance to PARPi and TGFβKi, with enhanced sensitivity to radiation and doxorubicin. In addition, a growing body of evidence has identified somatic and, more recently, germline mutations of DNA repair genes in PCa [64]. The ongoing identification and testing of additional genes governing DNA repair may enhance the precision of guided targeted therapies of PCa.
MATERIALS AND METHODS.
Genetic and Epigenetic Analysis.
To generate profiles of SNP6-based GISTIC2 G-scores along the GRCh37/hg19 human reference genome, and on chromosome 13, we downloaded a ‘scores.gistic’ file for n=492 TCGA legacy PCa (PRAD) primary tumors from firebrowse.org, and an hg19 chromosome length file from genome.ucsc.edu. We generated graphics with a custom R script, and used the R package karyoploteR v1.14.1 to generate the banded chromosome 13 graphic.
To compare DACH1 copy number gene status in between primary vs. metastatic lesions, homozygous and heterozygous deletions in primary and metastatic tumors in different datasets, we used cBioPortal (http://www.cbioportal.org/) [65].
To assess the relationship between DACH1 homozygous deletions and outcomes, we queried mutation, copy number and overall survival data for three cBioPortal cohorts (TCGA PanCancer Atlas 2018, SU2C 2019, and MCTP), which offered data for a total of n=667 tumor samples. Using a copy number threshold of −2 to segregate the data into samples with altered vs. unaltered DACH1, we generated a Kaplan-Meier plot for overall survival using R’s survival v3.2–7 package, and calculated median times for altered vs. unaltered DACH1 with a custom R script.
Comparative analysis of DACH1 and PTEN gene status in all PCa patients and between primary vs. metastatic lesions in different datasets was performed using data from cBioPortal (http://www.cbioportal.org/). Selected studies were identified based on query criteria and analyzed using default parameters [65]. Data for analysis of the association between low DACH1 expression with AR activity was derived from the supplementary data of [26], grouped by mutation status of ERG, ETV1/4/FLI1, FOXA1, “other” and adjacent tissue normal samples. Additional DACH1 deletion data for 333 samples from the study were downloaded from cBioPortal [65] and DACH1 homozygous deletions were defined as a thresholded GISTIC value of −2. All tumor and normal groups were tested for differences in AR score, AR mRNA, and AR protein levels, using two-sample two-tail Student’s t-test; results with p<0.05 were considered significant.
To assess the effect of DNA methylation on DACH1 gene expression, we downloaded from firebrowse.org the Illumina 450K DNA methylation data and RSEM gene expression data for n=498 TCGA ‘legacy’ PCa (PRAD) primary tumors and n=50 adjacent normal tissues. For the 26 DACH1 DNA methylation probes for which DNA methylation data was largely complete, we calculated Spearman correlations between DNA methylation beta values and gene-level RSEM expression in primary tumors with R’s cor.test, then used R’s p.adjust to correct the p-values for multiple hypothesis testing. We assessed scatterplots of DNA methylation beta vs. DACH1 gene expression for all 26 DNA methylation probes (data not shown). For each probe, we calculated median beta values for primary tumors and adjacent normal tissue. We compared locations of DNA methylation probes to the DACH1 gene structure by transforming the probe locations into a ‘bed’ file and the Spearman correlation coefficients into a ‘bedgraph’ file (genome.ucsc.edu/FAQ/FAQformat.html), and then displaying both in the UCSC hg19 genome browser (data not shown). This analysis identified that 15 of the 26 DACH1 DNA methylation probes were near that gene’s TSS, including probe cg13726218 (chr13:72438250). This probe had both the largest negative correlation with DACH1 gene expression, and a wide range of beta values (Figs. S3A, B). Together, these three factors suggested that this probe represents how DNA methylation can influence DACH1 gene expression, and this was the probe that cBioPortal reported for DACH1.
To assess the relationship between low expression of DACH1 and outcomes, we downloaded DACH1 RSEM expression Z-scores (considering all samples), for the TCGA cohort [26], i.e., for the n=290 of 333 tumor samples that had RSEM data. We defined samples with “altered” DACH1 expression as those in which the gene’s Z-score was below −2.0, which is the default threshold value at cBioPortal. Using the PanCancer outcomes [28], we segregated the samples by low vs not-low RSEM. The R survival v3.2–10 package returned a Kaplan-Meier log-rank P value of 0.028 for Progression Free Interval (PFI) outcome data.
For the Firehose Legacy cohort at cBioPortal (n=492 primary tumors), we downloaded thresholded copy number, mutations, and RSEM normalized gene expression data for DACH1, ERG, ETV1, FOXA1, GBF1, KMT2C, SPOP, TP53, and RPRD2. For n=360 records with homozygous DACH1 deletions (i.e., CNA=−2, n=51), and diploid DACH1 (i.e., CNA=0, n=309), we calculated the percent of mutated samples for SPOP, FOXA1, KMT2C, TP53 and GBF1 (excluding ERG because it had no mutations in this cohort, and ETV1 because it had only one mutation). For the five retained genes, we calculated p values with a Fisher exact test on count data and applied a Bonferroni (x5) correction for multiple hypothesis testing. For DACH1, ERG, FOXA1, SPOP, and RPRD2, we generated an oncoprint and reported cBioPortal mutual exclusivity and co-occurrence results for pairs of genes. From the RSEM normalized gene expression data, for the n=491 records for which DACH1 and SPOPL had no mutations, we generated a scatterplot of gene expression for these two genes and calculated Pearson and Spearman correlations between the two expression profiles.
Transgenics.
The Transgenic Adenocarcinoma Mouse Prostate (TRAMP) transgenic mice, Dach1fl/fl -Probasin-Cre [30], and ROSA26mT/mG transgenic mice were used to generate a prostate epithelial cell-specific Dach1 gene knockout mouse (Probasin-Cre-Dach1fl/fl ROSA26mT/mG-TRAMP) lines. The appropriate institutional committee-approved protocols were followed when working with these mice.
Gross anatomical analysis and immunohistochemistry.
Transgenic mice aged 15 weeks were euthanized by CO2 asphyxiation. Animals were dissected, and the following organs were removed: ventral, lateral and anterior prostate (AP) for hematoxylin & eosin (H&E) and ventro-dorsolateral (VDL) prostate for immunohistochemical (IHC) staining. Histopathological grading was undertaken in a blinded manner, comparing a total of 10 mice (5 of each genotype), analyzing sections from the anterior, ventral and lateral prostate in each animal (total N=15 in each group). DACH1 (#10914–1-AP, Proteintech), Ki-67 (#M7240, Dako), Beclin 1 (#11427, Santa Cruz Biotechnology), AR (N20) (#sc-816, Santa Cruz Biotechnology), SMAD2pSer465/467 (#44–244G, ThermoFisher Scientific), γH2AX (Ser139, 05–636, Millipore), 53BP1 (NB100–304, Novus Biologicals) were used as described [66]. ImageJ software was used in IHC quantification.
Microarray analysis.
Total RNA was prepared from ventral prostates of 3 Dach1 WT (Probasin-Cre-Dach1wt/wt ROSA26mT/mG-TRAMP) and 3 Dach1 KO (Probasin-Cre-Dach1fl/fl ROSA26mT/mG-TRAMP) (15w) using the RNeasy kit from Qiagen following the manufacturer’s instructions (Qiagen). RNA was labeled for hybridization with mouse Clariom D arrays (Applied Biosystems), and analysis was performed as previously described [30, 67]. Gene set enrichment analysis for upstream regulators responsible for a significant number of changed genes was done using QIAGEN’s Ingenuity® Pathway Analysis software (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity) with the “Upstream regulator” option; regulators that passed the P <0.01 threshold, had at least ten significantly affected target genes, and had predicted activation states (|Z|>0.5) were reported.
DACH1 and AR Immunohistochemical staining.
Immunostaining for DACH1 was performed on an Omnis autostainer (Agilent, Santa Clara, CA) on prostate cancer and matched normal/benign tissue in tissue microarray format (triplicate or quadruplicate 1.0 mm punch cores) from prostatectomies of a cohort of 71 cases[68]. For detection of DACH1 protein, antigen retrieval was done in Tris/EDTA buffer at pH 9 for 30 min at 97°C, followed by 30 min incubation with rabbit polyclonal DACH1 antibody (Cat. #10914–1-AP, Proteintech, Rosemont, IL; dilution 1:1,000)[30], HRP-conjugated polymer (Envision FLEX, Cat#GV80011–2, Agilent), and DAB chromogen deposition. After cover-slipping, the stained slides were scanned using a bright-field setting on a Pannoramic Flash 250 (3DHistech, Budapest, Hungary). Evaluable DACH1 staining was achieved for 68 cases after the exclusion of cases with missing prostate cancer cores. DACH1 status was considered positive if ≥2% of cancer cells showed detec staining. Scoring of DACH1 protein expression in prostate cancer cells was performed visually, based on the presence (positive) or absence (negative) of detectable DAB chromogen. Immunohistochemistry for the AR was performed using a Leica Bond Rx autostainer (Leica Biosystems, Deer Park, IL). Deparaffinized sections of prostate TMAs were performed at HpH for antigen retrieval, then incubated with a monoclonal mouse antibody to the Androgen Receptor (Leica Biosystems, catalog #AR-318-L-CE) at 1:50 dilution for 30 minutes and Bond Polymer Refine Detection kit (Leica Biosystems) was used with DAB (3,3′-Diaminobenzidine) chromogen for visualization. After coverslipping, the slides were scanned using Pannoramic 250 (3DHISTECH Ltd., Budapest, Hungary), and images were generated using Slideviewer software. Likelihood-ratio tests were used for statistical analysis of DACH1 status and Gleason score.
Cell culture, reagents, and plasmids.
LNCaP, LNCaP-LN3, C4, C4–2, C4–2B, PC-3M, PC-3M-LN4, PC-3M-Pro4, and MDA-MB453 cells were grown as previously described [30, 67]. The HEK293T, PC3, and C4–2 cells were cultured in DMEM supplemented with 10% fetal calf serum, 1% penicillin, and 1% streptomycin. The DNA methylase inhibitor 5-Aza-dC (10 μM) (#A3656, Sigma), the 26S proteasome inhibitors MG132 (20 μM) (#S2619, Selleckchem), or N-acetyl-L-leucyl-L-leucyl-L-nor leucinal (LLNL) (25 μM) (#A6185, Sigma), Doxorubicin (Sigma), Talazoparib (SeleckChem), NU7026 (Sigma) were used as described in the text. TGF-β receptor type I (TGF-βRI) kinase inhibitors LY2157299 and LY363947 were bought from Selleckchem.
The expression vectors encoding DACH1 [69], Ku70 (RFP-Ku70) and Ku80 (RFP-Ku80) [70], shDACH1[71] were previously described. The EGFP-DACH1 expression plasmid was made by inserting the human DACH1 cDNA into the HindIII and BamHI sites of the pEGFP-C1 vector.
Western blot analysis.
Whole-cell lysates, nuclear lysates, or cytoplasmic lysates were separated by 8%−11% SDS-PAGE gel and the proteins were transferred to a nitrocellulose membrane for Western blotting, as previously described [30, 67]. The bands were detected using the enhanced chemiluminescence detection system (Thermo Fisher Scientific #34578). The following antibodies were used: DACH1 (#10914–1-AP, Proteintech), γH2AX (#05–636, Millipore, or #80312, Cell Signaling Technology), Vimentin (#5741, Cell Signaling), cyclin D1 (sc-20044, Santa Cruz Biotechnology), p53 (#sc-6243, Santa Cruz Biotechnology), Vinculin (#V9131, Sigma), β-Actin (#sc-47778, Santa Cruz Biotechnology), β-Tubulin (#sc-9104, Santa Cruz Biotechnology), Lamin B1 (#ab16048, Abcam), and GAPDH (#sc-25778, Santa Cruz Biotechnology).
Cell Proliferation and Comet Assays.
Cells were seeded into 96 well plates in normal growth medium, and cell growth was measured daily by methylene blue assay [72]. Neutral pH comet assays were conducted as previously described [73] using the CometAssay Kit (Trevigen). After treatment with 2 μM doxorubicin or control for 18 hrs, cells were harvested and mixed with low-melting temperature agarose. After lysis, electrophoresis was conducted at 1V/cm for 20 minutes. Visualization involved SYBR Gold dye for 30 minutes and a Nikon C2+ Confocal Microscope with a 20× objective. Average tail moments from 55–72 cells per sample were obtained using OpenComet software (http://www.cometbio.org/index.html)[74].
Laser micro irradiation.
Cells were transfected with expression vectors encoding fusion proteins of DACH1 (EGFP-DACH1), Ku70 (EGFP-Ku70 or RFP-Ku70), or Ku80 (RFP-Ku80)[70], and, after 24 hrs, were treated with 100 μM 8-methoxypsoralen and subjected to 405 nm laser irradiation as previously described [70].
Identification of DACH1-associated proteins by mass spectrometry.
The DACH1 complex was purified from HEK 293T cells transiently transfected with expression vectors encoding FLAG-tagged DACH1, following the protocol described in Technical Bulletin (No. MB-925, Sigma-Aldrich, St. Louis, MO). Proteins were digested by the addition of 25 ng/μl sequence-grade modified trypsin (Promega, Madison, WI) in ammonium bicarbonate buffer for 16 h at 30 °C, with agitation. The digestion products were applied onto a MALDI plate as described[75].
DNA repair assays.
The DNA repair reporter assays for homologous repair (DR-GFP) assays in U2OS cells were conducted as previously described [33, 34]. pCAGGS-NZEGFP, a plasmid encoding expressed GFP, was a transfection efficiency control. The DNA repair activity was shown as (RSceI-RpCAGGS)/RNZEGFP. RI-SceI, RpCAGGS, and RNZEGFP represent the ratio of GFP positive cells in I-SceI, pCAGGS-BSKX (vector control for I-SceI expression plasmid), and NZEGFP transfected cells respectively.
SIGNIFICANCE.
Prostate-specific Dach1 deletion in OncoMice accelerated prostatic intraepithelial neoplasia. DACH1 decreased HR DNA repair via Ku70/Ku80 recruitment, determining sensitivity to PARP inhibitors and extending the repertoire of potential PCa theragnostics.
ACKNOWLEDGEMENTS
We gratefully acknowledge Dr. Andy Cherniack’s (The Broad Institute) assistance with Firehose GISTIC outputs, Dr. Lucia Languino (Thomas Jefferson University) for help in generating transgenic mice, Dr. Jeremy Stark (City of Hope Comprehensive Cancer Center) for technical help in DNA repair assays, and the thoughtful comments from Dr. Winfried Edelmann, Albert Einstein College of Medicine.
This work was partly supported by R01 CA132115, R21 CA235139–01-A1, and a Breakthrough Breast Cancer Research Program award # W81XWH1810605 (R.G.P). SJMJ acknowledges the support of the Canada Research Chairs program. This work was supported in part by funding from the Wisconsin Breast Cancer Showhouse (H.R, Y.S).
ABBREVIATIONS:
- PCa
Prostate cancer
- Pten
Phosphatase and tensin homolog gene
- TRAMP
the Transgenic Adenocarcinoma Mouse Prostate
- EGFR
epidermal growth factor receptor
- VDL
ventro-dorsolateral
- AP
prostate and anterior prostate
Footnotes
CONFLICT OF INTEREST:
The authors declare that they have no relevant financial conflicts of interest. R.G.P. holds ownership interests in CytoDyn, EcoGenome, StromaGenesis, and LightSeed, Inc. R.G.P. additionally holds ownership interests (value unknown) for several patents and submitted patent applications.
REFERENCES
- 1.Wang G, Zhao D, Spring DJ, DePinho RA. Genetics and biology of prostate cancer. Genes & Development 2018; 32: 1105–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dimakakos A, Armakolas A, Koutsilieris M. Novel tools for prostate cancer prognosis, diagnosis, and follow-up. Biomed Res Int 2014; 2014: 890697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Draisma G, Etzioni R, Tsodikov A, Mariotto A, Wever E, Gulati R et al. Lead time and overdiagnosis in prostate-specific antigen screening: importance of methods and context. J Natl Cancer Inst 2009; 101: 374–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li S, Silvestri V, Leslie G, Rebbeck TR, Neuhausen SL, Hopper JL et al. Cancer Risks Associated With BRCA1 and BRCA2 Pathogenic Variants. J Clin Oncol 2022; 40: 1529–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teyssonneau D, Margot H, Cabart M, Anonnay M, Sargos P, Vuong NS et al. Prostate cancer and PARP inhibitors: progress and challenges. J Hematol Oncol 2021; 14: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y et al. Punctuated evolution of prostate cancer genomes. Cell 2013; 153: 666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev 2010; 24: 1967–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005; 310: 644–648. [DOI] [PubMed] [Google Scholar]
- 9.Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N Engl J Med 2016; 375: 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong JT, Boyd JC, Frierson HF, Jr. Loss of heterozygosity at 13q14 and 13q21 in high grade, high stage prostate cancer. Prostate 2001; 49: 166–171. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y, Sadasivan SM, She R, Datta I, Taneja K, Chitale D et al. Breast and prostate cancers harbor common somatic copy number alterations that consistently differ by race and are associated with survival. BMC Med Genomics 2020; 13: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen C, Brabham WW, Stultz BG, Frierson HF Jr, Barrett JC, Sawyers CL et al. Defining a common region of deletion at 13q21 in human cancers. Genes Chromosomes Cancer 2001; 31: 333–344. [DOI] [PubMed] [Google Scholar]
- 13.Gandaglia G, Zaffuto E, Fossati N, Cucchiara V, Mirone V, Montorsi F et al. The role of prostatic inflammation in the development and progression of benign and malignant diseases. Curr Opin Urol 2017; 27: 99–106. [DOI] [PubMed] [Google Scholar]
- 14.Nelson WG, De Marzo AM, Isaacs WB . Prostate cancer. N Engl J Med 2003; 349: 366–381. [DOI] [PubMed] [Google Scholar]
- 15.Batlle E, Massague J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019; 50: 924–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song B, Park SH, Zhao JC, Fong KW, Li S, Lee Y et al. Targeting FOXA1-mediated repression of TGF-beta signaling suppresses castration-resistant prostate cancer progression. J Clin Invest 2019; 129: 569–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis AJ, Chen BP, Chen DJ. DNA-PK: a dynamic enzyme in a versatile DSB repair pathway. DNA Repair (Amst) 2014; 17: 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Difilippantonio MJ, Zhu J, Chen HT, Meffre E, Nussenzweig MC, Max EE et al. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 2000; 404: 510–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith GC, Jackson SP. The DNA-dependent protein kinase. Genes Dev 1999; 13: 916–934. [DOI] [PubMed] [Google Scholar]
- 20.Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev 2001; 15: 3237–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol 2004; 24: 9305–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mardon G, Solomon NM, Rubin GM. dachshund encodes a nuclear protein required for normal eye and leg development in Drosophila. Development 1994; 120: 3473–3486. [DOI] [PubMed] [Google Scholar]
- 23.Zhou J, Wang C, Wang Z, Dampier W, Wu K, Casimiro MC et al. Attenuation of Forkhead signaling by the retinal determination factor DACH1. Proc Natl Acad Sci U S A 2010; 107: 6864–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu K, Katiyar S, Witkiewicz A, Li A, McCue P, Song LN et al. The cell fate determination factor dachshund inhibits androgen receptor signaling and prostate cancer cellular growth. Cancer Res 2009; 69: 3347–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren S, Wei GH, Liu D, Wang L, Hou Y, Zhu S et al. Whole-genome and Transcriptome Sequencing of Prostate Cancer Identify New Genetic Alterations Driving Disease Progression. Eur Urol 2018; 73: 322–339. [DOI] [PubMed] [Google Scholar]
- 26.Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015; 163: 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerhauser C, Favero F, Risch T, Simon R, Feuerbach L, Assenov Y et al. Molecular Evolution of Early-Onset Prostate Cancer Identifies Molecular Risk Markers and Clinical Trajectories. Cancer Cell 2018; 34: 996–1011 e1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018; 173: 400–416 e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mo Q, Wang S, Seshan VE, Olshen AB, Schultz N, Sander C et al. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc Natl Acad Sci U S A 2013; 110: 4245–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen K, Wu K, Jiao X, Wang L, Ju X, Wang M et al. The endogenous cell-fate factor dachshund restrains prostate epithelial cell migration via repression of cytokine secretion via a cxcl signaling module. Cancer Res 2015; 75: 1992–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montanari M, Rossetti S, Cavaliere C, D’Aniello C, Malzone MG, Vanacore D et al. Epithelial-mesenchymal transition in prostate cancer: an overview. Oncotarget 2017; 8: 35376–35389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 2011; 13: 243–253. [DOI] [PubMed] [Google Scholar]
- 33.Gunn A, Stark JM. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol Biol 2012; 920: 379–391. [DOI] [PubMed] [Google Scholar]
- 34.Jiao X, Velasco-Velazquez MA, Wang M, Li Z, Rui H, Peck AR et al. CCR5 Governs DNA Damage Repair and Breast Cancer Stem Cell Expansion. Cancer Res 2018; 78: 1657–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018; 173: 321–337 e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Vizio D, Sotgia F, Williams TM, Hassan GS, Capozza F, Frank PG et al. Caveolin-1 is required for the upregulation of fatty acid synthase (FASN), a tumor promoter, during prostate cancer progression. Cancer Biol Ther 2007; 6: 1263–1268. [DOI] [PubMed] [Google Scholar]
- 37.Trerotola M, Ganguly KK, Fazli L, Fedele C, Lu H, Dutta A et al. Trop-2 is up-regulated in invasive prostate cancer and displaces FAK from focal contacts. Oncotarget 2015; 6: 14318–14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Di Vizio D, Morello M, Sotgia F, Pestell RG, Freeman MR, Lisanti MP. An absence of stromal caveolin-1 is associated with advanced prostate cancer, metastatic disease and epithelial Akt activation. Cell Cycle 2009; 8: 2420–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams TM, Hassan GS, Li J, Cohen AW, Medina F, Frank PG et al. Caveolin-1 promotes tumor progression in an autochthonous mouse model of prostate cancer: genetic ablation of Cav-1 delays advanced prostate tumor development in tramp mice. J Biol Chem 2005; 280: 25134–25145. [DOI] [PubMed] [Google Scholar]
- 40.Irshad S, Abate-Shen C. Modeling prostate cancer in mice: something old, something new, something premalignant, something metastatic. Cancer Metastasis Rev 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaplan-Lefko PJ, Chen TM, Ittmann MM, Barrios RJ, Ayala GE, Huss WJ et al. Pathobiology of autochthonous prostate cancer in a pre-clinical transgenic mouse model. Prostate 2003; 55: 219–237. [DOI] [PubMed] [Google Scholar]
- 42.Hurwitz AA, Foster BA, Allison JP, Greenberg NM, Kwon ED. The TRAMP mouse as a model for prostate cancer. Curr Protoc Immunol 2001; Chapter 20: Unit 20 25. [DOI] [PubMed] [Google Scholar]
- 43.Berman-Booty LD, Knudsen KE. Models of neuroendocrine prostate cancer. Endocr Relat Cancer 2015; 22: R33–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gelman IH. How the TRAMP Model Revolutionized the Study of Prostate Cancer Progression. Cancer Res 2016; 76: 6137–6139. [DOI] [PubMed] [Google Scholar]
- 45.Cerasuolo M, Maccarinelli F, Coltrini D, Mahmoud AM, Marolda V, Ghedini GC et al. Modeling Acquired Resistance to the Second-Generation Androgen Receptor Antagonist Enzalutamide in the TRAMP Model of Prostate Cancer. Cancer Res 2020; 80: 1564–1577. [DOI] [PubMed] [Google Scholar]
- 46.Grabowska MM, DeGraff DJ, Yu X, Jin RJ, Chen Z, Borowsky AD et al. Mouse models of prostate cancer: picking the best model for the question. Cancer Metastasis Rev 2014; 33: 377–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Augello MA, Liu D, Deonarine LD, Robinson BD, Huang D, Stelloo S et al. CHD1 Loss Alters AR Binding at Lineage-Specific Enhancers and Modulates Distinct Transcriptional Programs to Drive Prostate Tumorigenesis. Cancer Cell 2019; 35: 817–819. [DOI] [PubMed] [Google Scholar]
- 48.Augello MA, Liu D, Deonarine LD, Robinson BD, Huang D, Stelloo S et al. CHD1 Loss Alters AR Binding at Lineage-Specific Enhancers and Modulates Distinct Transcriptional Programs to Drive Prostate Tumorigenesis. Cancer Cell 2019; 35: 603–617 e608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baena E, Shao Z, Linn DE, Glass K, Hamblen MJ, Fujiwara Y et al. ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev 2013; 27: 683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kong D, Liu Y, Liu Q, Han N, Zhang C, Pestell RG et al. The retinal determination gene network: from developmental regulator to cancer therapeutic target. Oncotarget 2016; 7: 50755–50765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu K, Yang Y, Wang C, Davoli MA, D’Amico M, Li A et al. DACH1 inhibits transforming growth factor-beta signaling through binding Smad4. J Biol Chem 2003; 278: 51673–51684. [DOI] [PubMed] [Google Scholar]
- 52.Jiao X, Li Z, Wang M, Katiyar S, Di Sante G, Farshchian M et al. Dachshund Depletion Disrupts Mammary Gland Development and Diverts the Composition of the Mammary Gland Progenitor Pool. Stem Cell Reports 2019; 12: 135–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pal D, Pertot A, Shirole NH, Yao Z, Anaparthy N, Garvin T et al. TGF-beta reduces DNA ds-break repair mechanisms to heighten genetic diversity and adaptability of CD44+/CD24- cancer cells. Elife 2017; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Q, Helfand BT, Jang TL, Zhu LJ, Chen L, Yang XJ et al. Nuclear factor-kappaB-mediated transforming growth factor-beta-induced expression of vimentin is an independent predictor of biochemical recurrence after radical prostatectomy. Clin Cancer Res 2009; 15: 3557–3567. [DOI] [PubMed] [Google Scholar]
- 55.Wikstrom P, Stattin P, Franck-Lissbrant I, Damber JE, Bergh A. Transforming growth factor beta1 is associated with angiogenesis, metastasis, and poor clinical outcome in prostate cancer. Prostate 1998; 37: 19–29. [DOI] [PubMed] [Google Scholar]
- 56.Ao M, Williams K, Bhowmick NA, Hayward SW. Transforming growth factor-beta promotes invasion in tumorigenic but not in nontumorigenic human prostatic epithelial cells. Cancer Res 2006; 66: 8007–8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walker L, Millena AC, Strong N, Khan SA. Expression of TGFbeta3 and its effects on migratory and invasive behavior of prostate cancer cells: involvement of PI3-kinase/AKT signaling pathway. Clin Exp Metastasis 2013; 30: 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet 2008; 4: e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choi YE, Meghani K, Brault ME, Leclerc L, He YJ, Day TA et al. Platinum and PARP Inhibitor Resistance Due to Overexpression of MicroRNA-622 in BRCA1-Mutant Ovarian Cancer. Cell Rep 2016; 14: 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee EK, Matulonis UA. PARP Inhibitor Resistance Mechanisms and Implications for Post-Progression Combination Therapies. Cancers (Basel) 2020; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res 2006; 34: 6170–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Isabelle M, Moreel X, Gagne JP, Rouleau M, Ethier C, Gagne P et al. Investigation of PARP-1, PARP-2, and PARG interactomes by affinity-purification mass spectrometry. Proteome Sci 2010; 8: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paddock MN, Bauman AT, Higdon R, Kolker E, Takeda S, Scharenberg AM. Competition between PARP-1 and Ku70 control the decision between high-fidelity and mutagenic DNA repair. DNA Repair (Amst) 2011; 10: 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lozano R, Castro E, Aragon IM, Cendon Y, Cattrini C, Lopez-Casas PP et al. Genetic aberrations in DNA repair pathways: a cornerstone of precision oncology in prostate cancer. Br J Cancer 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pestell TG, Jiao X, Kumar M, Peck AR, Prisco M, Deng S et al. Stromal cyclin D1 promotes heterotypic immune signaling and breast cancer growth. Oncotarget 2017; 8: 81754–81775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Casimiro MC, Di Sante G, Ju X, Li Z, Chen K, Crosariol M et al. Cyclin D1 Promotes Androgen-Dependent DNA Damage Repair in Prostate Cancer Cells. Cancer Res 2016; 76: 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kravtsov O, Hartley CP, Comperat EM, Iczkowski KA. KIF3B protein expression loss correlates with metastatic ability of prostate cancer. Am J Clin Exp Urol 2019; 7: 178–181. [PMC free article] [PubMed] [Google Scholar]
- 69.Wu K, Chen K, Wang C, Jiao X, Wang L, Zhou J et al. Cell fate factor DACH1 represses YB-1-mediated oncogenic transcription and translation. Cancer Res 2014; 74: 829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ai J, Pascal LE, Wei L, Zang Y, Zhou Y, Yu X et al. EAF2 regulates DNA repair through Ku70/Ku80 in the prostate. Oncogene 2017; 36: 2054–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu Q, Li A, Yu S, Qin S, Han N, Pestell RG et al. DACH1 antagonizes CXCL8 to repress tumorigenesis of lung adenocarcinoma and improve prognosis. J Hematol Oncol 2018; 11: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oliver MH, Harrison NK, Bishop JE, Cole PJ, Laurent GJ. A rapid and convenient assay for counting cells cultured in microwell plates: application for assessment of growth factors. J Cell Sci 1989; 92 ( Pt 3): 513–518. [DOI] [PubMed] [Google Scholar]
- 73.Li Z, Jiao X, Wang C, Shirley LA, Elsaleh H, Dahl O et al. Alternative cyclin D1 splice forms differentially regulate the DNA damage response. Cancer Res 2010; 70: 8802–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, Clement MV. Corrigendum to OpenComet: An automated tool for comet assay image analysis [Redox Biol. Volume 2, 2014, Pages 457–465]. Redox Biol 2021; 40: 101876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou J, Liu Y, Zhang W, Popov VM, Wang M, Pattabiraman N et al. Transcription elongation regulator 1 is a co-integrator of the cell fate determination factor Dachshund homolog 1. J Biol Chem 2010; 285: 40342–40350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 2011; 12: R41. [DOI] [PMC free article] [PubMed] [Google Scholar]