

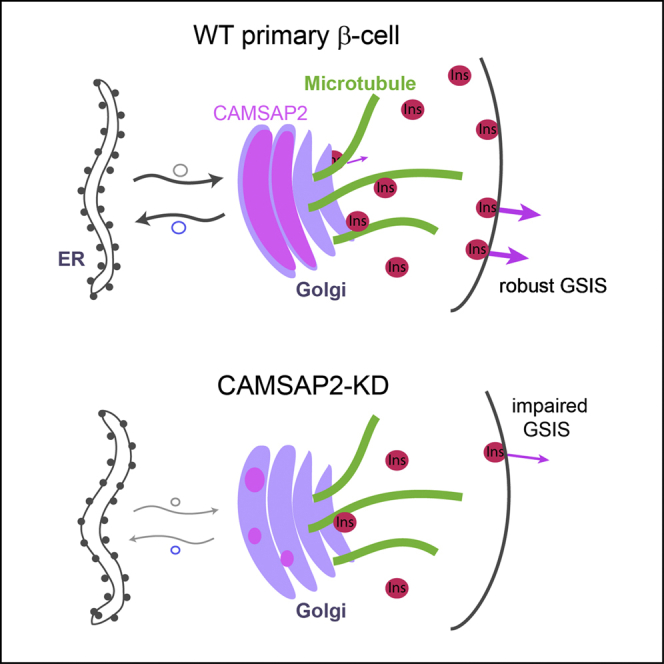
Summary
Glucose stimulation induces the remodeling of microtubules, which potentiates insulin secretion in pancreatic β-cells. CAMSAP2 binds to microtubule minus ends to stabilize microtubules in several cultured clonal cells. Here, we report that the knockdown of CAMSAP2 in primary β-cells reduces total insulin content and attenuates GSIS without affecting the releasability of insulin vesicles. Surprisingly, CAMSAP2 knockdown does not change microtubule stability. Unlike in cultured insulinoma cells, CAMSAP2 in primary β-cells predominantly localizes to the Golgi apparatus instead of microtubule minus ends. This novel localization is specific to primary β- but not α-cells and is independent of microtubule binding. Consistent with its specific localization at the Golgi, CAMSAP2 promotes efficient Golgi-ER trafficking in primary β-cells. Moreover, primary β-cells and insulinoma cells likely express different CAMSAP2 isoforms. We propose that a novel CAMSAP2 isoform in primary β-cells has a non-canonical function, which promotes Golgi-ER trafficking to support efficient production of insulin and secretion.
Subject areas: Biological sciences, Diabetology, Cell biology
Graphical abstract
Highlights
-
•
CAMSAP2 localizes to the Golgi apparatus in primary β-cells independent of microtubules
-
•
The localization of CAMSAP2 at the Golgi is specific to primary β- but not α-cells
-
•
CAMSAP2 facilitates Golgi-ER trafficking to support efficient insulin production
-
•
Knockdown of CAMSAP2 reduces total insulin and attenuates GSIS
Biological sciences; Diabetology; Cell biology
Introduction
Pancreatic islets are key regulators of glucose homeostasis. After a meal, glucose transported into pancreatic β-cells is metabolized to induce exocytosis of insulin secretory vesicles already existing in the cytoplasm.1,2,3 Insulin promotes the mobilization and usage of glucose in peripheral tissues to clear blood sugar.4,5 Thus, efficient biogenesis of insulin vesicles and proper stimulation-secretion coupling in β-cells are the prerequisite for avoiding diabetes, a disease featured by sustained high glucose in circulation.
The synthesis of insulin starts with the translation of its mRNA into preproinsulin, which is imported into the ER and processed to generate proinsulin.6 After proper folding and the formation of three intramolecular disulfide bonds, proinsulin is transported to the Golgi apparatus [referred to as Golgi in short].7 In the Golgi, proinsulin assembles into hexamers and is packed into secretory vesicles together with other vesicular proteins such as Ca2+ sensors.8,9 Further proteolytic cleavage generates C-peptide and mature insulin inside the secretory vesicles, which are thereafter stored in β-cell cytoplasm awaiting release.10,11,12
Islet β-cells store thousands of insulin vesicles, a portion of which is released during a biphasic response to glucose stimuli.13 The first phase of glucose-stimulated insulin secretion (GSIS) that lasts a few minutes utilizes mostly vesicles that are pre-docked or very close to the plasma membrane and can readily respond to calcium influx [the readily releasable pool, RRP].14,15,16 The second phase secretion utilizes both vesicles from RRP and ones stored deeper in the cytoplasm that needed mobilization to reach cell periphery [the reserved pool, RP].17,18 In both phases, “young” insulin granules generated within several hours before the stimulation are most efficiently utilized.19,20 Thus, renewal of stored insulin vesicles is critical to the ability of β-cells to function and survive under metabolic stress. To provide sustainable β-cell functions, glucose metabolism triggers insulin biosynthesis that replenishes and renews the insulin pools.21 Efficient processing of insulin through the ER-Golgi trafficking is necessary for this process.
Besides metabolic signals, GSIS is tightly regulated by the cytoskeleton. Both actin filaments and microtubules mediate the transportation of insulin vesicles by molecular motors and can restrict their docking to the plasma membrane.22,23,24,25 Microtubules are biological polymers comprise of directionally aligned α-/β-tubulin heterodimers forming an α-tubulin exposed (minus) end and a β-tubulin exposed (plus) end.26 The orientation of microtubules and positioning of their ends inside a cell determine the routes of vesicle transportation by plus or minus end-directed motors. Microtubules regulate both phases of GSIS by controlling the transportation of insulin vesicles toward/away from secretion sites and the selectivity of young over aged vesicles.23,27,28,29,30 Additionally, microtubule nucleation regulated by cAMP/EPAC signaling facilitates insulin vesicles budding from the Golgi.31 Although some mechanistic details of microtubule-mediated insulin vesicle biogenesis are known, the roles of many specific microtubule-binding proteins, which regulate fundamental properties of microtubules, have not been explored yet.
Microtubule plus ends can polymerize at relatively low tubulin concentrations, which allows its efficient elongation and stochastic dynamics (dynamic instability) in cells.32 Minus ends, in contrast, have a slower polymerization kinetics and require a higher tubulin concentration for growth. Accordingly, at low physiological tubulin concentrations in cells, minus ends are prone to depolymerization, unless protected. For long-lasting microtubules, minus ends are either capped by a γ-tubulin ring complex or stabilized by minus end-targeting proteins [-TIPs].33,34
CAMSAPs (CAlModulin-regulated Spectrin-Associated Proteins) are -TIPs presented by three paralogues in mammals, CAMSAP1, 2, and 3,35 which stabilize microtubule minus ends.36,37 CAMSAP1 tracks growing minus ends, whereas CAMSAP2 and CAMSAP3 remain bound to microtubule lattice of growing minus ends, which recruit more CAMSAPs and overtime form short stretches of CAMSAP-decoration.38,39 These properties lead to essential roles of CAMSAPs in organizing non-centrosomal microtubule arrays in epithelial cells and neurons.40,41,42,43 Specifically, CAMSAP2 promotes the anchoring of microtubule minus ends to the Golgi in retinal pigment epithelial cells through interacting with Golgi proteins AKAP450 and myomegalin.44 Intriguingly, polymorphisms in the CAMSAP2 locus are associated with type 1 diabetes in human.45 These findings, together with the established importance of microtubules in GSIS, led us to pursue its roles in islet β-cells. We report that CAMSAP2 exhibits a novel subcellular localization at the Golgi in primary β-cells independent of binding to microtubules. Further analyses revealed CAMSAP2 is required for efficient Golgi-ER trafficking, which facilitates robust insulin biosynthesis and GSIS.
Results
Knockdown of CAMSAP2 in primary islet β-cells impairs insulin secretion
To test whether CAMSAP2 is required for primary β-cells secretion, we used lentivirus to deliver CAMSAP2-targeting shRNAs for specific knockdown and examined stimulated secretion of these cells. To maximize the amount of islet cells accessible to lentivirus, isolated mouse islets were dissociated into individual cells, infected with lentivirus, and re-aggregated to form pseudoislets (Figures 1A and 1B). This process allows efficient infection of islet cells by lentivirus (60-70% of cells in a pseudoislet). The cell-cell interactions inside pseudoislets provide a niche similar to inside an intact islet and is a good platform to examine the secretion of β-cells.46 Compared to those infected with non-targeting control shRNA (control cells), islet cells infected with CAMSAP2-targeting shRNA (CAMSAP2-KD cells) show reduced CAMSAP2 immunofluorescence signal (Figures 1C and S1). These results demonstrate the specificity of CAMSAP2 antibodies while showing the feasibility of knocking down CAMSAP2 in primary islet β-cells. Importantly, pseudoislets comprise cells expressing either of the two CAMSAP2-targeting shRNAs that do not have altered basal insulin secretion, but exhibit significantly impaired GSIS when compared to the control (Figure 1D). These results suggest that CAMSAP2 is required for robust GSIS.
Figure 1.

Knockdown of CAMSAP2 in primary β-cells impairs glucose-stimulated insulin secretion but not cAMP- or potassium-stimulated secretion
(A and B) Representative fluorescence images of a pseudoislet generated from dissociated islet cells transduced with lentivirus containing shRNA and a tGFP expression cassette.
(C) Quantification of CAMSAP2 intensity of transduced primary β-cells in mouse islets. Dots represent individual β-cells. ∗p < 0.05, ∗∗∗p < 0.001 (Dunnett’s multiple comparisons test).
(D) Insulin secretion of pseudoislets expressing non-targeting (shCtrl) or CAMSAP2-targeting (shCAM) shRNA, shown as ng insulin secretion per IEQ within a 45 or 30 minutes window. Dots represent individual wells containing multiple pseudoislets. Columns represent the average of three animals ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (multiple comparisons corrected by the false discovery rate of Benjamini and Hochberg). Pseudoislets were incubated in 2.8 glucose for 60 min, in 16.5 mM glucose with or without 100 nM Liraglutide for 45 min, or in 2.8 mM glucose plus 30 mM KCl for 30 min. Secretion was normalized by the total pseudoislet volume presented in IEQ (defined as 1,767,146 μm3).
We next tested whether insulin vesicles in CAMSAP2-KD pseudoislets lack essential components for secretion. Because acute increase in cAMP or Ca2+ levels leads to the release of only secretion-competent insulin vesicles,9,47 we analyzed secretion responses to cAMP and Ca2+ in CAMSAP2-KD cells. We stimulated pseudoislets with Liraglutide, a GLP-1 analog that promotes GSIS mainly through the increasing cAMP level, and found that it enhances GSIS of both control and CAMSAP2-KD pseudoislets (Figure 1D). Elevated K+ depolarizes β-cell membrane to induce Ca2+ influx. We found that KCl induces similar levels of secretion in CAMSAP2-KD and in control pseudoislets (Figure 1D). These results suggest that CAMSAP2 does not regulate the responsiveness of insulin vesicle to cAMP or Ca2+ influx. We hypothesized that CAMSAP2 may influence GSIS by regulating the biogenesis of insulin vesicles and/or the replenishment of the releasable pool of insulin during secretion through modulating microtubule dynamics and tested them later in discussion.
CAMSAP2 expressed in primary islet β-cells does not regulate microtubule stabilization and architecture
CAMSAP2 is known as a microtubule minus end-binding protein and regulates microtubule dynamics and positioning.33 We first tested whether CAMSAP2 regulates the amount and stability of microtubules in islet β-cells. We incubated intact mouse islets with lentivirus carrying shRNAs and individual transduced β-cells on the surface of each islet were analyzed. To evaluate the level of microtubules in transduced cells, islets were first extracted with cold methanol to remove free tubulin in the cytosol and then fixed with paraformaldehyde. We compared CAMSAP2-KD to control β-cells and did not observe a clear difference in the organization of the microtubule network or a significant change in the amount of polymerized tubulin evaluated by mean tubulin intensity after methanol extraction (Figures 2A-2G).
Figure 2.

Knockdown of CAMSAP2 does not alter the amount and dynamics of microtubules in primary β-cells
(A-F) Immunofluorescence staining of tubulin (magenta) and insulin (white) in CAMSAP2-KD and control β-cells in mouse islets. Green, tGFP to indicate successful transduction. Dashed lines delineate transduced β-cells.
(G) Quantification of tubulin immunofluorescence intensity of CAMSAP2-KD and control β-cells in mouse islets. A.U., Arbitrary unit. Islets were extracted with methanol, which removed cytosolic free tubulin and the measured tubulin intensity represents polymerized tubulin. Dots represent individual β-cells. Bars represent the average of three animals (Dunnett’s multiple comparisons test).
(H) Quantification of detyrosinated tubulin immunofluorescence intensity in CAMSAP2-KD and control β-cells in mouse islets. Dots represent individual β-cells. Bars represent the average of three animals. ∗p < 0.05, ∗∗∗p < 0.001 (Dunnett’s multiple comparisons test).
(I-T) Immunofluorescence staining of detyrosinated tubulin (magenta), E-Cadherin (blue), and insulin (white) in CAMSAP2-KD and control β-cells in mouse islets incubated in media containing 2.8 or 11 mM glucose. Green, tGFP to indicate successful transduction. Dashed lines delineate transduced β-cells.
We next tested whether the lifetime of microtubules, a readout for their stability, is affected by CAMSAP2-KD. We measured the level of detyrosinated tubulin, which has the C-terminal tyrosine of α-tubulin removed, in microtubules. This modification occurs only to polymerized tubulin and its accumulation indicates long-lived microtubules.48 We found no difference in the levels of detyrosinated tubulin between CAMSAP2-KD and control β-cells incubated in low (2.8 mM) or high (11 mM) glucose concentrations (Figures 2H-2T). Moreover, CAMSAP2-KD does not affect the glucose-induced microtubule turnover, which we previously reported to be essential for robust GSIS,27 as evidenced by a significant reduction of detyrosinated tubulin level in both CAMSAP2-KD and control β-cells when stimulated with 11 mM glucose (Figures 2H-2T). These results suggest that CAMSAP2 is not involved in glucose-dependent microtubule destabilization and indicate that CAMSAP2 does not noticeably contribute to microtubule organization, lifetime, and stability in primary β-cells.
CAMSAP2 is enriched at the Golgi in primary β-cells
In contrast to the reported function of CAMSAP2 in stabilizing microtubule minus ends, CAMSAP2-KD in primary β-cells does not alter microtubule abundance and stability. We next examined the subcellular localization of CAMSAP2 in β-cells. MIN6 and INS1 cells are transformed β-cells with detectable but compromised GSIS. In interphase MIN6 cells, CAMSAP2 forms short stretches or puncta at microtubule ends (Figures S2A-S2D). In dividing MIN6 cells at telophase, we observed the CAMSAP2 signal at the distal ends of the mid-body, confirming the binding of CAMSAP2 to microtubule minus ends in MIN6 cells (Figure S2E). In INS-1 cells, we observed a similar pattern of CAMSAP2 puncta/stretches in the cytoplasm (S2F-G). These results are consistent with previously reported CAMSAP2 localization in cultured non-β-cells.38
Strikingly, we did not detect CAMSAP2 puncta/stretches in the cytoplasm of primary β-cells. Instead, the CAMSAP2 signal overlaps with Golgi markers, such as GM130 and GCC185 (Figures 3A-3F). This localization is specific to CAMSAP2 in contrast to the other two CAMSAP paralogues, CAMSAP1 and CAMSAP3, which are scattered in the cytoplasm of primary β-cells (Figures S3A and S3B). To confirm specific immuno-detection of CAMSAP2, we ectopically expressed EGFP-tagged CAMSAPs in MIN6 cells and compared the fluorescence signal with that from immunofluorescence staining. We observed strong co-localization of CAMSAP2 immunofluorescence signal with EGFP-CAMSAP2, but not with EGFP-CAMSAP1 or 3 (Figures S3C-S3E). These results, together with the reduced CAMSAP2 signals in CAMSAP2-KD cells, demonstrate the specificity of utilized antibodies.
Figure 3.

CAMSAP2 localizes to the Golgi in primary β-cells
(A-E) Immunofluorescence staining of GM130 (green), CAMSAP2 (magenta), and GCC185 (cyan) in primary β-cells in mouse islets. Blue, DAPI.
(F) Pearson’s correlation coefficient of the immunofluorescence signal of CAMSAP2 with GM130 (dark gray) and CAMSAP2 with insulin (light gray). Dots represent individual images of mouse islets. Columns represent the average of three animals. ∗∗∗p < 0.01 (t-test).
(G) Quantification of CAMSAP2 immunofluorescence intensity at the Golgi in mouse islets. Dots represent individual β-cells. Bars represent the average of three animals. ∗∗∗p < 0.001 (t test). A.U., Arbitrary unit.
(H) Pearson’s correlation coefficient of the immunofluorescence signal of CAMSAP2 with GM130 in primary β- and α-cells in mouse islets. Dots represent individual islet cells. Columns represent the average of three animals. ∗∗∗p < 0.01 (t test).
(I-N) Immunofluorescence staining of insulin (magenta), glucagon (blue), CAMSAP2 (red), and GM130 (green) in mouse islets.
(O-Q) Immunofluorescence staining of GM130 (green) and CAMSAP2 (magenta) in primary β-cells dissociated from mouse islets and cultured for one day.
Interestingly, the high level of CAMSAP2 at the Golgi is specific to primary β-cells but not to α-cells in the same islets (Figures 3G-3N). These results and the findings that CAMSAP2-KD compromises GSIS suggest that the localization of CAMSAP2 at the Golgi may be associated with its function to promote robust insulin secretion. Because CAMSAP2 exhibit different subcellular localization in primary β-cells and in MIN6 cells, we also tested whether CAMSAP2-KD in MIN6 affects GSIS. Unlike in primary β-cells, CAMSAP2-KD does not significantly impair GSIS in MIN6 (Figures 1D and S2H). Because CAMSAP2 in MIN6 cells localizes to microtubule minus ends, it is likely other minus end-binding proteins, such as CAMSAP3, may compensate for CAMSAP2-KD in MIN6 cells. It is noteworthy that MIN6 cells expressing one of the shCAMSAP2 constructs (#1) exhibit elevated basal secretion and significantly reduced fold change of stimulated over basal secretion when compared to control cells (Figure S2I). This phenotype is distinct from that in CAMSAP2-KD primary β-cells, which do not have altered basal secretion but significantly reduced stimulated secretion (Figure 1D). These different phenotypes of insulin secretion in CAMSAP2-KD primary β-cells and MIN6 cells support the hypothesis that the Golgi-localized CAMSAP2 in primary β-cells and microtubule minus end-binding CAMSAP2 in MIN6 may have differential functions.
Golgi association of CAMSAP2 in primary islet β-cells is microtubule-independent
We next asked why primary β-cells and immortalized β-cells have different CAMSAP2 localization. MIN6 and INS-1 do not have cell-cell interactions with other types of islet endocrine cells, which has been shown to influence β-cells secretion and cytoskeletal configuration.49,50 To test whether cell-cell interactions underlie the observed difference, we examined the subcellular localization of CAMSAP2 in primary β-cells dissociated from mouse islets. We found that CAMSAP2 still localizes to the Golgi In dissociated primary β-cells cultured for one day (Figures 3O-3Q). These results indicate that the unique localization of CAMSAP2 at the Golgi is intrinsic to primary β-cells.
The Golgi is the major microtubule nucleation site in primary β-cells.27 We next tested whether the accumulation of CAMSAP2 at the Golgi represents highly organized microtubule minus ends anchored to the Golgi. To test this possibility, isolated islets were treated with nocodazole or low temperature to induce microtubule disassembly. We found that neither perturbation alters the preferential association of CAMSAP2 with the Golgi, although the absence of microtubules led to the disassembly of the Golgi into smaller mini-stacks (Figures 4A-4F, S4A, and S4B). The removal of nocodazole allows microtubules to emerge from the Golgi, but it also does not change the localization of CAMSAP2 (Figures 4N-O). These results indicate that the localization of CAMSAP2 at the Golgi is independent of microtubules. In contrast, treating islets with Brefeldin A, which disrupts the Golgi due to inhibited ER-to-Golgi anterograde trafficking,51 abolishes detectable CAMSAP2 localization to a particular organelle, while not affecting microtubules (Figures 4G-4I and 4P-4Q). When we removed Brefeldin A in the presence of nocodazole to allow the anterograde trafficking and Golgi re-formation without microtubules, CAMSAP2 accumulates at nascent Golgi mini-stacks at a level comparable to that of untreated islet β-cells (Figures 4J-4M). We examined the protein levels of CAMSAP2 in MIN6 and in primary β-cells and found no change after incubation with different glucose concentrations, nocodazole, or Brefeldin A (Figures S4C-S4E). These results suggest that the disappearance of accumulated CAMSAP2 at the Golgi after Brefeldin A treatment is not due to its degradation. Collectively, these results indicate that microtubules do not influence CAMSAP2 localization at the Golgi in primary β-cells and suggest a yet-unknown function for CAMSAP2 at this location.
Figure 4.

The localization of CAMSAP2 to the Golgi in primary β-cells is independent of binding to microtubules
(A-L) Immunofluorescence staining of GM130 (green), CAMSAP2 (magenta), and insulin (white) in mouse islets treated with nocodazole (D-F), Brefeldin A [BfA] (G-I), or BfA and then removed BfA in the presence of nocodazole (J-L). Cyan, DAPI.
(M) Quantification of CAMSAP2 immunofluorescence intensity at the Golgi over that in the cytosol in primary β-cells in mouse islets. Dots represent individual β-cells. Columns represent the average of three animals (One-way ANOVA).
(N and O) Immunofluorescence staining of CAMSAP2 (magenta) and tubulin (green) in mouse islets. Islets treated with nocodazole were then incubated in media without nocodazole to allow microtubule growth. Panel O is a 3D reconstituted image of Golgi mini-stacks. Cyan, DAPI.
(P and Q) Immunofluorescence staining of tubulin (green), CAMSAP2 (magenta), and insulin (white) in mouse islets treated with Brefeldin A. Cyan, DAPI.
CAMSAP2 facilitates the Golgi-to-endoplasmic reticulum trafficking
Our data show that CAMSAP2 in primary β-cells does not regulate microtubule stability and its localization to the Golgi is independent of microtubules. We, therefore, explored other mechanisms by which CAMSAP2 may regulate GSIS. We first examined the total insulin content of CAMSAP2-KD β-cells. The average total insulin in CAMSAP2-KD β-cells is significantly lower than that in control β-cells (Figures S5A-S5H). In contrast, no difference in the level of proinsulin was observed amongst these cells (Figures S5I-S5P). These data suggest that CAMSAP2-KD impairs insulin biosynthesis after the stage of translation and production of proinsulin.
The conversion of proinsulin to insulin depends on ER-Golgi trafficking. Because CAMSAP2 is enriched at the Golgi in primary β-cells, we hypothesized that CAMSAP2 may regulate trafficking between the Golgi and ER. To test this hypothesis, we used Brefeldin A to inhibit the ER-to-Golgi anterograde trafficking. This is expected to reduce Golgi volume over time because of the constant retrograde transport and fusion of Golgi-derived vesicles to the ER. The cis-Golgi matrix protein, GM130, is not transported to the ER as the Golgi collapses and is released into the cytoplasm.52 We followed the level of GM130 in the cytoplasm as a readout of the rate of Golgi-to-ER retrograde trafficking in the presence of Brefeldin A (Figures 5A and 5B). The removal of Brefeldin A restores anterograde trafficking, which allows the incorporation of GM130 to the newly assembled Golgi and the reduction of its level in the cytoplasm (Figure 5C). Therefore, monitoring the disappearance of GM130 from the cytoplasm after Brefeldin A removal will reveal the rate of anterograde trafficking.
Figure 5.

Knockdown of CAMSAP2 impairs the ER-to-Golgi trafficking in primary islet β-cells
(A-C) Schematic of the dynamics of the Golgi and GM130 (magenta) localization before (A), after Brefeldin A addition (B), and after its removal (C).
(D-I) Immunofluorescence staining of GM130 (magenta) in Brefeldin A (BfA)-treated islets β-cells expressing shRNAs. Dashed lines delineate transduced cells. Cyan, Ins2 promoter-driven H2B-mApple to label β-cells in islets; Green, tGFP to indicate successful transduction. (J-K) Quantification of cytoplasmic GM130 immunofluorescence intensity of transduced β-cells in mouse islets treated with Brefeldin A (J) and after Brefeldin A removal (K). The average GM130 intensity of the entire cell was used to normalize data across different time points. Dots represent the average of three animals. Bars represent SD. ∗∗p < 0.01, ∗∗∗p < 0.001 (Dunnett’s multiple comparisons test). A.U., Arbitrary unit.
(L and M) Immunofluorescence staining of GM130 (magenta) in BfA-treated islets β-cells expressing shRNAs incubated in media without BfA for 90 min. Dashed lines delineate transduced cells. Cyan, Ins2 promoter-driven H2B-mApple to label β-cells in islets; Green, tGFP to indicate successful transduction.
(N) Quantification of β-COP immunofluorescence intensity of CAMSAP2-KD and control β-cells in mouse islets. Dots represent individual β-cells. Bars represent the average of three animals. ∗∗∗p < 0.001 (Dunnett’s multiple comparisons test).
(O-Q) Immunofluorescence staining of β-COP (magenta) in islets β-cells expressing shRNAs. Dashed lines delineate transduced cells. Blue, DAPI; green, tGFP to indicate successful transduction.
In the presence of Brefeldin A, the increase of cytoplasmic GM130 intensity in CAMSAP2-KD cells is significantly slower than that in control β-cells, suggesting a defective retrograde transport when CAMSAP2 level is reduced (Figures 5D-5J). When Brefeldin A is removed, CAMSAP2-KD β-cells exhibit a slower reduction of cytoplasmic GM130 intensity compared to that in control β-cells, suggesting that CAMSAP2-KD compromises efficient anterograde ER-to-Golgi trafficking (Figures 5K-5M).
We next examined the level of β-COP, a subunit of COPI, which mediates Golgi to ER retrograde as well as anterograde transport in CAMSAP2-KD cells. Compared to controls, primary β-cells expressing CAMSAP2-targeting shRNAs have significantly increased β-COP (Figures 5N-5Q). These results are consistent with our model that CAMSAP2-KD impairs the trafficking between Golgi and ER leading to the accumulation of β-COP. Together, these results suggest that CAMSAP2-KD compromises the trafficking between the Golgi and ER, which likely contributes to the reduced insulin content without changing proinsulin levels.
Primary β-cells produce different isoforms of CAMSAP2 compared to MIN6 cells
We last examined why CAMSAP2 localizes to the Golgi in primary islet β-cells but at microtubule minus ends in insulinoma cells. When compared to MIN6 cells, CAMSAP2 from primary β-cells exhibits increased electrophoretic mobility on an acrylamide gel with an estimated molecular weight difference of around 10.6 kDa (Figure 6A). These results suggest that primary β-cells may express a different CAMSAP2 variant or have different post-translational modifications on CAMSAP2 protein, which confers its unique localization to the Golgi.
Figure 6.

Primary islet β-cells express a different CAMSAP2 isoform compared to MIN6 cells
(A) Immunoblotting against CAMSAP2 and GM130 in lysates from islets and from MIN6. The dashed line indicates the position of CAMSAP2 from islet lysates.
(B) A fluorescence image of ectopically expressed EGFP-CAMSAP2 (human CAMSAP2 isoform 1, green) in mouse islets. Magenta, Ins2 promoter-driven H2B-mApple to label β-cells in islets.
(C-F) Immunofluorescence staining of GM130 (magenta) and insulin (white) in primary β-cells expressing EGFP-CAMSAP2 (human CAMSAP2 isoform 1, green) in mouse islets. Blue, DAPI. Open arrow heads, CAMSAP2 stretches in cell periphery; closed arrow heads, CAMSAP2 stretches associated with the Golgi. Panel C and D are at the same x-y position and 0.8 μm apart on the z axis.
(G) Schematic of the tested EGFP fusions of full-length or truncated CAMSAP2.
(H-P) Immunofluorescence images of GM130 (magenta) in MIN6 cells ectopically expressed truncated EGFP-CAMSAP2 (green). Blue, DAPI. Panel H-J and K-L are confocal images of the same cells at focal planes 0.75 μm apart on the z axis.
To distinguish these possibilities, we ectopically expressed an EGFP-tagged human CAMSAP2 in mouse primary islet β-cells. We used a full-length cDNA that is the longest one reported in GenBank (NM001297707.3, human CAMSAP2 transcript variant 1). It differs from other reported full-length cDNAs with two extra exons coding for 11 and 9 amino acid residues (residues 216-226 and 381-396 out of the 1489 amino acids in the CAMSAP2 protein), respectively. The homolog of this protein is reported in mice (NP001334038.1). The ectopically expressed EGFP-CAMSAP2 forms small stretches in the cytoplasm of primary islet β-cells, suggesting that it localizes to microtubule minus ends (Figure 6B). A large portion of these CAMSAP2 stretches localizes to the cell periphery instead of near the cell center, suggesting that mouse primary β-cells have abundant microtubule minus ends in the cell periphery. To further examine the subcellular localization of EGFP-CAMSAP2, we stained for GM130 in mouse primary β-cells. We found that a minor population of CAMSP2 stretches is associated with the Golgi, representing microtubule minus ends anchored to it (Figures 6C-6F). The localization and pattern of the human CAMSAP2 variant 1 expressed in mouse primary β-cells are very similar to that of endogenous CAMSAP2 in MIN6 cells (S2A-D). Because the expressed EGFP-CAMSAP2 from the human variant 1 cDNA does not localize to the Golgi, we conclude that mouse primary β-cells do not post-translationally modify CAMSAP2 protein to alter its subcellular localization from microtubule minus ends to the Golgi. These data favor the hypothesis that the different electrophoretic mobility between the endogenous CAMSAP2 in mouse primary β-cells and in MIN6 is likely due to alternatively splicing, which may confer the differential subcellular localization of CAMSAP2.
The linear structure of CAMSAP2 includes an N-terminal CH (calponin homology) domain and a C-terminal CKK (The C-terminal domain common to CAMSAP1, KIAA1078, and KIAA1543) domain. The CKK domain is essential for the recognition and binding to microtubule minus ends.35 We hypothesized that the putatively longer MIN6 CAMSAP2 variant (Compared to the variant in primary β-cells) may have extra residues that allow its localization to microtubule minus ends but not being recruited to the Golgi. Although the removal of the C-terminal 683 residues, including the entire CKK domain, compromises its localization to microtubule minus ends, the truncated protein is not specifically recruited to the Golgi in MIN6 cells (Figures 6G-6M). The function of the CH domain is not fully understood, but the human CAMSAP2 is reported to have N-terminal alternatively spliced variants.38,53 We tested an N-terminal truncation missing 292 residues, without the majority of the CH domain, but it still localizes to microtubule ends and is not restricted to the Golgi (Figures 6G and 6N-6O). Unfortunately, we are not able to obtain the full-length cDNA of CAMSAP2 from mouse primary β-cells, likely due to the rareness and expected long length (>8 kb) of its message that lessens the success of reverse transcription, PCR, and RACE (rapid amplification of cDNA ends). Nevertheless, our data indicate the likely existence of novel CAMSAP2 cDNA isoform(s) in primary islet β-cells that localize to the Golgi and have a function in the ER-Golgi trafficking.
Discussion
Regulated microtubule polymerization, disassembly, and anchorage are essential for the proper function of many cells. CAMSAP2 was shown to regulate cell morphology and organelle distribution through modulating microtubule organization and anchorage in specialized cell types such as neurons, glial, and polarized epithelia cells.40,43,54 Here, we report a novel function of CAMSAP2 in primary β-cells in facilitating the trafficking between the Golgi and ER and therefore promoting efficient insulin vesicle biogenesis and robust GSIS. Our studies, in combination with the established association between CAMSAP2 polymorphism and type 1 diabetes45 underscore the unique roles of this protein in pancreatic islet β-cell function. Our findings also establish an example wherein a well-established microtubule-associated protein localizes to a previously unreported subcellular localization primarily through microtubule-independent mechanisms to regulate cellular functions.
The function of CAMSAP in stabilizing microtubules, which has been studied in detail in cells and in vitro, are based on its recruitment to uncapped microtubule minus ends.36,53 CAMSAP2 and CAMSAP3 bind to elongating minus ends and form short stabilized microtubule stretches.40 These CAMSAP-coated regions promotes the positioning of minus ends to specific cellular structures including the Golgi,44 apical membrane in epithelial cells,41 and glial varicosities.54 To date, all cellular functions of CAMSAP2 have been attributed to microtubule stabilization and anchorage. Accordingly, we initially propose that CAMSAP2 may contribute to the regulation of β-cell function through modulating microtubule.
In contrast to our hypothesis, we found that CAMSAP2 does not noticeably regulate microtubule stability and abundance in primary β-cells. This finding might have been a manifestation of the redundancy of CAMSAP2 function, because in some specialized cells, CAMSAPs act redundantly with their paralogues or other microtubule stabilizing and anchoring proteins.55,56 Despite undisrupted microtubule network, we have determined that CAMSAP2-KD in primary β-cells compromises the trafficking between the Golgi and ER, which results in reduced total insulin content and impaired GSIS. This indicates that CAMSAP2 in primary β-cells has a unique function that cannot be compensated by its paralogues. Corresponding to this novel function and unlike other CAMSAP paralogues, CAMSAP2 in primary β-cells is largely restricted to the Golgi and this localization is independent of the presence of microtubules.
Intriguingly, the Golgi-localization of CAMSAP2 is only observed in primary β-cells, but not in MIN6, INS1, or non-β islet cells. The underlying reason for this difference is not known but it is likely related to the functional status of these cells. MIN6 and INS1 are immortalized insulinoma cells. They maintain detectable but relatively low levels of GSIS compared to primary β-cells57,58 and may not have efficient insulin vesicle biogenesis. While primary non-β islet cells have glucose-regulated hormone secretion, their response to glucose differs from β-cells (e.g., glucagon secretion is inhibited by high glucose). In addition, non-β islet cells do not experience the constant ER stress as β-cells caused by a high level of insulin mRNA translation and unfolded/misfolded proinsulin.59,60 Thus, it will be interesting in the future to examine whether the different CAMSAP2 localization is a cause for compromised insulin secretion in MIN6/INS1 cells and a factor that determines the biosynthesis and secretion properties of glucagon in α-cells.
We found that the knockdown of CAMSAP2 compromises both retrograde and anterograde Golgi-ER trafficking, which likely facilitates insulin biosynthesis at the stage of proinsulin transition to the Golgi. Because the anterograde transport requires tight coordination with the retrograde trafficking from the Golgi to the ER, CAMSAP2 localization to the Golgi supports the model that it facilitates ER-to-Golgi trafficking. Rab GTPases and their regulatory guanine nucleotide exchange factors (GEF) are essential for the targeting of vesicles in the Golgi-to-ER trafficking.61 Two large scale studies using affinity purification and proximity-dependent biotinylation respectively have identified several Rab GEF as CAMSAP2-interaction partners, including DENND1A, DENND4C,62 and DENND6A.63 We speculate that CAMSAP2 may facilitate the localization and/or functions of these GEFs at the Golgi to promote efficient Golgi-to-ER trafficking in primary β-cells, which can be explored in future studies.
Consistent with our model, we also found that the knockdown of CAMSAP2 in mouse primary β-cells increases the level of β-COP in the cytoplasm, suggesting a slower ER-Golgi trafficking. Importantly, COPI-mediated ER-Golgi trafficking depends on microtubules. Our data indicate that microtubules are not required for CAMSAP2 localization to the Golgi and CAMSAP2-KD does not significantly alter the amount and dynamics of total microtubules. However, because of the abundance of microtubules in β-cells, we may not be able to detect a significant reduction of microtubules even if CAMSAP2-KD destabilizes a sub-population of microtubules required for ER-Golgi trafficking. Therefore, we cannot rule out the possibility that CAMSAP2 may regulate the stability or positioning of a sub-population of microtubules between ER and Golgi required for trafficking. If this is the case, CAMSAP2 knockdown could compromise the Golgi-ER trafficking through both microtubule-independent and -dependent mechanisms.
Taking all findings together, we propose that CAMSAP2 is required for efficient insulin vesicle generation and secretion. Its knockdown impairs the Golgi-ER trafficking, reduce total insulin content and long-term β-cell functions. As a result, CAMSAP2-KD cells showed reduced secretion in response to glucose stimulation. This mechanism could contribute to the molecular connection between the reported CAMSAP2 polymorphism and human diabetes.45 Additionally, in some differentiated cell types, CAMSAP loss-of-function did not cause significant microtubule phenotypes,55 which may suggest unidentified microtubule-independent functions of CAMSAP in non-β-cells.
Limitations of the study
The underlying reason for the unique CAMSAP2 localization in primary β-cells is not known. Ectopically expressed human CAMSAP2 variant 1 cDNA construct in mouse primary β-cells forms stretches/puncta in the cytoplasm, suggesting this version of CAMSAP2 localizes to microtubule minus ends. These results also suggest that mouse primary β-cells do not post-translationally modify CAMSAP2 to alter its subcellular localization to the Golgi. Our western blot results show that CAMSAP2 from primary β-cells and from MIN6 cells have different electrophoretic mobility. Collectively, these data suggest that mouse primary β-cells may express an alternative CAMSAP2 transcript that is different from the reported human CAMSAP2 variant 1. Several versions of CAMSAP2 mRNAs have been reported in GenBank. Yet, testing the subcellular localization of truncated CAMSAP2 proteins could not identify the sequences that account for the Golgi-specific localization. Neither were we able to obtain the full-length CAMSAP2 cDNA from mouse primary islet cells. Thus, it remains possible that different mRNAs were transcribed or spliced to produce the Golgi-localized CAMSAP2 protein in primary β-cells. Although we favor this hypothesis, we cannot rule out the possibility that certain CAMSAP2-interacting proteins specifically expressed in primary β-cells may contribute to its localization to the Golgi. It is also unclear whether human primary islet β-cells express alternative CAMSAP2 variants localizing to the Golgi. A study compared human and mouse β-cells transcriptomes reports differential read coverage toward the 3′ end of CAMSAP2, suggesting the CAMSAP2 transcript in primary β-cells may also be different in these two species.64 While we don’t yet understand the mechanisms underlying the unique localization of CAMSAP2 in primary β-cells, it is likely to support the specialized function of CAMSAP2 to facilitate insulin secretion. Pursuing such a possibility in the future will be important to fully understand how cells use different isoforms of CAMSAP2 to perform different functions.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-α-Tubulin | Abcam (Cambridge, UK) | Cat# ab18251; RRID: AB_2210057 |
| Mouse monoclonal anti-α-Tubulin (clone DM1A) | MilliporeSigma, Burlington, MA | Cat# T9026; RRID: AB_477593 |
| Rabbit polyclonal anti-Detyrosinated tubulin | MilliporeSigma | Cat# AB3201; RRID: AB_177350 |
| Mouse monoclonal anti-E-Cadherin (clone 36) | BD Biosciences (San Jose, CA) | Cat# 610181; RRID: AB_397580 |
| Mouse monoclonal anti-Glucagon (clone K79bB10) | MilliporeSigma | Cat# G2654; RRID: AB_259852 |
| Guinea pig polyclonal anti-Insulin | Dako (Santa Clara, CA) | Cat# A0564; RRID: AB_10013624 |
| Mouse monoclonal anti-GM130 (Clone 35) | BD Biosciences | Cat# 610823; RRID: AB_398142 |
| Mouse monoclonal anti-Proinsulin | Developmental Studies Hybridoma Bank, Iowa City, IA | Cat# GS-9A8-C; RRID: AB_532383 |
| Rabbit polyclonal anti-CAMSAP1 | Novus Biologicals, Centennial, CO | Cat# NBP1-26645; RRID: AB_1852845 |
| Rabbit polyclonal anti-CAMSAP2 | Novus Biologicals | Cat# NBP1-21402; RRID: AB_1659977 |
| Rabbit polyclonal anti-CAMSAP3 | Thermo Fisher, Waltham, MA | Cat# PA5-48993; RRID: AB_2634449 |
| Guinea pig polyclonal anti-GCC185 | Covance research product Inc, Denver, PA | Cat# VU-140 (customized) |
| Mouse monoclonal anti-β-COP | MilliporeSigma | Cat# G6160; RRID: AB_477023 |
| Chemicals, peptides, and recombinant proteins | ||
| 0.25% Trypsin-EDTA | Gibco (Waltham, MA) | Cat# 25200 |
| Accumax | Innovative Cell Technologies (San Diego, CA) | Cat# AM-105 |
| Brefeldin A | MilliporeSigma, Burlington, MA | Cat# B5936 |
| CaCl2 | MilliporeSigma | Cat# C1016 |
| collagenase P | MilliporeSigma | Cat# C5138 |
| cOmplete™ protease inhibitor cocktail | Roche (Basel, Switzerland) | Cat# 04 693 124 001 |
| DAPI | Thermo Fisher, Waltham, MA) | Cat# 62247 |
| DMEM, high glucose medium | Thermo Fisher | Cat# 11995-065 |
| DMEM, no glucose medium | Thermo Fisher | Cat# 11966-025 |
| DMSO | MilliporeSigma | Cat# D8418 |
| Donor horse serum | Atlanta Biologicals (Flowery Branch, GA) | Cat# S12195 |
| Fetal Bovine Serum | Atlanta Biologicals | Cat# S11550 |
| Fibronectin | MilliporeSigma | Cat# F0162 |
| HBSS with Ca2+ & Mg2+ | Corning (Corning, NY) | Cat# 21-020-CV |
| HBSS without Ca2+ & Mg2+ | Corning | Cat# 21-022-CV |
| HEPES | Corning | Cat# 25-060-CI |
| Human extracellular matrix | Corning | Cat# 354237 |
| KCl | MilliporeSigma | Cat# PX1405 |
| KH2PO4 | Thermo Fisher | Cat# BP363 |
| Liraglutide | MedChemExpress (Monmouth Junction, NJ) | Cat# HY-P0014 |
| LiCl | Thermo Fisher | Cat# L121-100 |
| Methanol, anhydrous 99.8% | MilliporeSigma | Cat# 322415 |
| β-Mercaptoethanol | MilliporeSigma | Cat# M7522 |
| MgSO4 | MilliporeSigma | Cat# M7506 |
| NaCl | MilliporeSigma | Cat# S9625 |
| NaHCO3 | HyClone | Cat# SH30173.04 |
| Nocodazole | MilliporeSigma | Cat# M1404 |
| Opti-MEM® medium | Thermo Fisher | Cat# 31985-070 |
| Paraformaldehyde | MilliporeSigma | Cat# P6148 |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher | Cat# 15140-122 |
| Precision Plus Protein™ Dual Color Standards | Bio-Rad (Hercules, CA) | Cat# 1610374 |
| RPMI 1640 medium | Thermo Fisher | Cat# 11875-093 |
| RPMI 1640, no glucose medium | Thermo Fisher | Cat# 11879-020 |
| Saponin | MilliporeSigma | Cat# S4521 |
| SDS | MilliporeSigma | Cat# L3771 |
| TransIT®-Lenti | Mirus Bio (Madison, WI) | Cat# MIR 6606 |
| Triton X-100 | MilliporeSigma | Cat# T8787 |
| Trizma® | MilliporeSigma | Cat# T1503 |
| Tween® 20 | MilliporeSigma | Cat# P9416 |
| Vectashield mounting medium | Vactor Laboratories (Burlingame, CA) | Cat# H-1000 |
| Critical commercial assays | ||
| Coomassie Plus (Bradford) Assay | Thermo Fisher | Cat# 23236 |
| Mouse Ultrasensitive Insulin ELISA kit | ALPCO (Salem, NH) | Cat# 80-INSMSU |
| Deposited data | ||
| Original western blot and microscopy images | Mendeley Data | https://data.mendeley.com/datasets/x69hnss9ym/1 |
| Experimental models: Cell lines | ||
| INS1 | Asfari et al.58 | N/A |
| MIN6 | Miyazaki et al.67 | N/A |
| Experimental models: Organisms/strains | ||
| CD-1 (ICR) mice | Charles River Laboratories (Wilmington, MA) | N/A |
| Ins2H2B.Apple mice | Stancill et al.65 | N/A |
| Oligonucleotides | ||
| shCAM no #1 TRCN0000252418, 5′-TTGTCCGGCTAGAGGATATTT-3′ | MilliporeSigma | Gene ID: 67886 |
| shCAM no #2 TRCN0000252419, 5′-AGTTTCTCTGTCCGATTTAAA-3′ | MilliporeSigma | Gene ID: 67886 |
| shCtrl 5′-CCTAAGGTTAAGTCGCCCTCGCTC-3′] |
Addgene | 1864 |
| Recombinant DNA | ||
| pEGFP-C1-CAMSAP1 | This study | N/A |
| pEGFP-C1-CAMSAP2 | This study | N/A |
| pEGFP-CAMSAP3 | Jiang et al.38 | N/A |
| PCDH-EGFP-CAMSAP2 | This study | N/A |
| Software and algorithms | ||
| Nikon Elements | Nikon | N/A |
| Prism 9 | GraphPad | N/A |
| Other | ||
| 0.45 μm Cellulose Acetate Low Protein Binding filter | Corning | Cat# 430768 |
| Amaxa nucleofector II | Lonza (Walkersville, MD) | N/A |
| Amicon Ultra-15 Centrifugal Filter, Ultracel-100 regenerated cellulose membrane | MilliporeSigma | Cat# UFC910024 |
| GravityPLUS™ hanging drop 96-well microplate | PerkinElmer (Waltham, MA) | Cat# ISP-06-010 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Irina Kaverina irina.kaverina@vanderbilt.edu.
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mouse models
All Mouse experimentation followed protocols approved by the Vanderbilt University Institutional Animal Care and Use Committee. Euthanasia follows the standard protocol set by the AALAC and Isoflurane inhalation was used. Mice were fed a regular chow diet ad libitum and maintained on a 12-12h dark-light cycle. Wild-type CD-1 (ICR) mice were from Charles River Laboratories (Wilmington, MA). Ins2H2B.Apple mice have been described previously.65
Cell lines and primary cell culture
MIN667 and INS158 cells were grown in DMEM with 25 mM glucose (Gibco), 0.071 mM β-mercaptoethanol (MilliporeSigma, St Louis, MO), 10% HI-FBS, and 100 U/mL penicillin-100 μg/mL streptomycin. Primary β-cells were harvested from dissociation of isolated islets (see later in discussion in the islet isolation and in the pseudoislet generation procedure) and culture in islet media [RPMI-1640 media with 11 mM glucose (Gibco, Dublin, Ireland) plus 10% HI-FBS and 100 U/mL penicillin-100 μg/mL streptomycin (Gibco)] in a glass-bottom 35-mm dish (MatTek, Ashland, MA) coated with human extracellular matrix [ECM] (Corning). Primary β-cells were cultured over one night before fixation for imaging.
Method details
Small molecules, chemicals, and treatment
Liraglutide was from MedChemExpress (Monmouth Junction, NJ). Nocodazole, Brefeldin A, DMSO, KCl and other chemicals were from MilliporeSigma. For nocodazole treatment, isolated islets were incubated in islet media plus 5 mg/mL nocodazole for 2 h. For Brefeldin A treatment, isolated islets were incubated in islet media plus 10 μg/mL Brefeldin A for 2 h. To washout Brefeldin A, treated islets were transferred to islet media plus 5 mg/mL nocodazole three times and incubated for 2 h.
Plasmids, shRNA sequence, and lentivirus production
The CAMSAP2-targeting shRNA (shCAM) no #1 [TRCN0000252418, 5′-TTGTCCGGCTAGAGGATATTT-3'] and no #2 [TRCN0000252419, 5′-AGTTTCTCTGTCCGATTTAAA-3'] are in the plasmid pLKO.1-CMV-tGFP and are from MilliporeSigma. The non-targeting shRNA control (shCtrl) [5′-CCTAAGGTTAAGTCGCCCTCGCTC-3′] plasmid, pLKO.1-shCtrl-puro, is from Addgene (cat. # 1864). For expression of EGFP-CAMSAP fusion proteins, pEGFP-C1-CAMSAP2 and -CAMSAP1 plasmids were kind gifts from Dr. Stephen Norris (Calico Life Sciences) and pEGFP-CAMSAP3 was a kind gift from Dr. Anna Akhmanova (Utrecht University). For lentiviral packaging of EGFP-CAMSAP2, its cDNA was amplified with high-fidelity PCR from pEGFP-C1-CAMSAP2 and cloned into the multi-cloning site of pCDH-EF1 (Addgene, #72266). For EGFP fusion proteins with CAMSAP2 truncation, corresponding PCR fragments were obtained and ligated into the multi-cloning sites of pCDH-EF1 using the assembly approach. For lentivirus production, the package plasmid pMDL, pRSV, pVSV-G and the transfer plasmid were mixed in a 4:1:1:6 ratio and incubated with TransIT-Lenti reagent (Mirus Bio, Madison, WI) in Opti-MEM (Gibco). The assembled transfection complex was added to HEK293 cells at 80-95% confluency and incubated for 48 h. The harvested supernatant was precleared by filtering through a 0.45 μm-pore cellulose acetate membrane (Corning) and concentrated using Amicon Ultra-15 centrifugal filters (MilliporeSigma).
Islet isolation and culture
Islets were isolated from 8- to 12-week-old mice followed the published procedure.66 Briefly, pancreas was perfused through the common bile duct with 2 mL of 0.8 mg/mL collagenase P (Roche, Indianapolis, IN) in Hanks’ balanced salt solution [HBSS] with Ca2+ and Mg2+ (Corning, Corning, NY). The dilated pancreas was digested at 37°C for 20 min. The homogenate was washed with HBSS plus 10% heat-inactivated Fetal Bovine serum [HI-FBS] (Atlanta Biologicals, Flowery Branch, GA) four times and islets were handpicked into islet media (details see above in cell lines and primary cell culture) and cultured at 37°C with 5% CO2.
Islet dissociation, pseudoislets generation and transduction
To dissociate islets to primary cells, isolated mouse islets were recovered in islet media overnight, washed with HBSS without Ca2+ and Mg2+ for three times and incubated for 20 min, digested with Accumax (Innovative Cell Technologies, San Diego, CA) at 30°C for 10 min, and gently pipetted up and down for 10 times to dissociate cells. Dissociated islet cells were not sorted and the ratio of β and non-β endocrine cells were maintain as that in mouse islets. The cell suspension was spun at 1000g for 3 min, re-suspended in islet media, and mixed with concentrated lentivirus and 8 μg/mL DEAE-Dextran hydrochloride (MilliporeSigma). The mixture was loaded to the GravityPLUS hanging drop 96-well microplate (InSphero, Schlieren, Switzerland) and incubated for 4 days to generate pseudoislets. Pseudoislets were transferred to an ECM-coated 96-well plate and cultured for 1 day before analyses. For total pseudoislet volume calculation, two perpendicular diameters of each pseudoislet were measured and its volume was calculated using the equation 4/3π∗r1∗r2∗(r1+r2)/2, where r1 and r2 represent the two radii calculated from the measured diameters. The volume is converted to IEQ, which is defined as the volume of a spherical islet with a diameter of 150 μm and is equal to 1,767,146 μm3.68
GSIS assay
Pseudoislets were washed and conditioned in KRB buffer (111 mM NaCl, 4.8 mM KCl, 1.2 mM MgSO4, and 1.2 mM KH2PO4, 2.3 mM CaCl2, 25 mM NaHCO3, 10 mM HEPES and 0.2% BSA) plus 2.8 mM glucose (KRB-G2.8) for 1 h and incubated with fresh KRB-G2.8 for 1 h to assess basal secretion. After that, pseudoislets were incubated with KRB buffer plus 16.5 mM glucose (KRB-G16.5) for 45 min to assess glucose-stimulated secretion. Pseudoislets were then incubated with KRB-G16.5 plus 100 nM Liraglutide for 45 min, washed and conditioned in KRB-G2.8 for 45 min, and stimulated with KRB-G2.8 plus 30 mM KCl for 30 min. For GSIS in MIN6, cells were conditioned in KRB-G2.8 for 2 h, incubated with fresh KRB-G2.8 for 45 min and stimulated with KRB buffer plus 25 mM glucose for 45 min. Secreted insulin was quantified using ELISA kits from ALPCO (Salem, NH).
Immunofluorescence staining, microscopy, and quantification
For CAMSAP2 detection, samples were fixed with methanol at −20°C for 10 min. For the co-staining of CAMSAP2 with insulin or glucagon, samples were briefly fixed with 4% paraformaldehyde plus 0.1% saponin (MilliporeSigma) for 5 min and then fixed with methanol at −20°C for 10 min. For all other antigens, samples were fixed with 4% paraformaldehyde plus 0.1% saponin. Antibodies used are as follows: anti-α-tubulin (1:200, cat. #T9026, MilliporeSigma), anti-E-cadherin (1:250, #610181, BD Biosciences, San Jose, CA), CAMSAP1 (1:1000, #NBP1-26645, Novus Biologicals, Centennial, CO), CAMSAP2 (1:1000, #NBP1-21402, Novus Biologicals), CAMSAP3 (1:200 #PA5-48993, Thermo Fisher Scientific, Waltham, MA), anti-detyrosinated tubulin (1:250, #AB3201, MilliporeSigma), anti-glucagon (1:200, #G2654, MilliporeSigma), anti-GM130 (1:300, #610823, BD Biosciences), anti-insulin (1:500, #A0564, Dako, Santa Clara, CA), anti-proinsulin (1:30, #GS-9A8-C, Developmental Studies Hybridoma Bank, Iowa City, IA), anti-GCC185 (1:100, #VU-140 [customized], Covance research product Inc., Denver, PA), anti-β-COP (1:200, #G6160, MilliporeSigma). Secondary antibodies are from Invitrogen (1:200, Grand Island, NY). VECTASHIELD Mounting Medium was from Vector Labs (Burlingame, CA). All images, except in Figures 1A and 1B, were captured using Nikon Eclipse A1R laser scanning confocal microscope equipped with a CFI Apochromat TIRF 1003/1.45 oil objective. Images in Figures 1A and 1B were captured using EVOS FL microscope equipped with an LPlan PH2 203/0.4 objective. Images were quantified using Nikon NIS-Elements. The mean intensity (average fluorescence intensity per pixel after background subtraction) of the epitope in the region of interest (ROI) was measured. Background subtraction used the mean fluorescence intensity in a region without cells. For quantification of cytoplasmic GM130 immunofluorescence intensity in primary β-cell, a cell boarder ROI excluded the nucleus was first created to delineate a transduced β-cell on the tGFP (the transduction marker) channel. A second ROI was created on the GM130 channel to delineate GM130 positive area that is above a predetermined threshold that was kept consistent across cells expressing different shRNAs. Last, two small circular (measure) ROIs, 3 μm in diameter, were created between the first (cell boarder) and the second (Golgi) ROI on the tGFP channel. The average of the two measure ROIs on the GM130 channel was exported to represent the cytoplasmic GM130 intensity. For each β-cell, the average of three z-frames (+0.6 μm/-0.6 μm above and below the z-frame that shows the largest nuclear cross-section on the DAPI channel) was calculated for the statistical analysis.
Immunoblotting
Isolated mouse islets and MIN6 cells were lysed in Tris-lysis buffer (10 mM Tris-Cl pH 7.5, 100 mM NaCl, 1% Triton X-100, 10% glycerol, and cOmplete protease inhibitor cocktail [Roche, Basel, Switzerland]) on ice. For SDS-PAGE, 75 μg of total protein in the lysate was loaded per lane and was resolved in a 10% acrylamide gel. Antibodies used are as follows: anti-CAMSAP2 (1:500, #NBP1-21402, Novus Biologicals), anti-α-tubulin (1:1000, #ab18251, Abcam), anti-GM130 (1:200, #610823, BD Biosciences), IRDye 800 goat anti-rabbit IgG (1:5000, Rockland Immunochemicals, Pottstown, PA) and IRDye 700DX goat anti-mouse IgG (1:5000, LI-COR, Lincoln, NE). Blots were imaged using the Odyssey CLx imager (LI-COR).
Quantification and statistical analysis
All data represent three or more replicates of animals from separate experiments. Image quantification was performed in Nikon Elements and described in method details above. Statistical analyses were performed in GraphPad Prism. Student’s t test or ANOVA followed by multiple comparisons tests were used and specified in figure legends, with p < 0.05 considered significant.
Acknowledgments
This work is supported by National Institutes of Health, grants R01-DK106228 (to IK and GG), R35-GM127098 (to IK), DK72473, DK89523 (to MAM), P30-DK020593-44S1 (to KHH), CA-68485 and DK-20593 to the Vanderbilt Transgenic Mouse/ES Cell Shared Resource, and CA-68485 to the Vanderbilt Genome Sciences Resource. We thank Dr. Stephen Norris and Dr. Anna Akhmanova for kindly providing EGFP-CAMSAP plasmids for this study. We thank Hamida Ahmed for their supportive help.
Author contributions
KHH, IK, and GG conceptualized the work and designed the study. KHH designed and conducted the majority of experiments. AJ assisted the immunofluorescence, microscopy, and analysis of the islet detyrosinated tubulin assay. MY, AN, and GG conducted RT-PCR and RACE. ABO and MAM generated the Ins2H2B.Apple mice. KHH drafted the article. All authors contributed to writing the article.
Declaration of interests
The authors declare no conflicts of interest.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research.
Published: February 17, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.105938.
Contributor Information
Guoqiang Gu, Email: guoqiang.gu@vanderbilt.edu.
Irina Kaverina, Email: irina.kaverina@vanderbilt.edu.
Supplemental information
Data and code availability
-
•
This paper does not report original code.
-
•
This study did not generate new unique reagents, cell lines, or mouse lines.
-
•
Original western blot and microscopy images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
-
•
Plasmids generated in this study and any additional information required to reanalyze the data reported in this paper are available from the lead contact upon request.
References
- 1.Ashcroft F.M., Harrison D.E., Ashcroft S.J. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature. 1984;312:446–448. doi: 10.1038/312446a0. [DOI] [PubMed] [Google Scholar]
- 2.Cook D.L., Hales C.N. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature. 1984;311:271–273. doi: 10.1038/311271a0. [DOI] [PubMed] [Google Scholar]
- 3.Satin L.S., Cook D.L. Voltage-gated Ca2+ current in pancreatic B-cells. Pflügers Archiv. 1985;404:385–387. doi: 10.1007/BF00585354. [DOI] [PubMed] [Google Scholar]
- 4.Dean P.M., Matthews E.K. Electrical activity in pancreatic islet cells: effect of ions. J. Physiol. 1970;210:265–275. doi: 10.1113/jphysiol.1970.sp009208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malaisse W.J. Insulin secretion: multifactorial regulation for a single process of release. The Minkowski award lecture delivered on September 7, 1972 before the European Association for the study of Diabetes at Madrid, Spain. Diabetologia. 1973;9:167–173. doi: 10.1007/BF01219778. [DOI] [PubMed] [Google Scholar]
- 6.Chan S.J., Keim P., Steiner D.F. Cell-free synthesis of rat preproinsulins: characterization and partial amino acid sequence determination. Proc. Natl. Acad. Sci. USA. 1976;73:1964–1968. doi: 10.1073/pnas.73.6.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orci L. Macro- and micro-domains in the endocrine pancreas. Diabetes. 1982;31:538–565. doi: 10.2337/diab.31.6.538. [DOI] [PubMed] [Google Scholar]
- 8.Brill A.S., Venable J.H., Jr. The binding of transition metal ions in insulin crystals. J. Mol. Biol. 1968;36:343–353. doi: 10.1016/0022-2836(68)90160-5. [DOI] [PubMed] [Google Scholar]
- 9.Huang C., Walker E.M., Dadi P.K., Hu R., Xu Y., Zhang W., Sanavia T., Mun J., Liu J., Nair G.G., et al. Synaptotagmin 4 regulates pancreatic beta cell maturation by modulating the Ca(2+) sensitivity of insulin secretion vesicles. Dev. Cell. 2018;45:347–361.e5. doi: 10.1016/j.devcel.2018.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steiner D.F., Kemmler W., Tager H.S., Peterson J.D. Proteolytic processing in the biosynthesis of insulin and other proteins. Fed. Proc. 1974;33:2105–2115. [PubMed] [Google Scholar]
- 11.Hutton J.C. The internal pH and membrane potential of the insulin-secretory granule. Biochem. J. 1982;204:171–178. doi: 10.1042/bj2040171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orci L., Ravazzola M., Amherdt M., Madsen O., Perrelet A., Vassalli J.D., Anderson R.G. Conversion of proinsulin to insulin occurs coordinately with acidification of maturing secretory vesicles. J. Cell Biol. 1986;103:2273–2281. doi: 10.1083/jcb.103.6.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curry D.L., Bennett L.L., Grodsky G.M. Dynamics of insulin secretion by the perfused rat pancreas. Endocrinology. 1968;83:572–584. doi: 10.1210/endo-83-3-572. [DOI] [PubMed] [Google Scholar]
- 14.Ohara-Imaizumi M., Nishiwaki C., Kikuta T., Nagai S., Nakamichi Y., Nagamatsu S. TIRF imaging of docking and fusion of single insulin granule motion in primary rat pancreatic beta-cells: different behaviour of granule motion between normal and Goto-Kakizaki diabetic rat beta-cells. Biochem. J. 2004;381:13–18. doi: 10.1042/BJ20040434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shibasaki T., Takahashi H., Miki T., Sunaga Y., Matsumura K., Yamanaka M., Zhang C., Tamamoto A., Satoh T., Miyazaki J.I., Seino S. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc. Natl. Acad. Sci. USA. 2007;104:19333–19338. doi: 10.1073/pnas.0707054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohara-Imaizumi M., Fujiwara T., Nakamichi Y., Okamura T., Akimoto Y., Kawai J., Matsushima S., Kawakami H., Watanabe T., Akagawa K., Nagamatsu S. Imaging analysis reveals mechanistic differences between first- and second-phase insulin exocytosis. J. Cell Biol. 2007;177:695–705. doi: 10.1083/jcb.200608132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olofsson C.S., Göpel S.O., Barg S., Galvanovskis J., Ma X., Salehi A., Rorsman P., Eliasson L. Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B-cells. Pflügers Archiv. 2002;444:43–51. doi: 10.1007/s00424-002-0781-5. [DOI] [PubMed] [Google Scholar]
- 18.Rorsman P., Renström E. Insulin granule dynamics in pancreatic beta cells. Diabetologia. 2003;46:1029–1045. doi: 10.1007/s00125-003-1153-1. [DOI] [PubMed] [Google Scholar]
- 19.Rhodes C.J., Halban P.A. Newly synthesized proinsulin/insulin and stored insulin are released from pancreatic B cells predominantly via a regulated, rather than a constitutive, pathway. J. Cell Biol. 1987;105:145–153. doi: 10.1083/jcb.105.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yau B., Hays L., Liang C., Laybutt D.R., Thomas H.E., Gunton J.E., Williams L., Hawthorne W.J., Thorn P., Rhodes C.J., Kebede M.A. A fluorescent timer reporter enables sorting of insulin secretory granules by age. J. Biol. Chem. 2020;295:8901–8911. doi: 10.1074/jbc.RA120.012432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashcroft S.J., Bunce J., Lowry M., Hansen S.E., Hedeskov C.J. The effect of sugars on (pro)insulin biosynthesis. Biochem. J. 1978;174:517–526. doi: 10.1042/bj1740517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Varadi A., Ainscow E.K., Allan V.J., Rutter G.A. Involvement of conventional kinesin in glucose-stimulated secretory granule movements and exocytosis in clonal pancreatic beta-cells. J. Cell Sci. 2002;115:4177–4189. doi: 10.1242/jcs.00083. [DOI] [PubMed] [Google Scholar]
- 23.Varadi A., Tsuboi T., Johnson-Cadwell L.I., Allan V.J., Rutter G.A. Kinesin I and cytoplasmic dynein orchestrate glucose-stimulated insulin-containing vesicle movements in clonal MIN6 beta-cells. Biochem. Biophys. Res. Commun. 2003;311:272–282. doi: 10.1016/j.bbrc.2003.09.208. [DOI] [PubMed] [Google Scholar]
- 24.Thurmond D.C., Gonelle-Gispert C., Furukawa M., Halban P.A., Pessin J.E. Glucose-stimulated insulin secretion is coupled to the interaction of actin with the t-SNARE (target membrane soluble N-ethylmaleimide-sensitive factor attachment protein receptor protein) complex. Mol. Endocrinol. 2003;17:732–742. doi: 10.1210/me.2002-0333. [DOI] [PubMed] [Google Scholar]
- 25.Ivarsson R., Jing X., Waselle L., Regazzi R., Renström E. Myosin 5a controls insulin granule recruitment during late-phase secretion. Traffic. 2005;6:1027–1035. doi: 10.1111/j.1600-0854.2005.00342.x. [DOI] [PubMed] [Google Scholar]
- 26.Bergen L.G., Borisy G.G. Head-to-tail polymerization of microtubules in vitro. Electron microscope analysis of seeded assembly. J. Cell Biol. 1980;84:141–150. doi: 10.1083/jcb.84.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu X., Hu R., Brissova M., Stein R.W., Powers A.C., Gu G., Kaverina I. Microtubules negatively regulate insulin secretion in pancreatic beta cells. Dev. Cell. 2015;34:656–668. doi: 10.1016/j.devcel.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoboth P., Müller A., Ivanova A., Mziaut H., Dehghany J., Sönmez A., Lachnit M., Meyer-Hermann M., Kalaidzidis Y., Solimena M. Aged insulin granules display reduced microtubule-dependent mobility and are disposed within actin-positive multigranular bodies. Proc. Natl. Acad. Sci. USA. 2015;112:E667–E676. doi: 10.1073/pnas.1409542112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu R., Zhu X., Yuan M., Ho K.H., Kaverina I., Gu G. Microtubules and Galphao-signaling modulate the preferential secretion of young insulin secretory granules in islet beta cells via independent pathways. PLoS One. 2021;16 doi: 10.1371/journal.pone.0241939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trogden K.P., Lee J., Bracey K.M., Ho K.H., McKinney H., Zhu X., Arpag G., Folland T.G., Osipovich A.B., Magnuson M.A., et al. Microtubules regulate pancreatic beta-cell heterogeneity via spatiotemporal control of insulin secretion hot spots. Elife. 2021;10 doi: 10.7554/eLife.59912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trogden K.P., Zhu X., Lee J.S., Wright C.V.E., Gu G., Kaverina I. Regulation of glucose-dependent golgi-derived microtubules by cAMP/EPAC2 promotes secretory vesicle biogenesis in pancreatic beta cells. Curr. Biol. 2019;29:2339–2350.e5. doi: 10.1016/j.cub.2019.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burbank K.S., Mitchison T.J. Microtubule dynamic instability. Curr. Biol. 2006;16:R516–R517. doi: 10.1016/j.cub.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 33.Akhmanova A., Hoogenraad C.C. Microtubule minus-end-targeting proteins. Curr. Biol. 2015;25:R162–R171. doi: 10.1016/j.cub.2014.12.027. [DOI] [PubMed] [Google Scholar]
- 34.Akhmanova A., Steinmetz M.O. Microtubule minus-end regulation at a glance. J. Cell Sci. 2019;132 doi: 10.1242/jcs.227850. [DOI] [PubMed] [Google Scholar]
- 35.Baines A.J., Bignone P.A., King M.D.A., Maggs A.M., Bennett P.M., Pinder J.C., Phillips G.W. The CKK domain (DUF1781) binds microtubules and defines the CAMSAP/ssp4 family of animal proteins. Mol. Biol. Evol. 2009;26:2005–2014. doi: 10.1093/molbev/msp115. [DOI] [PubMed] [Google Scholar]
- 36.Goodwin S.S., Vale R.D. Patronin regulates the microtubule network by protecting microtubule minus ends. Cell. 2010;143:263–274. doi: 10.1016/j.cell.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meng W., Mushika Y., Ichii T., Takeichi M. Anchorage of microtubule minus ends to adherens junctions regulates epithelial cell-cell contacts. Cell. 2008;135:948–959. doi: 10.1016/j.cell.2008.09.040. [DOI] [PubMed] [Google Scholar]
- 38.Jiang K., Hua S., Mohan R., Grigoriev I., Yau K.W., Liu Q., Katrukha E.A., Altelaar A.F.M., Heck A.J.R., Hoogenraad C.C., Akhmanova A. Microtubule minus-end stabilization by polymerization-driven CAMSAP deposition. Dev. Cell. 2014;28:295–309. doi: 10.1016/j.devcel.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 39.Jiang K., Faltova L., Hua S., Capitani G., Prota A.E., Landgraf C., Volkmer R., Kammerer R.A., Steinmetz M.O., Akhmanova A. Structural basis of formation of the microtubule minus-end-regulating CAMSAP-katanin complex. Structure. 2018;26:375–382.e4. doi: 10.1016/j.str.2017.12.017. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka N., Meng W., Nagae S., Takeichi M. Nezha/CAMSAP3 and CAMSAP2 cooperate in epithelial-specific organization of noncentrosomal microtubules. Proc. Natl. Acad. Sci. USA. 2012;109:20029–20034. doi: 10.1073/pnas.1218017109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toya M., Kobayashi S., Kawasaki M., Shioi G., Kaneko M., Ishiuchi T., Misaki K., Meng W., Takeichi M. CAMSAP3 orients the apical-to-basal polarity of microtubule arrays in epithelial cells. Proc. Natl. Acad. Sci. USA. 2016;113:332–337. doi: 10.1073/pnas.1520638113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pongrakhananon V., Saito H., Hiver S., Abe T., Shioi G., Meng W., Takeichi M. CAMSAP3 maintains neuronal polarity through regulation of microtubule stability. Proc. Natl. Acad. Sci. USA. 2018;115:9750–9755. doi: 10.1073/pnas.1803875115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yau K.W., van Beuningen S.F.B., Cunha-Ferreira I., Cloin B.M.C., van Battum E.Y., Will L., Schätzle P., Tas R.P., van Krugten J., Katrukha E.A., et al. Microtubule minus-end binding protein CAMSAP2 controls axon specification and dendrite development. Neuron. 2014;82:1058–1073. doi: 10.1016/j.neuron.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 44.Wu J., de Heus C., Liu Q., Bouchet B.P., Noordstra I., Jiang K., Hua S., Martin M., Yang C., Grigoriev I., et al. Molecular pathway of microtubule organization at the Golgi apparatus. Dev. Cell. 2016;39:44–60. doi: 10.1016/j.devcel.2016.08.009. [DOI] [PubMed] [Google Scholar]
- 45.Onengut-Gumuscu S., Chen W.M., Burren O., Cooper N.J., Quinlan A.R., Mychaleckyj J.C., Farber E., Bonnie J.K., Szpak M., Schofield E., et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat. Genet. 2015;47:381–386. doi: 10.1038/ng.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reissaus C.A., Piston D.W. Reestablishment of glucose inhibition of glucagon secretion in small pseudoislets. Diabetes. 2017;66:960–969. doi: 10.2337/db16-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wiser O., Bennett M.K., Atlas D. Functional interaction of syntaxin and SNAP-25 with voltage-sensitive L- and N-type Ca2+ channels. EMBO J. 1996;15:4100–4110. [PMC free article] [PubMed] [Google Scholar]
- 48.Gundersen G.G., Khawaja S., Bulinski J.C. Postpolymerization detyrosination of alpha-tubulin: a mechanism for subcellular differentiation of microtubules. J. Cell Biol. 1987;105:251–264. doi: 10.1083/jcb.105.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu X., Yan F., Yao H., Chang M., Qin J., Li Y., Wang Y., Pei X. Involvement of RhoA/ROCK in insulin secretion of pancreatic beta-cells in 3D culture. Cell Tissue Res. 2014;358:359–369. doi: 10.1007/s00441-014-1961-2. [DOI] [PubMed] [Google Scholar]
- 50.Jain R., Lammert E. Cell-cell interactions in the endocrine pancreas. Diabetes Obes. Metabol. 2009;11:159–167. doi: 10.1111/j.1463-1326.2009.01102.x. [DOI] [PubMed] [Google Scholar]
- 51.Lippincott-Schwartz J., Donaldson J.G., Schweizer A., Berger E.G., Hauri H.P., Yuan L.C., Klausner R.D. Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell. 1990;60:821–836. doi: 10.1016/0092-8674(90)90096-w. [DOI] [PubMed] [Google Scholar]
- 52.Nakamura N., Rabouille C., Watson R., Nilsson T., Hui N., Slusarewicz P., Kreis T.E., Warren G. Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Atherton J., Jiang K., Stangier M.M., Luo Y., Hua S., Houben K., van Hooff J.J.E., Joseph A.P., Scarabelli G., Grant B.J., et al. A structural model for microtubule minus-end recognition and protection by CAMSAP proteins. Nat. Struct. Mol. Biol. 2017;24:931–943. doi: 10.1038/nsmb.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coquand L., Victoria G.S., Tata A., Carpentieri J.A., Brault J.B., Guimiot F., Fraisier V., Baffet A.D. CAMSAPs organize an acentrosomal microtubule network from basal varicosities in radial glial cells. J. Cell Biol. 2021;220 doi: 10.1083/jcb.202003151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sallee M.D., Zonka J.C., Skokan T.D., Raftrey B.C., Feldman J.L. Tissue-specific degradation of essential centrosome components reveals distinct microtubule populations at microtubule organizing centers. PLoS Biol. 2018;16 doi: 10.1371/journal.pbio.2005189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng Y., Buchwalter R.A., Zheng C., Wight E.M., Chen J.V., Megraw T.L. A perinuclear microtubule-organizing centre controls nuclear positioning and basement membrane secretion. Nat. Cell Biol. 2020;22:297–309. doi: 10.1038/s41556-020-0470-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng K., Delghingaro-Augusto V., Nolan C.J., Turner N., Hallahan N., Andrikopoulos S., Gunton J.E. High passage MIN6 cells have impaired insulin secretion with impaired glucose and lipid oxidation. PLoS One. 2012;7 doi: 10.1371/journal.pone.0040868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Asfari M., Janjic D., Meda P., Li G., Halban P.A., Wollheim C.B. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology. 1992;130:167–178. doi: 10.1210/endo.130.1.1370150. [DOI] [PubMed] [Google Scholar]
- 59.Harding H.P., Zeng H., Zhang Y., Jungries R., Chung P., Plesken H., Sabatini D.D., Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol. Cell. 2001;7:1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 60.Scheuner D., Vander Mierde D., Song B., Flamez D., Creemers J.W.M., Tsukamoto K., Ribick M., Schuit F.C., Kaufman R.J. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat. Med. 2005;11:757–764. doi: 10.1038/nm1259. [DOI] [PubMed] [Google Scholar]
- 61.Brandizzi F., Barlowe C. Organization of the ER-Golgi interface for membrane traffic control. Nat. Rev. Mol. Cell Biol. 2013;14:382–392. doi: 10.1038/nrm3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boldt K., van Reeuwijk J., Lu Q., Koutroumpas K., Nguyen T.M.T., Texier Y., van Beersum S.E.C., Horn N., Willer J.R., Mans D.A., et al. An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat. Commun. 2016;7 doi: 10.1038/ncomms11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Go C.D., Knight J.D.R., Rajasekharan A., Rathod B., Hesketh G.G., Abe K.T., Youn J.Y., Samavarchi-Tehrani P., Zhang H., Zhu L.Y., et al. A proximity-dependent biotinylation map of a human cell. Nature. 2021;595:120–124. doi: 10.1038/s41586-021-03592-2. [DOI] [PubMed] [Google Scholar]
- 64.Benner C., van der Meulen T., Cacéres E., Tigyi K., Donaldson C.J., Huising M.O. The transcriptional landscape of mouse beta cells compared to human beta cells reveals notable species differences in long non-coding RNA and protein-coding gene expression. BMC Genom. 2014;15:620. doi: 10.1186/1471-2164-15-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stancill J.S., Osipovich A.B., Cartailler J.P., Magnuson M.A. Transgene-associated human growth hormone expression in pancreatic β-cells impairs identification of sex-based gene expression differences. Am. J. Physiol. Endocrinol. Metab. 2019;316:E196–E209. doi: 10.1152/ajpendo.00229.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li D.S., Yuan Y.H., Tu H.J., Liang Q.L., Dai L.J. A protocol for islet isolation from mouse pancreas. Nat. Protoc. 2009;4:1649–1652. doi: 10.1038/nprot.2009.150. [DOI] [PubMed] [Google Scholar]
- 67.Miyazaki J., Araki K., Yamato E., Ikegami H., Asano T., Shibasaki Y., Oka Y., Yamamura K. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- 68.Ricordi C., Gray D.W., Hering B.J., Kaufman D.B., Warnock G.L., Kneteman N.M., Lake S.P., London N.J., Socci C., Alejandro R., et al. Islet isolation assessment in man and large animals. Acta Diabetol. Lat. 1990;27:185–195. doi: 10.1007/BF02581331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper does not report original code.
-
•
This study did not generate new unique reagents, cell lines, or mouse lines.
-
•
Original western blot and microscopy images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
-
•
Plasmids generated in this study and any additional information required to reanalyze the data reported in this paper are available from the lead contact upon request.