

Abstract
Background
Alterations of 11q23/KMT2A are the most prevalent cytogenetic abnormalities in acute myeloid leukemia (AML) and the prognostic significance of 11q23/KMT2A‐rearranged AML based on various translocation partners varies among different studies. However, few studies evaluated the molecular characteristics of 11q23/KMT2A‐rearranged pediatric AML. We aim to analyze the mutational landscape of 11q23/KMT2A‐rearranged AML and assess their prognostic value in outcomes.
Methods
The mutational landscape and clinical prognosis of 105 children with 11q23/KMT2A‐rearranged AML in comparison with 277 children with non‐11q23/KMT2A‐rearranged AML were analyzed using publicly accessible next‐generation sequencing data from Therapeutically Applicable Research to Generate Effective Treatments (TARGET) dataset.
Results
Pediatric AML patients with 11q23/KMT2A‐rearrangements harbored a low number of mutations (Median, 1 mutation/patient, range, 1‐22), 58% of which involved in RAS pathway mutations (KRAS, NRAS, and PTPN11) and 10.5% of which comprised of SETD2 mutations. Compared with non‐11q23/KMT2A‐rearranged AML, the incidence of KRAS (32.4% vs. 10.1%, P〈0.001) and SETD2 (10.5% vs. 1.4%, P=0.001) gene mutations in 11q23/KMT2A‐rearranged AML was significantly higher. Both KRAS and SETD2 mutations occurred more often in t(10;11)(p12;q23). KRAS mutations were correlated with worse 5‐year event‐free survival [EFS] (Plog‐rank = 0.001) and 5‐year overall survival [OS] (Plog‐rank = 0.009) and the presence of SETD2 mutations increases the 5‐year relapse rate (PGray = 0.004). Multivariate analyses confirmed KRAS mutations in 11q23/KMT2A‐rearranged AML as an independent predictor for poor EFS (hazard ratio [HR] = 2.10, P=0.05) and OS (HR = 2.39, P=0.054).
Conclusion
Our findings show that pediatric patients with 11q23/KMT2A rearrangements have characteristic mutation patterns and varying clinical outcomes depending on different translocation partners, which could be utilized to develop more accurate risk stratification and tailored therapies.
Keywords: 11q23/KMT2A , clinical outcome, gene mutations, pediatric acute myeloid leukemia, sequencing
For 11q23/KMT2A‐rearranged AML pediatric patients with FAB‐M5 morphology, patients with t(9;11)(p22;q23) demonstrated improved outcomes compared to those with other 11q23/KMT2A rearrangements. Patients with 11q23/KMT2A rearrangements have characteristic mutation patterns and varying clinical outcomes depending on different translocation partners.
1. INTRODUCTION
Acute myeloid leukemia (AML) is a heterogeneous malignancy frequently present with recurrent cytogenetic and genetic abnormalities. 1 , 2 AML with alterations of KMT2A gene (Lysine [K]‐specific methyltransferase 2A, also known as MLL gene), located at 11q23, accounts for 15% to 20% of all childhood leukemias and are of great interest because of their distinct clinical and biological features. 3 Morphologically, 11q23/KMT2A‐ rearranged AML is frequently classified as French‐American‐British (FAB) M4 and M5. To date, over 90 different 11q23/KMT2A rearrangements have been identified. The majority of 11q23/KMT2A rearrangements were seen in one of six common translocation partners: t(4;11)(q21;q23), t(6;11)(q27;q23), t(9;11)(p22;q23), t(10;11)(p12;q23), t(11;19)(q23;p13.1), t(11;19)(q23;p13.3). 4 , 5 Depending on the specific 11q23/KMT2A alterations, 11q23/KMT2A rearrangements correlate to a certain gene expression profile and prognoses. Fusion proteins of 11q23/KMT2A‐rearrangements dysregulate the expression of HOX cluster genes, contributing to self‐renewal potential in leukemic cells. 6 , 7 , 8
Over the past decades, only modest improvement in treatment outcomes for pediatric AML had been made in part because of a scarcity of targeted therapies and the slow development of immunotherapy. 9 Even though recent clinical trials have identified novel targets such as menin inhibitor in 11q23/KMT2A‐ rearranged AML to impair leukemia growth and proliferation, comprehensive genome analyses provide a more thorough biological understanding for 11q23/KMT2A‐rearranged AML, which contributes to the exploration of targeted therapy. 10 , 11 Recent studies by Lavallée et al. and Bill et al. identified a high frequency of RAS pathway mutations in adult AML with 11q23/KMT2A‐rearrangements. 12 , 13 However, due to the relatively low mutation rate in 11q23/KMT2A‐rearranged AML, mutational characteristics of pediatric AML patients with 11q23/KMT2A rearrangements are rarely studied and most collaborative cohort groups are too small for meaningful comprehensive molecular analyses of these patients. In addition, the prognostic value of gene mutations in 11q23/KMT2A‐rearranged AML is currently poorly understood.
Moreover, clinical evidence indicates that the translocation partners of 11q23/KMT2A are a primary determinant of outcome. Previous studies demonstrated t(9;11)(p22;q23) was correlated with an excellent prognosis albeit several studies could not validate these results. 14 , 15 , 16 , 17 , 18 , 19 Thus, there is a pressing need to identify the mutational landscape of 11q23/KMT2A‐rearranged pediatric AML and evaluate the prognostic effect of gene mutations and various 11q23/KMT2A translocation partners on a large scale.
To address this demand, we analyzed the baseline characteristics and mutational data of 105 pediatric AML patients with 11q23/KMT2A rearrangements. Further evaluation of gene mutations combined with clinical outcomes based on different 11q23/KMT2A translocation partners may also contribute to the refinement of risk stratification by genetics.
2. PATIENTS AND METHODS
2.1. Study population
Data on pediatric AML patients were collected from the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) program on 4 May 2021 (available at https://ocg.cancer.gov/programs/target/data‐matrix). In total, 493 pediatric patients under the age of 18 years with de novo AML between 2004 and 2010 were included in our study. Clinical features at the time of diagnosis (e.g., age, gender, white blood cell [WBC] count, cytogenetic abnormalities, and FAB classification) were obtained in clinical data file at TARGET data matrix. All pediatric patients were treated on the Children's Oncology Group (COG) protocol AAML03P1 and AAML0531. 20 , 21 Hematopoietic Stem Cell Transplantation (HSCT) was considered in the first complete remission for pediatric AML patients with matched‐related donors.
2.2. Data on cytogenetics and molecular characteristics
Cytogenetic analysis of pretreatment bone marrow samples was performed by G‐banded karyotyping; Fluorescence in situ hybridization (FISH) and reverse‐transcribed polymerase chain reaction (RT‐PCR). The International System for Human Cytogenetic Nomenclature (ISCN) was used to designate karyotypes. The mutational status of pediatric AML patients was determined centrally at the Complete Genomics sequencing platform employing high‐density DNA nanoarrays.
2.3. Statistical analyses
Chi‐square test was conducted to compare the frequencies of categorical variables and Mann–Whitney U test was used to compare continuous variables. Complete remission (CR) was defined as having fewer than 5% marrow blasts. Event‐free survival (EFS) was calculated from the initiation of primary treatment until the occurrence of the first event or loss to follow‐up. Overall survival (OS) was measured after the start of initial therapy to death from any cause or the point of last follow‐up. The cumulative incidence of relapse (CIR) was defined as the time from the achievement of CR until the occurrence of first relapse. The Kaplan–Meier model was applied to calculate 5‐year EFS and 5‐year OS, which were compared using log‐rank test. 5‐year CIR was estimated by the Fine‐Gray subdistribution hazard model and differences were analyzed by Gray's test. For multivariable analysis, the Cox proportional hazard regression model was utilized. The Cox regression model included factors that were clinically significant and statistically significant at the level of 0.05 in univariate analysis. p values of 0.05 or less were considered statistically significant. All analyses were carried out using R software version 4.0.5 (R Foundation for Statistical Computing).
3. RESULTS
3.1. Clinical characteristics
Our study cohort consists of 493 pediatric de novo AML patients. The clinical characteristics of 160 patients with 11q23/KMT2A rearrangements and 333 patients without 11q23/KMT2A rearrangements are shown in Table 1. There were no differences in gender distribution, WBC at diagnosis, treatment protocol, and complete remission achievement between patients with 11q23/KMT2A rearrangements and without 11q23/KMT2A rearrangements. Nevertheless, 11q23/KMT2A‐rearranged AML patients at diagnosis had significantly lower median age compared with non‐11q23/KMT2A‐rearranged AML patients (Median, 3.1 vs.10.4 years, p < 0.001). The prevalence of pediatric AML patients with 11q23/KMT2A rearrangements was highest among children under the age of 2 years. The median blast percentage in pretreatment bone marrow samples was significantly higher in 11q23/KMT2A‐rearranged AML patients (79.5% vs. 66.0%, 11q23/KMT2A‐rearranged AML vs. non‐11q23/KMT2A‐rearranged AML, p < 0.001). Patients with 11q23/KMT2A‐rearrangement was strongly correlated with FAB classification subtype M5 (71.5%, p < 0.001). Of the 160 cases, the most frequent translocation partners of 11q23/KMT2A abnormality were t(9;11)(p22;q23) (n = 58; 36.3%), t(10;11)(p12;q23) (n = 30; 18.8%); t(6;11)(q27;q23) (n = 16; 10.0%);t(11;19)(q23;p13.1) (n = 12; 7.5%); t(11;19)(q23;p13.3) (n = 8; 5.0%) (Figure 1). The presence of additional cytogenetic abnormalities (ACAs) is defined as additional chromosomal aberrations detected by conventional cytogenetic analysis. Even though no significant differences of additional abnormalities distribution were observed among different translocation partners, ACAs in t(9;11)(p22;q23) were higher than other groups and contain patients with Trisomy 21. The median age of children with the presence of ACAS is 3.4 years old (data not shown).
TABLE 1.
Clinical characteristics of pediatric patients with or without 11q23/KMT2A‐rearranged AML
| Total | With 11q23/KMT2A‐rearranged | Without 11q23/KMT2A‐rearranged | p‐value | |
|---|---|---|---|---|
| N | 493 | 160 | 333 | |
| Gender, n (% males) | 255 (51.7) | 77 (48.1) | 178 (53.5) | 0.268 |
| Age, median (year) | 8.7 | 3.1 | 10.4 | <0.001 |
| Less than 2 years, n (%) | 103 (20.9) | 67 (41.9) | 37 (10.8) | |
| 2 to 9 years, n (%) | 171 (34.7) | 51 (31.9) | 119 (36.0) | |
| 10 or more years, n (%) | 219 (44.4) | 42 (26.3) | 177 (53.2) | |
| WBC, ×109/L, Median (range) | 40.5 (0.8–610.0) | 42.9 (0.8–610.0) | 39.6 (1.0–447.3) | 0.647 |
| Median blast in BM, % (range) | 71.0 (3–100) | 79.5 (6–99) | 66.0 (3–100) | <0.001 |
| Central nervous system (CNS) involvement | 34 (6.9) | 7 (4.4) | 27 (8.1) | 0.126 |
| FAB classification, n (%) | <0.001 | |||
| M0 | 7 (1.6) | 2 (1.5) | 5 (1.7) | |
| M1 | 33 (7.7) | 5 (3.6) | 28 (9.6) | |
| M2 | 125 (29.2) | 5 (3.6) | 120 (41.2) | |
| M4 | 134 (31.3) | 23 (16.8) | 111 (38.1) | |
| M5 | 109 (25.5) | 98 (71.5) | 11 (3.8) | |
| M6 | 4 (0.9) | — | 4 (1.4) | |
| M7 | 16 (3.7) | 4 (2.9) | 12 (4.1) | |
| FLT3‐ITD, n (%) | <0.001 | |||
| Internal tandem duplication | 51 (10.3) | 4 (2.5) | 47 (14.1) | |
| Wild type | 440 (89.2) | 155 (96.9) | 285 (85.6) | |
| FLT3‐TKD, n (%) | 0.357 | |||
| Mutated | 27 (5.5) | 11 (6.9) | 16 (4.8) | |
| Wild type | 463 (93.9) | 149 (93.1) | 314 (94.3) | |
| WT1, n (%) | 0.004 | |||
| Mutated | 27 (5.5) | 2 (1.3) | 25 (7.5) | |
| Wild type | 461 (93.5) | 157 (98.1) | 304 (91.3) | |
| Treatment protocol, n (%) | 0.239 | |||
| AAML03P1 | 55 (11.2) | 14 (8.8) | 41 (12.3) | |
| AAML0531 | 438 (88.8) | 146 (91.3) | 292 (87.7) | |
| CR status at end of course 1 | 0.153 | |||
| CR, n (%) | 379 (76.9) | 118 (74.2) | 261 (78.4) | |
| Not CR, n (%) | 102 (20.7) | 35 (22.0) | 67 (20.1) | |
| Death, n (%) | 9 (1.8) | 6 (3.8) | 3 (0.90) | |
| CR status at end of course 2 | 0.465 | |||
| CR, n (%) | 433 (87.8) | 138 (87.9) | 295 (88.6) | |
| Not CR, n (%) | 39 (7.9) | 13 (8.3) | 26 (7.8) | |
| Death, n (%) | 11 (2.2) | 6 (3.8) | 5 (1.5) | |
| HSCT in 1st CR, n (%) | 0.101 | |||
| Yes | 45 (9.1) | 21 (13.1) | 24 (7.2) | |
| No | 411 (83.4) | 128 (80) | 283 (85.0) |
FIGURE 1.
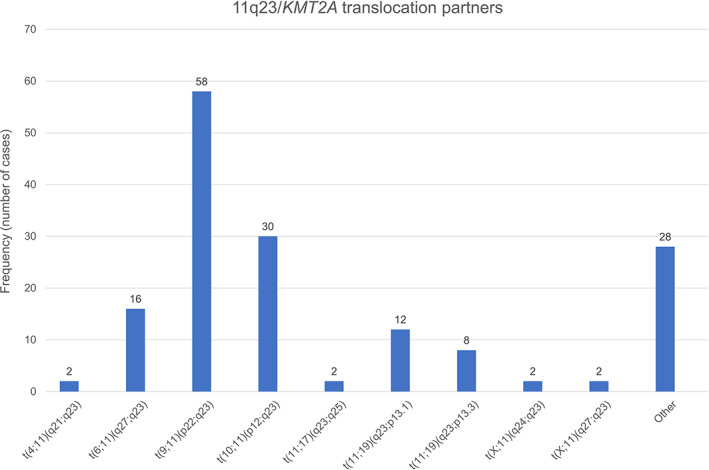
Frequency (number of cases) of pediatric patients with 11q23/KMT2A‐rearranged AML categorized by translocation partners.
3.2. Mutational landscape of pediatric 11q23/ KMT2A ‐rearranged AML
105 children with 11q23/KMT2A‐rearranged AML and 277 children with non‐11q23/KMT2A‐rearranged AML were available for comprehensive molecular analysis of 177 recurrently mutated genes by targeted capture sequencing. In 11q23/KMT2A‐rearranged AML, patients harbored at least one mutation (Median, 1 mutation/patient, range, 1–22) and mutations were found in 81 genes (Figure 2). The most common mutated genes were KRAS (n = 34, 32.4%), NRAS (n = 27, 25.7%), SETD2 (n = 11, 10.5%), FLT3‐TKD (n = 11, 10.5%), and PTPN11 (n = 7, 6.7%). Among non‐11q23/KMT2A‐rearranged AML, the recurrent gene mutations were NRAS (n = 105, 37.9%), KIT (n = 72, 26.0%), FLT3‐ITD (n = 47, 17.0%), WT1 (n = 36, 13.0%), and KRAS (n = 28, 10.1%). In summary, the incidence of KRAS (32.4% vs. 10.1%, p < 0.001) and SETD2 (10.5% vs. 1.4%, p = 0.001) gene mutations was substantially higher in 11q23/KMT2A‐rearranged AML pediatric patients than non‐11q23/KMT2A‐rearranged AML pediatric patients. However, mutations in KIT, FLT3‐ITD, and WT1 genes were less common in 11q23/KMT2A‐rearranged AML compared to non‐11q23/KMT2A‐rearranged AML (p < 0.001). No significant patterns of cooperative or mutually exclusive gene mutations were observed in 11q23/KMT2A‐rearranged AML cohort. Thus, our data indicated that mutations in RAS pathway involving KRAS, NRAS, and PTPN11 genes were the most common genetic alterations observed in 11q23/KMT2A‐rearranged AML pediatric patients.
FIGURE 2.

Mutational landscape of 11q23/KMT2A‐rearranged pediatric AML patients based on recurrent translocation partners. Each column represents a single patient, and each row represents a particular gene. Yellow, mutated; white, wild‐type. The frequencies of these gene mutations are provided on the left column.
Further, we compared the RAS pathway mutations and SETD2 mutations among various 11q23/KMT2A translocation partners and observed that KRAS mutations occurred with a significantly higher frequency in patients with t(10;11)(p12;q23) than in patients with other 11q23/KMT2A rearrangements (56.0% vs. 22.5%, p = 0.001) while NRAS and PTPN11 mutations were not significantly different. SETD2 gene was more frequently mutated in patients with t(10;11)(p12;q23) (5 of 25; 20.0%). To evaluate the pathogenesis of gene mutations during leukemogenesis, we investigated the variable allele frequencies (VAFs) of the 20 most prevalent gene mutations that occurred in 11q23/KMT2A‐rearranged AML (Figure 3). Among the recurrent gene mutations in children with 11q23/KMT2A‐rearranged AML, those with higher VAFs (>0.3) gene mutations were U2AF1, CCND3, STAG2, TET2, RYR1, and MKI67, which presumably occur earlier during leukemogenesis. On the contrary, low VAFs (<0.3) which may represent later events in a subset of leukemic cells and accelerate disease progression were found in RAS pathway genes, SETD2, and FLT3‐TKD.
FIGURE 3.
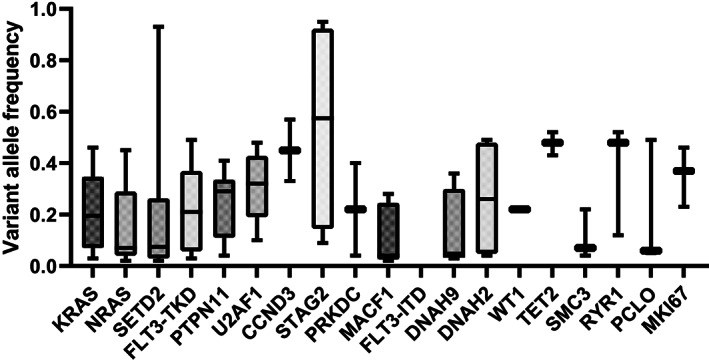
The variant allele frequencies (VAFs) of the 20 most common mutated genes in pediatric 11q23/KMT2A‐rearranged AML.
3.3. Clinical outcome of 11q23/ KMT2A ‐rearranged AML
All pediatric patients had a median follow‐up time of 4.7 years. Among 493 pediatric AML patients, 55 received protocol AAML03P1 and 438 were treated with protocol AAML0531 (11.2% vs. 88.8%, p = 0.239). For the cohort of 11q23/KMT2A‐rearranged AML pediatric patients, the overall CR rate was 74.2% at the end of the first phase of chemotherapy and 87.9% at the second phase of chemotherapy with no significant differences in CR rates between pediatric AML patients with 11q23/KMT2A‐rearrangements and without 11q23/KMT2A ‐rearrangements. Similarly, there were no significant differences observed in CR rates among recurrent 11q23/KMT2A translocation partners at the end of the first phase of chemotherapy (p = 0.352) and the second phase of chemotherapy (p = 0.628). Nonetheless, pediatric AML patients with 11q23/KMT2A‐rearrangements had worse outcomes with 5‐year EFS of 49%, 5‐year CIR of 38%, and 5‐year OS of 67% compared to pediatric AML patients without 11q23/KMT2A‐rearrangements (Table 2).
TABLE 2.
Survival estimates of prognostic factors in 11q23/KMT2A‐rearranged pediatric AML, including subgroups based on translocation partners
| % EFS (SE) | P (log‐rank) | % CIR (SE) | P (Gray) | % OS (SE) | P (log‐rank) | |
|---|---|---|---|---|---|---|
| All | 49 (23) | 38 (2) | 67 (2) | |||
| 11q23/KMT2A rearrangement | ||||||
| With 11q23/KMT2A‐rearranged | 40 (4) | <0.001 | 46 (4) | 0.004 | 59 (4) | 0.004 |
| Without 11q23/KMT2A‐rearranged | 54 (28) | 34 (3) | 70 (3) | |||
| Translocation partners | ||||||
| t(6;11)(q27;q23) | 13 (8) | <0.001 | 69 (13) | 0.015 | 25 (11) | 0.067 |
| t(9;11)(p22;q23) | 57 (7) | 33 (6) | 71 (6) | |||
| t(10;11)(p12;q23) | 23 (8) | 60 (9) | 57 (9) | |||
| t(11;19)(q23;p13.1) | 64 (14) | 17 (11) | 64 (15) | |||
| t(11;19)(q23;p13.3) | 13 (12) | 50 (21) | 50 (18) | |||
| Other 11q23/KMT2A rearrangements | 39 (8) | 53 (5) | 53 (9) | |||
| t(9;11)(p22;q23) subgroup | ||||||
| t(9;11)(p22;q23) | 57 (7) | <0.001 | 33 (6) | 0.003 | 71 (6) | 0.001 |
| Other 11q23/KMT2A rearrangements combined | 31 (5) | 53 (5) | 52 (5) | |||
| Without 11q23/KMT2A‐rearranged | 54 (28) | 34 (3) | 70 (3) | |||
| Age | ||||||
| Less than 2 years | 41 (6) | 0.154 | 42 (6) | 0.662 | 70 (6) | 0.024 |
| 2 to 9 years | 49 (7) | 45 (7) | 58 (7) | |||
| 10 or more years | 29 (7) | 52 (8) | 43 (8) | |||
| Additional cytogenetic aberrations | ||||||
| Absent | 42 (5) | 0.181 | 48 (4) | 0.313 | 65 (5) | 0.028 |
| Present | 37 (6) | 33 (11) | 51 (6) | |||
| KRAS mutation | ||||||
| 11q23/KMT2A‐rearranged AML with KRAS mutant | 21 (7) | 0.001 | 56 (9) | 0.080 | 41 (8) | 0.009 |
| 11q23/KMT2A‐rearranged AML with KRAS wildtype | 45 (5) | 41 (6) | 64 (4) | |||
| NRAS mutation | ||||||
| 11q23/KMT2A‐rearranged AML with NRAS mutant | 41 (10) | 0.827 | 52 (10) | 0.427 | 63 (9) | 0.577 |
| 11q23/KMT2A‐rearranged AML with NRAS wildtype | 40 (4) | 44 (5) | 58 (4) | |||
| SETD2 mutation | ||||||
| 11q23/KMT2A‐rearranged AML with SETD2 mutant | 18 (12) | 0.193 | 82 (13) | 0.004 | 46 (15) | 0.354 |
| 11q23/KMT2A‐rearranged AML with SETD2 wildtype | 42 (4) | 42 (5) | 60 (41) | |||
| Allogeneic HSCT | ||||||
| 11q23/KMT2A‐rearranged AML with HSCT | 52 (12) | 0.219 | 45 (12) | 0.284 | 63 (11) | 0.707 |
| 11q23/KMT2A‐rearranged AML without HSCT | 44 (5) | 53 (5) | 62 (44) | |||
Note: EFS, 5‐year event‐free survival estimates; CIR, indicates 5‐year cumulative incidence of relapse; OS, 5‐year overall survival estimate; P (Gray), P value from the Gray test; and P (log‐rank), P value from log‐rank test.
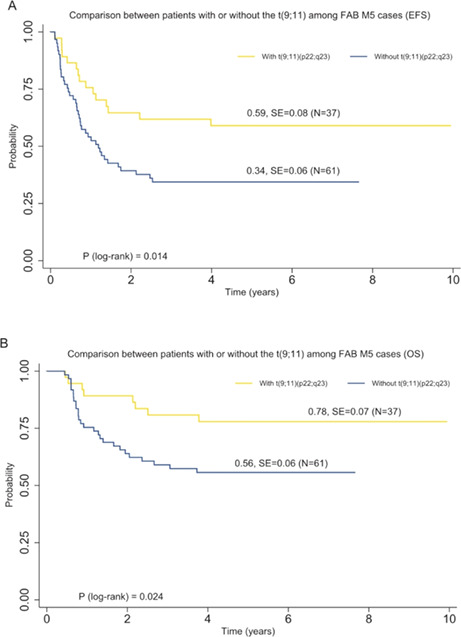
The outcome among various 11q23/KMT2A translocation partners differed significantly (Figure 4). Pediatric patients with t(9;11)(p22;q23) showed better EFS (5‐year EFS, 57% vs. 31% vs.54%, P log−rank < 0.001), CIR (5‐year CIR, 33% vs. 53% vs. 34%, P Gray = 0.003), and OS (5‐year OS, 71% vs. 52% vs. 70%, P log−rank = 0.001) compared with all other 11q23/KMT2A rearrangements combined and patients without 11q23/KMT2A rearrangements. Interestingly, we found that patients with t(11;19)(q23;p13.1) had a favorable outcome with a 5‐year EFS of 64% and a 5‐year CIR of 17%. For 11q23/KMT2A‐rearranged AML pediatric patients with FAB‐M5 morphology, patients with t(9;11)(p22;q23) demonstrated improved outcomes compared to those with other 11q23/KMT2A rearrangements (5‐year EFS, 59% vs. 34%, P log−rank = 0.014; 5‐year OS, 78% vs. 56%, P log−rank = 0.024; Figure 5).
FIGURE 4.
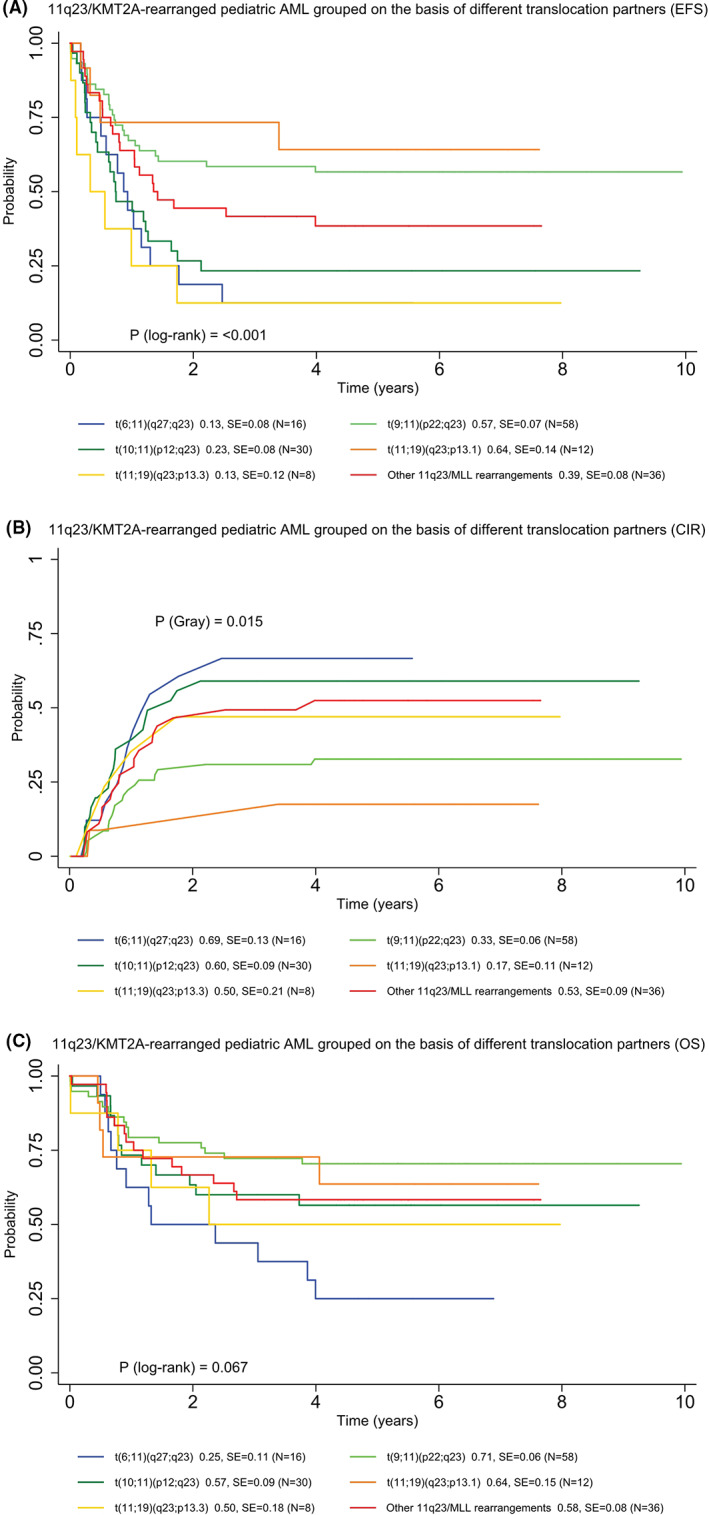
Clinical outcomes of 11q23/KMT2A‐rearranged pediatric AML grouped based on most common translocation partners. (A) 5‐year EFS. (B) 5‐year CIR. (C) 5‐year OS.
FIGURE 5.
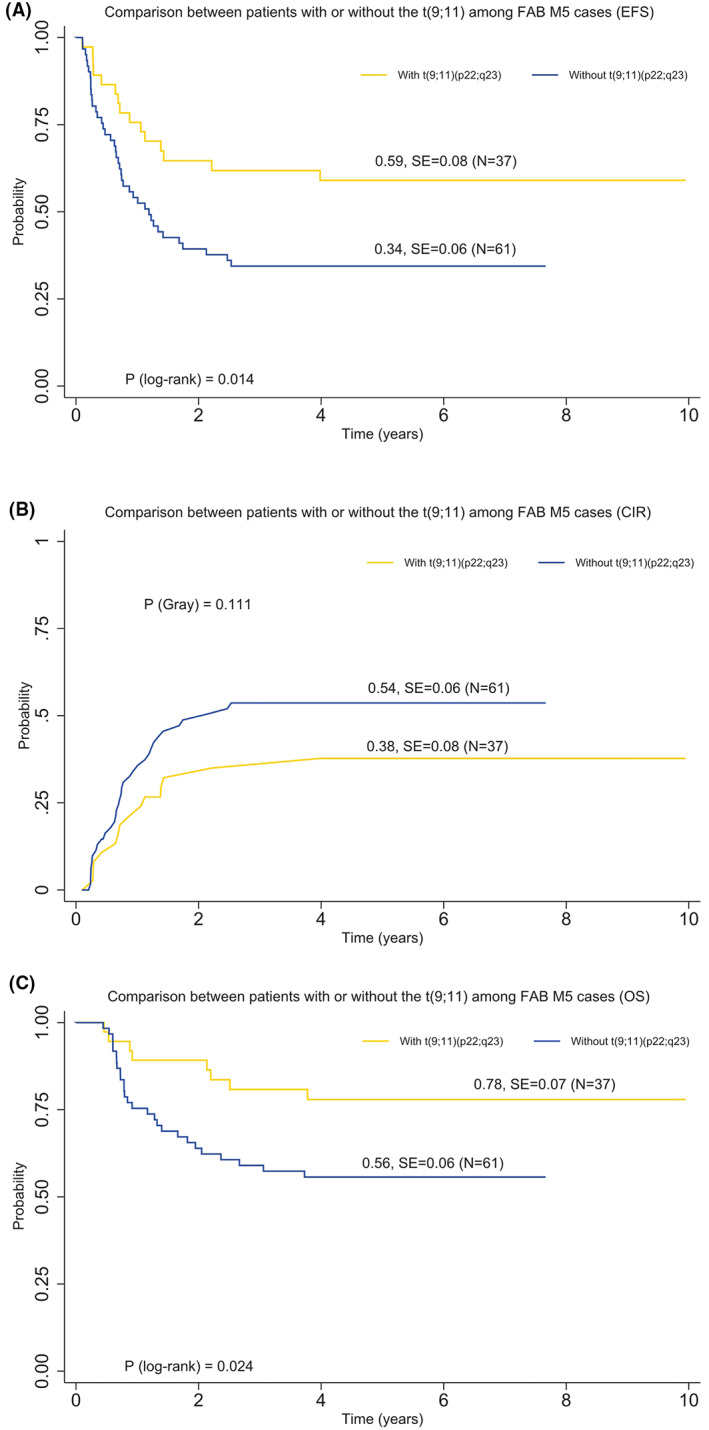
Comparison of clinical outcomes between pediatric patients with or without t(9;11)(p22;q23) among AML cases with FAB‐M5 morphology. (A) 5‐year EFS. (B) 5‐year CIR. (C) 5‐year OS.
3.4. Other prognostic factors
After classifying pediatric patients harboring 11q23/KMT2A‐rearrangements by age group, we found that patients older than 10 years old had a lower 5‐year OS when compared with patients younger than 2 years of age and patients between the ages of 2–9 years (43% vs. 70% vs. 58%, P log−rank = 0.024; Table 2). The worse 5‐year OS of patients older than 10 years old remained significant in t(9;11)(p22;q23) (P log−rank = 0.043). Moreover, we assessed the prognostic impact of addition cytogenetic aberrations on the outcome of 11q23/KMT2A‐rearranged AML pediatric patients. The presence of addition cytogenetic aberrations in pediatric patients tended to have a significantly shorter 5‐year OS than the other patients (51% vs. 65%, P log−rank = 0.028). Furthermore, the prognostic impact of RAS pathway mutations was investigated. Significant worse 5‐year EFS (21% vs. 45%, KRAS mutated vs. KRAS wildtype, Plog−rank = 0.001) and 5‐year OS (41% vs. 64%, KRAS mutated vs. KRAS wildtype, P log−rank = 0.009) were observed for KRAS mutated cases with 11q23/KMT2A‐rearrangements. However, no impact of mutation in the NRAS gene on survival was observed. Of interest, the presence of SETD2 mutations in 11q23/KMT2A‐rearranged AML showed a significant increased risk for relapse compared with SETD2 wildtype in 11q23/KMT2A‐rearranged AML (82% vs. 42%, P Gray = 0.004). Of the 160 pediatric patients with 11q23/KMT2A‐rearranged AML, 21 (13.1%) underwent HSCT in first remission. However, there was no significant difference seen in 5‐year EFS, 5‐year CIR, and 5‐year OS between patients who received HSCT and those who did not.
3.5. Multivariate analyses of pediatric patients with AML
We summarized the results of multivariate analyses for the complete cohort of patients with pediatric AML. In multivariable survival analysis for EFS, CIR, and OS using 11q23/KMT2A‐rearrangements, age, additional cytogenetic aberrations, FLT3‐ITD, KRAS mutation, NRAS mutation, and HSCT as covariates, we revealed t(9;11)(p22;q23) as an independent predictor with better EFS (hazard ratio [HR] = 0.56, p = 0.015) as well as lower CIR (HR = 0.50, p = 0.018). In addition, FLT3‐ITD mutation was found to be an independent predictor of poor EFS (HR = 2.01, p = 0.001) and higher incidence of relapse (HR = 2.23, p = 0.001). When the analyses were restricted to pediatric AML patients with 11q23/KMT2A rearrangements (Table 3), KRAS mutation was a significant poor prognostic factor for EFS and OS with an HR for EFS of 2.10 (p = 0.05) and OS of 2.39 (p = 0.054). We also identified pediatric patients older than 10 years old predicted poor OS (HR = 2.80, p = 0.001). The presence of additional cytogenetic aberrations was a significant independent predictor of OS (HR = 1.99, p = 0.008). Children with 11q23/KMT2A‐rearranged AML who underwent HSCT in the first remission tended to have longer EFS (HR = 0.58, p = 0.111) and OS (HR = 0.66, p = 0.289) than those who did not; although the difference was not statistically significant.
TABLE 3.
Multivariate survival analysis of prognostic factors in 11q23/KMT2A‐rearranged pediatric AML
| EFS | CIR | OS | ||||
|---|---|---|---|---|---|---|
| Hazard ratio (95% CI) | p | Hazard ratio (95% CI) | p | Hazard ratio (95% CI) | p | |
| 11q23/KMT2A rearrangement | ||||||
| t(9;11)(p22;q23) | 1.0 (Reference) | 1.0 (Reference) | 1.0 (Reference) | |||
| Other 11q23/KMT2A rearrangements | 1.56 (0.95–2.54) | 0.078 | 1.86 (1.06–3.26) | 0.030 | 1.63 (0.89–2.99) | 0.117 |
| Age | ||||||
| Less than 2 years | 1.0 (Reference) | 1.0 (Reference) | 1.0 (Reference) | |||
| 2 to 9 years | 0.96 (0.56–1.64) | 0.883 | 1.12 (0.62–2.01) | 0.700 | 1.88 (0.95–3.75) | 0.072 |
| 10 or more years | 1.46 (0.89–2.41) | 0.133 | 1.17 (0.67–2.03) | 0.580 | 2.80 (1.49–5.27) | 0.001 |
| Additional cytogenetic aberrations | ||||||
| Absent | 1.0 (Reference) | 1.0 (Reference) | 1.0 (Reference) | |||
| Present | 1.52 (0.99–2.33) | 0.053 | 1.14 (0.69–1.88) | 0.610 | 1.99 (1.19–3.31) | 0.008 |
| FLT3‐ITD | ||||||
| Wild type | 1.0 (Reference) | 1.0 (Reference) | 1.0 (Reference) | |||
| Internal tandem duplication | 2.32 (0.67–7.96) | 0.183 | 3.46 (1.26–9.51) | 0.016 | 1.73 (0.38–7.90) | 0.480 |
| KRAS mutation | ||||||
| Wild type | 1.0 (Reference) | 1.0 (Reference) | 1.0 (Reference) | |||
| Mutated | 2.10 (1.26–3.50) | 0.005 | 1.75 (0.98–3.15) | 0.060 | 2.39 (1.29–4.46) | 0.006 |
| NRAS mutation | ||||||
| Wild type | 1.0 (Reference) | 1.0 (Reference) | 1.0 (Reference) | |||
| Mutated | 0.97 (0.54–1.75) | 0.972 | 1.43 (0.76–2.71) | 0.270 | 0.89 (0.42–1.85) | 0.749 |
| SETD2 mutation | ||||||
| Wild type | 1.0 (Reference) | 1.0 (Reference) | 1.0 (Reference) | |||
| Mutated | 1.32 (0.61–2.86) | 0.480 | 1.99 (0.90–4.39) | 0.088 | 0.75 (0.26–2.15) | 0.588 |
| Allogeneic HSCT | ||||||
| No | 1.0 (Reference) | 1.0 (Reference) | 1.0 (Reference) | |||
| Yes | 0.58 (0.29–1.14) | 0.111 | 0.62 (0.34–1.14) | 0.130 | 0.66 (0.30–1.43) | 0.289 |
Note: CIR, indicates 5‐year cumulative incidence of relapse; EFS, 5‐year event‐free survival estimates; OS, 5‐year overall survival.
4. DISCUSSION
The present study analyzed a large cohort of children with de novo AML in the TARGET dataset, a collaborative initiative between the National Cancer Institute (NCI) and the Children's Oncology Group (COG). With the goal to discover novel therapeutic targets to facilitate the development of more effective treatment strategies, TARGET characterized the genomes, transcriptomes, and epigenomes of pediatric AML. Our study comprised 493 well‐annotated children from the TARGET AML dataset. Of these patients, 11.2% were enrolled onto pilot study AAML03P1 and 88.8% were randomized to the experimental arm of AAML0531. Both treatment regimens had an identical backbone, consisting of five‐course chemotherapy that includes cytarabine, daunomycin, etoposide, and Gemtuzumab Ozogamicin. AML with 11q23/KMT2A abnormalities is a heterogeneous subgroup of AML. It is reported that 11q23/KMT2A aberrations have more than 90 different translocation partners. 19 Our data showed that the most frequent translocation in pediatric AML is t(9;11)(p22;q23), followed by t(10;11)(p12;q23), t(6;11)(q27;q23), t(11;19)(q23;p13.1), and t(11;19)(q23;p13.3), which is consistent with previous studies. 18 This distribution is also analogous to adult AML. 16
This study presents the comprehensive mutational landscape of pediatric AML patients with 11q23/KMT2A rearrangements using next‐generation sequencing to elucidate distinctive characteristics of pediatric 11q23/KMT2A‐rearranged AML with the aim of improving clinical outcomes. Gilliland et al. have postulated that both type‐I aberrations and type‐II aberrations are required for the pathogenesis of AML. 22 Type‐I aberrations are mainly mutation hotspots involved in signaling and kinase pathway that provoke hyperproliferation of hematopoietic cells and type‐II aberrations occur as chromosomal rearrangements leading to maturation arrest. According to our data, Type‐I mutations were identified in all pediatric 11q23/KMT2A‐rearranged AML cases with a median of 1 mutation per patient, in line with the relatively low mutation rate in 11q23/KMT2A‐rearranged AML in comparison with other AML subtypes described in previous studies. 23 , 24 Notably, our mutational analysis revealed that 58% (61/105) of these mutations were RAS‐related mutations involved in signaling pathways (KRAS, NRAS, and PTPN11), similar to the high incidence of RAS mutations reported in adults. 12 , 13 , 25 Furthermore, we unraveled the distribution of RAS mutations among recurrent 11q23/KMT2A translocation partners in pediatric AML. In adult AML, the majority of KRAS mutations have been reported to be present in t(6;11)(q27;q23). 13 , 26 Whereas, our data showed that KRAS mutations were most frequently detected in pediatric AML patients with t(10;11)(p12;q23) while the mutation frequencies of NRAS and PTPN11 were similar among different translocation partners. It should be noted that few studies clearly demonstrated the prognostic value of gene mutations in 11q23/KMT2A‐rearranged AML. Through analyses of 160 pediatric patients with 11q23/KMT2A‐rearranged AML, we revealed KRAS mutations as an adverse prognostic predictor. The multivariate model of the complete cohort with AML identified that KRAS mutations were independent predictors of poor survival and remained independent prognostic significance in pediatric 11q23/KMT2A‐rearranged AML after adjustment for other risk factors. Recently, several clinical trials targeting the RAS signaling pathway with RAS inhibitors have been conducted, one of which indicated that RAS mutations with t(6;11)(q27;q23) may confer sensitivity to mitogen‐activated protein kinase kinase (MEK) inhibition. 27 , 28
In addition, we identified 10.5% of 11q23/KMT2A‐rearranged pediatric AML patients carried SETD2 mutations as opposed to only 1.4% of patients without 11q23/KMT2A‐rearrangements and SETD2 mutations were more prevalent in patients with t(10;11)(p12;q23). SETD2, known as an epigenetic tumor suppressor responsible for H3K36me3 modification, plays an important role in transcriptional regulation, DNA damage repair, and other cellular processes and is reported to be altered in a range of solid cancers. 29 , 30 , 31 In a study by Dong et al., loss‐of‐function SETD2 mutations in acute leukemia showed downregulation of genes involved in S and G2/M cell cycle progression, which conferred resistance to standard cytarabine‐based chemotherapy drugs. 32 Furthermore, when checkpoint inhibitors were added to Ara‐C treatment, SETD2 mutant AML cells resensitized to chemotherapy. Our finding of discriminative SETD2 mutations in 11q23/KMT2A‐rearranged pediatric AML suggested that cytarabine‐based chemotherapy in combination with checkpoint inhibitors might provide a promising therapeutic strategy for refractory 11q23/KMT2A‐rearranged pediatric AML patients with chemoresistance. Few studies have explored the prognostic value of SETD2 mutations in 11q23/KMT2A‐rearranged AML, particularly in pediatric patients. 33 In a recent publication from Wang et al., the genetic polymorphism of SETD2 in adult AML patients was evaluated for survival outcome and SETD2 rs76208147 TT genotype performed better in overall survival. 34 In our study, SETD2 mutations in 11q23/KMT2A‐rearranged AML were strongly related with a higher relapse rate, suggesting that a more aggressive approach such as allogeneic HSCT or novel therapeutic strategies should be considered in the early stage of treatment. Further studies should be conducted to clarify the biological background of SETD2 mutations and address its clinical application in pediatric AML.
The clinical outcome of 11q23/KMT2A rearrangements in AML has been investigated for more than 30 years with contradicting results reported in various studies. Nevertheless, there is consensus that the prognoses of 11q23/KMT2A‐rearranged AML are determined by various 11q23/KMT2A translocation partners. A meta‐analysis of the German AML Intergroup from eight collaborative trials suggested that t(v;11)(v;q23) other than t(9;11)(p22;q23) was associated with a poor prognosis. 17 Based on 2017 ELN risk stratification by genetics, 35 t(9;11)(p22;q23) was assigned to the intermediate‐risk group while other 11q23/KMT2A rearrangements were grouped as adverse risk. Our data confirmed that t(9;11)(p22;q23), distinct from other 11q23/KMT2A translocation partners, was an independent predictor of favorable survival in pediatric AML with a decreased rate of relapse. This was also noted in previous studies in large cohorts of adult patients 14 , 17 , 36 as well as pediatric patients. 15 In contrast to this finding, some studies showed that t(9;11)(p22;q23) did not differ substantially from other 11q23/KMT2A translocation partners. 18 , 37 These conflicting results may attribute to the heterozygosity of the population and the variety of treatment regimens for 11q23/KMT2A‐rearranged AML patients enrolled in different study groups. In our study, the median age of t(9;11)(p22;q23) was 3.4 years and treatment protocols AAML03P1 and AAML0531 were similarly split across different translocation partners. AML with 11q23/KMT2A‐rearrangement is primarily associated with M5 AML. Consistent with previous studies, 15 , 37 FAB‐M5 pediatric patients harboring t(9;11)(p22;q23) demonstrated a better outcome than those without the translocation. Kuipers et al. conducted extensive gene expression analysis in 11q23/KMT2A‐rearranged pediatric patients and elevated IGSF4 expression was detected in t(9;11)(p22;q23) patients with FAB‐M5. 38 IGSF4 overexpression in t(9;11)(p22;q23) patients with FAB‐M5, a cell adhesion molecule regulated by promoter methylation status, was correlated with a favorable outcome.
The present study had several limitations. Firstly, SETD2 mutations were highlighted in our study. According to prior studies, the loss of one allele of SETD2 does not significantly reduce H3K36me3 levels. 39 , 40 Hence, the clinical outcomes between monoallelic SETD2 mutations and biallelic SETD2 mutations may differ. However, the TARGET database does not provide detailed alterations of SETD2 mutations to specify the loss‐of‐function feature of SETD2 mutations. Secondly, owing to the low frequency of mutations in 11q23/KMT2A‐rearranged AML, the relatively small numbers of pediatric patients harboring molecular abnormalities we analyzed make validation of our results in a larger series necessary.
In conclusion, our comparisons of the mutational landscape between 11q23/KMT2A‐rearranged AML and non‐11q23/KMT2A‐rearranged AML revealed the relatively high frequency of mutations in the RAS signaling pathway and SETD2 in 11q23/KMT2A‐rearranged pediatric AML, suggesting that both gene mutations mentioned above should be incorporated into screening tests for prognostication. We also confirmed the independent prognostic influence of t(9;11)(p22;q23) on the clinical outcome and its correlation with FAB‐M5 morphology. Innovative treatment approaches should be explored based on the characteristics of 11q23/KMT2A‐rearranged pediatric AML found in our study.
AUTHOR CONTRIBUTIONS
Conceptualization, K.Y.Y., J.P.F., and L.H.X.; methodology, K.Y.Y. and Y.L.; data management/analysis/ visualization, K.Y.Y. and Y.L.; data analysis and visualization of cooccurrence/ mutual exclusivity, Y.Z.Z. and Y.W.; supervision of genetic analysis, D.H.Z.; supervision, J.P.F. and L.H.X.; writing—original draft preparation, K.Y.Y.; funding acquisition, J.P.F. and L.H.X. All authors have read and approved the published version of the manuscript.
FUNDING INFORMATION
This work was supported by the Guangdong Basic and Applied Basic Research Foundation (2021A1515010240 to J‐P F) as well as Guangdong Basic and Applied Basic Research Foundation (2020A1515010312 to L‐H X).
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This article does not contain any studies with human participants or animals performed by any of the authors.
ACKNOWLEDGMENTS
We sincerely acknowledge the help and consultation offered by Clinical Research Center of Sun Yat‐Sen Memorial Hospital, Sun Yat‐Sen University.
Yuen K‐Y, Liu Y, Zhou Y‐Z, et al. Mutational landscape and clinical outcome of pediatric acute myeloid leukemia with 11q23/ KMT2A rearrangements. Cancer Med. 2023;12:1418‐1430. doi: 10.1002/cam4.5026
Ka‐Yuk Yuen and Yong Liu have contributed equally to this work.
Contributor Information
Jian‐Pei Fang, Email: fangjpei@mail.sysu.edu.cn.
Lu‐Hong Xu, Email: xulhong@mail.sysu.edu.cn.
DATA AVAILABILITY STATEMENT
The datasets analyzed during the current study are available in the https://ocg.cancer.gov/programs/target/data‐matrix.
REFERENCES
- 1. Muntean AG, Chen W, Jones M, Granowicz EM, Maillard I, Hess JL. MLL fusion protein‐driven AML is selectively inhibited by targeted disruption of the MLL‐PAFc interaction. Blood. 2013;122(11):1914‐1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Foucar K, Anastasi J. Acute myeloid leukemia with recurrent cytogenetic abnormalities. Am J Clin Pathol. 2015;144(1):6‐18. [DOI] [PubMed] [Google Scholar]
- 3. Raimondi SC, Chang MN, Ravindranath Y, et al. Chromosomal abnormalities in 478 children with acute myeloid leukemia: clinical characteristics and treatment outcome in a cooperative pediatric oncology group study‐POG 8821. Blood. 1999;94(11):3707‐3716. [PubMed] [Google Scholar]
- 4. Meyer C, Burmeister T, Groger D, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32(2):273‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mercher T, Schwaller J. Pediatric acute myeloid leukemia (AML): from genes to models toward targeted therapeutic intervention. Front Pediatr. 2019;7:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ. Altered Hox expression and segmental identity in Mll‐mutant mice. Nature. 1995;378(6556):505‐508. [DOI] [PubMed] [Google Scholar]
- 7. Lawrence HJ, Helgason CD, Sauvageau G, et al. Mice bearing a targeted interruption of the homeobox gene HOXA9 have defects in myeloid, erythroid, and lymphoid hematopoiesis. Blood. 1997;89(6):1922‐1930. [PubMed] [Google Scholar]
- 8. Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J. 1998;17(13):3714‐3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rubnitz JE, Kaspers GJL. How I treat pediatric acute myeloid leukemia. Blood. 2021;138(12):1009‐1018. [DOI] [PubMed] [Google Scholar]
- 10. Kishtagari A, Levine RL, Viny AD. Driver mutations in acute myeloid leukemia. Curr Opin Hematol. 2020;27(2):49‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Winters AC, Bernt KM. MLL‐rearranged Leukemias‐an update on science and clinical approaches. Front Pediatr. 2017;5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lavallee VP, Baccelli I, Krosl J, et al. The transcriptomic landscape and directed chemical interrogation of MLL‐rearranged acute myeloid leukemias. Nat Genet. 2015;47(9):1030‐1037. [DOI] [PubMed] [Google Scholar]
- 13. Bill M, Mrozek K, Kohlschmidt J, et al. Mutational landscape and clinical outcome of patients with de novo acute myeloid leukemia and rearrangements involving 11q23/KMT2A. Proc Natl Acad Sci USA. 2020;117(42):26340‐26346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mrozek K, Heinonen K, Lawrence D, et al. Adult patients with de novo acute myeloid leukemia and t(9; 11)(p22; q23) have a superior outcome to patients with other translocations involving band 11q23: a cancer and leukemia group B study. Blood. 1997;90(11):4532‐4538. [PubMed] [Google Scholar]
- 15. Rubnitz JE, Raimondi SC, Tong X, et al. Favorable impact of the t(9;11) in childhood acute myeloid leukemia. J Clin Oncol. 2002;20(9):2302‐2309. [DOI] [PubMed] [Google Scholar]
- 16. Schoch C, Schnittger S, Klaus M, Kern W, Hiddemann W, Haferlach T. AML with 11q23/MLL abnormalities as defined by the WHO classification: incidence, partner chromosomes, FAB subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed AML cases. Blood. 2003;102(7):2395‐2402. [DOI] [PubMed] [Google Scholar]
- 17. Krauter J, Wagner K, Schafer I, et al. Prognostic factors in adult patients up to 60 years old with acute myeloid leukemia and translocations of chromosome band 11q23: individual patient data‐based meta‐analysis of the German acute myeloid leukemia intergroup. J Clin Oncol. 2009;27(18):3000‐3006. [DOI] [PubMed] [Google Scholar]
- 18. Balgobind BV, Raimondi SC, Harbott J, et al. Novel prognostic subgroups in childhood 11q23/MLL‐rearranged acute myeloid leukemia: results of an international retrospective study. Blood. 2009;114(12):2489‐2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Balgobind BV, Zwaan CM, Pieters R, Van den Heuvel‐Eibrink MM. The heterogeneity of pediatric MLL‐rearranged acute myeloid leukemia. Leukemia. 2011;25(8):1239‐1248. [DOI] [PubMed] [Google Scholar]
- 20. Cooper TM, Franklin J, Gerbing RB, et al. AAML03P1, a pilot study of the safety of gemtuzumab ozogamicin in combination with chemotherapy for newly diagnosed childhood acute myeloid leukemia: a report from the Children's oncology group. Cancer. 2012;118(3):761‐769. [DOI] [PubMed] [Google Scholar]
- 21. Gamis AS, Alonzo TA, Meshinchi S, et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event‐free survival by reducing relapse risk: results from the randomized phase III children's oncology group trial AAML0531. J Clin Oncol. 2014;32(27):3021‐3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100(5):1532‐1542. [DOI] [PubMed] [Google Scholar]
- 23. Cancer Genome Atlas Research N , Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059‐2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bolouri H, Farrar JE, Triche T Jr, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age‐specific mutational interactions. Nat Med. 2018;24(1):103‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grossmann V, Schnittger S, Poetzinger F, et al. High incidence of RAS signalling pathway mutations in MLL‐rearranged acute myeloid leukemia. Leukemia. 2013;27(9):1933‐1936. [DOI] [PubMed] [Google Scholar]
- 26. Issa GC, Zarka J, Sasaki K, et al. Predictors of outcomes in adults with acute myeloid leukemia and KMT2A rearrangements. Blood Cancer J. 2021;11(9):162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maiti A, Naqvi K, Kadia TM, et al. Phase II trial of MEK inhibitor Binimetinib (MEK162) in RAS‐mutant acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2019;19(3):142‐148 e141. [DOI] [PubMed] [Google Scholar]
- 28. Hou HA, Tien HF. Genomic landscape in acute myeloid leukemia and its implications in risk classification and targeted therapies. J Biomed Sci. 2020;27(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li J, Duns G, Westers H, Sijmons R, van den Berg A, Kok K. SETD2: an epigenetic modifier with tumor suppressor functionality. Oncotarget. 2016;7(31):50719‐50734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen R, Zhao WQ, Fang C, Yang X, Ji M. Histone methyltransferase SETD2: a potential tumor suppressor in solid cancers. J Cancer. 2020;11(11):3349‐3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kumari S, Muthusamy S. SETD2 as a regulator of N6‐methyladenosine RNA methylation and modifiers in cancer. Eur J Cancer Prev. 2020;29(6):556‐564. [DOI] [PubMed] [Google Scholar]
- 32. Dong Y, Zhao X, Feng X, et al. SETD2 mutations confer chemoresistance in acute myeloid leukemia partly through altered cell cycle checkpoints. Leukemia. 2019;33(11):2585‐2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhu X, He F, Zeng H, et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet. 2014;46(3):287‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang S, Yuan X, Liu Y, et al. Genetic polymorphisms of histone methyltransferase SETD2 predicts prognosis and chemotherapy response in Chinese acute myeloid leukemia patients. J Transl Med. 2019;17(1):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen Y, Kantarjian H, Pierce S, et al. Prognostic significance of 11q23 aberrations in adult acute myeloid leukemia and the role of allogeneic stem cell transplantation. Leukemia. 2013;27(4):836‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Coenen EA, Raimondi SC, Harbott J, et al. Prognostic significance of additional cytogenetic aberrations in 733 de novo pediatric 11q23/MLL‐rearranged AML patients: results of an international study. Blood. 2011;117(26):7102‐7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuipers JE, Coenen EA, Balgobind BV, et al. High IGSF4 expression in pediatric M5 acute myeloid leukemia with t(9;11)(p22;q23). Blood. 2011;117(3):928‐935. [DOI] [PubMed] [Google Scholar]
- 39. Duns G, van den Berg E, van Duivenbode I, et al. Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res. 2010;70(11):4287‐4291. [DOI] [PubMed] [Google Scholar]
- 40. Ho TH, Park IY, Zhao H, et al. High‐resolution profiling of histone h3 lysine 36 trimethylation in metastatic renal cell carcinoma. Oncogene. 2016;35(12):1565‐1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets analyzed during the current study are available in the https://ocg.cancer.gov/programs/target/data‐matrix.