

Abstract
Motivation
Several genomic databases host data and metadata for an ever-growing collection of sequence datasets. While these databases have a shared hierarchical structure, there are no tools specifically designed to leverage it for metadata extraction.
Results
We present a command-line tool, called ffq, for querying user-generated data and metadata from sequence databases. Given an accession or a paper’s DOI, ffq efficiently fetches metadata and links to raw data in JSON format. ffq’s modularity and simplicity make it extensible to any genomic database exposing its data for programmatic access.
Availability and implementation
ffq is free and open source, and the code can be found here: https://github.com/pachterlab/ffq.
1 Introduction
The extraordinarily large volume of user-generated sequencing data available in public databases is increasingly being utilized in research projects alongside novel experiments (Hippen and Greene, 2021; Huang et al., 2021; Kasmanas et al., 2021; Klie et al., 2021; Lung et al., 2020; Rajesh et al., 2021; Razmara et al., 2019; Simon et al., 2018; Booeshaghi et al., 2022; Wartmann et al., 2021). Collation of metadata is crucial for the effective use of publicly available data. Accurate metadata can provide information about the samples assayed and can facilitate the acquisition of raw data. For example, sra-tools enable users to query and download data from the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA), which currently hosts 13.67 PB of data. An alternative to sra-tools is the pysradb tool (Choudhary, 2019). pysradb was developed to access metadata from the SRA, using metadata obtained from the regularly updated SRAdb SQLite database (Zhu et al., 2013). MetaSRA adds additional standardized metadata on top of the SRAdb SQLite database (Bernstein et al., 2017) and also provides an application programming interface (API) for accessing them. While these and other tools (Bernstein et al., 2020; Eaton, 2020; Li et al., 2018; Mahi et al., 2019) have proven to be very useful, they provide access to a limited scope of databases. We developed ffq to facilitate metadata retrieval from a diverse set of databases, including
NCBI SRA and Gene Expression Omnibus (GEO),
European Molecular Biology Lab-European Bioinformatics Institute European Nucleotide Archive (EMBL-EBI ENA),
DNA Data Bank of Japan Gene Expression Archive (DDBJ GEA) and
Encyclopedia of DNA Elements (ENCODE) database (Davis et al., 2018; ENCODE Project Consortium, 2012).
In order to facilitate a modular architecture for ffq, we first studied the structure of these databases in detail to identify commonalities and relationships between them (Fig. 1).
Fig. 1.
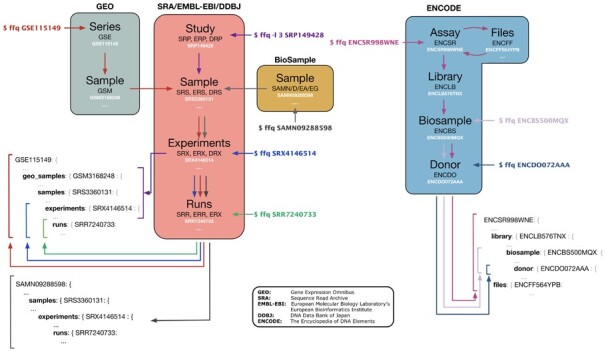
Metadata retrieval. ffq fetches and returns metadata as a JSON object by traversing the database hierarchy. Subsets of the database hierarchy can be returned by specifying -l [level]
The SRA, ENA and DDBJ databases all follow a similar hierarchical structure where studies are grouped into samples, experiments and runs, a shared architecture that is useful and likely the result of the longstanding International Nucleotide Sequence Database Collaboration (INSDC) between the ENA, NCBI and DDBJ. We note that the Genome Sequence Archive (Chen et al., 2021; CNCB-NGDC Members and Partners, 2022) is not a member of the INSDC. However, it also uses a similar hierarchical structure for its database and regularly ingests data from the SRA but does not expose its publicly available data for programmatic access.
The consistent database schemas used by members of the INSDC greatly simplify metadata retrieval for ffq. For example, GEO accession codes are grouped hierarchically through Series and Samples and have external relations to SRA accession codes for raw sequencing data submitted to the SRA. This enables ffq to fetch metadata and processed data from GEO that submitters have associated with raw sequencing data stored in the SRA.
2 Description
Based on the database architectures, we created ffq to fetch metadata using database accessions or paper DOIs as input. Importantly, ffq only fetches metadata and links to data files and does not offer data downloading. This deliberate design decision was motivated by the UNIX philosophy ‘Make each program do one thing well’ (McIlroy et al., 1978).
The ffq options are summarized below:
-
ffq [accession(s)]
Where [accession] can be any of the following: SR(R/X/S/P), ER(R/X/S/P), DR(R/X/S/P), GS(E/M), ENC(SR/BS/DO), CXR, SAM(N/D/EA/EG) and DOI.
-
ffq [-l level] [accession(s)]
Where [level] defines the hierarchy in the database to which data is subset data.
-
ffq [–ftp] [–aws] [–gcp] [–ncbi] [accession(s)]
Where the flags correspond to the types of data-storage links for the raw data.
-
ffq [-o out] [–split] [accession(s)]
Where [out] corresponds to a path on disk to save the JSON file and [–split] splits the metadata from multiple accessions into their own file.
The ffq codebase consists of 58 functions and 2198 lines of code across six files and relies on only four software dependencies. Users supply an accession or DOI and the tool returns metadata for the sequencing data associated with that accession or DOI.
Accession-based ffq metadata retrieval uses the NCBI Entrez programming utilities, ENA API, GEO FTP and ENCODE API to programmatically access metadata with HTTP requests. DOI-based metadata retrieval first converts the DOI to the manuscript title via the CrossRef API (Hendricks et al., 2020) and then retrieves all study accessions associated with the manuscript title with the ENA search API. The reliance on these external dependencies can make it challenging to track API updates that may break ffq functionality. To provide resilience to such changes, we have implemented extensive quality control via an automated testing framework that validates behavior against all external APIs and five Python versions (3.6, 3.7, 3.8, 3.9 and 3.10) that cover 78% of the code. This makes it easy to detect and address API updates within ffq.
Once fetched, metadata is returned as a Javascript Object Notation (JSON) object. Run times for metadata retrieval vary depending on database up-time, server connection speed and database rate-limiting, but generally, we find that ffq can download metadata at a rate of 10 s per sample. This rate includes short and deliberate delays we have added between HTTP requests to prevent a perceived Denial-of-Service.
External factors may impact ffq’s ability to fetch metadata that are independent of the tool. Internet connection, improperly formatted accessions, missing or incomplete metadata are some of the failure modes that users may face. To aid users in debugging missing or incomplete metadata, custom exceptions have been implemented and possible failure modes and caveats have been listed in the documentation.
3 Usage and documentation
The ffq tool is written in Python and can be installed with pip and conda. Users supply an accession or DOI and the tool returns metadata for the associated sequencing data. The JSON-return objects make ffq interoperable with other tools such as jq for easy command-line parsing. Additionally, ffq’s modularity and simplicity make it extensible to other genomic databases. By leveraging existing APIs, ffq offers a lightweight solution for querying data that is guaranteed to be more up-to-date than tools that rely on regular database builds.
These features enable researchers to use ffq to refine research questions. For example, ffq can be used to fetch publicly available scRNAseq data, which can be preprocessed with existing tools (Melsted, et al., 2021) and compared against newly generated data (Fig. 2). Alternatively, ffq can be used for sequencing quality control; sequencing reads can be fetched with ffq and piped into common command-line tools to count the number of reads or assess the per-base quality scores. These and other use cases are explained in the ffq documentation. The modularity of ffq makes possible streamwise processing of publicly available FASTQ files for any number of applications.
Fig. 2.
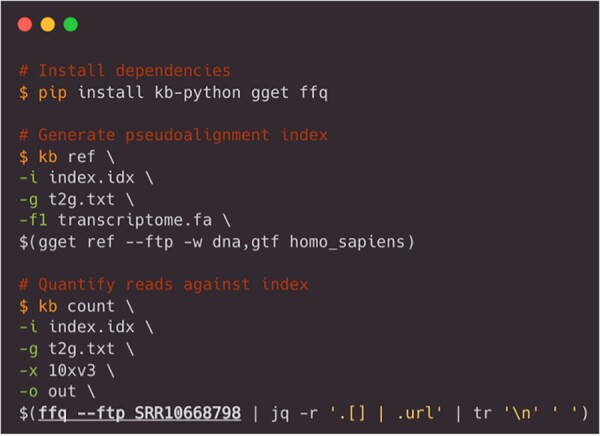
Example use case. Publicly available scRNAseq data are fetched with ffq and quantified with kb-python to generate a gene count matrix. The ffq command is underlined
4 Discussion
While ffq facilitates downloading of data from numerous genomic databases, the results retrieved are only useful to the extent that the metadata uploaded is meaningful and complete. Meaningful and complete user-generated data underlies the curation of genomic references essential for comparative genomic data analysis (Luebbert and Pachter, 2023). Unfortunately, there is little to no standardization of user-uploaded sequencing metadata (Rajesh et al., 2021; Wang et al., 2019), and metadata descriptions can become exceedingly complex for current multiplexed experiments, where different assays with distinct data types are combined. Improvement of metadata uploading in machine-readable standard formats is essential if publicly available genomic data are to be usable by scientists in the future.
Users who wish to refine research questions with complete and accurate publicly available data will benefit from ffq. By providing direct links to sequencing data and metadata, ffq allows any number of downstream procedures that operate on sequencing reads. Importantly, the modularity of ffq enables streamwise processing of data and metadata that obviates the need for large amounts of storage and lessens the cost of computing.
Acknowledgements
This work was motivated by the need to obtain metadata for Booeshaghi and Pachter (2020). We thank Ali Mortazavi for his suggestion to include ffq querying of the ENCODE database and Anders Goncalves da Silva, Andrea Telatin, Laura Luebbert and Phil Ewels for their contributions to the code base.
Contributor Information
Ángel Gálvez-Merchán, Division of Biology and Biological Engineering, California Institute of Technology, Pasadena, CA 91125, USA.
Kyung Hoi (Joseph) Min, Department of Computer Science and Electrical Engineering, Massachusetts Institute of Technology, Cambridge, MA 91125, USA.
Lior Pachter, Division of Biology and Biological Engineering, California Institute of Technology, Pasadena, CA 91125, USA; Department of Computing and Mathematical Sciences, Pasadena, CA 91125, USA.
A Sina Booeshaghi, Department of Mechanical Engineering, California Institute of Technology, Pasadena, CA 91125, USA.
Funding
This work was supported in part by National Institutes of Health (NIH) [U19MH114830].
Conflict of Interest: none declared.
Data availability
All data and code associated with this manuscript is available at https://github.com/pachterlab/ffq.
References
- Bernstein M.N. et al. (2020) Jupyter notebook-based tools for building structured datasets from the Sequence Read Archive. F1000Res., 9, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein M.N. et al. (2017) MetaSRA: normalized human sample-specific metadata for the Sequence Read Archive. Bioinformatics, 33, 2914–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T. et al. (2021) The genome sequence archive family: toward explosive data growth and diverse data types. Genomics Proteomics Bioinformatics, 19, 578–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary S. (2019) pysradb: a Python package to query next-generation sequencing metadata and data from NCBI Sequence Read Archive. F1000Res., 8, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CNCB-NGDC Members and Partners (2022) Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res., 50, D27–D38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis C.A. et al. (2018) The Encyclopedia of DNA Elements (ENCODE): data portal update. Nucleic Acids Res., 46, D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton K. (2020) NCBImeta: efficient and comprehensive metadata retrieval from NCBI databases. J. Open Source Softw., 5, 1990. [Google Scholar]
- ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks G. et al. (2020) Crossref: the sustainable source of community-owned scholarly metadata. Quant. Sci. Stud., 1, 414–427. [Google Scholar]
- Hippen A.A., Greene C.S. (2021) Expanding and remixing the metadata landscape. Trends Cancer Res., 7, 276–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.-N. et al. (2021) The systematic assessment of completeness of public metadata accompanying omics studies. bioRxiv, 2021.11.22.469640.
- Kasmanas J.C. et al. (2021) HumanMetagenomeDB: a public repository of curated and standardized metadata for human metagenomes. Nucleic Acids Res., 49, D743–D750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klie A. et al. (2021) Increasing metadata coverage of SRA BioSample entries using deep learning-based named entity recognition. Database, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. et al. (2018) GEOMetaCuration: a web-based application for accurate manual curation of Gene Expression Omnibus metadata. Database, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebbert L., Pachter L. (2023) Efficient querying of genomic reference databases with gget. Bioinformatics, 39(1), btac836. [DOI] [PMC free article] [PubMed]
- Lung P.-Y. et al. (2020) Maximizing the reusability of gene expression data by predicting missing metadata. PLoS Comput. Biol., 16, e1007450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahi N.A. et al. (2019) GREIN: an interactive web platform for re-analyzing GEO RNA-seq data. Sci. Rep., 9, 7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlroy M. et al. (1978) UNIX time-sharing system. Bell Syst. Techn. J., 57, 1899–1904. [Google Scholar]
- Melsted P. et al. (2021) Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat Biotechnol, 39, 813–818. [DOI] [PubMed]
- Rajesh A. et al. (2021) Improving the completeness of public metadata accompanying omics studies. Genome Biol., 22, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razmara A. et al. (2019) recount-brain: a curated repository of human brain RNA-seq datasets metadata. bioRxiv, 618025.
- Simon L.M. et al. (2018) MetaMap: an atlas of metatranscriptomic reads in human disease-related RNA-seq data, GigaScience, 7(6), giy070. [DOI] [PMC free article] [PubMed]
- Booeshaghi A.S. et al. (2022) Depth normalization for single-cell genomics count data. bioRxiv, 2022.05.06.490859.
- Booeshaghi A.S., Pachter L. (2020) Decrease in ACE2 mRNA expression in aged mouse lung. bioRxiv, 2020.04.02.021451.
- Wang Z. et al. (2019) Mining data and metadata from the gene expression omnibus. Biophys. Rev., 11, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartmann H. et al. (2021) Bias-invariant RNA-sequencing metadata annotation. Gigascience, 10, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y. et al. (2013) SRAdb: query and use public next-generation sequencing data from within R. BMC Bioinformatics, 14, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data and code associated with this manuscript is available at https://github.com/pachterlab/ffq.