

Summary
Recent advancements in computational tools have allowed protein structure prediction with high accuracy. Computational prediction methods have been used for modeling many soluble and membrane proteins, but the performance of these methods in modeling peptide structures has not yet been systematically investigated. We benchmarked the accuracy of AlphaFold2 in predicting 588 peptide structures between 10 and 40 amino acids using experimentally determined NMR structures as reference. Our results showed AlphaFold2 predicts α-helical, β-hairpin, and disulfide rich peptides with high accuracy. AlphaFold2 performed at least as well if not better than alternative methods developed specifically for peptide structure prediction. AlphaFold2 showed several shortcomings in predicting Φ/Ψ angles, disulfide bond patterns, and the lowest RMSD structures failed to correlate with lowest pLDDT ranked structures. In summary, computation can be a powerful tool to predict peptide structures, but additional steps may be necessary to analyze and validate the results.
Keywords: AlphaFold2, peptides, structure prediction, benchmark, protein folding, pLDDT, disulfide bonds
Graphical Abstract
eTOC Blurb
McDonald et al. investigate the performance of AlphaFold2 in comparison to other peptide and protein structure prediction methods. Root mean square deviation analyses show deep learning methods like AlphaFold2 and OmegaFold perform the best in most cases but have reduced accuracy with non-helical secondary structure motifs and solvent-exposed peptides.
Introduction
Peptides can be loosely defined as polyamides that consist of 2 to 50 amino acids, though this is an arbitrary definition and many molecules accepted to be peptides rather than proteins are larger than this cutoff, in particular if they fail to form tertiary structure 1. Often, the amount of secondary and tertiary structure formed influences whether a polyamide is classified as peptide (less structure) or protein (more structure), in addition to its length. A large number of peptides play important roles in nature as hormones 2,3, antimicrobials 4, and many synthetic peptides are being investigated as drug candidates 5. In addition to these roles, many toxins are peptides 6. While flexible, peptides adopt a variety of conformations on varying timescales that interact with binding partners and can be sensitive to environmental factors or interactions with lipids or proteins. Typically, solution or solid-state NMR spectroscopy are used for the determination of small peptide structures 7,8. NMR spectroscopy allows the determination of peptide structures under different experimental conditions, shedding light on how peptides may behave in different environments including different lipid compositions, temperatures, and pH values. However, variable environmental conditions may deviate NMR peptides models from functional conformations. Alternatively, computational methods pose a plausible approach for the prediction of the most stable potentially bioactive conformation of peptides. However, performance of prediction methods diminishes with peptide length. Considering these challenges, we benchmarked AlphaFold2 (AF2) on 588 peptides across six distinct groups of peptides to determine the utility and shortcomings of computational prediction on peptide structural biology.
There are multiple methods to predict peptide structures including de novo folding, homology modeling, molecular dynamics (MD) simulations, and deep-learning-based methods 9–11. PEP-FOLD3 is a peptide specific de novo folding based method that can be used to model peptides between 5 and 50 amino acids 12. APPTEST is a peptide specific protocol that utilizes a neural network architecture combined with Molecular Dynamics, which can be used to model peptides that are between 5 and 40 amino acids 13. AF2 is a deep learning-based protein prediction method that uses Multiple-Sequence Alignments (MSAs) to predict the structure of a protein based on co-evolving residues [24]. RoseTTAFold works through a similar logic but different deep learning arcitecture14. OmegaFold is a deep-learning-based method that uses only sequence and no MSAs for predictions based on a Natural Language Model 15. OmegaFold excelled in cases where high-quality MSAs cannot be obtained due to lack of close homolog sequences. In addition to these methods, homology modeling can be used with or without experimental restraints when there is a homologous peptide or protein structure available 16 or used to model protein mutant structure using wild-type experimental data 17. While this method is typically preferred for larger proteins, it was also applied to modeling peptide structures 18. However, homology modeling requires the presence of template structures with high sequence homology to the target protein. Considering that only sequence information is likely to be available for most natural peptides, sequence-based methods have advantages over structure-based methods. Understanding the weaknesses of current peptide prediction methods will guide development of future approaches by elucidating the contributions of specific training metrics (e.g. MSAs, sequence, experimental constraints) and informing on difficult-to-predict structural features that should be included in future training sets.
The availability of AF2 was a big step towards the prediction of protein structures with high accuracy. While AF2 can be used for the modeling of shorter peptides in theory, the benchmark set used to train AF2 excluded shortest peptide structures since the method of determination for these peptides is NMR spectroscopy in general, although it remains unclear why NMR structures were excluded from the AF2 training set. Some of the relatively poor predictions made by AF2 in CASP14 included protein structures determined by NMR 19, raising the question whether a similar pattern would be observed for flexible peptide structures as well. Therefore, comprehensive benchmarks are necessary to evaluate the utility of AF2 in modeling peptide structures. Although there are ongoing works on assessing the performance of AF2 in predicting peptide – protein complex structures 20–22, the performance of AF2 on small peptide structure prediction remains unexplored.
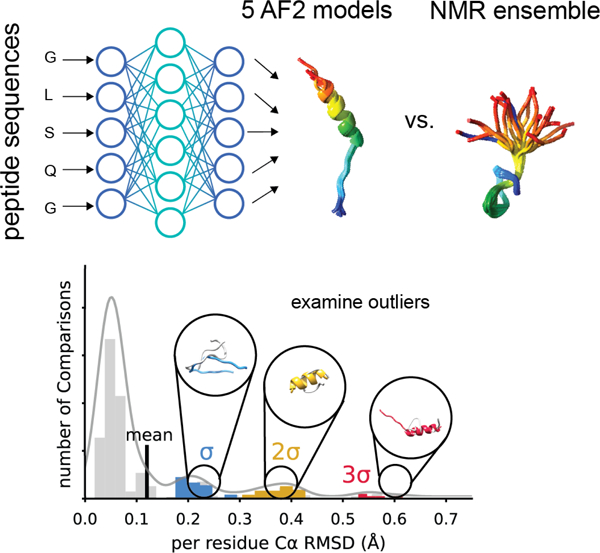
In this work, our aim was to lay the foundation for the use of AF2 to predict the structure of peptides that are between 10 and 40 amino acids long and compare AF2 performance with other peptide prediction methods. We sought to understand if AF2 was useful for large scale modeling of peptide structures that are hard to obtain through experimental methods. To achieve this goal, we selected 588 peptides from the Protein Data Bank (PDB) 23 and ran calculations to predict their structures with AF2 (Figure 1A). Next, we compared the predicted and experimental structures to calculate AF2 prediction performance as measured by root mean square deviation (RMSD) and Φ/Ψ angle recovery. We plotted the distribution of RMSD values to determine outliers where AF2 struggled to predict experiment accurately. We examined on case-by-case basis the inaccurately predicted AF2 models by looking at the lowest RMSD overlapping AF2 and NMR structures to better understand AF2 limitations in modeling peptides. Finally, alternative prediction methods PEPFOLD3, OmegaFold, RoseTTAFold, and APPTEST were used to predict the 588 peptide models and the performance of each method was evaluated on a statistical bases compared to AF2.
Figure 1. Workflow for benchmarking AlphaFold2 on peptide structure prediction.
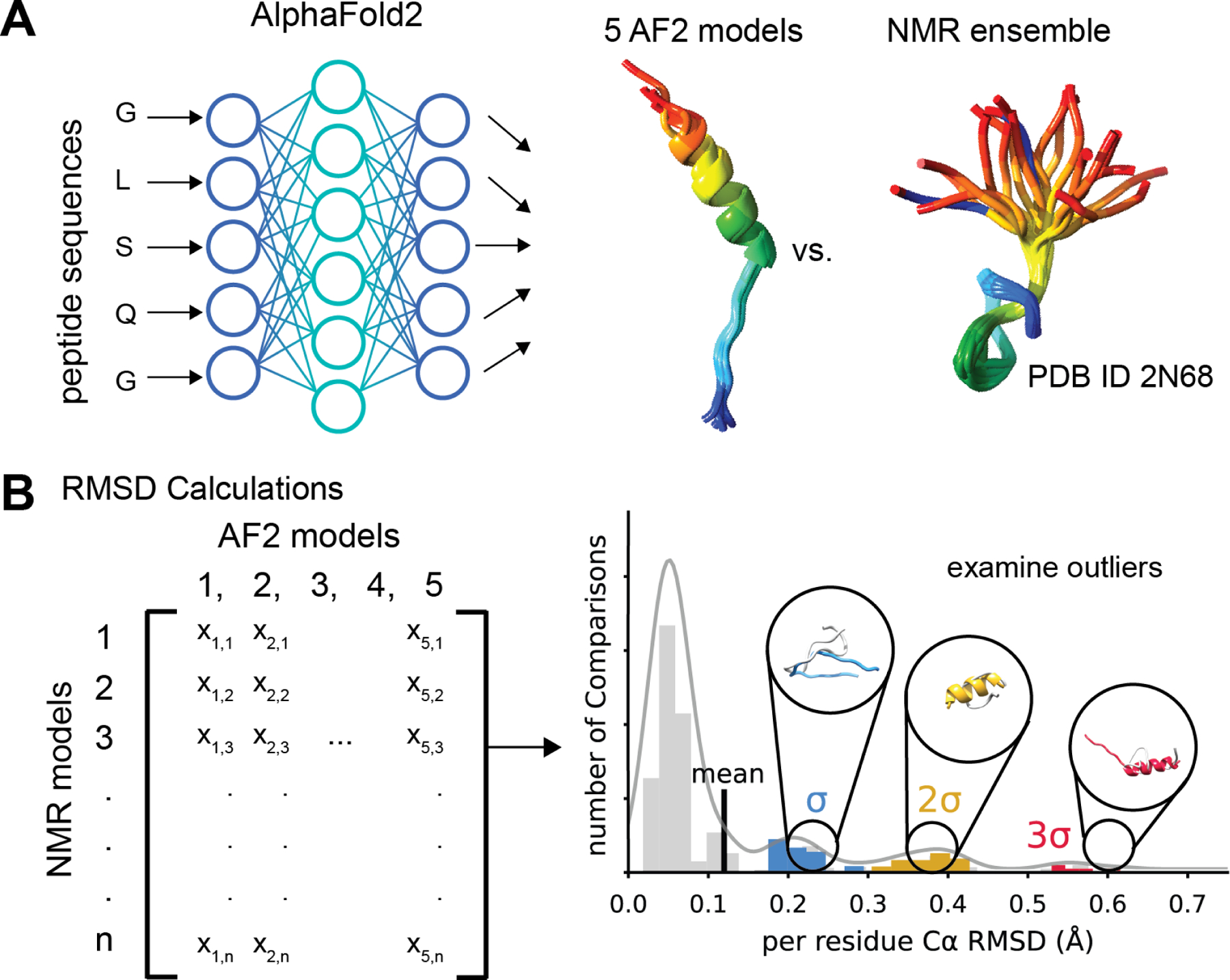
A. We used AlphaFold2 (AF2) to predict the structures of 588 peptide sequences across six peptides classes with experimentally determined NMR models. B. We compared the five AF2 output models to each NMR ensemble by calculating the Cα RMSD exhaustively pairwise. The distribution of RMSD for each class of peptides was then plotted as a histogram and models with an RMSD one standard deviations (σ), two standard deviations (σ), or three standard deviations (3 σ) above the mean were examined to understand how AF2 failed to predict the experimental model.
Results
Structure selection and statistical analysis
The structure selection and analysis are described in the Methods section. Briefly, we selected 588 peptides (Table S1) with experimentally determined NMR structures that included both well-defined secondary structure elements and disordered regions. The peptides were split into the following benchmark sets: α-helical membrane-associated peptides (AH MP), α-helical soluble peptides (AH SL), mixed secondary structure membrane-associated peptides (MIX MP), mixed secondary structure soluble peptides (MIX SL), β-hairpin peptides (BHPIN), and disulfide-rich peptides (DSRP). For each peptide, the ensemble of NMR structures was compared pairwise with all five AF2 structures and the distribution of all pairwise Cα RMSD was plotted to determine outliers and examine poorly predicted structures (Figure 1B). The Cα RMSD was calculated only for the secondary structural region of the peptide (Table S1) and normalized to the residue number within the region considered to prevent any biases caused by size variations of different peptides.
α-Helical membrane-associated peptides were predicted with good accuracy and very few outliers
These peptides are defined as polyamides that fold into a predominantly α-helical structure in the presence of a membrane environment. This group covers peptides including transmembrane helices, amphipathic helices, structures with a helix-turn-helix motif, and monotopic helices that partially span the membrane. This group was the second largest of all the investigated groups, consisting of 187 peptides. A histogram of normalized Cα RMSDs demonstrates a unimodal gaussian with a mean at 0.098 Å per residue for all pairwise comparisons between AF2 and NMR models (Figure 2A). We examined individual outliers based on the number of standard deviations (σ) above the mean to understand the structure shortcomings of AF2 predictions. In some cases, AF2 failed to predict the end of helices and helix-turn-helix for α-helical peptides (Figure 2B). Although AF2 predicted AH MPs well as measured by Cα RMSD, it failed to recover Φ and Ψ angles, especially for low Cα RMSD pairs (Figure S1A)
Figure 2. α-helical peptides predictions perform better for membrane associated peptides.
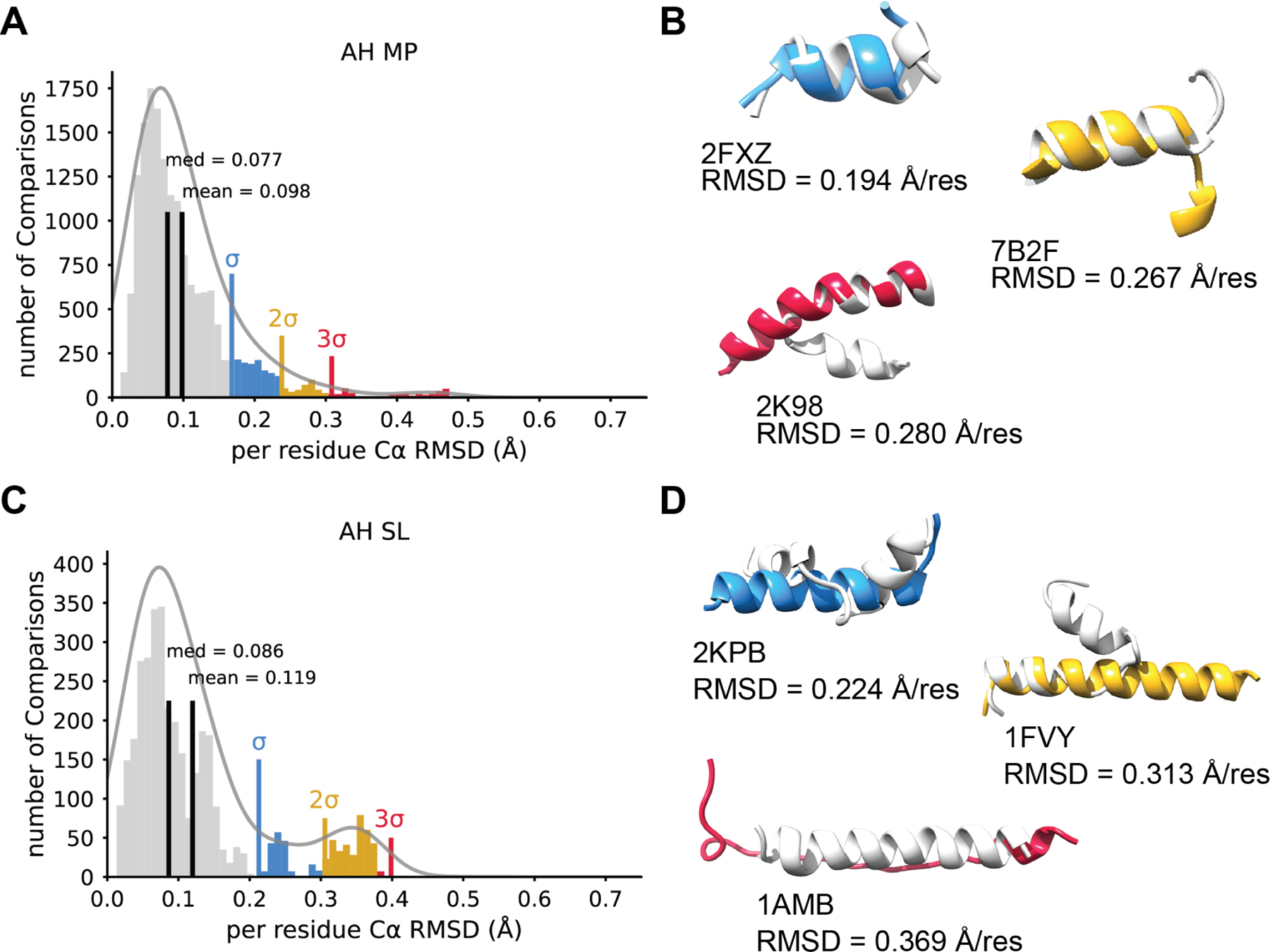
A. A histogram of Cα RMSDs for all comparisons between NMR ensembles and AF2 predictions for α-helical membrane peptides (AH MP) normalized to residue number. The mean and median are shown in black. A multimodal Gaussian was fit to the data using kernel density estimation. One, two and three standard deviations above the mean are shown in blue, yellow, and red respectively. B. Three example models of AF2 predictions that show an RMSD one, two, and three standard deviations above the mean. NMR models are shown in light grey and AF2 models depicted in blue (one standard deviation), yellow (two standard deviations), or red (near three standard deviations). The PDB ID and normalize RMSD value is provided for clarity. C. For comparison, the distribution of α-helical soluble peptides (AH SL) Cα RMSDs between AF2 and NMR plotted as a histogram and normalized to residue number. The mean and median are shown in black. Again, a multimodal Gaussian was fit to the data using kernel density estimation and one, two and three standard deviations above the mean are shown in blue, yellow, and red respectively. D. Example models portraying predictions one, two, and three standard deviation above the mean. The lowest pairwise RMSD is shown for each model. NMR model shown in light grey and the AF2 model is depicted in blue (σ), yellow (2σ), or red (3σ). Note, here 1AMB RMSD falls slightly below the 3σ, but represents the greatest outlier among the AH SL data.
α-Helical soluble peptides showed outliers and performed worse compared to their membrane-associated counterparts
The α-helical soluble peptide group was defined as α-helical peptides whose structures were not identified in a membrane environment, had no remarks regarding membrane interactions in the original publication, but fulfilled the remaining secondary structure conditions previously described for α-helical membrane peptides. 41 peptides belonged to this group. The distribution of normalized Cα RMSDs demonstrates a bimodal gaussian with a mean at 0.119 Å per residue and a second peak between 2σ and 3σ above the mean (Figure 2C). The outliers for soluble α-helical peptides again suggested AF2 struggled to predict helix-turn-helix structures (Figure 2D). Furthermore, AF2 failed to predict α-helical structure at all for 1AMB with an RMSD of 0.369 Å per residue (Figure 2D). Finally, AF2 also failed to recover Φ and Ψ angles for AH SL peptides, suggesting AF2 prediction lacked α-helical ideality (Figure S1B).
Mixed secondary structure membrane-associated peptides showed the largest variation and RMSD values among all the benchmark sets
The mixed secondary structure peptides were identified to interact with membranes like the α-helical membrane peptides, but they consisted of more than one secondary structure region (e.g., multiple α-helices separated by a turn, α/β or α/coil mixed secondary structure, etc.). 14 peptides belonged to this group and the normalized Cα RMSD histogram showed a multi-modal distribution with a mean of 0.202 Å per residue (Figure 3A). No AF2 models deviated from corresponding NMR models by more than 2σ above the mean, but rather the whole distribution skewed the mean with poor performing predictions. Illustrative models indicated AF2 predicted secondary structure correctly but failed to overlap with less structured regions of the peptides (Figure 3B). On the other hand, AF2 failed to predict any secondary structure for 1SOL (Figure 3B). In contrast to α-helical peptides, AF2 recovered Φ and Ψ angles for low Cα RMSD comparisons of mixed membrane peptides (Figure S2A).
Figure 3. AlphaFold2 poorly predicts mix membrane peptides and mix soluble peptides.
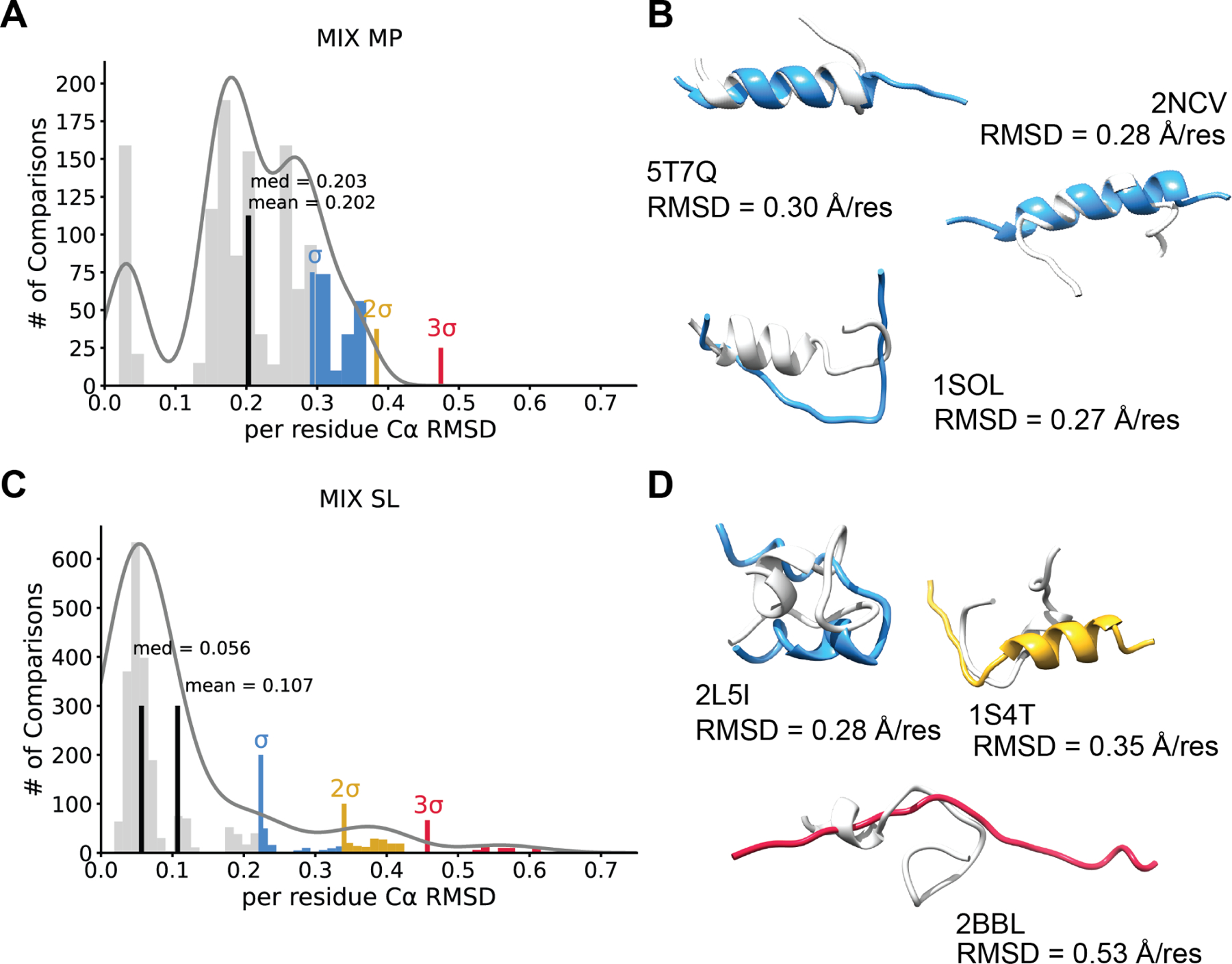
A. Mix membrane protein (MIX MP) histogram depicting the Cα RMSDs for all pairwise comparisons between AF2 and NMR models normalized to residue number. The mean and median are shown in black. A multimodal Gaussian was fit to the data using kernel density estimation. One, two and three standard deviations above the mean are shown in blue, yellow, and red respectively. Interestingly, no models fell above two standard deviations for this peptide class. B. Three example models of AF2 predictions that show an RMSD one standard deviation above the mean. NMR models are shown in light grey and AF2 models depicted in blue, and the PDB ID and normalize RMSD is shown. C. A histogram of mixed soluble peptides (MIX SL) residue number normalized Cα RMSDs between AF2 and NMR. The mean and median are shown in black. We fit a multimodal Gaussian to the data using kernel density estimation and one, two and three standard deviations above the mean are shown in blue, yellow, and red respectively. D. Illustrative models portraying predictions σ, 2σ, 3σ above the mean. The lowest pairwise RMSD is shown for each model. NMR model shown in light grey and the AF2 model is depicted in blue (one standard deviation), yellow (two standard deviations), or red (three standard deviations).
Mixed secondary structure soluble peptides showed moderate accuracy
Mixed secondary structure soluble peptides group was defined as peptides that have the same secondary structure properties as their membrane counterparts, but for the peptides whose structures were not identified in a membrane environment. 21 peptides belonged to this group with a normalized Cα RMSD histogram showing a modestly multi-modal gaussian with peaks at 1σ, 2σ, and 3σ above the mean of 0.107 Å per residue (Figure 3C). Outliers suggested AF2 failed to predict the orientation of secondary structure-unstructured boundary (Figure 3D). For example, AF2 predicted 2BBL as a completely unstructured peptide, despite the NMR model consisting of a well-defined compact structure throughout the entire ensemble (Figure 3D). Again, in contrast to α-helical peptides, AF2 Φ and Ψ angle RMSD compared to NMR structures correlated well with Cα RMSD for MH SL peptides (Figure S2B).
β-hairpin peptides were predicted with good accuracy for both stapled and non-stapled peptides
The β-hairpin peptides group includes peptides that have a single β-hairpin motif. Members of this group may or may not be stapled by the presence of a disulfide bond. There was a total of 59 peptides in this group (26 stapled by disulfide bonds and 33 non-stapled). Overall AF2 performed well on BHPIN peptides. A histogram of all pairwise Cα RMSD comparisons between AF2 and NMR models demonstrates a bimodal distribution with a peak at 1σ above the mean of 0.146 Å per residue (Figure 4A). Illustrative structures above the mean indicated AF2 predicts some BHPIN models as α-helical, subsequently leading to poor RMSD values (Figure 4B). Interestingly, all models above the mean were unstapled. Φ and Ψ angle recovery was predicted better than mixed secondary structure peptides but worse than α-helical peptides for BHPIN (Figure S3A) indicating AF2 has learned β-sheet Φ/Ψ ideality better than α-helix phi psi ideality.
Figure 4. AlphaFold2 predictions of β-hairpin and disulfide rich peptides.
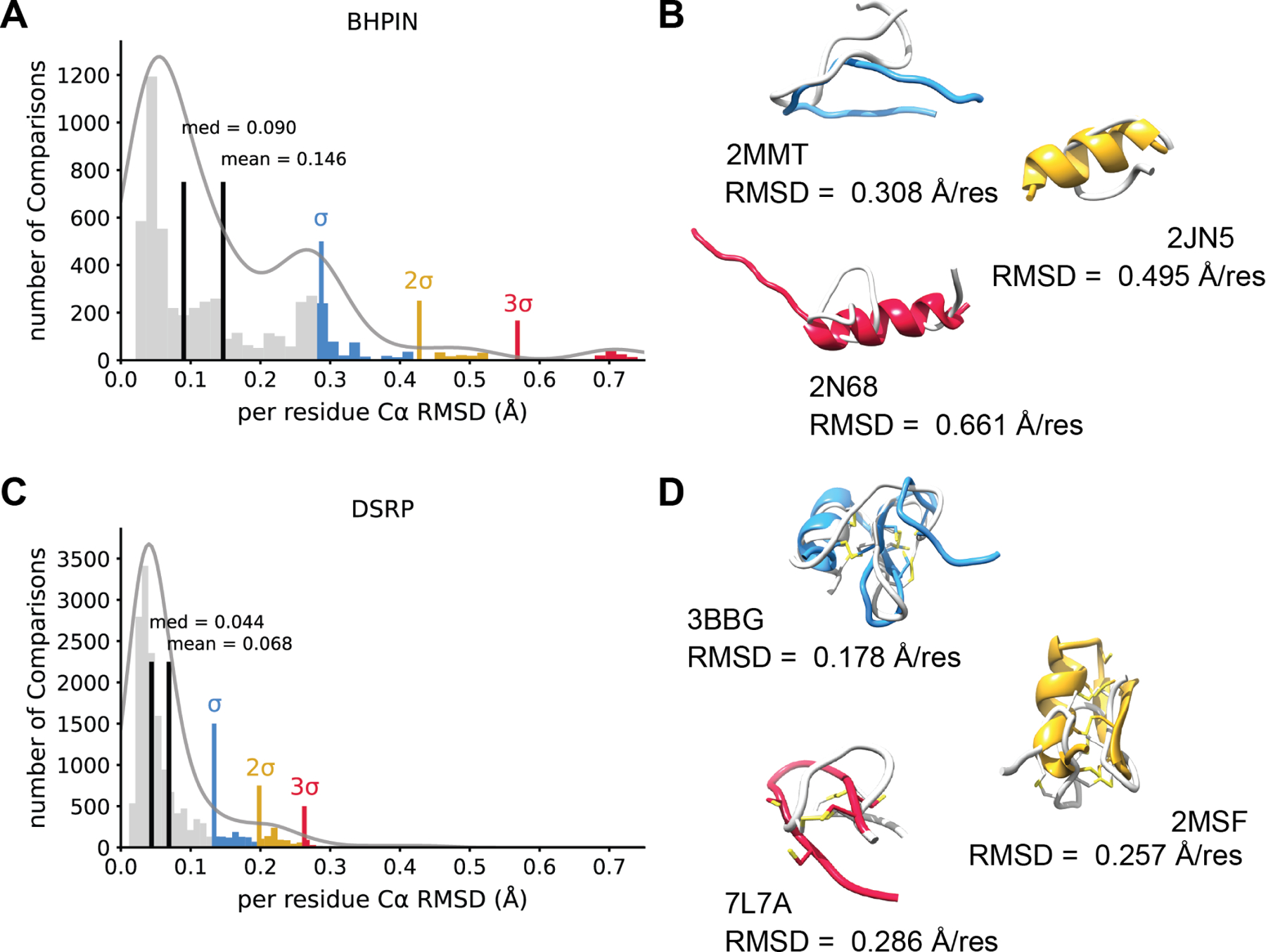
A. β-hairpin peptides (BHPIN) histogram showing the distribution the Cα RMSDs for all pairwise comparisons between AF2 and NMR models normalized to residue number. Mean and median are shown in black. A multimodal Gaussian was fit to the data using kernel density estimation and one, two and three standard deviations above the mean are shown in blue, yellow, and red respectively. B. Three example models of AF2 predictions that showed an RMSD at least one standard deviation above the mean for the BHPIN class. NMR models are shown in light grey and AF2 models depicted in blue (σ), yellow (2σ), and red (3σ). The PDB ID and normalize RMSD is shown. C. The distribution of disulfide residue number normalized Cα RMSDs normalized to residue number between AF2 and NMR plotted as a histogram. The mean and median are shown in black. We fit a multimodal Gaussian to the data using kernel density estimation and σ, 2σ, and 3σ above the mean are shown in blue, yellow, and red respectively. D. Illustrative models portraying predictions one, two, and three standard deviation above the mean (blue) The lowest pairwise RMSD is shown for each model. Cysteines shown in light yellow and disulfide bonds shown. NMR model shown in light grey and the AF2 model is depicted in blue (σ), yellow (2σ), or red (3σ).
Disulfide-rich peptide structures were predicted with high accuracy, but with variation in disulfide bonding patterns
Disulfide-rich peptides (DSRP) were defined in the context of this work as any peptide that had two or more disulfide bonds. This group included toxin peptides such as α-conotoxins, β-hairpins cyclized by multiple disulfide bonds, and some hormone peptides. DSRPs were the largest group in our benchmark set, containing a total of 266 peptides. DSRPs showed a tight, slightly bimodal gaussian histogram with a peak two standard deviations above mean of 0.068 Å per residue (Figure 4C) – the lowest mean of any group. Outliers failed to predict the correct disulfide bonding pattern. With 3BBG predicting one correctly and placing most remaining cysteine residues in proximity, 2MSF misplaced two disulfides and failed to predict bonding of another, and 7L7A failed to predict any disulfide bonding (Figure 4D). Like BHPIN, DSRPs recovered Φ/Ψ angles better than mixed and worse than α-helical (Figure S3B). Interestingly, AF2 failed to predict the correct disulfide bonding pattern of 25 DSRPs (Table S2). These peptides frequently contained consecutive cysteines or the predicted AF2 model showed a loss in disulfide bonding. Strangely, only 10 out of 25 were statistical outliers by normalized Cα RMSD (Table S2) mean AF2 predicted structure correctly while still missing the correct disulfide bonding pattern.
Statistical evaluation of computational structure prediction methods on peptides
We sought to understand whether AF2 has an advantage over other deep-learning and de novo protein/peptide prediction methods in predicting the experimental structure of peptides. Predictions were generated for all 588 peptides in our benchmark set using PEPFOLD3, OmegaFold, RoseTTAFold, and APPTEST (see Methods). PEPFOLD3 and APPTEST were designed for peptide structure prediction, whereas OmegaFold and RoseTTAFold were designed for general protein structure prediction. AF2 accurately predicted peptide structures better than all alternative methods for peptide groups AH MP, AH SL, BHPIN, and DSRP as measured by length normalized Cα RMSD (Figure 5A-D). Interestingly, AF2 only outperformed APPTEST on MIX MP (Figure 5E). Finally, AF2 outperformed PEPFOLD3, RoseTTAFold, and APPTEST, but performed just as well as OmegaFold for MIX SL peptides (Figure 5F).
Figure 5. AlphaFold2 predicts peptide structures better than alternative computational methods PEPFOLD-3 (PF), OmegaFold (OF), RoseTTAFold (RF), and APPTEST (AT).
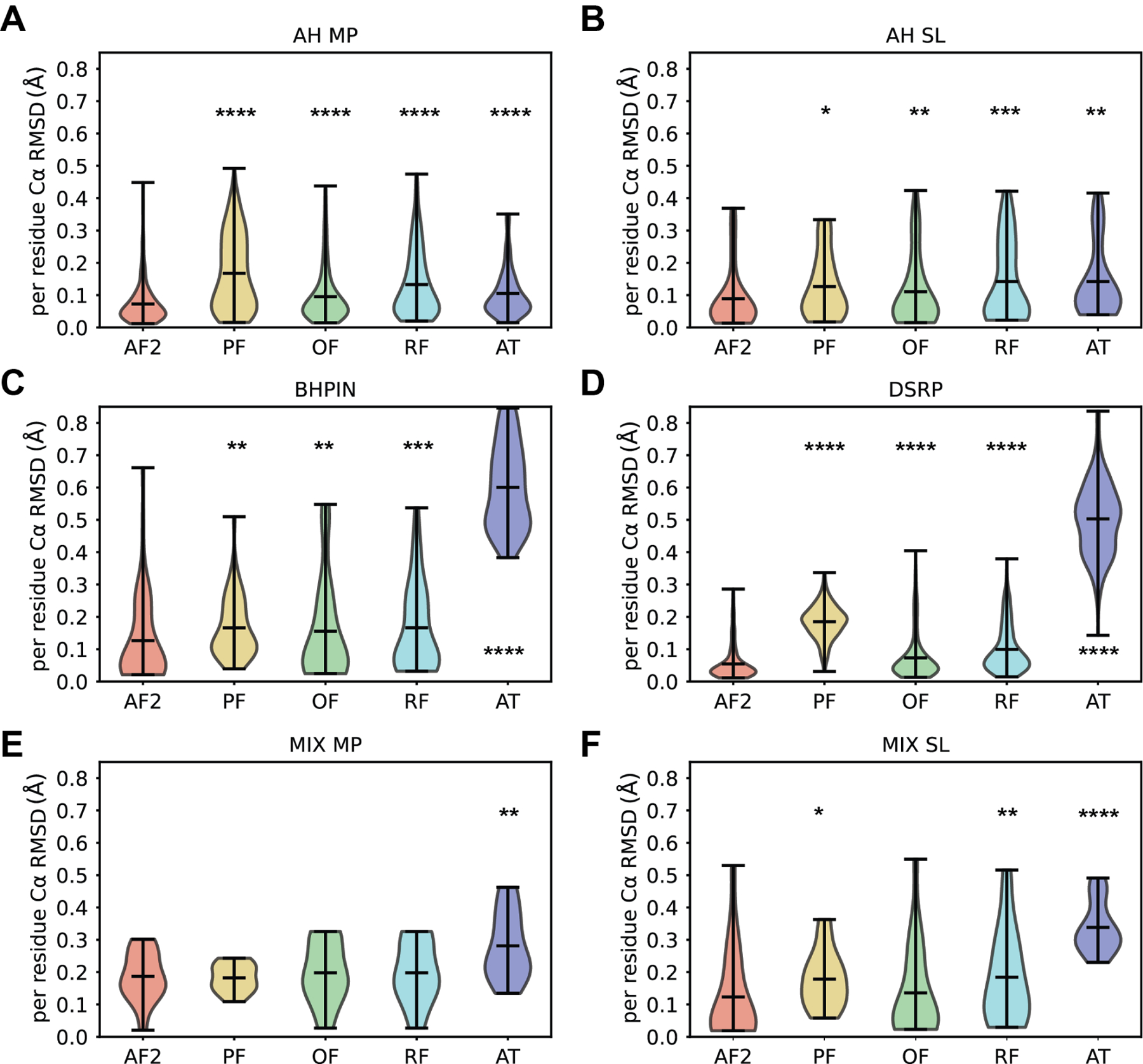
A-F. Distributions of average Cα RMSD per residue between the secondary structural regions of the NMR ensemble compared to each alternative computational prediction method. Peptide groups (A) α-helical membrane protein (AH MP), (B) α-helical soluble (AH SL), (C) β-hairpin peptides (BHPIN), (D) disulfide rich peptides (DSRP), (E) mixed membrane peptide (MIX MP), and (F) mixed soluble peptide (MIX SL) are shown. AlphaFold2 performed statistically significantly better than PEPFOLD3, OmegaFold, RoseTTFold and APPTEST, in all peptide classes except for MIX MP and MIX SL. For MIX MP, all methods perform with similar accuracy except for APPTEST. Interestingly, OmegaFold performed just as well as AF2 for MIX SL, and both outperformed the other methods. A paired two-side student t-test was used to calculate the statistical difference of each distribution with respect to AF2 and p value depicted by * < 0.05, ** < 0.01, *** < 0.001, and **** < 0.0001.
There is poor correlation between the predicted rank and prediction accuracy
A standard AF2 run generates five structures as output. These structures are ranked by the program based on their predicted local distance difference test (pLDDT) values, which is a metric that estimates how well a predicted structure would agree with an experimental structure based on backbone carbon coordinates 24. The lowest-ranking structure represents the best prediction of a given sequence. However, this assumption may not always be true. All five AF2 structures generated were compared pairwise with the corresponding NMR reference ensemble in this study to allow better structural sampling. In theory, the structures with lower ranks should give the lowest Cα RMSD values in our calculations. To test whether this assumption is true, we enumerated the lowest Cα RMSD pairwise comparisons of each rank (Figure S5). Based on these summations, there was no correlation between the first three ranks assigned by AF2 and the structure that gave the lowest Cα RMSD. However, ranks four and five were represented less in lowest Cα RMSD pairs (Figure S5). Therefore, our results suggest that the pLDDT metric that AF2 used to assess globular protein structures was not a meaningful metric to classify peptide conformations.
Limitations of the study
The first limitation of the study is caused by the intrinsic flexibility of many peptides in general. Peptides can have highly flexible regions including coils or turns that may result in multiple conformations for the same structure. NMR structures typically consist of an ensemble of conformations, which makes an exact comparison between the predicted and experimental structures challenging. To address this point, an ensemble of NMR models was compared pairwise to all predicted outputs for each method, resulting in a distribution of comparisons, and the lowest Cα RMSD was selected for illustrative purposes and statistical comparison between methods. Further, comparison of the NMR and X-ray structures of the peptides for which such data are available showed no significant difference in terms of AF2’s performance.
Another point related to flexibility is that alternative low-energy conformations to the structures identified by NMR may exist. Especially with helices that have multiple domains connected by turns or coils, the structures captured by experimental methods may represent only one of the multiple conformations the peptides. From that perspective, the structures predicted by AF2 may not necessarily be wrong, but they may simply correspond to an alternative conformation of the peptide, although AF2 predicted X-ray structures just as accurately (Figure S4). A study aimed at comparing the quality of AF2-predicted structures with experimental structures determined by NMR in solution identified that structures predicted by AF2 can be more energetically stable and therefore feasible compared to NMR structures in some cases 25. More systematic studies that focus on peptide dynamics through experimental and computational methods will be necessary to understand whether the AF2-predicted peptide structures that showed great variation from the experimental predictions are likelier to represent the actual structure of these peptides.
Finally, some peptides may fold into different secondary structures depending on the environmental conditions including pH, temperature, and the presence of a membrane. Because the structures from NMR experiments are obtained with a single set of experimental parameters, AF2 predictions may represent alternative conformations of the peptides that could have been obtained under different conditions. This may particularly be an issue when AF2 over- or underestimates the secondary structure of peptides, which may indeed be more or less disordered depending on environmental conditions. Therefore, it is necessary to investigate whether AF2 has a bias towards structures determined under particular conditions.
Discussion
Peptides comprise a class of 2 to 50 amino acid long polyamides that function naturally in signaling, toxicity, and hormones. Additionally, synthetic peptides are being pursued as drug candidates and cell biologists have probed subcellular localization with de novo designed peptides 26. Peptide structures were traditionally determined by NMR with draw backs in sensitivity to environmental conditions. Recent advances in protein structure prediction bore the opportunity to evaluate these methods in predicting NMR determined peptide models. We benchmarked 588 peptide across six groups and showed AF2 demonstrated strength in secondary structure predictions and peptide with increased residue contact, while demonstrating short comings in solvated peptides and helix-turn-helix structures. These shortcomings suggested AF2 peptide prediction may benefit from addition difficult to predict structures.
The α-helical membrane peptide structures were predicted overall with good success except for a few outliers. Outliers showed no single reason for the deviations, but the inability of AF2 to predict the secondary structure of some peptides and the inaccurate prediction of the kink angles accounted for most deviations. The mixed secondary structure peptides were also predicted with varying accuracy. In the case of the membrane-associated peptides, deviations were caused mostly by the errors in prediction of the angles between α-helical regions, but the secondary structure predictions were overall accurate with few exceptions.
When we compared α-helical membrane protein to α-helical solvated proteins, it was clear membrane solvated protein predicts performed better (RMSD average of 0.092 Å vs. 0.130 Å). By contrast, when we compared mixed membrane protein vs. mixed solvated proteins, the later performed better (RMSD 2.17 Å vs. 1.26 Å respectively). This suggested secondary structure drives AF2 accuracy more than solvation state. However, AF2 increased performance on less solvated BHPIN and DSRP supports the notion that highly solvated peptides remain difficult to predict (Figure 3 and 4). Solvated peptides may not require MSA based deep-learning models at all, as OmegaFold performance matched AF2 on soluble peptides and is solely sequenced based. This suggests solvated peptide structure prediction gains little benefit from evolutionary data inherent in MSAs.
On the other hand, β-hairpin peptides and DSRPs are more compact, less solvent exposed peptides and AF2 performance outstrips OmegaFold and all other alternatives in this group. DSRPs had the highest degree of accuracy among the tested peptides, potentially thanks to their constrained structures due to the presence of multiple disulfide bonds. These structures also had regions with well-defined secondary structures that make predictions easier for AF2 compared to coiled regions where multiple degrees of freedom are present. AF2 predicted DSRPs better statistically significantly better than any other computational method test. On the other hand, the results demonstrated that the lowest-RMSD AF2 predictions had different disulfide bonding patters or no disulfide bonding at all in some cases (Table S2). This suggested that special care must be shown when predicting DSRP structures that may have alternative disulfide bonding patterns, and the use of additional tools may be necessary to remodel the disulfide bonds following AF2 runs.
Poor Φ/Ψ angle recovery was observed for most the tested peptide groups except for solvated peptides. This is surprising considering the overall success of AF2 with globular proteins for which the calculated rotamer recoveries were over 80 % in another study 27. The reason for this discrepancy may be that the investigated peptides have mostly solvent-exposed residues with few buried amino acids due to the small and extended structures that prevent close interactions between the different domains. While this may be related to AF2’s inability to capture the right geometry of these flexible regions, it is also possible that the NMR structures were not solved under experimental conditions that would be more consistent with the geometries calculated by AF2. We looked for correlations between the calculated Φ/Ψ recovery percentages and parameters like RMSD (Figure S1-S3), but we only found correlation for solvent-exposed peptides (Figure S2). Considering the large deviation in Φ and Ψ angles in globular proteins, additional tools may be necessary to optimize the secondary structure regions or side chain conformations of the peptides generated through AF2. Additional tools such as Rosetta Disulfidize, side chain optimization, or simple potential energy function minimization28 may help optimize initial AF2 models.
Lastly, our analysis shows that pLDDT, which is the main metric used by AF2 to rank the generated structures is not a good measure of whether a peptide structure is accurately predicted. Rank one, two and three by pLDDT are represented relatively equally among lowest RMSD pairwise comparisons with NMR ensemble, while rank four and five were represented less (Figure S5). A study investigating multiple aspects of protein folding with AF2 involved testing of pLDDT values for proteins of varying lengths, which showed that shorter sequences tend to have larger pLDDT values in general 29. This may reduce the utility of this metric for small peptide structures by narrowing the gap between good and bad predicted structures. In the absence of a clear selection criterion for the AF2-generated structures, it may be necessary to increase the number of output structures generated and use approaches such as clustering to narrow down the conformations that are sampled more frequently or show more consistent patterns to select the accurate structure.
All these data raise the question of whether the issues regarding the shortcomings of AF2 identified in this study are unique to peptides, or are general issues associated with AF2 making predictions on structures on which it was not trained 30,31. It is unsurprising, AF2 demonstrates flaws with predicting for a class of systems e.g. peptide which were excluded from the initial training set and indicates that inclusion in future training data may improve performance on such systems. However, AF2 still outperformed peptide specific methods for high contact peptide categories. Strangely, AF2 performed better on short peptides than long peptides (Figure S6). Ever since the release of AF2, the focus of the benchmark studies has been what AF2 is capable of doing with little focus on its shortcomings. Our work presents a glimpse into limiting factors that should be considered when modeling peptides, but similar studies that investigate these elements in globular proteins are also necessary in the future.
Conclusions
AF2 can be used for the modeling of peptide structures smaller than 40 amino acids if the target peptide is anticipated to have a well-defined secondary structure and lacks multiple turn or flexible regions that may assume different conformations. AF2 is particularly successful in the prediction of α-helical membrane-associated peptides and DSRPs but has reduced accuracy in the cases with extended coiled or flexible regions. Even for the DSRPs, which predicted accurately overall, issues with disulfide bonding patterns may result in errors in modeling peptides. In addition, AF2 predictions showed no correlation between the predicted ranks and prediction accuracy, therefore alternative metrics to select from the AF2-generated structures may be necessary. Overall, use of AF2 for peptide structure prediction will require development of additional metrics and controls to improve their accuracy.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Dr. Alican Gulsevin (alican.gulsevin@vanderbilt.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All the PDB structures curated from the PDB for the calculations are available upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All data are generated from the datasets provided in the Key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| PDB structures and sequences used for the prediction and RMSD calculations | Protein Data Bank (PDB)32 | www.rcsb.org |
| Software and Algorithms | ||
| Software used for peptide structure prediction | AlphaFold224 | https://alphafold.ebi.ac.uk/ |
| Software used for peptide structure prediction | OmegaFold15 | https://github.com/HeliXonProtein/OmegaFold |
| Software used for peptide structure prediction | RoseTTAFold14 | https://github.com/RosettaCommons/RoseTTAFold |
| Software used for peptide structure prediction | PEP-FOLD312 | https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/ |
| Software used for peptide structure prediction | APPTEST13 | https://research.timmons.eu/apptest |
| Data processing and analysis | Python33 | https://www.python.org/ |
| RMSD calculations | PyRosetta34 | https://www.pyrosetta.org/ |
METHOD DETAILS
Structure selection criteria
A PDB search was done to select peptides smaller than 60 amino acids whose structures were determined through solid-state or solution NMR. Peptides cyclized through their backbones, that have floppy termini, predominantly lacking a secondary structure, lacking side chain conformations, and peptides bearing non-canonical amino acids were excluded from the set. For the peptides with multiple reported conformations in different media, the PDB entry that had the most secondary structure was selected if the conformations were similar, and the entry was excluded if the secondary structure elements showed variability among the different entries. A total of 588 peptides fit these criteria and were used for the benchmark calculations. The benchmark set was divided into further groups to facilitate analysis of the results. These groups are α-helical membrane peptides, α-helical soluble peptides, mixed secondary structure membrane peptides, mixed secondary structure soluble peptides, β-hairpin peptides, and disulfide-rich peptides (DSRP).
AF2 calculations
AF2 calculations were run locally in the monomer mode starting from the sequences of each entry as extracted from the PDB. Terminal modifications were removed from the sequence files prior to the calculations. For each entry, five structures were generated with AF2 and the best-ranking structure according to the LDDT scores was selected as the representative pose for the entry. All the analyses were done with these representative poses.
RoseTTAFold and OmegaFold Calculations
RoseTTAFold was run locally with default parameters for the MSA generation and secondary structure prediction steps, and the three-track prediction method for the structure prediction step. Individual calculations were set for each peptide for the RoseTTAFold calculations. A single structure was generated as the result of each calculation. A single sequence input file including all the peptide sequences were inputted for OmegaFold, and the resulting predicted structure file was used for all the comparisons with the AF2-predicted and experimental peptide structures.
APPTEST and PEPFOLD-3 Calculations
For predicting peptide structures with online servers, we used APPTEST and PEPFOLD3. For APPTEST (https://research.timmons.eu/apptest) we entered the peptide sequence and set simulated annealing + molecular dynamics to none - so just the neural network deep learning-based prediction produced results. APPTEST performance would be greatly improved with addition of Molecular Dynamics simulations steps, but at the cost of computational speed. Disulfide bonds was left blank, the job name was set to PDB ID and the prediction run and results downloaded. The PDB contained in folder cyana and subfolder with the sequence was used as the final prediction for APPTEST. For PEPFOLD3 found (https://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#forms::PEP-FOLD3), we pasted in the peptide sequence, changed the run label to PDB ID, set number of simulations to 100 (default), and sorted by sOPEP. No reference structure, mask, receptor structure, or residues defining an interaction patch with a receptor were entered. Demonstration mode was set to “No”. The prediction was run and resultant PDB downloaded and used as final prediction of PEPFOLD3.
QUANTIFICATION AND STATISTICAL ANALYSIS
RMSD calculations
RMSD values were calculated as the average root mean squared distance between the coordinates of Cα atoms of all five AF2-generated structures and each model contained in the corresponding PDB NMR ensemble. The structures were aligned based on Cα coordinates to eliminate translational differences between the reference and AF2 coordinates. A histogram of all pairwise Cα RMSD comparisons was plotted with 50 bins and fitted with a gaussian using kernel density estimation. The standard deviation (σ) was calculated for the distribution and histogram was colored blue (1σ above the mean), yellow (2σ above the mean), and red (3σ above the mean). AF2 predicts with RMSD values above the mean were selected for illustrative purposes to determine how AF2 failed with these structures. Only the lowest-RMSD AF2 structure was used illustrative purposes. Φ/Ψ angle recovery was calculated in the same manner using PyRosetta.
Supplementary Material
Highlights.
Performance of AlphaFold2 was compared with other protein or peptide folding methods.
Deep-learning-based methods outperformed dedicated peptide structure prediction tools.
Prediction accuracy is affected by peptide secondary structure.
Acknowledgements
This research was funded by NIH R01 HL144131 and NIH NIGMS R01 GM080403. The authors would like to thank Elleansar Okwei for her comments on the manuscript. EFM was funded by NIH #1F31HL162483-01A1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
Supplemental Table 1, in Relation to Star Methods: List of all the sequences, the PDB IDs and descriptions of the PDB entries corresponding to these sequences, peptide length information, and the list of the structured regions used for the RMSD calculations.
References
- 1.Otvos L, and Wade JD (2014). Current challenges in peptide-based drug discovery. Front. Chem 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Toprak U (2020). The Role of Peptide Hormones in Insect Lipid Metabolism. Front. Physiol 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirakawa Y, and Sawa S (2019). Diverse function of plant peptide hormones in local signaling and development. Curr. Opin. Plant Biol 51, 81–87. 10.1016/j.pbi.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 4.Yeaman MR, and Yount NY (2003). Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol. Rev 55, 27 LP – 55. 10.1124/pr.55.1.2. [DOI] [PubMed] [Google Scholar]
- 5.Lee ACL, Harris JL, Khanna KK, and Hong JH (2019). A comprehensive review on current advances in peptide drug development and design. Int. J. Mol. Sci 20, 1–21. 10.3390/ijms20102383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark GC, Casewell NR, Elliott CT, Harvey AL, Jamieson AG, Strong PN, and Turner AD (2019). Friends or Foes? Emerging Impacts of Biological Toxins. Trends Biochem. Sci 44, 365–379. 10.1016/j.tibs.2018.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Luca S, Heise H, and Baldus M (2003). High-Resolution Solid-State NMR Applied to Polypeptides and Membrane Proteins. Acc. Chem. Res 36, 858–865. 10.1021/ar020232y. [DOI] [PubMed] [Google Scholar]
- 8.Wüthrich K (2001). The way to NMR structures of proteins. Nat. Struct. Biol 8, 923–925. 10.1038/nsb1101-923. [DOI] [PubMed] [Google Scholar]
- 9.Kuczera K (2015). Molecular Modeling of Peptides BT - Computational Peptidology In, Zhou P and Huang J, eds. (Springer; New York: ), pp. 15–41. 10.1007/978-1-4939-2285-7_2. [DOI] [Google Scholar]
- 10.Thévenet P, Rey J, Moroy G, and Tuffery P (2014). De Novo Peptide Structure Prediction: An Overview. Comput. Pept 1268, 1–13. 10.1007/978-1-4939-2285-7_1. [DOI] [PubMed] [Google Scholar]
- 11.Yan J, Bhadra P, Li A, Sethiya P, Qin L, Tai HK, Wong KH, and Siu SWI (2020). Deep-AmPEP30: Improve Short Antimicrobial Peptides Prediction with Deep Learning. Mol. Ther. - Nucleic Acids 20, 882–894. 10.1016/j.omtn.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamiable A, Thévenet P, Rey J, Vavrusa M, Derreumaux P, and Tufféry P (2016). PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res 44, W449–54. 10.1093/nar/gkw329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Timmons PB, and Hewage CM (2021). APPTEST is a novel protocol for the automatic prediction of peptide tertiary structures. Brief. Bioinform 22, bbab308. 10.1093/bib/bbab308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minkyung B, Frank D, Ivan A, Justas D, Sergey O, Rie LG, Jue W, Qian C, N., K.L., Dustin SR, et al. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science (80-. ) 373, 871–876. 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu R, Ding F, Wang R, Shen R, Zhang X, Luo S, Su C, Wu Z, Xie Q, Berger B, et al. (2022). High-resolution de novo structure prediction from primary sequence. bioRxiv, 2022.07.21.500999. 10.1101/2022.07.21.500999. [DOI]
- 16.Nerli S, McShan AC, and Sgourakis NG (2018). Chemical shift-based methods in NMR structure determination. Prog. Nucl. Magn. Reson. Spectrosc 106–107, 1–25. 10.1016/j.pnmrs.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDonald EF, Woods H, Smith ST, Kim M, Schoeder CT, Plate L, and Meiler J (2022). Structural Comparative Modeling of Multi-Domain F508del CFTR. Biomol 12. 10.3390/biom12030471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gulsevin A, and Meiler J (2020). An investigation of three-finger toxin-nachr interactions through rosetta protein docking. Toxins (Basel) 12. 10.3390/toxins12090598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. (2021). Applying and improving AlphaFold at CASP14. Proteins Struct. Funct. Bioinforma 89, 1711–1721. 10.1002/prot.26257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsaban T, Varga JK, Avraham O, Ben-Aharon Z, Khramushin A, and Schueler-Furman O (2022). Harnessing protein folding neural networks for peptide–protein docking. Nat. Commun 13, 1–12. 10.1038/s41467-021-27838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johansson-Akhe I, and Wallner B (2021). Benchmarking Peptide-Protein Docking and Interaction Prediction with AlphaFold-Multimer. bioRxiv, 2021.11.16.468810.
- 22.Lee J (2021). C an a Lpha F Old 2 Predict Protein - Peptide Complex Structures Accurately ? bioRxiv, 2–7.
- 23.Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, Feng Z, Gilliland GL, Iype L, Jain S, et al. (2002). The Protein Data Bank. Acta Crystallogr. Sect. D 58, 899–907. 10.1107/S0907444902003451. [DOI] [PubMed] [Google Scholar]
- 24.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fowler NJ, and Williamson MP (2022). The accuracy of protein structures in solution determined by AlphaFold and NMR. bioRxiv, 2022.01.18.476751. 10.1101/2022.01.18.476751. [DOI] [PMC free article] [PubMed]
- 26.Rhys GG, Cross JA, Dawson WM, Thompson HF, Shanmugaratnam S, Savery NJ, Dodding MP, Höcker B, and Woolfson DN (2022). De novo designed peptides for cellular delivery and subcellular localisation. Nat. Chem. Biol 18, 999–1004. 10.1038/s41589-022-01076-6. [DOI] [PubMed] [Google Scholar]
- 27.Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Žídek A, Bridgland A, Cowie A, Meyer C, Laydon A, et al. (2021). Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596. 10.1038/s41586-021-03828-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, et al. (2011). Rosetta3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol 487, 545–574. 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monzon V, Haft DH, and Bateman A (2022). Folding the unfoldable: using AlphaFold to explore spurious proteins. Bioinforma. Adv 2, 1–6. 10.1093/bioadv/vbab043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gulsevin A, Han B, Porta JC, Mchaourab HS, Meiler J, and Kenworthy AK (2022). Template-free prediction of a new monotopic membrane protein fold and oligomeric assembly by Alphafold2. bioRxiv, 2022.07.12.499809. 10.1101/2022.07.12.499809. [DOI] [PMC free article] [PubMed]
- 31.Xiong H, Han L, Wang Y, and Chai P (2022). Evaluating the Reliability of AlphaFold 2 for Unknown Complex Structures with Deep Learning. bioRxiv, 2022.07.08.499384. 10.1101/2022.07.08.499384. [DOI]
- 32.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, and Bourne PE (2000). The Protein Data Bank. Nucleic Acids Res 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Rossum G, and Drake FL (2009). Python/C Api Manual - Python 3 (CreateSpace).
- 34.Chaudhury S, Lyskov S, and Gray JJ (2010). PyRosetta: A script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 26, 689–691. 10.1093/bioinformatics/btq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the PDB structures curated from the PDB for the calculations are available upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.