

Abstract
At its basic conceptualization, photoclick chemistry embodies a collection of click reactions that are performed via the application of light. The emergence of this concept has had diverse impact over a broad range of chemical and biological research due to the spatiotemporal control, high selectivity and excellent product yields afforded by the combination of light and click chemistry. While the reactions designated as “photoclick” have many important features in common, each has its own particular combination of advantages and shortcomings. A more extensive realization of the potential of this chemistry requires a broader understanding of the physical and chemical characteristics of the specific reactions. This review discusses the features of the most frequently employed photoclick reactions reported in the literature: photomediated azide alkyne cycloadditions, other 1,3-dipolarcycloadditions, Diels Alder and inverse electron demand Diels Alder additions (IEDA), radical alternating addition chain transfer additions and nucleophilic additions (e.g. thiol-ene and thiol-Michael), among others. Applications of these reactions in a variety of chemical syntheses, materials chemistry and biological contexts are surveyed with particular attention paid to the respective strengths and limitations of each reaction and how that reaction benefits from its combination with light. Finally, challenges to broader employment of these reactions are discussed along with strategies and opportunities to mitigate such obstacles.
Graphical Abstract

1. Introduction
Photoclick reactions combine high yields and selectivity with the advantages of light-triggered conjugation. While these chemistries fall into just a handful of diverse reactions, they do not necessarily share much in common with regards to mechanism for either their respective photochemical processes or their subsequent coupling. It is rather their broad applicability to diverse chemical requirements and their ease of use that defines the category of “photoclick chemistry.” While this classification originated in 2008 with the rise in popularity of tetrazole-ene couplings, many reactions described currently as “photoclick” have been in use for decades earlier, and in 2013, Tasdelen et al. reviewed this nascent field.1 Here this review expands upon and updates that survey to address recent advances in the field. A brief introduction to click concepts and reactions as well as photochemistry is presented along with a general description of those features common to photoclick reactions. The chemistry of those reactions most frequently described as “photoclick” is then presented followed by a description of the various fields and applications in which those reactions have been used most effectively. Particular attention is given to the specific features of each reaction and how those characteristics are exploited for the respective applications of each chemistry. The review concludes with a discussion of obstacles to broader application of photoclick chemistry and an outlook for the future of this category of reactions.
1.1. The Click Chemistry Paradigm
The concept of “Click Chemistry” is at once potentially transformative and tantalizingly simple.2 At its most fundamental this philosophy could be summarized as: chemists should use reactions that work well. While this may seem obvious, it is instructive for the community of researchers to identify and give preference to those reactions that are typically more “fool proof” than average so that those who seek to replicate and build on others’ past achievements will start their endeavors with the greatest potential for success.
When Sharpless and coworkers elucidated the philosophy of “Click Chemistry,” they established criteria requisite for inclusion in this elite class of chemical conjugations. Such reactions, they proposed, should be “modular, wide in scope, give very high yields, generate only inoffensive byproducts that can be removed via nonchromatographic methods” and be carried out via processes that include “simple reaction conditions…should be insensitive to oxygen and water…, readily available starting materials and reagents, the use of no solvent or a solvent that is benign or easily removed…, and simple product isolation.”2 These criteria are summarized in Table 1. Notable is the requirement of “modularity” which indicates the potential of the reactions to be used as part of a strategy for generating complex molecular structures by the independent synthesis and subsequent assembly of smaller subunits. This requirement favors reactions between complementary reactive groups; many dimerization reactions may be extremely efficient, but do not otherwise result in modularity. Modularity is also favored by reactions that are orthogonal to other functional groups potentially present in the environment in which the reaction is employed. While all of these click chemistry characteristics are self-evidently beneficial to the ease and success of synthesis of complex, conjugated products, many of them are dependent on the context in which they are employed. What may be an inoffensive, easily removed byproduct in the synthesis of a small molecule, for example, may be detrimental to the application of that chemistry in the presence of living cells or organisms or polymeric species. Conversely, those cells may well tolerate byproducts (thus rendering them “inoffensive”) that would otherwise require chromatographic separation.
Table 1.
Original criteria for click chemistry as defined by Kolb et al.,2 supplementary criteria for designation of click reactions, as proposed by Barner-Kowollik et al.,3 in the context of polymer chemistry and supplementary criteria for designation of photoclick chemistry recommended in this work.
| 2001 | 2011 | 2021 |
|---|---|---|
| Original Criteria for Click Reactions | Additional Criteria for Polymer Click Reactions | Proposed Criteria for Photoclick Reactions |
| Kolb et al. 2 | Barner-Kowollik et al. 3 | This work |
| • Modular • Wide scope • High yielding • Inoffensive byproducts • Stereospecific where applicable • Simple reaction conditions (e.g. O2, H2O insensitive) • Readily available materials • Amenable to solvent-free or easily-removed solvent • Simple product isolation |
• Scalable purification • Fast reaction timescales • Successful reactions under equimolar conditions • Suited to polymer-polymer conjugation |
• Can be performed with easily obtained light • exposure equipment • Avoids use of high energy wavelengths of light • Triggerable by relatively low light intensities • Fast reaction timescales • High specificity |
As an illustration of the context-dependence of the utility of click reactions, one may consider the challenge of conjugating or functionalizing macromolecules.3, 4 In some cases, “click” criteria are more easily met in the application of reactions to the coupling of such compounds. A coupling between two small molecules may result in a byproduct that is difficult to separate from the desired small molecule product; the same chemistry applied to the conjugation of two macromolecules may result in the same undesired byproduct which, in this latter case, is easily removed via a quick precipitation of the desired high MW species in a poor solvent (e.g. methanol for highly hydrophobic polymers or diethyl ether for hydrophilic polymers). More frequently, however, the reactions often referred to as “click” in the context of small molecule synthesis, are less effective in macromolecular modification. The high molecular weights of these compounds and the necessity of significant amounts of solvent to dissolve them often preclude the performing of any conjugations in anything but dilute solutions.4 As a consequence, the polymer research community has designated additional criteria for the designation of “click reactions” for the purposes of polymer modification.3 These are summarized in Table 1 and include amenability to scalable purification, fast reaction timescales, successful reactions under equimolar conditions, and suitability for generation of polymer-polymer conjugates.
1.2. Photochemistry Introduction
The reactions designated in the literature as “photoclick” proceed via various mechanisms; therefore, it is advantageous to review the fundamentals of photochemistry to establish a solid grounding for subsequent discussion of these reactions.
The electromagnetic spectrum encompasses radiative energy with wavelengths from as small as one picometer to 100,000 kilometers - a range of 17 orders of magnitude. This energy is distributed in quanta referred to as “photons,” yet by convention, the term “photochemistry” does not generally include those reactions mediated by highly energetic ionizing radiation (below about 150 nm). Very long wavelengths provide inadequate energy to initiate many chemical reactions at all, wavelengths exceeding that of the near infrared region of the spectrum are generally considered less consequential to the study of photochemistry.
Whereas catalysts decrease the activation energy barrier for a reaction to occur, (Figure 1) photochemical reactions are achieved by photoactivation of species “R,” providing the reactant alterantive reaction pathways potentially exhibiting lower energy barriers. For more detailed discussion of reaction energy coordinate diagrams of photochemical processes, the reader is directed to this 2016 review by Kärkäs et al.5 and this book on the principles of photochemistry by Turro, Ramamurthy, and Scaiano.6 This lends to some significant advantageous features of photochemical reactions. Many reactions that would otherwise be extremely slow, with the exposure to an appropriate wavelength of light, occur at relatively low temperatures. This is of particular relevance in the management of synthetic polymer waste; solar radiation of polymers such as polybutadiene and polystyrene accelerates their degradation.7 Another pertinent example especially relevant to the discussion of photoclick reactions, is the decomposition of tetrazoles to nitrile imides, which can be accomplished thermally, but may progress much more quickly and with much lower energy input with exposure to appropriate wavelengths of light.8
Figure 1.
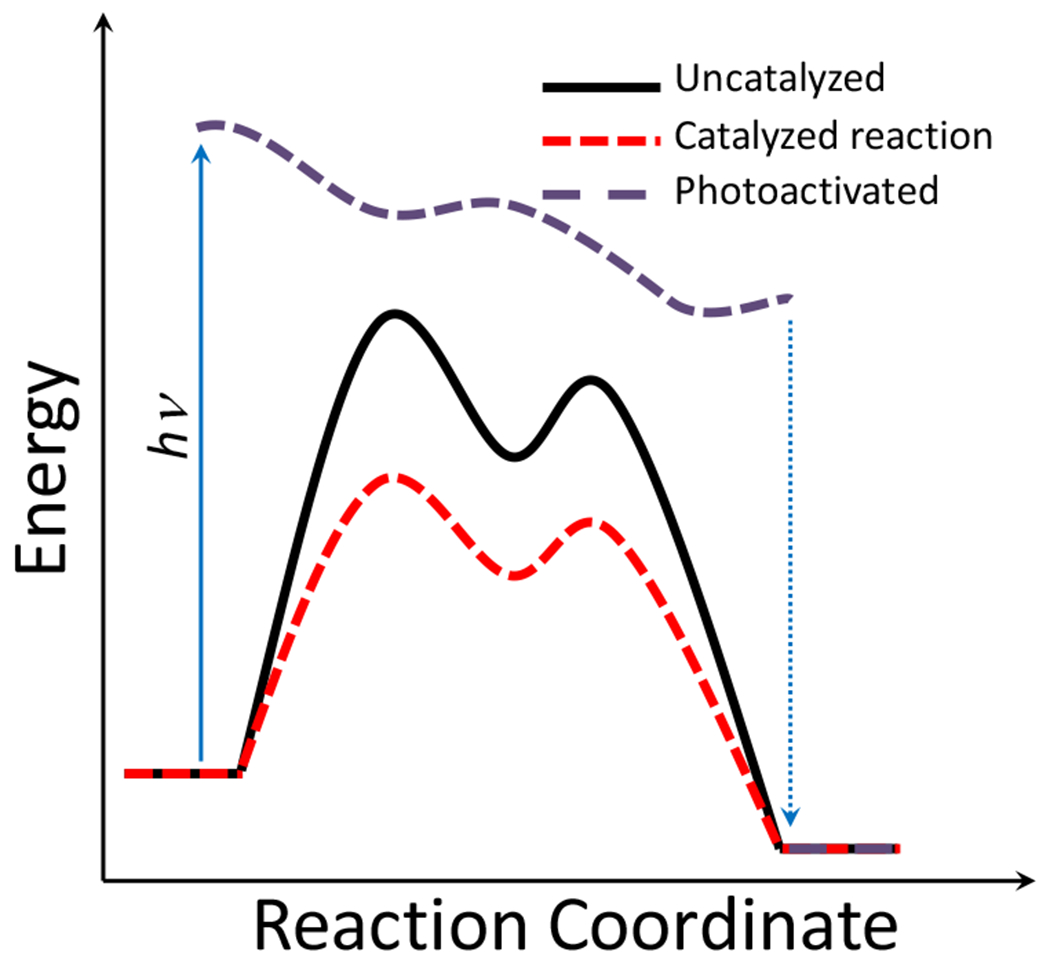
Reaction coordinate plotted against energy for uncatalyzed, catalyzed and photo-mediated chemical reactions. Catalysts lower activation energy barriers, facilitating increased reaction rates. In photochemical reactions, absorbed photons put the absorbing species in a higher energy state from which alternative chemical reaction pathways may effectively proceed.
Thus, the application of photochemistry can be exceptionally energy efficient, but the advantages extend well beyond this. The fact that chemical reactions can be performed at lower temperatures and with the selection of particular, appropriate spectra of light exposure provides a specific selectivity or orthogonality to the performing of such reactions; photoreactions can be performed with exposure at one wavelength of light in the presence of other chemical moieties that might be sensitive to the input of thermal energy or even to other wavelengths of light.
The mediation of chemical reactions via light also provides for temporal control. It should be noted that while a specific photochemical reaction proceeds only upon exposure and is arrested by the removal of the stimulus, downstream chemical reactions may persist, potentially mitigating the absolute temporal control over the reaction. For example, light may release a photolatent catalyst that proceeds to facilitate the reaction. When the light is turned off, the catalyst remains potent. In such a case, temporal control only over commencement of the desired reaction is realized, and cessation of the reaction is determined by other aspects of the reaction conditions.
A similar relationship with spatial control is also realized by the application of photochemistry. As with temporal control, spatial occlusion of light exposure provides dimensional control over where the reaction takes place. This restriction is most easily done in two dimensions, at interphases or with three dimensional projections of two dimensional images. Sophisticated manipulation of light by focusing, multiphoton excitement, interference, etc., however, has allowed true three dimensional control over exposure in general and photoclick chemistry in particular. As with temporal control, however, spatial control may be limited by the persistence of products from the photochemical reaction; photoactivated catalysts may diffuse beyond the spatial limits of exposure, decreasing the resolution of the physical boundaries of the reaction space.
In order for a photochemical reaction to take place, there must be a species that absorbs photons of the wavelength of light to which it is exposed. This guideline is often referred to as the first law of photochemistry or the Grothuss-Draper law.9 The second law of photochemistry or the Stark-Einstein law states that one absorbed photon can excite only one molecule.10 Typically, one absorbed photon excites exactly one molecule, though multiphoton activation, wherein the quick succession of sequential absorption of lower energy photons activates a single absorbing species, is an exception to this.
For single photon activation, however, it is the light dose, rather than intensity, that dictates how many molecules will become photoactivated. For the initial stage of the primary photochemical processes, longer exposure times at lower intensity are equivalent to shorter times at a higher intensity. This outcome is limited though because reactions subsequent to the primary photochemical event may have nonlinear relationships with the concentration of light activated species. As such, the end result often does not result in a proportional relationship with light dose. The relationship describing the amount of light of a particular wavelength (A) absorbed by a chromophore is known as the Beer Lambert law (Equation 1), wherein refers to the concentration of the absorbing species, refers to the pathlength of the light and ε refers to the wavelength (λ) dependent extinction coefficient, a standard measurement of how much light is absorbed by a given concentration of absorbing species at a standard pathlength. The extinction coefficient typically has units of L/(mole cm).
| Equation 1. The Beer Lambert law |
While the Beer Lambert law indicates a linear relationship between absorption and concentration, it should be noted that this linearity does not always extend indefinitely. A variety of factors cause nonlinear deviation at higher concentrations.11
The intramolecular or atomic photophysical processes that occur following absorption of light are often portrayed in what is known as a Jablonski diagram. A general Jablonski diagram portrays the possible energy processes a light absorbing molecule may undergo (Figure 2). Upon absorption of a photon, a molecule or atom may move from its ground state, So, to an excited singlet state, Sn. From higher singlet states (n>1) the excited species relaxes to its lowest excited singlet state, S1, via a radiationless decay process designated “internal conversion” or “IC.” The exitation energy lost in this process is converted to thermal energy via vibrational relaxation. From the S1 state, the remaining energy from the absorbed photon may be dissipated via a number of means as the molecule returns to its ground state. It may be released as a photon of lower energy in the process of fluorescence, or it may be lost via internal conversion to thermal energy. Alternatively, it may undergo another radiationless decay process, “intersystem crossing” or “ISC” that moves the excited species to a triplet state (Tn) from which it undergoes internal conversion to reach its lowest excited triplet state (T1). This ISC process results in the excited state molecule possessing two unpaired electrons with the same spin. As returning to ground state So from T1 requires a change in electron spin, this state generally takes more time and because of the longer triplet state lifetime, many photoreactions occur from the excited triplet state. Indicative of this phenomenon is the difference between radiative decay from the excited singlet state (fluorescence; lifetime from <10−10 to 10−7 s)12 which occurs very quickly following irradiation, and the radiative decay from the excited triplet state (phosphorescence; lifetime from 10−5 to >103 s),13 which persists, frequently for minutes, following the cessation of photoexposure.
Figure 2.
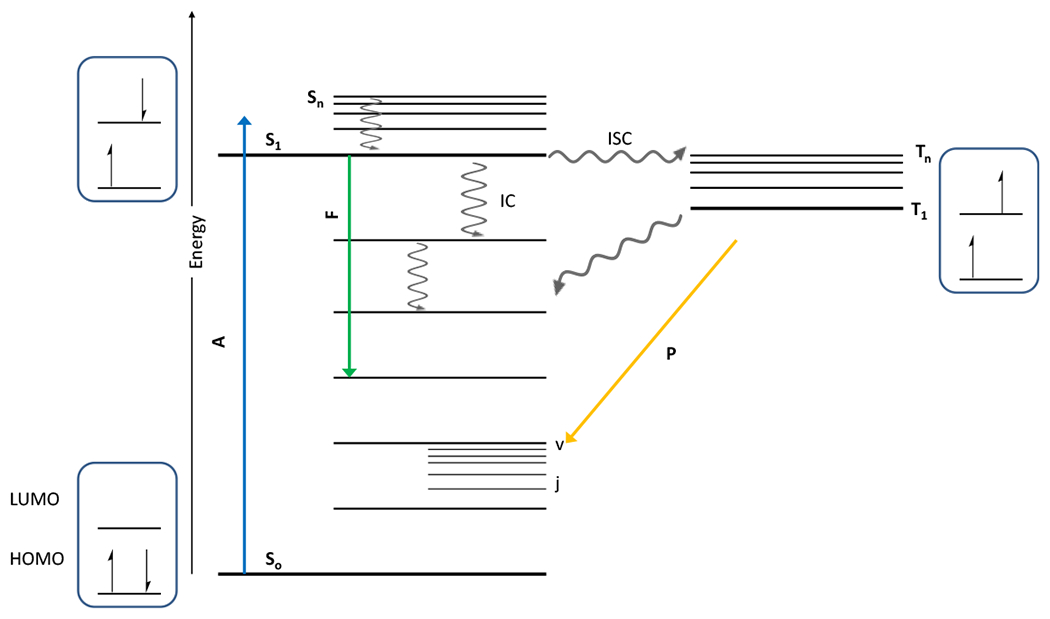
General Jablonski diagram for the possible fates of photonic energy absorbed by a photoactivated molecule. Internal conversion is denoted by IC; intersystem crossing, ISC; fluorescence, F; phosphorescence, P; absorbance, A. Also shown are the electron orbitals and spins. Highest occupied molecular orbital is abbreviated HOMO and lowest unoccupied molecular orbital is abbreviated LUMO.
An excited atom or molecule can undergo a combination of radiative (fluorescence, phosphorescence) and non-radiative (internal conversion and intersystem crossing) processes in its pathway to return to the ground state. In addition, it may undergo bond rearrangement, either intramolecularly or intermolecularly, to generate new products. The fraction of excited molecules that undergoes a particular process is referred to as the quantum yield (ϕ) and is a measure of the efficiency of a given photochemical process relative to the light absorbed. A quantum yield exists for every potential photochemical process; however, some are frequently negligible. Quantum yield can be defined in terms of the respective first order reaction rate constants “k” (Figure 3) and the lifetime of the excited states “τ”, which is the inverse of the sum of all possible reactions from that state (Equation 2 and Equation 3 where subscripts S, and T correspond to the singlet and triplet states, respectively, and subscripts r1, ISC1, F, IC, r2, ISC2, P and Tr correspond to the processes of reaction from the singlet state, intersystem crossing to the triplet state, fluorescence, internal conversion, reaction from the triplet state, intersystem crossing to the ground state, phosphorescence and energy transfer, respectively). The quantum yield for any process is the reaction rate constant of that process multiplied by the lifetime of the excited state from which it occurs whether for fluorescence (Equation 4), a photochemical reaction from the triplet state (Equation 5) or any of the processes shown in Figure 3.
| Equation 2 |
| Equation 3 |
| Equation 4 |
| Equation 5 |
Figure 3.
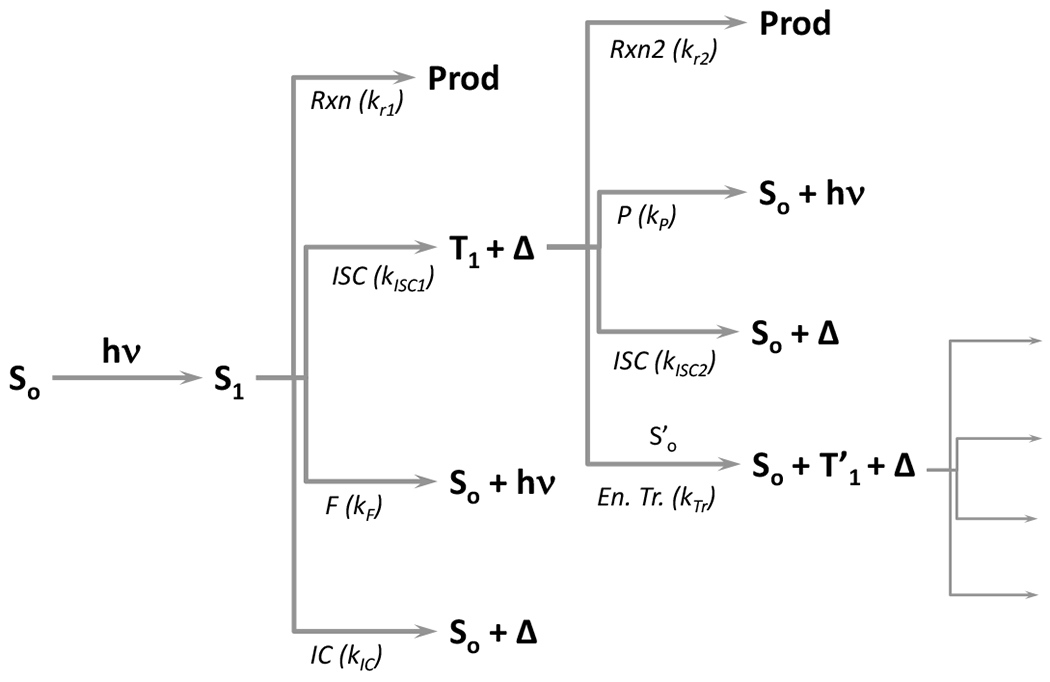
Flow chart showing the processes that an absorbing molecule may take with the accompanying potential products and the reaction constants.
In addition to the photochemical processes mentioned above, an excited atom or molecule can transfer energy to another molecule or atom, resulting in that species being elevated to a higher energy state despite having not directly absorbed any photons (Figure 4). The newly excited atom or molecule can undergo similar radiative and nonradiative decay processes from its own excited state (Figure 3).
Figure 4.
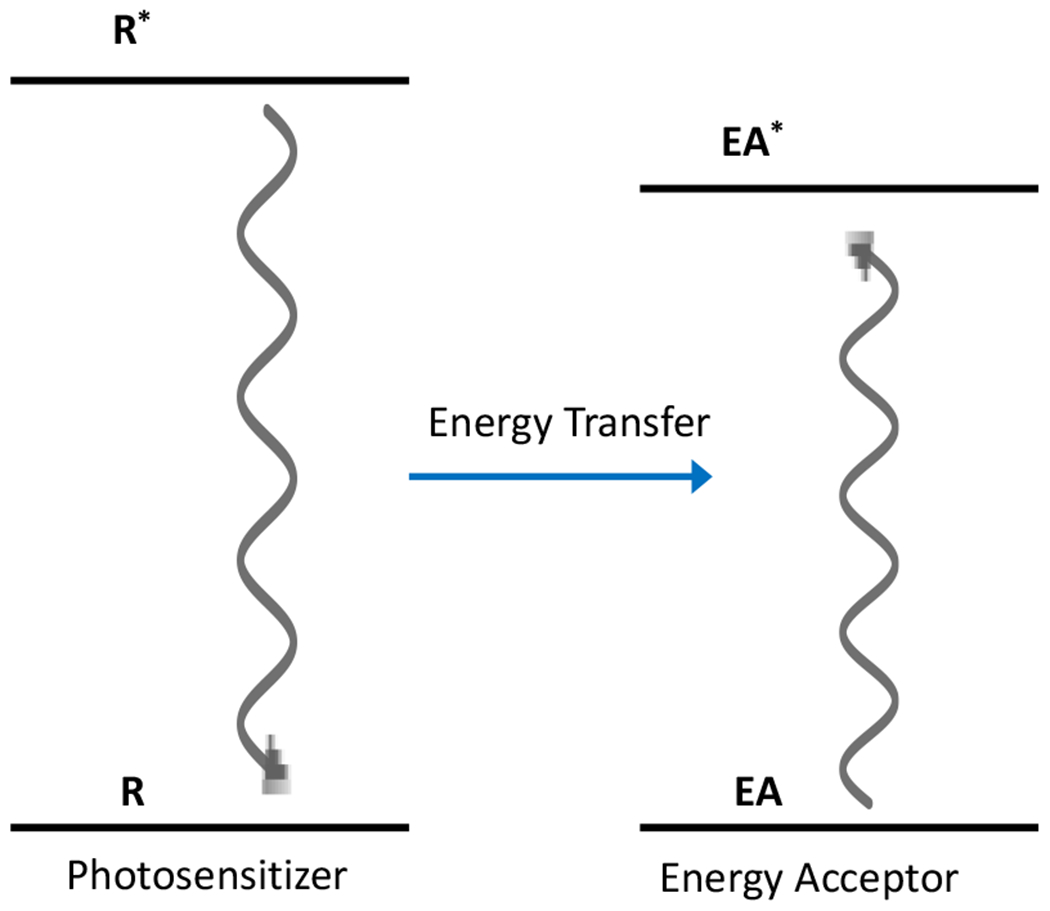
Jablonski diagram for photosensitization wherein an excited molecule transfers energy from its high energy, excited state, to another molecule, placing it in an excited state while the original molecule relaxes to ground state.
Photochemical reactions are broadly categorized into six non-discrete categories (Figure 5). Excited species undergo bond breaking wherein the compound generates one or more chemically distinct products. Examples of such reactions are abundant in both the lab and in many day to day experiences. Photolabile groups on reactive side groups during solid phase peptide synthesis have been used for decades as orthogonal protecting groups (Figure 6). Cleavage-type radical photoinitiators are common both in research and in consumer products (Figure 6). Beer and red wines are frequently stored in green or amber tinted bottles to mitigate the UV-mediated degradation of flavor compounds.
Figure 5.
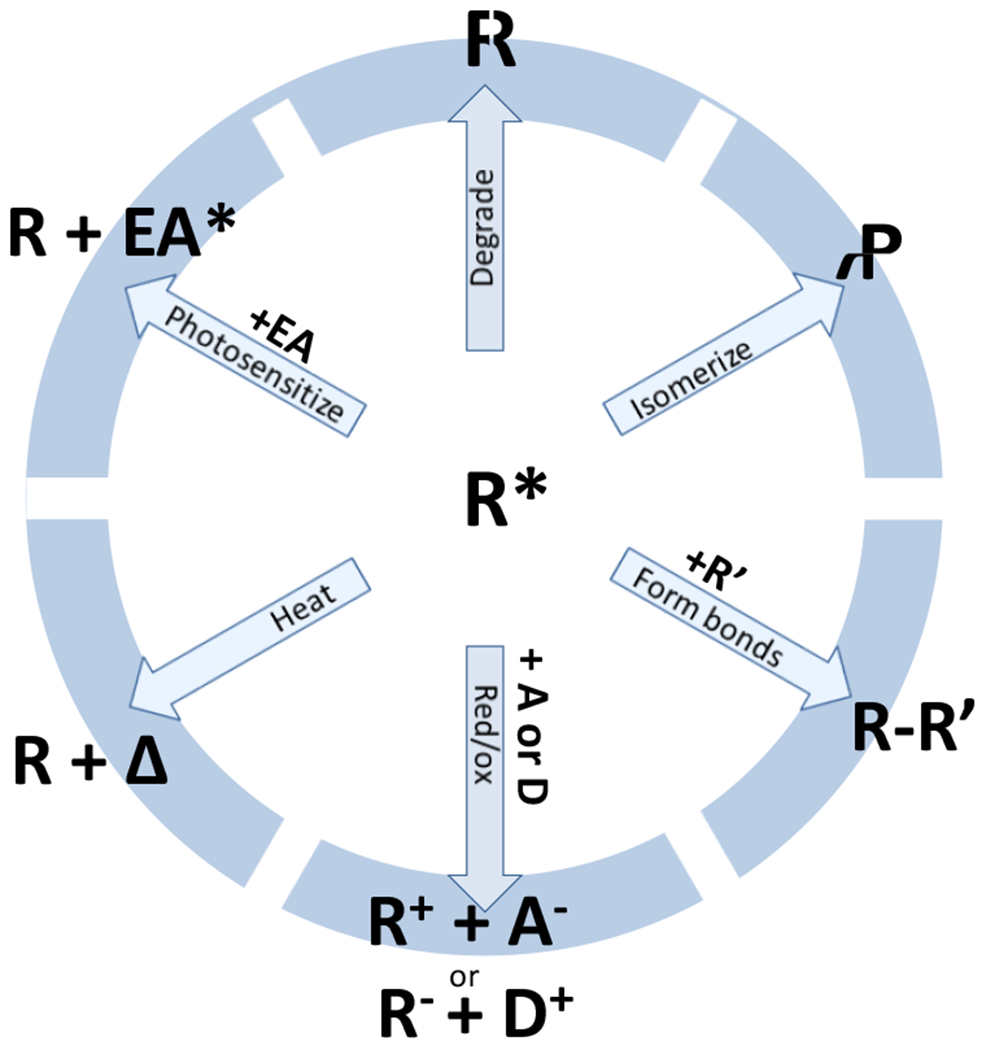
Photoactivated species can participate or initiate chemical reactions via one of these six general mechanisms.
Figure 6.
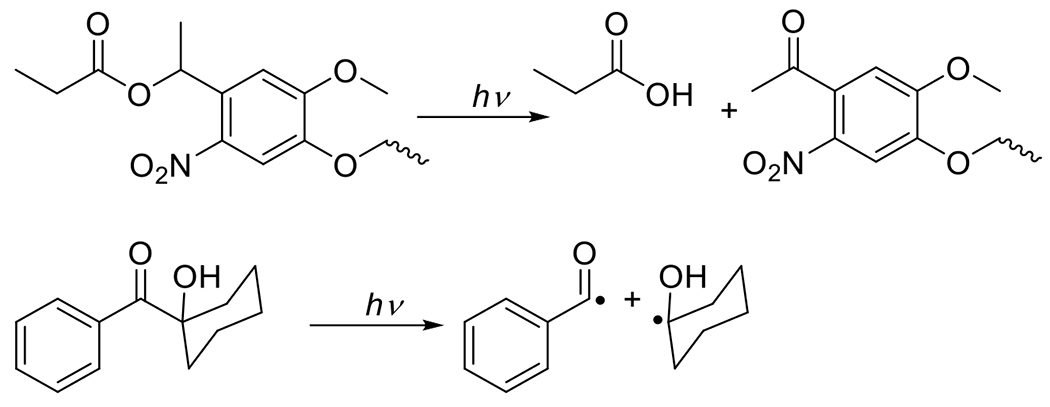
Two examples of photomediated degradation including photodeprotection of a carboxylic acid (top) and photomediated cleavage of 1-hydroxycyclohexyl phenyl ketone.
Bond breaking that results in subsequent intramolecular reorganization of those bonds rather than the more permanent scission thereof creates isomers of the ground state compound in photoisomerization reactions. While azobenzene and its derivatives are among the most widely studied compounds prone to photoisomerization (Figure 7), many people are familiar with photochromic eyeglasses that change absorbance characteristics in sunlight due to photoisomerization. Phototherapy to treat neonatal jaundice employs blue and green lights to photoisomerize bilirubin to a more soluble form for more efficient clearing from the bloodstream.14
Figure 7.

Photoisomerization of azobenzene wherein high energy UV light isomerizes the trans form to the cis form. Exposure to visible light isomerizes the cis form to the trans form.
In the presence of photoexcited species, bonds may break and reform with other molecules to create new bonds in photoinduced reactions. While many photoreactions lead indirectly to bond formation (e.g. frequently cited as an example of photochemistry in introductory chemistry textbooks, the chlorination of methane begins with the photocleavage of Cl2 to Cl radicals), these are examples of bonds forming as a result of the reactions of photo-generated species rather than the reactions of photoexcited species themselves. Among this class of directly photoactivated bond forming cyclization reactions is the [2+2] cycloaddition. First observed in 1908, this reaction consists of the reaction of a photon-absorbing enone with an alkene.15 Frequently the enone provides the alkene in the reaction as well with the resulting product being the dimer. The photomediated [2+2] cycloaddition of pyrimidine nucleobases (thymine and cytosine)16 is one of the mutagenic reactions in the DNA of epidermal cells that has been implicated in the development of melanomas.17
Not all photochemical processes that effect subsequent chemical reactions involve the breaking or rearrangement of covalent bonds within the chromophore itself. Molecules in the S1 or T1 state have unpaired electrons in both higher and lower energy orbitals (Figure 2), they are more prone than their ground state counterparts to participate in electron transfer processes, either as an oxidant or reductant (Figure 9). The donation of electrons or acceptance of electrons results in the formation of a radical ion species without necessarily breaking any chemical bonds. Although subsequent bond breaking is a frequently observed consequence of the photoinduced electron transfer, the photocatalysts is often regenerated by downstream electrochemical processes.18
Figure 9.
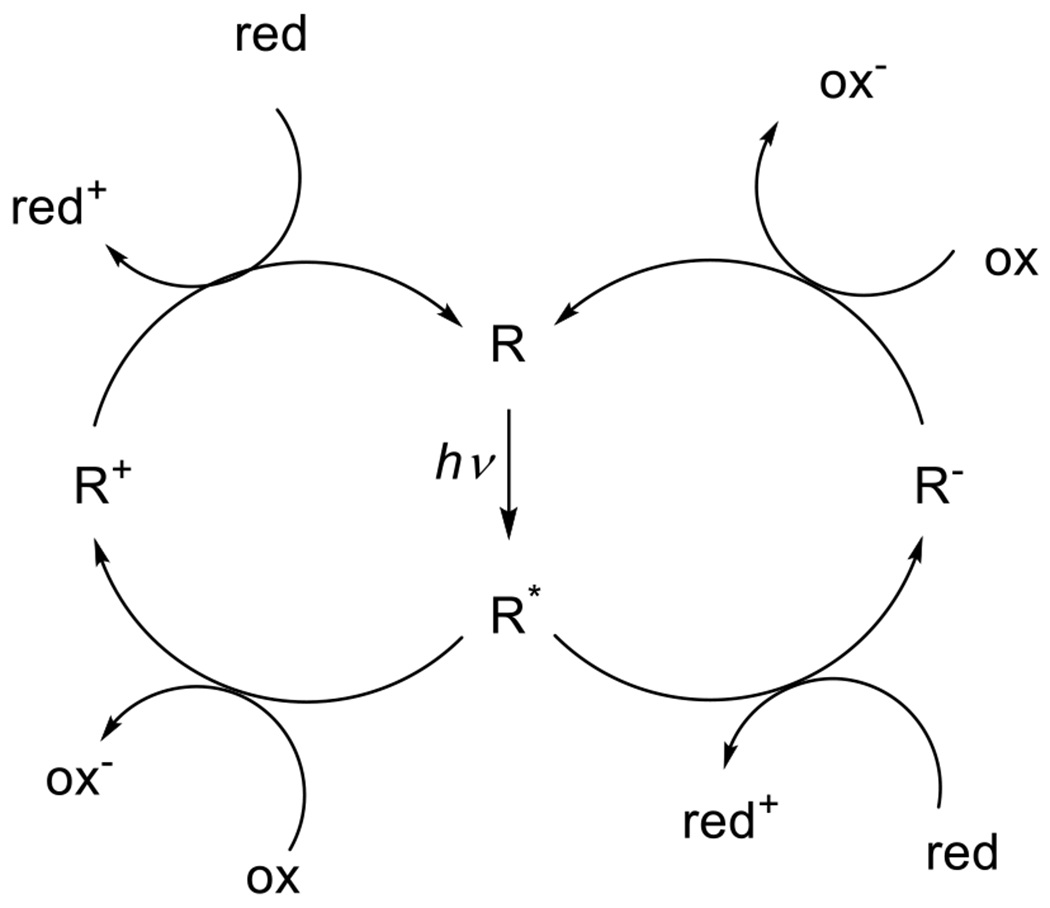
General scheme for photoredox reactions with oxidative quenching shown on the left and reductive quenching shown on the right. In oxidative quenching, the light-activated species loses an electron to an oxidizer, gaining a net positive charge. The photoactive species is regenerated by reduction. In the reductive quenching mechanism, the photoactivated species accepts an electron from a reducer, gaining a net negative charge. The photoactive species is regenerated by oxidation.
The internal conversion of photonic energy to heat (photothermal) may also mediate chemical reactions among other molecules within proximity without any chemical change of the absorbing species. A common application of photothermal effects involves the introduction of gold or silver nanoparticles with high infrared and near infrared absorbances (Figure 10). Upon irradiation with NIR and IR light, the nanoparticles absorb photons and release the acquired energy via internal conversion to heat. While this heats up the average temperature of the surrounding environment, the local responses of temperature sensitive polymers (e.g. poly(N-isopropylacrylamide)) have exhibited temperature dependent responses corresponding to temperatures substantially higher than that of the bulk medium, indicating highly localized thermal effects of the light activated nanoparticles. Similar effects are achieved with IR dyes. In general, photothermal effects provide a means for initiating thermally sensitive reactions via exposure to light.
Figure 10.
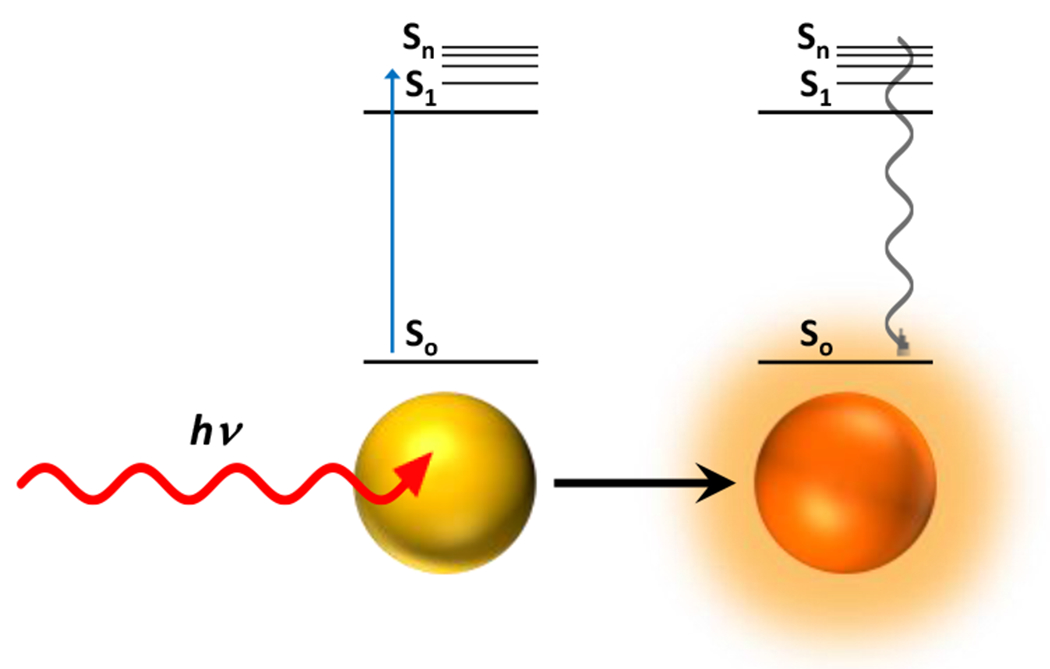
Photothermal relaxation of gold nanoparticles, transitioning absorbed photonic energy into thermal energy via internal conversion.
Finally, as noted above, excited state species may transfer that excitement to other molecules via photosensitization whereupon those newly excited molecules may undergo any of the above mentioned photochemical and photophysical reactions.
1.3. The Concept of Photoclick Chemistry
While conventional click reactions are often mediated by the addition of catalysts or moderate heating, temporal control over the reaction is frequently limited; once the reactants are all in the reaction vessel, the reaction begins. Photomediated reactions allow at least one element of temporal control over the reaction and frequently, depending on the particular reaction employed, a very high degree of both spatial and temporal control over the reaction.
This combination of advantages leads to photomediated click reactions being of particular benefit to a wide variety of applications, including 3D printing, patterned surface chemistry modification, in situ hydrogel synthesis and many more described in the applications section of this review. It is not the spatial and temporal control only, however, that dictates the applications of photoclick reactions. High rates relative to non-photomediated reactions may also be reason to employ photoclick chemistry in particular applications. There are, however, contexts in which photoclick reactions may not be appropriately suited and a better understanding of the features of those reactions will foster informed decisions as to when they can be effectively utilized.
1.3.1. One Photon, One Event vs One Photon Many Events Reactions
In many photomediated reactions, photolatent catalysts are released upon exposure to an appropriate wavelength of light, initiating the reaction. As one photon is capable of activating one catalyst molecule which is in turn capable of facilitating many reactions, this amplification is often referred to as a “one photon, many events” chemistry. Without a mechanism to consume or poison the catalyst, the reaction will continue to completion even after the light is turned off. In some recent examples, photolatent acids have been used to deactivate base catalysts.19 Similarly, acid-catalyzed reactions could be deactivated upon light exposure of endogenous photolatent bases, thus providing alternative or additional photoelements of temporal control.
Other photomediated reactions occur upon intramolecular activation of the reacting species itself. This might be via deprotection of a reactive functional group, allowing it to participate in an otherwise precluded reaction, or via direct absorbance and excitation of that species (e.g. via 2, 2-cycloaddition). In these cases, one photon leads directly to at most one reaction (a “one photon, one event” chemistry). As might be expected, absent the photoamplification of a one photon-many events type reaction, a one-photon, one event reaction necessarily requires a higher concentration of light-absorbing, photoreactive species and therefore frequently requires higher intensities and significantly longer exposure times. Such reactions are also often subject to significant light attenuation through the thickness of the exposed sample.20 As a result, “one photon, one event” chemistries are frequently limited to applications with thin geometries and frequently at surfaces. There are advantages, however, to these one-photon, one event chemistries. The desired reaction occurs only upon photon absorption so that the removal of the light stimulus causes the reaction to cease, providing a high degree of temporal and spatial control (subject to the practical depth of cure limitations brought about by the relatively high concentration of chromophores).
Radically mediated photoreactions, however, occupy somewhat of a middle ground in this tradeoff between photoamplification of reaction and spatial and temporal control. A single initiating radical in a chain reaction may propagate hundreds or thousands of times, but its lifetime is functionally short. While anionic or cationic catalysts and reaction intermediates will not rapidly recombine with other molecules of similar charge, radicals are prone to termination by recombination and disproportionation. Whereas an anionic, photogenerated catalyst, in the absence of a poison, will continue to facilitate reactions indefinitely, actively propagating radicals will terminate and the reaction will cease rapidly unless radicals are replenished. In this way, radical reactions exhibit the photoamplification effect of a one photon, many events chemistry but with limited dark cure and maintenance of a greater degree of spatiotemporal control.
Some related reviews on the subject of photomediated click chemistry in particular fields of research have preferred discussion primarily of those reactions that are self-sufficiently photoreactive.21–23 Conversely others have adopted a broader view of the chemistry.1, 24 This review favors the inclusion of reactions that take advantage of the photoamplification of self-perpetuating, photomediated reactions for their obvious benefits in the contexts in which they are used. Click reactions as a whole are put in the click category because of their performance and utility. Similarly, with photoclick reactions, their defining characteristic, provided that that they meet the criteria for click reactions generally, is that they are mediated by the application of light.
1.3.2. Light Attenuation
Despite the significant advantages of photochemical reactions, there are significant intrinsic challenges to effectively and practically conducting such chemistry. Some of these challenges to the broad application of photochemistry include the inescapable problem that the absorbance of photons requisite for photoactivation prevent photoactive molecules farther away from the incident light from being activated themselves, creating a light gradient through the exposed sample. While this is especially consequential, as discussed above, in one-photon, one event reactions, it is an issue with one photon many events reactions as well. Light attenuation may cause inadequate light penetration through the depth of a sample, preventing anything but superficial conversion (Figure 11). Furthermore, as chromophores are reacted, their absorption profiles often change in disadvantageous ways; photodegradation products may absorb more light than the species from which they originated, blocking even more light from reaching the reactive molecules below. This outcome makes scalability of reactions a challenge. Options to mitigate such light attenuation include stirring the reaction mixture to distribute either photoactivated catalyst or to increase the median residence time near the incident light of all molecules in the vessel. Flow photochemistry, wherein the reactants are flowed through a thin, transparent polymer tube while exposed to light, also increase the time that molecules spend in proximity to the photoexposed surfaces. Frequently, however, mechanical agitation is not an option. The choice, therefore, of excitation light exposure is not trivial; sometimes choosing a light with an emission that matches an absorption maximum is counterproductive. A light source with a wavelength that overlaps with the absorption spectrum of the photoactive molecule less than perfectly may serve to better provide effective reaction through the depth of the sample. While the attenuation of light may be a difficult problem to overcome, it should be noted that because of their high efficiency, click reactions are frequently applied to situations where the complementary reactive groups are in very dilute conditions diminishing the light attenuated by absorbing species. Reaction conditions, therefore, that would disfavor reactions mediated via other means, may promote effective photomediated reactions. For a more in-depth description of the relationship between irradiation/absorption overlap and depth of penetration, the reader is referred to recent work by Barner-Kowolik and coworkers.25, 26
Figure 11.

Hypothetical light intensity vs sample depth diagrams for a reaction with high absorbance (left) and low absorbance (right) and intermediate absorbance (middle). Linearity as described by the Beer Lambert is often not observed at high absorptions.11
1.3.3. Other General Challenges in Photoclick Chemistry and Proposed Criteria for Designation
Atom economy is defined as the ratio of the molecular weight of the product of a chemical reaction to the sum of the molecular weights of the reactants. Relatively high atom economy, while not explicitly described in the seminal paper describing click chemistry, is often intuited as a requisite or at least preferred feature, given the established criteria related to byproducts.27, 28 The applicability of this metric varies with context. In many applications of click chemistry (e.g. polymeric and biomacromolecular conjugations) reactants have substantial molecular weights. So long as the molecular weight of the conjugated macromolecules is many times that of any byproducts, the atom economy will necessarily be high, even with a sizeable number of unutilized atoms. While atom economy, as defined, may not reflect actual atom utilization efficiency in such contexts, the general feature of generating smaller (or zero) byproducts is a useful one. Yet another important, and related, consideration is the size of the reactive moieties and their contribution to the molecular weight of the product. This is particularly true in the case of photochemical reactions where, in order to modulate light absorbance characteristics, red-shifting substituents are appended to chromophores, considerably increasing the molecular weight of the reactive group. Reactive handles that make up significant fractions of the molecular weight of the reactants can change the physical and biochemical properties of those compounds themselves-physical and biochemical properties, the maintenance of which may be integral to their function in the desired context. Solubility of polymers and proteins may change; new interactions with other components in complex systems may be introduced. Additionally, it should be noted, that typically, if not always, larger and more complex reactive moieties represent a more significant synthetic challenge. For these reasons, the size of reactive functional groups is a valid consideration for designation as photoclick chemistry.
Another challenge to the effective application of photochemical techniques is the availability of instrumentation required for carrying them out. Hot plates and thermometers are ubiquitous in chemistry laboratories. High powered light sources of discrete or variable wavelengths and radiometers are less so. Though as LEDs of various wavelengths and intensities have become more available in recent years, the difficulty in obtaining adequate photochemistry instrumentation has been substantially mitigated.
High energy (low wavelength) photons can have deleterious health effects on researchers exposed to them including acute injury (e.g. sunburns, temporary blindness) and disease due to cumulative exposure (e.g. development of macular degeneration, skin cancers). As a result of these hazards, additional PPE is recommended for those employing light-mediated chemistry, including tinted safety glasses, face shields, photoexposure cabinets, etc., but even with such supplementary protection, chemists who perform photochemical reactions should be cognizant of the specific hazards of the particular wavelengths and intensities of light that they employ. It should also be noted that it is not only the chemist who is susceptible to deleterious effects of light. High doses of high energy light can damage other chemical compounds and moieties present in the reaction mixture as well. For these reasons reactions that require low wavelengths of UV light, high intensities, or long exposure times, may not adequately meet the spirit of the original click classification criteria.
The incorporation of photochemistry into the philosophical framework of “click” chemistry introduces new considerations, it is beneficial to consider how photoclick may correlate with the original criteria of “click chemistry.” While the application of light generally has proven an invaluable tool in synthetic organic chemistry, polymer chemistry and biochemistry, stringent application of standards must be applied to the characterization of “photoclick” chemistry to prevent the phrase from becoming just another synonym for “useful reaction.” Just as click reactions are supposed to avoid hazardous reactants, which increase the likelihood of undesired side reactions and present a hazard to the chemist performing the reaction, photoclick chemistry should similarly avoid hazardous reaction conditions like high doses of low wavelength light. Additionally, click reactions originally were intended to employ widely available or easily synthesized reagents and catalysts. If a corollary between the initiating chemical and physical stimuli can be made, then broad accessibility of light instrumentation and mild exposure conditions to carry out the reaction would be necessary for a reaction to qualify as “photoclick.” For these reasons, it is proposed that additional criteria be met for a reaction to qualify as a “photoclick” reaction (Table 1).
In the present body of literature on the subject, photoclick reactions generally fall into four general categories: 1,3-dipolarcycloadditions, Diels Alder and inverse electron demand Diels Alder additions, radical alternating propagation and chain transfer reactions, and nucleophilic addition reactions. Figure 12 demonstrates the increasing popularity of photoclick reactions generally and of the specific subdivisions as well. This plot was generated from results by the SciFinder (American Chemical Society) search feature with queries of ‘“[chemistry]” + “photo” + “click” to generate an index of publications, which were then individually examined to verify applicability and to avoid duplicates in counting. As there may be a good number of publications with these various chemistries that may not explicitly employ the term “photoclick,” the numbers represented here are necessarily an undercount. However, it is believed that the trends are accurate. With this increase in use and research into these reactions, a thorough examination of their relative strengths and limitations should serve to direct their application to those contexts to which each reaction is most suited.
Figure 12.
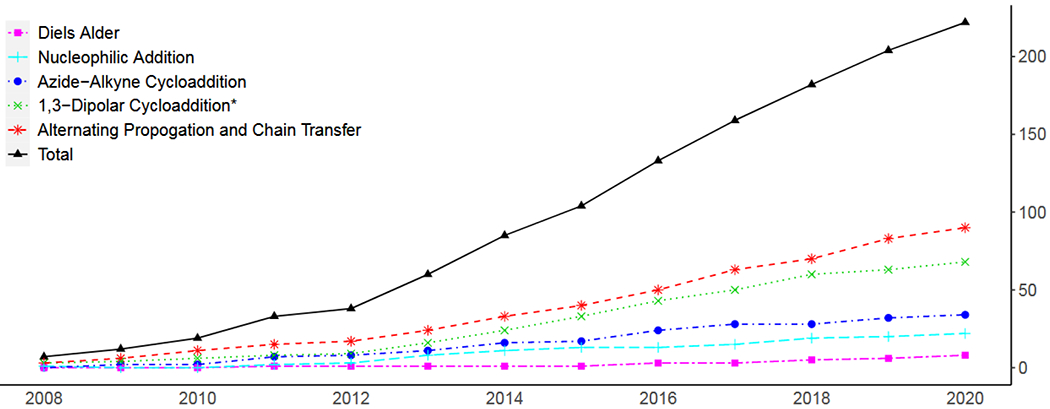
Number of journal articles published by year on photoclick chemistry, divided by class of reaction. *indicates 1,3-dipolarcylcoadditions other than azide alkyne cycloadditions.
2. Photoclick Reactions
The characterization of “photoclick chemistry” was first applied to the tetrazole-ene reaction in 2008.29 Since then, the designation has been applied to a number of other photomediated reactions. Table 2 shows the general reaction schemes for the most common photoclick reactions along with relevant features of those reactions, such as the role the chromophore plays in the reaction, typical wavelengths and reaction durations. The table also indicates whether the nature of each reaction is one-photon, one event or photoamplified (one photon, many events), which, to a great extent, determines appropriate applications for these reactions. The remainder of this section follows the organization of this table by discussing each reaction in detail and how it meets or fails to meet the photoclick classification, depending on the reaction and the conditions under which it is implemented.
Table 2.
A list of general reaction schemes of photomediated reactions designated as “photoclick” in the literature, grouped by reaction mechanism. The chromophore in each reaction is indicated in blue. Included in table are relevant features: typical wavelengths at which the corresponding reaction is conducted, typical duration of reaction and whether the reaction is photoamplified (PA), photocatalyzed but not amplified (PC), or whether the reaction is one photon-one event.
| Reaction | Name | Typical λ | Typical Rxn Time | Amplification |
|---|---|---|---|---|
| 1,3-dipolarcycloadditions | ||||
|
Photo-CuAAC30, 31 | 330-500 | 30 s – 1 h | PA |
|
|
PcAAC32 | Visible light | >1 h | PC |
|
|
PhotoSPAAC33, 34 | 350-420 | 1-10 m | 1P1E |
|
Tetrazole-ene8, 29, 35 | 300-405 NIR with TPA | 1-10 m | 1P1E |
|
|
Sydnone-ene36 | 300-405 | 1-10 m | 1P1E |
|
|
Azirine-ene37 | 302-420 | 1-10 m | 1P1E |
| Diels Alder and inverse electron demand Diels Alder | ||||
|
|
Photoenol DA38 | 320-350 | 100 m | 1P1E |
|
|
hetero DA39 | 320-350 | 100 m | 1P1E |
|
|
Photoenol alkyne DA40 | 320-350 | 15-60 m | 1P1E |
|
|
Naphtoquin-one methide41 IEDDA | 320-350 | 15 m | 1P1E |
|
|
Photo Tetrazine IEDDA42, 43 | 600-900 | 60 s | PC |
|
|
Tetrazine Photocyclo-alkyne IEDDA44 | 350-420 | 5-10 m | 1P1E |
| Radical alternating propagation chain transfer reactions | ||||
|
|
Thiol-ene45, 46 | 320-700 | 30 s – 30 m | PA |
|
|
Thiol-yne47, 48 | 320-500 | 5 m – 30 m | PA |
| Nucleophilic additions | ||||
|
|
Photolatent base-catalyzed thiol-epoxide49 | 320-400 | 5 m -30 m | PA |
|
|
Photolatent base-catalyzed thiol-isocyanate50, 51 | 365 | 1 m -30 m | PA |
|
|
Photolatent base-catalyzed thiol-Michael52, 53 | 320-500 | 5 m – 30 m | PA |
|
|
Photouncaged thiol-Michael54, 55 | 320-740 | 1 m – 60 m | 1P1E |
|
Photomediated hydrazone and Oxime ligation56, 57 | 355-370 | 2 m – 30 m | 1P1E |
2.1. Dipolar Cycloadditions
Among click reactions generally and photoclick reactions more specifically, 1,3-dipolar cycloadditions constitute a significant number of reactions so categorized. The generalized mechanism of a 1,3-dipolar cycloaddition involves electron rearrangement of a 1,3-dipole and an unsaturated dipolarophile resulting in the generation of a five membered ring (Figure 13a).
Figure 13.
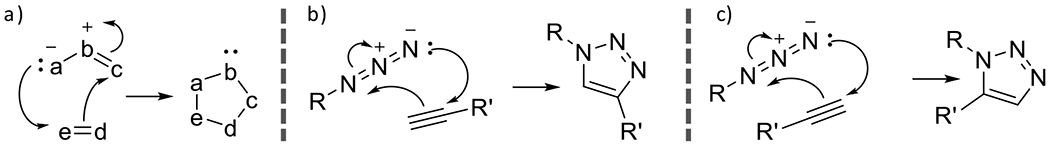
a) general mechanism for 1,3-dipolar cycloaddition wherein rearrangement of electrons between a 1,3-dipole and dipolarophile results in generation of five membered ring structure. b) 1,3-dipolar cycloaddition between an azide (1,3-dipole) and alkyne (dipolarophile) (Huisgen azid alkyne cycloaddition), resulting in 1,4 regioisomer and c) the cycloaddition resulting in the 1,5-regioisomer.
2.1.1. Azide alkyne cycloaddition
The classic example of such a reaction is the 1,3-dipolar cycloaddition between an azide dipole and an alkyne dipolarophile. This reaction is also known as the Huisgen azide-alkyne cycloaddition in recognition of the work performed by Rolf Huisgen on these cycloaddition reactions.58 In the absence of a catalyst, the azide-alkyne cycloaddition is accomplished by overcoming the activation barrier thermally. The elevated temperatures required for reaction and the non-regiospecificity of the product (thermal cycloadditions result in approximately equivalent yields of 1,4-substituted and 1,5-substituted products, Figure 13b and Figure 13c), however, disqualify the solely thermally mediated reaction from a click designation.
The copper catalyzed variant of the 1,3-dipolar cycloaddition between an azide and an alkyne (CuAAC) is currently probably the most recognized click reaction. Terminal alkynes and azides are frequently readily available or easily prepared (though caution is recommended in the synthesis of organic azides as low C:N ratio compounds can pose an explosion hazard). Whereas the thermal Huisgen azide alkyne cycloaddition is relatively sluggish and results in a mixture of regioisomers, catalysis by copper(I) results exclusively in the 1,4-product with a rate of reaction 107 or 108 times as high as that of the uncatalyzed reaction.59 Since its introduction in 2001,59 the CuAAC reaction has been a flagship for click reactions, having been successfully employed in a wide variety of conditions and frequently resulting in high conversions. Moreover, as azides and alkynes are generally not particularly reactive towards many of the most common functional groups employed in the biochemical and materials laboratories, the CuAAC reaction is tolerant of many reaction environments that might preclude the performance of other coupling chemistries.
While copper(I) salts can be used directly as catalysts, the in situ reduction of copper(II) allows for facile aqueous reactions via the inclusion of water soluble copper salts (most frequently CuSO4) and a reducing agent (e.g. sodium ascorbate). Frequently, the reducing agent is included in large excesses to disfavor the otherwise rapid oxidation of the Cu(I) back to Cu(II). For a similar reason, chelating ligands have been used to maintain effective Cu(I) concentrations. These strategies greatly mitigate the reactions’ otherwise sensitivity to oxygen. While a common class of Cu(I) ligands for the CuAAC reaction include multifunctional tertiary amines, triazole rings have proven among the more effective ligands. Indeed, the products of CuAAC reactions may serve as ligands during the progression of the reaction. Despite its seeming ubiquity, the precise mechanism for the copper catalyzed cycloaddition remains somewhat of a mystery. The mechanism proposed by Rostovtsev et al. in 2002 suggests a single copper atom facilitates the formation of cyclic intermediates and ultimately the triazole (Figure 14a).60 Subsequent experiments by Worrell, Malik and Fokin,61 however, demonstrated that copper coordination to the alkyne was insufficient to explain the increased cycloaddition rates by measuring reaction kinetics of a preformed s-bound copper(I)-acetylide with an organic azide in the presence or absence of an exogenously added copper(I) catalyst. The reaction occurred to an appreciable rate only in the presence of catalysts suggested a two-copper atom mechanism for the cycloaddition (Figure 14b).
Figure 14.

a) Original proposed mechanism for Cu(I) catalysis of azide alkyne cycloaddition. Adapted, with permission, from reference 60. Copyright 2002 John Wiley and Sons. b) Updated mechanism for Cu(I) catalyzed azide alkyne cycloaddition indicating concerted effect of two copper atoms reflecting low experimental yields of copper(I)-acetylide in reactions without exogenous Cu(I). Adapted from reference 61, with permission, from AAAS, Copyright 2013.
Azide-alkyne cycloadditions may also be catalyzed with ruthenium(II) complexes, resulting in the 1,5-substituted triazoles rather than the 1,4-substituted triazoles of the copper catalyzed cycloadditions or the mixture resulting from thermally mediated reactions.62 Furthermore, whereas the CuAAC reactions are exclusive to terminal alkynes, the RuAAC reaction are performed with internal alkynes. However, the RuAAC reaction may require elevated temperatures or longer reaction times and the ruthenium catalysts necessary for these reactions are more expensive or difficult to acquire than are the simple copper salts and vitamin C so frequently used in CuAAC reactions.62 For these reasons, and perhaps suffering from generally unfavorable comparisons to its copper catalyzed counterpart, RuAAC reactions are seldom referred to as “click reactions”.
One additional advantage of employing a copper(I) catalyst is the copper(II) species serving as a latent catalyst, activated by the application of a reducing agent. As described prior, this is typically achieved by physically mixing in the exogenous electron donor, though latent catalyst may also be activated electrochemically.63
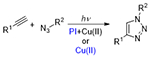
2.1.1.1. Photo CuAAC and PcAAC
By photochemical means, however, the reducing agent can be generated in situ by activating endogenous photoactive species. In 2006, Ritter and König reported the photoreduction of Cu(II) with the visible light exposure of riboflavine tetraacetate in the presence of an electron donor trimethylamine.64 To an equimolar solution of alkyne and organic azide (200 mM in acetonitrile) was added 8 mol% CuCl2, 10 mol% TEA and 4 mol% of the photoactive riboflavine and exposed to visible light irradiation of unspecified intensity. The resulting Cu(I) was capable of catalyzing the CuAAC reaction between an alkyne and an organic azide to above 80% conversion in twenty minutes. Aqueous reactions were similarly performed with CuSO4 in place of the CuCl2 and benzyl alcohol replacing the triethylamine, achieving lower conversions with longer exposure, reportedly due to the water-promoted disproportionation of Cu(I) to Cu(0) and Cu(II). This was supported by the observation that in aqueous systems, the reaction was arrested upon termination of light exposure, indicating consumption of the catalytic species.
Tasdelen and Yagci were able to generate Cu(I) catalysts by the direct photoactivation of a Cu(II)Cl2 PMDETA (pentamethyldiethylenetriamine) ligand complex.30 This reaction was achieved with exposure to low intensity (3 mW/cm2), low energy ultraviolet radiation (350 nm). Despite relatively low concentrations of reactants (0.2 mM in DMSO) and Cu(II) (20 μM), high conversions (93-99%) were reached for a variety of alkynes with benzyl azide at room temperature and open to air. Reaction times varied from 120 to 600 minutes.
Adzima et al. took a different approach to the photoreduction of Cu(II), taking advantage of the vast work that had been completed in the fine tuning of radical-generating photoinitiators.31 Specifically, they employed a cleavage-type radical phosphine oxide photoinitiator (phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide) to generate radical fragments that subsequently reduced Cu(II) by direct electron donation from a radical initiator (Figure 15).
Figure 15.
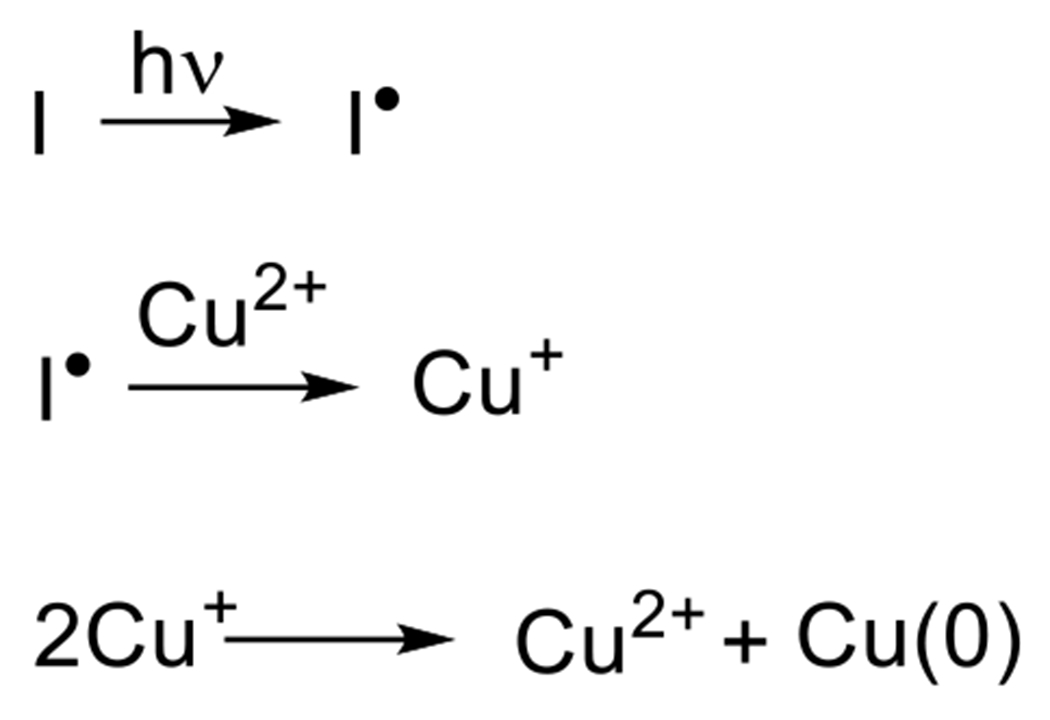
Photogeneration of Cu(I) via direct donation of electron from photogenerated radicals. Also depicted is the disproporationation resulting in regeneration of Cu(II) and the formation of Cu(0).31
This reaction was performed both in model, monofunctional studies, as well as with multifunctional alkynes and azides, resulting in network photopolymerization. The authors noted two seemingly contradictory observations. First, relatively short irradiation times generated enough catalyst to continue the reaction for hours following the shuttering of the light. For example, in one reaction that exhibited only 5% conversion when exposure ceased at 5 minutes reached approximately 75% after 100 minutes indicating the presence of a long-lived catalytic Cu(I) species. Secondly, the authors demonstrated a relatively high degree of spatial control over photomasked reactions of multifunctional reactants on surfaces with features as small as 4 μm obtained. While the observation of the long dark-reaction of previously irradiated reaction would suggest that the Cu(I) would have sufficient lifetime to diffuse away from the photoexposed regions into masked regions and catalyze reaction there as well, this “dark reaction” apparently did not happen. The failure to do so was explained by the formation of Cu(I) ligands with the triazole products of the photoCuAAC reaction, which remained effective catalysts, but within a 3D network, prevented the diffusion of Cu(I) outside of the irradiated regions. The autoligandization then demonstrated benefits of photo generated radical and general photolatent catalyst mediated reactions, high resolution spatial control and post-irradiation dark reaction. While initial studies of this photo CuAAC reaction reported reaction times on the order of one hour, subsequent papers have demonstrated much improved kinetics on the order of seconds to minutes under modest irradiation conditions (e.g. 10 mW cm−2, 365 nm).65 The copper ligand and anion have significant effects over the reaction kinetics.66 While preligation of copper with multifunctional aliphatic tertiary amines, such as tetramethylenediamene (TMEDA), greatly improved reaction rates, pyridine based ligands showed little improvement. Meanwhile, copper salts with halide anions proved substantially more effective in initiating systems than those with organic anions such as triflates and bistriflimides. Initiator and copper concentrations also have significant effects on the reaction rate, although recent reports have indicated limits to this relationship. El-Zaatari et al. noted that by increasing the copper and initiator concentrations, the reaction reached a threshold at which it transitioned from a first order to a zero order reaction with respect to copper and initiator concentrations. This outcome indicated that side reactions, such as the disproportionation of copper (Figure 15), that are negligible at relatively low catalyst concentrations, likely become significant as those concentrations increase.67 The properties of the radical photoinitiator exhbit significant influence over this reaction, which have been explored in great depth in the context of radical chain addition polymerizations and in radical addition, chain transfer reactions. More attention is given to these compounds in Section 2.3.
The reduction of copper was also carried out with photoinduced electron transfer (PET) catalysts.68 Cleavage type photoinitiators are generally limited to UV and lower wavelength visible light initiators; however, the application of PET catalysts, with their more varied absorption spectra, provides the benefit of being able to conduct the photoinitiated CuAAC reaction with substantially longer wavelength exposure. Kutahya et al. demonstrated effective catalysis of the CuAAC reaction using a NIR cyanine-based PET catalyst with a barbital group in the meso position (Figure 16).68 Exposure of the PET catalyst to 790 nm light at 100 mW/cm2 in the presence of a phenyl acetylene and benzyl azide in equimolar ratios and with PMDETA, achieved complete conversion within two hours. Effective polymer-polymer coupling was also demonstrated via this NIR PET-catalyzed CuAAC in the reaction between a 3000 g/mol polystyrene and a 4000 g/mol polycarpolactone. The product, analyzed by gel permeation chromatography, demonstrated the expected molecular weight and a low dispersity, indicating efficient coupling with relatively little unreacted starting material.
Figure 16.
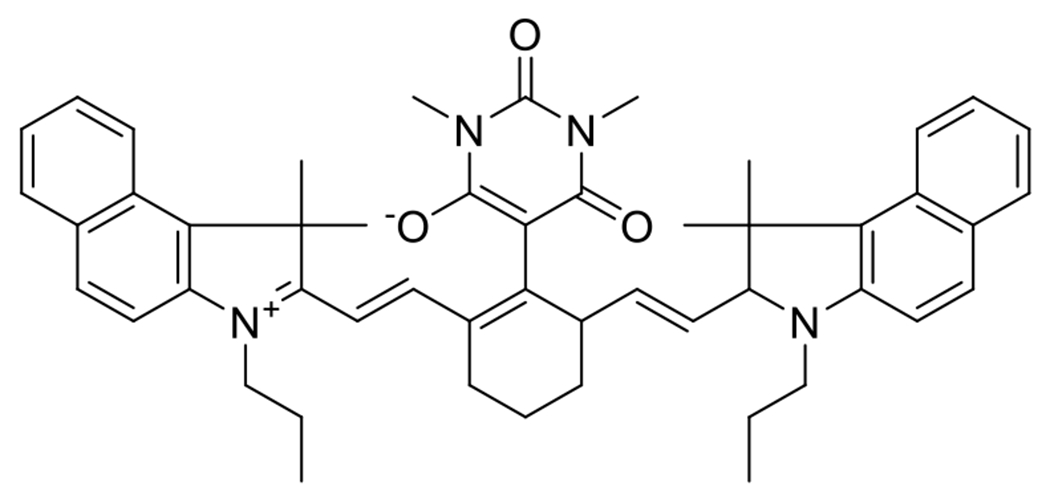
Cyanine-based PET catalyst with a barbital group in the meso position employed in the NIR mediated reduction of Cu(II) to Cu(I) and successful catalysis of CuAAC reaction.68
Another means by which the CuAAC reaction may be photomediated is by the photodeprotection of an otherwise unreactive alkyne. Because only terminal alkynes are reactive in CuAAC reactions, reversible protection of the terminal alkyne with a photoremovable group (generally a nitrobenzyl moiety69) allows researchers to conduct photomediated CuAAC reactions. Tebikachew et al. generated terminal alkynes by the photolysis of nitrobenzyl arylpropiolates and subsequent decarboxylation in the presence of copper catalysts.70 While the description of this reaction as “photoclick” may not be accurate (the wavelength of light employed in this reaction was very low, thermal input was needed for decarboxylation, and the atom economy is poor), it does represent an unusual approach to carrying out these conjugations and deserves mention.
Copperless photoredox azide alkyne cycloaddition has been recently demonstrated by Wu et al. in a process they have called “photocatalyzed azide-alkyne cycloaddition” or PcAAC.32 While photoredox catalysts are used in this PcAAC reaction, rather than the photoreduction of copper(II) to copper(I), the excited photocatalysts oxidizes the alkyne, generating a radical cation, which is a much more reactive dipolarophile than the original alkyne that reacts with the azide to form a radical cation triazole that is subsequently reduced, regenerating the photocatalyst (Figure 17). While the reaction rates achieved were modest compared to those in which photoreduced copper served as the catalyst, near quantitative yields in model reactions between phenylacetylene and benzyl azide were achieved in as little as six hours under visible light irradiation. Sunlight also proved an effective light source. In contrast to thermally-mediated azide alkyne cycloadditions, complete regioselectivity was observed with only the 1,4-disubstituted triazole recovered. The reaction was carried out in DCM and DMF as well as in aqueous solutions and open to air, demonstrating insensitivity toward oxygen and water. According to the authors’ data, the most effective photocatalyst screened for the PcAAC was bis(1-phenylisoquinoline)(acetylacetonate)iridium(III), though significant reaction was achieved with 2,4,6-tris(4-methoxyphenyl)pyrylium tetrafluoroborate as well, demonstrating for the first time a completely metal free photomediated azide alkyne cycloaddition without employing ring strained cycloalkynes.
Figure 17.
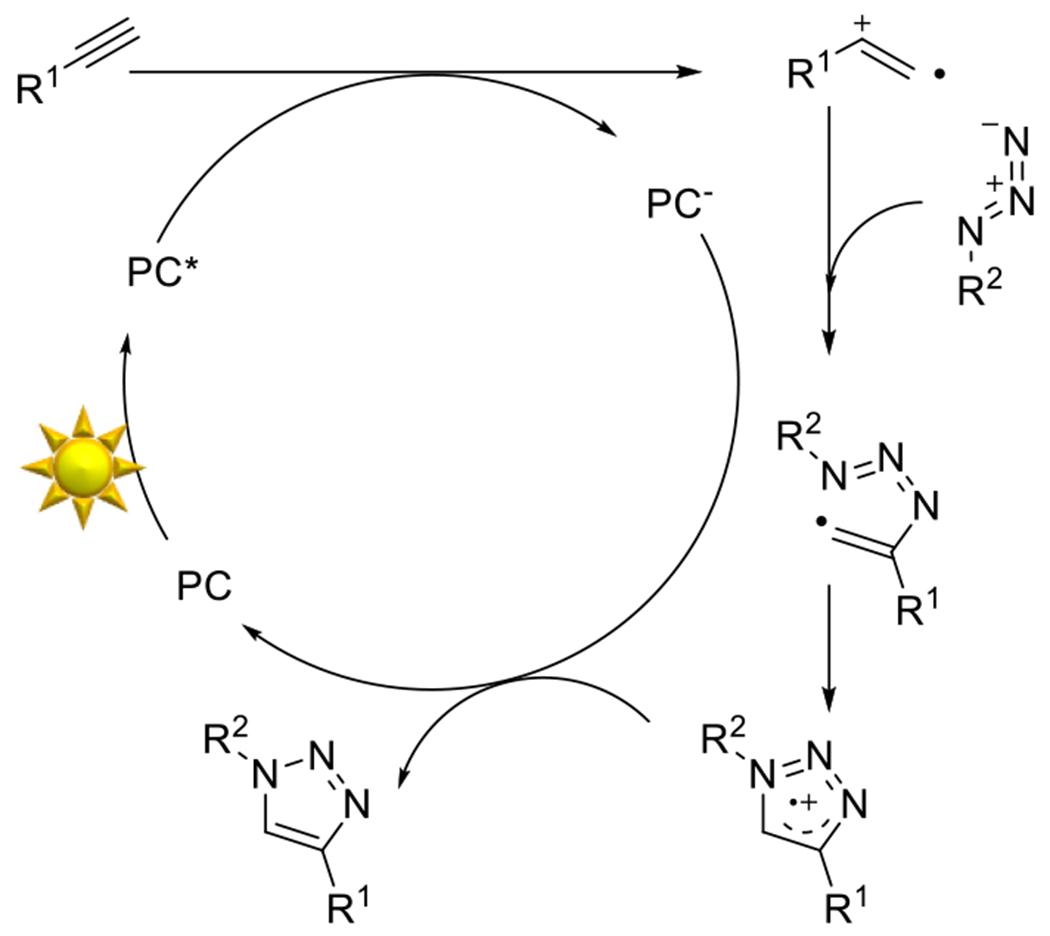
PcAAC mechanism-the photomediated azide alkyne cycloaddition by direct photooxidation of alkyne and subsequent 1,3-dipolarcycloaddition and regeneration of photocatalyst. Figure adapted, with permission, from reference 32. Copyright 2020 John Wiley and Sons.
Photothermal initiation of click reactions is not especially common in the literature. A likely reason for this is the avoidance of relying on highly thermally dependent reactions. Typically, in order for any reaction to be considered “click” it must exhibit a considerable thermodynamic driving force, a feature even suggested by the moniker “click” as if a gentle push over the energy barrier causes all reactants to simply click into place. Thus, any more than a little added energy to overcome an activation barrier would seem to exclude reactions from a designation as “click” altogether. Photothermal effects, however, potentially being extremely localized, may provide the thermal energy necessary for overcoming the activation barrier of a reaction while heating the bulk reaction medium less than would otherwise be required. While photothermal-mediated click reactions are sparsely reported, there are examples. Sun et al. demonstrated a synergistic photothermal and hot electron mechanism for the reduction of Cu(II) to Cu(I) sufficient to catalyze the azide alkyne cycloaddition between benzyl azide and phenylacetylene within 90 minutes with exposure to visible light (420-780 nm).71 Hot electrons refer to electrons that have a higher kinetic energy and therefore a higher effective temperature of the electrons despite a lower bulk temperature of the material.
2.1.1.2. Photomediated Strain Promoted Azide Alkyne Cycloaddition (SPAAC)
In no small part because of the desire to perform click reactions in the presence of living cells and because of the toxicity of copper, strain promoted azide alkyne cycloadditions have received considerable attention over the last decade.72–74 Cyclooctyne, the smallest, stable, isolable, cycloalkyne, undergoes significant release of ring strain when the alkyne, with a preferred bond angle of 180 degrees, but exhibiting a bond angle of 163 in its eight membered ring, is converted to a triazole ring, eliminating the triple bond (Figure 18).75
Figure 18.
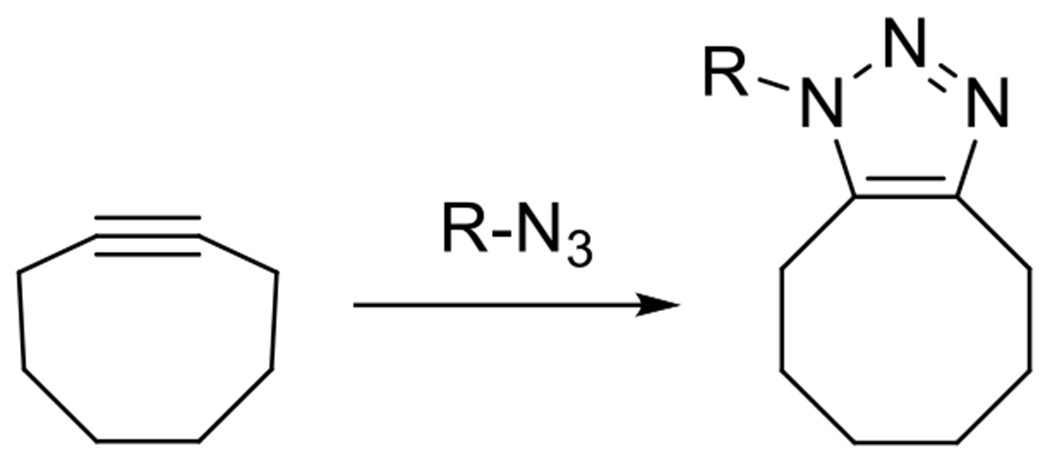
Ring strain in cyclooctyne facilitates fast, room temperature, copperless cycloaddtion.
Electron withdrawing fluorine atoms further destabilized the alkyne, contributing to greater reactivity in the absence of any catalyst. Greater destabilization still was achieved via a series of dibenzoannulated cyclooctynes with additional ring strain imparted by the presence of multiple sp2 hybridized atoms in the cycloalkyne ring (Figure 19). Azides and alkynes are generally less reactive towards many functional groups found in the biological context. For this reason, the azide alkyne cycloaddition reactions have found substantial application in selective labeling of biological structure and macromolecules. Diminishing the stability of alkynes by introducing ring strain, for example, not only increases its reactivity towards azides but towards other functional groups as well. Thiols, for example, are slow to react with most linear, terminal alkynes in the absence of radicals, catalysts, or elevated temperatures, but will readily and spontaneously react with cyclooctyne at room temperature.47 This example demonstrates the difficult tradeoff between reactivity and selectivity. Click reactions are supposed to be both fast and specific. Achieving both is not a trivial matter.
Figure 19.
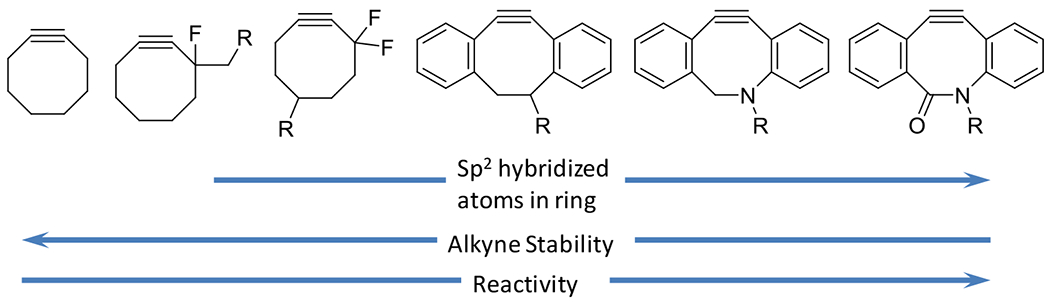
The propensity for cycloaddition with azides is increased in cycloalkynes by the destabilization of the alkyne by proximal electron withdrawing groups (e.g. fluorine atoms) and by the increase of sp2 hybridized atoms in the ring.
Cyclopropenones, upon exposure to UV light, decompose into one molecule of carbon monoxide and an alkyne. Highly ring-strained themselves, the conversion of a cyclopropenone to the strained triple bond of a cyclooctyne is still an energetically favorable reaction, allowing for the photodeprotection of strained cycloalkynes for subsequent, spontaneous reaction in azide alkyne cycloaddition (Figure 20).33 It should be noted that as in the case of the thermally mediated Huisgen azide alkyne cycloaddition, a mixture of regioisomers is obtained from the copper-free SPAAC reaction, whether photomediated or spontaneous.
Figure 20.
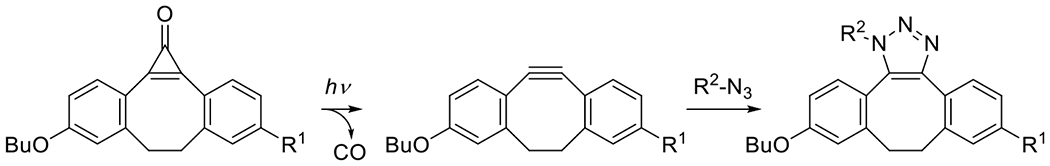
Photoduncaging of cyclooctyne for subsequent strain promoted cycloaddition with organic azide.
Generally, cyclooctynes are considered the smallest stable cycloalkynes that can be isolated and stored without decomposing. Cyclopropenones serve as a temporary protecting group, the removal of which generates, in situ, cycloalkynes that may be relatively short-lived. In the presence of their reactive azide complement, however, the rate of cycloaddition far exceeds the rate of decomposition, producing the desired product in high yields. A particularly effective example of this ability to generate protected, otherwise unstable cycloalkynes was demonstrated by Sutton et al. in their synthesis of a photocaged cyclooctadiyne (the Sondheimer diyne, Figure 21).64 This protected diyne is, as would be expected, difunctional, though the quantum yield for the first and subsequent cyclopropenone decompositions are not equal; that for the first decarbonylation is approximately ten times that of the second at a wavelength of 350 nm. This allows for the selective, sequential reaction of the resulting alkynes with complementary azides.76, 77
Figure 21.
Other notable cyclopropenone-protected strained alkynes including a photocaged dibenzocyclooctadiyne (dibenzo[a,e]dicyclopropa[c,g][8]annulene-1,6-dione; left)64 and a photocaged dibenzosilacycloheptyne (6,6-dimethyldibenzo[b,f]cyclopropa[d]silepin-1(6H)-one; right).34
Cycloheptynes, (employing heteroatoms such as sulfur, selenium, or silicon, in the alkynyl ring) can also be synthesized to yield even more reactive alkynes. However, working with the highly unstable cycloheptynes can be difficult. Similar to the work with the cyclopropenone-protected octadiyne, cyclopropenone-protected dibenzosilacycloheptynes have been synthesized by Martínek et al. that exhibit very good quantum yield (>0.50) and subsequent cycloaddition reaction rates (Figure 21).34
The direct photolysis of cyclopropenones is a one photon, on event reaction; the generation of every reactive cylcoalkyne requires absorption of light. Under dilute reaction conditions, this is not an obstacle to carrying out the reaction. In more concentrated conditions, however, the necessary light will become more attenuated in thicker geometries, preventing reaction from occurring within the bulk of the sample. This is in contrast to photo CuAAC reactions wherein a small amount of photogenerated Cu(I) may catalyze thousands of cycloadditions, allowing the researcher to perform the reaction with very low concentrations of light absorbing chromophore. This limitation has resulted in the photoSPAAC reaction to find particular application in highly diluted reactions including biological labeling as described subsequently in this review. In such context, however, the potential deleterious effects of significant doses of lower wavelength UV light may be a concern.
In order to address this requirement for relatively low wavelength light exposure, recently Mishiro et al. reported an indirect photolysis of cyclopropenones to generate the ring strained cyclooctyne.78 Prior studies had indicated that the photomediated decarbonylation of cyclopropenone occurred from the excited singlet state and so photosensitization via energy transfer from a lower energy, visible-light sensitizer to a UV-absorbing chromophore would not be possible. The indirect photolysis would, then, necessarily proceed via a wholly different decarbonylation mechanism. The authors demonstrated a photoredox catalyst abstracting an electron from the cyclopropenone, generating an unstable radical cation, opening the ring structure that could then remove an electron from the reduced photocatalyst, regenerating it in the process and subsequently decomposing into carbon dioxide and the desired alkyne.
Although the use of cyclopropenones as photocaging groups for cycloalkynes is favored given its relatively high quantum yields and inoffensive byproducts, there are other potential strategies for the photogeneration of ring strained cycloalkynes. Jedináková et al. have reported photoSPAAC reactions via the synthesis of cyclooctynes via the photodecomposition of cycloota-1,2,3-selenadiazole (see Figure 22).79 This reaction, however, exhibits a relatively poor quantum yield. Moreover, though selenium is an essential nutrient, in elevated concentrations it may have unpleasant or toxic physiological effects, making the broader application of its use in click reactions problematic. In this respect, selenium is similar to copper, which is also an essential nutrient in trace quantities but toxic at higher concentrations.
Figure 22.
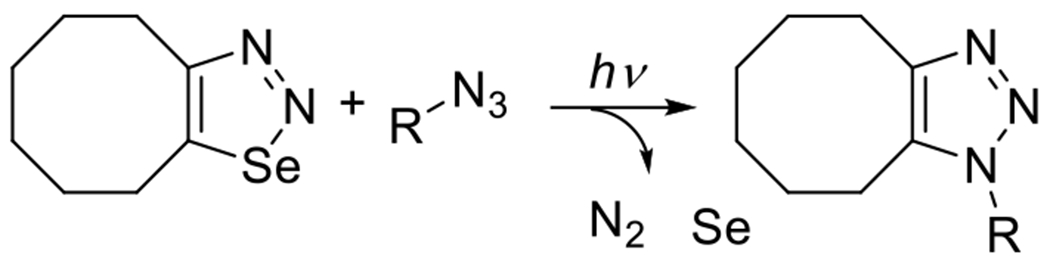
PhotoSPAAC reaction using selenadiazole photocaging group.79
A final photoSPAAC reaction that deserves some attention is the photogeneration of benzyne and subsequent reaction with organic azides. Rather than the decarbonylation of cyclopropenone, however, Gann et al. reported the photodecomposition of 2-(3-acetyl-3-methyltriaz-1-en-1-yl)benzoic acid to produce the strained cycloalkyne (Figure 23).80 This photoreaction was accomplished in just a few minutes at 365 nm irradiation. The subsequent reaction with organic azides was rapid and high yielding. Although this reaction provided an efficient and rapid reaction, benzyne is a very reactive compound, particularly towards nucleophiles generally. For this reason, there is concern regarding the specificity and orthogonality of this reaction under a variety of conditions in which photoclick reactions may be employed.
Figure 23.
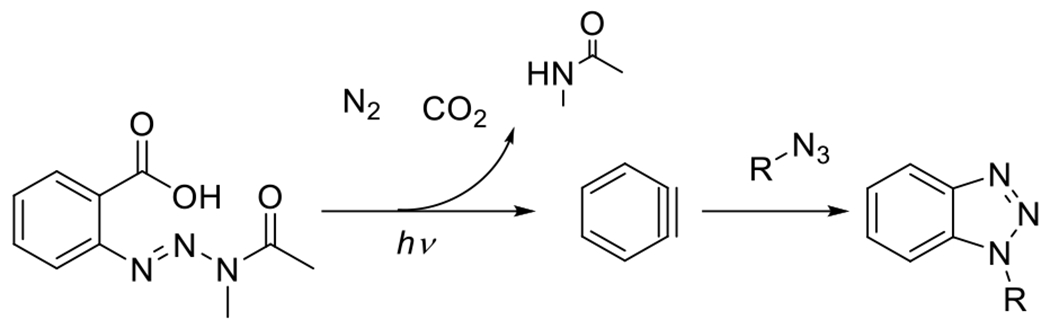
Photogeneration of benzyne via decomposition of 2-(3-acetyl-3-methyltriaz-1-en-1-yl)benzoic acid and subsequent conjugation to organic azide.
2.1.1.3. Strain Promoted Azide Alkene Cycloaddition
While most attention in azide dipolarcycloaddition reactions has been focused on the employment of alkyne dipolarophiles, alkenes are frequently the coreactant of choice for other reactions in this category. Alkenes are capable of participating in cycloadditions with azides and that reaction is favored, as it is with alkynes, by strained alkenes. Huisgen noted a hundred fold increase of the reaction of ring strained norbornene with benzyl azide.81 In 2018, Singh et al. demonstrated the photosensitization of the isomerization of a cyclooctene to a more highly ring strained conformation and the subsequent reaction with benzyl azide in a 1,3-dipolarcycloaddition.82 The photosensitizer was a derivative of fac-tris-[2-phenylpyridinato-C2,N]iridium(III) (Figure 24) and the sensitization was performed with blue light. While the reaction required 16 hours for completion and a slightly elevated temperature (30 °C), near quantitative conversion was achieved (Figure 24). While the conditions of this reaction were not what might be considered sufficient for a “click” reaction, this demonstration of photogenerated ring-strain to promote dipolarcycloadditions has proven consequential in the development of other photoisomerization-promoted click reactions.
Figure 24.
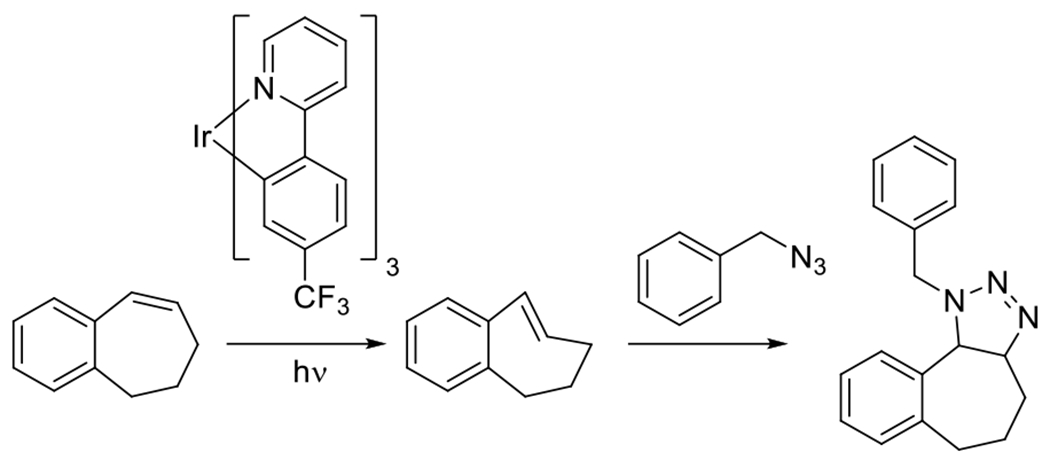
Photosensitized ring-strain generation in cycloheptene, promoting subsequent reaction with benzyl azide.82
2.1.2. Photocaged Nitrile Imines and Nitrile Ylides
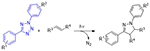
2.1.2.1. Tetrazole and Sydnone Reactions
Nitrile imines were first obtained by the thermal decomposition of tetrazoles, releasing N2 during the process.83 Exposure to appropriate wavelengths of light (dictated by chromophore substituents), accomplishes the same decomposition, yielding the same reactive intermediate. In the presence of alkenes the nitrile imine may undergo 1,3-dipolar cycloaddition to form the conjugated product linked by a five membered pyrazoline ring (Figure 25). An interesting and useful feature of the reaction between alkenes and nitrile imines is that the generation of the fluorophore pyrazoline can serve as an in situ reaction monitoring system, absorbing UV light and emitting visible light as an indication of successful reaction. While a variety of alkenes (e.g. acrylates, styrene derivatives) participate in this reaction, the dipolarophilicity of alkenes is substantially increased by conjugation or by proximal electron withdrawing substituents.58 This photodecomposition of the tetrazole to the nitrile imine has the advantageous feature of generating only inert N2 as a byproduct. As is the case with most dipolar cycloadditions, the nitrile imine-alkene reaction is amenable to a variety of solvents including aqueous media. Also, like other 1,3-dipolar cycloadditions, the reaction may not be regiospecific.84
Figure 25.
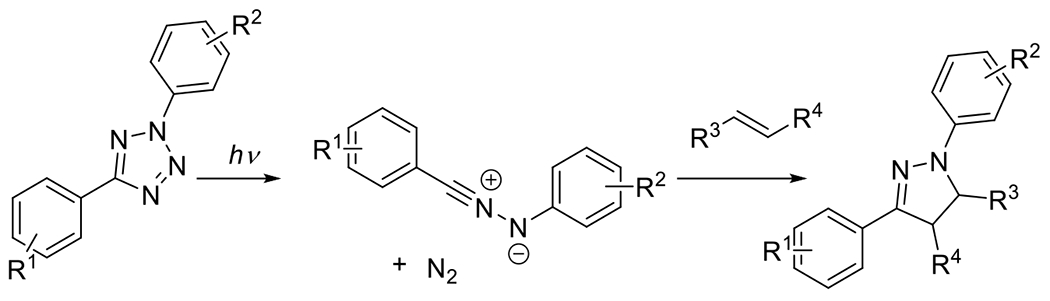
Photomediated decomposition of bisaryl tetrazole to nitrile imine for subsequent participation in 1,3-dipolarcycloaddition.
The wavelengths at which the reaction is performed are tunable via the choice of substituents. The reaction has been performed from 300 nm to 420 nm by single photon excitation and at 700 nm with multiphoton excitation.85 The red shifting that allowed the 405 nm exposure was accomplished by replacing the benzene ring substituents on either side of the tetrazole with oligothiophene substituents, greatly increasing the absorbance in the visible spectrum (Figure 26).86 Lederhose et al accomplished a similar shift in absorbance via synthesis of a pyrene-substituted aryl tetrazole, permitting reaction with exposure to light at wavelengths of 410-420 nm.87
Figure 26.
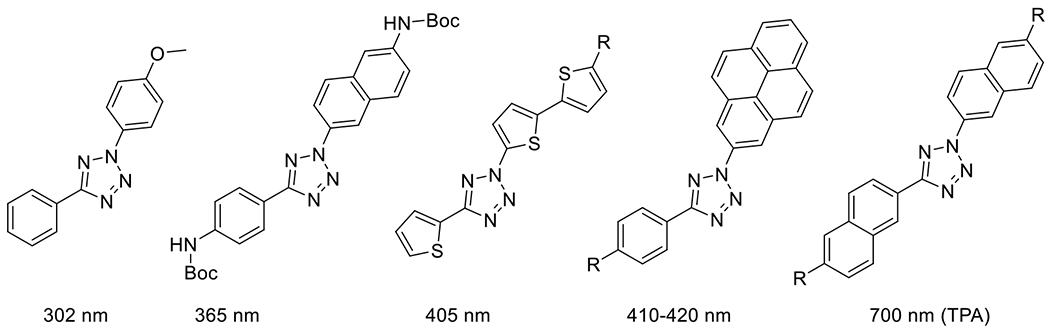
Tuning of photoabsorption profiles can be accomplished by choice of tetrazole substituents.
One interesting strategy to allow longer wavelengths of light in the performance of tetrazole-alkene photoclick chemistry was the employment of upconverting nanoparticles to generate light capable of photoconversion of a pyrene aryl tetrazole (λmax=346 nm) upon exposure to NIR light. Photon upconversion is a process distinct from two photon absorption but with significant similarities. Upconverting nanoparticles absorb photons of a particular wavelength and emit photons of a shorter wavelength. Thus, the researchers were able to effect a UV-photomediated reaction with NIR exposure.88 In contrast to previously described two photon absorption strategies, the aryl tetrazole chromaphore in this example is not absorbing the NIR photons, but rather UV photons emitted by the upconverting nanoparticles; this reaction is still mediated by UV radiation. The potential benefit to employing photon upconversion here, isn’t avoiding exposure to UV light, but rather the avoidance of light attenuation of shorter wavelength photons.
Diaryl tetrazoles are not unique in their photodecomposition to dipolar nitrile imines. Diaryl sydnones similarly form the highly reactive intermediates upon light activation. Zhang et al. in 2018, synthesized a library of diaryl sydnones and noted higher absorbance of the sydnone chromophores at longer wavelengths relative to their tetrazole counterparts, portending beneficial application in biological research where visible light excitation is preferred.36 Rather than releasing nitrogen, photoconversion of the sydnone, which involves a flipping of the aryl moieties, results in the release of carbon dioxide (Figure 27).
Figure 27.
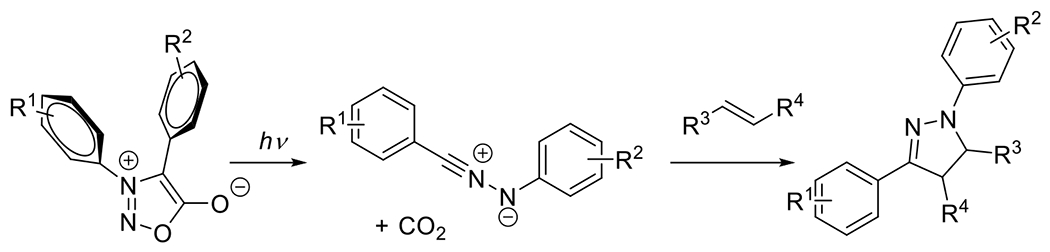
Photoconversion of diaryl sydnone to nitrile imine and subsequent 1,3-dipolarcycloaddition with alkene.36
Despite the advantageous features of the photo tetrazole-alkene and sydnone-alkene reactions, the reactive intermediates exhibit a significant lack of reaction selectivity. Recently, researchers have recommended the application of the reaction as a general photocrosslinking reagent rather than for biorthogonal labeling protocols, citing the relative preference of the photogenerated nitrile imine to react with biologically present functional groups,89 in particular carboxylic acids,90 relative to their experimental, more aliphatic alkene moieties. They noted significant reaction with acetonitrile as well, when used as a cosolvent for the reaction.
While reactive towards nucleophiles generally, nitrile imides are particularly reactive towards thiols,91 first observed by Rolf Huisgen in 1962.92 This reaction can be so fast that the photomediated, addition of thiols to nitrile imides has been compared to other “photoclick” reactions such as the tetrazole-alkene and thiol-ene conjugations. Feng et al. used the photodecomposition of tetrazoles in the presence of thiols to perform polymer-polymer conjugation reactions as well as surface functionalization-both frequent applications of many click chemistries.93 The authors suggested that the biological availability of thiols (e.g. in reduced disulfides within proteins) could make the photo tetrazole thiol reaction an effective tool for the selective labeling of biomolecules, noting a much higher rate of reaction of the nitrile imine with thiol than with amines.
Additional dipolarophiles that may react with nitrile imines include alkynes, carbon dioxide, isocyanates, thiocarbonyls, amines, alcohols and more, although the rates of reaction vary greatly.84 Yu and coworkers have examined the photoreactions of diaryl tetrazoles and diaryl sydnones with various alkynes.94, 95 While the linear, terminal alkynes yielded no observable product under dilute conditions (0.5-500 μM), ring strained alkynes were converted in high fractions with an excess of coreactants.
As indicated by these results, in addition to conjugation and electron deficiency, ring strain can also improve the reactivity of dipolarophiles to promote the 1,3-dipolarcycloaddition to nitrile imines. Fascinating recent examples of ring-strain promoted reaction were demonstrated by Yu and coworkers who employed the photoisomerizable cyclic azobenzene-containing compounds such as dibenzo[b,f][1,4,5]thiadiazepine (DBTD, Figure 29) as a photoactivatable dipolarophile in reactions with nitrile imines generated by the photodecomposition of either tetrazoles or sydnones.96, 97 In the cis configuration, DBTD exhibits little ring strain energy. Upon irradiation with 405 nm light, the DBTD is isomerized to a predominantly trans configuration, increasing ring strain and increasing the dipolarophilicity of the nitrogen-nitrogen double bond. Cycloaddition is followed by opening of the N-N bridge, resulting in a ten-membered ring (Figure 29). Noteworthy in this reaction is the double role that the light may play in the reaction, serving both in the generation of the reactive nitrile imine and in creating the ring strain via photoisomerization in the DBTD. In a subsequent report, the same team demonstrated a similar ring-strain by photoisomerization in carbon-bridged octocyclic azobenzene (CBOA) to promote the 1,3-dipolarcycloaddition.53 In this later study, nitrile imides were synthesized independently and delivered exogenously to isolate the photoisomerization effects independent from the photogeneration of the reactive dipole. Both of these photochemical transformations, (the photoisomerization and the photodecomposition of the tetrazole/sydnone) are one-photon, one event type reactions, meaning that the concentration of light absorbing chromophores is necessarily twice what would otherwise be necessary for the reaction to take place with a different dipolarophile, recommending applications where low concentrations of reactants are required. Near quantitative conversions were achieved.
Figure 29.
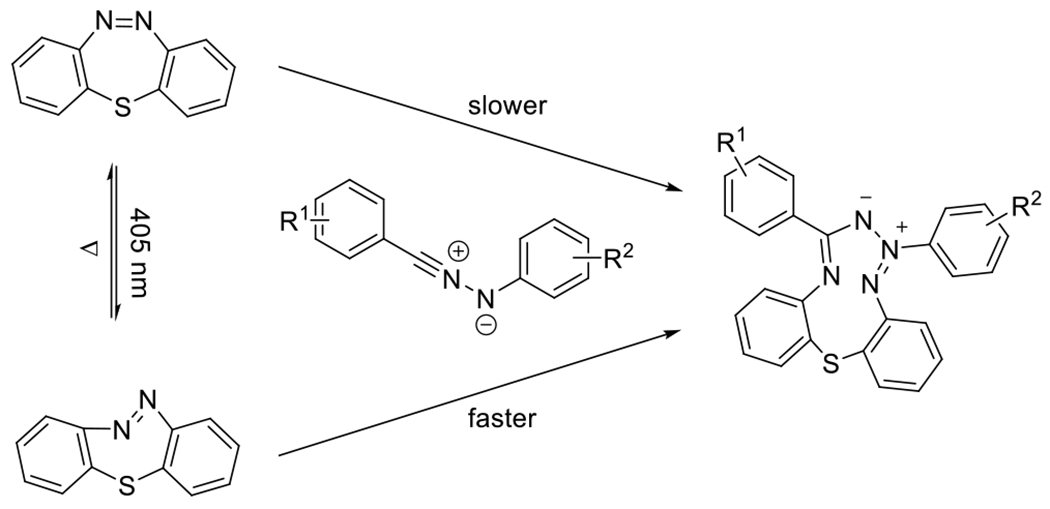
Photoisomerization of cis configuration of DBTD to trans configuration of DBTD by irradiation with 405 nm light permits the addition to photogenerated nitrile imine. In contrast to cis DBTD, trans DBTD proves an effective dipolarophile in the reaction.81, 82
The apparent lack of specificity of the tetrazole-ene photoclick reaction could be a cause for concern for those seeking to employ it to precisely modify their particular compounds of interest. As with other “click” reactions, however, the utility of such reactions must take into account the relative rates of competing reactions for participating reactive moieties generally, and not only for those conditions understood to be far from ideal. The side reaction of a photogenerated nitrile imide with acetonitrile cosolvent may be significant when the intended coreactant is a poor dipolarophile e.g. an alkyl ethylene compound. However, a fumarate used in place of the less effective dipolarophile, will likely improve the selectivity of the dipolar cycloaddition.91 The alkene selection is not the only way in which selectivity of the tetrazole alkene reaction can be improved. A recent report has demonstrated a particularly effective strategy for obtaining higher selectivity for alkenes over carboxylic acids, alcohols, amines and other potentially nitrile imide-reactive nucleophiles. This was accomplished by the employment of tetrazoles with bulky substituent groups that sterically hinder the nucleophilic addition reaction but not the 1,3-dipolar cycloaddition with alkenes. An et al. synthesized a range of tetrazoles and reported increases in selectivity of the 1,3-dipolar cycloaddition adducts with a variety of alkenes relative to the nucleophilic adducts formed with glutathione (Figure 30).91 Selectivity was demonstrated in all eight of the alkene substrates examined, but the reaction with styrene is a particularly good example of the effect steric hindrance had on the progression of the reaction. While in the presence of equimolar styrene and glutathione, the non-sterically hindered tetrazole showed a preference for the thiol-adduct of 91% to 5%. The sterically hindered tetrazole, in contrast, showed a preference for the styrene cycloadduct, 96% to 2%.
Figure 30.
Photoreactive tetrazole (left) and bulky substituent analog (right). Pendant groups sterically hinder nucleophilic additions and improve selection of 1,3-dipolarcycloaddition adduct with a variety of alkenes.91
While strategies are available to address the broad reactivity of nitrile imides resulting from the photodecomposition of tetrazoles, these may introduce limitations into the general application of this reaction (e.g. by inclusion of a large chemical moiety that may influence physical as well as chemical properties of the reactant and by requiring additional synthetic steps). Whereas the azide alkyne chemistries are often touted for their orthogonality, those who employ the tetrazole-alkene photoclick reaction should be cognizant that, in the absence of adequate concentrations of a good dipolarophile, the photogenerated nitrile imine is not functionally inert and will frequently react with other reactive groups present.
2.1.2.2. Azirine-Alkene Reactions
Like tetrazoles, aryl azirines undergo photomediated decomposition to generate a reactive dipole (nitrile ylide moieties in the case of azirines) prone to 1,3-dipolar cycloaddition.37 As is the case with the tetrazole-alkene reaction, this reaction is favored by strong dipolarophiles including alkenes with proximal electron withdrawing substituents.
The photoactivation of bisaryl azirines as shown in Figure 31 requires exposure to relatively low wavelengths of light (320 nm). However, the pyrene-bound azirine exhibited significant red-shifted absorbance, thus being activatable with visible light at wavelengths from 400 nm to 410 nm (Figure 32).98 This reaction was used to link pyrene to a variety of small molecule and electron-deficient alkene terminal polymers with yields ranging from 50% to quantitative.
Figure 31.
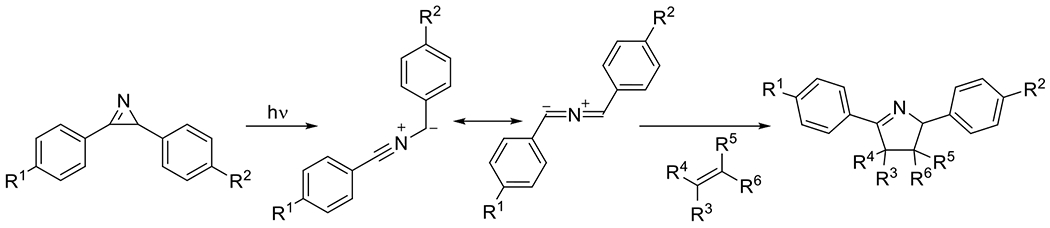
Photogeneration of nitrile ylide from bisaryl azirine and subsequent 1,3-dipolarcycloaddition. 37
Figure 32.
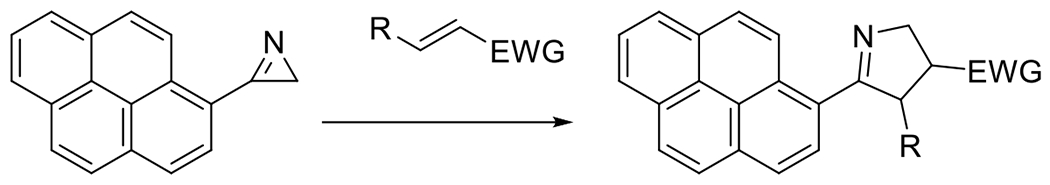
Visible light activation of pyrene azirine and conjugation to electron deficient alkenes. EWG = electron withdrawing group. R = H, COOEt, PEG.98
Despite the similarities between the analogous reactions, azirine-alkene photoclick reactions are used much less frequently than the tetrazole-alkene reaction.
2.2. Diels Alder Reactions
Among the original reactions described by Kolb et al. in their seminal paper laying out the qualifications for click chemistry was the Diels Alder reaction.2 Typically, in this cycloaddition an electron deficient dienophile adds to a relatively electron rich diene. As the pi bonds in both the diene and the dienophile are higher energy than the resulting, created sigma bonds, the reaction is energetically favorable and is often performed at relatively low temperatures and in the absence of catalysts (though catalysts can be employed to achieve improved rates of reaction). While examples of this reaction are abundant, a very simple case is the reaction of cyclopentadiene with an acrylate (Figure 33), generating a norbornene. Participating pi bonds on either the diene or dienophile need not necessarily be carbon-carbon double bonds. Diels Alder reactions in which atoms other than carbon are involved in the cycloaddition are referred to as “hetero Diels Alder” reactions. Inverse electron demand Diels Alder reactions (IEDDA) are analogous to Diels Alder reactions but where the electron densities of the diene and dienophiles are reversed. The classic example of an IEDDA is the reaction of 2-propenal with a vinyl ether (Figure 33).
Figure 33.
a) Generalized Diels Alder reaction between electron rich diene and electron deficient dienophile and example of cyclopentadiene reaction with methyl acrylate. b) Generalized inverse electron demand Diels Alder reaction and example of reaction between propenal and methyl vinyl ether.
2.2.1. Photoenolization DA reactions
Photoconversion of ortho-methyl phenyl ketones and aldehydes to form highly reactive enol dienes via a diradical intermediate was first described many decades ago, achieving high conversions with dienophilic compounds.99, 100 In 2011 Gruendling et al. used this photoenolization to conjugate an ortho-methyl phenyl ketone-terminated polycaprolactone to a maleimide-terminated poly(methyl methacrylate) with irradiation at 320 nm for 100 minutes.38 While the light source chosen produced a maximum emission at a low wavelength, the authors noted some conversion under exposure to 365 nm or upon exposure to sunlight after “a few hours reaction time.” Subsequent reports show effective conjugation with exposure to 382 nm LEDs.101 A scheme for the photoDA reaction is shown in Figure 34.
Figure 34.
Photoenolization of ortho-methyl phenyl aldehyde and subsequent Diels Alder cycloaddition with dienophilic maleimide (top) or hetero Diels Alder cycloaddition with dithiobenzoate (bottom). 38
Photoenols also readily react with a variety of dienophiles with heteroatoms. For example, Oehlenschlaeger et al. have demonstrated the reactivity of dithiobenzoate with photoenols,39 a particularly advantageous reaction considering the use of dithiobenzoates and other thiocarbonylthio groups in RAFT chain transfer agents and their consequent presence as RAFT polymer termini. This reaction then allows for direct conjugation to such polymers without any additional modification. In another example of hetero Diels Alder reactions involving photoenols, triazolinediones have proven effective dienophiles as well. Recently, the orthogonality of this reaction was highlighted in the utilization of light as a reaction-selection tool or “gate” to generate specific products from a mixture of reactants, including triazolinediones and tetrazoles, by selective application of particular stimuli including thermal and multiple wavelengths of light.101, 102
Coordinated light exposure can provide another aspect of control to the photoenol-Diels Alder reaction. Photoenolization of o-methyl benzaldehydes with NIR light via two photon absorption results in a mixture of isomers that, in the absence of a complementary reactive moiety, reketonize via reverse hydrogen transfer to the starting material. Differences in rates of reketonization allow the E form of the photoenol, which has a relatively long lifetime (10 μs), to participate in DA addition reactions while the Z form, with a lifetime two orders of magnitude shorter (0.1 μs) is effectively prevented from doing so. Simultaneous exposure with 440 nm light, photoisomerizes the E photoenol to the Z form, which rapidly reverts to starting material (Figure 35). Researchers have employed this feature to spatially narrow the photoenolization of a-methyl benzaldehydes on surfaces in the presence of a maleimide dienophile via stimulated-emission depletion (STED) excitation, creating laser written features on size scales below the diffraction limit.103
Figure 35.
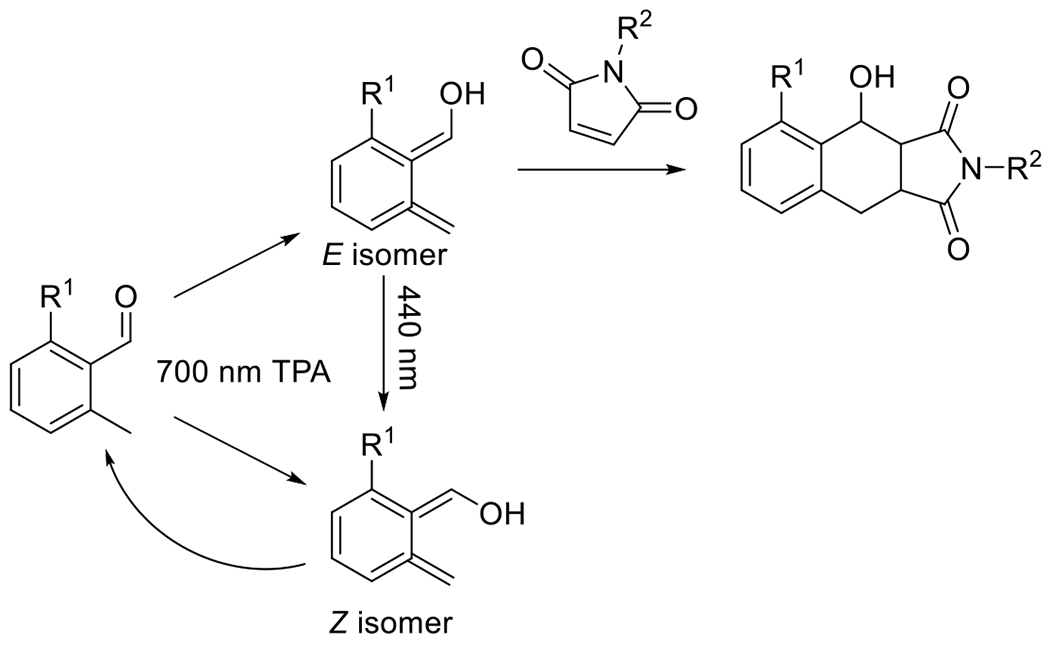
Dual wavelength control over the photoenol DA reaction. Two photon absorption at 700 nm generates a mixture of isomers with differing lifetimes while simultaneous exposure to 440 nm converts a long-lived isomer to a short-lived isomer, preventing effective cycloaddition. Figure adapted, with permission, from reference 103. Copyright 2017 American Chemical Society.
While most common dienophiles employed in Diels Alder reactions possess double bonds, there is no requirement that precludes the pi-bonds in triple bonds from participation. The electron deficient triple bond in dimethyl acetylene dicarboxylate has been shown to be reactive with 2,5-dibenzoyl-p-xylene under 24 hours of irradiation.25, 104, 105 More recently, Feist et al. have performed Diels Alder reaction with photogenerated o-quinodimethanes and electron deficient alkynes within 15–30 minutes of light exposure, noting that subsequent addition of a catalytic amount of acid resulted in the elimination of the alcohol and the generation of highly fluorescent naphthalene derivatives.40 A general scheme for these reactions is shown in Figure 36. Notable here is the absorption profile of the non-fluorescent naphthole Diels Alder product, which does not exhibit significant absorption at those wavelengths most conducive the photogeneration of the diene reactant, absorption which would otherwise attenuate the light necessary to carry out the photoclick reaction. In contrast to the other photoclick reactions described previously that result in the generation of new fluorophores or chromophores, this approach yields such only following the hydroxyl elimination. In addition to preventing attenuation of the requisite photostimulus, this also mitigates the potential photobleaching of the formed fluorophores and other deleterious light-induced side reactions.
Figure 36.
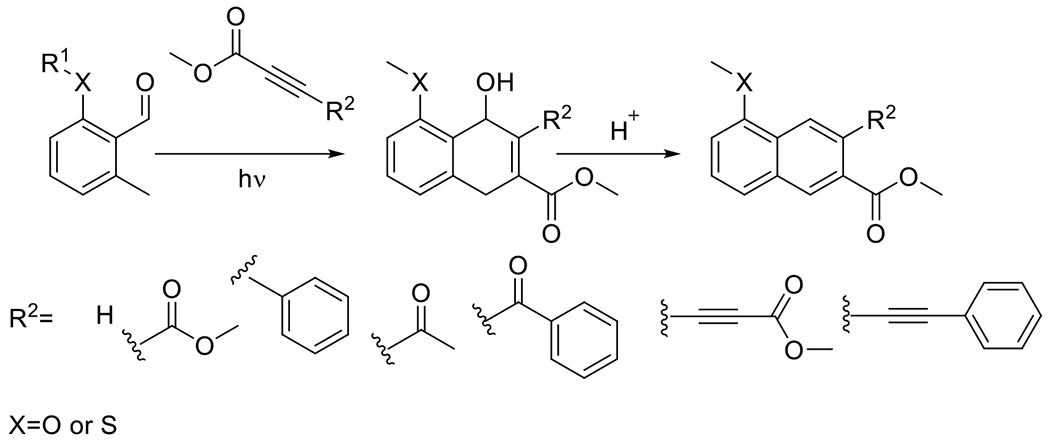
General scheme for the photomediated Diels Alder reaction with electron deficient alkynes and subsequent hydroxyl elimination to yield fluorescent naphthalene derivative.
2.2.2. Inverse Electron Demand Diels Alder
In 2011, Arumugam et al. demonstrated the photoconversion of 3-(hydroxymethyl)-2-naphtol derivatives to 2-naphotoquinone-3-methides, eliminating water and producing effective dienes for IEDDA reactions with appropriate electron deficient alkenes.41 In an aqueous environment, this photogenerated compound quickly converts back to the starting material (Figure 37). There are two advantageous consequences of the short lived (τ = 7 μs in water) diene intermediate in this photoclick reaction. First, the reversion to starting material prevents reaction with all but the most IEDDA-reactive alkenes, vinyl ethers and eneamines. In contrast to, for example, a tetrazole-alkene reaction, there is diminished opportunity for the activated species to react with unintended moieties. With a tetrazole-alkene reaction, even if the dipolarophile is highly reactive and dominates in a competitive reaction with other dipolarophiles and nucleophiles, when consumed, any remaining activated nitrile imide will be free to react with other functional groups. For this particular photo-IEDDA reaction, however, the rate of reversion to starting material outcompetes the IEDDA reaction with any but the most reactive alkenes, preventing off-target conjugations even in the absence of vinyl ethers or eneamines. The second consequence of this short lifetime of the 2-napthoquinone-3-methides is the prevention of significant diffusion of the reactive intermediate, improving spatial control over the reaction; IEDDA occurs only in areas that are exposed to light.
Figure 37.
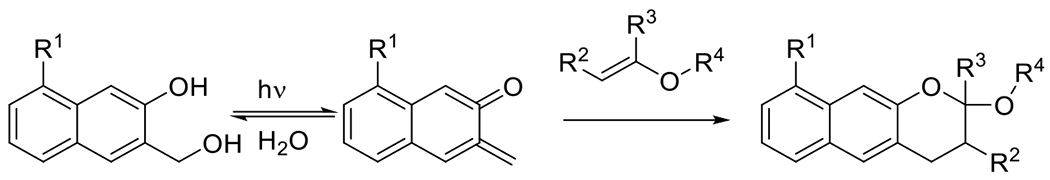
Photodegradation of 3-(hydroxymethly)-2-naphtol to 2-naphtoquinone-3-methide and subsequent IEDDA reaction with vinyl ether. 41
However, the product is not stable (Figure 37), because it contains acetals and hemiaminal ethers which are prone to hydrolytic degradation (Figure 38). While the acetal product shows relative stability under neutral pH aqueous conditions, it degrades in the presence of acid. The hemiaminal ether product is even more susceptible to hydrolysis. It should be noted that conjugation of R3 is maintained even following hydrolysis, indicating that appropriate design of the dienophile results in effective coupling despite subsequent hydrolysis of the product.
Figure 38.
Hydrolysis of IEDDA adducts proceeds in either acidic (acetal, top) or even neutral (hemiaminal ether, bottom) aqueous conditions.
As with other functional groups described previously (e.g. ring strained alkynes, nitrile imides) naphtoquinone methides react with nucleophiles (via a Michael-type addition) in alternative conjugation reactions. Thiols are particularly reactive with a reaction rate approximately five times that of the Diels Alder reaction (Figure 39). Given this preference, the naphtoquinone methide-thiol reaction has been described in a previous review as a “photoclick” chemistry.1 This reaction, however, is reversible in the presence of water and under exposure to the same wavelength of light that produces the naphtoquinone methide in the first place, whereas the Diels Alder reaction product is stable. When the photomediated reaction is conducted in the presence of both a vinyl ether and a thiol, the sulfide product is first produced in high yield, though under continued exposure the fraction of Diels Alder product increases until all sulfide is consumed. Arumugam et al. have taken advantage of this reversibility to photochemically pattern a thiolated surface by the selective conjugation and replacement of 3-(hydroxymethly)-2-naphtol derivatives.106
Figure 39.
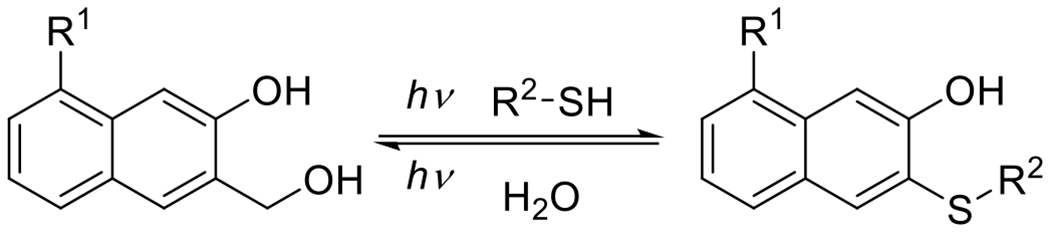
Naphtoquinone methides are also highly reactive toward thiols, though the reaction is reversible.106
Among the numerous examples of IEDDA, tetrazine reactions stand out for their specificity and high reaction rates. In 2016, Zhang et al. described the photomediated conversion of a dihydrotetrazine to tetrazine via photooxidation with methylene blue as the photocatalyst under irradiation at 660 nm. The tetrazines subsequently reacted with endogenous trans-cyclooctene in an IEDDA reaction.43 Truong et al. employed the same photooxidation of dihydrotetrazine in the presence of the norbornene functional group to accomplish the IEDDA coupling.42 In contrast to other IEDDA photoreactions, this strategy was able to take advantage of the long wavelength absorbance (600 – 900 nm) of the catalyst to conduct the reaction with tissue-penetrating red and NIR light and avoid wavelengths that could damage the more light-sensitive biomolecules. Employing this chemistry, the authors performed both polymer conjugation reactions and hydrogel crosslinking, as discussed in section 3.4.5 of this paper. Notable in this report was the relative thickness of the cure itself, 0.3 mm for rheometry experiments. While 0.3 mm may not seem like a particularly thick sample, in those reactions employing a one-photon, one event mechanism, even such thicknesses are often difficult to react thoroughly or uniformly. The employment of photocatalysts that are regenerated allows researchers to use lower concentrations of the light absorbing species and mitigate light attenuation. This reaction remains a one-photon, one event reaction, but in contrast to those IEDDA reactions in which the photochemistry is performed directly on the reactants, the concentration of chromophores is kept well below the reactant concentration.
Ring strained cycloalkynes may also participate in IEDDA reactions as dienophiles. While described previously in the context of photoSPAAC reactions, the photocaged, bisaryl cycloheptyne shown in Figure 41 was employed in a photomediated IEDDA reaction as a dienophile with a tetrazine moiety, with the reaction exhibiting a rate increase of an order of magnitude greater than that with the unsubstituted cyclooctyne.34
Figure 41.
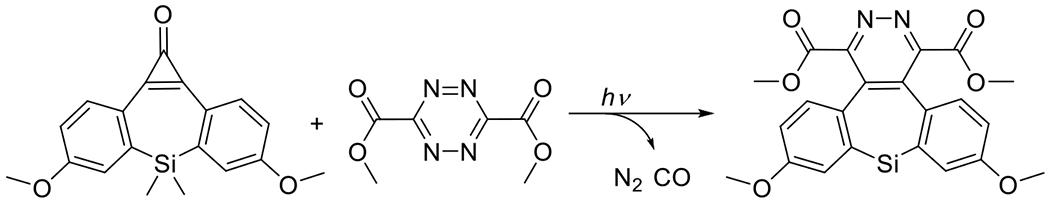
IEDDA reaction between tetrazine and photogenerated cycloheptyne dienophile.34
Another effective dienophile in IEDDA reactions with tetrazine derviatives is the ring strained trans-cyclooctene. While synthesis of trans-cyclooctenes from their cis-analogs is a viable and efficient production strategy, the conditions under which that photoisomerization takes place are typically not conducive to in situ generation.107, 108 One example that may show promise is the iridium(III) photocatalyzed isomerization of cyclooctene described previously (section 2.1.1.3).82 It is possible that employment of such a photosensitizer could facilitate an IEDDA reaction by in situ generation of a reactive dienophile, rather than the diene shown in the previous example.
2.3. Alternating Radical Propagation Chain Transfer Reactions
Radical chain addition reactions occur when a radical propagates across a pi bond, generating a new bond and a radical that subsequently propagates across another pi bond. This cycle continues until the radical is terminated by recombination or disproportionation. In contrast, radical alternating propagation chain transfer (APT) reactions result from the addition of a radical across a pi bond and subsequent atom abstraction by the resulting radical, regenerating the first radical species (typically intermolecularly).109 Examples of these reactions include thiol-ene,45 thiol-yne,110 iodo-ene,109 silane-ene,111 and phosphane-ene112 reactions. It should be noted here that the term “thiol-ene” and, to a lesser extent, “thiol-yne” are used in the literature to refer to both the radically-mediated reactions between thiols and their unsaturated reaction complements, as well as to the anionically-mediated reactions between thiols and Michael acceptors. To avoid confusion, the terms “thiol-ene” and “thiol-yne” will be used here to refer only to the radical-mediated reactions and “thiol-Michael” will be used to refer to the Michael-type reactions as has been done previously.46 As these addition/chain transfer cycles regenerate participating radical species, one initiating radical results in potentially hundreds or thousands of subsequent reactions. However, as radicals are prone to termination, a continuous source of reinitiating radicals is necessary to reach maximum conversion. As these reactions are radically-mediated, they may be initiated by nearly any method typically employed to generate appropriate radicals (e.g. thermally with a temperature sensitive initiator, via an electron beam, with a redox initiator, or with a photoinitiator to render the reaction light sensitive). Despite sharing a general mechanism (Figure 42), these distinct APT reactions may vary greatly in terms of efficiency and versatility.
Figure 42.
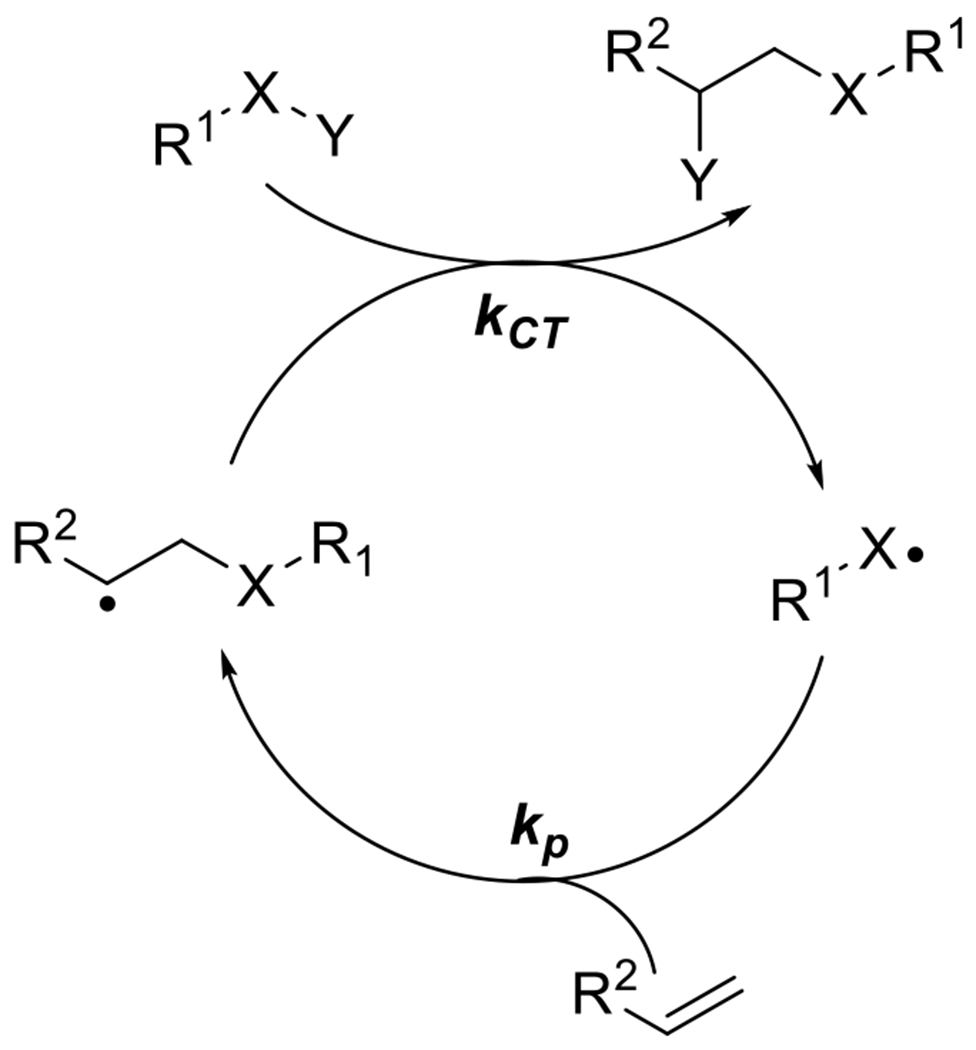
General mechanism for radical alternating propagation, chain transfer reactions wherein radical propagation across a pi bond results in generation of a radical that abstracts an atom from a donor molecule to generate the reaction product.
2.3.1. Thiol-Ene
While originally reported in 1905,113 the radical addition of thiols to alkenes has only in the last couple decades been recognized as a particularly powerful tool for the construction of carbon-sulfur bonds. Such conjugations are particularly important because organosulfur moieties are widely represented within natural products, pharmaceutical and polymeric materials.114–116 The utility of the reaction comes, in no small part, from the simplicity of its execution, mild reaction conditions, 100% atom economy, high yields and orthogonality with many other reactions.
The reaction frequently proceeds by the activation of a photoinitiator which abstracts a hydrogen radical from a thiol to form the thiyl radical, which adds across the alkene carbon-carbon double bond, forming a new carbon-sulfur bond and a carbon-centered radical. Abstraction of the hydrogen from another thiol regenerates the thiyl radical for the cycle to repeat (Figure 43). Alternatively, the initiator may produce a radical that adds across an alkene carbon-carbon double bond first, generating a carbon centered radical that then abstracts a hydrogen atom from the thiol.
Figure 43.

The general mechanism of the photoinitiated thiol–ene coupling, consisting of alternating radical propagation and chain transfer reactions, leading to a one-to-one addition between thiols and alkenes.46
The overall rate of reaction in the thiol-ene addition depends on the rate of propagation (rP with rate constant kp) and on the rate of chain transfer (rCT with rate constant kCT) with the overall rate (r) necessarily dictated by the slower of the two constitutive reactions-the rate limiting step-as described by Equation 6–Equation 8117:
| Equation 6. |
| Equation 7. |
| Equation 8. |
Although the thiol-ene reaction generally proceeds nearly quantitatively, the characterization of the thiol-ene coupling as a “click” reaction has been challenged due to poor yields in some polymer conjugation reactions under certain reaction conditions.118 Furthermore, while the relative orthogonality of the reaction has been touted in several reports, the potential reactivity of the nucleophilic thiols with a variety of other functional groups (e.g. Michael acceptors, halides, epoxides) must be a consideration in the application of the thiol-ene coupling reaction, albeit one that is greatly mitigated in the absence of base. Perhaps most significant is the reaction of thiols with themselves under oxidizing conditions to generate disulfides. Nevertheless, radical thiol-ene reactions have been used frequently for effective, selective conjugation reactions under a variety of reaction conditions,4 in water and open to air, and requiring as little as a few seconds for near quantitative conversions. It is the very high potential reaction rates that allow the thiol-ene reaction to be successfully employed as a click reaction, avoiding undesired competitive reactions. It should be noted that the efficiency of the thiol-ene reaction depends on multiple variables. Reaction parameters that play a particularly influential role in the efficiency of the thiol-ene coupling reaction include the selection of solvent, initiator, thiol, and alkene. This latter factor may not influence just the rate and ultimate conversion, but also the degree of participation in competing chain-addition reactions (e.g. acrylate homopolymerization).
The solvent in which the reaction occurs may influence the rate of thiyl radical generation. Although the thiol-ene reaction has been performed in a wide variety of solvents, there are few studies explicitly examining the solvent effects on the reaction.119 Munar et al. utilized conceptual DFT to model the reactivity of the carbon and sulfur radicals in a thiol-ene reaction in different solvents to elucidate the role of solvent on the propagation and chain transfer steps. The authors noted that solvent ultimately has a negligible influence on the sulfur-centered radical, thus exhibiting little effect on propagation rates. 120 The chain transfer step, however, was significantly more sensitive to the reaction environment, purportedly due to stabilization of the carbon-centered radical intermediate in the presence of a polar solvent.
2.3.1.1. Radical Initiation
Thiol-ene reactions have been initiated via a variety of means. Thermal and redox radical initiation strategies have been employed to conduct thiol-ene couplings. Thiol-ene reactions without traditional radical initiators have also been carried out by generating hydroxyl radicals upon sonication in water.121 Typically it has been photoinitiated reactions that have been most advantageous in the application of the chemistry.4 While the use of photoinitiators to facilitate the reaction is the most commonly employed practice, the photoinitiated reaction can be conducted without initiator by direct photolysis of reactants. In this way the thiol-ene photoreaction between alkenes and mercaptopropionates has been initiated with both 254 nm and 365 nm light exposure, despite very low absorbance of reactants at that higher wavelength. In such reactions, however, considerably higher light intensities are required and the rates of reaction are considerably lower than in those reactions employing photoinitiators. As described subsequently, however, employment of aryl substituents greatly improves the absorption characteristics of thiols in a thiol-ene reaction to allow more effective initiatorless reaction.122 The selection of photoinitiators is one of the most consequential in determining the overall rate of reaction in all APT as well as in the previously described photoinitiated photoCuAAC reactions. Therefore a thorough understanding of the available initiators and their salient features is recommended for successful application of such reactions in the various contexts in which they may be employed.
Norrish type I photoinitiators such as dimethoxyphenylacetophenone (DMPA), and bis(2,4,6-trimethylbenzoyl)-phenylphosphineoxide (BAPO), are the most frequently used class of initiators in thiol-ene and other APT reactions. These initiators, upon absorption of photons, decompose into two or more radical fragments that initiate the reaction as previously described. The initiation rate in a radical polymerization of an optically thin film where the light intensity is nearly constant throughout the film depth is described by Equation 9.
| Equation 9 |
In this expressions ϕ is the quantum yield of radical generation, f is the efficiency or the fraction of those radicals that initiate reaction, ε is the molar extinction coefficient at the wavelength of exposure, Io is the intensity of light exposure, [In] is the concentration of photoinitiator, NA is Avogodro’s number, and hν is the photonic energy. It is important to highlight the difference between quantum yield of photolysis and efficiency as not all radicals are equally likely to initiate the reaction and so some radical initiators are better or more poorly suited to thiol-ene reactions regardless of their respective quantum yields. Uygun et al. evaluated a series of photoinitiators and thermal initiators for initiation of thiol-ene reactions and concluded that the type I or cleavage type initiators were most effective. A particularly interesting demonstration of the relative efficiencies of radicals generated by different photoinitiators is the case of hexaarylbiimidazoles (HABI), which are poor initiators for (meth)acrylate chain addition reactions, but effective in thiol-ene reactions.123 Moreover, in contrast to most radicals generated by photodecomposition of an initiator, HABI-based radicals are moderately stable, recombining over the course of several minutes, permitting significant dark cure. HABI is also noteworthy in its high absorption at longer wavelengths of light allowing initiation with 469 nm exposure. Most type I initiators absorb primarily in the UV region of the spectrum, although phosphine oxide,124 acyl germane,125 and acylstannane126 photoinitiators (see Figure 44) have extended the absorption spectra of cleavage type radical initiators progressively deeper into the visible spectrum past 500 nm. Despite these advances, use of acylgermane and acylstannane photoinitiators remains somewhat uncommon.
Figure 44.
Structures of representative radical photoinitiators that may be used in thiol-ene and other radical APT reactions: 1) hydrophobic type I photoinitiator, 2,2-dimethoxy-2-phenylacetophenone (DMPA); 2) hydrophobic, type I, bis(2,4,6-trimethylbenzoyl)-phenylphosphineoxide (BAPO, I819); 3) hydrophobic type I photoinitiator benzoyltrimethylgermane (BTG); 4) hydrophobic type I photoinitiator tetrakis(2,4,6-trimethylbenzoyl)stannane; 5) hydrophilic, type I, 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (I2959); 6) hydrophilic, type I, lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP); 7) hydrophobic type I hexaarylbiimidazole (HABI) photoinitiator; 8) hydrophobic, type II, benzophenone, 9) hydrophilic type II initiator, eosin Y.
Emerging applications of thiol-ene reactions frequently require performing the reaction in an aqueous environment. While most commercial radical photoinitiators are intended for use in hydrophobic environments and are accordingly poorly water soluble, a few hydrophilic initiators, such as lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) and 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone, have been successfully applied to the aqueous thiol-ene reaction (Figure 44).127–133
As alluded to previously, direct photolysis of aryl thiols allows for the generation of thiyl radicals with milder wavelengths and intensities. The use of disulfides as type I photoinitiators has been well established.134 The photoactivation of thiols has received less attention. Such strategy results not only in the direct coupling of aryl thiols to alkenes, but also in the photoamplified coupling of other thiols as well, serving the function of effective photoinitiators. Love et al.122 evaluated the initiating potential for various aromatic thiols, demonstrating near quantitative conversions with divinyl ether under irradiation with modest intensity (10 mW/cm2) of 405 nm visible light. Reactions reached completion in as little as 5 to 10 minutes. The same aromatic thiols were effectively employed to initiate reactions between norbornenes and mercaptopropionates in dilute aqueous reactions. Ortho and para substituted mercaptobenzoic acids and trifluoromethyl thiophenols were particularly effective at initiating the reaction.
One particularly interesting type I, thiol-ene initiating system was developed by Li et al. who attached oxime-ester coumarin groups to the surface of upconverting nanoparticles.135 Upon absorption of NIR light at 974 nm, these nanoparticles would, through a combination of FRET and photon upconversion, cause cleavage of the oxime ester bond resulting in the release of phenyl radicals to initiate bulk thiol-ene reaction. The authors compared this release of reactive radicals to the dispersion of dandelion seeds (see Figure 45).
Figure 45.

Initiation of thiol-ene reactions with NIR exposure is accomplished by release of initiating radical from upconverting nanoparticles, which, upon absorption of photons, cause the decomposition of the pendant oxime ester coumarin via a combination of FRET and photon upconversion.135
Type II initiating systems, in which the initiator, from an excited state, undergoes a multistep electron transfer reaction with a coinitiator (frequently an amine) to generate the initiating radical species, are also frequently used in thiol-ene reactions. In this case, the thiol itself may serve as a coinitiator, donating a hydrogen atom to the excited chromophore, obviating any need for the presence of other coinitiating species. Substantial variability in initiator efficiency when employing type II initiators has been reported, however, with benzophenone, for example, and its derivatives providing for efficient reactions but camphorquinone considerably less so.136, 137
As cleavage type radical photoinitiators typically absorb in the UV and shorter wavelength visible spectrum, in order to conduct the radical thiol-ene reaction at higher wavelengths, other initiating strategies have been and continue to be developed. This can be accomplished by the use of transition metal photoredox catalysts as type II photoinitiators. Upon exposure to visible light, the thiol-ene reaction proceeds by the formation of a strong metal-ligand charge transfer excited state, which undergoes reductive quenching by a thiol to generate the thiyl radical cation. The deprotonation of the radical cation then generates a thiyl radical which then participates in the thiol-ene reaction cycle as described previously. For example, the most efficient ruthenium catalysis for the thiol-ene click reaction, using Ru(bpz)3(PF6)2, was found to occur in the presence of an excess of thiol; however, bulkier thiols such as cyclohexyl and tert-butyl mercaptan required longer reaction times.132 Addition of p-toluidine, which exhibited thiol-activating capabilities, either via direct hydrogen atom abstraction or sequential electron- and proton-transfer steps, to afford the chain-propagating thiyl radical species, improved the rate of reaction by circumventing the slow direct photooxidation of thiol with Ru*(bpz)32+.132, 138 Various other metal based photoredox catalysts, like, Ru(bpy)3Cl2, fac-Ir(ppy)3, and Zinc tetraphenylporphyrin (Zn-TPP) have been similarly exploited, but are often associated with low catalytic efficacy and lower conversion, which has led researchers to explore a metal-free organic photoredox catalyst, Eosin-Y.138,139,140 An organic photocatalyst, 9-mesityl-10-methylacridinum tetrafluoroborate designed by Zhao et al. resulted in efficient thiol-ene conjugation involving biomolecules.127 Although this visible-light-mediated photoredox catalysis under neutral conditions resulted in high yield and few side products, it did require long irradiation times (1–24 h) and high light intensities.
2.3.1.2. Thiol Influence on Rate
The dependence on the rate of reaction of the thiol’s molecular structure, although incompletely examined, is significant. Among the most frequently used thiols in thiol-ene reactions are mercaptopropionates, mercaptoacetate, alkyl and aromatic thiols. Mercaptopropionates are more reactive than mercaptoacetates which, in turn, are more reactive than aliphatic thiols. This relationship is attributed to the weakening of the S-H bond by intramolecular hydrogen bonding between the thiol proton and the carbonyl that is facilitated by the favorable formation of a six-membered structure in mercaptopropionates and similar thiols.141, 142 Such hydrogen bonding would be considerably less effective in mercaptoactates and absent entirely from the alkyl thiols.
While secondary and tertiary thiols are infrequently used in thiol-ene reactions, their employment may provide advantages in terms of formulation stability. Varying the thiol substitution from primary to secondary to tertiary has substantial effect on the reaction kinetics due to steric hindrance, though the effect itself is highly dependent on the rate limiting step of the reaction, which varies from alkene to alkene.143 As increased sterics increase the activation energy more significantly for the chain transfer step, relative to the propagation step, those reactions that are limited by the rate of chain transfer (e.g. reactions with allyl ethers) are particularly affected. However, under most commonly used conditions for thiol-ene reactions, secondary thiols generally perform adequately. Examination of rates in more dilute conditions, however, have not been reported. While the degree of substitution of the thiol has been shown to affect the rate of chain transfer, other evidence indicates that the propagation step can also be negatively influenced by electron withdrawing group substituents proximal to the thiol.144
2.3.1.3. Alkene Influence on Rate
Apart from initiator, the most widely explored influence on reaction rate may be the selection of an alkene. Early experimental evaluations of the rates of reaction with various alkenes indicated that ring strained (e.g. norbornenes) and electron rich (e.g. vinyl ethers) alkenes reacted most rapidly, while electron poor alkenes such as acrylates and maleimides react more slowly (Figure 46).145 Subsequently propagation rate was shown to be directly correlated to the electron density of the alkene, with the chain transfer rate being inversely proportional to the stability of the carbon-centered radical.146 Computational modeling studies have elucidated the relationship that exists between alkene structure and the activation barrier to propagation and chain-transfer. This kinetic analysis demonstrates the importance of the stability of the carbon-centered radical intermediate, which exhibits direct influence on the chain-transfer activation barrier, as well as the reversibility of the propagation step. It is evident that a small change to the alkene structure would significantly impact the rates of propagation and chain-transfer, as well as the stability of the carbon-centered radical intermediate, which would then directly influence the relative rate constants kP and kCT.147,148
Figure 46.
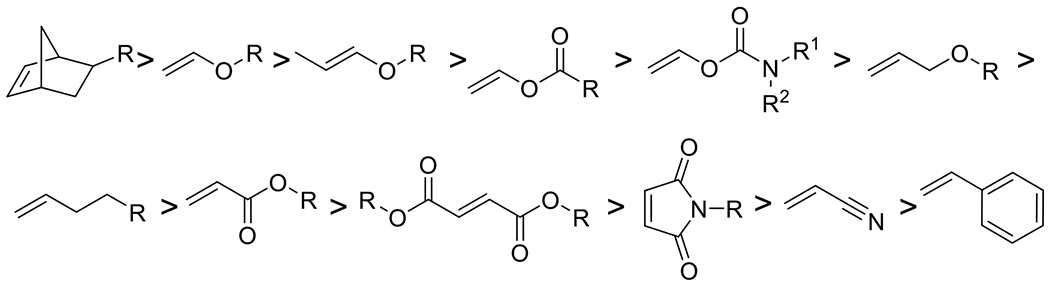
Relative reaction rates of various alkenes as determined experimentally and reported by Roper and Hoyle.145
There are other factors that may influence the rate of the thiol-ene coupling reaction. The presence of radical inhibitors or retarding agents, as would be expected, may interfere with the initiation or other steps of the reaction. Interestingly, however, inhibitory effects of oxygen (a considerable problem in acrylic radical chain addition reactions) is muted in most thiol-ene couplings. Additionally, in contrast to thiol-Michael additions, thiol-ene reactions proceed quite rapidly in neutral or acidic conditions. Thiol-ene coupling reactions can also be performed in mildly basic conditions, though deprotonation of the thiol precludes effective chain transfer, so the reaction will not proceed in strongly basic environments. Moreover, even small quantities of generated thiolate can result in the formation of metastable disulfide radical anion species that effectively retard the thiol-ene reaction.149 Because their generation requires the presence of thiolate anions, the propensity for base-mediated retardation of the thiol-ene reaction is largely dependent on the pKa of the respective thiols as well as the strength and concentration of the base in the system. In general, the thiol-ene reaction is extremely accommodating to a variety of reaction conditions.
2.3.2. Thiol-yne
The radical thiol-yne reaction is analogous to the thiol-ene click chemistry although considerably less well explored. Recently the reaction has received significant attention, particularly in its suitability for synthesis of dendrimers, modification of peptides and protein as well as for surface functionalization. An important and distinguishing feature of the thiol-yne reaction compared to thiol-ene reactions, is the ability of one equivalent of the alkyne to react with two equivalents of thiol, resulting in a double addition product with 1,2-regioselectivity. As with the radical thiol-ene reaction, radical thiol–yne reactions may be performed open to ambient atmosphere, and in aqueous conditions and, like other click reactions, it can be performed at room temperature with relatively high reaction rates. The radical thiol-yne reaction proceeds similarly to two consecutive thiol-ene reactions (Figure 47) and is initiated similarly to the thiol-ene reactions. An initiating radical is generated that abstracts a hydrogen atom from a thiol, resulting in the formation of thiyl radical. The sulfur centered radical adds to the carbon-carbon triple-bond, resulting in the formation of a carbon-centered vinyl sulfide radical. Abstraction of a hydrogen from another thiol (chain-transfer) yields the intermediate vinyl sulfide and regenerates the thiyl radical. The vinyl sulfide subsequently undergoes a second addition of thiyl radical yielding a carbon centered radical which then undergoes a second chain transfer reaction with thiol producing the double addition product and another thiyl radical. Fairbanks et al. demonstrated that for the reaction of mercaptopropionates with aliphatic alkynes the second addition of a thiol to the vinyl-sulfide was approximately three times faster than the initial thiol-yne reaction150 an observation that has subsequently been computationally validated.148 This relationship (i.e. the rate of the second addition exceeding the rate of the first) was observed for other thiol-yne pairs, though not universally; reactions with methyl propiolate and propargylamine showed slower thiol-vinyl sulfide reactions than initial thiol-yne reactions. It was also shown that the ring strained cyclooctyne efficiently and quickly undergoes addition by thiyl radical, as a consequence of releasing ring-strain associated with the cyclic alkyne species. However, the addition of a second equivalent of thiol to the cyclic vinyl-sulfide is restricted due to steric hindrance.47, 151 Additionally, the spontaneous reaction between the cyclooctyne and thiol were observed in the absence of any light exposure indicating limits to the orthogonality of both the thiol-yne and SPAAC reactions. Structures of various alkynes with their relative order of reactivity towards mercaptopropionate are shown in Figure 48.
Figure 47.
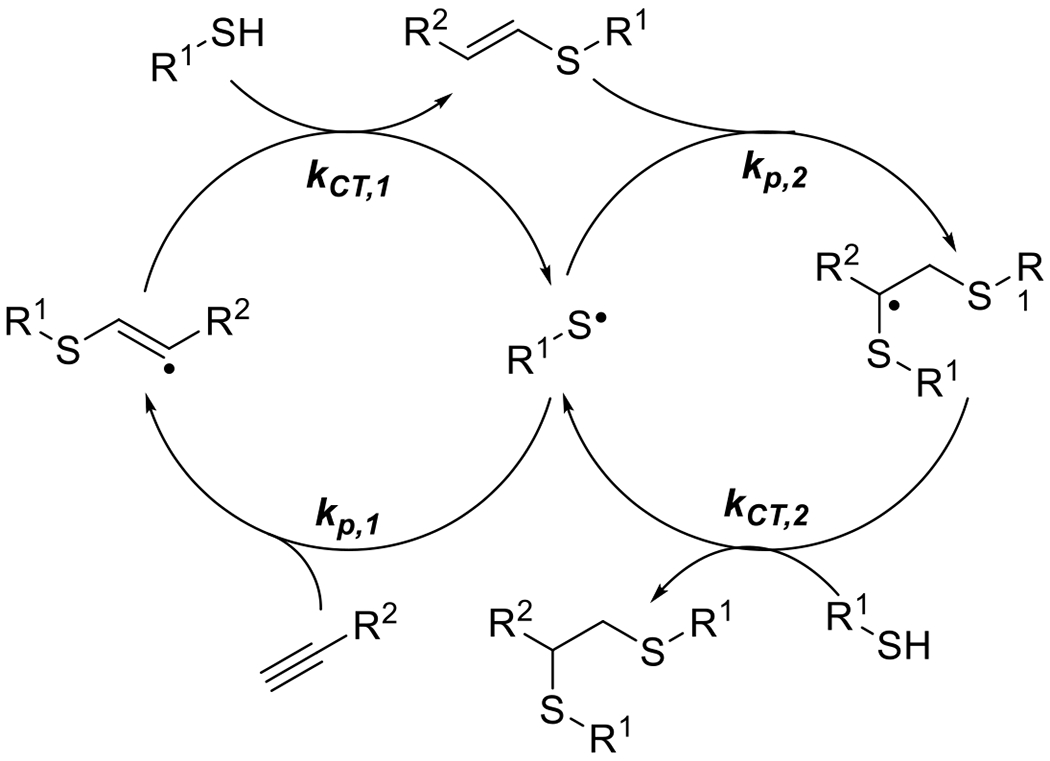
The simplified mechanism of the radical thiol–yne coupling reaction consisting of sequential propagation, chain transfer cycles.150
Figure 48.

Order of alkyne reactivity toward mercaptopropionates. Notable is that while the initial addition rate is fastest in the ring strained cyclooctyne, subsequent addition was not observed.36
Similar to thiol-ene reactions, thiol-yne chemistry has been successfully accomplished using UV and visible light photoinitiators which is the most commonly employed methodology. Kritchenkov and co-workers implemented an initiator-free thiol-yne click reaction using the ultrasound-catalysis to develop antibacterial chitosan derivatives, but only one substitution of thiol was observed, though the authors do not speculate as to why this is the case.152 Similarly, upon use of photoredox catalyst monoadduct vinyl sulfide products predominate, usually as a mixture of E:Z anti-Markonikov form.153,154 One plausible explanation for the reaction regioselectivity and stereoselectivity is the concerted effects of single electron oxidation of the thiol moiety from the excited state of Eosin Y and the steric influence of the substituent. However, the organic photocatalyst, 9-mesityl-10-methylacridinum tetrafluoroborate produced diadducts when less sterically hindered thiols were used, providing corresponding adducts in good yields.155
Like the thiol-ene reaction, the thiol-yne reaction can be performed open to the air and presumably is similarly affected by the presence of base that results in the generation of disulfide radical anion retardants. While examination of the effects of the thiol substitution on reaction rate have yet to be conducted, given the chain transfer rate dependence of the reported thiol-yne reactions, it may be speculated that secondary and tertiary thiols would have adverse effects on the overall reaction rates.
2.3.3. Other APT Reactions: Iodo-ene, Silane-ene and Phosphane-ene
Thiols are particularly effective chain transfer agents, making them especially suited to APT reactions. Other chain transfer moieties, however, may also participate in such similar mechanisms. Although these reactions are generally not considered “click” reactions, they warrant mention, in no small part because their examination and applications have been so heavily informed by the study of thiol-ene couplings.
One such example is the iodo-ene reaction wherein iodo compounds add radically to alkenes, the iodine atom filling the role in chain transfer that the hydrogen atom fills in thiol-ene reactions. Obstacles to the wide adoption of the iodo-ene reaction include the relatively poor reactivity of non-fluorinated iodocompounds and the propensity for side reactions of the iodides generally.156 The presence of fluorine adjacent to iodide results in the enhancement of the chain transfer of iodine due to the electron withdrawing ability of the fluorine. Amongst many other perfluoro derivatives, perfluoroiodide has been observed to exhibit high efficiency and reactivity. For example, perfluorohexyl iodide exhibits better efficiency as transfer agent when compared to 1,1,2,2-tetrafluoro-3-iodopropane, 1,1,1,3,3-pentafluorobutane which resulted in lower conversion.157–159 Though not widely used, the iodo-ene reaction imparts various advantageous properties, depending on the desired application, to materials thus formed due to the presence of the fluorine atoms and conservation of the iodo moiety, which can participate in subsequent reactions. 160,161
The silane-ene reaction shares many of the features of the other APT reactions described previously including its insensitivity towards oxygen and the same one-to-one reactivity of the thiol-ene and iodo-ene reactions.111, 162, 163 However, the relatively high bond dissociation energy of Si-H bonds disfavor chain transfer reactions to the silane resulting in relatively slow reactions. This drawback can be overcome by employing silanes with low bond dissociation energies but availability of such suitable silanes is severely limited.
Finally, one more reaction that should be noted is the radical reaction between a primary or secondary phosphine and an alkene.112, 164 Via a mechanism similar to but inverted from that of the thiol-yne reaction, primary phosphines may react consecutively with two alkenes. The limited availability of primary phosphines and their sensitivity to air may prevent more frequent employment of this reaction, although air stable phosphines have been synthesized by Guterman et al.165 and polymerized via photoinitiation with a phosphine oxide initiator.
2.4. Photoinduced Click Reactions Based on Thiolate Nucleophiles
In addition to the radical mediated thiol-ene click reactions, thiols are particularly effective functional groups for nucleophilic addition click reactions.45, 46, 166–168 Chemically, the high reactivity of these soft nucleophiles towards various complementary reactive moieties, high conversions in short time and their broad substrate scope make these click reactions superior over other click chemistries. The very same feature makes them susceptible to multiple simultaneous reactions and thus may be unsuited for some orthogonal strategies. Since the reactivity of thiolate nucleophiles varies based on the nature of the thiolate, a fine tuning of the reactivity is also possible by wise choice of the substrates. Among the nucleophilic thiolate mediated click strategies those which are interesting to both chemists and biologists are the thiol-epoxy, thiol-isocyanate and thiol-Michael additions and are discussed in the following sections with particular emphasis on the versatile photoinduced thiol-Michael click reactions (Figure 49).
Figure 49.
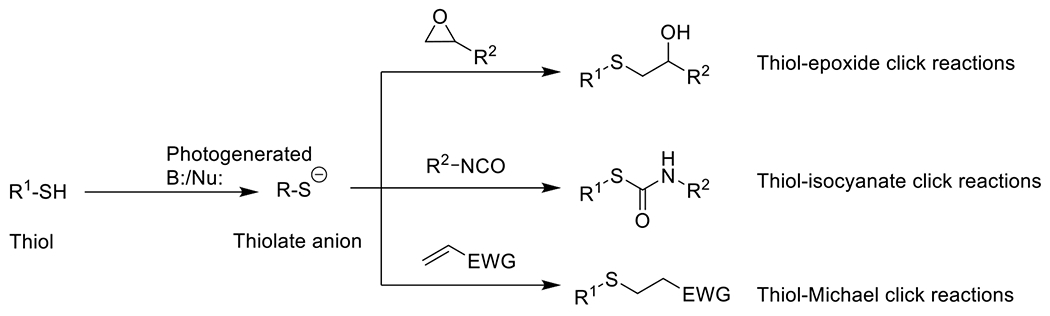
Various photolatent thiolate nucleophile mediated click reactions.
2.4.1. Photolatent Base/Nucleophile Catalyzed Thiol-Epoxide Click Reactions
Addition of a thiolate nucleophile to a strained ring such as oxirane is among one of the most prominent thiolate-mediated click reactions known.49, 166, 169 In general, these ring opening reactions are catalyzed by appropriate strong bases that are capable of deprotonating the thiol more efficiently. In order to achieve spatial-temporal control, several photobase generators have been developed and their performance in various thiolate mediated click reactions have been extensively studied in recent years.170, 171 Ideally an efficient photobase generator should have a suitable chromophore for good absorption at a desired wavelength and should be able to produce active species in high yield upon photoexcitation. Thioxanthone,172 trans-o-coumaric acid,173 o-acyloxyime,174 pthalimidocarbamates,175 α-ketocarbamates,176 and o-nitrobenzyl carbamates177 are a few among the well-known chromophores used for this purpose. In general, highly efficient photobase generators are often necessary to achieve the thiolate mediated click reactions efficiently. However, there are very few reports on the photobase generators that are capable of releasing robust and stable tertiary amine bases suitable for nucleophilic ring opening click reactions.172, 178, 179 For example, Salmi et al. developed a series of photobase generators in the form of a quaternary ammonium salts of phenylglyoxalate that are capable of releasing strong bases such as DBU, DBN and TMG upon exposure to UV light via photodecarboxylation and were successfully applied in the photocrosslinking of epoxide-based resins.178 Thus, by using an appropriate base, near quantitative conversion could be easily achieved in these reactions without forming any byproducts. Tetraphenyl borate salts of bicyclic guanidinium is another family of short-wave photobase generators that are capable to generate a much stronger base than the strongest base DBU.180 Upon photoexcitation, the BPh4− ion undergoes rearrangement which can abstract a proton from the neighboring TBD•H+ cation to release the free base (TBD) (Figure 50). The trivalent arylborane then decomposes further to aromatic biproducts.
Figure 50.
Proposed mechanism of the photogeneration of TBD from TBD.HBPh4. Adapted, with permission, from reference 180. Copyright 2008 American Chemical Society.
An interesting feature of the base catalyzed thiol-epoxide click reaction is that the reduced volumetric shrinkage which are caused by the reduction in free volume during the formation of covalent bonds.181 Thus, the thiol-epoxy/thiolacrylate hybrid photopolymer produced using a combination of free radical photoinitiator (ITX) and photobase generator (TBD.HBPh4) system exhibited low shrinkage along with enhanced mechanical and physical properties upon increasing the thiol-epoxy content.182 In another example, Chen et al. used a photogenerated TBD base for delayed photopolymerization of the thiol-epoxy resin to demonstrate an efficient photopolymerization of pigmented or thick composites.183 In their work, the thiol-epoxy resin formulated with photoinitiator (ITX) and photobase generator (TBD.HBPh4) system was exposed to UV to release the active tertiary amine base at room temperature. After the release, the resin mixture was mechanically stirred to cause the diffusion of the active base species and then heated at elevated temperature to complete the thiol-epoxy click polymerization. Since no polymerization occurred at room temperature, the liquid resin formulation could be further processed to the desired size and shape.
The general mechanism of photoinduced thiol-epoxide click reactions involve the deprotonation of a thiol by a photogenerated strong base followed by ring opening of the epoxide via the nucleophilic addition of the thiolate anion on the less hindered and most electrophilic site on the epoxide ring. As the secondary alkoxide is the most basic component generated, it autocatalyzes the polymerization via a proton transfer from a new thiol to the secondary alkoxide producing a new thiolate anion for subsequent reactions. During this process the secondary alkoxide is quenched by a proton transfer from the thiol to the alkoxide resulting in the quenching of the alkoxide to a secondary hydroxyl group. Reaction rates depend on the reaction conditions and the nature of the thiol and epoxide structure (Figure 51). However, the absence of any acidic protons or strong nucleophiles like a thiol in the system results in homopolymerization of the epoxy groups. The structure of the polymer backbone is thus determined by the proton transfer to alkoxide ion as reported by Frèchet and coworkers and coined the name “proton transfer polymerization.”184–186
Figure 51.
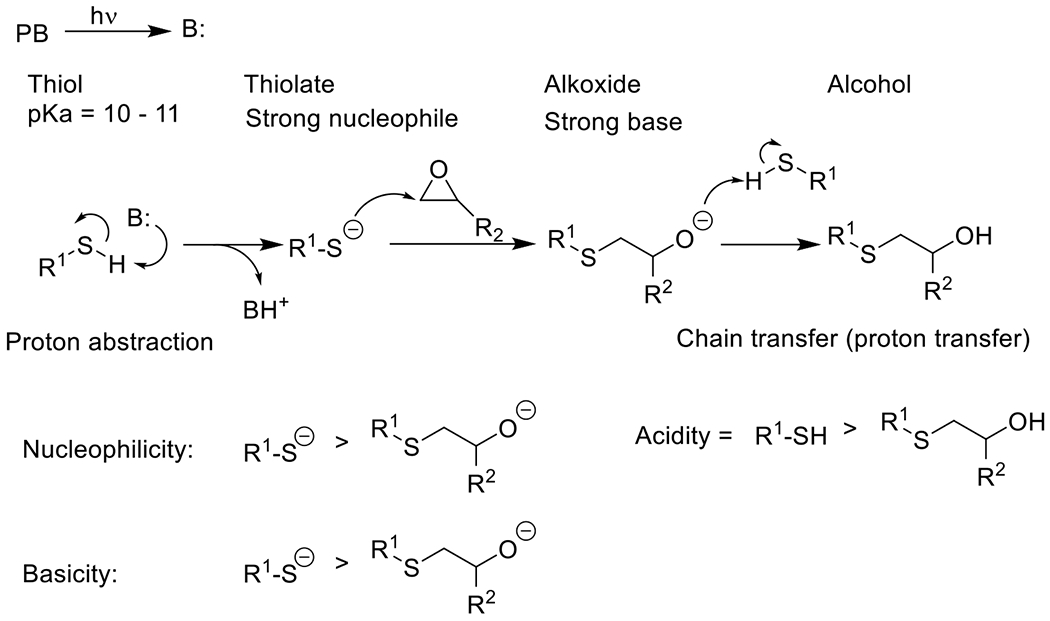
Mechanism and functional group reactivity consideration for base catalyzed thiolate mediated epoxide ring opening click reaction. Figure adapted, with permission, from reference 187. Copyright 2020 American Chemical Society.
The key benefit of using the thiol-epoxide click reaction in micro and nanopatterning is the reduced polymerization induced shrinkage, providing dimensional stability especially at lower length scales. Recently, Khan et al. demonstrated the photoinduced proton transfer thiol-epoxide polymerization for micro and nanopatterning using a photolabile salt prepared by the supramolecular complexation of a guanidine base, DBU with benzoylphenyl propionic acid (ketoprofen).187 The authors have successfully demonstrated the thiol-epoxide ring opening polymerization and further polymer modification for micro and macro patterning. The imprinted features were further modified with fluorescent dyes via the AgBF4 mediated alkylation of thioether linkages.
Another key benefit of thiol-epoxide based click reaction is the formation of β-hydroxythio-ether groups with high adhesion properties which further enable the postpolymerization modification for polymer surfaces. Coupled with photolabile o-nitrobenzene chemistry Romano et al. used photoclick thiol-epoxide reaction to alter the bulk and surface properties of thiol-epoxy based network polymers. For this purpose, the authors used a difunctional epoxy monomer containing o-ntirobenzylester groups in combination with multifunctional thiol to form the thiol-epoxide network. The thiol-epoxide ring opening was initiated with photo latent 1,5-diazabicyclo-[4.3.0]non-5-ene (DBN). To avoid the interference of the photoexcitation of the base with o-NB absorption, the photosensitivity of the base was further extended to the visible region by incorporating the photosensitizer ITX in the resin formulations. Nearly complete conversion of the thiol and epoxide functionalities was achieved upon exposure to the visible light at 400 nm without any damage to the imbedded o-NB groups. After the network formation the wettability, solubility and the cross-link density of the bulk film were altered via the photo cleavage of the imbedded o-NB groups which upon UV exposure cleaves the ester linkage to release the carboxylate groups.188
In summary, the photolatent base catalyzed thiol-epoxide click reaction is certainly a valuable tool for researchers working in both chemistry and biological fields. The commercial availability of a large number of small molecule epoxides and thiols and their ability to reach higher conversion in both aqueous and solvent free conditions make them a frequently employed chemistry for biomedical applications,189–191 suggesting a potential role for the photomediated reaction. Moreover, the enhancement in material properties such as low polymerization shrinkage and strong adhesion, coupled with spatio-temporal control achieved by photochemical methods and tolerance towards radical reactions are the key benefits of the photoinduced thiol-epoxide click reaction. However, the requirements for highly efficient photobase generators and the nature of thiol structures are few hindrances to this approach.
2.4.2. Photolatent Base Catalyzed Thiol-Isocyanate Click Reactions
Thiourethanes, the sulfur analogue of urethane, are another prominent class of high yielding click reactions that does not form any byproduct.166, 192 By using appropriate catalysts such as tertiary amines or tryalkyl phosphines, excellent conversions are achieved in short times with no other side reactions. The incorporation of thiourethane groups in the network polymer results in polymeric materials with significantly enhanced material properties through extensive hydrogen bonding interactions. Other key benefits of using thiourethane click chemistry are the significantly higher refractive index and enhanced uniformity leading to a narrow mechanical transition than the corresponding polyurethanes which makes them suitable for various optical materials development.50 The mechanism of photoinduced thiol-isocyanate click reactions follows a two-step process identical to that of the radical thiol-ene click reaction involving the attachment of photogenerated thiolates with carbonyl carbon followed by a chain transfer to a thiol or any proton donating species in the system (Figure 52).
Figure 52.
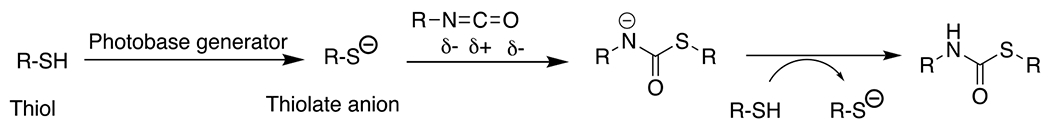
Mechanism of the photolatent base-catalyzed thiol-isocyanate click reaction.
The reaction kinetics of the thiol-isocyanates click reaction are typically fast with primary and secondary thiols adding across aryl isocyanates, but at a slower rate with alkyl isocyanates.166 Another advantage of using the thiol-isocyanate click reaction is the orthogonality of the isocyanates group towards radical polymerization conditions. This key feature has been used in formulating segmented linear polymers193 and network polymers194 via the sequential radical thiol-ene and thiol-isocyante click chemistries and more recently in two stage holographic photopolymers195 and surface modification of functional polymeric surfaces.196 In one recent example reported by Shin et al. fine tuning of the poly thiourethane network was achieved when thiol-isocyanate click polymerization was initiated by using photolytically generated tributylamine.51 The photolytically generated tertiary amine was able to initiate the thiol-isocyanate click reaction reaching nearly quantitative conversion in minutes to form highly elastic polymers with varying cross-linking densities. Furthermore, these materials showed excellent energy damping performance due to a narrow glass transition coupled with the extensive hydrogen bonding interactions.
To summarize, the photoinduced thiol-isocyanate click reaction is an emerging, highly efficient click reaction that has large potential in both synthetic chemistry and biology fields due to its fast reaction kinetics, excellent conversion and orthogonality towards radical reactions. Moreover, their ability to achieve higher refractive index than comparable polyurethane analogues makes them most suitable for optical materials development. However, the competing reactivity of isocyanates in protic media and low shelf life stability might be the few shortcomings of this click strategy.
2.4.3. Photolatent Base/Nucleophile Catalyzed Thiol-Michael Addition
Although many photolatent base/nucleophile mediated click reactions found in the literature are effective in thiol-epoxide, thiol-isocyante reactions, they have limited potential in thiol–Michael addition click reactions mediated small molecule synthesis. The reason for this lack of preference is presumably due to the undesired formation of radical intermediates during the photoinduced dissociation that trigger competitive radical reactions or homopolymerization of activated alkenes such as the common acrylate Michael acceptors.
The Michael addition click reaction involves the facile and selective addition of a nucleophile to an α,β-unsaturated carbonyl, resulting in a Michael adduct with a wide variety of C-X bonds such as C-C, C-N, C-S and C-O.197, 198 Among these, the Michael addition of a thiolate anion to the α,β-unsaturated carbonyl is considered one of the most explored and widely implemented click reaction particularly in network polymerization, polymer functionalization and bioconjugation.166, 168, 199 Unlike the radical mediated thiol-ene reaction, the thiol-Michael addition requires an activated carbon-carbon double bond typically employing an electron withdrawing group in conjugation with the double bond. The ability to perform this robust reaction with high efficiency under simple, highly regio-specific, high yielding conditions qualifies the criteria for nearly ideal click reaction. The thiol-Michael addition click reaction occurs typically through one of two distinct pathways: traditional base catalyzed or nucleophile catalyzed. In the base catalyzed reaction protocol, a base such as Et3N is typically used to deprotonate the thiol generating a thiolate anion. The strongly nucleophilic thiolate anion then reacts with the electron deficient carbon-carbon double bond producing a carbanion intermediate followed by subsequent deprotonation of next thiol or the conjugate acid of the base leading to propagation.199 Whereas in the nucleophilic mechanism, the nucleophile first attacks the electron deficient carbon-carbon double bond generating a carbanion which then deprotonates the thiol instigating the reaction cycle (Figure 53).
Figure 53.
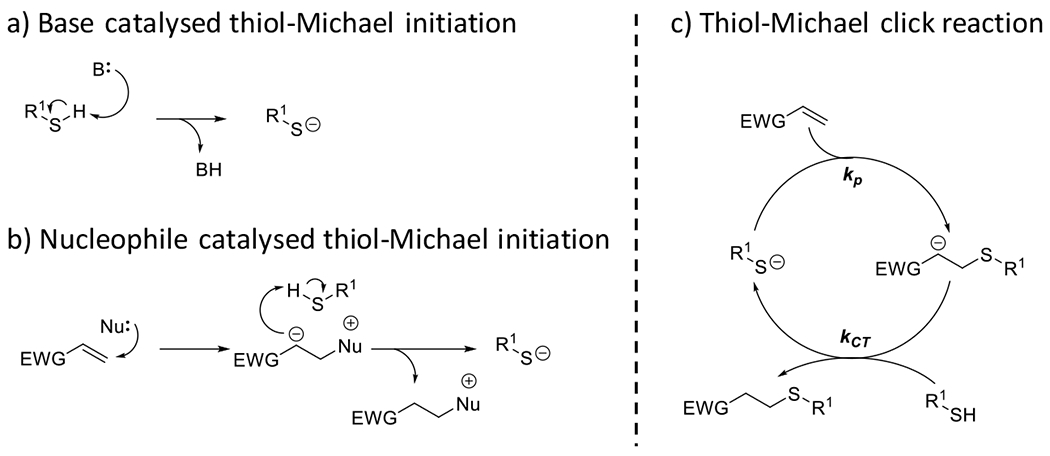
Mechanism of a) base and b) nucleophile catalyzed c) thiol-Michael addition click reaction cycle. In the case of photomediated thiol-Michael addition, the base species, B:, is most often generated by photodecomposition of a photolatent base.
The efficiency of the reaction depends on the nature of the thiols, electron deficient bonds and chemical environment such as solvent polarity and strength and concentration of the base or the nucleophile catalyst.200, 201 The reactivity of the thiols may vary based on their pKa and the chemical environment. Similarly, the structure of the ene also plays an important role in the kinetic properties of the click reaction. It has been found that the more electron deficient C-C double bonds are more susceptible to Michael addition click reactions (Figure 54).168, 202 Apart from these prominent electron deficient vinyl functionalities, aklynones203 have also emerged as efficient and promising Michael acceptors for multiple thiol-Michael addition click reactions. Quite recently, Van Herck et al. explored the utility of this chemistry as a versatile crosslinker for the thiol-yne Michael addition based covalent adaptable polymer network.204 The kinetic model studies using small thiol-Michael-yne system revealed that the exchange rates are significantly varied depending on the steric and electronic nature of the alkynone cross-linker. Moreover, this thiol-Michael-yne system has a large potential for photoinduced thiol-yne dynamic systems in the near future.
Figure 54.
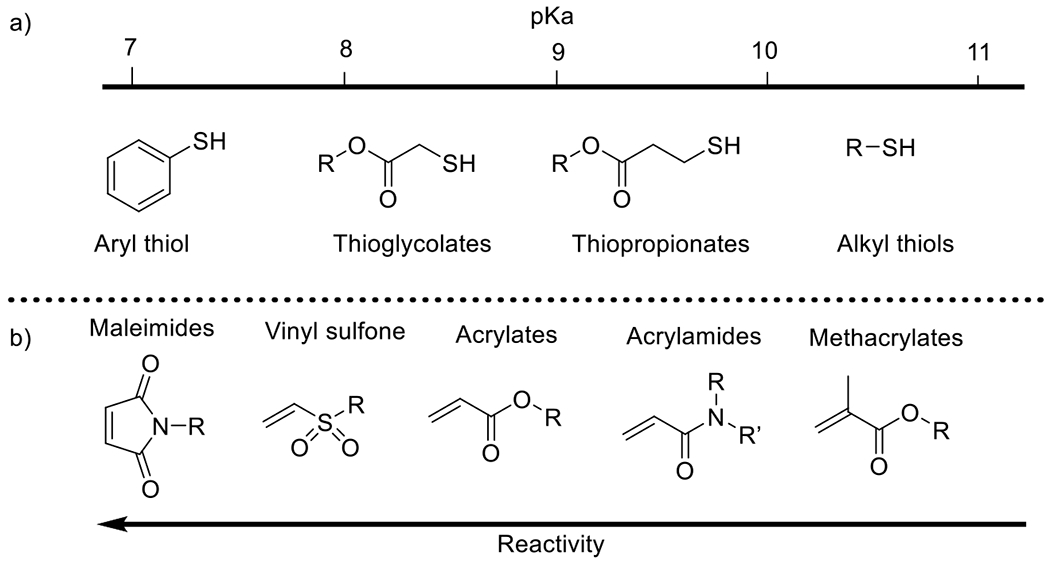
a) pKa values of commonly used thiols in Michael addition click reaction, b) Reactivities of commonly used vinyl groups in thiol-Michael addition click reactions. 152, 186
A general kinetic mechanism supported with mathematical modeling for non-radical 2-(2-nitrophenyl)propyloxycarbon tetramethyl guanidine (NPPOC-TMG, see Figure 55) mediated thiol-Michael click reaction, GDMP and ethyl vinyl sulfone model monomers showed an increase in the reaction kinetics upon increasing the light intensity and/or initial photobase concentration.205 The reaction kinetics were also found to be more sensitive to the intensity changes at low light intensity when compared to the changes in light intensity at high intensities. Furthermore, minor variations in the initial photobase concentration have a significant impact on the reaction kinetics. These modeling studies indicate that the overall polymerization kinetics follow a pseudo-first-order reaction rate equation in base and coreactant concentrations, determined by kp/kCT which in turn depends on the rate limiting step. One of the key features of such photobase catalyzed step-growth thiol-Michal addition click reactions is its unique living nature enabling an extended dark-cure reaction, resulting from the lack of radical mediated termination.
Figure 55.
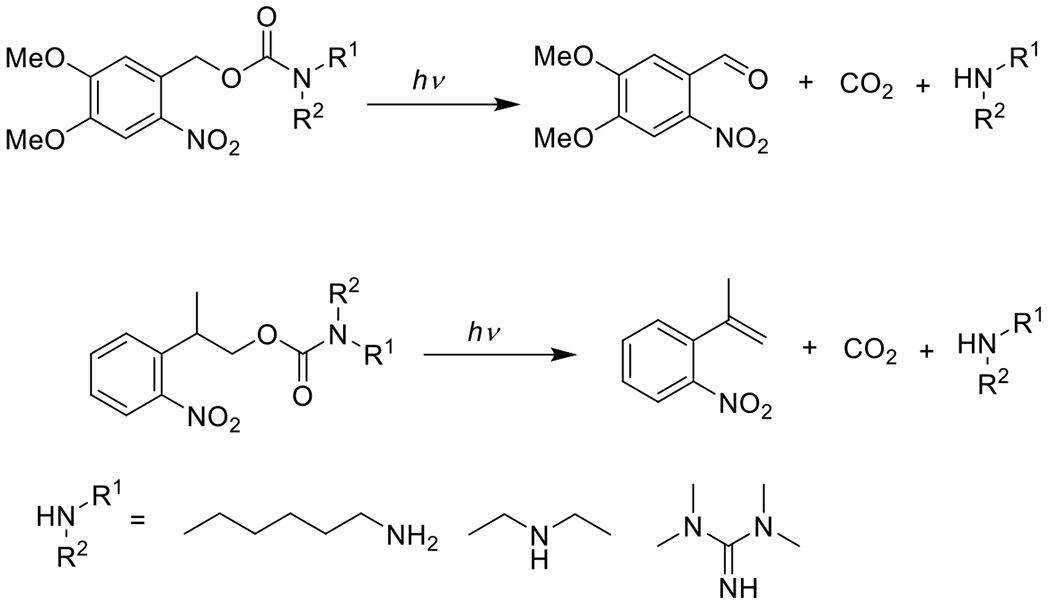
Photolytic Pathway of NVOC–Amine and NPPOC–Amine Compounds.206
Although there exists earlier reports on photo latent base induced thiol-epoxide click chemistry, the first photo triggered thiol-Michael click chemistry was introduced by Xi et al. in 2013.52 In their work, 2-(2-nitrophenyl)propyloxycarbonyl (NPPOC) caged photolabile primary amine (hexylamine) was used as the nucleophilic catalyst. Upon irradiation with 320–390 nm UV light, the photocleavage of the NPPOC group released hexylamine via an aci-nitro intermediate mediated pathway. The released hexylamine initiated the thiol-Michael addition click reaction of thiol glycolate and methyl acrylate model monomers efficiently with up to 90% conversion in 30 min. Real time FTIR revealed that the photoinduced catalysis proceeds in two stages with a delayed upturn nearly at 6 min, which is presumably attributed to the slow formation of hexylamine and anionic intermediates at the initial stages. Furthermore, this photoinduced strategy was utilized in the formation of network polymers by using multifunctional thiols and acrylates. A year later the same authors expanded the photocaged amines catalysts with more efficient photolabile species.206 For this purpose a series of NVOC and NPPOC photocaged amines (Figure 55) was synthesized using hexylamine, diethylamine and TMG as bases. Among those, amines caged with NPPOC exhibited higher quantum yields than that of NVOC caged ones presumably due to the quenching of released amines with the redox byproduct o-nitrobenzaldehyde of NVOC upon irradiation.
The thiol Michael addition of the model compounds n-butylthiolglycolate and ethyl acrylate using photolabile TMG exhibited over 90% conversion within several minutes, exceeding that observed with photoinitiation with latent primary and secondary amines. This behavior was consistent with the higher pKa of TMG. Additionally, the spatial and temporal control of these catalysts was demonstrated in photopatterning and kinetically controlled two stage polymer network formation. It should be noted that resolution in photopatterning was achieved by precluding diffusion of catalyst in rapidly cured, glassy networks. Photopatterning would presumably be less effective in rubbery and low crosslink-density materials.
Further to these investigations, Chatani et al. reported visible light initiated thiol-Michael addition polymerization using ITX/tetraphenylborate system in combination with catalytic amounts TEMPO (Figure 56).53 In this approach, ITX that possess longer wavelength absorption (>400 nm) was used as a photosensitizer to push the performance of PGBs to longer wavelengths via the triplet energy transfer from ITX to PGBs. Such energy transfer process and the degradation products of tetraphenylborate often involve the generation of radical intermediates causing the unwanted radical mediated acrylate-acrylate chain growth homopolymerizations to occur.182, 194
Figure 56.
Photosensitization system for photobase-catalyzed thiol-Michael additions with photosensitizer (ITX) photolatent base (TBD-HBPH4) and radical scavenger (TEMPO) to mitigate homopolymerization of acrylate Michael acceptor.53
The incorporation of catalytic amount of the radical inhibitor, TEMPO, in combination with the photo initiating system resulted in significantly reduced radical reactions.
Coupling strong bases such as TMG with visible light responsive protecting groups such as coumarin derivatives, serves as a highly efficient photobase generator for thiol-Michael and other thiolate anion mediated click reactions. In 2016, Zhang et al. explored the visible light induced photolysis of coumarin-TMG photo base and successfully demonstrate its catalytic activity in thiol-Michael addition click reactions in small molecules as well as in polymer settings (Figure 57).207
Figure 57.
Proposed photolysis mechanism of Coumarin-TMG upon visible light irradiation.207
This photobase generator with a quantum yield of 0.044 was able to catalyze the Michael addition of various monothiols and DVS to nearly quantitative conversions upon irradiating with 400–500 nm light in a very short period of time (as little as 5 minutes). Moreover, the thiol-Michael photopolymerization of PETMP and DVS showed faster reaction rates by using coumarin-TMG photobase compared to that of coumarin-hexylamine analogue which is attributed to the difference in the basicity of the released base.208
One major challenge associated with the photobase induced click reaction is that low quantum yields result in a low concentration of photobase generated via dissociation. One approach that developed to circumvent this shortcoming is the photo triggered base proliferation system.209 The base proliferation mechanism involves autocatalytic decomposition of a base amplifier, triggered by the catalytic amine generated from a photobase generator, resulting in a dramatic increase in the base concentration. In 2017 Xu et al. reported the use of NPPOC caged hexylamine and Fmoc protected hexyl amine as a photobase and base amplifier, respectively.210 Upon irradiation with 365 nm UV light, the n-hexylamine liberated from NPPOC-hexylamine triggered the base proliferation reaction. Thus, the liberated hexylamine nucleophile diffused further into non-irradiated regions to activate base proliferation and effectively catalyze the thiol-Michael addition reaction of monothiols with acrylic acid to achieve >90% conversion.
Another interesting aspect of the photobase induced thiol-Michael addition click reaction is the possibility for dark curing, i.e., the continuation of the click reaction or curing process even after the light is turned off. Unlike radicals which usually have very short life times due to recombination and disproportionation, the released base/nucleophile remains active throughout the completion of the reaction and therefore facilitates high conversions, even following termination of light exposure. To this end, Sinha et al. demonstrated an effective, light-activated dark curing possibility of photo triggered base amplification in thiol-Michael click addition (Figure 58).208 In their work, various combinations of photocaged amine (NPPOC-amine) and base amplifier (Fmoc-amine) were evaluated for the thiol-Michael addition reaction between various thiols and two different Michael acceptors, an acrylate and a vinyl sulfone, and exhibited noticeably higher final conversions for photoinduced thiol-Michael addition reactions. Furthermore, this enhancement in reaction kinetics and conversion was implemented in the preparation of crosslinked network polymers with efficient dark cure rates with limited light exposure.
Figure 58.
Mechanism of Phototriggered Base Proliferation System for Initiating the Thiol-Michael Addition Reaction.208
In summary, photoinduced thiol-Michael click reactions proceed with high efficiency and follow the majority of the criteria recommended for an ideal click reaction, excellent conversions and fast reaction kinetics. The reactivity of various substrates used in this click reaction can be finely tuned by varying various parameters such as the nature of the thiol, electron beneficent vinyl groups, the light intensity and pH. However, implementation of the photoinduced thiol-Michal addition is challenging due to undesired photogeneration of radical species along with the intended base catalyst, which often leads to homopolymerization of electron deficient carbon-carbon double bonds such as acrylates and, in biological applications, may adversely affect sensitive biomolecules. The future development of effective and selective photolatent bases and nucleophilic catalysts is therefore an area of continued research.
2.4.4. Click Reactions Based on Photolatent Thiols and Other Nucleophiles
2.4.4.1. Photolatent Thiol Mediated Michael Addition Click Reactions
Photomediated thiol-Michael addition can also be facilitated and controlled by employing photoremovable protecting groups or caging groups that have been developed to mask specific click functionalities such as thiols.211–213 Upon irradiation, with light of suitable wavelength, controlled release of highly nucleophilic thiolate anions is achieved followed by subsequent clicking of the released thiols with their reactive complements (Figure 59). Unlike other photoclick reactions described herein, these reactions frequently release relatively large byproducts from the photodecomposition of the photoprotected thiol. Tetrazole-alkene reactions release only nitrogen, sydnone-alkene reactions release carbon dioxide and cyclopropenone-based photoSPAAC reactions release carbon monoxide. By contrast, photodeprotection of thiols may release compounds with molecular masses in the hundreds. As the criteria for click chemistry requires the generation of easily removed and inoffensive byproducts, it is justified to question the categorization of such reactions as “photoclick.” Their description is included here because nucleophilic Michael additions were among the original click reactions described by Kolb et al.2 and in the applications in which these photodeprotection Michael reactions are employed, (e.g. in the context of macromolecular coupling and surface modification), the photocleaved byproducts are frequently small relative to the reactants and either easily removed or tolerated.
Figure 59.
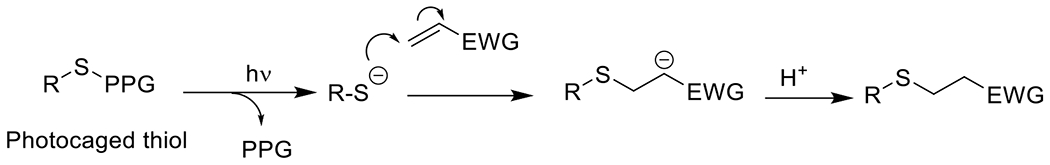
Photouncaging thiol and subsequent thiol-Michael addition click reaction.
The use of photocaged thiols has emerged as an interesting strategy particularly in the field of biomaterial development and photopatterning. Due to its high nucleophilicity and reactivity towards various click reactions, researchers preferred thiols as photolatent functionality over ubiquitous amine and relatively less reactive alcohols.212
The first and the most widely used photo protecting group for most of the click functionalities including thiols was the o-nitrobenzyl derivatives that was developed by Barltrop et al. in the 1960’s.214 These caging groups can be cleaved upon irradiation with UV light at 365 nm and has been proposed as a versatile caging group since the 1970’s.215 The photorelease mechanism of o-nitrobenzyl protected groups has been studied by many researchers using time-resolved spectroscopy on the parent o-nitro toluene and several other o -nitrobenzyl derivatives.216–219 The primary photoreaction mechanism for the release of o-nitrobenzyl protected functional groups proceeds via aci-nitro tautomer intermediates in the ground state formed by the hydrogen transfer from the o-alkyl substituent to the nitro group. The decay of these aci-nitro transient intermediates follows a biexponential rate law due to the formation of both tautomers and vary strongly with substitution, solvent, and pH in aqueous solution.218 The thus formed o-quinonoid intermediate undergoes cyclization to N-hydroxybenzisoxazolidine followed by a ring chain tautomerism to release the caged functionality and nitrosobenzaldehyde byproduct (Figure 60).220, 221
Figure 60.
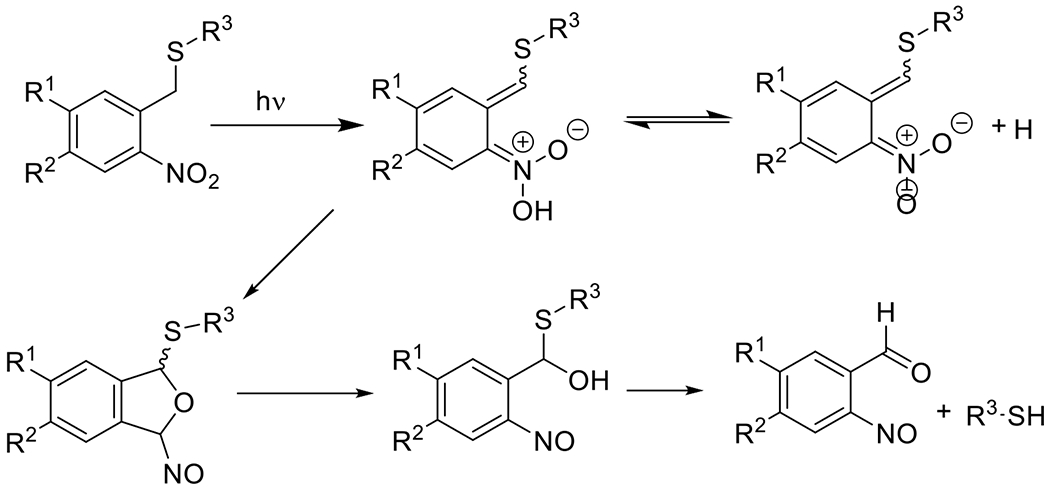
Although there exist several variations in o-nitrobenzyl caging groups with varying substituents on the benzene ring as well as on the benzylic methylene, the generic form of this photoprotection group (PPG) with no substitution is usually preferred for thiols in which the leaving thiol is directly attached to the benzylic site. However, for alcohols and amines the carbonic acid derivatives are preferred as this forms the better leaving group and is easily protected by using commercially available chloroformates. One of the key features of this deprotection-click strategy is that its orthogonality, where the deprotection and the click reaction could be performed in the presence of other catalysts that could be used in the next stage. This versatile deprotection strategy has been used to achieve excellent spatial and temporal in the development advanced biomaterial with tunable material properties.222, 223
Recently, Pauloehrl et al. explored the feasibility of this highly efficient strategy in ambient temperature polymer end functionalization and backbone modification.54 The authors attempted the deprotection of ONB capped thiol end functionalized PEG methyl ether in the presence of initiators/catalysts such as α,α-dimethoxy-α-phenylacetophenone (DMPA) for radical thiol-ene and triethylamine (TEA) and dimethylphenylphosphine (DMPP) for thiol-Michael click reaction. Although a complete retardation of thiol-uncaging was observed when the deprotection was carried out in the presence of TEA at 320 nm UV light, a quantitative formation of the desired thiol-end capped species was achieved in the presence of catalytic amounts of DMPA or DMPP with no noticeable disulfide formation. Moreover, the authors were able to demonstrate a nearly complete backbone modification of an ATRP derived polyacrylate containing thiol protected pendent groups.
Although o-nitrobenzyl derivatives are by far the most commonly used photocaging groups for thiols and other click functionalities, the photolysis of these compounds forms potentially toxic and strongly absorbing byproducts such as o-nitrosobenzaldehyde. Another major drawback of this strategy is the formation of nitrosobenzaldehyde derived side products via the condensation of released amines with nitrosobenzaldehyde. The brown-colored byproduct formed via the decomposition of nitrosobenzaldehyde acts as an optical filter by absorbing the incident light. Moreover, these conventional photocaging groups were found to be inefficient in releasing the substrates and lack two photon sensitivity.55, 224 In this context, the coumarin-derived photocaging groups discovered by Roger Furuta et al. that could be efficiently cleaved by two photon irradiation have become more popular in recent years.225
A general uncaging mechanism of coumarin derivatives starts with the relaxation to the lowest excited singlet state (S1) 1[CM-X]*. The decay of the 1[CM-X]* singlet state involved three different processes: fluorescence, non-radiative processes and a competing heterolytic C-X bond cleavage to form an ion pair intermediate of coumarinylmethyl cation and leaving group conjugate.226, 227 Subsequent separation of the base ion pair by polar solvents followed by the trapping of coumarinylmethyl cation with the polar solvent leads to a new stable coumarylmethyl product and along with uncaged functional groups (Figure 61). Time-resolved absorption studies showed that the heterolytic bond cleavage is the fastest among the most rapid photo release with a rate constant reaching 2 × 1010 s−1. However, the recombination of the tight ion pair which leads to the regeneration of the ground-state caged derivative occurs with a reaction rate of 2.3 × 109 s−1 and is 10 times faster than the hydrolysis rate of the separated ion pair by polar solvent molecules.228 It has been found that the rate of heterolytic bond cleavage and therefore the efficiency of photo release depends on the stability of the singlet ion pair formed. If both the components of the singlet ion pair are stabilized, a significant weakening of the C-X bond occurs during the excitation, which in turn depends on the nature of the substituent at the 6,7 position of the coumarin group. Therefore, the substitution of the coumarin ring with electron donating groups to stabilize the cation and the stabilization of released anion by lowering the basicity should enhance the heterolytic cleavage and therefore the uncaging efficiency. In addition, the factors that accelerate the heterolysis also retard the recombination process. For the same reason the poor leaving groups such as alcohols, phenols, and thiols facilitate heterolysis. In such cases more efficient release is achieved through the introduction of the carbonate linkages. Heterolysis of these moieties give initially unstable carbonate or thiocarbonates which undergo decarboxylation to give free alcohol or thiol.
Figure 61.
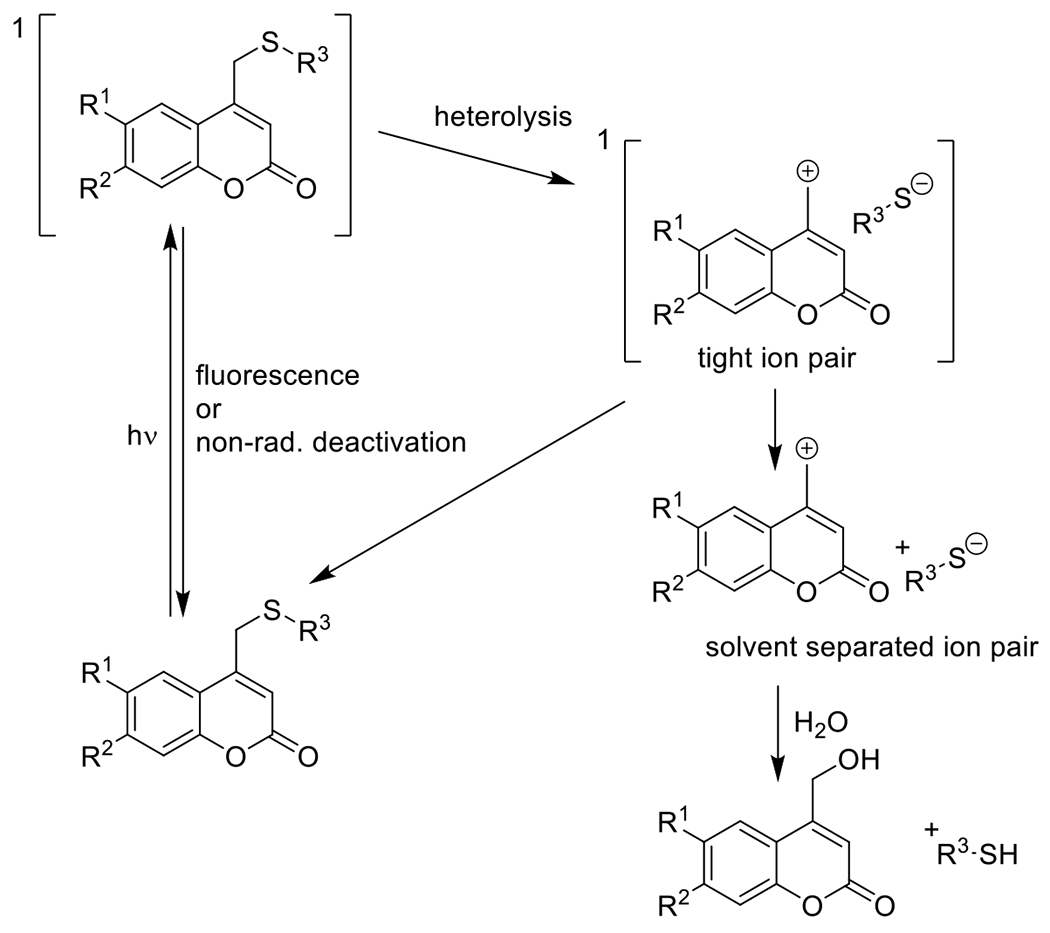
General uncaging mechanism of coumarin derived PPGs.210, 211
Some of the key features of the coumarin based photocaging are the large molar absorption coefficients at longer wavelengths and rapid release rates. To improve its absorption maximum, stability and water solubility, researchers made several modifications at C6 and C7 substituents on the coumaryl group. Among these, the 6-bromo, 7-hydroxy coumarin was found to be the most efficient for photocaging thiols suitable for thiol-Michael type click reaction.
In an interesting inversion of the photoclick paradigm, the nucleophilic thiol-Michael addition may be used to allow subsequent photochemistry to take place. The high regioselectivity of thiol-Maleimide click reaction coupled with the fluorescence quenching ability of maleimide has been used to control the photocleavage of the coumarin phototrigger.229 The phototrigger design involves a photo induced electron transfer quencher (maleimide) coupled to 7-amino coumarin dye (Figure 62). In the absence of any thiols, the PET takes place from coumarin to the conjugated maleimide moiety deactivating the S1 exited state preventing photocleavage.230, 231 The Michael addition of a thiol to an electron deficient maleimide restores the fluorescence and photocleavage pathways, unlocking the photo trigger to release the click functionality (thiol). The photo release of the thus released biotin was monitored by HPLC and UV–vis absorption and fluorescence emission. The authors also examined the target activation using various biologically relevant compounds including amino acids as routine.
Figure 62.
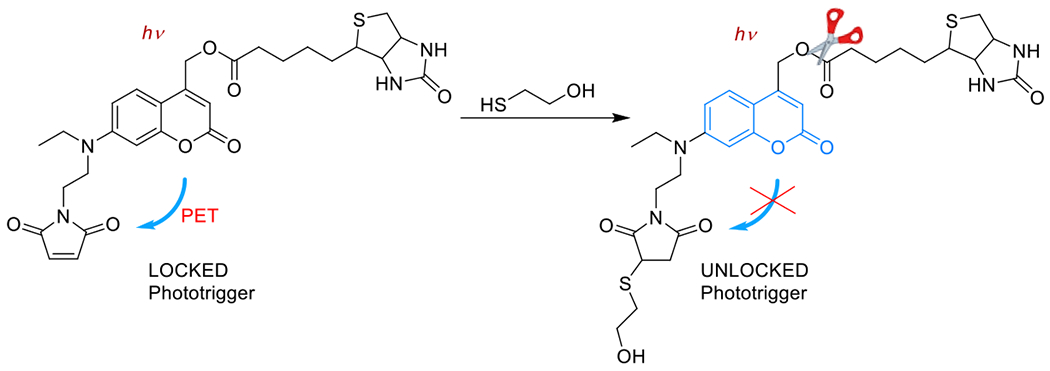
Activation of a locked phototrigger via the reaction with thiol to form an unlocked phototrigger. The reaction of the maleimide substituent with β-mercaptoethanol prevents PET quenching of the phototrigger, allowing subsequent photochemistry to occur. Adapted, with permission, from reference 229. Copyright 2012 American Chemical Society.
Although the coumarin based thiol protecting groups such as 7-amino coumarin and 6-bromo-7-hydroxycoumarin are potentially useful and easy to synthesize, their uncaging efficiency is often hampered by the undesired photoisomerization. To address this issue, Distefano et al. developed 6-bromo-7-hydroxy-3-methylcoumarin-4-ylmethyl as an alternative coumarin-based caging group for thiols, in which one of the hydrogens at the C-3 position was replaced by a methyl group. Based on the photoisomarization mechanism, the presence of alkyl substituent at the 3-position would render the photoisomer formation by blocking the re-aromatization of intermediate 1 and the migration of sulfur to migrate to the C-3 position due to the syn pentane type interaction between the sulfur and the C-3 methyl group (Figure 63). The one- and two-photon photo uncaging efficiency demonstrated by using mBhc-caged thiol containing peptide showed a clean and efficient photo-cleavage upon irradiation without any detectable formation of the photoisomer.55
Figure 63.
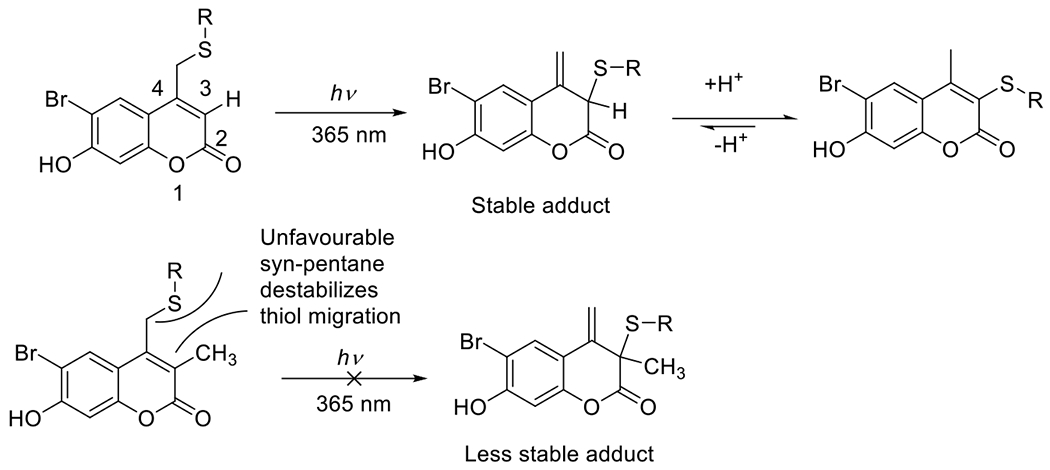
Illustration of potential effects of C-3 substitution on photoisomerization process.55 A methyl group installed in the C-3 position improves photolysis by preventing the migration of the thioether.
Nitrodibenzofuran (NDBF) is another thiol photocaging molecule that does not undergo photoisomerization and is capable of uncaging thiol-containing peptides efficiently with one- and two-photon irradiation.232 Recently, Fisher et al. compared photuncaging efficiency of NDBF, Bhc, and mBhc and demonstrated the superior efficiency of NDBF biomolecule photopatterning.233
Bimane is another interesting compact photocaging group with qualities that are advantageous for biology related studies such as good water solubility, high stability in biological media and low toxicity to cells. Photocaging of the thiol group is achieved by efficient reaction of bromobimane in the presence of a base. The high reactivity of bromobimane towards free thiols has been used for fluorescent tagging of proteins.234–236 Recently Truong et al. utilized this thioether bimane photouncaging strategy for the crosslinking polymerization of multiarm-PEG functionalized with various Michael acceptors such as acrylates, maleimide and propargyl esters.237 Upon irradiation with visible light at 420 nm, efficient release of anionic thiols was achieved which spontaneously react with various Michael acceptors efficiently in biological media without the requirement of an additional catalyst (Figure 65). Crosslinking of the photouncaged multiarm thiol occurred even at a pH of 6.9 indicative of the anionic form of the released thiol.
Figure 65.
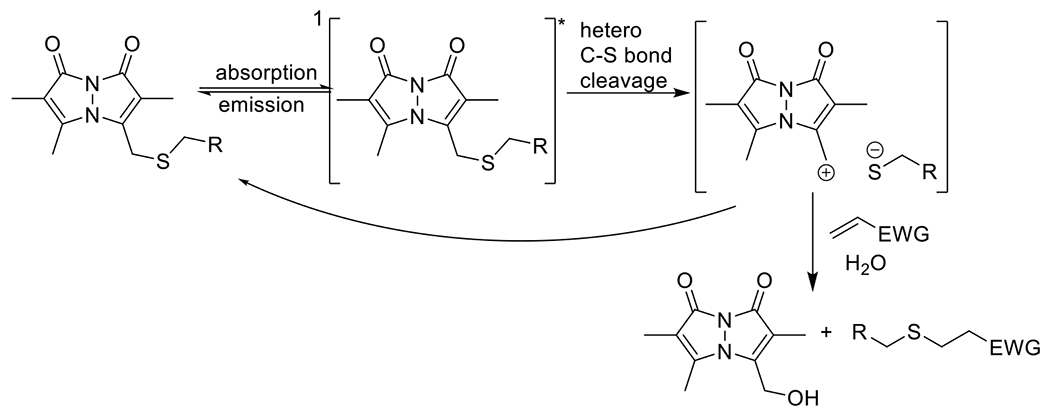
Mechanism for the photo-Uncaging of thiol-bimane in the presence of an electrophile.237
The temporal control of the bimane thioether photodeprotection strategy was further demonstrated by stopping and resuming photo-uncaging and trapping process by turning the light on and off. Spatial control over the conjugation process was demonstrated by photopatterning the biotin-maleimide and was confirmed by the specific reaction of biotin with TRITC-streptavidin.
Photoinduced thiol-isocynate click reactions have also been achieved via use of photocaged thiol. Hensarling et al. employed photocaged thiol strategy to enable postpolymerization modification of pendent thiol polymer brushed using thiol-isocyanate click chemistry.238 Upon irradiation with UV light the photodeprotection of caged thiols released thiolate anions for subsequent reaction with small molecule isocyanates for surface modification of RAFT derived polymer brushes.
Modification of coumarin photocaging groups with an electron rich styryl moiety at the 3-postion enabled further enhancement in long wavelength abortion, two photon absorption and quantum yields resulting in rapid photocleavage.239 This enhancement was attributed to the large π-conjugated structure offered by the styryl moiety and stabilization of the carbocation intermediate by electron donors para to the styryl group. A comparative study of these styryl modified PPGs displayed an uncaging quantum yield of 0.19-0.7, which is approximately 3 to 10 times higher than that of the parent diethyl aminocoumarin (0.07). After the photolysis, the carbocation intermediate undergoes intramolecular cyclization rearrangement forming a five membered ring (Figure 66). As a result, the conjugation at 3-postion is blocked allowing the byproducts to photobleach after the photolysis. These modified PPGs are therefore well suited for high concentration samples and thick specimens. The role of electron donating and electron withdrawing substituents on the styryl groups were supported by Density Functional Theory (DFT) calculations showing that the electron-rich effect significantly stabilizes the carbocation intermediate and enables the styryl conjugated coumarin to undergo intramolecular cyclization rearrangement to enable faster photolysis. In addition, a futher enhancement in the photolysis rate was observed for the thione-modified PPGs due to the enhanced intramolecular charge-transfer through the 3d empty atomic orbitals of the sulfur atom.
Figure 66.
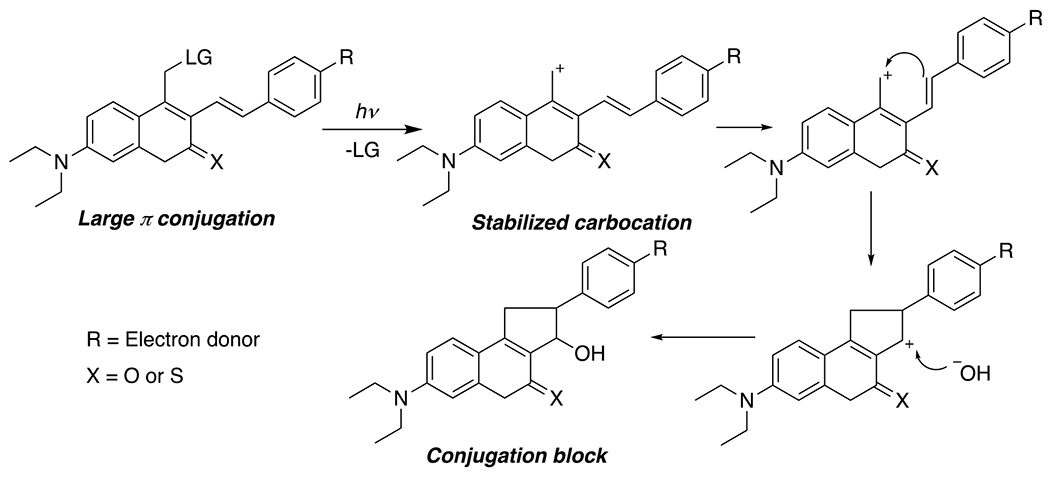
Proposed photolysis mechanism for the electron donor-styryl-conjugated coumarin-based PPGs as reported in reference.239
While discussed previously in connection with the photoclick IEDDA reactions, the photoreversible Michael addition between o-naphthoquinone methide and thiols bears mentioning here as well.106 In contrast to the previously described photomediated thiol-Michael additions, it is the Michael acceptor that bears the chromophore and is generated by absorption of photons rather than the thiol.
2.4.4.2. Other Photolatent Nucleophiles Mediated Click Reactions
Photouncaging strategies have also been used extensively with other functionalities such as aldehydes and amines for subsequent photomediated coupling reactions. One such reaction is the oxime ligation that involves the reaction between an alkoxyamine (−ONH2) and an aldehyde (–CHO) or ketone to form oxime linkages.240, 241 The rare occurrence of these two complementary click functional groups in living organisms provides excellent bioorthogonality under physiological conditions. Moreover, the kinetics of oxime ligation are easily controlled by the pH and catalyst concentrations.240 Two main photouncaging approaches have been employed to afford spatiotemporal control over such ligation involving the photoinduced liberation of one of the complementary reactive species. In the first approach in situ photogenerated o-nitrobenzaldehyde from o-nitrobenzyl alcohol was reacted with free hydrazines or amines.242, 243 For example, in 2016, Azagarsamy et al. reported the utilization of phototriggered hydrazone chemistry for the formation of cytocompatibile hydrogels using mutliarm-PEG postfunctionalized with hydrazine and o-nitrobenzyl alcohol functionalities. (Figure 67).242
Figure 67.
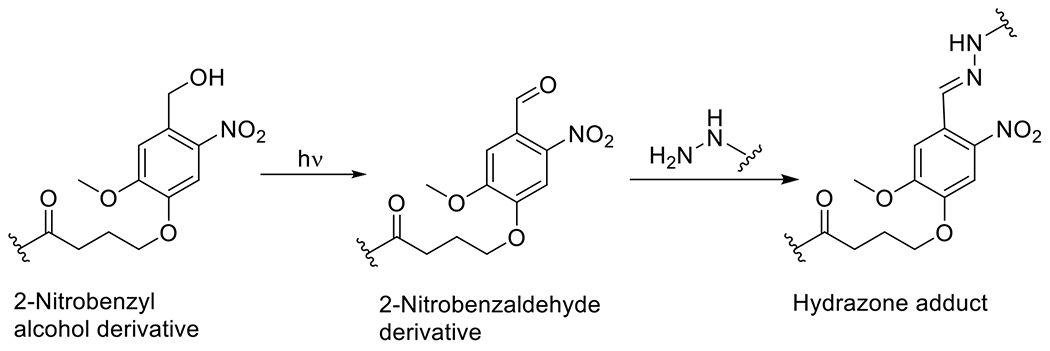
Schematic representation of photoinduced hydrazone reaction.242
Although hydrazone/imine-based click reactions are efficient under physiological conditions, the reversible nature of these chemistries leads to hydrolytic cleavage in just a few days under physiological pH.244, 245 This hydrolytic cleavage is eliminated using an alternative approach. With oxime ligation the photodeprotected aldehyde reacts with an alkoxy amine forming an irreversible oxime functionality. In 2012, Pauloehrl et al. implemented this strategy employing the photouncaging of an o-nitrobenzyl acetal under mild irradiation conditions using 370 nm light to achieve rapid, quantitative photouncaging of o-nitrobenxzaldehyde patterned on silicon wafers followed by the oxime ligation reaction with amino oxy functionalized biomolecules with excellent spatiotemporal control (Figure 68).56
Figure 68.
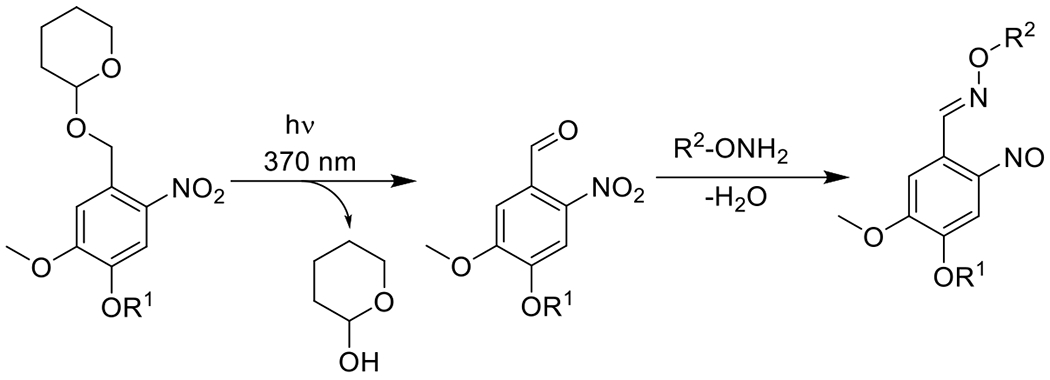
Photoinduced cleavage of a 2-[(4,5-dimethoxy-2-nitrobenzyl)oxy]tetrahydro-2H-pyranyl derivative and subsequent oxime ligation with hydroxylamine derivatives. R=C3H6COOH.56
In a second approach the photocaging of alkoximes with NPPOC was utilized. The alkoxyamines generated in situ upon photoirradiation directly participate in oxime bond formation with the aldehydes.
2.4.5. Photoinduced Native Chemical Ligation
Native chemical ligation (NCL) introduced by Kent and co-workers in 1994 is one of the more powerful and widely used methodologies for protein chemical synthesis.246 This reaction involves an initial chemoselective transthioesterification between N-terminal cysteinyl and C-terminal thioester fragments, followed by a rapid intramolecular S to N acyl transfer, to generate a native peptide bond under mild conditions. The ability of NCL to proceed under mild conditions with a wide range of substrates and high chemoselectivity makes it an attractive ligation strategy for highly specific conjugation of biomolecules. The very same features qualify NCL as a click reaction. Beyond the total chemical synthesis of peptides and proteins, NCL has also been recognized as a straightforward pathway to bioconjugation and labeling. A review detailing the mechanism, scope, limitations on NCL and extended methods is available.247 In 2005 Ueda et al. reported purification free, one pot sequential native chemical ligation using 4-(dimethylamino)phenacyl-type photolabile amine and thiol protecting groups248 where the photocaged amines exhibited adequate stability towards acidic and basic reagents commonly used in protein ligation chemistry. Along with the NCL chemistry the phototriggered amine release enabled efficient, one-pot sequential NCL reactions under mild conditions without the requirements for purification of the intermediates or change in the ligation conditions. In another example, Aihara et al. used the photocaged N-sulfanylethylanilide peptide to synthesize a 41-residue SNX-482 by a one-pot, sequential four-fragment ligation in an N to C-direction as illustrated in Figure 69. Upon thiol photo uncaging, the inactive amide form undergoes intramolecular rearrangement to an active thioester form in the presence of phosphate salt and subsequent NCL.
Figure 69.
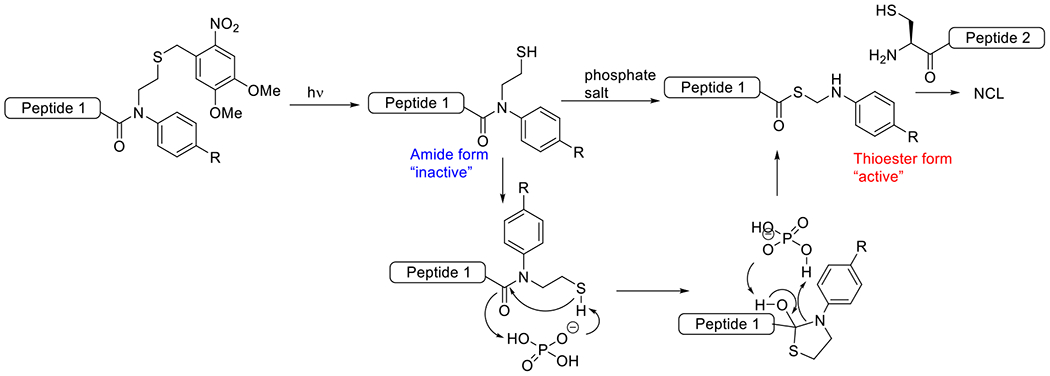
Mechanism and strategy of peptide/protein synthesis using photocaged N-sulfanylethylanilide peptide. Adapted, with permission, from reference 249. Copyright 2016 American Chemical Society.
To summarize, photouncaging click functionalities is an established concept with continuing improvements moving towards increased efficiencies and implementation as a promising tool for various biological, materials, and synthesis applications. This approach is well suited for temporarily inactivating click functionalities, particularly in the presence of biologically active molecules. Upon light irradiation stoichiometric uncaging of the functional group facilitates the subsequent click reaction with excellent spatial and temporal control.
3. Applications
As click reactions generally have been employed broadly across a wide variety of synthetic applications photoclick reactions specifically have similarly proven advantageous in addressing challenges in diverse fields of (bio)chemical research and development, albeit with distinct advantages and challenges relative to non-photomediated chemistries. Despite many similar and overlapping benefits that span across nearly all of the reactions discussed herein, photoclick reactions are far from interchangeable; each has its own strengths and weaknesses that determine the applications for which they are best suited. Reactants bearing highly conjugated chromophores may permit the reactions to take place at higher wavelengths of light, but the increase in molecular weight may be deleterious to the intended function of the resulting product. Reactants that are activated by direct photon absorption may benefit from no requirement for photolatent catalyst or photoinitiator, but the requisite, relatively high concentration of the chromophore may hinder scalability of the reaction due to depleted transmittance of light through optically thick samples. Photoamplified reactions, on the other hand, may be conducted on much larger scales but may lack an ability for true spatiotemporal control, depending on the photocatalytic mechanism.
Self-reporting systems, reaction which produce fluorescent products, have recently been shown to be a beneficial feature of some photoclick reactions with obvious advantages in a variety of applications (e.g., fluorescent probes, labeling and conversion indicators). These reactions represent a significant fraction of present photoclick reactions in many emerging fields and are particularly well represented in biological applications where live cell imaging has become an important focus and biorthorthogonal, spatial and temporal reactions are key. Self-reporting systems have been highlighted in the relevant application section.
Despite the simplified, popular conception of “click” chemistry as reactions that should work virtually all the time and under any conditions, these practical considerations dictate where and how photoclick reactions can benefit the user. This section of the review surveys the applications of photoclick reactions organized according to the context of the molecular architecture in which these disparate reactions are employed: small molecule synthesis, soluble macromolecule synthesis and modification, functionalization of materials’ surfaces or the development of network polymers.
3.1. Drug Development and Small Biomimetic Molecules
Applications of photoclick reactions are more abundant in those areas of research that frequently require precise management over where and when a desired reaction should occur, i.e, where the most immediate and apparent advantage of photo-mediated chemistry would have the greatest impact. Typically, these features are not considered of particular importance in the synthesis of small molecules, a situation wherein there is minimal need to spatially limit the reaction within the predetermined confines of the reaction vessel. Additionally, due to the large substituents used to shift absorbance of functional groups, “small” molecule synthesis via many photoclick reactions is often impractical if not impossible. Yet, there are several significant examples of photoclick reactions being employed in drug development and the synthesis of small, biomimetic molecules though the advantages that necessitate such approaches vary. Photoclick reactions are often simply faster and more efficient than alternative synthetic strategies. Temporal control may allow dictation of the order of multiple consecutive reactions and can be used effectively to limit reaction rates for click reactions that may otherwise proceed too rapidly. Ultimately, photoclick reactions are used in small molecule synthesis because they have been proven effective in that arena.
Among photoclick reactions, the thiol-ene and thiol-yne reactions employ perhaps the simplest of reactive groups, resulting in very few superfluous atoms in the resulting linkage. This, along with the insensitivity to water and oxygen, makes these reactions particularly useful in the modification and mimicry of oligomeric biomolecules and their synthetic analogs.
These reactions have been used as a tool for site specific glycosylation for cysteine containing peptides.250 Dondoni and Marra produced an excellent review on glycoconjugation via thiol-ene chemistry.250 In brief, the reaction has been used to modify carbohydrates, for amino acid synthesis,251 assembly of glyoclusters and peptide and protein glycosylation.252 In particularly, this reaction has been explored in the construction of carbohydrate mimics such as S-glycoside, an unnatural carbohydrate, from readily available peracetylated glucosyl thiol and the diacetonide-protected galactose-derived alkene, yielding abundant and highly diastereoselective 1,6-linked S-disaccharides.253, 254 Not limited to carbohydrates, a broad range of 2,3-unsaturated sugars including O-, C-, S-, and N-glycosides have been successfully generated. However, different anomeric heteroatoms profoundly affected the reactivity of the 2,3-unsaturated sugars. Hydrothiolation of 2,3-dideoxy O-glycosyl enosides efficiently produced the axially C2-S-substituted addition products with high regioselectivity but 2,3-unsaturated N-glycosides resulted in a varying regioselectivity and stereoselectivity. No addition was observed onto the endocyclic double bond of C-glycosides and addition to 2,3-unsaturated S-glycosides resulted in 3-S-substituted glycals (Figure 70).251, 255, 256 Researchers have envisioned the application of thiol-ene chemistry to successfully synthesize nucleoside enofuranoside derivatives to produce an array of thio-substituted D-ribo, -arabino, -xylo and L-lyxo-configured pyrimidine nucleosides, which are prime synthetic targets for anticancer and antiviral drug development.257
Figure 70.
Products generated via thiol-ene coupling of various thiols with unsaturated glycosides.257
Thiol-ene conjugations provide a versatile strategy to synthesize various models of complex biological systems like cyclic peptides, which have been shown to exhibit improved bioactivity and stability to proteolytic degradation relative to their linear counterparts. Aimetti et al. exploited the use allyl ester-containing building block, Fmoc-Lys(Alloc)-OH, to synthesize peptides with pendant, thiol-reactive alkenes and pendant thiols resulting from incorporated cysteine residues. Subsequent photoinitiation resulted in the intramolecular thiol-ene conjugation and formation of cyclic peptides. This strategy clearly demonstrated that cyclic peptides could be generated using commercially-available amino acids without significant post-synthetic modification, leading to a straightforward synthetic pathway for an important class of biomolecules.258
While cyclic peptides may exhibit profound differences in bioactivity relative to their linear analogs, secondary structure within those cyclic peptides also exerts substantial influence over the function thereof. In order to elucidate the influence of this secondary structure on cyclic peptides, achiral centers in the tether of a stapled peptide were introduced. By utilizing thiol-ene chemistry, optimally-tethered cyclic peptides and non-tethered linear peptides were synthesized successfully starting from cyclo-Ac-CAAAS5(2-Me)-NH2 (S5((S)pentenylglycine), 2-Me (the methyl group located at the b-position with respect to the a-carbon of the amino acid). While the cyclic peptide was found to be a random coil, the linear peptide was found to be in helical form, due to a substituent at the tethered chiral center, suggesting that the substitution group also interacts directly with the binding groove.259 This simple strategy enabled access to thioether-based constrained peptides, peptide stapling, precisely-positioned chiral centers on thioether-based tethers, and thioether-constrained pentapeptides.260–262 These facile, one-pot reactions recently led to the synthesis of truncated S-lipidated teixobactin analogs from commercially sources.263, 264 In one particularly interesting application of the thiol-ene reaction for peptide stapling, Hoppmann et al. employed photoisomerization of a vinyl azobenzene amino acid to facilitate a photocontrollable bridge with the pendant thiol of a cysteine residue.265
The thiol-yne reaction has not been employed to the same extent as has the thiol-ene. While this may, in part, be due to the more limited application of the diaddition of thiols to alkynes, this feature of the thiol-yne reaction is advantageous in the synthesis of clustered cyclic peptides. For example, various cyclic and multivalent alkene and alkyne derivatives of Arg-Gly-Asp (RGD) were reacted via the radically photoinitiated thiol-yne chemistry, forming clustered peptides which inhibited the binding of fibrinogen to GPIIb/IIIa relative to linear RGD (Figure 71).266
Figure 71.
Cyclic peptides formed via intramolecular thiol-ene (left) and thiol-yne (right) reactions.266
While the thiol-ene and thiol-yne reactions have been successfully employed in the synthesis of small molecules in large part because of the relatively atomically dense reactive functional group structures, few other photoclick reactions exhibit similarly small reactive moieties and so their applications for synthesis on this molecular scale more considerably limited. Another such example is the photoCuAAC and PcAAC reactions. As described previously, Wu et al. implemented a copperless, visible light, photoredox-mediated azide alkyne cycloaddition for the synthesis of 1,4-disubstituted 1,2,3-triazole derivatives, including bioactive molecules with high regioselectivity and good yield.267 In another example of implementation of the azide-alkyne cycloaddition employed for the potential synthesis of small therapeutic molecules, Beniazza et al.employed photoCuAAC to drive the alkyne-azide reaction with a variety of protected and non-protected azide and alkyne-functionalized monosaccharides generating di- and tetrasaccharides in high yield within 10 min of UV irradiation and ~100min under daylight illumination (Figure 72).268
Figure 72.

Examples of bioactive molecules and potential drug candidates generated via the PhotoCuAAC reaction (top) and via the PcAAC reaction (bottom).268
In limited contexts, spatial and temporal control are advantageous in small molecule synthesis (e.g. the in situ synthesis of self-assembling amphiphilic compounds). Konetski et al. successfully employed photoinitiated CuAAC to produce self-assembled phospholipid vesicles under exposure to visible light. Higher vesicle density via the photomediated reaction was observed relative to that obtained via catalysis with sodium ascorbate and copper sulfate. Additionally, the rate of reaction, tunable via the intensity of light exposure, had a profound impact on the morphology of the self-assembled vesicle structures as shown in Figure 73.269
Figure 73.
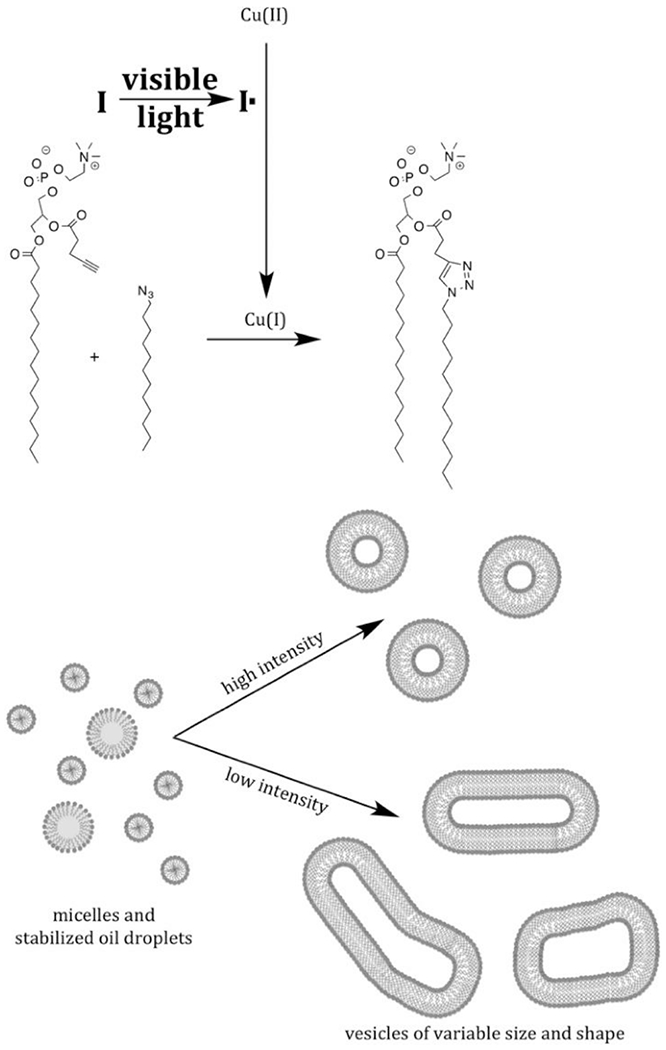
PhotoCuAAC reaction results in in situ synthesized phospholipids that self assemble into vesicles. Morphology of the resulting vesicles depends on exposure conditions, with high intensity resulting in smaller and more uniform structures. Figure reprinted, with permission, from reference 269. Copyright 2016 American Chemical Society.
3.2. Oligomerization and Polymerization Reactions
There are two general mechanisms for polymerization: chain addition, wherein monomers add to the active center of a growing chain, and step-growth wherein complementary functional groups on multifunctional monomers react with each other to form ever larger molecules. While the rapidly increasing molecular weight development in the former may allow for tolerance of some impurities, inefficiencies, or other nonidealities during the polymerization, step-growth polymerizations require highly effective and selective reactions to obtain high molecular weight species. For this reason, click reactions are well suited for step-growth polymerizations.
Several examples of photoclick reactions employed for linear polymerizations have been reported. In one example, Porel and Alabi employed sequential photoinitiated thiol-ene and nucleophile-initiated thiol-Michael reactions to build, unit by unit, a sequence controlled oligomer taking advantage of the semiorthogonality of the two reactions.270 Difunctional thiols were reacted with heterobifunctional allyl acrylamides (the allyl being radically reactive with thiols and the acrylamide providing the Michael acceptor), bearing various pendant moieties. Initial reactions with the Michael acceptor allowed for subsequent chain extension radically via the allyl group without the need for deprotection between additions. The reactions were performed in the solution phase; the high efficiency of each reaction and employing a fluorous tag as a liquid phase reaction support allowed for simplified chromatographic purification between each unit extension.
Photoinitiated thiol-ene photoclick chemistry has been used in the development of oligomeric nucleic acid mimics. In 2015, clickable nucleic acids (CNAs), which are single stranded DNA-mimicking oligonucleotides, were synthesized employing thiol-ene click reactions. Both homooligomers and oligomers of sequence-controlled, periodic repeats were obtained (Figure 74).271 In contrast to the solid-phase, sequential addition-based synthesis of oligomers, which may take many hours to obtain the desired product, oligomers of up to 15 nucleobase residues were synthesized in a matter of seconds via the photoinitiated reaction between thiols and eneamides on heterobifunctional monomers. Subsequently, longer sequential patterns such as the precursors of tetramer, pentamer, hexamer, etc. were efficiently synthesized in a simple and scalable manner, leading to highly selective hybridization between these CNA-2G polymers and complementary ssDNA.272, 273 The versatility of this new generation of CNA molecules and the ease of synthesis afforded by the thiol-ene reaction portends applications of artificial nucleic acids and related synthetic bioactive molecules across a wide variety of research fields.
Figure 74.
Examples of Click Nucleic Acid (CNA) polymerizable nucleobase monomers including first generation thymine monomer (top left), 271 first generation polymerizable TA dimer (top right), 271 second generation thymine monomer (bottom left) and polymerizable TTA trimer (bottom right). 255, 256
Consecutive photoclick reactions have been used to obtain ordered oligomeric and polymeric structures with high precision. Barner-Kowollik et al.274 achieved sequence-defined macromolecular assembly using a photoenol Diels-Alder reaction between monomers carrying both the photolatent diene (benzaldehyde) and dienophilic alkenes. Sequentially varying introductions of monomer pairs, the authors obtained precise homopolymers, copolymers, and block copolymers. By controlling the structure of the introduced monomers, the physical properties of the resulting macromolecules were able to be tuned, affording user-defined materials with potential use as a molecular information storage system. The efficiency and orthogonality of the photo DA reaction has allowed researchers to employ the reaction in conjunction with other reactions (e.g., the Passerini three component reaction) to generate monodisperse oligomers with precise, alternating sequences via consecutive additions of monomers.275
Photoclick reactions have been employed toward the development of linear polymers with advantageous mechanical properties as well. For such application, reactions requiring relatively large functional groups are limited by the influence of those groups on the physical properties of the resulting materials. Photoclick reactions that employ relatively small functional groups are better suited to allow more precise tuning of those properties by monomer design without the concern for disproportionate influence from the reactive groups themselves. For this reason, thiol-ene reactions are particularly suited to this application. Recently, utilizing the thiol-ene reaction, high molecular weight semicrystalline linear and loosely crosslinked polymers were formed from 1,6-hexanedithiol and diallyl terephthalate monomers within a few seconds of irradiation with low intensity light and with demonstrated excellent mechanical properties (tensile strengths of 24 MPa at failure elongations of >800% ).276, 277
Among the described photoclick reactions, the thiol-yne reaction is unusual in its difunctionality. This has allowed it to become particularly useful in the synthesis of dendrimers, highly branched, regularly structured, frequently fractal macromolecules. The most straightforward synthesis of dendrimers or dendrimer-like materials was conducted by the homopolymerization of a monothiol monoalkyne, which yielded a hyperbranched, polysulfide with many terminal alkynes.278 The resulting product, however, was polydisperse. Moreover, given the observed homopolmyerization of alkynes upon depletion of thiols, the hyperbranched product may have exhibited considerable structural nonidealities.
By alternating thioglycerol-yne reactions with an orthogonal alkyne coupling reaction, researchers were able to geometrically increase the functionality of each successive generation of dendrimer discretely, generating effectively monodisperse dendrimeric species (Figure 75).279, 280 This strategy had been employed previously with thiol-ene photoclick reactions, employing a reaction to couple pentenoic acid to the hydroxy groups that resulted from coupling of thioglycerol.281 While the thiol-ene strategy resulted in a doubling of functionality with completion of each set of coupling reactions, the thiol-yne strategy resulted in a theoretical quadrupling of functionality. Such materials have demonstrated utility in drug delivery and as stabilizing contrast agents for CT imaging.
Figure 75.
Dendrimer made via alternating thioglyerol-yne and alkyne conjugation reactions. The thioglycerol-yne coupling results in the introduction of two hydroxyl groups per reaction while the subsequent alkyne coupling replaces the alcohol with the difunctional yne for the next thioglycerol coupling reaction. Each successive reaction pair results in a quadrupling of functionality as each alkyne is difunctional.279
3.3. Macromolecular modification
3.3.1. Biolabeling
The labeling of biologics in vivo and in vitro allows investigators to better understand structural and functional relationships via the visualization of complex molecular interactions within biological systems.282, 283 Labeling cells, organelles, and biopolymers with fluorescent molecules via biorthogonal click reactions is a popular strategy as it provides generally high sensitivity, minimal invasiveness and frequently the potential for real-time observation in living systems. The ease and degree of control provided by photomediated reactions with mild reaction conditions, high yields and high selectivity has incentivized the development of ever new photoactive compounds to label biological polymers and higher order structures to track and elucidate cellular processes.
While CuAAC reactions have long been the gold standard for click reactions, cytotoxicity and other deleterious biological interactions associated with the use of copper, have necessitated development of alternative click strategies. Photoactivation of cycloalkynes for copperless azide-alkyne cycloaddition reactions has been particularly successful when applied to labeling in the biological sciences. In an early example, this strategy was employed for the efficient labeling of living cells expressing glycoproteins containing N-azidoacetyl-sialic acid.33 The authors highlight many advantages of using this photo-triggered click reactions for labeling, including the ability to allow for a homogeneous concentration of reagents in the cells before the click reaction is initiated. The relatively high quantum yield for this reaction also improves the biological compatibility of its utilization.
This approach has also been used for the fluorescent labeling of BSA with a Rhodamine B dye by using a bis-cyclopropenone to link the two molecules (BSA-DIBOD-Rhodamine B). By exploiting the advantages of the photoSPAAC reaction, the labeling occurred in a one-pot reaction with only a few minutes of light exposure (Figure 76).284 In similar applications, SPAAC photoclick reactions were employed as light-responsive photoaffinity probes for detecting carbohydrate-binding proteins and antibodies.285–287
Figure 76.
a) Sequential photo-labeling of azido-BSA with photogenerated ring-strained cyclooctadiyne. b) Selective light-directed Immobilization of rhodamine B on a 96-well plate with photogenerated cyclooctadiyne. In these examples, the photocaged cyclooctadiyne serves as an exogenously activatable crosslinker, permitting the coupling of two azide-containing species. Reprinted, with permission, from reference 284. Copyright 2016 Royal Society of Chemistry.
Photo-activated, ring stained alkynes have also been used in IEDDAC reactions with tetrazines. As demonstrated by Mayer et al. this reaction is efficient for labeling proteins by using a photo-activatable bicyclononyne (BCN) probe, offering spatial and temporal control over the labeling of tetrazine labeled proteins.44 The authors were able to conduct this photo-triggered reaction in E.coli demonstrating the biorthogonal nature of this reaction (Figure 77). The photo-IEDDAC reaction proceeded with an on-protein rate constant of 50 m−1 s−1 on par with the fastest biorthogonal photo-induced reactions. This enables protein labeling within minutes at low concentrations. Although this technique has yet to be explored further, sustained improvements in the design of complementary strained alkenes288 and new synthetic approaches for derivatives of the tetrazine moiety289 hold great promise for the future of the photoinduction of tetrazine ligation reactions, particularly in intracellular labeling and conjugation applications by virtue of the bioorthogonality of the reactant chemical species and inverse electron demand cycloaddition mechanism.
Figure 77.

Photo-IEDDAC labeling on living cells. a) labeling of sfGFP-N150mTetK with photo-11. b) Structures of water-soluble photo-DMBO conjugates. Both photo-11 and the Cy5-conjugate photo-14 are decarbonylated by short irradiation at 365 nm to quantitatively form 11 and 14, respectively. c) Fluorescence imaging and fluorescence microscopy show efficient and light-induced labeling of a cell-surface protein in living E. coli. Reproduced, with permission, from reference 44. Copyright 2019 John Wiley and Sons.
Initial work by Lin and co-workers pioneered the tetrazole-ene reaction as a photoclick reaction for biological applications and as a fluorescent self-reporting system.290 In one approach to the application of this chemistry in the labeling of biopolymers, the tetrazole or alkene moiety was introduced into the protein via genetic encoding of functional group-bearing, non-natural amino acids.85, 291 A fluorescent moiety was then incorporated in a site selective manner. This was first achieved with a genetically encoded alkene reporter, O-allyl tyrosine, for protein labeling in bacteria.29 However, this reaction was slow (k2 = 0.00202 ± 0.0007 M−1 s−1) because of the low reactivity of the alkene on the reporter. Various routes have been explored to increase the reactivity including the use of an electron-deficient acrylamide-modified lysine side chain. The use of an acrylamide is advantageous as it is relatively small and stable under biological conditions. It was successfully used in a number of cases including the labeling of the membrane protein OmpX on the surface of Escherichia coli and successfully incorporated into GFP-TAG-mCherry-HA protein in Arabidopsis thaliana, a plant model.292, 293 Norbornene and cyclopropene functional groups have also been incorporated into proteins to increase the reaction rate with tetrazoles.294–296 For example a cyclopropene-modified lysine was used to direct site specific modifications of green fluorescent protein inside HEK293T cells. The reaction rate increased five orders of magnitude to k2 = 58 ± 16 M−1s−1 for cyclopropene which was reported to be twice as fast as norbornene as a consequence of the increased ring strain.297 Spiro[2.3]hex-1-ene has also been recognized as another strained alkene that readily reacts with tetrazoles for bioorthogonal protein labeling, despite its 3,3-disubstituted conformation.298
DNA has also been successfully labeled through the tetrazole-ene reaction. Wagenknecht’s group was the first to present tetrazole-modified building blocks for solid-state DNA synthesis and a nucleoside triphosphate for the polymerase-based DNA preparation. The ligation with maleimide dyes occurred with LED excitation at 365 nm (k = 23 ± 7 M−1s−1).299, 300 The group developed this technique further by exploiting the fluorescent pyrazoline product that is produced via the tetrazole-ene reaction.301 By synthesizing tetrazole functionalized 2’-deoxyuridine, a modified DNA structure formed which could then react with a maleimide functional dye for site specific labeling. The researchers also exploited the use of the fluorescent pyrazoline, formed in the tetrazine-ene reaction, and enhanced the fluorescent intensity by enabling energy transfer to the labeled dye. Lehmann et al. indicate that the energy transfer would work on a multitude of dyes and could be tuned to the desired wavelength of choice, demonstrating the versatility of the pyrazoline product. In addition, Yu et al. also exploited the use of the fluorescent pyrazoline product in intramolecular tetrazole-ene cycloaddition.251 This single reagent approach was demonstrated to be successful as a probe in cancer cells.290, 302 Pre-fluorescent naphthalene-tetrazole conjugates induced by two-photon excitation at λ=700 nm for fluorogenic labeling of olefin-tagged intracellular structures have been formed via this photoclick reaction.303, 304
More recently the reaction has been used in multicellular organisms. Wu et al. incorporated vinyluracil into the DNA of zebrafish to allow for spatially controlled imaging of DNA through the use of a coumarin-linked tetrazole in real time without DNA denaturation (Figure 78).305 The authors highlight that through the use of the tetrazole-ene reaction a “switch-on” labeling approach was achieved.
Figure 78.
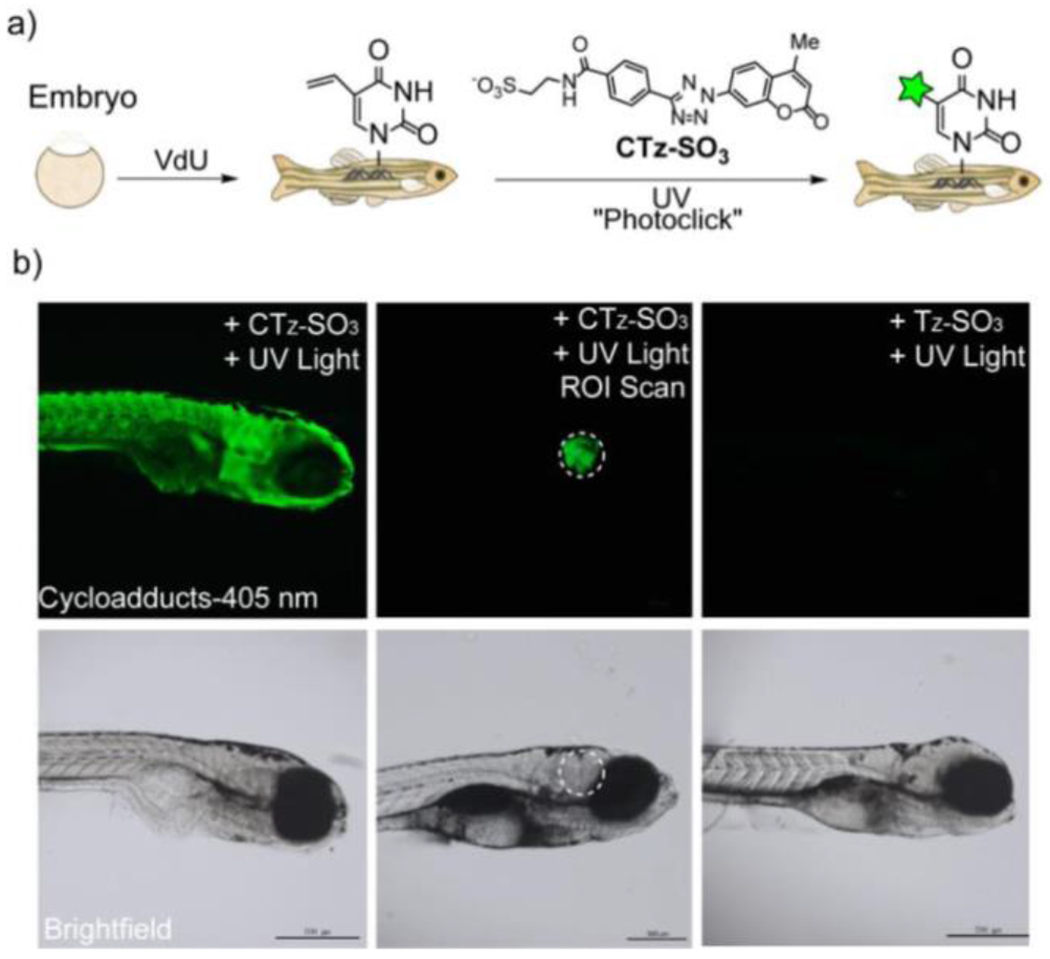
a) schee for labeling DNA of zebrafish via tetrazole-ene photoclick reaction and b) fluorescence microscopy images of cellular DNA spatially labeled through the tetrazine-ene reaction. Reproduced, with permission, from reference 305. Copyright 2019 American Chemical Society.
Despite these examples of relative bioorthogonality, the high reactivity of the intermediate nitrile imine with many common biological nucleophiles89, 90, 306 (e.g. thiols, amines, and acids) may limit its application, though strategies have been developed to improve specificity.91, 307 Utility in this case, however, would effectively be limited to those situations in which labeling with a substantial photoactivatable probe is practical, in contrast to previous examples where biomacromolecules bearing small, alkene functional groups are selectively labeled via photoactivated tetrazoles. Ultimately, the numerous examples of tetrazole-ene photochemistry for labeling reflects its broad utility for that purpose and suggests extensive continued and future application in biolabeling.
The photomediated 1,3-dipolarcycloaddition via uncaging of nitrile-imine from sydnones form the same pyrazoline cycloaddition adduct as does the reaction with the tetrazole and thus has found similar labeling applications as a fluorescent functional group.36 Since its introduction in 2018, several examples of labeling reactions between substituted sydnones and variable alkenes and alkynes have been demonstrated.94, 308, 309 Included in these was the use of photoisomerizable dipolarophiles, such as dibenzothiadiazepine, the photoinduced ring strain of which allowed dual wavelength control over labeling biomacromolecular structures in live cells.95
The azirine-alkene reaction was also used as a bioorthogonal label for lysozyme, although this application required 302-nm UV light, posing a challenge to implementation in living cells.37 However, this limitation was overcome through the synthesis of an azirine moiety coupled to the visible-light-absorbing chromophore pyrene, resulting in a single molecule for facilitating the azirine-alkene cycloaddition reaction at wavelengths greater than 400 nm. The small size of the functional groups of the bioorthogonal azirine-based photochemistries, in addition to their high reactivity, make them as an attractive albeit underutilized alternative to other photoclick reactions.310
Photocycloadditions of 9, 10-phenathrenequinones with electron rich alkenes to form fluorogenic [4+2] cycloadducts have also been utilized for labeling proteins in live cells.311 Li et al. demonstrated the use of a fluorescent self-reporting system that enabled the temporal and spatial surface labeling of A549 cells in a biorthogonal manner using visible light.
Although the photo catalyzed thiol-ene reaction has been used extensively as a bioorthogonal reaction, it has not received the same interest for biolabeling that other photoclick reactions (e.g. tetrazole-ene, photo SPAAC) have. The advantage of using the thiol-ene reaction for labeling proteins is the natural occurrence of cysteine residues that readily react with an alkene functional label.312 Despite the lack of spatiotemporal control, the thiol-maleimide Michael addition reaction has been a far more popular route for labeling proteins, as it does not require light exposure or the presence of radical initiators. There are, however, examples of modifying biopolymers with the radical thiol-ene reaction. Early examples of using the thiol-ene reaction to react with naturally occurring cysteine residues focused on the glycosylation of proteins in a non-site specific manner. Dondoni et al. glycosylated BSA that contains one disulfide bond and one free cysteine using 365 nm light and demonstrated that all three cysteine residues were glycosylated.250, 313 However if site specific labeling is required, other cysteine groups require modification to avoid also participating in the reaction and thus leading to heterogeneous protein mixtures. More recently, photo-catalyzed thiol-ene reactions have been used for paramagnetic tagging of proteins for use in NMR imaging where site specific labeling is not required. Denis et al. have demonstrated, by using an alkene functionalized paramagnetic tag, proteins of interest are labeled through the use of thiol-ene reactions with cysteines present on the protein of interest.314 The thiol-ene reaction offers high yields and short reaction times enabling stable thioether bonds to form, demonstrating the benefits of the reaction as a route to label proteins with paramagnetic tags.
To allow for site specific labeling of proteins, alkene handles have been incorporated into proteins allowing for reactions with thiol functional labels upon exposure to 365 nm light.315 This was first demonstrated by Li et al. who synthesized an alkene functional amino acid to genetically incorporate into proteins using alkenyl-pyrrolysine analogues.316 These proteins demonstrated site specific ligation with thiol modified dyes upon irradiation with 365 nm light. Weinrich et al. used a similar method to immobilize proteins on a thiol functionalized surface.317 The group functionalized proteins with farnesyl pyrophosphate and were able to immobilize the protein by exposure to UV light demonstrating a simple method for immobilizing proteins with a high yield using mild conditions.
Although these conditions have been shown to maintain protein structure and activity, relatively few researchers have used photointiated radical thiol-ene reactions for protein modification and labeling. In contrast to other photoclick reactions more frequently employed in the labeling of biopolymers and other biological structures, the thiol-ene reaction employs a photoinitiator for the photoamplification of the thiol-ene cycle. Such a mechanism is advantageous in applications wherein the concentration of chromophores is a concern. While photoSPAAC and tetrazole-ene reactions employ functional groups that bear their own chromophores, the concentrations of those groups in labeling reactions is generally so low as to guarantee optically thin samples despite the one-photon, one-event nature of the reaction. Thus, one of the otherwise primary advantages of the radical thiol-ene reaction is not particularly useful in the context of labeling.
Like the thiol-ene reaction, the thiol-yne reaction can exploit the use of naturally occurring cysteines and disulfides present in proteins to modify and label proteins.315, 318 Dondoni and co-workers demonstrate the ability of attaching two modifications at a single site on BSA using the thiol-yne reaction.252 They demonstrate the modular addition of alkene functionality that allows for glycosylation and fluorescent labeling at the thiol containing sites on BSA (Figure 79). However, the researchers note that unanticipated side-reactions occurred when the thiol-yne reaction was used in conjugation with other click reactions, most notably cycloalkynes. To enable the thiol-yne reaction for site specific labeling, alkyne handles have been genetically incorporated into proteins. Li et al. demonstrated the use of alkyne-containing pyrrolysine derivatives genetically encoded into proteins for site specific labeling with bi-dansyl-cystamine.319 They demonstrated that the modification has little effect on the enzyme activity or conformation of the protein and could be expanded on as a dual labeling strategy.
Figure 79.
Thiol-yne modification of BSA. Addition of propargyl 1-thio-β-D- glucopyranoside at thiol present sites on BSA to form alkene functional handles which then react with thiol functional peptide chains and fluorescein. Reproduced, with permission, from reference 252. Copyright 2011 Royal Society of Chemistry.
More recently, the thiol-yne reaction has been used in vivo to crosslink proteins. Liu et al. genetically incorporated an alkyne functional amino acid, S-propargyl-cysteine (SprC) into a GDT protein in E. coli.320 Through irradiation at 302 nm a thiol-yne reaction took place between the SprC modified GST and a cysteine residue on the interface of another GST protein, because of the compact size of SprC the modification and crosslinking reaction did not affect the protein folding or interactions. Western blot analysis showed covalent crosslinking of the GST dimer demonstrating an application of the thiol-yne reaction for probing protein interactions in living cells. The photocatalyzed thiol-yne reaction has been applied less than other methods to label proteins potentially as a consequence of the research lacking into the effect radical initiator and irradiation have on protein structure and activity as well as the multifunctional nature of the alkyne itself.
In another example, Luo et al. developed a general strategy that demonstrated the utility of these photoactive and bio-orthogonal chemistries via liposome fusion to cell surfaces for on-demand microtissue assembly and disassembly. In this work, the phototriggered oxime ligation coupled with o-nitobenzyl chemistry allowed for the remote-controlled disassembly upon irradiation with UV light and demonstrated the potential of these chemistries for creating engineered photoswitchable cell surfaces for tissue engineering applications (Figure 80).321
Figure 80.
Schematic representation of the molecular level control of tissue assembly and disassembly via the chemoselective, bio-orthogonal and photoswitchable cell surface engineering approach employing photoinduced oxime ligation and o-nitrobenzyl chemistry. Reproduced, with permission, from reference 321. Copyright 2014 Springer Nature.
3.3.2. Polymer-Polymer Conjugation
Block copolymers have a wide variety of applications, both commercially and in research. They are used as thermoplastic elastomers, surfactants, and adhesives.322 Amphiphilic block copolymers continue to be of great interest for drug delivery, and surface self-assembly of block copolymers results in topographical features of extremely small size (e.g. < 10 nm) with great potential for the advance of microchip manufacturing, semiconductor development and digital media storage.323
Such block copolymers are frequently produced via sequential living polymerizations (e.g. anionic polymerization, ROMP, etc.) or controlled radical polymerizations (e.g. RAFT, ATRP) changing constitutive monomers for each successive polymer block. Conjugation of preformed polymers via click reactions is a strategy that obviates the need for blocks to consist of monomers with compatible polymerization mechanisms.
Polymer-polymer conjugation is also a route for designing more complex polymer architectures such as bottle brushes, hyperbranched and star polymers. Some challenges of polymer-polymer conjugation, include the necessarily dilute conditions required and the potential steric hindrance. In spite of these challenges, macromolecular conjugations require near full conversion between precursors in equimolar conditions, as unconsumed reactants are generally difficult to separate from the conjugated product.
Doran et al.324 demonstrated sequential photoclick reactions to perform polymer-polymer conjugation and to modify the terminus of a newly formed diblock copolymer. A telechelic poly(caprolactone) with methacrylate and alkyne termini was placed in a flask with an azide-terminal PMMA and an initiating system of copper (I) bromide and radical photoinitiator TPO. Exposure to 350 nm light resulted in the formation of the methacrylate-terminal diblock copolymer. Subsequent addition of N-Acetyl-L-cysteine and more radical photoinitiator resulted in the conjugation of the cysteine derivative to the free terminus via radical thiol-ene coupling. It should be noted that the methacrylate used here is generally a less reactive alkene in such reactions, perhaps explaining, in part, the abnormally long reaction times employed in this one pot synthesis (16 hours).
Though thiol-ene and thiol-yne reactions have been employed for polymer-polymer conjugation reactions, under some reaction conditions, byproducts are obtained in significant and even predominant yields. This undesirable outcome is the result of the radical initiation mechanism and the propensity of thiyl radicals to recombine into disulfides. Other byproducts may result from radical termination with radical initiator fragments. Typically, in thiol-ene and thiol-yne coupling reactions, photoinitiators are in such low concentrations and initiate the thiol-ene and thiol-yne reactions so efficiently that any byproduct is negligible. When using low concentrations of thiols and enes such as those found in polymer-polymer conjugation, the concentration of initiators relative to thiol and ene functional groups is generally high and these side reactions become more significant. Another factor that may mitigate the efficacy of polymer-polymer conjugations via thiol-ene reactions is the limited solubility of the polymer with the reactive species.118, 325, 326 These limitations are readily addressed with the appropriate selection of reactive alkenes, (e.g. norbornenes), and more reactive thiols (e.g. mercaptopropionate) along with an efficient initiator to ensure that the rate of the thiol-ene reaction exceeds that of competing side reactions. Polymer-polymer conjugations were performed with good yields and relatively high purity employing mercaptopropionate-terminated PEGs and norbornene-terminated PEGs in both organic and aqueous conditions.4, 327, 328 Allyl ether-terminated PEGs, however, resulted in significantly poorer conjugation. Sequential thiol-ene photoclick reactions were conducted to form a three-arm star copolymer. While all polymers in this study were PEG-based, the results indicated the potential to generate heteroarm star copolymers. In the same study, pentynoate-terminal PEG was reacted with the PEG-thiol, resulting in a higher fraction of conjugated polymers than did the reaction with the allyl ether. However, the monoaddition product dominated, indicating, contrary to couplings with smaller reactants, that the second addition was significantly less efficient that the first, thereby potentially demonstrating the influence of steric hindrance on the reaction.
Photo Diels Alder reactions have been used to conjugate dienophile-terminated polymers to o-methylbenzaldehyde thioether terminated polymers via photoenolization of the latter to the reactive diene ortho-quinodimethane. Feist et al. used illumination with either 365 nm or 410 nm light in a photoflow reactor to couple hexaethylene glycols with maleimide and methylbenzaldehyde thioether termini within ten or twenty minutes (for the UV and visible light irradiation respectively).329 The thioether, in place of an ether, improved the absorbance of the benzaldehyde chromophore in the visible region relative to earlier work with this reaction. While the reaction in this example did not employ polymers of high molecular weight, the results suggests that possibility. The authors did, however, employ a similar reaction to generate diblock copolymers via the photo Diels Alder polymer-polymer conjugation.
Among the most popular current polymerization technologies is the RAFT-mediated polymerization that allows controlled radical polymerization of a wide variety of radically polymerizable monomers. With appropriate RAFT agents, users obtain narrowly dispersed polymers with predefined molecular weight and terminal groups. Polymers synthesized via RAFT-mediated polymerization contain, on one terminus, the thiocarbonylthio moiety common among RAFT agents. Some of these, the dithiobenzoate in particular, have proven effective dienophiles. Generating a poly(methyl methacrylate) via RAFT mediated polymerization with a dithiobenzoate RAFT agent, the authors obtained a polymer with a dienophile terminal functional group that was effectively conjugated to a methylbenzaldehyde thioether-terminated poly(caprolactone) in acetonitrile with 410 nm light exposure with a residence time of 20 minutes in a photoflow reactor.
3.3.3. Biomacromolecule-Polymer Conjugation
An important subclass of polymer-polymer conjugates are those involving natural biomacromolecules. Protein-polymer conjugates in particular are increasingly utilized in biomedical fields as they exhibit advantageous properties derived from their hybrid composition (e.g. increased stability, improved pharmacokinetics, etc.). Wholly synthetic diblock copolymers are, as stated previously, most frequently synthesized via consecutive living polymerizations with different monomers. Such a synthetic approach is not applicable to biomacromolecule-polymer conjugates. Although the “grafting from” approach employed via ATRP and RAFT is a popular synthetic strategy as reported in the literature for development of protein-polymer conjugates,330 some researchers have used photoclick reactions to facilitate polymer-biomolecule coupling. This post polymerization coupling is frequently referred to as “grafting to” and offers the advantage of decoupling the polymerization conditions from the requirements of an environment conducive to maintaining the bioactivity of potentially sensitive biomacromolecules.
PEGylation, in particular, increases the hydrodynamic volume of protein-based therapeutics to mitigate their depletion via renal filtration. However traditional PEGylation reactions are generally not site-specific. Indiscriminate conjugation may result in a heterogeneous mixture of conjugate products and potentially a loss of bioactivity if the polymer chain obstructs or otherwise interferes with the protein’s active sites. Purification of the mixed products is time and labor intensive. Incorporating non-natural amino acids with biorthogonal handles allows for site specific PEGylation, potentially increasing the efficiency of the production of their corresponding conjugates. This technique has frequently been used with CuAAC and SPAAC reactions owing to the efficiency and selectivity of these reactions. However the thiol-ene reaction has also been used as a successful method for PEGylating proteins.
Ye et al. used a reductant to liberate cysteine thiols from their respective disulfide bridges and then grafted mPEG-methacrylate onto the protein via photoinitated thiol-ene coupling.331 As an alternative to using native cysteine residues in the reactions, thiol-ene conjugations may use non-natural alkene functional amino acids to produce alkene-modified proteins. A thiol functional polymer is then used to react with the alkenes on the protein. This latter approach is advantageous as the thiols on the native cysteine residues are most frequently bound up in disulfide bridges, the reduction of which may interfere with protein structure and function. While reactive in thiol-ene reactions, other thiols demonstrate improved reactivity and less likelihood of participation in side-reactions such as disulfide formation.
Siti et al. incorporated allyl-containing nonnative amino acids into bovine beta-lactoglobulin (BLG) for photoconjugation of the protein to a diaryl tetrazole-modified PEG.332 While conjugation was successful, nonspecific coupling was observed and subsequently identified as resulting from the cycloaddition with tryptophan residues on the protein. Additional, non-allyl bearing proteins were reacted with the tetrazole-PEG and conjugation was observed in every case where there was a tryptophan and at rates and yields comparable to reactions with the allyl, demonstrating the ability to take advantage of native amino acid residues for photoclick conjugations (Figure 81).
Figure 81.
Structure of tryptophan (left) with reactive alkene indicated in red. Proposed conjugate linkage (right) between tryptophan-containing protein and tetrazole-terminated PEG. 332
While thiol-ene and nitrile imine reactions may take advantage of native amino acid residues for conjugation reactions, in general, the successful application of site-specific protein-polymer coupling relies on the incorporation of non-natural amino acids into the protein structure. The library of reported non-natural amino acids includes those with a variety of pendant moieties capable of participating in photoclick reactions, including alkynes, alkenes, azides and tetrazoles (Figure 82).
Figure 82.
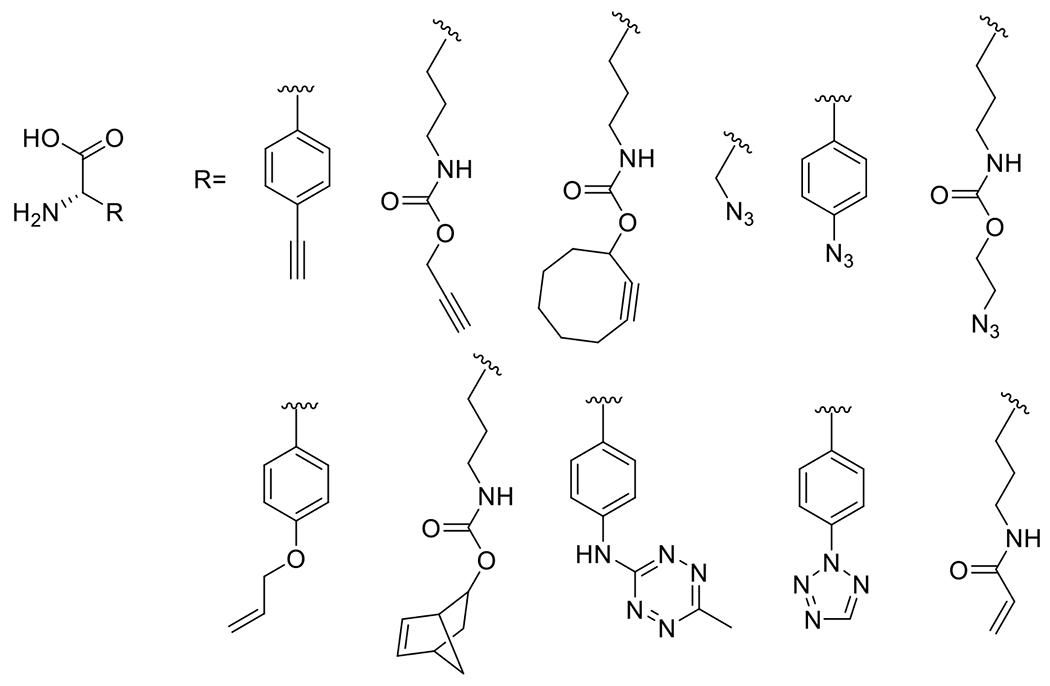
Examples of nonnative amino acids capable of participating in various photoclick reactions.
Photoclick reactions have also been used to generate conjugates of different biopolymers. Fruk and coworkers333, 334 used the photo Diels Alder reaction to couple DNA to proteins by modifying, on CPG, oligomeric single stranded DNA with a photoenol. Once cleaved from the solid phase, the ssDNA was irradiated in the presence of a maleimide-modified protein (myoglobin and horseradish peroxidase) to generate the biopolymer-biopolymer conjugate.
3.4. Surface Modification and Patterning
Material surface modification provides independent control of the bulk physical properties and interfacial interactions between materials. For example, the hydrophobicity of a material is readily controlled to improve surface fouling resistance or to change the interaction of a substrate with a contacting solution. In biomedical applications, the surface of implant materials that might otherwise elicit a foreign body response may be modified with functional groups that mitigate such a reaction from the host. Alternatively, materials’ surfaces may be modified to promote specific in vivo interactions intended to better integrate the material with the host tissue. In diagnostics, surface modification may be desirable to attach a substrate for subsequent detection of an analyte. The spatial control offered by many photoclick reactions makes them particularly well suited to the spatially heterogeneous requirements for many of these surface-level applications.
While photoclick reactions are certainly amenable to the stringent requirements of surface reactions, one challenge to such utility is obtaining surfaces with the requisite functional groups present. For this reason, most surface modifications via photoclick chemistry first require a priming treatment to establish one or the other reactive moieties on the surface. Such surfaces may be thus generated via one or a combination of techniques such as chemical vapor deposition (CVD),335 layer by layer deposition,336 polymer grafting via RAFT337 or ATRP338 mediated polymerization, or any of many others. Jonkheijm et al. employed plasma enhanced chemical vapor deposition onto silicon oxide surfaces followed by successive coupling reactions to generate a thiolated surface from which the authors were able to write, via 411 nm scanning laser, nanoscale features of covalent attachment of an alkene-modified biotin for subsequent immobilization of streptavidin.339 Subsequent experiments with alkene-bearing phosphopeptide demonstrated the bioorthgonal patterning of biomacromolecules.
In a demonstration of a particularly versatile surface priming technique, Wu et al. employed a reactive CVD to coat a variety of material surfaces (silver, polystyrene, stainless steel, titanium, PMMA, and PDMS rubber) with alkene and alkyne-pendant polymers onto which were photopatterned either cell-adhesion reducing PEG thiol or cell-adhesion-promoting GRGDYC peptides (see Figure 83).340 Masked exposure of the surface and respective thiols under 365 nm light in the presence of the radical photoinitiator I2959 resulted in controlled cellular adhesion.
Figure 83.
Reactive chemical vapor deposition to prime surface for thiol-ene and thiol-yne surface patterning reactions with thiol-bearing molecules. Reproduced, with permission, from reference 340. Copyright 2012 John Wiley and Sons.
While the versatility and availability of alkenes is a particular advantage of this reaction, there are situations in which thiol-yne surface functionalization may be preferred. Bhairamadgi et al. ran a comparative study of surface modification of thiol-ene and thiol-yne reactions and, by XPS, noted a faster rate of reaction at the surface and a higher density of reaction products with the thiol-yne reaction when employing aliphatic, surface bound alkenes and alkynes.341 While a more reactive alkene might yield a faster reaction than reported, the authors noted 1.5 times as much sulfur bound to the surface with the thiol-yne reaction as with the thiol-ene at reaction completion.
Surface priming reactions are not always necessary for the covalent modification of materials’ surfaces. Endogenous functional groups may result from incomplete conversion or stoichiometric imbalance in the material synthesis itself. This approach is typified by Ma et al. who employed a thiol-Michael reaction to generate a thiolated surface on the surface of a polymeric material formed with an excess of acrylate moieties.342 Radical thiol-ene photoclick chemistry was then used to pattern peptides on the surface to encourage cell adhesion. Thiol-ene and thiol-yne reactions are particularly amenable to this surface treatment approach as a small stoichiometric imbalance can be well tolerated for maintenance of bulk material properties while providing ample surface-level functionality for subsequent reaction. Alternatively, some materials intrinsically display surface functional groups amenable to photoclick modification directly (e.g. the grafting of thiol-terminated polystyrene to ethylene propylene diene monomer rubber via radical thiol-ene photoclick coupling).343
The 2D geometry of surface functionalization reactions permits extremely thin samples, which diminishes the concern of light attenuation that may be significant for some photoclick reactions in bulk curing applications. For this reason, reactions involving a one photon, one event type mechanism do not suffer as much from this potential drawback that may mitigate their use in other contexts.
For example, the photoenol DA reaction has been employed for surface functionalization with a variety of surface priming techniques and subsequent surface treatment. In one example, Barner Kowolik and coworkers344 synthesized a photoreactive DOPA molecule capable of non-photomediated polymerization on a variety of surfaces. Subsequent photoenolization in the presence of maleimide-terminal complements resulted in patternable surface attachment. Inverting the surface and soluble reagent functional groups, Vigovskaya et al. modified the 5’ terminus of oligonucleotides with the photocaged diene and patterned surface modification by masked exposure to a maleimide-modified surface.334
Many natural materials display abundant functional groups that can be used as functional handles to attach photoclick moieties to their surfaces. Proteins in silk and wool, for example, display a variety of such groups including amines, alcohols, carboxylic acids and disulfides that have been modified to permit subsequent click and photoclick modifications.345–347 Cellulose, a polysaccharide with a high concentration of pendant alchohols, is a particularly attractive candidate for such applications given its low cost of production and near ubiquity in consumer goods in the form of paper, cotton, and wood products. In one particularly interesting demonstration of cellulose surface patterning, Wilke et al. employed laser printing to selectively introduce maleimide groups bound to modified cellulose-binding peptides (CBP).348 Treating the printed paper with toner binding peptides (TBP) occluded maleimide attachment to printed regions. Subsequent photoclick reaction with bisaryl tetrazoles allowed for the patterned attachment of other peptides, suggesting the potential for this chemistry as a means of developing economical medical testing devices.
In addition to the functionalization of flat surfaces,349 the tetrazole-ene reactions have also been used to surface-functionalize as well as to synthesize particles. Delafresnaye et al. used miniemulsion polymerization of tetrazole-containing acrylic monomers to generated microspheres with surface-pendant tetrazoles allowing for the subsequent modification with alkenes or other nitrile imide-reactive groups.350
A significant challenge in modification of surfaces is obtaining a high surface density of the attached molecule. This is particularly difficult when attaching macromolecular structures to the surface. Increasing the functionality at the surface can often improve the density of attached groups following conjugation. That is, effectively, what employing thiol-yne chemistry accomplished in the previous example due to the higher effective functionality of the yne as compared to the ene. Another option is grafting pendant-functional polymers from the surface to provide a greater number of click groups relative to the area occupied. For example, ATRP has been used to graft a polytetrazole copolymer to the surface of gold, from which either maleimide modified biotin or additional ATRP initiators could be photoclicked to the backbone polymer for subsequent processes (streptavidin immobilization in the former case and ATRP grafting of bottle brush polymers in the latter).351
In one interesting example, of PhotoSPAAC-mediated surface modification, Arumugam and Popik used a heterobifunctional SPAAC and photoSPAAC reactive linker to perform consecutive click reactions (one spontaneous and one photomediated) to selectively attach BSA to the surface of azide-modified silicon dioxide particles.352 In the first click reaction, the cyclooctyne reacts with surface azides, following which the cyclopropenone-protected cyclooctynes is irradiated to promote the coupling of the azide-modified BSA to the surface.
Also worth mentioning, the photochemically generated o-naphthoquinone methides have been employed in surface modification via one of two reactions: IEDDA with appropriate dienophile353 or photoreversible Michael addition with a thiol.106 Photomediated Michael additions have also been carried out at interfaces of materials via photogeneration of the Michael donor thiols as well. Hensarling et al. grafted functional polymers on silicon dioxide surfaces and then modified pendant groups with o‐nitrobenzyl and p‐methoxyphenacyl protected thiols.238 Photodeprotection and reaction with maleimide or isocyanates was then carried out.
It may be desirable to generate complex, 3D structures with photopatterned or otherwise user-directed hetergenous surface chemistry. This may be the case particularly when attempting to mimic or recreate naturally occurring materials such as biological tissue that exhibits spatially complex architecture on multiple size scales, from the macroscale, to the microscale and molecular scale. Here, again, employing photoclick chemistry allows unmatched control over not only the size and shape of the synthesized material, but also over its surface chemistry. Richter et al. demonstrated this by the direct laser surface modification by TPA-mediated photoenol DA click reaction on TPA-generated microstructures.354
3.5. Network Polymers
While photopolymerization is infrequently used (previously discussed exceptions notwithstanding) for the generation of linear macromolecules, it is a common method for the generation of crosslinked materials. While acrylic networks comprise the majority of thermosets generated via a photochemical means, some photoclick reactions do exhibit advantages for particular applications of photopolymerized thermosets. These benefits are intrinsically related to those features that make these reactions “click,” namely their efficiency and their specificity. The one-to-one reactivity of functional groups dictates a delayed gelation, as described by Flory and Stockmayer.355 Chain-addition polymerized thermosets may form gels at very low conversions. As soon as the gel point is reached, stress associated with shrinkage due to the reaction begins to accumulate while the diminished diffusion of reactants may result in a significant fraction of unreacted monomer that may subsequently be released from the material. Alternatively, networks polymerized via photoclick reaction between multifunctional thiols and alkenes, for example, form gels at conversions typically much higher in accordance with the functionalities of each reactant. A larger fraction of the reaction occurring prior to gelation allows materials to easily relax and accommodate any shrinkage associated with that conversion, resulting in materials with lower internal stress relative to comparable chain-addition crosslinked materials. At the same time, the delayed gelation and efficiency of the reaction mitigates the quantity of wholly unreacted comonomers, decreasing the leachable fraction of the resulting materials.
However, not all photoclick reactions are well suited for network polymerization. In order to take advantage of these features, appropriate photoclick reactions generally must share additional characteristics as well. These include relatively small reactive groups, which otherwise may exert a disproportionate effect on the material properties, and a mechanism for photoamplification, which allows minimal light exposure to initiate the greatest number of reactions while mitigating the attenuation of light penetration through thick samples. Among photoclick reactions, the radically mediated thiol-ene, thiol-yne, photolatent base-catalyzed thiol-Michael and photoCuAAC in particular exhibit these attributes.
3.5.1. Dental Restoratives
One application of photopolymerizations in the medical industry currently is the curing of dental restorative materials. While most commonly used resins are methacrylate-based, the use of step growth photoinitiated reactions, such as thiol-ene, thiol-yne, thiol-Michael, and photoCuAAC reactions have been studied as potential alternatives.356–361
Due to the high conversions of monomers and the regular network organization that their step-growth reaction provides, thiol-ene, thiol-yne and thiol-Michael resins exhibit several features advantageous to their potential application in dental restoratives including reduced volumetric shrinkage, low development of shrinkage stress, delayed gelation, rapid reaction rates, formation of homogeneous polymer network with advantageous mechanical properties, narrow glass transition and low leachable fractions. In dental restoratives, stress accompanying polymerization may cause cracking within the materials and is one of the major causes of failure in dental composites.362 The delayed gelation and reduced volumetric shrinkage per bond reacted, both contribute to the significant reduction in the stress that arises in thiol-ene polymerization.358, 363–365 Despite these advantages, thiol derivatives are often accompanied by a disagreeable odor, which makes its use in this field problematic. Researchers have tried to overcome by preparing thiol functional oligomers and by employing secondary thiol derivatives, which reportedly lack the offensive odor of primary thiols.363, 366
Another advantage in the application of thiol-ene chemistry for dental materials is the versatility of available chemistries. Water sorption and subsequent degradation of dental restoratives is a concern for materials made with hydrolytically susceptible methacrylate and other ester-containing monomers. The broad availability and ease of synthesis of thiol-ene monomers allows for the formulation of restoratives without such problematic bonds. A few examples of these include ester-free vinyl silanes, cyclotetrasiloxane based thiol derivatives or vinyl sulfonamide, and vinyl sulfone based alkene derivatives.358, 361, 364, 366, 367 Although methacrylate systems are associated with high shrinkage and stress, the mechanical properties of the resulting resins are difficult to match and research is still underway to take advantage of thiol-ene, thiol-yne and photolatent catalyzed thiol-Michael additions reactions for dental restoratives.
PhotoCuAAC reactions have also been explored for their potential in dental restorative materials. The reaction leads to materials with substantially increased material toughness relative to acrylic systems with similar Young’s modulus, possibly due, in part, to hydrogen bonding and weak pi stacking of the triazole rings.368 The heat of reaction, however, is significant, and the reaction itself, while complete in as few as three minutes, lags behind that of thiol-ene reactions and the industry standard methacrylic systems, proving yet unresolved obstacles to the application of the chemistry in such a context.
3.5.2. Photolithography and Additive Manufacturing
In contrast to the photoCuAAC and photolatent-base catalyzed thiol-Michael reactions, the radically mediated thiol-ene and thiol-yne reactions are not dependent upon the generation of a catalyst, the persistence and diffusion of which continues to activate reactions even following termination of light exposure. The radicals generated from photoinitiation are, with minimal exceptions, short lived and therefore reaction outside the limits of light exposure is greatly reduced. This behavior lends these reactions particular efficacy in light-patterned applications including photolithography and additive manufacturing.
Additive manufacturing technologies that employ photochemistry and photopolymerization include stereolithography (SLA) and digital light processing (DLP). In both cases light exposure is used to polymerize monomer from a reservoir, layer by layer, to build up the desired or preprogrammed structure. While SLA employs a laser to write lines of cured resins and generally provides better spatial resolution, DLP writes plane by plane via projection of two dimensional images and typically allows for faster printing. As with microfluidic devices and dental restoratives, low shrinkage stress and higher conversion (lower leachables) observed with step growth reactions are advantages to employing photoclick reactions, imparting better control over final dimensions of the printed structure.369 Another advantage over the use of acrylic resins for 3D printed structures is the ability to generate materials capable of accepting post-printing modifications by the use of off-stoichiometric formulations. Curing with an excess of either reactant results in materials with residual functional groups that may be subsequently reacted with other compounds bearing their respective complementary reactive moieties. This approach enbles the production of materials where the bulk and surface properties may be independently and easily tuned for particular applications. Because the reactions are highly efficient, leaving a fraction of the unreacted moieties does not necessarily greatly increase the fraction of leachables in the printed materials. Ropollo et al. used this strategy while printing thiol-yne formulations to subsequently modify unreacted alkyne groups on the surface with an azide-modified dye.370 While this demonstration only proved the concept, this approach could be used, for example, to apply a nonfouling surface treatment to printed biomaterials or to attach analytes to surfaces for biodetection or diagnostic purposes.
Achieving highly ductile 3D printed materials has proved challenging. As described previously, recently semicrystaline linear polymers have been prepared via thiol-ene polymerization. The high ductility exhibited by the bulk-polymerized material was, however, diminished in the printed materials. This drawback was overcome by generating low crosslinking density, semicrystaline networks made primarily with hexanedithiol and diallyl terephthalate with small fractions of crosslinkers, yielding 3D printed materials with superior ductility.371
The fast reaction kinetics of thiol-ene and thiol-yne photoclick reaction permits the retension of otherwise unstable materials’ features. Complex microscopic and macroscopic heterogeneous features generated by phase separation in emulsions and foams can be preserved to create porous structures with high surface area to volume ratios.372, 373 Bubbles, for example, generated in more slowly-reacting monomer formulations, may merge, or pop changing the volume and density of bubbles in the final, polymerized material. The ability to quickly trap the bubble in place is especially advantageous to the 3D printing of foams, as demonstrated by Amato, et al., who used direct writing of multifunctional thiol-ene monomer and initiator and a custom-designed shell-core nozzle to print foams with user-dictated cell size and density.374 While photopolymerization of larger foam structures may be adversely affected by the scattering and diffusion of light due to the bubbles, this phenomenon is much less significant in the thin streams produced from the nozzle in this direct bubble writing setup.
Photopolymerized emulsions materials have been used in conjunction with additive manufacturing and stereolithographic techniques to generate microfluidic devices with porous, high surface area open cell scaffolds.375, 376
The relatively low degree of light attenuation achievable with one photon, many event radical polymerization allows researchers and technicians precise control over the absorptive properties of liquid monomer formulations where depth of cure must be precisely tuned. While the photoamplification and small reactive moieties make thiol-ene and thiol-yne particularly attractive reactions for additive manufacturing, other photoclick reactions have also been employed.
In the case of multiphoton absorption-mediated reactions light attenuation is less consequential. The generally very low absorbance of the chromophores at the wavelengths employed generally allow for much greater depth of penetration of the high intensity IR or NIR light and effective reaction in only a minimal volume surrounding the laser focal point. This results in the ability to directly write, within monomers in a liquid or solution state, complex solid, 3D materials with extremely small features. This can be accomplished with photoinitiated systems (two photon absorption has been employed in the direct writing of microstructures using the thiol-ene reaction377–379 and acrylate chain addition polymerizations380–382) as well as with one-photon, one event reactions such as the photo Diels Alder reaction. In one representative example, Quick et al. employed direct laser writing of microstructures as described by using two photon absorption at 700 nm to mediate the reaction between multifunctional o-quinodimethanes and maleimides.383 This allowed the researchers to generate structures with features with spacing of 500 nm and allowed for subsequent surface modification via reaction with residual o-quinodimethanes.
The temporal control imparted by photo-mediated reactions can also allow for the generation of regular features while taking advantage of other pattern-generating phenomena. In one such example, Dickey et al. photopolymerized spontaneously generated pillars of thiol-ene monomer formulation, resulting from an electric field-induced force imbalance at the liquid-air interface.384 While this is not an example of photolithography, this work does present an interesting related strategy, the use of patterned electrodes, to provided spatial control of the generation of the thiol-ene generated pillars.
While the topic of “bioprinting” certainly falls under the category of “additive manufacturing,” due to its combining 3D printing technologies with hydrogel chemistry and techniques (described in some detail in Section 3.5.5), this subject is presented subsequently in Section 3.5.5.3.
3.5.3.1. Microfluidic Applications
A subset of additive manufacturing and photolithography that requires particular mention is microfluidics. While the field of microfluidics involves the study and manipulation of flow of very small volumes and has diverse applications from the design of fuel cells, inkjet printheads and medical diagnostic devices, the fabrication of those devises is frequently accomplished via additive manufacturing and photolithographic techniques. Microfluidic devices themselves may be tools for the fabrication of monodisperse microspheres or microshells. Capable of mimicking the microfluidic capillary vascularization found in many organs, microfluidic devices have been especially valuable tools in biological research and tissue engineering.
The development of microfluidic devices requires the fabrication of microscale features (e.g. chambers of various dimension, narrow channels that branch or combine) to accommodate the precise transportation, mixing, and splitting of the fluids in question. Thiol-ene coupling reactions have been particularly effective in this area of research. An excellent review by Sticker et al. has recently been published describing the advantages and applications of thiol-ene coupling in microfluidic devices.385 While there are several techniques for microfluidic fabrication, including casting386 and 3D printing,387 one of the most common is the transfer of patterns from a photomask to the device via photolithography (Figure 84).
Figure 84.

General procedure for photomasked image transfer for microfluidic device fabrication. A mask is placed over monomer mixture and exposed to light. Masked regions form channels and other negative features in the generated device.
Unlike many other photoclick reactions, thiol-ene coupling employs small functional groups that exert minimal influence on the mechanical properties of the resulting materials, allowing researchers to dictate those properties via inclusion of soft or rigid monomer cores or pendant groups to generate soft elastomeric materials or hard glassy materials tailored to the particular application of the device.388–390 Additionally, as the thiol-ene reaction is performed with a diverse selection of thiols and alkenes, development of monomers with desirable attributes may be accomplished with more flexibility in the synthetic routes.
There are other reasons that thiol-ene reactions are particularly suited to microfluidic device fabrication. The photoamplification of radical initiation mitigates light attenuation while radical termination minimizes the reaction rate of light-activated processes outside areas of illumination enabling precise spatial control. In addition, the delayed gelation of thiol-ene and thiol-yne networks, relative to (meth)acrylate networks, for example, mitigates the development of internal stresses in the material associated with shrinkage, which could cause deformation of device features.389 These qualities have allowed researchers to generate high resolution channels, pillars and holes with high aspect ratios (~14), a substantial improvement over acrylic counterparts.391 Even higher resolution features are obtained with off-stoichiometric formulations as image transfer from the mask is improved by the diffusion of limiting reactant into the exposed area during curing, resulting in a depletion of that reactant in unexposed areas that reduces feature broadening.392
Another advantage of employing an excess of one reactant in the monomer formulation is the maintaining of unreacted functional groups following complete cure. These reactive groups bound to the network can be subsequently modified post device fabrication either by diffusing in compounds with the reactive complements or by conducting reactions at the surface of the material (discussed subsequently in section 3.4).389 It should be noted, however, that in masked exposure, diffusion of the deficient reactant from unexposed to exposed regions can have a substantial effect on the concentration of functional groups at and near the surface.392 In addition to allowing modification with chemically dissimilar compounds, the presence of reactive groups at the surface and in the bulk of microfluidic devices may facilitate more effective bonding of subsequent layers during device fabrication, mitigating the chance for solvent or stress mediated delamination.386, 393 This advantage can also be obtained by employing the temporal control of the photoclick reaction to arrest stoichiometrically balanced reactions before full conversion is reached.
Among the many various applications in this field, thiol-ene microfluidic devices have been used for the high throughput screening of immune cells,394 the culturing of breast cancer spheroids,395 mimicking intestinal barrier,396 and the mechanical lysis of cells.397 In a recent illustration of the versatility of the thiol-ene coupling reaction for microfluidic devices, ene-functionalized temperature-responsive NIPAM was grafted to the surface of a thiol-modified substrate to generate thermally actuatable gates within microfluidics channels for controlled sequestration and release in individual channels.398 More recently, this versatility was demonstrated in the fabrication of an all-thiol-ene microfluidic enzyme reactor and electrophoretic separator for continuous sample preparation and direct analysis by mass spectrometry,399 with these applications showing significant promise for thiol-ene photopolymers as materials for numerous lab-on-a-chip technologies with stimuli-responsive behavior to multiple inputs.
Although it shares many features with the thiol-ene reaction, thiol-yne couplings have been used considerably less in the field of microfluidics. This may be because of the moderately slower reaction. One potential advantage of the use of thiol-yne reactions in this area, however, would be the ability to maintain high crosslinking density in off-stoichiometric formulations. Moreover, with a faster second thiol addition, a large fraction of any excess alkyne would likely be maintained at full conversion, leaving a versatile reactive handle for post-fabrication modification with another thiol-yne or azide alkyne cycloaddition reaction.
3.5.4. Optical Materials
Incorporation of more polarizable atoms or groups into the polymer network is one of the well-established strategies to produce high refractive index polymeric materials.400, 401 Owing to the high intrinsic molar refraction of the sulfur atom, the polymers prepared by thiol mediated click reactions received considerable attention in the field of optical material development. Rapid and efficient thiol-X click polymerizations allow for the uniform incorporation of a large number of high refractive index sulfide linkages throughout the polymer network resulting in photopolymers with high optical quality, thermal stability, and mechanical strength. Advantages of these polymers such as light weight, low cost, ease of processing and high impact resistance made them an interesting alternative to silicon and glass for optoelectronic applications. Furthermore, the step-growth mechanism of thiol-X photopolymerization results in polymer network with low intrinsic shrinkage stress with minimum leachable monomers and exceptional resistance to oxygen inhibition. While there exist several recent reports on high refractive index polymers based on radical thiol-ene reactions, a majority of these strategies rely on thermal or self-initiated polymerizations.402–406 Incorporation of more polarizable groups such as P=S, P=Se and highly polarizable main group elements such as Si, Ge, Sn etc. enabled these polymers to achieve a refractive index as high as 1.75 with excellent optical transparency.
Use of photoinduced thiol-X click polymerizations allows for the reaction to be performed under ambient conditions to achieve spatial and temporal control over various mechanical properties. For example, in 2003, Natarajan et al. demonstrated the formation of holographic reflection grafting in polymer-dispersed liquid crystals (H-PDLC), offering lower switching fields, higher diffraction efficiencies, better optical properties, and higher thermal stabilities. Upon forming refraction gratings at several wavelengths across the visible spectrum, diffraction efficiency reached up to ~70% in the blue and green spectral region with no significant polarization dependence. Additionally, implementation of the thiol-ene chemistry reduced the nonuniform shrinkage of the film as a result of higher reactant conversions at much lower viscosities.407, 408
Peng et al. explored the utilization of a unique sequential orthogonal strategy for two-stage thiol-ene click polymerization, in which an initial thiol–acrylate Michael reaction produced a loosely-crosslinked photopolymeric substrate with excess thiol-allyl reactive species.409 The presence of high-refractive index allyl monomer, 9-(2,3-bis(allyloxy)propyl)- 9H-carbazole (BAPC), in the matrix allowed for large-index gradients to be developed within the network upon subsequent exposure to coherent laser beams and initiation of the radical-mediated thiol–ene reaction. Upon further investigation, allyl conversion during the thiol-ene reaction was attributed to relatively low volume shrinkage of only ~3%. Moreover, such shrinkage is further alleviated by introducing additives such as cyclic allyl sulfides or nanoparticles. This novel two-stage system resulted in a holographic material with high Tg, high modulus, diffraction efficiency as high as 82%, and refractive index modulation of 0.004. The novelty and advantage of this two-stage approach lies in the elimination of traditional solvent processing methodologies; these reactions are also rapid and oxygen-tolerant without light scattering or microphase separation, resulting in good optical transparency.
Photoinduced thiol-ene click reactions have been used in UV nanoimprinting lithography application using high refractive index polythioethers prepared via the radical mediated thiol-ene click reaction of synthesized fluorene based dithiol monomer with various ene monomers. The nanoimprinted patterns with various features on the order of 100–500 nm were successfully fabricated and characterized by SEM analysis.410 More recently, McClain et al. carried out a viability study of thiol-one click derived photopolymers in optical coating applications. A comparative study of optical, thermal and hydrophilic character of these photopolymers formed by using a common tetrathiol (PETMP) with various multi-vinyl monomers via photoinduced thiol-ene polymerization resulted in transparent, high refractive index, hydrophilic materials that could be polymerized and molded. Within the comonomer tested, the thiol-ene polymers fabricated using divinyl comonomers showed greater optical transmission but low in refractive index than that of the thiol-ene polymers that contained tetravinyl comonomers.
Incorporation of nanoparticles such as silica nanoparticles in the holographic recording media greatly improved the optical, mechanical and electrical properties along with leading to a significant reduction in the polymerization shrinkage. To this end, Hata et al. reported the volume holographic recording characteristics of silica nanoparticle-polymer composites using thiol-ene and thiol-yne photopolymer systems.411, 412 While these nanoparticle-(thiol-ene)polymer composites exhibited a high transparency and low shrinkage as low as 0.4–1%, their saturated refractive index modulation were found to be 0.01 and 0.008 for thiol-ene and thiol-yne systems, respectively, and meet the acceptable values for holographic data storage media.
More recently, Alim et al. developed a highly modular synthetic strategy to produce multifunctional thiol and ene monomers with high refractive index cores containing aryl and sulfide groups.413 The advantage of these low viscosity liquid monomers (<500 cP) with high refractive indices (nD/20°C ~1.64) and high optical quality have been demonstrated by fabricating aspherized plano-convex lens by an overmolding procedure to increase the optical power and numerical aperture. Furthermore, a high-performance holographic recording medium developed by these thiol and ene monomer formulations resulted in a holographic material with high dynamic range of 0.04 with haze as low as 0.2%.195
As the refractive index of the thiol-X network is directly proportional to the sulfur content, incorporation of more sulfide linkages by using analogous thiol-yne systems significantly enhances the refractive index development.414 In combination with orthogonal thiol-Michael addition click reactions, Peng et al. employed the sequential photoinduced thiol-Michael/thiol-yne click polymerization approach to implement direct image patterning with high diffraction efficiency (96%), index modulation of 0.004 with low volume shrinkage (1.1%).415
3.5.5. Hydrogels and Tissue Engineering
3.5.5.1. Hydrogel Synthesis
Hydrogels are highly swollen polymer networks that have become useful platforms to study a wide variety of biological systems in vitro and as injectable scaffolds in vivo.416 The use of light-activated click reactions has allowed for control over the rapid synthesis of hydrogel networks, enabling precise spatiotemporal control over network formation and for the introduction of biomolecules.417 One of the most popular uses of photoclick hydrogels is for the three-dimensional (3D) encapsulation of cells permitting them to experience a 3D environment which more closely resembles their native extracellular matrix (ECM) allowing researchers to have a better model to understand cell-cell and cell-matrix interactions. With the use of low energy wavelengths of light, photoclick hydrogels have also become popular vehicles for the delivery of biologicals and as platforms to precisely pattered hydrogels with both peptides and proteins. Spatiotemporally controlled hydrogels and their applications in regenerative medicine have been recently comprehensively reviewed by Brown and Anseth.418 Here, we describe various photoclick reactions that have been used for hydrogel synthesis (e.g. thiol-ene, thiol-yne, thiol-Michael and tetrazole-ene) and highlight the power of light for their fabrication and for photopattering.
The photoclick thiol-ene reaction has become one of the most popular methods for the synthesis and post modification of hydrogels over the last decade as demonstrated by these recent reviews.417, 419–421 The popularity of the reaction is owed to its many advantages; (1) the thiol-ene step-growth polymerization reaction is frequently preferred over chain-growth polymerization reactions as it allows for the formation of more homogeneous network and is not inhibited by the presence of oxygen.45, 422 Therefore, minimal photointiator concentrations may be employed leading to a lower concentration of radicals in the presence of cells, improving cytocompatibility and enabling the encapsulation of cells, tissues and proteins.423 (2) The hydrogel formation rate is tuned in various ways; using a range of different photointiators, wavelengths of light, light intensities and photoinitiator concentrations. (3) Facile incorporation of peptides and proteins as crosslinks and pendant bioactive moieties is possible via utilization of cysteine thiols. Through the use of photoclick reactions these fabrication methods are highly tunable and applicable to ranges of cell lines and applications. These advantages allow for milder conditions to be used for encapsulation and lower cell densities compared to chain growth reactions. Recent work using thiol-ene hydrogels has demonstrated the use of these materials for a variety of exciting applications for cartilage development, antimicrobial surfaces and as controlled drug delivery vehicles (Figure 85).424–428
Figure 85.).
VonKossa MacNeil stained longitudinal sections of the femur defect area at 12 weeks. A sample bridged partially by fibrous tissue (left). A sample bridged by combination of bone and cartilage (right). The poly(propylene fumarate) scaffold stains light blue (indicated by yellow arrows), mineralized tissue stains black (indicated by pink arrow), cartilaginous material stains blue/purple (indicated by orange arrows) and fibrous tissue stains a light blue (red arrow). At the 12 week timepoint there was no evidence of the thiol-ene hydrogel macroscopically or microscopically. Endochondral ossification of cartilage is highlighted in the expanded view of the sample on the right. Reproduced, with permission, from reference 426. Copyright 2019 John Wiley and Sons.
It should be noted that the thiol-ene reaction cannot strictly be considered perfectly biorthogonal as the alkene readily reacts with a thiol-containing natural amino acid, cysteine. However, the natural abundance of cysteine is low and is normally present as disulfides so does not interfere significantly with hydrogel synthesis.429
In comparison to its thiol-ene counterpart, the photo-triggered radical thiol-yne reaction has received dramatically less attention as a route to synthesize biomaterials. Through the use of the thiol-yne reaction the alkyne is able to undergo two consecutive thiol additions to form a dithiol adduct. There are a few accounts of the thiol-yne reaction for hydrogel synthesis. One example demonstrated by Daniele et al. shows the synergistic effects of the step-growth and chain growth polymerization reactions.430 They use a tetrafunctional PEG thiol-yne system with a gelatin methacrylate to tune the characteristics of the interpenetrating network. They state that this system allows for a more representative mimic of the ECM as it mimics the more complex structure and behavior. Although this system does not exploit the advantages of using photoclick reactions it highlights the ability for this reaction to work alongside other photopolymerization reactions.
Recent advances in the Michael-type thiol-yne reaction have demonstrated the nucleophilic addition reaction with activation using green light. Truong and coworkers have utilized boron-dipyrromethane (BODIPY) moieties with pendant thioether groups that undergo photolysis in the presence of green light (λ = 530 nm) excitation.431 The liberated free thiolate ion can then undergo a nucleophilic addition with an activated alkyne to form an alkene thioether with controlled stereochemistry. The authors demonstrated this photoclick reaction for hydrogel synthesis using star PEG macromers functionalized with either the BODIPY thioether or alkyne groups, which rapidly forms a hydrogel upon irradiation of light (λ = 530 nm, 30 mW cm−2). The green light-induced ligation has been shown to pattern hydrogels and for cell encapsulation demonstrating the low toxicity of green light.
Although the tetrazole-ene reaction has been used extensively for labeling proteins and for molecular probes, it has not been significantly used for hydrogel fabrication. Zhong and co-workers used the reaction to synthesize hydrogels demonstrating rapid gelation times with low energy light, an advantage of this photoclick reaction.432 A unique characteristic of these hydrogels is their fluorescent nature through the formation of a pyrazoline ring, aiding the imaging of photopattered regions but could hinder the imaging of cellular structures. The reaction has also been used to modulate self-assembled hydrogels through stacking of short peptide chains with tetrazole functionality.433 In this study, the tetrazole-ene reaction is used to modulate the properties of the hydrogel with encapsulated hMSCs.
Although most photoclick reactions utilize UV light, it is not the optimal wavelength to use for biological applications as the high energy wavelengths are damaging to peptides, enzymes and DNA. For photoclick reactions to become more widely used in biomedical applications, the low-energy long wavelengths of light should be used. The optical therapeutic window is between 600–900 nm where there is minimal adsorption of light by water and tissue. Truong et al. addressed this issue by using 625 nm light for the catalytic activation of IEDDA under physiological conditions (Figure 86).42 They used multiarm PEGs functionalized with tetrazines and norbornenes and demonstrated the in situ hydrogel formation behind dermal tissue models suggesting that the system could be used as a controlled injectable hydrogel model into skin. The group stated that this method would allow for better control over gelation than spontaneously forming gels as the longer wavelengths of light could penetrate deeper into the tissue without damaging the tissue.
Figure 86.
a) iEDDAC network formation using red light (b) Picture of agarose gels used as a dermal model. (c) Change in the storage modulus with the exposure to light. Reproduced, with permission, from reference 42. Copyright 2017 American Chemical Society.
Adzima et al. were the first to demonstrate the synthesis of hydrogels using photointiated copper catalyzed azide-alkyne cycloaddition reaction (photo-CuAAC).434 Cu(II) was reduced to Cu(I) through radicals generated by a photo-activated initiator. Gelation between a multi arm PEG azide and PEG dialkyne then took place in a controlled manner only in areas which had been exposed to light. By using an excess of alkynes in network formation the researchers demonstrated in situ fluorescent photopattering through the addition an azide fluorophore. Using this technique, they determined that 25 um features could be formed with this technique with 50 seconds of irradiation. This photoclick reaction combined the advantages of CuAAC reactions (i.e., bioorthognal and fast reaction kinetics) and may mitigate its significant drawback, the toxicity of Cu(I), by photo-generating Cu(I) only in user-designated regions. However, as the reaction still relies on the use of Cu(I) catalyst other photoclick reactions with lower toxicity levels are still preferential for biomaterial applications. This preference notwithstanding, this photoclick reaction has recently been implemented in the synthesis of shape memory hydrogels.435 The click nature of the reaction allowed for facile control over the thermoresponsive units feed ratio, controlling the mechanical properties of the hydrogel above and below the LCST.
3.5.5.2. Post-cure Modification of Hydrogels
An important application of synthetic hydrogel scaffolds is to create ECM mimics. The ECM is dynamic and responsive to external stresses from the surrounding environment including increased stiffness with disease progression. To observe these dynamic processes in vitro, hydrogels have been created that respond to external stimuli (e.g. the stiffening of a scaffold to mimic disease progression). Light has been used as an external stimulus to increase the stiffness in hydrogels by increasing crosslinking density through various light activated reactions. In particular the radical thiol-ene reaction has been used to in situ stiffen hydrogel materials to mimic disease progression.436–438 Mabry et al. showed that in situ stiffening hydrogels using subsequent thiol-ene crosslinking enabled a better understanding of myofibroblast to fibroblast transition.439 Discrete regions of stiffness are also created in hydrogels to further explore medchanobiology.440–442 The discrete nature of the light activated reaction allows for remarkable control over the stiffness of the material.443 The popularity of the thiol-ene reaction for postmodification of hydrogels reflects the reputation of this reaction for hydrogel fabrication and the subsequent ease of using these reactive handles for postmodifications.
In situ generation of reactive thiolate functionalities coupled with subsequent thiol-Michael click reactions has been used to control local stiffness in space, time and intensity of hydrogel matrix.444, 445 To this end, Mosiewicz et al. combined phototriggered uncaging chemistry and subsequent Michael-addition click reaction to spatiotemporally control the physical properties by tuning the crosslinking density of a poly(ethylene glycol) (PEG) hydrogel network, which directly translated to highly localized stiffness patterns of broadly tunable shape and stiffness range (Figure 87). Using this approach, user-defined stiffness patterns in a range of soft tissue microenvironments (i.e. between 3–8 kPa) were obtained and shown to influence the migratory behavior of primary human mesenchymal stem cells (hMSC).
Figure 87.
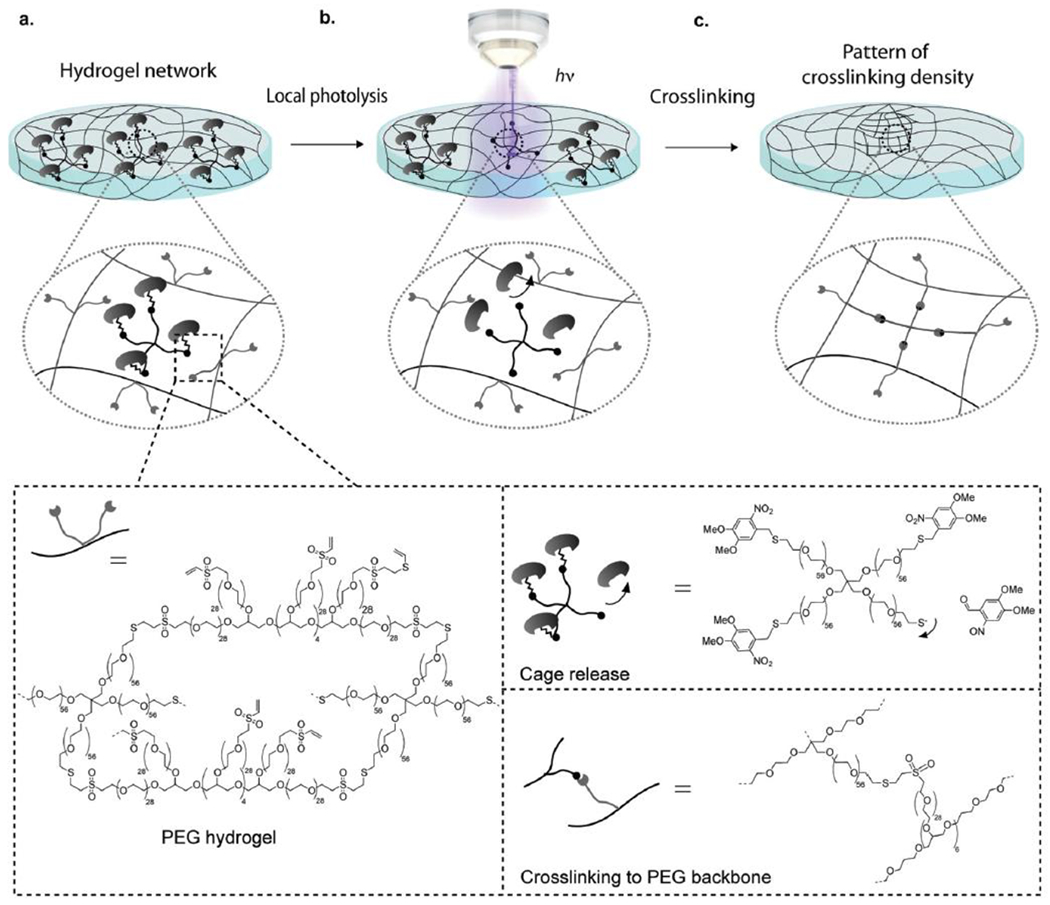
Concept of light-mediated Michael-type addition for spatiotemporally controlled hydrogel crosslinking; a, A thiol-containing macromer is protected with photo-labile caging group and is dissolved in a hydrogel network comprising free vinyl sulfone groups; b, localized laser illumination causes photocleavage of the caging groups reactivating thiols; c, activated thiol-containing PEGs react to vinyl sulfone via Michael-type addition reaction creating localized stiffness patterns. Reproduced, with permission, from reference 445. Copyright 2012 Royal Society of Chemistry.
Perhaps one of the most popular and valuable postmodification applications of hydrogels is the ability to photopattern proteins and small molecules into the network after formation. The use of photoclick reactions for this process affords spatial control of ligand immobilization through the use of photomasks and the discrete nature of light. In addition, the use of photoclick reaction after crosslinking allows for attachment of sensitive moieties in a homogeneous manner and with high resolution control over patterning.43 As mentioned in the hydrogel fabrication section the ability to add cysteine pendant peptides into thiol-ene hydrogel networks has been demonstrated many times, to allow for cell adhesion and motility.446 The use of the thiol-ene reaction for the controlled presentation of biomolecules in hydrogels was reviewed by Grim et al. and highlights the ability to use the reaction for biomolecule patterning and the subsequent photocleavage patterning and reversible patterning that can achieved with these hydrogels.417
Hydrogels can also be photopattered through the combination of two photoclick reactions. Batchelor used a thiol-ene reaction to form a hydrogel network and a subsequent tetrazole-ene reaction to further functionalize the hydrogel with peptides to encourage cell attachment (Figure 88).447 They exploit the use of the self-reporting fluorescent pyrazole ring to show successful ligation of the peptide. The researchers also used riboflavin instead of LAP as the photoinitator in the thiol-ene reaction to enable the use of visible light (455 nm) to initiate the reaction and UV light (326 nm) to activate the tetrazole-ene reaction. Through the use of lithographic techniques, the hydrogels were functionalized in a spatially resolved fashion allowing selective cell adhesion in the irradiated sections.
Figure 88.
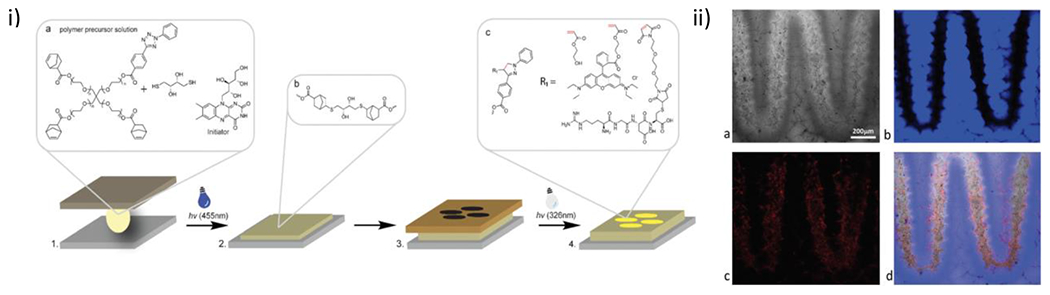
i) General procedure for the fabrication and functionalization of the PEG based hydrogels. (1) Precursor solution placed between a methacrylate functional glass slide and PDMS cover. (2) Irradiated under blue light to obtain the crosslinked gel. (3) Gel swollen with acrylate solution and photomask placed on top. (4) Irradiated under UV light to obtain patterned sections. (a) Hydrogel precursor solution. 1 : 1 equivalent of DTT and norbornene moieties. (b) Crosslinks consist of a double thioether linkage. (c) Chemistry of the photopatterned areas. ii)Surface adhesion of NHDF cells. (a) DIC image of NHDF cells after 24 h incubation on the hydrogel surface. (b) Fluorescence confocal image of the photopatterned section of the gel (photopatterned areas appear black). (c) Fluorescence confocal image of NHDF cells stained with propidium iodide. (d) Merged image of (b) and (c). Reproduced, with permission, from reference 447. Copyright 2018 Royal Society of Chemistry.
Recently in 2020 Ma et al. have used the thiol-yne reaction alongside SPAAC and iEDDA photoclick reactions to photopattern proteins into hydrogel networks.448 The system was used to study the interactive effects of proteins, in particularly TGF-B, BMP-2 and FGF-2, on valvular institute cells (VICs) activation as a model platform to further understand the aortic valve niche. The authors were able to use the photointiated thiol-yne reaction to sequentially incorporate moieties for protein immobilization. The thiol-yne reaction was the only choice for this work to allow for the reaction to take place without interfering with the other click reactions, SPAAC and IEDDA.
Spatiotemporally controlled release of caged thiols also promoted specific immobilization of biomolecules including photopatterning.449 For example, in their earlier studies Shoichet and coworkers used S-2-nitrobenzyl protected cysteine to create spatially defined protein patterns in agarose-based 3D hydrogels by selectively activating the thiol functional groups for thiol-maleimide mediated biomolecule conjugation.450, 451 For this purpose, the pendent primary hydroxyl groups on the dissolved agarose were modified with o-nitrobenzyl protected cysteine using carbodiimide coupling chemistry. After the gelation, the photocaged agarose gels were irradiated with 266 nm UV light to release free thiolate functionalities, which were used to immobilize maleimide-functionalized peptides or proteins via a subsequent via thiol-Michael click reactions only at the irradiated regions.
Shoichet and coworkers used 6-bromo,7-hydroxycoumarin sulfide modified agarose hydrogel for highly efficient 3D patterning of maleimide modified Alexa Fluor 488 and other biomolecules without causing any cross-linking or change in physical properties of the hydrogel (Figure 89).452 The uncaging of the nucleophilic thiolate functionalities was achieved upon irradiation with UV or pulsed multiphoton excitation.
Figure 89.
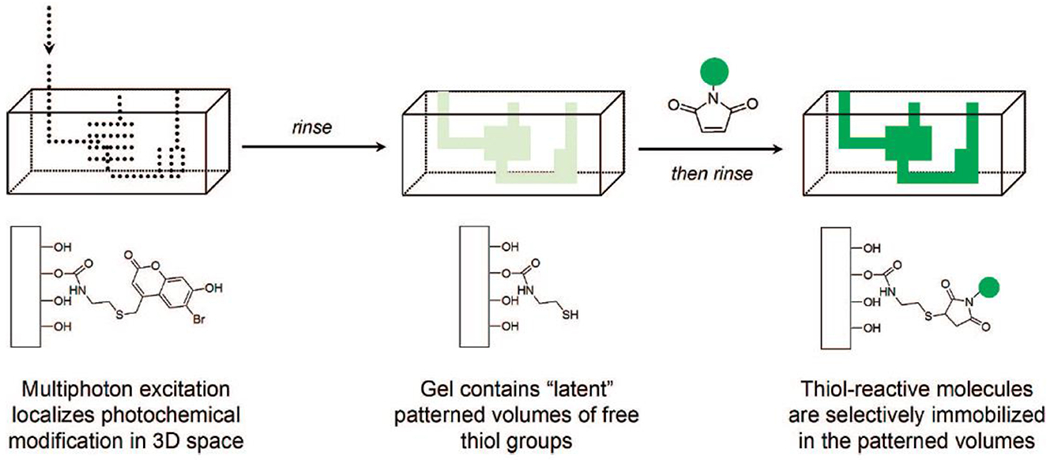
Schematic representation of multiphoton chemical patterning in hydrogel. Reproduced from reference 452. Copyright 2008 American Chemical Society.
In other work the authors modified an agarose matrix with the same photolabile 6-bromo-7-hydroxycoumarin-protected thiols in order to photopattern the conjugation of maleimide modified human serum albumin (HSA-maleimide).453 After binding the HSA to the matrix, fibroblast growth factor-2 (FGF2) was immobilized by forming stable complex with ABD fusion protein with the photopatterned HSA.
In another example, Liu et al. demonstrated a highly cytocompatible gelation approach to fabricate 3D ECMs with precisely controlled size, shape, and stiffness via light-controlled thiol-Michael addition cross-linking.454 In this approach, the authors used a methacrylate copolymer containing pendent macrocyclic 7-amino coumarin-caged thiol and polyethylene glycol side chains. Upon photo-excitation, intramolecular photouncaging of macrocyclic coumarin caged thiol and subsequent reaction of liberated free thiols with multifunctional PEG maleimide produced 3D hydrogels. The combined use of 7-amino coumarin and nitrobenzyl caging groups for thiols was thus used for wavelength selective release of free thiols in a one pot reaction approach. For example, Liu et al. used the different excitation of coumarin and nitrobenzyl phototriggers at 420 nm and at 365 nm, respectively, to achieve wavelength-selective photodeprotection of thiols at different locations on the same polymer backbone.455 The different thiols generated this way were used for the bioconjugation of two different functional materials (QDs and proteins) on the same polymer chain to produce QDs–polymer–protein conjugates in one pot (Figure 90).
Figure 90.
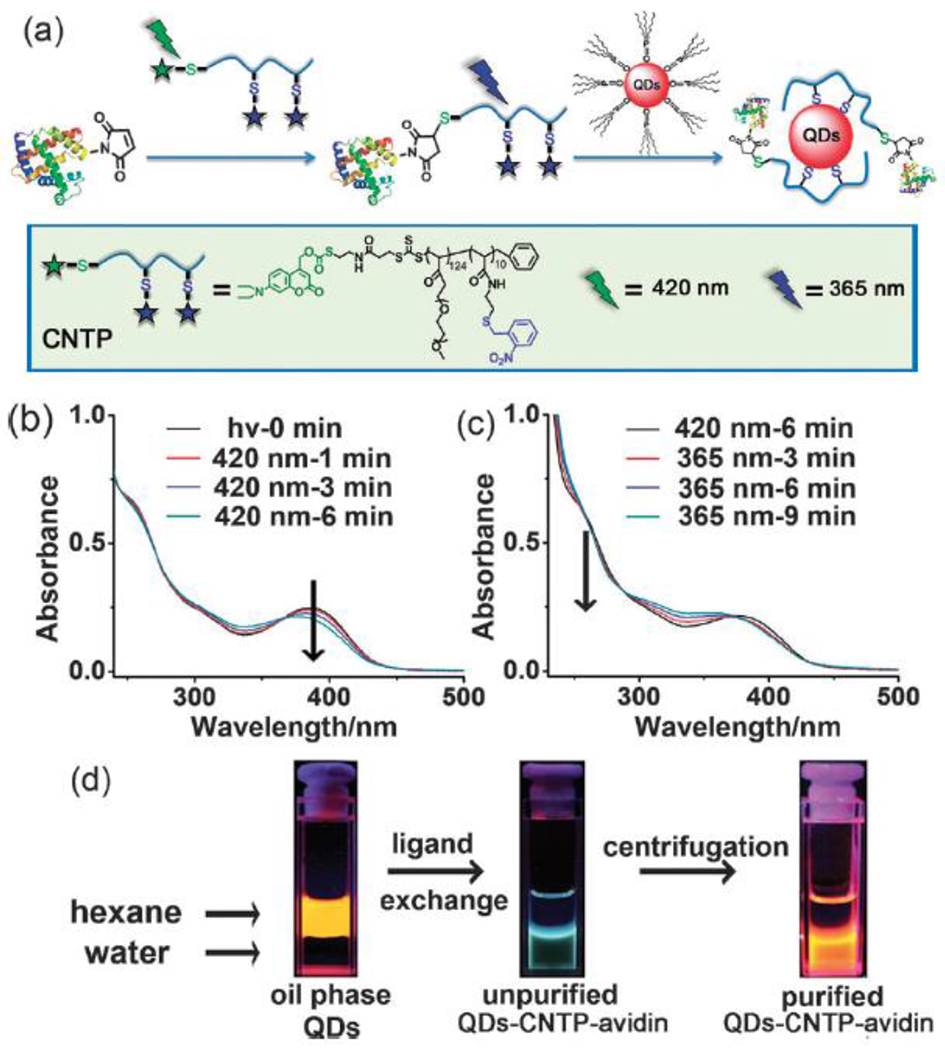
a) Schematic representation of sequential photorelease of thiols and bioconjugations with protein and QDs in one-pot process. (b, c) UV-vis spectra of CNTP (PBS, pH = 7.4) upon 420 nm irradiation and followed 365 nm irradiation. (d) Pictures are TOPO-QDs, unpurified QDs–CNTP–avidin and purified QDs–CNTP–avidin after centrifugation (from left to right) Reproduced from reference 455. Copyright 2014 Royal Society of Chemistry.
The authors used a well-defined, hydrophilic polymethacrylate block co-polymer in which 7-amino-coumarin and o-nitrobenzyl phototriggers were used to protect the end- and pendant-thiols, respectively. Upon excitation at 420 nm, the absorption of the 7-amino-coumarin phototrigger caused the release of thiol at the end of the polymer chain to trigger the subsequent reaction with maleimide functionalized protein, whereas the o-nitrobenzyl phototrigger remained intact. Following this reaction, the exposure to 365 nm light photo released the pendent thiol functionalities via niitrobenzyl photocleavage and subsequently captured the QDs from the oil phase by ligand exchange.
Surface patterning of hydrogels has also been shown using the photoinitated CuAAC reaction. Chen et al. used Irgacure 2959, CuSO4∙5H2O to surface pattern a PEG hydrogel on an azide-functionalized surface.456 This approach allowed the researchers to control the thickness of the hydrogel and modulate cell attachment using a photomask and UV irradiation.
The photoSPAAC reaction has also been used to pattern hydrogels. Bjerknes et al. selectively labelled hydrogels through incorporation of a cyclopropenone-caged cyclooctyne.457 The authors demonstrated the uniformed labelling of the network using 365nm light and an azide functionalized dye displaying the reaction as a powerful photopatterning tool.
Photopattering hydrogels with photoclick reactions has been demonstrated for a range of applications and cell types, albeit with a significant focus on thiol-ene reactions. Although the area has focused on this reaction, as a consequence of their accessibility and ease of synthesis, the full spectrum of photoclick reactions could be applied to this application. It is envisioned that as the full range of photoclick reactions are increasingly used in hydrogel synthesis, they will also find applications in the orthogonal control of heterogeneous mechanical and biochemical properties of biomimetic hydrogels.
3.5.5.3. Bioprinting
Photoclick reactions lend themselves to applications in bioprinting as a consequence of their biorthogonal nature and spatiotemporal control. Over the last decade, chain growth polymerization reactions have dominated the field especially through the use of gelatin methacrylate. However, this type of polymerization is affected by oxygen inhibition, heterogeneous polymer networks and complex polymerization kinetics.458 In recent years photoclick reaction have been seen as an alternative route for bioprinting. The thiol-ene, thiol-yne and thiol-Michael reactions have been used for extrusion based printing and stereolithography as the step-growth nature of the reactions forms homogeneous polymer networks at high yields with rapid reaction rates.156
The thiol-ene reaction is becoming commonly adopted to bioprinting as its significant advantages over radical chain polymerizations are becoming evident in various applications. Blood vessel-like structures have been printed through crosslinking thiolated hylaronic acid with 4 arm PEG tetra acrylates. The authors used extrusion based printing enabling the encapsulated of NIH 3T3 cells within the printed tubular tissue constructs. The culture was viable for 4 weeks and demonstrated the feasibility of building controlled bioartificial constructs through the use of light.459 Norbornene functionalized alginate crosslinked with PEG dithiol has also been used to print scaffolds with ATDC5 and L929 cells.428 Through the use of the thiol-ene system they were able to also incorporate a thiol functionalized RGD sequence to improve cell adhesion within the construct.
More recently, Bertlein et al. and Stichler et al. have used thiol-ene biofabrication technology to create scaffolds.460, 461 Stichler used a synthetic poly(allyl glycidyl ether-co-glycidyl) (P(AGE-co-G)) and poly(glycidol) to create hydrogel precursors with alkene and thiol groups showing an example of biofabrication with a PEG based system. The structures were cytocompatible through the use of the photoclick reaction and showed good reproducibility. In 2018 the Burdick group demonstrated 3D extrusion bioprinting of single and double network hydrogels with dynamic covalent crosslinks.462 The authors successfully printed a hyaluronic hydrogel with shear thinning behavior owing to the hydrazine chemistry of the network. The second network used a photoclick reaction and was composed of a norbornene functional hyaluronic acid combined with a tetrathiol crosslinker. This thiol-ene system allowed for patterning and increased mechanics of the original network as a consequence of the spatiotemporal control that the photoclick reaction enables. Although these techniques demonstrate a range of materials and applications of 3D bioprinting they are restricted to low resolution. Liska and co-workers demonstrate the versatile of alkenes and thiols for 2 photon microfabrication demonstrating that gelatine-vinyl esters, reduced bovine serum albumin (BSA), DTT or PVA-thiols can be used for biofabrication (Figure 91).463–465 Recently Dobos et al. reported the use of a photoclick reaction for lithography-based approaches to enable the production of intrinsic architectures and complex geometries.466 The group used two photon polymerizations for sub-micrometer spatial resolution to print norbornene functionalized gelatin crosslinked with dithiolthreitol (DTT). This was the first example of 2 photon polymerization processing while maintaining cell viability which had previously only been obtained with gelatin methacrylate.
Figure 91.
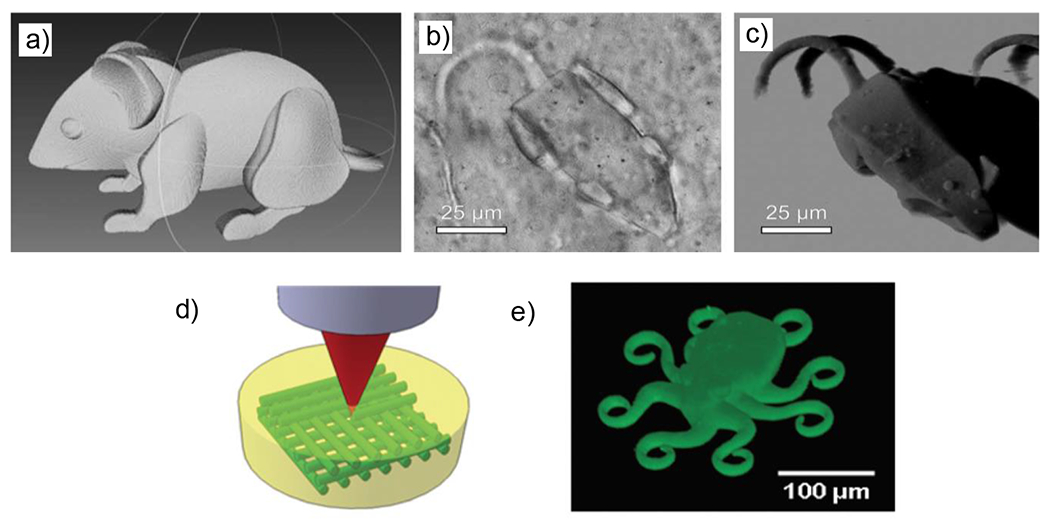
Mouse as a test structure: (a) CAD model, 3D printed N19T06–060 visualized by (b) light microscope and (c) laser scanning microscope using PVA thiol-ene material. Adapted, with permission, from reference 465. Copyright 2016 John Wiley and Sons. d) schematics of the 2PP microfabrication; (e) z-stacked laser scanning microscope image of a 3D “Octopus” construct produced by 2PP of HA-VE/DTT. Reproduced, with permission, from reference 464. Copyright 2014 Royal Society of Chemistry.
Overall the photoclick thiol-ene systems offers many advantages to biofabrication including the ability to form more homogenous networks in comparison to chain growth polymerization reactions which are currently widely used in this area. The benefits of this system suggest that it will be used more widely in the future especially for cell laden materials.
The thiol-yne reaction has not been used to its full potential in bioprinting. One rare example was reported by Skardal et al. who used thiol-yne chemistry to form a secondary crosslinking network to form a stable hydrogel structure after printing.467 The group first used a thiol-acrylate system that reacted spontaneously to form a soft extrudable gel to bioprint. They then added a PEG alkyne and used UV light to photoclick the networks together and increase the stiffness of the network to match tissue (100 Pa to 20 kPa). The use of the thiol-yne reaction here exploits the bifunctionality of each alkyne, enabling a rapid controlled increase in strength. They highlight that the modular nature of this hydrogel bioink could be adapted for multiple tissue types.
Although most of the photoclick reactions have been employed in hydrogel fabrication they have not been applied to 3D biofabrication. This includes thiol-Michael, photo- strained promoted azide-alkyne cycloadditions and tetrazole-ene.468 While diffusion of photoliberated base catalyst in thiol-Michael reactions may mitigate, in some bioprinting applications, the spatial control over the reaction. The spatial resolution offered by non-photoamplified reactions would seem to be a good fit for such photo-mediated 3D printing techniques, providing adequate depth of cure for the printing techniques employed. In contrast to the fabrication of thicker materials, the relatively high chromophore concentrations and consequent light attenuation would not preclude the application of such chemistries in thin layers. It is speculated that these reactions have not been used for biofabrication because of the synthetic challenges the precursors present. Many of these photoclick reactions are not commercially available and require many synthetic steps to reach the desired product. The cost and yield of these reaction also prevents large quantities of these reagents to be synthesized. This limits the reaction’s use in bioprinting as this application often requires large amounts of reagents. With the increase in popularity and the improvement in commercial availability of easily coupled photoclick moieties, the spatiotemporal advantages of these reactions may be more fully recognized in 3D bioprinting.
3.5.6. Microparticles and Nanoparticles
Polymeric microparticles have broad utility in materials science. They are used in paints and coatings, adhesives, cosmetics, solid supports for chemical syntheses, biomedical diagnostics, drug delivery as well as many other fields.469 As described previously, photoclick reactions, including thiol-ene and tetrazole-ene reactions, have been used effectively to modify the surface of microparticles, allowing independent dictation of the surface and bulk chemistry respectively. Photoclick reactions may also be used to generate microparticles. There are a variety of means of synthesizing microparticles, including emulsion polymerization and microfluidic formation of monomer microdroplets. The quality of the microparticles obtained via these methods is diminished if the polymerization is slow, allowing droplets to fuse or emulsions to break. Rapid rates of reaction make thiol-ene and thiol-yne reactions especially effective in such processes. Microfluidic devices, discussed previously, also provide a means to generate highly uniform droplets of monomer that can be rapidly photocured to create highly uniform microparticles.
This discussion of microparticles and nanoparticles is divided into a description of those materials with relatively high crosslinking density and highly swollen microgels and nanogels.
3.5.6.1. Highly Crosslinked
Some of the earliest work on thiol-ene and thiol-yne microparticles was performed by Prasath et al. who synthesized a variety microparticles by photopolymerization of monomer droplets passing through microfluidic channels.470 Monomer formulations included a tetrathiol and mono and multifunctional alkenes or alkynes along with a photoinitiator and, in the synthesis of porous microparticles, a porogen. Hydroxy and amine containing alkenes and alkynes permitted subsequent surface attachment suggesting potential for use in solid-supported chemical synthesis. Similar microparticles with thiol-ene and thiol-yne photoclick reactions have been obtained by Shipp and coworkers,471, 472 as well as Gao and coworkers473 via suspension polymerization, a process generally perceived as more scalable than microfluidic fabrication. Alimohammadi et al. have also used dispersion polymerization, in which a homogenous solution of monomers phase separates into solvent and microparticles during polymerization, to generate microspheres via thiol-ene and thiol-yne photoclick reactions.474 Photobase mediated thiol-Michael click reactions have similarly been used to generate very narrowly disperse microparticles via dispersion polymerization.475
Tetrazole-ene reactions have also been used to synthesize particles which can be modified post-synthesis. For example, Hooker et al. used tetrazole-alkene reactions to generate surface functionalizable microspheres via the off-stoichiometric crosslinking reaction between complementary tetrazole and acrylate-bearing polystyrene chains,476 an extension of the groups work generated surface-funtionalizable nanospheres via radical polymerization of acrylic-modified, photoclickable tetrazoles, discussed earlier.350
Recently, Barner-Kowollik and coworkers have also developed highly uniform microparticles using Diels-Alder photoclick chemistries (section 2.2.1).477 The group has also utilized a self-reporting Diels-Alder photoclick reaction between ortho-methylbenzaldehydes and electron deficient alkynes using UV light (λ = 320 nm) to generate fluorescent microparticles478 a system which that group has pioneered.40 By using visible light (λ = 525 nm) they were also able to synthesize narrow disperse microparticles using a naphthalene- triazolinedione [2+4] photocycloaddition reaction.479 The group has highlighted the flexibility of the Diels-Alder reaction for the synthesis of materials with small dimensions using a range of different wavelengths of light highlighting the versatility of these Diels-Alder cycloaddition reaction.
3.5.6.2. Microgels and Nanogels
Water swollen microgels (typically 1-350 μm) and nanogels (typically 20-250 nm) have become popular biomaterials for applications in drug delivery, bionanotechnology and biomedical implants.480 Due to their hydrophilic network, nanogels and microgels exhibit high swelling, good biocompatibility and defined mechanical and diffusional properties.481 This allows for precise loading and release profiles which can be further tuned by degradation and postmodification reactions.
The thiol-ene reaction was first used to make nanoscale hydrogels in 2014 using the aqueous cavity of a liposome to act as a nanoreactor.482 Hachet et al. demonstrated the synthesis using norbornene functionalized hyaluronic acid and PEG dithiol to form uniform nanogels with tunable swelling properties. Since then, radical thiol-ene reactions have also been used to form peptide functionalized microgels to sequester and release vascular endothelial growth factor (VEGF).483 This approach, using a water-in-water emulsion to control the size, relies on the rapid kinetics and efficiency of the photoinitiated thiol-ene reaction for batch microparticle synthesis. Building on this work Alge and co-workers used the same photoclick chemistry to form microgels with excess norbornene groups that could be used to functionalize the microgels with tetrazine functionalized proteins.484 Alkaline phosphatase and glucose-oxidase were used to demonstrate the utility of this process with bioactive proteins and quantitively assess microparticle bioactivity. Recently Xin et al. have developed this work further, using the same thiol-ene chemistry to synthesize microgels but through a microfluidic device to create gradients in microporous annealed particle (MAP) hydrogels.485 These gradients allowed for a unique insight into cell-material interactions as demonstrated with hMSCs that had differential spreading depending on the degradability and stiffness gradient of the particles (Figure 92).
Figure 92.
(a) Schematic of the microfluidic device-based microgel production procedure. (b) Schematic illustrating microgel extrusion into a 3 mm rectangular mold and microgel annealing into scaffolds. (c) Image showing gradient MAP scaffolds injected into a mouse femoral defect. (d) Z projection images of hMSC proliferation and spreading in MAP scaffolds with either a stiffness gradient (top) or a degradability gradient (bottom). Green represents F-actin and blue represents nuclei. Reproduced, with permission, from reference 485. Copyright 2020 John Wiley and Sons.
Thiol-ene microgels have also been postmodified using tetrazole-ene chemistry through utilization of norbornene functionality. Michel et al. synthesized microgels by crosslinking norbornene functionalized chitosan with a PEG dithiol in a low-energy water-in-oil nanoemulsion templating method.486 They demonstrated that the microgels were nontoxic to human dermofibroblasts (HDF). The researchers then exploited the tetrazole-ene reaction to react unreacted norbornenes with a tetrazine coumarin to increase the fluorescence for ease of tracking. The researchers state that the coumarin increases the florescence of the pyrazoles ring by up to 10 times making the microgels ideal for cell targeting moieties. Alternatively, the thiol-ene reaction can be used to postfunctionalize nanogels and microgels that have been synthesized through other methods. Huang et al. synthesized PMMA microgels using a RAFT-assisted dispersion photopolymerization technique to control the number of vinyl groups present on the particles.487 A thiolated biotin was conjugated to the surface of the microgels through a photoinduced radical thiol-ene reaction. The biotinylated PMMA microgels could then effectively immobilize fluorescent streptavidin demonstrating the ability to make controlled monodispersed functional microgels through thiol-ene reactions.
Nanogel postmodification has also been demonstrated using the thiol-yne reaction to form a nanogel coated monolayer.488 The PEG nanogels were synthesized through a CuAAC crosslinking reaction with excess alkyne groups. A thiol-yne reaction was then carried out between a thiolated surface and the nanogels with exposure to UV light and I-2959 to form a 100% PEG monolayer. The formed layer was superior for producing a protein resistant coting compared to other reaction because of the bioorthogonal nature of the reactions used.
An ideal nanogel application is as drug delivery vehicles with controlled and targeted release.489 Most drugs are hydrophobic and therefore are cleared out of the body quickly. By trapping drugs within hydrophilic nanogels, they can deliver their load efficiently to an area of interest and release in a controlled manner through a range of degradation routes built into the nanogel structure. This has been demonstrated numerous times in the literature with a range of techniques.480 However, as clearly demonstrated by Lochhart et al. photoclick reactions enable a facile way to synthesize and load nanogels.490 Very recently Graf et al. have also demonstrated this with PEG microgels to deliver therapeutic proteins to the alveolar sacs. Here PEG microgels offer the correct size and hydrophilic surface to prevent a macrophage response enabling efficient delivery. Thiol-ene reactions enabled fast, specific crosslinking in the presence of a therapeutic protein. Furthermore, this photoclick reaction has been used to encapsulate antibodies and growth factors into nanogels for controlled, target delivery.491, 492
For the production of fluorescent nanogels the self-reporting tetrazole-ene reaction has the obvious advantage of intrinsic fluorescence. This facilitates tracking nanogels and microgels in vivo without additional synthesis steps to label the material. This photoclick reaction has particularly found an application as a synthetic route to form fluorescent drug delivery vehicles owing to its fast reaction kinetics and specificity, enabling drug encapsulation without side reactions. Zhong and co-workers have used tetrazole-ene nanogels to improve the intracellular target of cancer drugs.493 The formation of a fluorescent pyrazoles ring through crosslinking, enabled the enhanced CT imaging of MCF-7 breast tumor in nude mice compared to iodoxanol, a clinically used contrast agent with non-specific distribution. The study demonstrated the excellent biocompatibility, high stability and effective targetability to CD44 overexpressing cells with these tetrazole-ene nanogels and presented the system in vivo for high tumor accumulation and improved survival rate. By loading PTX into the nanogels the systemic toxicity of the drug was reduced compared to free PTX and the fluorescent nanogels significantly improved imaging.
Photo-triggered thiol-Michael reactions have also been used for nanogel synthesis. Gao et al. demonstrated the compatibility of using 400–500 nm light to form nanostructures and 365 nm light to allow for bond exchange.494 In another example, Liang et al. utilized photo-triggered Michael-type additions of o-nitrobenzyl caged multi-arm PEG thiol and maleimides for liposome-templated production of lipid-coated polymer nanogels.495 Photo triggered release of PEG–thiols upon irradiation of 365 nm UV light and subsequent crosslinking permitted the formation of well-defined nanogels by providing temporal control of the reaction avoiding the macroscale gelation. The photo triggered formation of nanogels was monitored by oscillatory rheology experiments of bulk hydrogels, and the production of nanogels was confirmed via dynamic light scattering and transmission electron microscopy (Figure 93).
Figure 93.
Schematic representation of liposome templated synthesis of PEG-based nanogels via photo triggered thiol-maliemide click reaction in aqueous media (1) DI water, polycarbonate membrane; (2) 365 nm @ 10 mW/cm2, 2 h. and TEM images of the lipid-coated nanogels dried from aqueous solution. Reproduced, with permission, from reference 495. Copyright 2014 Royal Society of Chemistry.
Overall, the synthesis of nanogels and microgels using photoclick reactions is a growing area of research with exciting and impactful applications. The area is still new with many papers only published in the last several years. Therefore, the current area focuses heavily on thiol-ene and tetrazole-ene reactions because of their ease of synthesis and rapid reaction rates. There is a plethora of alkene and thiol functional groups that can be functionalized onto natural and synthetic polymers enabling the growth of these two reactions for the synthesis of nano- and micro- hydrogels. The advantages of photo-initiated CuAAC and SPAAC reactions are less beneficial to the synthesis of nanogels and microgels as many of the synthetic methods used require oil-water phases and high shear rates. The advantages of discrete areas of a liberated catalyst or ring strained alkyne are less beneficial for nano- and micro- hydrogel synthesis in comparison to other photoclick reactions.
3.6. Obstacles in Application of Photoclick reactions
Despite the successful application of photoclick reactions in such disparate fields of research, there are obstacles to the implementation of these reactions more broadly.Limited scalability of the reaction has been described briefly, and the requirement for exposure to light in any reaction may mean less effective mediation of that reaction where light cannot penetrate or where it is otherwise obscured by refraction. This may not be as big of an issue where photolatent catalysts, that persist in the absence of light are employed (e.g. with photo thiol-Michael reactions), but most of the reactions described herein cease completely in the absence of light. Moreover, light-absorbing chromophores on one or both reactive groups limit the ability to obtain optically thin reactions without extremely low concentrations or very small volumes. Flow reactors, as compared to batch systems, avoid constraints associated with light attenuation and allow for simple scale-up of photoinitiated polymerizations,496 which can be combined with orthogonal reactions (e.g. thermally driven polymer modifications) within the reactor to produce complex products in unprecedented yield and in essentially one step.497 Using a photoinduced Diels-Alder cycloaddition between o-quinodimethane thioether and maleimide, Feist et al. demonstrated full conversion of starting materials in a photoflow polymerization in aqueous solution and catalyzed by visible light from LEDs, modeling remarkable reagent and catalyst sustainability in a highly efficient polymerization system.329
Another obstacle to effective use of photoclick chemistry results from inadequate reporting of the reaction conditions as they apply to light exposure. It is not uncommon for papers to omit relevant information that would assist replication of experimental conditions. Such omissions might consist of the particular exposure wavelength and characteristics of the light source. Instead, general terms like “blue” or “UV light” are often used, in the methods description as well as in other sections of the paper. More frequently, however, it is the light intensity (with units of energy per area that is incident on the top surface of the sample) that is omitted. Lamp power may be noted, but that is entirely unuseful for repeating an experiment, hindering reproduction of reported results. Where low intensity, near UV light is employed in photoclick reactions, mass-produced black lights may be effectively used as a cheap alternative to specialized illumination products. However, the fluorescent tubes of such black lights may use one of two phosphores which, upon excitation, emit light centered at either 352 nm or 368 nm. Such a difference in emission may not be significant in recreational applications, but can be consequential when used to conduct photochemical reactions – as of course is the incident intensity at the sample which must be measured by a reliable radiometer tuned to the wavelengths of light used and reported. Further, reports of solar-initiated reactions frequently omit pertinent wavelength and intensity information. Solar output in terms of intensity and spectral output are subject to season and geography. In addition, details about the sample geometry are also critical as the sample thickness in the direction of exposure, the initiator concentration, and the presence of any other attenuating species must be included.
4. Conclusions
Rather than having a common mechanism, photoclick reactions are so categorized based on common features that enable their application in a wide variety of chemical and biological contexts: efficiency, selectivity, exogenous control, and so forth. Yet the commonality among these reactions can be overstated. Different photoclick reactions exhibit distinct advantages and disadvantages, the better understanding of which allows more effective implementation of such reactions to address the synthetic and materials challenges at hand for a particular reaction. Perhaps the most fundamental parameter that divides photoclick reactions is the role the chromophore plays in the reaction, whether its activation leads to the generation of the reactive species directly or whether its activation participates in a catalytic or otherwise photoamplified mechanism with the reactive species.
The general orthogonality of click reactions and photoclick reactions drives, to a considerable degree, the varied applications in which they may be employed. While there are many significant advantages to the use of photomediated click reactions, one that holds particular potential, particularly given the recent advances in discreet spectrum LED sources, is the utilization of multiple wavelengths as independent exogenous stimuli. Selective initiation of specific orthogonal photoclick reactions in a predetermined order via consecutive irradiation with different wavelengths has allowed the modular synthesis of complex chemical architectures and holds potential for the development of higher order structures as well. Expansion of this utility relies on development of chromophores for such reactions with non-overlapping regions of their respective absorbance spectra. While examples of photoclick reactions conducted with longer wavelengths of light are not exactly rare, it is the near UV and low wavelength, blue regions of the spectra (i.e. 350 nm – 420 nm) that are most heavily utilized. Furthermore, many of those reactions conducted at higher wavelengths employ inefficient multiphoton absorption or upconversion techniques that are impractical for all but the most niche applications. Continued development of highly efficient, longer wavelength photolatent catalysts, photoinitiators and reactive chromophores is key to the broader exploitation of the orthogonality of the sundry photoclick reactions in combination one with another as well as in situations requiring biologically most benign photoexposure.
The concept of photoclick chemistry has expanded the synthetic, polymeric, and biological research toolbox immensely in just a few short years and its continued relevance is anticipated well into the future.
Figure 8.
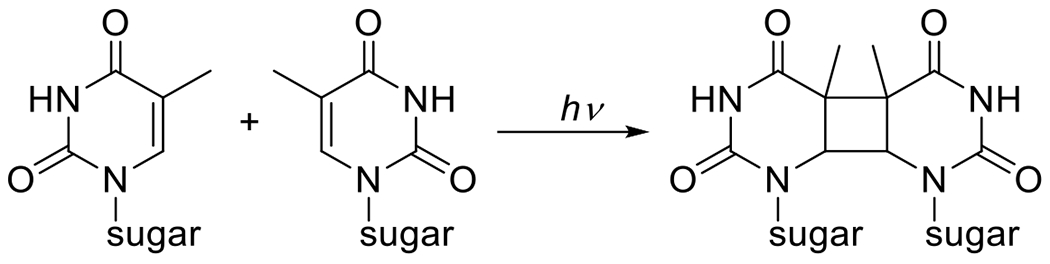
Photodimerization of thymine (shown) and cytosine in DNA has been implicated in the development of skin cancer.
Figure 28.
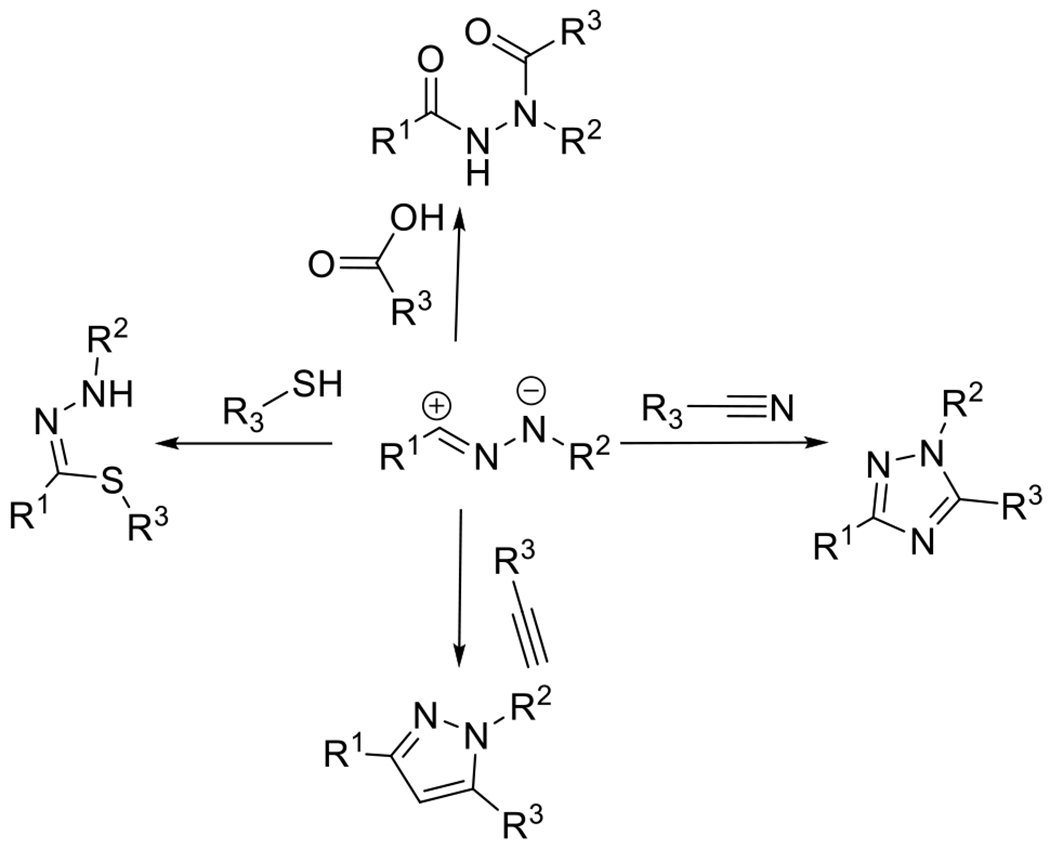
Reactions of nitrile imine 1,3-dipole with thiols and carboxylates as well as other dipolarophiles including nitriles and alkynes.
Figure 40.
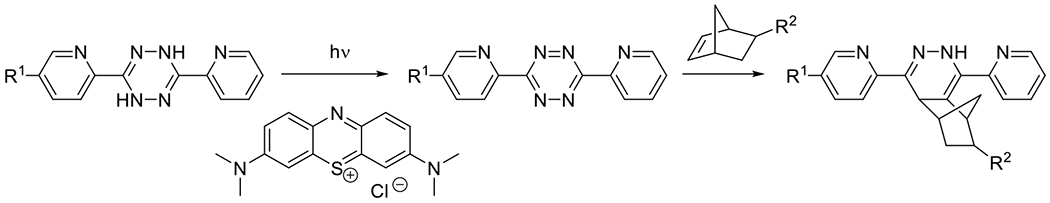
Conversion of dihydrotetrazine to tetrazine via methylene blue-photocatalyzed oxidation and subsequent IEDDA reaction with norbornene.42
Figure 64.
Nitrodibenzofuran has been shown to be an effective photocaging group for thiols.233
Acknowledgemetns
This work was supported by grants from the United States Defense Advanced Research Projects Agency (DARPA, grant W911NF-19-2-0024), from the National Science Foundation (NSF, grant CHE 1808484) and from the US National Institutes of Health (NIH, grant R01 DE016523). Graphical abstract created with Biorender.com
Abbreviations
- ABD
albumin binding domain
- AgBF4
silver tetrafluoroborate
- APT
alternating propagation and chain transfer
- ATRP
atom transfer radical polymerization
- BAPC
9-(2,3-bis(allyloxy)propyl)- 9H-carbazole
- BAPO
bis(2,4,6-trimethylbenzoyl)-phenylphosphineoxide
- BCN
bicyclononyne
- BLG
bovine beta-lactoglobulin
- BSA
bovine serum albumin
- CBOA
carbon-bridged octocyclic azobenzene
- CNA
click nucleic acid
- CNA-2G
click nucleic acid second generation
- Coumarin-TMG
coumarin-1,1,3,3-tetramethylguanidine
- CPG
controlled pore glass
- CT
computed tomography
- DA
diels alder
- DBN
1,5-diazabicyclo[4.3.0]non-5-en
- DBTD
dibenzo[b,f][1,4,5]thiadiazepine
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DCM
dichloromethane
- DFT
density functional theory
- DIBOD
dibenzo [a,e] cyclooctadiyne
- DLP
digital light processing
- DMBO
dibenzoannulated bicyclononynes
- DMF
dimethylformamide
- DMPA
2,2-dimethoxy-2-phenylacetophenone
- DMPP
dimethylphenylphosphine
- DMSO
dimethyl sulfoxide
- DNA
deoxyribonucleic acid
- DTT
dithiolthreitol
- DVS
divinyl sulfone
- ECM
extracellular matrix
- EGFR
epidermal growth factor receptor
- EWG
electron withdrawing group
- fac-Ir(ppy)3
(fac-tris(2-phenylpyridine)iridium)
- Fmoc
9-fluorenylmethyl carbamate
- GDMP
glycol dimercaptopropionate
- GST
glutathione S-transferase
- HA
hyaluronic acid
- HDF
human dermofibroblasts
- HOMO
highest occupied molecular orbital
- HPLC
high performance liquid chromatography
- HSA
human serum albumin
- IEDDA
inverse electron demand Diels–Alder
- ISC
intersystem crossing
- ITX
isopropylthioxanthone
- LAP
lithium phenyl-2,4,6-trimethylbenzoylphosphinate
- LED
light emitting Diode
- LUMO
lower unoccupied molecular orbital
- MAP
microporous annealed particle
- MSC
mesenchymal stem cells
- NCL
native chemical ligation
- NDBF
nitrodibenzofuran
- NIR
near-infrared
- NMR
nuclear magnetic resonance
- NPPOC
2-(2-nitrophenyl)propyloxycarbonyl
- NPPOC-TMG
2-(nitrophenyl)propoxycarbonyl-1,1,3,3-tetramethylguanidine
- NVOC
nitroveratryloxycarbonyl
- o-NB
o-nitrobenzylester
- PBS
phosphate buffered saline
- PcAAC
photocatalyzed azide-alkyne cycloaddition
- PCL
polycaprolactone
- PDLC
polymer-disperses liquid crystals
- PEG
Poly(ethylene glycol)
- PET
photoinduced electron transfer
- PETMP
pentaerythritol tetrakis(3-mercaptopropionate)
- PGB
photogenerated base
- PMDETA
N, N, N′, N″, N″- pentamethyldiethylenetriamine
- PMMA
poly(methyl methacrylate)
- pNIPAM
poly(n-isopropylacrylamide)
- PPE
personal protective equipment
- PPG
photoremovable protecting group
- PTX
paclitaxel
- PVA
poly(vinyl alcohol)
- QDs
quantum dots
- RAFT
reversible addition-fragmentation chain-transfer
- ROMP
ring-opening metathesis polymerization
- Ru(bpy)3Cl2
tris(2,2′-bipyridyl)-ruthenium(II) chloride
- Ru(bpz)3[PF6]2
Tris(2,2′-bipyrazine)ruthenium bis(hexafluorophosphate)
- RuAAC
ruthenium-catalyzed alkyne-azide cycloaddition
- SLA
stereolithography
- SPAAC
strain-promoted azide–alkyne click chemistry
- ssDNA
single stranded deoxyribonucleic acid
- TBD
1,5,7-triazabicyclo[4.4.0]dec-5-ene
- TBD.HBPh4
1,5,7-triazabicyclo[4.4.0]dec-5-ene tetraphenylborate
- TEA
triethylamine
- TEMPO
2,2,6,6-tetramethyl-1-piperidinyloxy
- TMEDA
N, N, N′, N′ - tetramethylethylenediamene
- TMG
1,1,3,3-tetramethylguanidine
- TPO
diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide
- TRITC
tetramethylrhodamine
- UV
ultraviolet
- VEGF
vascular endothelial growth factor
- VIC
valvular institute cells
- YAP/TAZ
yes-associated protein/transcriptional co-activator with pdz-binding motif
- Zn-TPP
zinc tetraphenylporphyrin
Biographies
Biographies
Benjamin D. Fairbanks received his BS in Chemical Engineering from the University of Oklahoma in 2004 and his PhD in Chemical Engineering from the University of Colorado in 2009, studying thiol-ene and thiol-yne polymerizations for biomedical applications. He worked on RAFT polymerization as postdoctoral research fellow and subsequently as a research scientist at CSIRO in Victoria, Australia until 2015, when he returned to the University of Colorado as a Senior Research Associate.
Laura Macdougall received her Ph.D. in Chemistry from the University of Warwick, UK, in 2018. She earned her Master’s degree from the University of York and is currently a postdoctoral researcher in Chemical and Biological Engineering at the University of Colorado Boulder under the mentorship of Professor Kristi Anseth. Her research interests are in the development of click hydrogel biomaterials for tissue engineering.
Sudheendran Mavila received his Ph.D. degree in Polymer Chemistry from the University of Stuttgart, Germany in 2011. He holds Masters degree in Organic Chemistry from Mahatma Gandhi University, Kerala, India and worked as Senior Scientist in Synthetic Organic Chemistry for four years at Syngene International Ltd, India. After completing the doctoral studies in Germany, he moved to Israel in the early 2012 as a postdoctoral fellow at Department of Chemistry at Ben-Gurion University of the Negev. Since January 2016, he has been working as a Postdoctoral Research Associate at the Department of Chemical and Biological engineering, University of Colorado-Boulder, USA. His research interests lie at the interface of Synthetic Organic Chemistry and Material Science.
Jasmine Sinha received her M.Sc. in chemistry in 2005 from IIT, Guwahati, and Ph.D. in chemistry in 2011 from the IIT, Bombay, India, studying clickable thiophene and amplified fluorescent polymers for its applications in sensing. After receiving her Ph.D., she moved to Johns Hopkins University as postdoctoral fellow under the direction of Prof. Howard Katz and focused on organic, hybrid, and interfacial materials for electronic devices. She is currently a postdoctoral associate in Department of Chemical and Biological Engineering at the University of Colorado (Boulder). Her current research efforts focus on designing monomers for crosslinked polymerizations.
Bruce E. Kirkpatrick is an M.D.-Ph.D. student in the Anseth Research Group at the University of Colorado. He received his B.S. in Chemical and Biological Engineering in 2017 from the University of Colorado Boulder, where he worked with Prof. Andrew Goodwin on the synthesis of mechanoresponsive click monomers and with Dr. Kavita Jeerage at NIST on quantification assays for biological applications of gold nanoparticles. His research interests involve the design and implementation of engineered systems to stabilize and preserve biological materials.
Kristi Anseth is the Tisone Professor of Chemical and Biological Engineering and Associate Director of the BioFrontiers Institute at the University of Colorado at Boulder. Dr Anseth earned her BS degree from Purdue University, her PhD degree from the University of Colorado, and completed postdoctoral research at MIT as an NIH fellow. Her research interests lie at the interface between biology and engineering where she designs new biomaterials for applications in drug delivery and regenerative medicine. She is an elected member of the National Academy of Engineering (2009), National Academy of Medicine (2009), National Academy of Science (2013)s, and National Academy of Inventors (2016). Dr. Anseth was also recognized with the L’Oreal-UNESCO for Women in Science Award in the Life Sciences in 2020.
Professor Christopher N. Bowman received his B.S. and Ph.D. in Chemical Engineering from Purdue University in 1988 and 1991, respectively. After receiving his Ph.D., he began his academic career at the University of Colorado in January of 1992 as an Assistant Professor. Since that time Professor Bowman has built a program focused on the fundamentals and applications of crosslinked polymers formed via photopolymerization reactions. He works in the broad areas of the fundamentals of polymerization reaction engineering, polymer chemistry, crosslinked polymers, photopolymerizations and biomaterials. Professor Bowman is currently the Patten Endowed Chair of the Department of Chemical and Biological Engineering as well as a Clinical Professor of Restorative Dentistry at the University of Colorado at Denver.
Footnotes
The authors declare no competing financial interest.
Contributor Information
Benjamin D. Fairbanks, Department of Chemical and Biological Engineering, University of Colorado, Boulder
Laura J. Macdougall, Department of Chemical and Biological Engineering, University of Colorado, Boulder; The BioFrontiers Institute, University of Colorado, Boulder
Sudheendran Mavila, Department of Chemical and Biological Engineering, University of Colorado, Boulder.
Jasmine Sinha, Department of Chemical and Biological Engineering, University of Colorado, Boulder.
Bruce E. Kirkpatrick, Department of Chemical and Biological Engineering, University of Colorado, Boulder; The BioFrontiers Institute, University of Colorado, Boulder; Medical Scientist Training Program, School of Medicine, University of Colorado, Aurora CO
Kristi S. Anseth, Department of Chemical and Biological Engineering, University of Colorado, Boulder; The BioFrontiers Institute, University of Colorado
Christopher N. Bowman, Department of Chemical and Biological Engineering, University of Colorado, Boulder; Materials Science and Engineering Program, University of Colorado, Boulder
5. References
- 1.Tasdelen MA and Yagci Y, Light-Induced Click Reactions, Angew. Chem. Int. Ed, 2013, 52, 5930–5938. [DOI] [PubMed] [Google Scholar]
- 2.Kolb HC, Finn MG, and Sharpless KB, Click Chemistry: Diverse Chemical Function from a Few Good Reactions, Angew. Chem. Int. Ed, 2001, 40, 2004-+. [DOI] [PubMed] [Google Scholar]
- 3.Barner-Kowollik C, Du Prez FE, Espeel P, Hawker CJ, Junkers T, Schlaad H, and Van Camp W, “Clicking” Polymers or Just Efficient Linking: What Is the Difference?, Angew. Chem. Int. Ed, 2011, 50, 60–62. [DOI] [PubMed] [Google Scholar]
- 4.Fairbanks BD, Love DM, and Bowman CN, Efficient Polymer-Polymer Conjugation Via Thiol-Ene Click Reaction, Macromol. Chem. Phys, 2017, 218. [Google Scholar]
- 5.Kärkäs MD, Porco JA, and Stephenson CRJ, Photochemical Approaches to Complex Chemotypes: Applications in Natural Product Synthesis, Chem. Rev, 2016, 116, 9683–9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turro NJ, Ramamurthy V, and Scaiano JC, Principles of Molecular Photochemistry, an Introduction. Sausalito, CA: University Science Books, 2009. [Google Scholar]
- 7.Yousif E and Haddad R, Photodegradation and Photostabilization of Polymers, Especially Polystyrene: Review, SpringerPlus, 2013, 2, 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song W, Wang Y, Qu J, Madden MM, and Lin Q, A Photoinducible 1,3-Dipolar Cycloaddition Reaction for Rapid, Selective Modification of Tetrazole-Containing Proteins, Angew. Chem. Int. Ed, 2008, 47, 2832–2835. [DOI] [PubMed] [Google Scholar]
- 9.Albini A, Some Remarks on the First Law of Photochemistry, Photochem. Photobiol, 2016, 15, 319–324. [DOI] [PubMed] [Google Scholar]
- 10.Einstein A, Über Die Von Der Molekularkinetischen Theorie Der Wärme Geforderte Bewegung Von in Ruhenden Flüssigkeiten Suspendierten Teilchen, Ann. Phys, 1905, 322, 549–560. [Google Scholar]
- 11.Mayerhöfer TG, Höfer S, and Popp J, Deviations from Beer’s Law on the Microscale – Nonadditivity of Absorption Cross Sections, PCCP, 2019, 21, 9793–9801. [DOI] [PubMed] [Google Scholar]
- 12.Berezin MY and Achilefu S, Fluorescence Lifetime Measurements and Biological Imaging, Chem. Rev, 2010, 110, 2641–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fleming KG, Fluorescence Theory, in Encyclopedia of Spectroscopy and Spectrometry (Third Edition), Lindon JC, Tranter GE, and Koppenaal DW, Editors. Academic Press: Oxford. 647–653, 2017. [Google Scholar]
- 14.McDonagh A, Palma L, and Lightner D, Blue Light and Bilirubin Excretion, Science, 1980, 208, 145–151. [DOI] [PubMed] [Google Scholar]
- 15.Büchi G and Goldman IM, Photochemical Reactions. Vii.1 the Intramolecular Cyclization of Carvone to Carvonecamphor2, J. Am. Chem. Soc, 1957, 79, 4741–4748. [Google Scholar]
- 16.Khorana HG, Razzell WE, Gilham PT, Tener GM, and Pol EH, Syntheses of Dideoxyribonucleotides, J. Am. Chem. Soc, 1957, 79, 1002–1003. [Google Scholar]
- 17.Schreier WJ, Schrader TE, Koller FO, Gilch P, Crespo-Hernández CE, Swaminathan VN, Carell T, Zinth W, and Kohler B, Thymine Dimerization in DNA Is an Ultrafast Photoreaction, Science (New York, N.Y.), 2007, 315, 625–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Romero NA and Nicewicz DA, Organic Photoredox Catalysis, Chem. Rev, 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- 19.Worrell BT, McBride MK, Lyon GB, Cox LM, Wang C, Mavila S, Lim C-H, Coley HM, Musgrave CB, Ding Y, et al. , Bistable and Photoswitchable States of Matter, Nat. Commun, 2018, 9, 2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown TE, Marozas IA, and Anseth KS, Amplified Photodegradation of Cell-Laden Hydrogels Via an Addition–Fragmentation Chain Transfer Reaction, Adv. Mater, 2017, 29, 1605001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Kong H, Zhu C, and Zhang Y, Photo-Controllable Bioorthogonal Chemistry for Spatiotemporal Control of Bio-Targets in Living Systems, Chem. Sci, 2020, 11, 3390–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar GS and Lin Q, Light-Triggered Click Chemistry, Chem. Rev, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herner A and Lin Q, Photo-Triggered Click Chemistry for Biological Applications, Top. Curr. Chem, 2015, 374, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaur G, Singh G, and Singh J, Photochemical Tuning of Materials: A Click Chemistry Perspective, Mater. Today Chem, 2018, 8, 56–84. [Google Scholar]
- 25.Menzel JP, Noble BB, Lauer A, Coote ML, Blinco JP, and Barner-Kowollik C, Wavelength Dependence of Light-Induced Cycloadditions, J. Am. Chem. Soc, 2017, 139, 15812–15820. [DOI] [PubMed] [Google Scholar]
- 26.Marschner DE, Kamm PW, Frisch H, Unterreiner A-N, and Barner-Kowollik C, Photocycloadditions in Disparate Chemical Environments, Chem. Commun, 2020, 56, 14043–14046. [DOI] [PubMed] [Google Scholar]
- 27.Arseneault M, Wafer C, and Morin J-F, Recent Advances in Click Chemistry Applied to Dendrimer Synthesis, Molecules, 2015, 20, 9263–9294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fahrenbach AC and Stoddart JF, Reactions under the Click Chemistry Philosophy Employed in Supramolecular and Mechanostereochemical Systems, Chem. Asian J, 2011, 6, 2660–2669. [DOI] [PubMed] [Google Scholar]
- 29.Song W, Wang Y, Qu J, and Lin Q, Selective Functionalization of a Genetically Encoded Alkene-Containing Protein Via “Photoclick Chemistry” in Bacterial Cells, J. Am. Chem. Soc, 2008, 130, 9654–9655. [DOI] [PubMed] [Google Scholar]
- 30.Tasdelen MA and Yagci Y, Light-Induced Copper(I)-Catalyzed Click Chemistry, Tetrahedron Lett., 2010, 51, 6945–6947. [Google Scholar]
- 31.Adzima BJ, Tao YH, Kloxin CJ, DeForest CA, Anseth KS, and Bowman CN, Spatial and Temporal Control of the Alkyne-Azide Cycloaddition by Photoinitiated Cu(Ii) Reduction, Nat. Chem, 2011, 3, 256–259. [DOI] [PubMed] [Google Scholar]
- 32.Wu Z-G, Liao X-J, Yuan L, Wang Y, Zheng Y-X, Zuo J-L, and Pan Y, Visible-Light-Mediated Click Chemistry for Highly Regioselective Azide–Alkyne Cycloaddition by a Photoredox Electron-Transfer Strategy, Chem. Eur. J, 2020, 26, 5694–5700. [DOI] [PubMed] [Google Scholar]
- 33.Poloukhtine AA, Mbua NE, Wolfert MA, Boons G-J, and Popik VV, Selective Labeling of Living Cells by a Photo-Triggered Click Reaction, J. Am. Chem. Soc, 2009, 131, 15769–15776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martínek M, Filipová L, Galeta J, Ludvíková L, and Klán P, Photochemical Formation of Dibenzosilacyclohept-4-Yne for Cu-Free Click Chemistry with Azides and 1,2,4,5-Tetrazines, Org. Lett, 2016, 18, 4892–4895. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Hu WJ, Song W, Lim RKV, and Lin Q, Discovery of Long-Wavelength Photoactivatable Diaryltetrazoles for Bioorthogonal 1,3-Dipolar Cycloaddition Reactions, Org. Lett, 2008, 10, 3725–3728. [DOI] [PubMed] [Google Scholar]
- 36.Zhang L, Zhang X, Yao Z, Jiang S, Deng J, Li B, and Yu Z, Discovery of Fluorogenic Diarylsydnone-Alkene Photoligation: Conversion of Ortho-Dual-Twisted Diarylsydnones into Planar Pyrazolines, J. Am. Chem. Soc, 2018, 140, 7390–7394. [DOI] [PubMed] [Google Scholar]
- 37.Lim RKV and Lin Q, Azirine Ligation: Fast and Selective Protein Conjugation Via Photoinduced Azirine-Alkene Cycloaddition, Chem. Commun, 2010, 46, 7993–7995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gruendling T, Oehlenschlaeger KK, Frick E, Glassner M, Schmid C, and Barner-Kowollik C, Rapid Uv Light-Triggered Macromolecular Click Conjugations Via the Use of O-Quinodimethanes, Macromol. Rapid Commun, 2011, 32, 807–812. [DOI] [PubMed] [Google Scholar]
- 39.Oehlenschlaeger KK, Mueller JO, Heine NB, Glassner M, Guimard NK, Delaittre G, Schmidt FG, and Barner-Kowollik C, Light-Induced Modular Ligation of Conventional Raft Polymers, Angew. Chem. Int. Ed, 2013, 52, 762–766. [DOI] [PubMed] [Google Scholar]
- 40.Feist F, Rodrigues LL, Walden SL, Krappitz TW, Dargaville TR, Weil T, Goldmann AS, Blinco JP, and Barner-Kowollik C, Light-Induced Ligation of O-Quinodimethanes with Gated Fluorescence Self-Reporting, J. Am. Chem. Soc, 2020, 142, 7744–7748. [DOI] [PubMed] [Google Scholar]
- 41.Arumugam S and Popik VV, Light-Induced Hetero-Diels–Alder Cycloaddition: A Facile and Selective Photoclick Reaction, J. Am. Chem. Soc, 2011, 133, 5573–5579. [DOI] [PubMed] [Google Scholar]
- 42.Truong VX, Tsang KM, Ercole F, and Forsythe JS, Red Light Activation of Tetrazine–Norbornene Conjugation for Bioorthogonal Polymer Cross-Linking across Tissue, Chem. Mater, 2017, 29, 3678–3685. [Google Scholar]
- 43.Zhang H, Trout WS, Liu S, Andrade GA, Hudson DA, Scinto SL, Dicker KT, Li Y, Lazouski N, Rosenthal J, et al. , Rapid Bioorthogonal Chemistry Turn-on through Enzymatic or Long Wavelength Photocatalytic Activation of Tetrazine Ligation, J. Am. Chem. Soc, 2016, 138, 5978–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mayer SV, Murnauer A, Wrisberg v.M.-K., Jokisch M-L, and Lang K, Photo-Induced and Rapid Labeling of Tetrazine-Bearing Proteins Via Cyclopropenone-Caged Bicyclononynes, Angew. Chem. Int. Ed, 2019, 58, 15876–15882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoyle CE, Lee TY, and Roper T, Thiol–Enes: Chemistry of the Past with Promise for the Future, J. Polym. Sci., Part A: Polym. Chem, 2004, 42, 5301–5338. [Google Scholar]
- 46.Hoyle CE and Bowman CN, Thiol–Ene Click Chemistry, Angew. Chem. Int. Ed, 2010, 49, 1540–1573. [DOI] [PubMed] [Google Scholar]
- 47.Fairbanks BD, Sims EA, Anseth KS, and Bowman CN, Reaction Rates and Mechanisms for Radical, Photoinitated Addition of Thiols to Alkynes, and Implications for Thiol-Yne Photopolymerizations and Click Reactions, Macromolecules, 2010, 43, 4113–4119. [Google Scholar]
- 48.Prasath RA, Gokmen MT, Espeel P, and Du Prez FE, Thiol-Ene and Thiol-Yne Chemistry in Microfluidics: A Straightforward Method Towards Macroporous and Nonporous Functional Polymer Beads, Polym. Chem, 2010, 1. [Google Scholar]
- 49.Stuparu MC and Khan A, Thiol-Epoxy “Click” Chemistry: Application in Preparation and Postpolymerization Modification of Polymers, J. Polym. Sci., Part A: Polym. Chem, 2016, 54, 3057–3070. [Google Scholar]
- 50.Nakayama N and Hayashi T, Synthesis of Novel Uv-Curable Difunctional Thiourethane Methacrylate and Studies on Organic–Inorganic Nanocomposite Hard Coatings for High Refractive Index Plastic Lenses, Prog. Org. Coat, 2008, 62, 274–284. [Google Scholar]
- 51.Shin J, Lee J, and Jeong HM, Properties of Polythiourethanes Prepared by Thiol–Isocyanate Click Reaction, J. Appl. Polym. Sci, 2018, 135, 46070. [Google Scholar]
- 52.Xi W, Krieger M, Kloxin CJ, and Bowman CN, A New Photoclick Reaction Strategy: Photo-Induced Catalysis of the Thiol-Michael Addition Via a Caged Primary Amine, Chem. Commun, 2013, 49, 4504–4506. [DOI] [PubMed] [Google Scholar]
- 53.Chatani S, Gong T, Earle BA, Podgórski M, and Bowman CN, Visible-Light Initiated Thiol-Michael Addition Photopolymerization Reactions, ACS Macro Lett, 2014, 3, 315–318. [DOI] [PubMed] [Google Scholar]
- 54.Pauloehrl T, Delaittre G, Bastmeyer M, and Barner-Kowollik C, Ambient Temperature Polymer Modification by in Situ Phototriggered Deprotection and Thiol–Ene Chemistry, Polym. Chem, 2012, 3, 1740–1749. [Google Scholar]
- 55.Mahmoodi MM, Fisher SA, Tam RY, Goff PC, Anderson RB, Wissinger JE, Blank DA, Shoichet MS, and Distefano MD, 6-Bromo-7-Hydroxy-3-Methylcoumarin (Mbhc) Is an Efficient Multi-Photon Labile Protecting Group for Thiol Caging and Three-Dimensional Chemical Patterning, Org. Biomol. Chem, 2016, 14, 8289–8300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pauloehrl T, Delaittre G, Bruns M, Meißler M, Börner HG, Bastmeyer M, and Barner-Kowollik C, (Bio)Molecular Surface Patterning by Phototriggered Oxime Ligation, Angew. Chem. Int. Ed, 2012, 51, 9181–9184. [DOI] [PubMed] [Google Scholar]
- 57.Farahani PE, Adelmund SM, Shadish JA, and DeForest CA, Photomediated Oxime Ligation as a Bioorthogonal Tool for Spatiotemporally-Controlled Hydrogel Formation and Modification, J. Mater. Chem. B, 2017, 5, 4435–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huisgen R, Kinetics and Mechanism of 1,3-Dipolar Cycloadditions, Angew. Chem. Int. Ed, 1963, 2, 633–645. [Google Scholar]
- 59.Demko ZP and Sharpless KB, A Click Chemistry Approach to Tetrazoles by Huisgen 1,3-Dipolar Cycloaddition: Synthesis of 5-Sulfonyl Tetrazoles from Azides and Sulfonyl Cyanides, Angew. Chem. Int. Ed, 2002, 41, 2110–2113. [PubMed] [Google Scholar]
- 60.Rostovtsev VV, Green LG, Fokin VV, and Sharpless KB, A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes, Angew. Chem. Int. Ed, 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- 61.Worrell BT, Malik JA, and Fokin VV, Direct Evidence of a Dinuclear Copper Intermediate in Cu(I)-Catalyzed Azide-Alkyne Cycloadditions, Science, 2013, 340, 457–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boren BC, Narayan S, Rasmussen LK, Zhang L, Zhao H, Lin Z, Jia G, and Fokin VV, Ruthenium-Catalyzed Azide–Alkyne Cycloaddition: Scope and Mechanism, J. Am. Chem. Soc, 2008, 130, 8923–8930. [DOI] [PubMed] [Google Scholar]
- 63.Hong V, Udit AK, Evans RA, and Finn MG, Electrochemically Protected Copper(I)-Catalyzed Azide-Alkyne Cycloaddition, Chembiochem, 2008, 9, 1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ritter SC and Konig B, Signal Amplification and Transduction by Photo-Activated Catalysis, Chem. Commun, 2006, 4694–4696. [DOI] [PubMed] [Google Scholar]
- 65.Song HB, Baranek A, and Bowman CN, Kinetics of Bulk Photo-Initiated Copper(I)-Catalyzed Azide-Alkyne Cycloaddition (Cuaac) Polymerizations, Polym. Chem, 2016, 7, 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.El-Zaatari BM, Cole SM, Bischoff DJ, and Kloxin CJ, Copper Ligand and Anion Effects: Controlling the Kinetics of the Photoinitiated Copper(I) Catalyzed Azide-Alkyne Cycloaddition Polymerization, Polym. Chem, 2018, 9, 4772–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.El-Zaatari BM, Shete AU, Adzima BJ, and Kloxin CJ, Towards Understanding the Kinetic Behaviour and Limitations in Photo-Induced Copper(I) Catalyzed Azide-Alkyne Cycloaddition (Cuaac) Reactions, PCCP, 2016, 18, 25504–25511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kutahya C, Yagci Y, and Strehmel B, Near-Infrared Photoinduced Copper-Catalyzed Azide-Alkyne Click Chemistry with a Cyanine Comprising a Barbiturate Group, ChemPhotoChem , 2019, 3, 1180–1186. [Google Scholar]
- 69.Gschneidtner TA and Moth-Poulsen K, A Photolabile Protection Strategy for Terminal Alkynes, Tetrahedron Lett., 2013, 54, 5426–5429. [Google Scholar]
- 70.Tebikachew BE, Börjesson K, Kann N, and Moth-Poulsen K, Release of Terminal Alkynes Via Tandem Photodeprotection and Decarboxylation of O-Nitrobenzyl Arylpropiolates in a Flow Microchannel Reactor, Bioconjugate Chem., 2018, 29, 1178–1185. [DOI] [PubMed] [Google Scholar]
- 71.Sun X, Zou Y, and Jiang J, Surface Plasmon Resonances Enhanced Click Chemistry through Synergistic Photothermal and Hot Electron Effects, Chem. Commun, 2019, 55, 4813–4816. [DOI] [PubMed] [Google Scholar]
- 72.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, and Bertozzi CR, Copper-Free Click Chemistry for Dynamic <Em>in Vivo</Em> Imaging, PNAS, 2007, 104, 16793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Codelli JA, Baskin JM, Agard NJ, and Bertozzi CR, Second-Generation Difluorinated Cyclooctynes for Copper-Free Click Chemistry, J. Am. Chem. Soc, 2008, 130, 11486–11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gordon CG, Mackey JL, Jewett JC, Sletten EM, Houk KN, and Bertozzi CR, Reactivity of Biarylazacyclooctynones in Copper-Free Click Chemistry, J. Am. Chem. Soc, 2012, 134, 9199–9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jewett JC and Bertozzi CR, Cu-Free Click Cycloaddition Reactions in Chemical Biology, Chem. Soc. Rev, 2010, 39, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sutton DA and Popik VV, Sequential Photochemistry of Dibenzo[a,E]Dicyclopropa[C,G][8]Annulene-1,6-Dione: Selective Formation of Didehydrodibenzo[a,E][8]Annulenes with Ultrafast Spaac Reactivity, J. Org. Chem, 2016, 81, 8850–8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sutton DA, Yu S-H, Steet R, and Popik VV, Cyclopropenone-Caged Sondheimer Diyne (Dibenzo[a,E]Cyclooctadiyne): A Photoactivatable Linchpin for Efficient Spaac Crosslinking, Chem. Commun, 2016, 52, 553–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mishiro K, Kimura T, Furuyama T, and Kunishima M, Phototriggered Active Alkyne Generation from Cyclopropenones with Visible Light-Responsive Photocatalysts, Org. Lett, 2019, 21, 4101–4105. [DOI] [PubMed] [Google Scholar]
- 79.Jedináková P, Šebej P, Slanina T, Klán P, and Hlaváč J, Study and Application of Noncatalyzed Photoinduced Conjugation of Azides and Cycloocta-1,2,3-Selenadiazoles, Chem. Commun, 2016, 52, 4792–4795. [DOI] [PubMed] [Google Scholar]
- 80.Gann AW, Amoroso JW, Einck VJ, Rice WP, Chambers JJ, and Schnarr NA, A Photoinduced, Benzyne Click Reaction, Org. Lett, 2014, 16, 2003–2005. [DOI] [PubMed] [Google Scholar]
- 81.Huisgen R, Ooms PHJ, Mingin M, and Allinger NL, Exceptional Reactivity of the Bicyclo[2.2.1]Heptene Double Bond, J. Am. Chem. Soc, 1980, 102, 3951–3953. [Google Scholar]
- 82.Singh K, Fennell CJ, Coutsias EA, Latifi R, Hartson S, and Weaver JD, Light Harvesting for Rapid and Selective Reactions: Click Chemistry with Strain-Loadable Alkenes, Chem, 2018, 4, 124–137. [Google Scholar]
- 83.Huisgen R, Seidel M, Sauer J, McFarland J, and Wallbillich G, Communications: The Formation of Nitrile Imines in the Thermal Breakdown of 2,5-Disubstituted Tetrazoles, J. Org. Chem, 1959, 24, 892–893. [Google Scholar]
- 84.Jamieson C and Livingstone K, The Reactivity of Nitrile Imines, in The Nitrile Imine 1,3-Dipole: Properties, Reactivity and Applications. Springer International Publishing: Cham. 37–97, 2020. [Google Scholar]
- 85.Ramil CP and Lin Q, Photoclick Chemistry: A Fluorogenic Light-Triggered in Vivo Ligation Reaction, Curr. Opin. Chem. Biol, 2014, 21, 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.An P, Yu Z, and Lin Q, Design of Oligothiophene-Based Tetrazoles for Laser-Triggered Photoclick Chemistry in Living Cells, Chem. Commun, 2013, 49, 9920–9922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lederhose P, Wüst KNR, Barner-Kowollik C, and Blinco JP, Catalyst Free Visible Light Induced Cycloaddition as an Avenue for Polymer Ligation, Chem. Commun, 2016, 52, 5928–5931. [DOI] [PubMed] [Google Scholar]
- 88.Lederhose P, Chen Z, Müller R, Blinco JP, Wu S, and Barner-Kowollik C, Near-Infrared Photoinduced Coupling Reactions Assisted by Upconversion Nanoparticles, Angew. Chem. Int. Ed, 2016, 55, 12195–12199. [DOI] [PubMed] [Google Scholar]
- 89.Li Z, Qian L, Li L, Bernhammer JC, Huynh HV, Lee J-S, and Yao SQ, Tetrazole Photoclick Chemistry: Reinvestigating Its Suitability as a Bioorthogonal Reaction and Potential Applications, Angew. Chem. Int. Ed, 2016, 55, 2002–2006. [DOI] [PubMed] [Google Scholar]
- 90.Zhao S, Dai J, Hu M, Liu C, Meng R, Liu X, Wang C, and Luo T, Photo-Induced Coupling Reactions of Tetrazoles with Carboxylic Acids in Aqueous Solution: Application in Protein Labelling, Chem. Commun, 2016, 52, 4702–4705. [DOI] [PubMed] [Google Scholar]
- 91.An P, Lewandowski TM, Erbay TG, Liu P, and Lin Q, Sterically Shielded, Stabilized Nitrile Imine for Rapid Bioorthogonal Protein Labeling in Live Cells, J. Am. Chem. Soc, 2018, 140, 4860–4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huisgen R, Grashey R, Seidel M, Knupfer H, and Schmidt R, 1.3-Dipolare Additionen, Iii. Umsetzungen Des Diphenylnitrilimins Mit Carbonyl Und Thiocarbonyl-Verbindungen, Liebigs Ann. Chem, 1962, 658, 169–180. [Google Scholar]
- 93.Feng W, Li L, Yang C, Welle A, Trapp O, and Levkin PA, Uv-Induced Tetrazole-Thiol Reaction for Polymer Conjugation and Surface Functionalization, Angew. Chem. Int. Ed, 2015, 54, 8732–8735. [DOI] [PubMed] [Google Scholar]
- 94.Zhang X, Wu X, Jiang S, Gao J, Yao Z, Deng J, Zhang L, and Yu Z, Photo-Accelerated “Click” Reaction between Diarylsydnones and Ring-Strained Alkynes for Bioorthogonal Ligation, Chem. Commun, 2019, 55, 7187–7190. [DOI] [PubMed] [Google Scholar]
- 95.Gao J, Xiong Q, Wu X, Deng J, Zhang X, Zhao X, Deng P, and Yu Z, Direct Ring-Strain Loading for Visible-Light Accelerated Bioorthogonal Ligation Via Diarylsydnone-Dibenzo[B,F ][1,4,5]Thiadiazepine Photo-Click Reactions, Commun. Chem, 2020, 3, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jiang S, Wu X, Liu H, Deng J, Zhang X, Yao Z, Zheng Y, Li B, and Yu Z, Ring-Strain-Promoted Ultrafast Diaryltetrazole–Alkyne Photoclick Reactions Triggered by Visible Light, ChemPhotoChem, 2020, 4, 327–331. [Google Scholar]
- 97.Deng J, Wu X, Guo G, Zhao X, and Yu Z, Photoisomerization-Enhanced 1,3-Dipolar Cycloaddition of Carbon-Bridged Octocyclic Azobenzene with Photo-Released Nitrile Imine for Peptide Stapling and Imaging in Live Cells, Org. Biomol. Chem, 2020, 18, 5602–5607. [DOI] [PubMed] [Google Scholar]
- 98.Mueller JO, Schmidt FG, Blinco JP, and Barner-Kowollik C, Visible-Light-Induced Click Chemistry, Angew. Chem. Int. Ed, 2015, 54, 10284–10288. [DOI] [PubMed] [Google Scholar]
- 99.Yang NC and Rivas C, A New Photochemical Primary Process, the Photochemical Enolization of O-Substituted Benzophenones, J. Am. Chem. Soc, 1961, 83, 2213–2213. [Google Scholar]
- 100.Sammes PG, Photoenolisation, Tetrahedron, 1976, 32, 405–422. [Google Scholar]
- 101.Menzel JP, Feist F, Tuten B, Weil T, Blinco JP, and Barner-Kowollik C, Light-Controlled Orthogonal Covalent Bond Formation at Two Different Wavelengths, Angew. Chem. Int. Ed, 2019, 58, 7470–7474. [DOI] [PubMed] [Google Scholar]
- 102.Houck HA, Du Prez FE, and Barner-Kowollik C, Controlling Thermal Reactivity with Different Colors of Light, Nat. Commun, 2017, 8, 1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mueller P, Zieger MM, Richter B, Quick AS, Fischer J, Mueller JB, Zhou L, Nienhaus GU, Bastmeyer M, Barner-Kowollik C, et al. , Molecular Switch for Sub-Diffraction Laser Lithography by Photoenol Intermediate-State Cis–Trans Isomerization, ACS Nano, 2017, 11, 6396–6403. [DOI] [PubMed] [Google Scholar]
- 104.Porter G and Tchir MF, Photoenolization of Ortho-Substituted Benzophenones by Flash Photolysis, Journal of the Chemical Society A: Inorganic, Physical, Theoretical, 1971, 3772–3777. [Google Scholar]
- 105.Tyson DS, Carbaugh AD, Ilhan F, Santos-Pérez J, and Meador MA, Novel Anthracene Diimide Fluorescent Sensor, Chem. Mater, 2008, 20, 6595–6596. [Google Scholar]
- 106.Arumugam S and Popik VV, Attach, Remove, or Replace: Reversible Surface Functionalization Using Thiol–Quinone Methide Photoclick Chemistry, J. Am. Chem. Soc, 2012, 134, 8408–8411. [DOI] [PubMed] [Google Scholar]
- 107.Billaud EMF, Shahbazali E, Ahamed M, Cleeren F, Noël T, Koole M, Verbruggen A, Hessel V, and Bormans G, Micro-Flow Photosynthesis of New Dienophiles for Inverse-Electron-Demand Diels–Alder Reactions. Potential Applications for Pretargeted in Vivo Pet Imaging, Chem. Sci, 2017, 8, 1251–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Svatunek D, Denk C, Rosecker V, Sohr B, Hametner C, Allmaier G, Fröhlich J, and Mikula H, Efficient Low-Cost Preparation Of trans-Cyclooctenes Using a Simplified Flow Setup for Photoisomerization, Monatshefte für Chemie - Chem. Mon, 2016, 147, 579–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Scott TF, Furgal JC, and Kloxin CJ, Expanding the Alternating Propagation–Chain Transfer-Based Polymerization Toolkit: The Iodo–Ene Reaction, ACS Macro Lett., 2015, 4, 1404–1409. [DOI] [PubMed] [Google Scholar]
- 110.Fairbanks BD, Scott TF, Kloxin CJ, Anseth KS, and Bowman CN, Thiol–Yne Photopolymerizations: Novel Mechanism, Kinetics, and Step-Growth Formation of Highly Cross-Linked Networks, Macromolecules, 2009, 42, 211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.El-Roz M, Lalevée J, Allonas X, and Fouassier J-P, The Silane-Ene and Silane-Acrylate Polymerization Process: A New Promising Chemistry?, Macromol. Rapid Commun, 2008, 29, 804–808. [Google Scholar]
- 112.Alvey LJ, Rutherford D, Juliette JJJ, and Gladysz JA, Additions of Ph3 to Monosubstituted Alkenes of the Formula H2cch(Ch2)X(Cf2)Ycf3: Convenient, Multigram Syntheses of a Family of Partially Fluorinated Trialkylphosphines with Modulated Electronic Properties and Fluorous Phase Affinities, J. Org. Chem, 1998, 63, 6302–6308. [DOI] [PubMed] [Google Scholar]
- 113.Posner T, Beiträge Zur Kenntniss Der Ungesättigten Verbindungen. Ii. Ueber Die Addition Von Mercaptanen an Ungesättigte Kohlenwasserstoffe, Ber. Dtsch. Chem. Ges, 1905, 38, 646–657. [Google Scholar]
- 114.Chauhan P, Mahajan S, and Enders D, Organocatalytic Carbon-Sulfur Bond-Forming Reactions, Chem. Rev, 2014, 114, 8807–8864. [DOI] [PubMed] [Google Scholar]
- 115.Clayden J and MacLellan P, Asymmetric Synthesis of Tertiary Thiols and Thioethers, Beilstein J. Org. Chem, 2011, 7, 582–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tucker-Schwartz AK, Farrell RA, and Garrell RL, Thiol-Ene Click Reaction as a General Route to Functional Trialkoxysilanes for Surface Coating Applications, JACS, 2011, 133, 11026–11029. [DOI] [PubMed] [Google Scholar]
- 117.Cramer NB, Reddy SK, O'Brien AK, and Bowman CN, Thiol–Ene Photopolymerization Mechanism and Rate Limiting Step Changes for Various Vinyl Functional Group Chemistries, Macromolecules, 2003, 36, 7964–7969. [Google Scholar]
- 118.Koo SPS, Stamenovic MM, Prasath RA, Inglis AJ, Du Prez FE, Barner-Kowollik C, Van Camp W, and Junkers T, Limitations of Radical Thiol-Ene Reactions for Polymer-Polymer Conjugation, J. Polym. Sci., Part A: Polym. Chem, 2010, 48, 1699–1713. [Google Scholar]
- 119.Zgrzeba A, Andrzejewska E, and Marcinkowska A, Ionic Liquid – Containing Ionogels by Thiol–Ene Photopolymerization. Kinetics and Solvent Effect, RSC Adv, 2015, 5, 100354–100361. [Google Scholar]
- 120.Munar I, Findik V, Degirmenci I, and Aviyente V, Solvent Effects on Thiol-Ene Kinetics and Reactivity of Carbon and Sulfur Radicals, Journal of Physical Chemistry A, 2020, 124, 2580–2590. [DOI] [PubMed] [Google Scholar]
- 121.Skinner EK, Whiffin FM, and Price GJ, Room Temperature Sonochemical Initiation of Thiol-Ene Reactions, Chem. Commun, 2012, 48, 6800–6802. [DOI] [PubMed] [Google Scholar]
- 122.Love DM, Fairbanks BD, and Bowman CN, Evaluation of Aromatic Thiols as Photoinitiators, Macromolecules, 2020, 53, 5237–5247. [Google Scholar]
- 123.Ahn D, Sathe SS, Clarkson BH, and Scott TF, Hexaarylbiimidazoles as Visible Light Thiol–Ene Photoinitiators, Dent. Mater, 2015, 31, 1075–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kolczak U, Rist G, Dietliker K, and Wirz J, Reaction Mechanism of Monoacyl- and Bisacylphosphine Oxide Photoinitiators Studied by 31p-, 13c-, and 1h-Cidnp and Esr, J. Am. Chem. Soc, 1996, 118, 6477–6489. [Google Scholar]
- 125.Ganster B, Fischer UK, Moszner N, and Liska R, New Photocleavable Structures. Diacylgermane-Based Photoinitiators for Visible Light Curing, Macromolecules, 2008, 41, 2394–2400. [Google Scholar]
- 126.Mitterbauer M, Knaack P, Naumov S, Markovic M, Ovsianikov A, Moszner N, and Liska R, Acylstannanes: Cleavable and Highly Reactive Photoinitiators for Radical Photopolymerization at Wavelengths above 500 Nm with Excellent Photobleaching Behavior, Angew. Chem. Int. Ed, 2018, 57, 12146–12150. [DOI] [PubMed] [Google Scholar]
- 127.Zhao G, Kaur S, and Wang T, Visible-Light-Mediated Thiol-Ene Reactions through Organic Photoredox Catalysis, Org. Lett, 2017, 19, 3291–3294. [DOI] [PubMed] [Google Scholar]
- 128.Bhat VT, Duspara PA, Seo S, Abu Bakar NSB, and Greaney MF, Visible Light Promoted Thiol-Ene Reactions Using Titanium Dioxide, Chem. Commun, 2015, 51, 4383–4385. [DOI] [PubMed] [Google Scholar]
- 129.Keylor MH, Park JE, Wallentin C-J, and Stephenson CRJ, Photocatalytic Initiation of Thiol-Ene Reactions: Synthesis of Thiomorpholin-3-Ones, Tetrahedron, 2014, 70, 4264–4269. [Google Scholar]
- 130.Limnios D and Kokotos CG, Photoinitiated Thiol-Ene "Click" Reaction: An Organocatalytic Alternative, Adv. Synth. Catal, 2017, 359, 323–328. [Google Scholar]
- 131.Tyson EL, Ament MS, and Yoon TP, Transition Metal Photoredox Catalysis of Radical Thiol-Ene Reactions, J. Org. Chem, 2013, 78, 2046–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tyson EL, Niemeyer ZL, and Yoon TP, Redox Mediators in Visible Light Photocatalysis: Photocatalytic Radical Thiol-Ene Additions, J. Org. Chem, 2014, 79, 1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wimmer A and Koenig B, Photocatalytic Formation of Carbon-Sulfur Bonds, Beilstein J. Org. Chem, 2018, 14, 54–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Koyama D and Orr-Ewing AJ, Photochemical Reaction Dynamics of 2,2′-Dithiobis(Benzothiazole): Direct Observation of the Addition Product of an Aromatic Thiyl Radical to an Alkene with Time-Resolved Vibrational and Electronic Absorption Spectroscopy, PCCP, 2016, 18, 12115–12127. [DOI] [PubMed] [Google Scholar]
- 135.Li Z, Zou X, Shi F, Liu R, and Yagci Y, Highly Efficient Dandelion-Like near-Infrared Light Photoinitiator for Free Radical and Thiol-Ene Photopolymerizations, Nat. Commun, 2019, 10, 3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Burget D, Mallein C, and Fouassier JP, Photopolymerization of Thiol-Allyl Ether and Thiol-Acrylate Coatings with Visible Light Photosensitive Systems, Polymer, 2004, 45, 6561–6567. [Google Scholar]
- 137.Uygun M, Tasdelen MA, and Yagci Y, Influence of Type of Initiation on Thiol-Ene "Click" Chemistry, Macromol. Chem. Phys, 2010, 211, 103–110. [Google Scholar]
- 138.Xu J and Boyer C, Visible Light Photocatalytic Thiol-Ene Reaction: An Elegant Approach for Fast Polymer Postfunctionalization and Step-Growth Polymerization, Macromolecules, 2015, 48, 520–529. [Google Scholar]
- 139.Guerrero-Corella A, Maria Martinez-Gualda A, Ahmadi F, Ming E, Fraile A, and Aleman J, Thiol-Ene/Oxidation Tandem Reaction under Visible Light Photocatalysis: Synthesis of Alkyl Sulfoxides, Chem. Commun, 2017, 53, 10463–10466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shih H and Lin C-C, Cross-Linking and Degradation of Step-Growth Hydrogels Formed by Thiol-Ene Photoclick Chemistry, Biomacromolecules, 2012, 13, 2003–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Morgan CR, Magnotta F, and Ketley AD, Thiol-Ene Photo-Curable Polymers, J. Polym. Sci., Part A: Polym. Chem, 1977, 15, 627–645. [Google Scholar]
- 142.Wutticharoenwong K and Soucek MD, Influence of the Thiol Structure on the Kinetics of Thiol-Ene Photopolymerization with Time-Resolved Infrared Spectroscopy, Macromol. Mater. Eng, 2008, 293, 45–56. [Google Scholar]
- 143.Long KF, Bongiardina NJ, Mayordomo P, Olin MJ, Ortega AD, and Bowman CN, Effects of 1 Degrees, 2 Degrees, and 3 Degrees Thiols on Thiol-Ene Reactions: Polymerization Kinetics and Mechanical Behavior, Macromolecules, 2020, 53, 5805–5815. [Google Scholar]
- 144.Findik V, Degirmenci I, Catak S, and Aviyente V, Theoretical Investigation of Thiol-Ene Click Reactions: A Dft Perspective, Eur. Polym. J, 2019, 110, 211–220. [Google Scholar]
- 145.Hoyle CE, Lee TY, and Roper T, Thiol-Enes: Chemistry of the Past with Promise for the Future, J. Polym. Sci., Part A: Polym. Chem, 2004, 42, 5301–5338. [Google Scholar]
- 146.Cramer NB, Reddy SK, O'Brien AK, and Bowman CN, Thiol-Ene Photopolymerization Mechanism and Rate Limiting Step Changes for Various Vinyl Functional Group Chemistries, Macromolecules, 2003, 36, 7964–7969. [Google Scholar]
- 147.Northrop BH and Coffey RN, Thiol-Ene Click Chemistry: Computational and Kinetic Analysis of the Influence of Alkene Functionality, JACS, 2012, 134, 13804–13817. [DOI] [PubMed] [Google Scholar]
- 148.Stolz RM and Northrop BH, Experimental and Theoretical Studies of Selective Thiol-Ene and Thiol-Yne Click Reactions Involving N-Substituted Maleimides, Journal of Organic Chemistry, 2013, 78, 8105–8116. [DOI] [PubMed] [Google Scholar]
- 149.Love DM, Kim K, Goodrich JT, Fairbanks BD, Worrell BT, Stoykovich MP, Musgrave CB, and Bowman CN, Amine Induced Retardation of the Radical-Mediated Thiol–Ene Reaction Via the Formation of Metastable Disulfide Radical Anions, J. Org. Chem, 2018, 83, 2912–2919. [DOI] [PubMed] [Google Scholar]
- 150.Fairbanks BD, Scott TF, Kloxin CJ, Anseth KS, and Bowman CN, Thiol-Yne Photopolymerizations: Novel Mechanism, Kinetics, and Step-Growth Formation of Highly Cross-Linked Networks, Macromolecules, 2009, 42, 211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.van Geel R, Pruijn GJM, van Delft FL, and Boelens WC, Preventing Thiol-Yne Addition Improves the Specificity of Strain-Promoted Azide-Alkyne Cycloaddition, Bioconjugate Chem, 2012, 23, 392–398. [DOI] [PubMed] [Google Scholar]
- 152.Kritchenkov AS, Egorov AR, Artemjev AA, Kritchenkov IS, Volkova OV, Kurliuk AV, Shakola TV, Rubanik VV Jr., Rubanik VV, Tskhovrebov AG, et al. , Ultrasound-Assisted Catalyst-Free Thiol-Yne Click Reaction in Chitosan Chemistry: Antibacterial and Transfection Activity of Novel Cationic Chitosan Derivatives and Their Based Nanoparticles, Int. J. Biol. Macromol, 2020, 143, 143–152. [DOI] [PubMed] [Google Scholar]
- 153.Zalesskiy SS, Shlapakov NS, and Ananikov VP, Visible Light Mediated Metal-Free Thiol-Yne Click Reaction, Chem. Sci, 2016, 7, 6740–6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Burykina JV, Shlapakov NS, Gordeev EG, König B, and Ananikov VP, Selectivity Control in Thiol–Yne Click Reactions Via Visible Light Induced Associative Electron Upconversion, Chem. Sci, 2020, 11, 10061–10070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kaur S, Zhao G, Busch E, and Wang T, Metal-Free Photocatalytic Thiol-Ene/Thiol-Yne Reactions, Org. Biomol. Chem, 2019, 17, 1955–1961. [DOI] [PubMed] [Google Scholar]
- 156.Boyer C, Ameduri B, and Hung MH, Telechelic Diiodopoly(Vdf-Co-Pmve) Copolymers by Iodine Transfer Copolymerization of Vinylidene Fluoride (Vdf) with Perfluoromethyl Vinyl Ether (Pmve), Macromolecules, 2010, 43, 3652–3663. [Google Scholar]
- 157.Boyer C, Valade D, Sauguet L, Ameduri B, and Boutevin B, Iodine Transfer Polymerization (Itp) of Vinylidene Fluoride (Vdf). Influence of the Defect of Vdf Chaining on the Control of Itp, Macromolecules, 2005, 38, 10353–10362. [Google Scholar]
- 158.David G, Boyer C, Tonnar J, Ameduri B, Lacroix-Desmazes P, and Boutevin B, Use of Iodocompounds in Radical Polymerization, Chem. Rev, 2006, 106, 3936–3962. [DOI] [PubMed] [Google Scholar]
- 159.Valade D, Boyer C, Ameduri B, and Boutevin B, Poly(Vinylidene Fluoride)-B-Poly(Styrene) Block Copolymers by Iodine Transfer Polymerization (Itp): Synthesis, Characterization, and Kinetics of Itp, Macromolecules, 2006, 39, 8639–8651. [Google Scholar]
- 160.Scott TF, Furgal JC, and Kloxin CJ, Expanding the Alternating Propagation-Chain Transfer-Based Polymerization Toolkit: The Iodo-Ene Reaction, ACS Macro Lett., 2015, 4, 1404–1409. [DOI] [PubMed] [Google Scholar]
- 161.Sinha J, Fairbanks B, Chen M, Song HB, and Bowman C, Phosphate-Based Crosslinked Polymers from Iodo-Ene Photopolymerization, Abst. Pap. Am. Chem. S, 2019, 257. [DOI] [PubMed] [Google Scholar]
- 162.Steindl J, Svirkova A, Marchetti-Deschmann M, Moszner N, and Gorsche C, Light-Triggered Radical Silane-Ene Chemistry Using a Monosubstituted Bis(Trimethylsilyl)Silane, Macromol. Chem. Phys, 2017, 218. [Google Scholar]
- 163.Steindl J, Koch T, Moszner N, and Gorsche C, Silane-Acrylate Chemistry for Regulating Network Formation in Radical Photopolymerization, Macromolecules, 2017, 50, 7448–7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pellon J, Phosphines: Chain Transfer Studies with Styrene and Methyl Methacrylate, Journal of Polym. Sci, 1960, 43, 537–548. [Google Scholar]
- 165.Guterman R, Rabiee Kenaree A, Gilroy JB, Gillies ER, and Ragogna PJ, Polymer Network Formation Using the Phosphane–Ene Reaction: A Thiol–Ene Analogue with Diverse Postpolymerization Chemistry, Chem. Mater, 2015, 27, 1412–1419. [Google Scholar]
- 166.Hoyle CE, Lowe AB, and Bowman CN, Thiol-Click Chemistry: A Multifaceted Toolbox for Small Molecule and Polymer Synthesis, Chem. Soc. Rev, 2010, 39, 1355–1387. [DOI] [PubMed] [Google Scholar]
- 167.Kade MJ, Burke DJ, and Hawker CJ, The Power of Thiol-Ene Chemistry, J. Polym. Sci., Part A: Polym. Chem, 2010, 48, 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lowe AB, Thiol-Ene “Click” Reactions and Recent Applications in Polymer and Materials Synthesis, Polym. Chem, 2010, 1, 17–36. [Google Scholar]
- 169.Fringuelli F, Pizzo F, Tortoioli S, and Vaccaro L, Thiolysis of 1,2-Epoxides by Thiophenol Catalyzed under Solvent-Free Conditions, Tetrahedron Lett, 2003, 44, 6785–6787. [Google Scholar]
- 170.Dietliker K, Hüsler R, Birbaum JL, Ilg S, Villeneuve S, Studer K, Jung T, Benkhoff J, Kura H, Matsumoto A, et al. , Advancements in Photoinitiators—Opening up New Applications for Radiation Curing, Prog. Org. Coat, 2007, 58, 146–157. [Google Scholar]
- 171.Suyama K and Shirai M, Photobase Generators: Recent Progress and Application Trend in Polymer Systems, Prog. Polym. Sci, 2009, 34, 194–209. [Google Scholar]
- 172.Dong X, Hu P, Zhu G, Li Z, Liu R, and Liu X, Thioxanthone Acetic Acid Ammonium Salts: Highly Efficient Photobase Generators Based on Photodecarboxylation, RSC Adv., 2015, 5, 53342–53348. [Google Scholar]
- 173.Arimitsu K, Oguri A, and Furutani M, 365nm-Light-Sensitive Photobase Generators Derived from Trans-O-Coumaric Acid, Mater. Lett, 2015, 140, 92–94. [Google Scholar]
- 174.Ning X and Ishida H, Phenolic Materials Via Ring-Opening Polymerization: Synthesis and Characterization of Bisphenol-a Based Benzoxazines and Their Polymers, J. Polym. Sci., Part A: Polym. Chem, 1994, 32, 1121–1129. [Google Scholar]
- 175.Chae KH, Cho HI, Kim YH, and Yang UC, Photo-Crosslinking and Negative-Type Micropattern Formation of a Polymeric Photobase Generator Containing Phthalimido Carbamate Groups, Eur. Polym. J, 2012, 48, 1186–1194. [Google Scholar]
- 176.Cameron JF, Willson CG, and Fréchet JMJ, Photogeneration of Amines from Α-Keto Carbamates: Photochemical Studies, J. Am. Chem. Soc, 1996, 118, 12925–12937. [Google Scholar]
- 177.Cameron JF and Frechet JMJ, Photogeneration of Organic Bases from O-Nitrobenzyl-Derived Carbamates, J. Am. Chem. Soc, 1991, 113, 4303–4313. [Google Scholar]
- 178.Salmi H, Allonas X, Ley C, Defoin A, and Ak A, Quaternary Ammonium Salts of Phenylglyoxylic Acid as Photobase Generators for Thiol-Promoted Epoxide Photopolymerization, Polym. Chem, 2014, 5, 6577–6583. [Google Scholar]
- 179.Katogi S and Yusa M, Photobase Generation from Amineimide Derivatives and Their Use for Curing an Epoxide/Thiol System, J. Polym. Sci., Part A: Polym. Chem, 2002, 40, 4045–4052. [Google Scholar]
- 180.Sun X, Gao JP, and Wang ZY, Bicyclic Guanidinium Tetraphenylborate: A Photobase Generator and a Photocatalyst for Living Anionic Ring-Opening Polymerization and Cross-Linking of Polymeric Materials Containing Ester and Hydroxy Groups, J. Am. Chem. Soc, 2008, 130, 8130–8131. [DOI] [PubMed] [Google Scholar]
- 181.Ye S, Cramer NB, Smith IR, Voigt KR, and Bowman CN, Reaction Kinetics and Reduced Shrinkage Stress of Thiol–Yne–Methacrylate and Thiol–Yne–Acrylate Ternary Systems, Macromolecules, 2011, 44, 9084–9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jian Y, He Y, Sun Y, Yang H, Yang W, and Nie J, Thiol–Epoxy/Thiol–Acrylate Hybrid Materials Synthesized by Photopolymerization, J. Mater. Chem. C, 2013, 1, 4481–4489. [Google Scholar]
- 183.Chen L, Zheng Y, Meng X, Wei G, Dietliker K, and Li Z, Delayed Thiol-Epoxy Photopolymerization: A General and Effective Strategy to Prepare Thick Composites, ACS Omega, 2020, 5, 15192–15201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chang H-T and Fréchet JMJ, Proton-Transfer Polymerization: A New Approach to Hyperbranched Polymers, J. Am. Chem. Soc, 1999, 121, 2313–2314. [Google Scholar]
- 185.Gong C and Fréchet JMJ, Proton Transfer Polymerization in the Preparation of Hyperbranched Polyesters with Epoxide Chain-Ends and Internal Hydroxyl Functionalities, Macromolecules, 2000, 33, 4997–4999. [Google Scholar]
- 186.Emrick T, Chang H-T, and Fréchet JMJ, An A2 + B3 Approach to Hyperbranched Aliphatic Polyethers Containing Chain End Epoxy Substituents, Macromolecules, 1999, 32, 6380–6382. [Google Scholar]
- 187.Yeo H and Khan A, Photoinduced Proton-Transfer Polymerization: A Practical Synthetic Tool for Soft Lithography Applications, J. Am. Chem. Soc, 2020, 142, 3479–3488. [DOI] [PubMed] [Google Scholar]
- 188.Romano A, Roppolo I, Giebler M, Dietliker K, Možina Š, Šket P, Mühlbacher I, Schlögl S, and Sangermano M, Stimuli-Responsive Thiol-Epoxy Networks with Photo-Switchable Bulk and Surface Properties, RSC Adv., 2018, 8, 41904–41914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mühlman A, Classon B, Hallberg A, and Samuelsson B, Synthesis of Potent C2-Symmetric, Diol-Based Hiv-1 Protease Inhibitors. Investigation of Thioalkyl and Thioaryl P1/P1‘ Substituents, J. Med. Chem, 2001, 44, 3402–3406. [DOI] [PubMed] [Google Scholar]
- 190.Basuki JS, Esser L, Duong HTT, Zhang Q, Wilson P, Whittaker MR, Haddleton DM, Boyer C, and Davis TP, Magnetic Nanoparticles with Diblock Glycopolymer Shells Give Lectin Concentration-Dependent Mri Signals and Selective Cell Uptake, Chem. Sci, 2014, 5, 715–726. [Google Scholar]
- 191.Buerkli C, Lee SH, Moroz E, Stuparu MC, Leroux J-C, and Khan A, Amphipathic Homopolymers for Sirna Delivery: Probing Impact of Bifunctional Polymer Composition on Transfection, Biomacromolecules, 2014, 15, 1707–1715. [DOI] [PubMed] [Google Scholar]
- 192.Arnold RG, Nelson JA, and Verbanc JJ, Recent Advances in Isocyanate Chemistry, Chem. Rev, 1957, 57, 47–76. [Google Scholar]
- 193.Shin J, Matsushima H, Chan JW, and Hoyle CE, Segmented Polythiourethane Elastomers through Sequential Thiol–Ene and Thiol–Isocyanate Reactions, Macromolecules, 2009, 42, 3294–3301. [Google Scholar]
- 194.Shin J, Matsushima H, Comer CM, Bowman CN, and Hoyle CE, Thiol–Isocyanate–Ene Ternary Networks by Sequential and Simultaneous Thiol Click Reactions, Chem. Mater, 2010, 22, 2616–2625. [Google Scholar]
- 195.Hu Y, Kowalski BA, Mavila S, Podgórski M, Sinha J, Sullivan AC, McLeod RR, and Bowman CN, Holographic Photopolymer Material with High Dynamic Range (Δn) Via Thiol–Ene Click Chemistry, ACS Appl. Mater. Interfaces, 2020, 12, 44103–44109. [DOI] [PubMed] [Google Scholar]
- 196.Hensarling RM, Rahane SB, LeBlanc AP, Sparks BJ, White EM, Locklin J, and Patton DL, Thiol–Isocyanate “Click” Reactions: Rapid Development of Functional Polymeric Surfaces, Polym. Chem, 2011, 2, 88–90. [Google Scholar]
- 197.Mather BD, Viswanathan K, Miller KM, and Long TE, Michael Addition Reactions in Macromolecular Design for Emerging Technologies, Prog. Polym. Sci, 2006, 31, 487–531. [Google Scholar]
- 198.Nising CF and Bräse S, The Oxa-Michael Reaction: From Recent Developments to Applications in Natural Product Synthesis, Chem. Soc. Rev, 2008, 37, 1218–1228. [DOI] [PubMed] [Google Scholar]
- 199.Nair DP, Podgórski M, Chatani S, Gong T, Xi W, Fenoli CR, and Bowman CN, The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry, Chem. Mater, 2014, 26, 724–744. [Google Scholar]
- 200.Desmet GB, Sabbe MK, D'Hooge DR, Espeel P, Celasun S, Marin GB, Du Prez FE, and Reyniers M-F, Thiol-Michael Addition in Polar Aprotic Solvents: Nucleophilic Initiation or Base Catalysis?, Polym. Chem, 2017, 8, 1341–1352. [Google Scholar]
- 201.Chan JW, Hoyle CE, Lowe AB, and Bowman M, Nucleophile-Initiated Thiol-Michael Reactions: Effect of Organocatalyst, Thiol, and Ene, Macromolecules, 2010, 43, 6381–6388. [Google Scholar]
- 202.Chan JW, Yu B, Hoyle CE, and Lowe AB, The Nucleophilic, Phosphine-Catalyzed Thiol–Ene Click Reaction and Convergent Star Synthesis with Raft-Prepared Homopolymers, Polymer, 2009, 50, 3158–3168. [Google Scholar]
- 203.Joshi G and Anslyn EV, Dynamic Thiol Exchange with Β-Sulfido-Α,Β-Unsaturated Carbonyl Compounds and Dithianes, Org. Lett, 2012, 14, 4714–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Van Herck N, Maes D, Unal K, Guerre M, Winne JM, and Du Prez FE, Covalent Adaptable Networks with Tunable Exchange Rates Based on Reversible Thiol–Yne Cross-Linking, Angew. Chem. Int. Ed, 2020, 59, 3609–3617. [DOI] [PubMed] [Google Scholar]
- 205.Claudino M, Zhang X, Alim MD, Podgórski M, and Bowman CN, Mechanistic Kinetic Modeling of Thiol–Michael Addition Photopolymerizations Via Photocaged “Superbase” Generators: An Analytical Approach, Macromolecules, 2016, 49, 8061–8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Xi W, Peng H, Aguirre-Soto A, Kloxin CJ, Stansbury JW, and Bowman CN, Spatial and Temporal Control of Thiol-Michael Addition Via Photocaged Superbase in Photopatterning and Two-Stage Polymer Networks Formation, Macromolecules, 2014, 47, 6159–6165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhang X, Xi W, Wang C, Podgórski M, and Bowman CN, Visible-Light-Initiated Thiol-Michael Addition Polymerizations with Coumarin-Based Photobase Generators: Another Photoclick Reaction Strategy, ACS Macro Lett., 2016, 5, 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sinha J, Podgórski M, Tomaschke A, Ferguson VL, and Bowman CN, Phototriggered Base Amplification for Thiol-Michael Addition Reactions in Cross-Linked Photopolymerizations with Efficient Dark Cure, Macromolecules, 2020, 53, 6331–6340. [Google Scholar]
- 209.Arimitsu K, Miyamoto M, and Ichimura K, Applications of a Nonlinear Organic Reaction of Carbamates to Proliferate Aliphatic Amines, Angew. Chem. Int. Ed, 2000, 39, 3425–3428. [DOI] [PubMed] [Google Scholar]
- 210.Xu R, Guan X, He M, and Yang J, Phototriggered Base Proliferation: A Powerful 365 nm Led Photoclick Tool for Nucleophile-Initiated Thiol-Michael Addition Reaction, RSC Adv., 2017, 7, 914–918. [Google Scholar]
- 211.Bao C, Zhu L, Lin Q, and Tian H, Building Biomedical Materials Using Photochemical Bond Cleavage, Adv. Mater, 2015, 27, 1647–1662. [DOI] [PubMed] [Google Scholar]
- 212.Klán P, Šolomek T, Bochet CG, Blanc A, Givens R, Rubina M, Popik V, Kostikov A, and Wirz J, Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy, Chem. Rev, 2013, 113, 119–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kotzur N, Briand B, Beyermann M, and Hagen V, Wavelength-Selective Photoactivatable Protecting Groups for Thiols, J. Am. Chem. Soc, 2009, 131, 16927–16931. [DOI] [PubMed] [Google Scholar]
- 214.Barltrop JA, Plant PJ, and Schofield P, Photosensitive Protective Groups, Chem. Commun, 1966, 822–823. [Google Scholar]
- 215.Patchornik A, Amit B, and Woodward RB, Photosensitive Protecting Groups, J. Am. Chem. Soc, 1970, 92, 6333–6335. [Google Scholar]
- 216.Gravel D, Giasson R, Blanchet D, Yip RW, and Sharma DK, Photochemistry of the Ortho-Nitrobenzyl System in Solution - Effects of O-H Distance and Geometrical Constraint on the Hydrogen Transfer Mechanism in the Excited-State, Can. J. Chem, 1991, 69, 1193–1200. [Google Scholar]
- 217.Takezaki M, Hirota N, and Terazima M, Nonradiative Relaxation Processes and Electronically Excited States of Nitrobenzene Studied by Picosecond Time-Resolved Transient Grating Method, J. Phys. Chem. A, 1997, 101, 3443–3448. [Google Scholar]
- 218.Schwörer M and Wirz J, Photochemical Reaction Mechanisms of 2-Nitrobenzyl Compounds in Solution, I. 2-Nitrotoluene: Thermodynamic and Kinetic Parameters of the Aci-Nitro Tautomer, Helv. Chim. Acta, 2001, 84, 1441–1458. [Google Scholar]
- 219.Schmierer T, Laimgruber S, Haiser K, Kiewisch K, Neugebauer J, and Gilch P, Femtosecond Spectroscopy on the Photochemistry of Ortho-Nitrotoluene, PCCP, 2010, 12, 15653–15664. [DOI] [PubMed] [Google Scholar]
- 220.Il'ichev YV, Schwörer MA, and Wirz J, Photochemical Reaction Mechanisms of 2-Nitrobenzyl Compounds: Methyl Ethers and Caged Atp, J. Am. Chem. Soc, 2004, 126, 4581–4595. [DOI] [PubMed] [Google Scholar]
- 221.Walker JW, Reid GP, McCray JA, and Trentham DR, Photolabile 1-(2-Nitrophenyl)Ethyl Phosphate Esters of Adenine Nucleotide Analogs. Synthesis and Mechanism of Photolysis, J. Am. Chem. Soc, 1988, 110, 7170–7177. [Google Scholar]
- 222.Kloxin AM, Kasko AM, Salinas CN, and Anseth KS, Photodegradable Hydrogels for Dynamic Tuning of Physical and Chemical Properties, Science, 2009, 324, 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Tsurkan MV, Wetzel R, Pérez-Hernández HR, Chwalek K, Kozlova A, Freudenberg U, Kempermann G, Zhang Y, Lasagni AF, and Werner C, Photopatterning of Multifunctional Hydrogels to Direct Adult Neural Precursor Cells, Adv. Healthcare Mater, 2015, 4, 516–521. [DOI] [PubMed] [Google Scholar]
- 224.Brown EB, Shear JB, Adams SR, Tsien RY, and Webb WW, Photolysis of Caged Calcium in Femtoliter Volumes Using Two-Photon Excitation, Biophys. J, 1999, 76, 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Furuta T, Wang SS-H, Dantzker JL, Dore TM, Bybee WJ, Callaway EM, Denk W, and Tsien RY, Brominated 7-Hydroxycoumarin-4-Ylmethyls: Photolabile Protecting Groups with Biologically Useful Cross-Sections for Two Photon Photolysis, PNAS, 1999, 96, 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Schmidt R, Geissler D, Hagen V, and Bendig J, Mechanism of Photocleavage of (Coumarin-4-Yl)Methyl Esters, J. Phys. Chem. A, 2007, 111, 5768–5774. [DOI] [PubMed] [Google Scholar]
- 227.Schade B, Hagen V, Schmidt R, Herbrich R, Krause E, Eckardt T, and Bendig J, Deactivation Behavior and Excited-State Properties of (Coumarin-4-Yl)Methyl Derivatives. 1. Photocleavage of (7-Methoxycoumarin-4-Yl)Methyl-Caged Acids with Fluorescence Enhancement, J. Org. Chem, 1999, 64, 9109–9117. [DOI] [PubMed] [Google Scholar]
- 228.Schmidt R, Geissler D, Hagen V, and Bendig J, Kinetics Study of the Photocleavage of (Coumarin-4-Yl)Methyl Esters, J. Phys. Chem. A, 2005, 109, 5000–5004. [DOI] [PubMed] [Google Scholar]
- 229.Lin Q, Bao C, Cheng S, Yang Y, Ji W, and Zhu L, Target-Activated Coumarin Phototriggers Specifically Switch on Fluorescence and Photocleavage Upon Bonding to Thiol-Bearing Protein, J. Am. Chem. Soc, 2012, 134, 5052–5055. [DOI] [PubMed] [Google Scholar]
- 230.Girouard S, Houle M-H, Grandbois A, Keillor JW, and Michnick SW, Synthesis and Characterization of Dimaleimide Fluorogens Designed for Specific Labeling of Proteins, J. Am. Chem. Soc, 2005, 127, 559–566. [DOI] [PubMed] [Google Scholar]
- 231.Chen X, Zhou Y, Peng X, and Yoon J, Fluorescent and Colorimetric Probes for Detection of Thiols, Chem. Soc. Rev, 2010, 39, 2120–2135. [DOI] [PubMed] [Google Scholar]
- 232.Mahmoodi MM, Abate-Pella D, Pundsack TJ, Palsuledesai CC, Goff PC, Blank DA, and Distefano MD, Nitrodibenzofuran: A One- and Two-Photon Sensitive Protecting Group That Is Superior to Brominated Hydroxycoumarin for Thiol Caging in Peptides, J. Am. Chem. Soc, 2016, 138, 5848–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Fisher SA, Tam RY, Fokina A, Mahmoodi MM, Distefano MD, and Shoichet MS, Photo-Immobilized Egf Chemical Gradients Differentially Impact Breast Cancer Cell Invasion and Drug Response in Defined 3d Hydrogels, Biomaterials, 2018, 178, 751–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Hu LQ and Colman RF, Monobromobimane as an Affinity Label of the Xenobiotic Binding-Site of Rat Glutathione-S-Transferase-3–3, J. Biol. Chem, 1995, 270, 21875–21883. [DOI] [PubMed] [Google Scholar]
- 235.Lavis LD and Raines RT, Bright Ideas for Chemical Biology, ACS Chem. Biol, 2008, 3, 142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kosower NS, Kosower EM, Newton GL, and Ranney HM, Bimane Fluorescent Labels: Labeling of Normal Human Red Cells under Physiological Conditions, PNAS, 1979, 76, 3382–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Truong VX, Li F, and Forsythe JS, Visible Light Activation of Nucleophilic Thiol-X Addition Via Thioether Bimane Photocleavage for Polymer Cross-Linking, Biomacromolecules, 2018, 19, 4277–4285. [DOI] [PubMed] [Google Scholar]
- 238.Hensarling RM, Hoff EA, LeBlanc AP, Guo W, Rahane SB, and Patton DL, Photocaged Pendent Thiol Polymer Brush Surfaces for Postpolymerization Modifications Via Thiol-Click Chemistry, J. Polym. Sci., Part A: Polym. Chem, 2013, 51, 1079–1090. [Google Scholar]
- 239.Lin Q, Yang L, Wang Z, Hua Y, Zhang D, Bao B, Bao C, Gong X, and Zhu L, Coumarin Photocaging Groups Modified with an Electron-Rich Styryl Moiety at the 3-Position: Long-Wavelength Excitation, Rapid Photolysis, and Photobleaching, Angew. Chem. Int. Ed, 2018, 57, 3722–3726. [DOI] [PubMed] [Google Scholar]
- 240.Lin F, Yu J, Tang W, Zheng J, Defante A, Guo K, Wesdemiotis C, and Becker ML, Peptide-Functionalized Oxime Hydrogels with Tunable Mechanical Properties and Gelation Behavior, Biomacromolecules, 2013, 14, 3749–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Grover GN, Lam J, Nguyen TH, Segura T, and Maynard HD, Biocompatible Hydrogels by Oxime Click Chemistry, Biomacromolecules, 2012, 13, 3013–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Azagarsamy MA, Marozas IA, Spaans S, and Anseth KS, Photoregulated Hydrazone-Based Hydrogel Formation for Biochemically Patterning 3d Cellular Microenvironments, ACS Macro Lett., 2016, 5, 19–23. [DOI] [PubMed] [Google Scholar]
- 243.Yang Y, Zhang J, Liu Z, Lin Q, Liu X, Bao C, Wang Y, and Zhu L, Tissue-Integratable and Biocompatible Photogelation by the Imine Crosslinking Reaction, Adv. Mater, 2016, 28, 2724–2730. [DOI] [PubMed] [Google Scholar]
- 244.Boehnke N, Cam C, Bat E, Segura T, and Maynard HD, Imine Hydrogels with Tunable Degradability for Tissue Engineering, Biomacromolecules, 2015, 16, 2101–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Kalia J and Raines RT, Hydrolytic Stability of Hydrazones and Oximes, Angew. Chem. Int. Ed, 2008, 47, 7523–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Dawson P, Muir T, Clark-Lewis I, and Kent S, Synthesis of Proteins by Native Chemical Ligation, Science, 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]
- 247.Agouridas V, El Mahdi O, Diemer V, Cargoët M, Monbaliu J-CM, and Melnyk O, Native Chemical Ligation and Extended Methods: Mechanisms, Catalysis, Scope, and Limitations, Chem. Rev, 2019, 119, 7328–7443. [DOI] [PubMed] [Google Scholar]
- 248.Ueda S, Fujita M, Tamamura H, Fujii N, and Otaka A, Photolabile Protection for One-Pot Sequential Native Chemical Ligation, ChemBioChem, 2005, 6, 1983–1986. [DOI] [PubMed] [Google Scholar]
- 249.Aihara K, Yamaoka K, Naruse N, Inokuma T, Shigenaga A, and Otaka A, One-Pot/Sequential Native Chemical Ligation Using Photocaged Crypto-Thioester, Org. Lett, 2016, 18, 596–599. [DOI] [PubMed] [Google Scholar]
- 250.Dondoni A and Marra A, Recent Applications of Thiol–Ene Coupling as a Click Process for Glycoconjugation, Chem. Soc. Rev, 2012, 41, 573–586. [DOI] [PubMed] [Google Scholar]
- 251.Fiore M, Lo Conte M, Pacifico S, Marra A, and Dondoni A, Synthesis of S-Glycosyl Amino Acids and S-Glycopeptides Via Photoinduced Click Thiol–Ene Coupling, Tetrahedron Lett., 2011, 52, 444–447. [Google Scholar]
- 252.Conte ML, Staderini S, Marra A, Sanchez-Navarro M, Davis BG, and Dondoni A, Multi-Molecule Reaction of Serum Albumin Can Occur through Thiol-Yne Coupling, Chem. Commun, 2011, 47, 11086–11088. [DOI] [PubMed] [Google Scholar]
- 253.Fiore M, Marra A, and Dondoni A, Photoinduced Thiol–Ene Coupling as a Click Ligation Tool for Thiodisaccharide Synthesis, J. Org. Chem, 2009, 74, 4422–4425. [DOI] [PubMed] [Google Scholar]
- 254.Limnios D and Kokotos CG, Photoinitiated Thiol-Ene “Click” Reaction: An Organocatalytic Alternative, Adv. Synth. Catal, 2017, 359, 323–328. [Google Scholar]
- 255.Kelemen V, Csávás M, Hotzi J, Herczeg M, Poonam, Rathi B, Herczegh P, Jain N, and Borbás A, Photoinitiated Thiol–Ene Reactions of Various 2,3-Unsaturated O-, C- S- and N-Glycosides – Scope and Limitations Study, Chem. Asian J, 2020, 15, 876–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Lázár L, Csávás M, Herczeg M, Herczegh P, and Borbás A, Synthesis of S-Linked Glycoconjugates and S-Disaccharides by Thiol–Ene Coupling Reaction of Enoses, Org. Lett, 2012, 14, 4650–4653. [DOI] [PubMed] [Google Scholar]
- 257.Bege M, Bereczki I, Herczeg M, Kicsák M, Eszenyi D, Herczegh P, and Borbás A, A Low-Temperature, Photoinduced Thiol–Ene Click Reaction: A Mild and Efficient Method for the Synthesis of Sugar-Modified Nucleosides, Org. Biomol. Chem, 2017, 15, 9226–9233. [DOI] [PubMed] [Google Scholar]
- 258.A. Aimetti A, K. Shoemaker R, Lin C-C, and S. Anseth K, On-Resin Peptide Macrocyclization Using Thiol – Ene Click Chemistry, Chem. Commun, 2010, 46, 4061–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Hu K, Geng H, Zhang Q, Liu Q, Xie M, Sun C, Li W, Lin H, Jiang F, Wang T, et al. , An in-Tether Chiral Center Modulates the Helicity, Cell Permeability, and Target Binding Affinity of a Peptide, Angew. Chem. Int. Ed, 2016, 55, 8013–8017. [DOI] [PubMed] [Google Scholar]
- 260.Wang Y and Chou DH-C, A Thiol–Ene Coupling Approach to Native Peptide Stapling and Macrocyclization, Angew. Chem. Int. Ed, 2015, 54, 10931–10934. [DOI] [PubMed] [Google Scholar]
- 261.Tian Y, Yang D, Ye X, and Li Z, Thioether-Derived Macrocycle for Peptide Secondary Structure Fixation, Chem. Rec, 2017, 17, 874–885. [DOI] [PubMed] [Google Scholar]
- 262.Choi H, Kim M, Jang J, and Hong S, Visible-Light-Induced Cysteine-Specific Bioconjugation: Biocompatible Thiol-Ene Click Chemistry, Angew. Chem. Int. Ed n/a. [DOI] [PubMed] [Google Scholar]
- 263.Yim VV, Cameron AJ, Kavianinia I, Harris PWR, and Brimble MA, Thiol-Ene Enabled Chemical Synthesis of Truncated S-Lipidated Teixobactin Analogs, Front. Chem, 2020, 8, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.V. Yim V, Kavianinia I, J. Cameron A, R. Harris PW, and A. Brimble M, Direct Synthesis of Cyclic Lipopeptides Using Intramolecular Native Chemical Ligation and Thiol–Ene Clippa Chemistry, Org. Biomol. Chem, 2020, 18, 2838–2844. [DOI] [PubMed] [Google Scholar]
- 265.Hoppmann C, Schmieder P, Heinrich N, and Beyermann M, Photoswitchable Click Amino Acids: Light Control of Conformation and Bioactivity, Chembiochem, 2011, 12, 2555–2559. [DOI] [PubMed] [Google Scholar]
- 266.A. Aimetti A, R. Feaver K, and S. Anseth K, Synthesis of Cyclic, Multivalent Arg-Gly-Asp Using Sequential Thiol – Ene / Thiol – Yne Photoreactions, Chem. Commun, 2010, 46, 5781–5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Wu Z-G, Liao X-J, Yuan L, Wang Y, Zheng Y-X, Zuo J-L, and Pan Y, Visible-Light-Mediated Click Chemistry for Highly Regioselective Azide-Alkyne Cycloaddition by a Photoredox Electron-Transfer Strategy, Chemistry-a European Journal, 2020, 26, 5694–5700. [DOI] [PubMed] [Google Scholar]
- 268.Beniazza R, Lambert R, Harmand L, Molton F, Duboc C, Denisov S, Jonusauskas G, McClenaghan ND, Lastecoueres D, and Vincent J-M, Sunlight-Driven Copper-Catalyst Activation Applied to Photolatent Click Chemistry, Chemistry-a European Journal, 2014, 20, 13181–13187. [DOI] [PubMed] [Google Scholar]
- 269.Konetski D, Gong T, and Bowman CN, Photoinduced Vesicle Formation Via the Copper-Catalyzed Azide-Alkyne Cycloaddition Reaction, Langmuir, 2016, 32, 8195–8201. [DOI] [PubMed] [Google Scholar]
- 270.Porel M and Alabi CA, Sequence-Defined Polymers Via Orthogonal Allyl Acrylamide Building Blocks, J. Am. Chem. Soc, 2014, 136, 13162–13165. [DOI] [PubMed] [Google Scholar]
- 271.Xi W, Pattanayak S, Wang C, Fairbanks B, Gong T, Wagner J, Kloxin CJ, and Bowman CN, Clickable Nucleic Acids: Sequence-Controlled Periodic Copolymer/Oligomer Synthesis by Orthogonal Thiol-X Reactions, Angew. Chem. Int. Ed, 2015, 54, 14462–14467. [DOI] [PubMed] [Google Scholar]
- 272.Han X, Domaille DW, Fairbanks BD, He L, Culver HR, Zhang X, Cha JN, and Bowman CN, New Generation of Clickable Nucleic Acids: Synthesis and Active Hybridization with DNA, Biomacromolecules, 2018, 19, 4139–4146. [DOI] [PubMed] [Google Scholar]
- 273.Han X, Fairbanks BD, Sinha J, and Bowman CN, Sequence-Controlled Synthesis of Advanced Clickable Synthetic Oligonucleotides, Macromol. Rapid Commun, 2020, 41, 2000327. [DOI] [PubMed] [Google Scholar]
- 274.Zydziak N, Konrad W, Feist F, Afonin S, Weidner S, and Barner-Kowollik C, Coding and Decoding Libraries of Sequence-Defined Functional Copolymers Synthesized Via Photoligation, Nat. Commun, 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Konrad W, Bloesser FR, Wetzel KS, Boukis AC, Meier MAR, and Barner-Kowollik C, A Combined Photochemical and Multicomponent Reaction Approach to Precision Oligomers, Chem. Eur. J, 2018, 24, 3413–3419. [DOI] [PubMed] [Google Scholar]
- 276.Alim MD, Childress KK, Baugh NJ, Martinez AM, Davenport A, Fairbanks BD, McBride MK, Worrell BT, Stansbury JW, McLeod RR, et al. , A Photopolymerizable Thermoplastic with Tunable Mechanical Performance, Mater. Horiz, 2020, 7, 835–842. [Google Scholar]
- 277.Childress KK, Alim MD, Hernandez JJ, Stansbury JW, and Bowman CN, Additive Manufacture of Lightly Crosslinked Semicrystalline Thiol-Enes for Enhanced Mechanical Performance, Polym. Chem, 2020, 11, 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Konkolewicz D, Gray-Weale A, and Perrier S, Hyperbranched Polymers by Thiol–Yne Chemistry: From Small Molecules to Functional Polymers, J. Am. Chem. Soc, 2009, 131, 18075–18077. [DOI] [PubMed] [Google Scholar]
- 279.Li N, Tsoi T-H, Lo W-S, Gu Y-J, Wan H-Y, and Wong W-T, An Efficient Approach to Synthesize Glycerol Dendrimers Via Thiol–Yne “Click” Chemistry and Their Application in Stabilization of Gold Nanoparticles with X-Ray Attenuation Properties, Polym. Chem, 2017, 8, 6989–6996. [Google Scholar]
- 280.Chen G, Kumar J, Gregory A, and Stenzel MH, Efficient Synthesis of Dendrimersvia a Thiol–Yne and Esterification Process and Their Potential Application in the Delivery of Platinum Anti-Cancer Drugs, Chem. Commun, 2009, 6291–6293. [DOI] [PubMed] [Google Scholar]
- 281.Killops KL, Campos LM, and Hawker CJ, Robust, Efficient, and Orthogonal Synthesis of Dendrimers Via Thiol-Ene “Click” Chemistry, J. Am. Chem. Soc, 2008, 130, 5062–5064. [DOI] [PubMed] [Google Scholar]
- 282.F. Debets M, Hest v.J.C.M., and T. Rutjes FPJ, Bioorthogonal Labelling of Biomolecules: New Functional Handles and Ligation Methods, Org. Biomol. Chem, 2013, 11, 6439–6455. [DOI] [PubMed] [Google Scholar]
- 283.Ivancová I, Leone D-L, and Hocek M, Reactive Modifications of DNA Nucleobases for Labelling, Bioconjugations, and Cross-Linking, Curr. Opin. Chem. Biol, 2019, 52, 136–144. [DOI] [PubMed] [Google Scholar]
- 284.A. Sutton D, Yu S-H, Steet R, and V. Popik V, Cyclopropenone-Caged Sondheimer Diyne (Dibenzo[ a , E ]Cyclooctadiyne): A Photoactivatable Linchpin for Efficient Spaac Crosslinking, Chem. Commun, 2016, 52, 553–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Chang T-C, Adak AK, Lin T-W, Li P-J, Chen Y-J, Lai C-H, Liang C-F, Chen Y-J, and Lin C-C, A Photo-Cleavable Biotin Affinity Tag for the Facile Release of a Photo-Crosslinked Carbohydrate-Binding Protein, Biorg. Med. Chem, 2016, 24, 1216–1224. [DOI] [PubMed] [Google Scholar]
- 286.Arts R, Ludwig SKJ, van Gerven BCB, Estirado EM, Milroy L-G, and Merkx M, Semisynthetic Bioluminescent Sensor Proteins for Direct Detection of Antibodies and Small Molecules in Solution, ACS Sens., 2017, 2, 1730–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Sadiki A, Kercher EM, Lu H, Lang RT, Spring BQ, and Zhou ZS, Site-Specific Bioconjugation and Convergent Click Chemistry Enhances Antibody–Chromophore Conjugate Binding Efficiency, Photochem. Photobiol, 2020, 96, 596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Ramil CP, Dong M, An P, Lewandowski TM, Yu Z, Miller LJ, and Lin Q, Spirohexene-Tetrazine Ligation Enables Bioorthogonal Labeling of Class B G Protein-Coupled Receptors in Live Cells, J. Am. Chem. Soc, 2017, 139, 13376–13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Wang L, Zhang J, Zhao J, Yu P, Wang S, Hu H, and Wang R, Recent Synthesis of Functionalized S-Tetrazines and Their Application in Ligation Reactions under Physiological Conditions: A Concise Overview, Catal. Rev, 2020, 0, 1–42. [Google Scholar]
- 290.Yu Z, Ho LY, and Lin Q, Rapid, Photoactivatable Turn-on Fluorescent Probes Based on an Intramolecular Photoclick Reaction, J. Am. Chem. Soc, 2011, 133, 11912–11915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Wang J, Zhang W, Song W, Wang Y, Yu Z, Li J, Wu M, Wang L, Zang J, and Lin Q, A Biosynthetic Route to Photoclick Chemistry on Proteins, J. Am. Chem. Soc, 2010, 132, 14812–14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Lee Y-J, Wu B, Raymond JE, Zeng Y, Fang X, Wooley KL, and Liu WR, A Genetically Encoded Acrylamide Functionality, ACS Chem. Biol, 2013, 8, 1664–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Li F, Zhang H, Sun Y, Pan Y, Zhou J, and Wang J, Expanding the Genetic Code for Photoclick Chemistry in E. Coli, Mammalian Cells, and A. Thaliana, Angew. Chem. Int. Ed, 2013, 52, 9700–9704. [DOI] [PubMed] [Google Scholar]
- 294.Yu Z, Lim RKV, and Lin Q, Synthesis of Macrocyclic Tetrazoles for Rapid Photoinduced Bioorthogonal 1,3-Dipolar Cycloaddition Reactions, Chem. Eur. J, 2010, 16, 13325–13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Lang K, Davis L, Torres-Kolbus J, Chou C, Deiters A, and Chin JW, Genetically Encoded Norbornene Directs Site-Specific Cellular Protein Labelling Via a Rapid Bioorthogonal Reaction, Nat. Chem, 2012, 4, 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Kaya E, Vrabel M, Deiml C, Prill S, Fluxa VS, and Carell T, A Genetically Encoded Norbornene Amino Acid for the Mild and Selective Modification of Proteins in a Copper-Free Click Reaction, Angew. Chem. Int. Ed, 2012, 51, 4466–4469. [DOI] [PubMed] [Google Scholar]
- 297.Yu Z, Pan Y, Wang Z, Wang J, and Lin Q, Genetically Encoded Cyclopropene Directs Rapid, Photoclick-Chemistry-Mediated Protein Labeling in Mammalian Cells, Angew. Chem. Int. Ed, 2012, 51, 10600–10604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Yu Z and Lin Q, Design of Spiro[2.3]Hex-1-Ene, a Genetically Encodable Double-Strained Alkene for Superfast Photoclick Chemistry, J. Am. Chem. Soc, 2014, 136, 4153–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Arndt S and Wagenknecht H-A, “Photoclick” Postsynthetic Modification of DNA, Angew. Chem. Int. Ed, 2014, 53, 14580–14582. [DOI] [PubMed] [Google Scholar]
- 300.Merkel M, Arndt S, Ploschik D, Cserép GB, Wenge U, Kele P, and Wagenknecht H-A, Scope and Limitations of Typical Copper-Free Bioorthogonal Reactions with DNA: Reactive 2′-Deoxyuridine Triphosphates for Postsynthetic Labeling, J. Org. Chem, 2016, 81, 7527–7538. [DOI] [PubMed] [Google Scholar]
- 301.Lehmann B and Wagenknecht H-A, Fluorogenic “Photoclick” Labelling of DNA Using a Cy3 Dye, Org. Biomol. Chem, 2018, 16, 7579–7582. [DOI] [PubMed] [Google Scholar]
- 302.Zhou Z, Xie X, Yi Q, Yin W, Kadi AA, Li J, and Zhang Y, Enzyme-Instructed Self-Assembly with Photo-Responses for the Photo-Regulation of Cancer Cells, Org. Biomol. Chem, 2017, 15, 6892–6895. [DOI] [PubMed] [Google Scholar]
- 303.Yu Z, Ohulchanskyy TY, An P, Prasad PN, and Lin Q, Fluorogenic, Two-Photon-Triggered Photoclick Chemistry in Live Mammalian Cells, J. Am. Chem. Soc, 2013, 135, 16766–16769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Zhou M, Hu J, Zheng M, Song Q, Li J, and Zhang Y, Photo-Click Construction of a Targetable and Activatable Two-Photon Probe Imaging Protease in Apoptosis, Chem. Commun, 2016, 52, 2342–2345. [DOI] [PubMed] [Google Scholar]
- 305.Wu Y, Guo G, Zheng J, Xing D, and Zhang T, Fluorogenic “Photoclick” Labeling and Imaging of DNA with Coumarin-Fused Tetrazole in Vivo, ACS Sens, 2019, 4, 44–51. [DOI] [PubMed] [Google Scholar]
- 306.Zhu B, Ge J, and Yao SQ, Developing New Chemical Tools for DNA Methyltransferase 1 (Dnmt 1): A Small-Molecule Activity-Based Probe and Novel Tetrazole-Containing Inhibitors, Biorg. Med. Chem, 2015, 23, 2917–2927. [DOI] [PubMed] [Google Scholar]
- 307.An P and Lin Q, Sterically Shielded Tetrazoles for a Fluorogenic Photoclick Reaction: Tuning Cycloaddition Rate and Product Fluorescence, Org. Biomol. Chem, 2018, 16, 5241–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Yao Z, Wu X, Zhang X, Xiong Q, Jiang S, and Yu Z, Synthesis and Evaluation of Photo-Activatable Β-Diarylsydnone- L -Alanines for Fluorogenic Photo-Click Cyclization of Peptides, Org. Biomol. Chem, 2019, 17, 6777–6781. [DOI] [PubMed] [Google Scholar]
- 309.Favre C and Friscourt F, Fluorogenic Sydnone-Modified Coumarins Switched-on by Copper-Free Click Chemistry, Org. Lett, 2018, 20, 4213–4217. [DOI] [PubMed] [Google Scholar]
- 310.Krell K, Harijan D, Ganz D, Doll L, and Wagenknecht H-A, Postsynthetic Modifications of DNA and Rna by Means of Copper-Free Cycloadditions as Bioorthogonal Reactions, Bioconjugate Chem., 2020, 31, 990–1011. [DOI] [PubMed] [Google Scholar]
- 311.Li J, Kong H, Huang L, Cheng B, Qin K, Zheng M, Yan Z, and Zhang Y, Visible Light-Initiated Bioorthogonal Photoclick Cycloaddition, J. Am. Chem. Soc, 2018, 140, 14542–14546. [DOI] [PubMed] [Google Scholar]
- 312.Jung S and Kwon I, Expansion of Bioorthogonal Chemistries Towards Site-Specific Polymer–Protein Conjugation, Polym. Chem, 2016, 7, 4584–4598. [Google Scholar]
- 313.Dondoni A, Massi A, Nanni P, and Roda A, A New Ligation Strategy for Peptide and Protein Glycosylation: Photoinduced Thiol–Ene Coupling, Chem. Eur. J, 2009, 15, 11444–11449. [DOI] [PubMed] [Google Scholar]
- 314.Denis M, Softley C, Giuntini S, Gentili M, Ravera E, Parigi G, Fragai M, Popowicz G, Sattler M, Luchinat C, et al. , The Photocatalyzed Thiol-Ene Reaction: A New Tag to Yield Fast, Selective and Reversible Paramagnetic Tagging of Proteins, ChemPhysChem, 2020, 21, 863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Gunnoo SB and Madder A, Chemical Protein Modification through Cysteine, ChemBioChem, 2016, 17, 529–553. [DOI] [PubMed] [Google Scholar]
- 316.Li Y, Yang M, Huang Y, Song X, Liu L, and Chen PR, Genetically Encoded Alkenyl–Pyrrolysine Analogues for Thiol–Ene Reaction Mediated Site-Specific Protein Labeling, Chem. Sci, 2012, 3, 2766–2770. [Google Scholar]
- 317.Weinrich D, Lin P-C, Jonkheijm P, Nguyen UTT, Schröder H, Niemeyer CM, Alexandrov K, Goody R, and Waldmann H, Oriented Immobilization of Farnesylated Proteins by the Thiol-Ene Reaction, Angew. Chem. Int. Ed, 2010, 49, 1252–1257. [DOI] [PubMed] [Google Scholar]
- 318.Minozzi M, Monesi A, Nanni D, Spagnolo P, Marchetti N, and Massi A, An Insight into the Radical Thiol/Yne Coupling: The Emergence of Arylalkyne-Tagged Sugars for the Direct Photoinduced Glycosylation of Cysteine-Containing Peptides, J. Org. Chem, 2011, 76, 450–459. [DOI] [PubMed] [Google Scholar]
- 319.Li Y, Pan M, Li Y, Huang Y, and Guo Q, Thiol–Yne Radical Reaction Mediated Site-Specific Protein Labeling Via Genetic Incorporation of an Alkynyl-L-Lysine Analogue, Org. Biomol. Chem, 2013, 11, 2624–2629. [DOI] [PubMed] [Google Scholar]
- 320.Liu J, Cheng R, Wu H, Li S, Wang PG, DeGrado WF, Rozovsky S, and Wang L, Building and Breaking Bonds Via a Compact S-Propargyl-Cysteine to Chemically Control Enzymes and Modify Proteins, Angew. Chem. Int. Ed, 2018, 57, 12702–12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Luo W, Pulsipher A, Dutta D, Lamb BM, and Yousaf MN, Remote Control of Tissue Interactions Via Engineered Photo-Switchable Cell Surfaces, Sci. Rep, 2014, 4, 6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Lodge TP, Block Copolymers: Past Successes and Future Challenges, Macromol. Chem. Phys, 2003, 204, 265–273. [Google Scholar]
- 323.Hu H, Gopinadhan M, and Osuji CO, Directed Self-Assembly of Block Copolymers: A Tutorial Review of Strategies for Enabling Nanotechnology with Soft Matter, Soft Matter, 2014, 10, 3867–3889. [DOI] [PubMed] [Google Scholar]
- 324.Doran S, Murtezi E, Barlas FB, Timur S, and Yagci Y, One-Pot Photo-Induced Sequential Cuaac and Thiol-Ene Click Strategy for Bioactive Macromolecular Synthesis, Macromolecules, 2014, 47, 3608–3613. [Google Scholar]
- 325.Derboven P, D'Hooge DR, Stamenovic MM, Espeel P, Marin GB, Du Prez FE, and Reyniers M-F, Kinetic Modeling of Radical Thiol-Ene Chemistry for Macromolecular Design: Importance of Side Reactions and Diffusional Limitations, Macromolecules, 2013, 46, 1732–1742. [Google Scholar]
- 326.Colak B, Da Silva JCS, Soares TA, and Gautrot JE, Impact of the Molecular Environment on Thiol-Ene Coupling for Biofunctionalization and Conjugation, Bioconjugate Chemistry, 2016, 27, 2111–2123. [DOI] [PubMed] [Google Scholar]
- 327.Stamenovic MM, Espeel P, Van Camp W, and Du Prez FE, Norbornenyl-Based Raft Agents for the Preparation of Functional Polymers Via Thiol-Ene Chemistry, Macromolecules, 2011, 44, 5619–5630. [Google Scholar]
- 328.Walker CN, Sarapas JM, Kung V, Hall AL, and Tew GN, Multiblock Copolymers by Thiol Addition across Norbornene, ACS Macro Lett., 2014, 3, 453–457. [DOI] [PubMed] [Google Scholar]
- 329.Feist F, Menzel JP, Weil T, Blinco JP, and Barner-Kowollik C, Visible Light-Induced Ligation Via O-Quinodimethane Thioethers, J. Am. Chem. Soc, 2018, 140, 11848–11854. [DOI] [PubMed] [Google Scholar]
- 330.Niu J, Lunn DJ, Pusuluri A, Yoo JI, O'Malley MA, Mitragotri S, Soh HT, and Hawker CJ, Engineering Live Cell Surfaces with Functional Polymers Via Cytocompatible Controlled Radical Polymerization, Nat. Chem, 2017, 9, 537–545. [DOI] [PubMed] [Google Scholar]
- 331.Ye X, Yuan J, Jiang Z, Wang S, Wang P, Wang Q, and Cui L, Thiol-Ene Photoclick Reaction: An Eco-Friendly and Facile Approach for Preparation of Mpeg-G-Keratin Biomaterial, Eng. Life Sci, 2020, 20, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Siti W, Khan AK, de Hoog H-PM, Liedberg B, and Nallani M, Photo-Induced Conjugation of Tetrazoles to Modified and Native Proteins, Org. Biomol. Chem, 2015, 13, 3202–3206. [DOI] [PubMed] [Google Scholar]
- 333.Bauer DM, Rogge A, Stolzer L, Barner-Kowollik C, and Fruk L, Light Induced DNA–Protein Conjugation, Chem. Commun, 2013, 49, 8626–8628. [DOI] [PubMed] [Google Scholar]
- 334.Vigovskaya A, Abt D, Ahmed I, Niemeyer CM, Barner-Kowollik C, and Fruk L, Photo-Induced Chemistry for the Design of Oligonucleotide Conjugates and Surfaces, J. Mater. Chem. B, 2016, 4, 442–449. [DOI] [PubMed] [Google Scholar]
- 335.Asatekin A, Barr MC, Baxamusa SH, Lau KKS, Tenhaeff W, Xu J, and Gleason KK, Designing Polymer Surfaces Via Vapor Deposition, Mater. Today, 2010, 13, 26–33. [Google Scholar]
- 336.Decher G and Hong JD, Buildup of Ultrathin Multilayer Films by a Self-Assembly Process: Ii. Consecutive Adsorption of Anionic and Cationic Bipolar Amphiphiles and Polyelectrolytes on Charged Surfaces, Ber. Bunsen Ges. Phys. Chem, 1991, 95, 1430–1434. [Google Scholar]
- 337.Ohno K, Ma Y, Huang Y, Mori C, Yahata Y, Tsujii Y, Maschmeyer T, Moraes J, and Perrier S, Surface-Initiated Reversible Addition–Fragmentation Chain Transfer (Raft) Polymerization from Fine Particles Functionalized with Trithiocarbonates, Macromolecules, 2011, 44, 8944–8953. [Google Scholar]
- 338.Yan W, Dadashi-Silab S, Matyjaszewski K, Spencer ND, and Benetti EM, Surface-Initiated Photoinduced Atrp: Mechanism, Oxygen Tolerance, and Temporal Control During the Synthesis of Polymer Brushes, Macromolecules, 2020, 53, 2801–2810. [Google Scholar]
- 339.Jonkheijm P, Weinrich D, Köhn M, Engelkamp H, Christianen PCM, Kuhlmann J, Maan JC, Nüsse D, Schroeder H, Wacker R, et al. , Photochemical Surface Patterning by the Thiol-Ene Reaction, Angew. Chem. Int. Ed, 2008, 47, 4421–4424. [DOI] [PubMed] [Google Scholar]
- 340.Wu J-T, Huang C-H, Liang W-C, Wu Y-L, Yu J, and Chen H-Y, Reactive Polymer Coatings: A General Route to Thiol-Ene and Thiol-Yne Click Reactions, Macromol. Rapid Commun, 2012, 33, 922–927. [DOI] [PubMed] [Google Scholar]
- 341.Bhairamadgi NS, Gangarapu S, Caipa Campos MA, Paulusse JMJ, van Rijn CJM, and Zuilhof H, Efficient Functionalization of Oxide-Free Silicon(111) Surfaces: Thiol–Yne Versus Thiol–Ene Click Chemistry, Langmuir, 2013, 29, 4535–4542. [DOI] [PubMed] [Google Scholar]
- 342.Ma SJ, Ford EM, Sawicki LA, Sutherland BP, Halaszynski NI, Carberry BJ, Wagner NJ, Kloxin AM, and Kloxin CJ, Surface Chemical Functionalization of Wrinkled Thiol–Ene Elastomers for Promoting Cellular Alignment, ACS Appl. Bio Mater, 2020, 3, 3731–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Liang H, Gagné JD, Faye A, Rodrigue D, and Brisson J, Ground Tire Rubber (Gtr) Surface Modification Using Thiol-Ene Click Reaction: Polystyrene Grafting to Modify a Gtr/Polystyrene (Ps) Blend, Prog. Rubber Plast. Recycl. Technol, 2019, 36, 81–101. [Google Scholar]
- 344.Preuss CM, Tischer T, Rodriguez-Emmenegger C, Zieger MM, Bruns M, Goldmann AS, and Barner-Kowollik C, A Bioinspired Light Induced Avenue for the Design of Patterned Functional Interfaces, J. Mater. Chem. B, 2014, 2, 36–40. [DOI] [PubMed] [Google Scholar]
- 345.Zhang XN, Liang JW, Chen ZY, Donley C, Zhang XL, and Cheng GT, Surface Modification of Bombyx Mori Silk Fibroin Film Via Thiol-Ene Click Chemistry, Processes, 2020, 8. [Google Scholar]
- 346.Zhang P, Wang Q, Shen J, Wang P, Yuan J, and Fan X, Enzymatic Thiol–Ene Click Reaction: An Eco-Friendly Approach for Mpegma-Grafted Modification of Wool Fibers, ACS Sustainable Chem. Eng, 2019, 7, 13446–13455. [Google Scholar]
- 347.Hu Y, Wang W, Xu L, and Yu D, Surface Modification of Keratin Fibers through Step-Growth Dithiol-Diacrylate Thiol-Ene Click Reactions, Mater. Lett, 2016, 178, 159–162. [Google Scholar]
- 348.Wilke P, Abt D, Große S, Barner-Kowollik C, and Börner HG, Selective Functionalization of Laser Printout Patterns on Cellulose Paper Sheets Coated with Surface-Specific Peptides, J. Mater. Chem. A, 2017, 5, 16144–16149. [Google Scholar]
- 349.Blasco E, Piñol M, Oriol L, Schmidt BVKJ, Welle A, Trouillet V, Bruns M, and Barner-Kowollik C, Photochemical Generation of Light Responsive Surfaces, Adv. Funct. Mater, 2013, 23, 4011–4019. [Google Scholar]
- 350.Delafresnaye L, Zaquen N, Kuchel RP, Blinco JP, Zetterlund PB, and Barner-Kowollik C, A Simple and Versatile Pathway for the Synthesis of Visible Light Photoreactive Nanoparticles, Adv. Funct. Mater, 2018, 28, 1800342. [Google Scholar]
- 351.de los Santos Pereira A, Kostina NY, Bruns M, Rodriguez-Emmenegger C, and Barner-Kowollik C, Phototriggered Functionalization of Hierarchically Structured Polymer Brushes, Langmuir, 2015, 31, 5899–5907. [DOI] [PubMed] [Google Scholar]
- 352.Arumugam S and Popik VV, Sequential “Click” – “Photo-Click” Cross-Linker for Catalyst-Free Ligation of Azide-Tagged Substrates, J. Org. Chem, 2014, 79, 2702–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Arumugam S, Orski SV, Locklin J, and Popik VV, Photoreactive Polymer Brushes for High-Density Patterned Surface Derivatization Using a Diels–Alder Photoclick Reaction, J. Am. Chem. Soc, 2012, 134, 179–182. [DOI] [PubMed] [Google Scholar]
- 354.Richter B, Pauloehrl T, Kaschke J, Fichtner D, Fischer J, Greiner AM, Wedlich D, Wegener M, Delaittre G, Barner-Kowollik C, et al. , Three-Dimensional Microscaffolds Exhibiting Spatially Resolved Surface Chemistry, Adv. Mater, 2013, 25, 6117–6122. [DOI] [PubMed] [Google Scholar]
- 355.Stockmayer WH, Theory of Molecular Size Distribution and Gel Formation in Branched Polymers Ii. General Cross Linking, The Journal of Chemical Physics, 1944, 12, 125–131. [Google Scholar]
- 356.Lu H, Carioscia JA, Stansbury JW, and Bowman CN, Investigations of Step-Growth Thiol-Ene Polymerizations for Novel Dental Restoratives, Dent. Mater, 2005, 21, 1129–1136. [DOI] [PubMed] [Google Scholar]
- 357.Reinelt S, Tabatabai M, Moszner N, Fischer UK, Utterodt A, and Ritter H, Synthesis and Photopolymerization of Thiol-Modified Triazine-Based Monomers and Oligomers for the Use in Thiol-Ene-Based Dental Composites, Macromol. Chem. Phys, 2014, 215, 1415–1425. [Google Scholar]
- 358.Podgorski M, Becka E, Claudino M, Flores A, Shah PK, Stansbury JW, and Bowman CN, Ester-Free Thiol-Ene Dental Restoratives-Part A: Resin Development, Dent. Mater, 2015, 31, 1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Song HB, Sowan N, Shah PK, Baranek A, Flores A, Stansbury JW, and Bowman CN, Reduced Shrinkage Stress Via Photo-Initiated Copper(I)-Catalyzed Cycloaddition Polymerizations of Azide-Alkyne Resins, Dent. Mater, 2016, 32, 1332–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.El-Zaatari B, Shete A, and Kloxin C, Photo-Cuaac Applications in Dental Restorative Materials, Abst. Pap. Am. Chem. S, 2017, 253. [Google Scholar]
- 361.Huang S, Podgorski M, Zhang X, Sinha J, Claudino M, Stansbury JW, and Bowman CN, Dental Restorative Materials Based on Thiol-Michael Photopolymerization, J. Dent. Res, 2018, 97, 530–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Lovell LG, Berchtold KA, Elliott JE, Lu H, and Bowman CN, Understanding the Kinetics and Network Formation of Dimethacrylate Dental Resins, Polym. Adv. Technol, 2001, 12, 335–345. [Google Scholar]
- 363.Carioscia JA, Lu H, Stanbury JW, and Bowman CN, Thiol-Ene Oligomers as Dental Restorative Materials, Dent. Mater, 2005, 21, 1137–1143. [DOI] [PubMed] [Google Scholar]
- 364.Podgorski M, Becka E, Claudino M, Flores A, Shah PK, Stansbury JW, and Bowman CN, Ester-Free Thiol-Ene Dental Restoratives-Part B: Composite Development, Dent. Mater, 2015, 31, 1263–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Ye S, Azarnoush S, Smith IR, Cramer NB, Stansbury JW, and Bowman CN, Using Hyperbranched Oligomer Functionalized Glass Fillers to Reduce Shrinkage Stress, Dent. Mater, 2012, 28, 1004–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Roth M, Oesterreicher A, Mostegel FH, Moser A, Pinter G, Edler M, Piock R, and Griesser T, Silicon-Based Mercaptans: High-Performance Monomers for Thiol-Ene Photopolymerization, J. Polym. Sci., Part A: Polym. Chem, 2016, 54, 418–424. [Google Scholar]
- 367.Sinha J, Dobson A, Bankhar O, Podgorski M, Shah PK, Zajdowicz SLW, Alotaibi A, Stansbury JW, and Bowman CN, Vinyl Sulfonamide Based Thermosetting Composites Via Thiol-Michael Polymerization, Dent. Mater, 2020, 36, 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Song HB, Wang X, Patton JR, Stansbury JW, and Bowman CN, Kinetics and Mechanics of Photo-Polymerized Triazole-Containing Thermosetting Composites Via the Copper(I)-Catalyzed Azide-Alkyne Cycloaddition, Dent. Mater, 2017, 33, 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Leonards H, Engelhardt S, Hoffmann A, Pongratz L, Schriever S, Blasius J, Wehner M, and Gillner A, Advantages and Drawbacks of Thiol-Ene Based Resins for 3d-Printing, in Laser 3d Manufacturing Ii, Helvajian H, et al. , Editors. Spie-Int Soc Optical Engineering: Bellingham. 2015. [Google Scholar]
- 370.Roppolo I, Frascella F, Gastaldi M, Castellino M, Ciubini B, Barolo C, Scaltrito L, Nicosia C, Zanetti M, and Chiappone A, Thiol–Yne Chemistry for 3d Printing: Exploiting an Off-Stoichiometric Route for Selective Functionalization of 3d Objects, Polym. Chem, 2019, 10, 5950–5958. [Google Scholar]
- 371.Childress KK, Alim MD, Hernandez JJ, Stansbury JW, and Bowman CN, Additive Manufacture of Lightly Crosslinked Semicrystalline Thiol–Enes for Enhanced Mechanical Performance, Polym. Chem, 2020, 11, 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Lovelady E, Kimmins SD, Wu J, and Cameron NR, Preparation of Emulsion-Templated Porous Polymers Using Thiol–Ene and Thiol–Yne Chemistry, Polym. Chem, 2011, 2, 559–562. [Google Scholar]
- 373.Chen C, Eissa AM, Schiller TL, and Cameron NR, Emulsion-Templated Porous Polymers Prepared by Thiol-Ene and Thiol-Yne Photopolymerisation Using Multifunctional Acrylate and Non-Acrylate Monomers, Polymer, 2017, 126, 395–401. [Google Scholar]
- 374.Amato DN, Amato DV, Sandoz M, Weigand J, Patton DL, and Visser CW, Programmable Porous Polymers Via Direct Bubble Writing with Surfactant-Free Inks, ACS Appl. Mater. Interfaces, 2020, 12, 42048–42055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Svejdal RR, Dickinson ER, Sticker D, Kutter JP, and Rand KD, Thiol-Ene Microfluidic Chip for Performing Hydrogen/Deuterium Exchange of Proteins at Subsecond Time Scales, Anal. Chem, 2019, 91, 1309–1317. [DOI] [PubMed] [Google Scholar]
- 376.Jönsson A, Svejdal RR, Bøgelund N, Nguyen TTTN, Flindt H, Kutter JP, Rand KD, and Lafleur JP, Thiol-Ene Monolithic Pepsin Microreactor with a 3d-Printed Interface for Efficient Uplc-Ms Peptide Mapping Analyses, Anal. Chem, 2017, 89, 4573–4580. [DOI] [PubMed] [Google Scholar]
- 377.Quick AS, Fischer J, Richter B, Pauloehrl T, Trouillet V, Wegener M, and Barner-Kowollik C, Preparation of Reactive Three-Dimensional Microstructures Via Direct Laser Writing and Thiol-Ene Chemistry, Macromol. Rapid Commun, 2013, 34, 335–340. [DOI] [PubMed] [Google Scholar]
- 378.Kumpfmueller J, Stadlmann K, Li Z, Satzinger V, Stampfl J, and Liska R, Two-Photon-Induced Thiol-Ene Polymerization as a Fabrication Tool for Flexible Optical Waveguides, Des. Monomers Polym, 2014, 17, 390–400. [Google Scholar]
- 379.Griesser T, Wolfberger A, Daschiel U, Schmidt V, Fian A, Jerrar A, Teichert C, and Kern W, Cross-Linking of Romp Derived Polymers Using the Two-Photon Induced Thiol–Ene Reaction: Towards the Fabrication of 3d-Polymer Microstructures, Polym. Chem, 2013, 4, 1708–1714. [Google Scholar]
- 380.Nguyen LH, Straub M, and Gu M, Acrylate-Based Photopolymer for Two-Photon Microfabrication and Photonic Applications, Adv. Funct. Mater, 2005, 15, 209–216. [Google Scholar]
- 381.Wu S, Straub M, and Gu M, Single-Monomer Acrylate-Based Resin for Three-Dimensional Photonic Crystal Fabrication, Polymer, 2005, 46, 10246–10255. [Google Scholar]
- 382.Batchelor R, Messer T, Hippler M, Wegener M, Barner-Kowollik C, and Blasco E, Two in One: Light as a Tool for 3d Printing and Erasing at the Microscale, Adv. Mater, 2019, 31, 1904085. [DOI] [PubMed] [Google Scholar]
- 383.Quick AS, Rothfuss H, Welle A, Richter B, Fischer J, Wegener M, and Barner-Kowollik C, Fabrication and Spatially Resolved Functionalization of 3d Microstructures Via Multiphoton-Induced Diels–Alder Chemistry, Adv. Funct. Mater, 2014, 24, 3571–3580. [Google Scholar]
- 384.Dickey MD, Collister E, Raines A, Tsiartas P, Holcombe T, Sreenivasan SV, Bonnecaze RT, and Willson CG, Photocurable Pillar Arrays Formed Via Electrohydrodynamic Instabilities, Chem. Mater, 2006, 18, 2043–2049. [Google Scholar]
- 385.Sticker D, Geczy R, Häfeli UO, and Kutter JP, Thiol–Ene Based Polymers as Versatile Materials for Microfluidic Devices for Life Sciences Applications, ACS Appl. Mater. Interfaces, 2020, 12, 10080–10095. [DOI] [PubMed] [Google Scholar]
- 386.Tähkä SM, Bonabi A, Jokinen VP, and Sikanen TM, Aqueous and Non-Aqueous Microchip Electrophoresis with on-Chip Electrospray Ionization Mass Spectrometry on Replica-Molded Thiol-Ene Microfluidic Devices, J. Chromatogr. A, 2017, 1496, 150–156. [DOI] [PubMed] [Google Scholar]
- 387.Waheed S, Cabot JM, Macdonald NP, Lewis T, Guijt RM, Paull B, and Breadmore MC, 3d Printed Microfluidic Devices: Enablers and Barriers, Lab Chip, 2016, 16, 1993–2013. [DOI] [PubMed] [Google Scholar]
- 388.Mongkhontreerat S, Öberg K, Erixon L, Löwenhielm P, Hult A, and Malkoch M, Uv Initiated Thiol–Ene Chemistry: A Facile and Modular Synthetic Methodology for the Construction of Functional 3d Networks with Tunable Properties, J. Mater. Chem. A, 2013, 1, 13732–13737. [Google Scholar]
- 389.Carlborg CF, Haraldsson T, Öberg K, Malkoch M, and van der Wijngaart W, Beyond Pdms: Off-Stoichiometry Thiol–Ene (Oste) Based Soft Lithography for Rapid Prototyping of Microfluidic Devices, Lab Chip, 2011, 11, 3136–3147. [DOI] [PubMed] [Google Scholar]
- 390.Good BT, Reddy S, Davis RH, and Bowman CN, Tailorable Low Modulus, Reversibly Deformable Elastomeric Thiol–Ene Materials for Microfluidic Applications, Sens. Actuators, B, 2007, 120, 473–480. [Google Scholar]
- 391.Ashley JF, Cramer NB, Davis RH, and Bowman CN, Soft-Lithography Fabrication of Microfluidic Features Using Thiol-Ene Formulations, Lab Chip, 2011, 11, 2772–2778. [DOI] [PubMed] [Google Scholar]
- 392.Hillmering M, Pardon G, Vastesson A, Supekar O, Carlborg CF, Brandner BD, van der Wijngaart W, and Haraldsson T, Off-Stoichiometry Improves the Photostructuring of Thiol–Enes through Diffusion-Induced Monomer Depletion, Microsyst. Nanoeng, 2016, 2, 15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Zhou XC, Sjöberg R, Druet A, Schwenk JM, van der Wijngaart W, Haraldsson T, and Carlborg CF, Thiol–Ene–Epoxy Thermoset for Low-Temperature Bonding to Biofunctionalized Microarray Surfaces, Lab Chip, 2017, 17, 3672–3681. [DOI] [PubMed] [Google Scholar]
- 394.Zurgil N, Afrimzon E, Deutsch A, Namer Y, Shafran Y, Sobolev M, Tauber Y, Ravid-Hermesh O, and Deutsch M, Polymer Live-Cell Array for Real-Time Kinetic Imaging of Immune Cells, Biomaterials, 2010, 31, 5022–5029. [DOI] [PubMed] [Google Scholar]
- 395.Markovitz-Bishitz Y, Tauber Y, Afrimzon E, Zurgil N, Sobolev M, Shafran Y, Deutsch A, Howitz S, and Deutsch M, A Polymer Microstructure Array for the Formation, Culturing, and High Throughput Drug Screening of Breast Cancer Spheroids, Biomaterials, 2010, 31, 8436–8444. [DOI] [PubMed] [Google Scholar]
- 396.Tan H-Y, Trier S, Rahbek UL, Dufva M, Kutter JP, and Andresen TL, A Multi-Chamber Microfluidic Intestinal Barrier Model Using Caco-2 Cells for Drug Transport Studies, PLOS ONE, 2018, 13, e0197101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Burke JM, Pandit KR, Goertz JP, and White IM, Fabrication of Rigid Microstructures with Thiol-Ene-Based Soft Lithography for Continuous-Flow Cell Lysis, Biomicrofluidics, 2014, 8, 056503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.D'Eramo L, Chollet B, Leman M, Martwong E, Li M, Geisler H, Dupire J, Kerdraon M, Vergne C, Monti F, et al. , Microfluidic Actuators Based on Temperature-Responsive Hydrogels, Microsyst. Nanoeng, 2018, 4.31057894 [Google Scholar]
- 399.Lu N, Sticker D, Kretschmann A, Petersen NJ, and Kutter JP, A Thiol-Ene Microfluidic Device Enabling Continuous Enzymatic Digestion and Electrophoretic Separation as Front-End to Mass Spectrometric Peptide Analysis, Anal. Bioanal. Chem, 2020, 412, 3559–3571. [DOI] [PubMed] [Google Scholar]
- 400.Macdonald EK and Shaver MP, Intrinsic High Refractive Index Polymers, Polym. Int, 2015, 64, 6–14. [Google Scholar]
- 401.Liu J. g. and Ueda M, High Refractive Index Polymers: Fundamental Research and Practical Applications, J. Mater. Chem, 2009, 19, 8907–8919. [Google Scholar]
- 402.Bhagat SD, Filho EBDS, and Stiegman AE, High Refractive Index Polymer Composites Synthesized by Cross-Linking of Oxozirconium Clusters through Thiol-Ene Polymerization, Macromol. Mater. Eng, 2015, 300, 580–585. [Google Scholar]
- 403.Su Y, Filho EBDS, Peek N, Chen B, and Stiegman AE, High Refractive Index Polymers (N > 1.7), Based on Thiol–Ene Cross-Linking of Polarizable P═S and P═Se Organic/Inorganic Monomers, Macromolecules, 2019, 52, 9012–9022. [Google Scholar]
- 404.Fang L, Sun J, Chen X, Tao Y, Zhou J, Wang C, and Fang Q, Phosphorus- and Sulfur-Containing High-Refractive-Index Polymers with High Tg and Transparency Derived from a Bio-Based Aldehyde, Macromolecules, 2020, 53, 125–131. [Google Scholar]
- 405.Chen X, Fang L, Wang J, He F, Chen X, Wang Y, Zhou J, Tao Y, Sun J, and Fang Q, Intrinsic High Refractive Index Siloxane–Sulfide Polymer Networks Having High Thermostability and Transmittance Via Thiol–Ene Cross-Linking Reaction, Macromolecules, 2018, 51, 7567–7573. [Google Scholar]
- 406.Bhagat SD, Chatterjee J, Chen B, and Stiegman AE, High Refractive Index Polymers Based on Thiol–Ene Cross-Linking Using Polarizable Inorganic/Organic Monomers, Macromolecules, 2012, 45, 1174–1181. [Google Scholar]
- 407.Natarajan LV, Shepherd CK, Brandelik DM, Sutherland RL, Chandra S, Tondiglia VP, Tomlin D, and Bunning TJ, Switchable Holographic Polymer-Dispersed Liquid Crystal Reflection Gratings Based on Thiol–Ene Photopolymerization, Chem. Mater, 2003, 15, 2477–2484. [Google Scholar]
- 408.Natarajan LV, Brown DP, Wofford JM, Tondiglia VP, Sutherland RL, Lloyd PF, and Bunning TJ, Holographic Polymer Dispersed Liquid Crystal Reflection Gratings Formed by Visible Light Initiated Thiol-Ene Photopolymerization, Polymer, 2006, 47, 4411–4420. [Google Scholar]
- 409.Peng H, Nair DP, Kowalski BA, Xi W, Gong T, Wang C, Cole M, Cramer NB, Xie X, McLeod RR, et al. , High Performance Graded Rainbow Holograms Via Two-Stage Sequential Orthogonal Thiol–Click Chemistry, Macromolecules, 2014, 47, 2306–2315. [Google Scholar]
- 410.Nakabayashi K, Sobu S, Kosuge Y, and Mori H, Synthesis and Nanoimprinting of High Refractive Index and Highly Transparent Polythioethers Based on Thiol-Ene Click Chemistry, J. Polym. Sci., Part A: Polym. Chem, 2018, 56, 2175–2182. [Google Scholar]
- 411.Hata E, Mitsube K, Momose K, and Tomita Y, Holographic Nanoparticle-Polymer Composites Based on Step-Growth Thiol-Ene Photopolymerization, Opt. Mater. Express , 2011, 1, 207–222. [Google Scholar]
- 412.Mitsube K, Nishimura Y, Nagaya K, Takayama S, and Tomita Y, Holographic Nanoparticle-Polymer Composites Based on Radical-Mediated Thiol-Yne Photopolymerizations: Characterization and Shift-Multiplexed Holographic Digital Data Page Storage, Opt. Mater. Express , 2014, 4, 982–996. [Google Scholar]
- 413.Alim MD, Mavila S, Miller DB, Huang S, Podgórski M, Cox LM, Sullivan AC, McLeod RR, and Bowman CN, Realizing High Refractive Index Thiol-X Materials: A General and Scalable Synthetic Approach, ACS Materials Letters, 2019, 1, 582–588. [Google Scholar]
- 414.Chan JW, Zhou H, Hoyle CE, and Lowe AB, Photopolymerization of Thiol-Alkynes: Polysulfide Networks, Chem. Mater, 2009, 21, 1579–1585. [Google Scholar]
- 415.Peng H, Wang C, Xi W, Kowalski BA, Gong T, Xie X, Wang W, Nair DP, McLeod RR, and Bowman CN, Facile Image Patterning Via Sequential Thiol–Michael/Thiol–Yne Click Reactions, Chem. Mater, 2014, 26, 6819–6826. [Google Scholar]
- 416.Slaughter BV, Khurshid SS, Fisher OZ, Khademhosseini A, and Peppas NA, Hydrogels in Regenerative Medicine, Adv. Mater, 2009, 21, 3307–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Grim JC, Marozas IA, and Anseth KS, Thiol-Ene and Photo-Cleavage Chemistry for Controlled Presentation of Biomolecules in Hydrogels, J. Controlled Release, 2015, 219, 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Brown T and Anseth K, Spatiotemporal Hydrogel Biomaterials for Regenerative Medicine, Chem. Soc. Rev, 2017, 46, 6532–6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Lin C-C, Ki CS, and Shih H, Thiol–Norbornene Photoclick Hydrogels for Tissue Engineering Applications, J. Appl. Polym. Sci, 2015, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Kharkar PM, Rehmann MS, Skeens KM, Maverakis E, and Kloxin AM, Thiol–Ene Click Hydrogels for Therapeutic Delivery, ACS Biomater. Sci. Eng, 2016, 2, 165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Kharkar PM, Kiick KL, and Kloxin AM, Designing Degradable Hydrogels for Orthogonal Control of Cell Microenvironments, Chem. Soc. Rev, 2013, 42, 7335–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Polizzotti BD, Fairbanks BD, and Anseth KS, Three-Dimensional Biochemical Patterning of Click-Based Composite Hydrogels Via Thiolene Photopolymerization, Biomacromolecules, 2008, 9, 1084–1087. [DOI] [PubMed] [Google Scholar]
- 423.McCall JD and Anseth KS, Thiol–Ene Photopolymerizations Provide a Facile Method to Encapsulate Proteins and Maintain Their Bioactivity, Biomacromolecules, 2012, 13, 2410–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Shih H and Lin C-C, Photoclick Hydrogels Prepared from Functionalized Cyclodextrin and Poly(Ethylene Glycol) for Drug Delivery and in Situ Cell Encapsulation, Biomacromolecules, 2015, 16, 1915–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Cleophas RTC, Riool M, Quarles van Ufford HC, Zaat SAJ, Kruijtzer JAW, and Liskamp RMJ, Convenient Preparation of Bactericidal Hydrogels by Covalent Attachment of Stabilized Antimicrobial Peptides Using Thiol–Ene Click Chemistry, ACS Macro Lett, 2014, 3, 477–480. [DOI] [PubMed] [Google Scholar]
- 426.Wojda SJ, Marozas IA, Anseth KS, Yaszemski MJ, and Donahue SW, Thiol-Ene Hydrogels for Local Delivery of Pth for Bone Regeneration in Critical Size Defects, J. Orthop. Res, 2020, 38, 536–544. [DOI] [PubMed] [Google Scholar]
- 427.Hunckler MD, Medina JD, Coronel MM, Weaver JD, Stabler CL, and García AJ, Linkage Groups within Thiol–Ene Photoclickable Peg Hydrogels Control in Vivo Stability, Adv. Healthcare Mater, 2019, 8, 1900371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Ooi HW, Mota C, ten Cate AT, Calore A, Moroni L, and Baker MB, Thiol–Ene Alginate Hydrogels as Versatile Bioinks for Bioprinting, Biomacromolecules, 2018, 19, 3390–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Deng J-R, Chung S-F, Leung AS-L, Yip W-M, Yang B, Choi M-C, Cui J-F, Kung KK-Y, Zhang Z, Lo K-W, et al. , Chemoselective and Photocleavable Cysteine Modification of Peptides and Proteins Using Isoxazoliniums, Commun. Chem, 2019, 2, 1–10. [Google Scholar]
- 430.Daniele MA, Adams AA, Naciri J, North SH, and Ligler FS, Interpenetrating Networks Based on Gelatin Methacrylamide and Peg Formed Using Concurrent Thiol Click Chemistries for Hydrogel Tissue Engineering Scaffolds, Biomaterials, 2014, 35, 1845–1856. [DOI] [PubMed] [Google Scholar]
- 431.Li M, Dove AP, and Truong VX, Additive-Free Green Light-Induced Ligation Using Bodipy Triggers, Angew. Chem. Int. Ed, 2020, 59, 2284–2288. [DOI] [PubMed] [Google Scholar]
- 432.Fan Y, Deng C, Cheng R, Meng F, and Zhong Z, In Situ Forming Hydrogels Via Catalyst-Free and Bioorthogonal “Tetrazole–Alkene” Photo-Click Chemistry, Biomacromolecules, 2013, 14, 2814–2821. [DOI] [PubMed] [Google Scholar]
- 433.He M, Li J, Tan S, Wang R, and Zhang Y, Photodegradable Supramolecular Hydrogels with Fluorescence Turn-on Reporter for Photomodulation of Cellular Microenvironments, J. Am. Chem. Soc, 2013, 135, 18718–18721. [DOI] [PubMed] [Google Scholar]
- 434.Adzima BJ, Tao Y, Kloxin CJ, DeForest CA, Anseth KS, and Bowman CN, Spatial and Temporal Control of the Alkyne–Azide Cycloaddition by Photoinitiated Cu(Ii) Reduction, Nat. Chem, 2011, 3, 256–259. [DOI] [PubMed] [Google Scholar]
- 435.Maiti B, Abramov A, Franco L, Puiggalí J, Enshaei H, Alemán C, and Díaz DD, Thermoresponsive Shape-Memory Hydrogel Actuators Made by Phototriggered Click Chemistry, Adv. Funct. Mater, 2020, 30, 2001683. [Google Scholar]
- 436.Arkenberg MR, Nguyen HD, and Lin C-C, Recent Advances in Bio-Orthogonal and Dynamic Crosslinking of Biomimetic Hydrogels, J. Mater. Chem. B, 2020, 8, 7835–7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Munoz-Robles BG, Kopyeva I, and DeForest CA, Surface Patterning of Hydrogel Biomaterials to Probe and Direct Cell–Matrix Interactions, Adv. Mater. Interfaces, 2020, 7, 2001198. [Google Scholar]
- 438.Petrou CL, D’Ovidio TJ, Bölükbas DA, Tas S, Brown RD, Allawzi A, Lindstedt S, Nozik-Grayck E, Stenmark KR, Wagner DE, et al. , Clickable Decellularized Extracellular Matrix as a New Tool for Building Hybrid-Hydrogels to Model Chronic Fibrotic Diseases in Vitro, J. Mater. Chem. B, 2020, 8, 6814–6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Mabry KM, Lawrence RL, and Anseth KS, Dynamic Stiffening of Poly(Ethylene Glycol)-Based Hydrogels to Direct Valvular Interstitial Cell Phenotype in a Three-Dimensional Environment, Biomaterials, 2015, 49, 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Aziz AH, Wahlquist J, Sollner A, Ferguson V, DelRio FW, and Bryant SJ, Mechanical Characterization of Sequentially Layered Photo-Clickable Thiol-Ene Hydrogels, J. Mech. Behav. Biomed. Mater, 2017, 65, 454–465. [DOI] [PubMed] [Google Scholar]
- 441.Hui E, Gimeno KI, Guan G, and Caliari SR, Spatiotemporal Control of Viscoelasticity in Phototunable Hyaluronic Acid Hydrogels, Biomacromolecules, 2019, 20, 4126–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Tong X, Jiang J, Zhu D, and Yang F, Hydrogels with Dual Gradients of Mechanical and Biochemical Cues for Deciphering Cell-Niche Interactions, ACS Biomater. Sci. Eng, 2016, 2, 845–852. [DOI] [PubMed] [Google Scholar]
- 443.Carberry BJ, Rao VV, and Anseth KS, Phototunable Viscoelasticity in Hydrogels through Thioester Exchange, Ann. Biomed. Eng, 2020, 48, 2053–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Mosiewicz KA, Kolb L, van der Vlies AJ, Martino MM, Lienemann PS, Hubbell JA, Ehrbar M, and Lutolf MP, In Situ Cell Manipulation through Enzymatic Hydrogel Photopatterning, Nature Materials, 2013, 12, 1072–1078. [DOI] [PubMed] [Google Scholar]
- 445.Mosiewicz KA, Kolb L, van der Vlies AJ, and Lutolf MP, Microscale Patterning of Hydrogel Stiffness through Light-Triggered Uncaging of Thiols, Biomater. Sci, 2014, 2, 1640–1651. [DOI] [PubMed] [Google Scholar]
- 446.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, and Anseth KS, A Versatile Synthetic Extracellular Matrix Mimic Via Thiol-Norbornene Photopolymerization, Adv. Mater, 2009, 21, 5005–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Batchelor RR, Blasco E, Wuest KNR, Lu H, Wegener M, Barner-Kowollik C, and Stenzel MH, Spatially Resolved Coding of Λ-Orthogonal Hydrogels by Laser Lithography, Chem. Commun, 2018, 54, 2436–2439. [DOI] [PubMed] [Google Scholar]
- 448.Ma H, Caldwell AS, Azagarsamy MA, Gonzalez Rodriguez A, and Anseth KS, Bioorthogonal Click Chemistries Enable Simultaneous Spatial Patterning of Multiple Proteins to Probe Synergistic Protein Effects on Fibroblast Function, Biomaterials, 2020, 255, 120205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Horn-Ranney EL, Khoshakhlagh P, Kaiga JW, and Moore MJ, Light-Reactive Dextran Gels with Immobilized Guidance Cues for Directed Neurite Growth in 3d Models, Biomater. Sci, 2014, 2, 1450–1459. [DOI] [PubMed] [Google Scholar]
- 450.Luo Y and Shoichet MS, A Photolabile Hydrogel for Guided Three-Dimensional Cell Growth and Migration, Nature Materials, 2004, 3, 249–253. [DOI] [PubMed] [Google Scholar]
- 451.Luo Y and Shoichet MS, Light-Activated Immobilization of Biomolecules to Agarose Hydrogels for Controlled Cellular Response, Biomacromolecules, 2004, 5, 2315–2323. [DOI] [PubMed] [Google Scholar]
- 452.Wosnick JH and Shoichet MS, Three-Dimensional Chemical Patterning of Transparent Hydrogels, Chem. Mater, 2008, 20, 55–60. [Google Scholar]
- 453.Wylie RG and Shoichet MS, Three-Dimensional Spatial Patterning of Proteins in Hydrogels, Biomacromolecules, 2011, 12, 3789–3796. [DOI] [PubMed] [Google Scholar]
- 454.Liu Z, Lin Q, Sun Y, Liu T, Bao C, Li F, and Zhu L, Spatiotemporally Controllable and Cytocompatible Approach Builds 3d Cell Culture Matrix by Photo-Uncaged-Thiol Michael Addition Reaction, Adv. Mater, 2014, 26, 3912–3917. [DOI] [PubMed] [Google Scholar]
- 455.Liu Z, Liu T, Lin Q, Bao C, and Zhu L, Photoreleasable Thiol Chemistry for Facile and Efficient Bioconjugation, Chem. Commun, 2014, 50, 1256–1258. [DOI] [PubMed] [Google Scholar]
- 456.Chen RT, Marchesan S, Evans RA, Styan KE, Such GK, Postma A, McLean KM, Muir BW, and Caruso F, Photoinitiated Alkyne–Azide Click and Radical Cross-Linking Reactions for the Patterning of Peg Hydrogels, Biomacromolecules, 2012, 13, 889–895. [DOI] [PubMed] [Google Scholar]
- 457.Bjerknes M, Cheng H, McNitt CD, and Popik VV, Facile Quenching and Spatial Patterning of Cylooctynes Via Strain-Promoted Alkyne–Azide Cycloaddition of Inorganic Azides, Bioconjugate Chem, 2017, 28, 1560–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Yu C, Schimelman J, Wang P, Miller KL, Ma X, You S, Guan J, Sun B, Zhu W, and Chen S, Photopolymerizable Biomaterials and Light-Based 3d Printing Strategies for Biomedical Applications, Chem. Rev, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Skardal A, Zhang J, and Prestwich GD, Bioprinting Vessel-Like Constructs Using Hyaluronan Hydrogels Crosslinked with Tetrahedral Polyethylene Glycol Tetracrylates, Biomaterials, 2010, 31, 6173–6181. [DOI] [PubMed] [Google Scholar]
- 460.Stichler S, Jungst T, Schamel M, Zilkowski I, Kuhlmann M, Böck T, Blunk T, Teßmar J, and Groll J, Thiol-Ene Clickable Poly(Glycidol) Hydrogels for Biofabrication, Ann. Biomed. Eng, 2017, 45, 273–285. [DOI] [PubMed] [Google Scholar]
- 461.Bertlein S, Brown G, Lim KS, Jungst T, Boeck T, Blunk T, Tessmar J, Hooper GJ, Woodfield TBF, and Groll J, Thiol–Ene Clickable Gelatin: A Platform Bioink for Multiple 3d Biofabrication Technologies, Adv. Mater, 2017, 29, 1703404. [DOI] [PubMed] [Google Scholar]
- 462.Wang LL, Highley CB, Yeh Y-C, Galarraga JH, Uman S, and Burdick JA, Three-Dimensional Extrusion Bioprinting of Single- and Double-Network Hydrogels Containing Dynamic Covalent Crosslinks, Journal of Biomedical Materials Research Part A, 2018, 106, 865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Qin X-H, Torgersen J, Saf R, Mühleder S, Pucher N, Ligon SC, Holnthoner W, Redl H, Ovsianikov A, Stampfl J, et al. , Three-Dimensional Microfabrication of Protein Hydrogels Via Two-Photon-Excited Thiol-Vinyl Ester Photopolymerization, J. Polym. Sci., Part A: Polym. Chem, 2013, 51, 4799–4810. [Google Scholar]
- 464.Qin X-H, Gruber P, Markovic M, Plochberger B, Klotzsch E, Stampfl J, Ovsianikov A, and Liska R, Enzymatic Synthesis of Hyaluronic Acid Vinyl Esters for Two-Photon Microfabrication of Biocompatible and Biodegradable Hydrogel Constructs, Polym. Chem, 2014, 5, 6523–6533. [Google Scholar]
- 465.Baudis S, Bomze D, Markovic M, Gruber P, Ovsianikov A, and Liska R, Modular Material System for the Microfabrication of Biocompatible Hydrogels Based on Thiol–Ene-Modified Poly(Vinyl Alcohol), J. Polym. Sci., Part A: Polym. Chem, 2016, 54, 2060–2070. [Google Scholar]
- 466.Dobos A, Hoorick JV, Steiger W, Gruber P, Markovic M, Andriotis OG, Rohatschek A, Dubruel P, Thurner PJ, Vlierberghe SV, et al. , Thiol–Gelatin–Norbornene Bioink for Laser-Based High-Definition Bioprinting, Adv. Healthcare Mater, 2020, 9, 1900752. [DOI] [PubMed] [Google Scholar]
- 467.Skardal A, Devarasetty M, Kang H-W, Mead I, Bishop C, Shupe T, Lee SJ, Jackson J, Yoo J, Soker S, et al. , A Hydrogel Bioink Toolkit for Mimicking Native Tissue Biochemical and Mechanical Properties in Bioprinted Tissue Constructs, Acta Biomater., 2015, 25, 24–34. [DOI] [PubMed] [Google Scholar]
- 468.Lee M, Rizzo R, Surman F, and Zenobi-Wong M, Guiding Lights: Tissue Bioprinting Using Photoactivated Materials, Chem. Rev, 2020. [DOI] [PubMed] [Google Scholar]
- 469.Campos E, Branquinho J, Carreira AS, Carvalho A, Coimbra P, Ferreira P, and Gil MH, Designing Polymeric Microparticles for Biomedical and Industrial Applications, Eur. Polym. J, 2013, 49, 2005–2021. [Google Scholar]
- 470.Prasath RA, Gokmen MT, Espeel P, and Du Prez FE, Thiol-Ene and Thiol-Yne Chemistry in Microfluidics: A Straightforward Method Towards Macroporous and Nonporous Functional Polymer Beads, Polym. Chem, 2010, 1, 685–692. [Google Scholar]
- 471.Durham OZ, Norton HR, and Shipp DA, Functional Polymer Particles Via Thiol–Ene and Thiol–Yne Suspension “Click” Polymerization, RSC Adv., 2015, 5, 66757–66766. [Google Scholar]
- 472.Durham OZ, Krishnan S, and Shipp DA, Polymer Microspheres Prepared by Water-Borne Thiol–Ene Suspension Photopolymerization, ACS Macro Lett., 2012, 1, 1134–1137. [DOI] [PubMed] [Google Scholar]
- 473.Cai S, Weng Z, Zheng Y, Zhao B, Gao Z, and Gao C, High Porosity Microspheres with Functional Groups Synthesized by Thiol–Yne Click Suspension Polymerization, Polym. Chem, 2016, 7, 7400–7407. [Google Scholar]
- 474.Alimohammadi F, Wang C, Durham OZ, Norton HR, Bowman CN, and Shipp DA, Radical Mediated Thiol-Ene/Yne Dispersion Polymerizations, Polymer, 2016, 105, 180–186. [Google Scholar]
- 475.Wang C, Zhang X, Podgórski M, Xi W, Shah P, Stansbury J, and Bowman CN, Monodispersity/Narrow Polydispersity Cross-Linked Microparticles Prepared by Step-Growth Thiol–Michael Addition Dispersion Polymerizations, Macromolecules, 2015, 48, 8461–8470. [Google Scholar]
- 476.Hooker JP, Delafresnaye L, Barner L, and Barner-Kowollik C, With Polymer Photoclicks to Fluorescent Microspheres, Mater. Horiz, 2019, 6, 356–363. [Google Scholar]
- 477.Hooker JP, Feist F, Delafresnaye L, Barner L, and Barner-Kowollik C, Precisely Controlled Microsphere Design Via Visible-Light Cross-Linking of Functional Prepolymers, Adv. Funct. Mater, 2020, 30, 1905399. [Google Scholar]
- 478.Hooker JP, Feist F, Delafresnaye L, Cavalli F, Barner L, and Barner-Kowollik C, On-Demand Acid-Gated Fluorescence Switch-on in Photo-Generated Nanospheres, Chem. Commun, 2020, 56, 4986–4989. [DOI] [PubMed] [Google Scholar]
- 479.Schmitt CW, Walden SL, Delafresnaye L, Houck HA, Barner L, and Barner-Kowollik C, The Bright and the Dark Side of the Sphere: Light-Stabilized Microparticles, Polym. Chem, 2021, 12, 449–457. [Google Scholar]
- 480.Jiang Y, Chen J, Deng C, Suuronen EJ, and Zhong Z, Click Hydrogels, Microgels and Nanogels: Emerging Platforms for Drug Delivery and Tissue Engineering, Biomaterials, 2014, 35, 4969–4985. [DOI] [PubMed] [Google Scholar]
- 481.Gruber A, Navarro L, and Klinger D, Reactive Precursor Particles as Synthetic Platform for the Generation of Functional Nanoparticles, Nanogels, and Microgels, Adv. Mater. Interfaces, 2020, 7, 1901676. [Google Scholar]
- 482.Hachet E, Sereni N, Pignot-Paintrand I, Ravaine V, Szarpak-Jankowska A, and Auzély-Velty R, Thiol-Ene Clickable Hyaluronans: From Macro-to Nanogels, J. Colloid Interface Sci, 2014, 419, 52–55. [DOI] [PubMed] [Google Scholar]
- 483.Belair DG, Khalil AS, Miller MJ, and Murphy WL, Serum-Dependence of Affinity-Mediated Vegf Release from Biomimetic Microspheres, Biomacromolecules, 2014, 15, 2038–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Jivan F, Yegappan R, Pearce H, Carrow JK, McShane M, Gaharwar AK, and Alge DL, Sequential Thiol–Ene and Tetrazine Click Reactions for the Polymerization and Functionalization of Hydrogel Microparticles, Biomacromolecules, 2016, 17, 3516–3523. [DOI] [PubMed] [Google Scholar]
- 485.Xin S, Dai J, Gregory CA, Han A, and Alge DL, Creating Physicochemical Gradients in Modular Microporous Annealed Particle Hydrogels Via a Microfluidic Method, Adv. Funct. Mater, 2020, 30, 1907102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Michel SES, Dutertre F, Denbow ML, Galan MC, and Briscoe WH, Facile Synthesis of Chitosan-Based Hydrogels and Microgels through Thiol–Ene Photoclick Cross-Linking, ACS Appl. Bio Mater, 2019, 2, 3257–3268. [DOI] [PubMed] [Google Scholar]
- 487.Huang Y, Yuan J, Tang J, Yang J, and Zeng Z, One Step Synthesis of Monodisperse Thiol-Ene Clickable Polymer Microspheres and Application on Biological Functionalization, Eur. Polym. J, 2019, 110, 22–30. [Google Scholar]
- 488.Donahoe CD, Cohen TL, Li W, Nguyen PK, Fortner JD, Mitra RD, and Elbert DL, Ultralow Protein Adsorbing Coatings from Clickable Peg Nanogel Solutions: Benefits of Attachment under Salt-Induced Phase Separation Conditions and Comparison with Peg/Albumin Nanogel Coatings, Langmuir, 2013, 29, 4128–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Huang DC, Qian HL, Qiao HS, Wei C, Jan FJ, and Zhong ZY, Bioresponsive Functional Nanogels as an Emerging Platform for Cancer Therapy, Expert Opin. Drug Delivery, 2018, 15, 703–716. [DOI] [PubMed] [Google Scholar]
- 490.Lockhart JN, Beezer DB, Stevens DM, Spears BR, and Harth E, One-Pot Polyglycidol Nanogels Via Liposome Master Templates for Dual Drug Delivery, J. Controlled Release, 2016, 244, 366–374. [DOI] [PubMed] [Google Scholar]
- 491.Impellitteri NA, Toepke MW, Lan Levengood SK, and Murphy WL, Specific Vegf Sequestering and Release Using Peptide-Functionalized Hydrogel Microspheres, Biomaterials, 2012, 33, 3475–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Gregoritza M, Abstiens K, Graf M, and Goepferich AM, Fabrication of Antibody-Loaded Microgels Using Microfluidics and Thiol-Ene Photoclick Chemistry, Eur. J. Pharm. Biopharm, 2018, 127, 194–203. [DOI] [PubMed] [Google Scholar]
- 493.Chen J, Zou Y, Deng C, Meng F, Zhang J, and Zhong Z, Multifunctional Click Hyaluronic Acid Nanogels for Targeted Protein Delivery and Effective Cancer Treatment in Vivo, Chem. Mater, 2016, 28, 8792–8799. [Google Scholar]
- 494.Gao G, Han X, Sowan N, Zhang X, Shah PK, Chen M, Bowman CN, and Stansbury JW, Stress Relaxation Via Covalent Dynamic Bonds in Nanogel-Containing Thiol–Ene Resins, ACS Macro Lett, 2020, 9, 713–719. [DOI] [PubMed] [Google Scholar]
- 495.Liang Y and Kiick KL, Multifunctional Lipid-Coated Polymer Nanogels Crosslinked by Photo-Triggered Michael-Type Addition, Polym. Chem, 2014, 5, 1728–1736. [Google Scholar]
- 496.Janssens P, Debrouwer W, Van Aken K, and Huvaere K, Thiol-Ene Coupling in a Continuous Photo-Flow Regime, ChemPhotoChem, 2018, 2, 884–889. [Google Scholar]
- 497.Junkers T and Wenn B, Continuous Photoflow Synthesis of Precision Polymers, React. Chem. Eng, 2016, 1, 60–64. [Google Scholar]