

Abstract
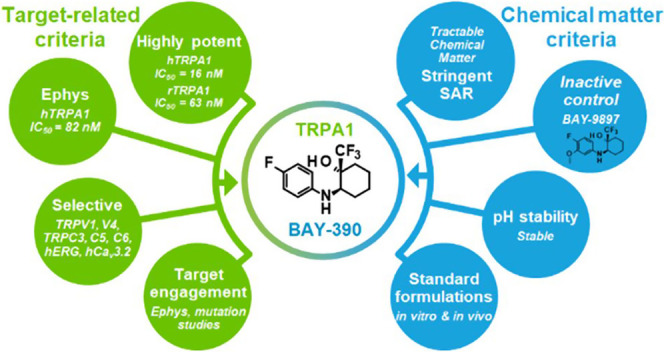
Transient receptor potential ankyrin 1 (TRPA1) is a voltage-dependent, ligand-gated ion channel, and activation thereof is linked to a variety of painful conditions. Preclinical studies have demonstrated the role of TRPA1 receptors in a broad range of animal models of acute, inflammatory, and neuropathic pain. In addition, a clinical study using the TRPA1 antagonist GRC-17536 (Glenmark Pharmaceuticals) demonstrated efficacy in a subgroup of patients with painful diabetic neuropathy. Consequently, there is an increasing interest in TRPA1 inhibitors as potential analgesics. Herein, we report the identification of a fragment-like hit from a high-throughput screening (HTS) campaign and subsequent optimization to provide a novel and brain-penetrant TRPA1 inhibitor (compound 18, BAY-390), which is now being made available to the research community as an open-source in vivo probe.
Introduction
Transient receptor potential (TRP) channels are a superfamily of nonselective cation channels that are involved in a diverse range of physiological processes. Transient receptor potential ankyrin 1 (TRPA1) is one of the 28 members of the TRP channel family and the sole member of the TRPA subfamily in mammals. TRPA1 is a voltage-dependent, ligand-gated channel and is permeable to both monovalent and divalent cations.1−5
TRPA1 is expressed in the nervous system within small- and medium-sized peptidergic neurons present in the dorsal root ganglia (DRGs) and was reported to be expressed in both peripheral and central terminals. Within the CNS, TRPA1 has been identified in glial cells such as astrocytes and oligodendrocytes.6−9 TRPA1 is also expressed in various other tissues and cells including immune cells (e.g., mast cells), vascular endothelium, and all barrier tissues such as skin (e.g., keratinocytes, epithelial melanocytes), lung cells (fibroblast), gut, joint cells (e.g., synoviocytes, chondrocytes), and cancer cells.3,4,10,11 Activation and modulation of TRPA1 occur in multiple ways, either directly or indirectly, by the action of endogenous and exogenous reactive and nonreactive ligands as well as by changes in pH and/or temperature.12,13 Endogenous mediators such as oxidized lipids (i.e., 4-hydroxy-2-nonenal), prostaglandins, metabolites, and reactive molecules (i.e., H2O2) have been shown to activate TRPA1 in vitro, thus initiating Ca2+ signaling in the cells in which it is expressed.
TRPA1 has been linked to several pathological conditions such as painful diseases, skin diseases, lung diseases, urogenital disorders, and gastrointestinal-tract disorders.1,2,4,5 In particular, the prominent role of TRPA1 receptors in acute nociception and in chronic inflammatory and neuropathic pain has been widely reported and studied in the literature.14,15
Numerous preclinical studies have demonstrated the key role played by TRPA1 receptors in a broad range of acute nociception, inflammatory pain, and neuropathic pain animal models.14,15 In humans, a gain of function mutation in the human TRPA1 gene was shown to be responsible for the condition known as familial episodic pain syndrome-15,16 and a clinical study demonstrated the efficacy of the TRPA1 antagonist 2 (GRC-17536; Glenmark Pharmaceuticals) in a subgroup of patients with painful diabetic neuropathy.17 More recently, the TRPA1 antagonist GDC-0334 was able to reduce TRPA1 agonist-induced dermal blood flow, pain, and itch in a healthy volunteer phase 1 study.18,19
In recent years, a variety of chemotypes have been identified as TRPA1 antagonists in the literature (Figure 1) but only a few compounds (cpds) have progressed to clinical development due to several well-recognized challenges in this field of drug discovery.13 Among them, notable species differences in potency are often observed leading to the inactivity of compounds at the rodent TRPA1 channels. In addition, challenging physicochemical features of several existing compounds have been described such as poor solubility, low metabolic stability, and limited blood–brain barrier permeability.
Figure 1.
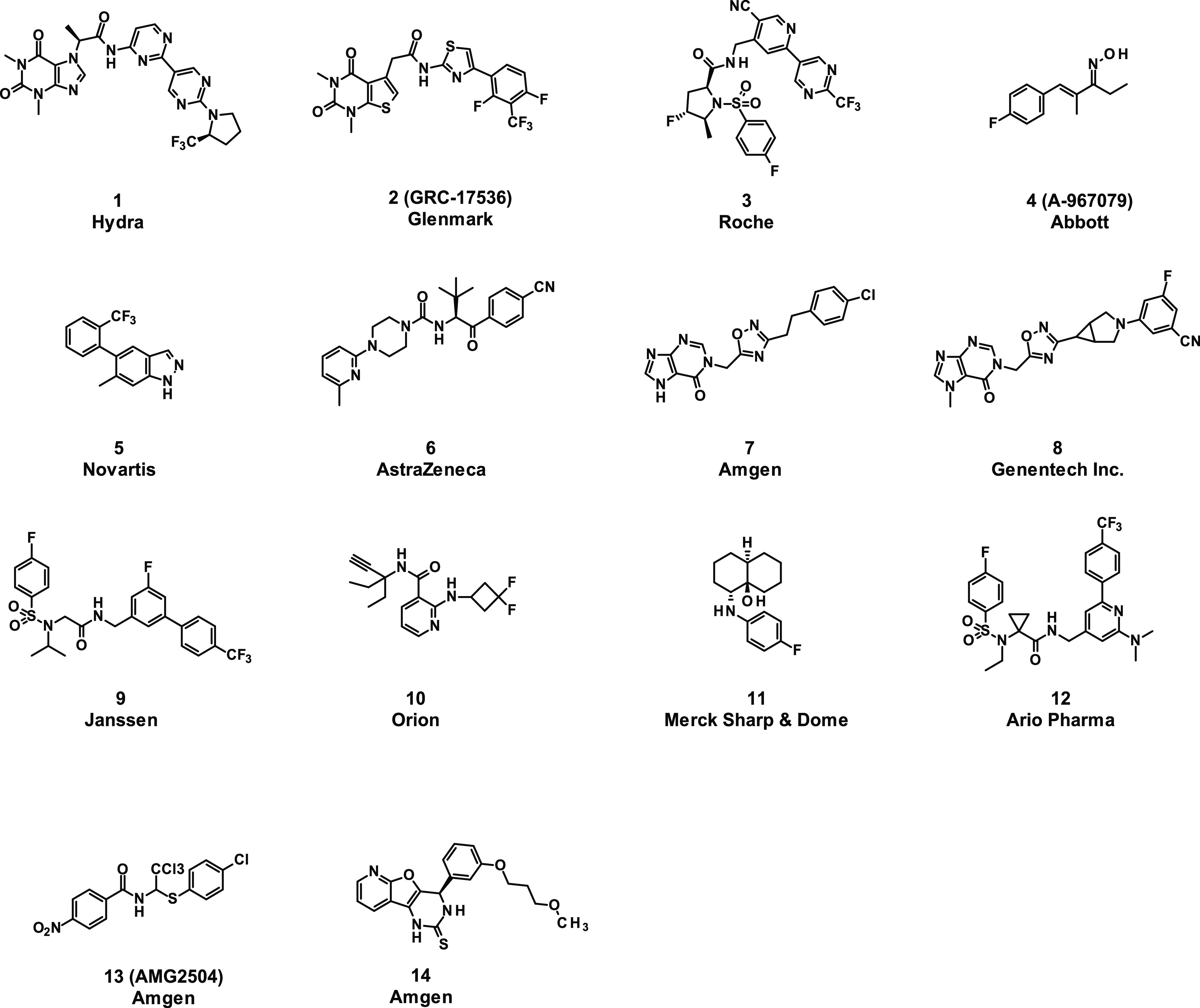
Overview of TRPA1 inhibitors reported in the literature. 1,202,21,223,234,24,255,266,277,28,298,309,3110,3211,3312,3413,35 and 14(36) (for a patent review covering years 2015–2019, see ref (37)).
Herein, we describe the identification and optimization of a novel and centrally active series of TRPA1 antagonists, culminating in the discovery of BAY-390, a useful compound for further probing the role of TRPA1 in preclinical rodent models.
Results and Discussion
A high-throughput screening (HTS)-based campaign was initiated aiming at identifying novel antagonists of TRPA1 with physicochemical properties commensurate with daily oral application. Four million compounds were screened at 5 μM against human TRPA1 in a calcium fluorescence assay using the reactive agonist cinnamaldehyde (CA).38 The assay was run in the 1536 microtiter plate format using a recombinant Chinese hamster ovary (CHO) cell line expressing hTRPA1 and the genetically encoded calcium sensor GCaMP6 to measure hTRPA1-mediated calcium influx on an in-house MTP fluorescence imager (Fluobox). Given that the TRPA1 channel can be activated by numerous reactive and nonreactive agonists such as zinc39 and PF-484015440 hit activity was further confirmed using zinc chloride as a nonreactive endogenous agonist.41 As described in the literature in many cases,35,42 we observed significant species differences with only a few hits having comparable potency at both human and rodent TRPA1.
Hit clusters with reasonable cross-species (human and rat) activity and with low similarity to known problematic chemotypes (refer to Figure 1) were prioritized, and this led us to the identification of singleton compound 15 as a promising starting point for follow-up hit validation and hit-expansion activities.
The initial hit compound comprised a diastereomeric mixture (mixture of two isomers, unknown ratio) with high antagonistic potency at the human TRPA1 channel (hIC50 = 14 nM) and moderate rat TRPA1 potency (rTRPA1 IC50 = 1370 nM). In addition to being only moderately active at the rat TRPA1 channel, the compound is also rather lipophilic (log D 4.1) and features an undesirable nitro-substituted phenyl group. First, the nitro group was replaced by a fluorine atom by chemical modification and the enantiomers were separated. Only one enantiomer (compound 16, undefined absolute stereochemistry) showed activity. Subsequent hit-to-lead activities resulted in the identification of compound 17 with improved human and much improved rat TRPA1 potency but with only a marginal improvement in the measured log D value (3.8). Further exploration of structure–activity relationship (SAR) and optimization of properties led to the identification of BAY-390 (compound 18), which combined high potency across species with a decreased and more acceptable level of lipophilicity (log D 3.0), having some structural similarity to compound 11(33) (Figures 1 and 2).
Figure 2.

Key compounds identified in the hit-to-lead phase.
Chemistry
The synthesis of close analogues of the hit compound 15 started from commercially available 5,5-dimethylcyclohexane-1,3-dione (19, Scheme 1), which was dissolved in ethanol and heated at 130 °C with para-toluene sulfonic acid (PTSA) to yield 3-ethoxy-5,5-dimethylcyclohex-2-en-1-one (20). Subsequent reduction with lithium aluminum hydride led to intermediate 21. In the next step, lithium diisopropylamide (LDA) was generated in situ; then, the addition of intermediate 21 followed by the addition of substituted benzyl bromides yielded compounds of general formula 22a–i.
Scheme 1.

The double bond was reduced by the addition of Wilkinson’s catalyst under a hydrogen atmosphere to yield compounds of formula 23a–i. In the final step, the conversion of the ketone was accomplished in good yields by the addition of the Ruppert–Prakash reagent to give final compounds (rac)-16 and 24a–h (diastereomeric ratio (dr) cis to trans 10:1). During the course of trifluoromethyl addition to gem-dimethyl cyclohexanone motifs (23a–i), it is expected that the addition to the ketone occurs from the equatorial position to minimize the 1,3-diaxial interaction with the axial-methyl group on the ring. As a consequence, the predominant formation of cis-isomer can be expected.43 Final compounds 24a–h were in most cases tested in the primary assay as diastereomeric mixtures.
Further analogues were prepared, as outlined in Scheme 2.
Scheme 2.

Commercially available cyclohexanone 25 was alkylated by reaction with LDA and substituted benzyl bromides to give intermediates 26a–i, as shown in Scheme 3. Subsequent reaction with the Ruppert–Prakash reagent yielded a 2:1 ratio of cis-to-trans stereoisomers. After separation of the cis/trans isomers, chiral separation was performed and one of the trans isomers was determined as the eutomer, yielding final compounds 17 and 27a–h.
Scheme 3.
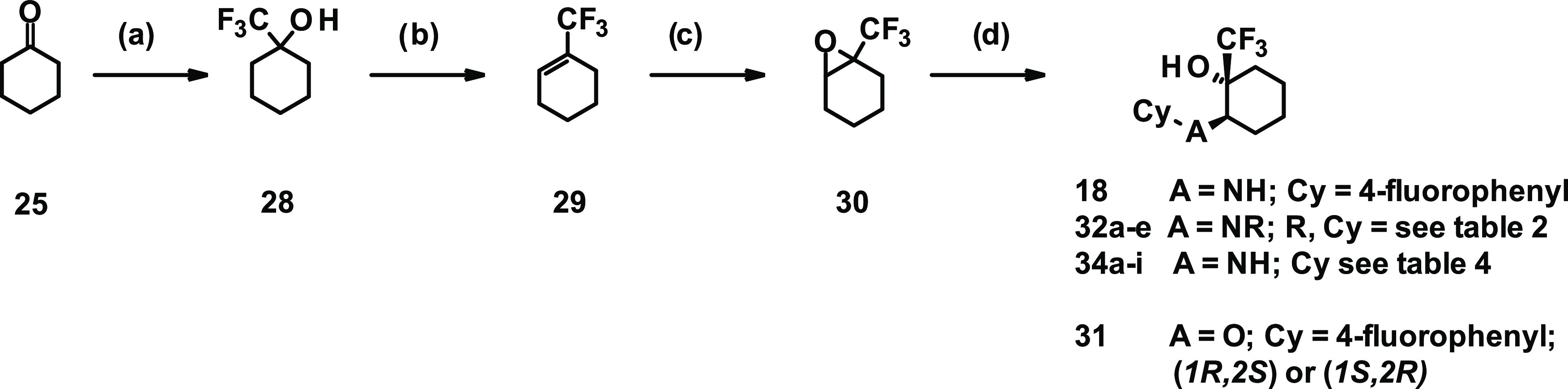
For the synthesis of compounds 18 and 31–34 displaying alternative linker types connecting the cyclohexane to the cyclic substituents Cy, a different approach was applied. Converting cyclohexanone 25 into 1-(trifluoromethyl)cyclohexanol (28) followed by dehydration gave 1-(trifluoromethyl)cyclohexene (29). Subsequent oxidation with mCPBA to epoxide 30 followed by reaction with various nucleophiles yielded compounds 18, 31, and 32a–f. In the case of compound 33, the sequence was slightly different, starting instead from the commercially available (rac)-2-(methylamino)cyclohexanone and reaction under standard sulfonylating conditions with 4-fluorobenzenesulfonyl chloride (not depicted in the scheme).
Hit-to-Lead Optimization
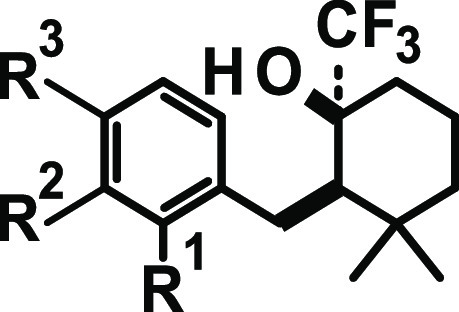
Attracted by the small, almost fragment-like hit 15, initial SAR investigations were directed toward the modification of the aromatic substitution pattern (Table 1). We started by replacing the nitro-substituent with halogen substituents (compounds (rac)-16 and 24a) as well as with a para-nitrile group (compound 24b). While compound (rac)-16 was almost equipotent to compound 15, the introduction of a para chloro (compound 24a) or para-nitrile substituent (compound 24b) led to a drop in activity, especially at the rodent receptor. Introduction of electron-donating substituents, e.g., para methyl (compound 24c) or para methoxy (compound 24d), as well as introduction of amides (compound 24e), resulted in a loss of activity. Neither a shift of para substituent to the meta position (compounds 24f and 24g) nor the introduction of additional substituents (e.g., in the ortho position to the linker) was well tolerated (compound 24h).
Table 1. SAR of Aromatic Substitution, h/rTRPA1 IC50 [nM] Potency, for Compounds 15, 16, and 24a–h.
| compounda | R1 | R2 | R3 | hTRPA1 IC50 [nM] | rTRPA1 IC50 [nM] |
|---|---|---|---|---|---|
| 15 | H | H | NO2 | 14 | 1370 |
| (rac)-16 | H | H | F | 36 | 1560 |
| eutomer 16a | H | H | F | 33 | 860 |
| 16b | H | H | F | >25 000 | >25 000 |
| 24a | H | H | Cl | 340 | 11 500 |
| 24b | H | H | CN | 390 | 7570 |
| 24c | H | H | Me | 1270 | >12 500 |
| 24d | H | H | OMe | >125 00 | >12 500 |
| 24e | H | H | –C(O)NHEt | >12 500 | >12 500 |
| 24f | H | F | H | 750 | 3800 |
| 24g | H | CN | H | 5550 | 13 000 |
| 24h | F | H | F | 1000 | >10 000 |
Compounds are diastereomeric mixtures unless stated otherwise (#4, ratio >10:1 cis/trans).
As the relatively steep SAR outlined above provided few opportunities for improving the overall compound properties, we turned our focus to selected variations of the cyclohexyl core. Replacement of the gem-dimethyl group with cyclopropane or cyclobutane was not tolerated (data not shown). To our surprise, the removal of the gem-dimethyl substituents followed by separation of the single isomers yielded compound 17 with improved human and much improved rat TRPA1 activity (hTRPA1 IC50 = 4.0 nM, rTRPA1 IC50 = 24 nM). It is interesting to note that the eutomer had relative trans-stereochemistry opposite to that observed for compound 16a.
With this compound in hand and going forward with respect to further characterization in secondary assays and in vivo, we faced a challenge in that we were unable to quantify the compound in plasma or blood samples by liquid chromatography mass spectrometry (LC-MS) analysis. Alternative methods such as 19F NMR and gas chromatography (GC)-MS proved equally unsuitable for bioanalysis, and consequently, we were unable to quantify compound concentrations in key in vitro and in vivo experiments. It became evident that additional optimization to increase polarity and the ionization potential of our compounds was necessary for the series to be progressed further.
Initially, we introduced heterocycles or polar groups on the side of the aryl substituent in 17 (referred to as “Cy” in Table 2). Chloro-substituted thiophene-based compound 27a showed similar potency in the human-based assay but significantly lower potency in the rodent-based assay. An increase of polarity led to a potency drop in the human-based assay and stronger loss of activity in the rodent-based assay (compounds 27b–h; Table 2).
Table 2. Influence of Heterocycles and Linker Variations on Human and Rat TRPA1 Potency.


Unless stated otherwise, compounds are single enantiomers.
Next, we investigated potential changes to the CH2-linker yielding the N- and O-linked compounds 18 and 31, respectively. Determination of the absolute stereochemistry of compound 18 using vibrational circular dichroism (VCD) measurements (refer to S28 in the Supporting Information) showed that the eutomer had the R,R-configuration. Removing the p-fluoro (32c) yielded a loss in activity. Further derivatization of the NH linker, for example, by alkylation (compounds 32a and 32b) or cyclization (compounds 32e, 32f), resulted in reduced potency.
Ether-linked compound 31 (hTRPA1 IC50 = 20 nM, rTRPA1 IC50 = 120 nM) and amino-linked compound 18 (hTRPA1 IC50 = 16 nM, rTRPA1 IC50 = 63 nM) were highly potent across species and were somewhat less lipophilic than earlier compounds (log D = 3.5 and 3.0, respectively). However, compound 31 was still difficult to detect by LC-MS and presented significant challenges with regard to quantification of the compound in biological samples. We therefore turned our attention to BAY-390 (compound 18), which ionized well and could easily be detected using standard analytical techniques. In addition, this compound also showed improved TRPA1 activity across species as well as a good level of solubility (56 mg/L in phosphate-buffered saline (PBS), pH 6.5).
Mutation Studies
In the absence of in-house structural information to identify the TRPA1 binding site, we sought to characterize our compounds further by determining their activity at human TRPA1 mutants, which are known to affect the binding of previously reported chemotypes. Several mutation studies have been published previously,44 but the binding of the chemotypes used in these different studies had not been directly compared to each other against all of the mutants. We therefore selected representative compounds that had been reported to bind differentially to specific TRPA1 mutants and the wild-type channel and characterized these and our own compound more broadly across a number of mutant channels.
The antagonist binding sites are shown in Figure 3 using a cryogenic electron microscopy (cryoEM) structure with bound A-967079 (compound 4).45 The F909T46 and T874V47 mutants were referred to as the binding site 1a. Similarly, the M911A mutant,48 due to the proximity to F909, was referred to as binding site 1b, and the N855S mutant42 and G238K/N249S/K270N mutant49 were referred to as binding sites 2 and 3, respectively. Using transient transfections of the above-mentioned mutants, we looked for the loss of activity compared to the wild-type channel using a fluorescent imaging plate reader (FLIPR) calcium assay. The results, represented by the ratio of mutant potency to wild-type potency, are shown in Table 3, with any shifts greater than 3-fold being considered significant.
Figure 3.
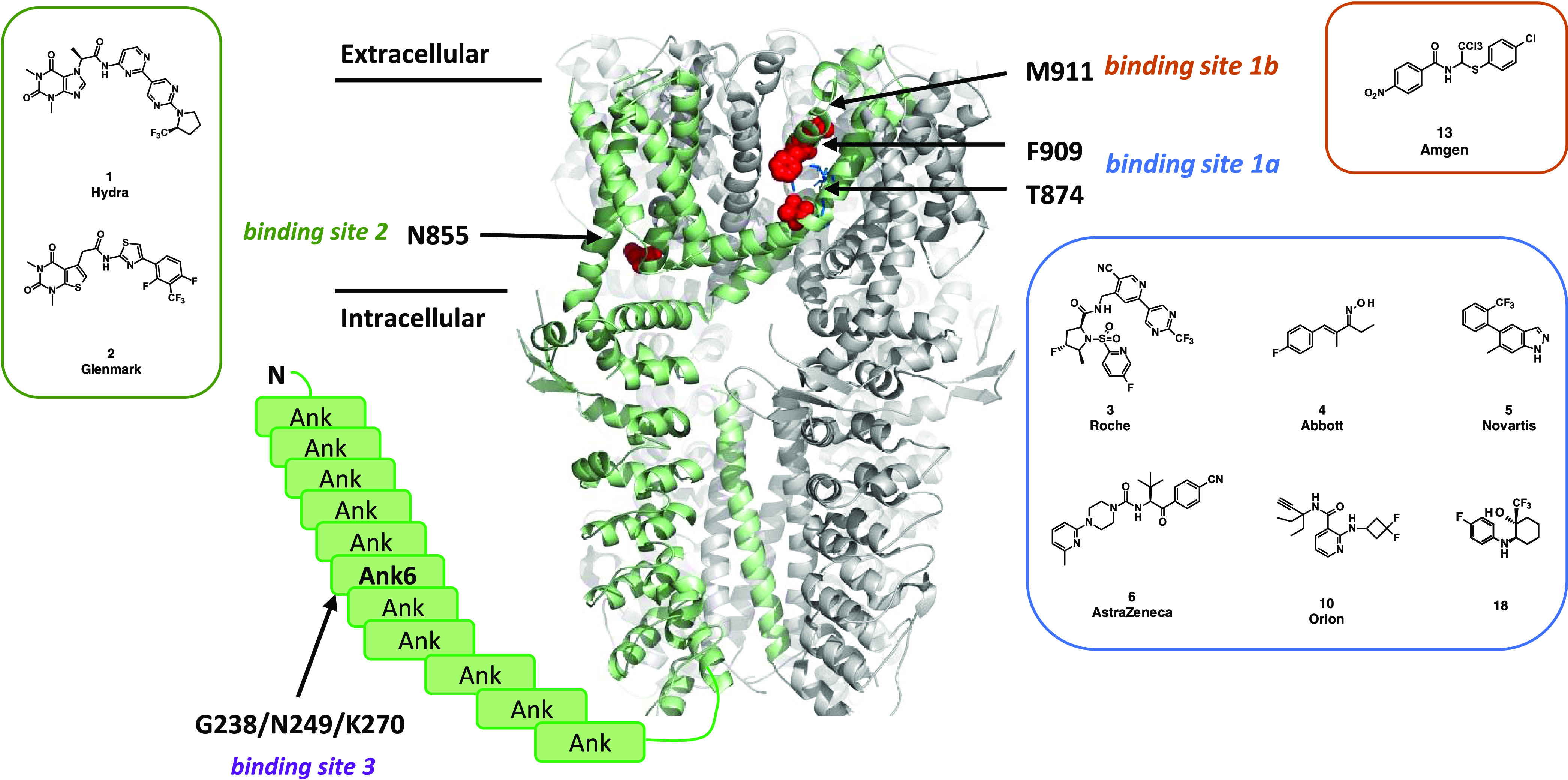
TRPA1 antagonist binding sites. Side view of hTRPA1 using a cryoEM structure (PDB: 6V9Y). Unresolved Ankyrin repeats 1–11 are shown as boxes. The mutated residues are shown in red when resolved and indicated by arrows. The different sites with the respective mutations are labeled as binding site 1a (F909T and T874V) with bound A-967079 (compound 4) in blue, binding site 1b (M911A), binding site 2 (N855S), and binding site 3 (G238K/N249S/K270N).
Table 3. Ratio of Mutant and Wild-Type IC50s for Selected TRPA1 Inhibitors.

Compound-induced fluorescence interfered with IC50 determination.
Significant shifts in potency are observed with the T874V and F909T mutants clearly grouped BAY-390, compound 4 (A-96707946,47), compound 5 (Novartis;44 rat T877 corresponds to human T874), compound 10 (Orion, no mutation data), compound 3 (Roche), and compound 13 (AMG2504) together and suggested that these are binding to site 1a.46 In addition, compound 6(39) showed smaller shifts with the M911A mutant and it may be that the double mutation (M911A/M912A)48 is required to see more pronounced effects. Finally, the N855S mutation resulted in significant changes in potency for both compound 1 (Hydra) and compound 2,42 confirming their binding to site 2. None of the compounds tested were affected by the G238K/N249S/K270N mutations (the Pfizer compound itself was not tested). No loss of potency for either compound 14, which is speculated to covalently bind to cysteines present in the protein,1,36 or compound 7,2828 for which no binding mode data have been published, was observed against any of the mutants. The outcome of the shift experiment of compound 7 is in line with the recently published protein–ligand complex with a structurally related analogue showing no interaction with N855.29
While only the structure of the apo protein46 was available when the HTS campaign was performed, a number of cryoEM structures with bound ligands have been published in recent years. Binding sites for a covalent agonist JT01 near C62150 and noncovalent agonist GNE551 near Tyr 84051 have been characterized but are distinct from the inhibitor binding sites 1–3 studied here. The mutation data suggest that compounds 4 and 3 bind to site 1a, which is in agreement with cryoEM structures for A-967079 (ref (45)) and GDC-0334, a close analogue of compound 3.29 In summary, the results from described mutation studies are in agreement with published data and show that BAY-390 binds to site 1a.
Pharmacodynamics
Although the in vitro metabolic stability of compound 18 was only moderate in rat hepatocytes (Clblood 2.4 [L/h/kg]), its high permeability and low efflux (CaCo-2 A–B: 293 nm/s; efflux ratio = 0.6) together with reasonable solubility resulted in relatively good free exposure after oral (p.o.) application in rats. The compound demonstrated robust and good efficacy in various animal models upon achieving minimum free exposures at least equivalent to the in vitro IC50 (fu in rats = 6%).
BAY-390 Reduces Cinnamaldehyde (CA)-Induced Nocifensive Behaviors in Rats
Intraplantar injection of the TRPA1 agonist cinnamaldehyde (CA) in rats results in a TRPA1-mediated nocifensive response characterized by robust flinching and licking behaviors (Figure 4A). The use of this animal model allows the direct investigation of a TRPA1-mediated response and evaluation of on-target effects of TRPA1 antagonists. In this experiment, BAY-390 was administrated per os (p.o.) at doses of 3, 10, and 30 mg/kg. Administration of BAY-390 resulted in a significant reduction of both the number of flinches and duration of paw licking (Figure 4A), therefore confirming the ability of BAY-390 to inhibit TRPA1 activation in vivo. Measured free plasma concentrations of BAY-390 1 h after dosing (10, 35, and 115 nM after 3, 10, and 30 mg/kg, respectively) indicate that the in vitro IC50 needs to be reached at minimum to observe the strongest peripheral TRPA1 inhibition.
Figure 4.

Pharmacological efficacy of BAY-390 in in vivo pain models in rats. (A) Cinnamaldehyde (CA)-induced nocifensive behaviors. (B) Complete Freund’s adjuvant (CFA)-induced inflammatory pain. (C, D) Spinal nerve ligation (SNL)-induced neuropathic pain.
BAY-390 Reduces Complete Freund’s Adjuvant (CFA)-Induced Inflammatory Pain in Rats
Inflammatory pain can be induced in rodents by intraplantar injection in one hind paw of complete Freund’s adjuvant (CFA). This peripheral inflammatory pain model is characterized by a robust mechanical hyperalgesia, and acute inflammation is well described in the literature and is routinely used for assessing antinociceptive and anti-inflammatory effects of novel compounds.52,53 As previously described, a mechanical hyperalgesia reflected by a reduction of the paw withdrawal threshold (PWT) was observed following intraplantar CFA injection (Figure 4B). Oral administration of BAY-390, 24 h after intraplantar CFA, resulted in a significant reduction of CFA-induced mechanical hyperalgesia at 2 h postdose after 10 and 30 mg/kg doses and also at 4 h post the 30 mg/kg dose (Figure 4B). Plasma unbound levels of BAY-390 were measured 4.5 h after dosing (7, 10, and 103 nM after 3, 10, and 30 mg/kg, respectively) and confirmed that the in vitro rat IC50 needed to be reached to achieve relevant peripheral TRPA1 inhibition.
BAY-390 Reverses Mechanical Allodynia in the Spinal Nerve Ligation (SNL) Neuropathic Pain Model in Rats
BAY-390 or vehicle was dosed orally (p.o.), twice daily (b.i.d.), over a period of 10 consecutive days from day 15 to day 24 post SNL surgery. Pain testing was performed before (day 11, post SNL baseline) and during chronic treatment (day 15, 18, 21, and 24 post SNL surgery), 2 h after the morning dosing (Figure 4C). In SNL animals, baseline withdrawal thresholds (WTs) of injured (ipsilateral) hind paws were significantly lower compared to those in the noninjured (contralateral) hind paws 11 days after spinal nerve ligation, indicating the development of a robust mechanical allodynia. Oral administration of vehicle had no effect on the withdrawal threshold of the injured hind paws during the 10 day dosing period (Figure 4D). Oral administration of BAY-390 at 90 mg/kg initially only produced a moderate reversal of mechanical allodynia, with significant effects only starting to be apparent after 6 days of daily treatment (p < 0.05), and these improvements were maintained after a further 10 days (p < 0.05) (Figure 4D). No effect was observed when BAY-390 was administered at 30 mg/kg. As a positive control, orally administered gabapentin, at 30 mg/kg daily, produced a time-dependent increase in paw withdrawal threshold compared to vehicle-treated animals after 4 days of daily treatment (p < 0.05) which was maintained after a further 10 days of dosing (p < 0.001). Unbound plasma levels of BAY-390 were measured 2.5 h after the last repeated dose (115 and 201 nM after 30 and 90 mg/kg, respectively) and indicated, together with the absence of efficacy at 30 mg/kg, that free plasma levels exceeding the biochemical assay IC50 were not sufficient to observe TRPA1-mediated efficacy in this neuropathic pain model. Based on the experimentally measured brain/plasma ratio of BAY-390 in rats (Kp,uu = 0.29; refer to Supporting Information S32–S34), the estimated unbound brain levels of BAY-390 were 33 and 61 nM after 30 and 90 mg/kg, respectively. Consequently, the collective data suggest that IC50 needs to be exceeded in the CNS to achieve TRPA1-mediated efficacy in the SNL neuropathic pain model, confirming the involvement of TRPA1 in central pain pathways.
Lead Optimization
Encouraged by the excellent in vivo data obtained with BAY-390, we embarked on a lead optimization phase to mitigate the possible risk of Ames-positive aniline-derived metabolites being formed in vivo. The parent compound 18 itself was Ames-negative and we did not observe the release of para-fluoroaniline in Met-ID studies. However, the close analogue 34a did show the trace formation of 4-chloroaniline in Met-ID studies in hepatocytes. To de-risk this potential mutagenic risk, we focused on SAR optimization with Ames-negative anilines. At the same time, changes that could lead to improvements in metabolic stability were also explored. The SAR of the amino-linked series was mostly consistent with that of the aforementioned −CH2-linked series, where, for example, the replacement of a para-fluorine substituent on the aromatic ring with a para-nitrile group (compound 34b) led to a significant loss in potency. Interestingly, and contrary to data obtained with the −CH2-linked series, the replacement of a para-fluorine substituent with a chlorine atom (compound 34a) was well tolerated in this series. We also observed that the para substituent is crucial, and any attempt to remove or shift this substituent to a meta- or ortho position diminished the potency, especially toward rat TRPA1 (e.g., compound 34c; Table 4).
Table 4. SAR of Anilino Substituents and Assessment of Mutagenicity Risk Associated with Possible Metabolites.
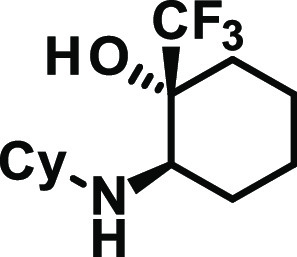

Based on the confirmed stereoconfiguration of compound 18, compounds synthesized according to GP1 are assumed to have the same (R,R)-stereoconfiguration.
Starting with small changes to the aryl substitution pattern, it was found that the 4-chloro-3-fluoro (compound 34e) modification maintained good human and moderate rat TRPA1 potency (Table 4). The metabolic stability remained unchanged (rHeps, Clblood 2.3 [L/h/kg]; Table 5), but a low-dose pharmacokinetics (PK) study in rats revealed a decrease in free exposure (fu% rat = 0.36). In the case of 4-fluoro-3-chloro and 3,4-dichloro substitution (compounds 34f, 34g), the rodent potency dropped significantly, making both compounds less well suited for subsequent in vivo studies and toxicological evaluation. Unfortunately, either removing the para-fluoro substituent (compound 34c) or substitution with a nitrile group (compound 34b) led to a significant decrease in rat potency. The introduction of heterocycles such as pyridyl (34d) was not well tolerated (hTRPA1 IC50 = 440 nM, rTRPA1 IC50 = 6300 nM).
Table 5. TRPA1 Potency and Metabolic Stability of Compounds 18 and 34e–ga.
| compound | hTRPA1 IC50 [nM] | rTRPA1 IC50 [nM] | rHeps [L/h/kg] | hLMs [L/h/kg] | log D (HPLC) |
|---|---|---|---|---|---|
| 18 | 16 | 63 | 2.4 | 0.38 | 3.0 |
| 34e | 21 | 200 | 2.3 | 0.48 | n.d. |
| 34f | 55 | 680 | 3.2 | 0.68 | 3.4 |
| 34g | 41 | 650 | 1.8 | 0.44 | 3.8 |
Compound 18: fu% rat = 6%; brain= 4.3%; mouse = 8.1%; human = 12%, guinea pig = 9.7%.
Following extensive SAR exploration, we unfortunately failed to improve upon the overall compound property profile of BAY-390 while maintaining good levels of on-target activity. Further changes to the linker portion, in an attempt to avoid the inclusion of aniline motifs, also proved unsuccessful as the decrease in potency was dramatic in such cases (Table 2; compounds 32e, 32f, 33).
Ultimately, we had to revise our strategy and concentrate efforts on further, more synthetically challenging changes to the cyclohexyl ring core. These results will be reported in a subsequent publication.
Conclusions
Starting from a fragment-like singleton hit, we were able to optimize the potency and physicochemical properties to obtain a brain-penetrant compound that was suitable for evaluating TRPA1-related effects in in vivo models of inflammatory and neuropathic pain. Although we were not able to optimize lead compound BAY-390 sufficiently to be able to nominate a clinical candidate, the compound identified (BAY-390) is very well suited as an in vitro and in vivo probe to investigate the central and peripheral roles played by TRPA1 in rodent preclinical models. Consequently, BAY-390 has been provided as a probe, along with BAY-9897 as a corresponding inactive antiprobe, to the SGC consortium for use within the wider scientific community. Both compounds were further characterized by collecting additional off-target activity data within the TRP family and more widely as well. These data are available in the information pack held by the SGC (https://www.sgc-ffm.uni-frankfurt.de/#!specificprobeoverview/BAY-390).54
Experimental Section
Chemistry
General
All reagents and solvents were used as purchased unless otherwise specified. Intermediates 19 (CAS 126-81-8), 20 (CAS 6267-39-6), 21 (CAS 4694-17-1), and 25 (CAS 108-94-1) were purchased; all others were prepared as described below. All air- and moisture-sensitive reactions were carried out in oven-dried (at 120 °C) glassware under an inert atmosphere of argon or nitrogen. The purity of most final compounds was ≥95%, determined by liquid chromatography mass spectrometry (LC-MS) and 1H NMR. Reactions were monitored by thin-layer chromatography (TLC) and ultraperformance liquid chromatography (UPLC) analysis with a Waters Acquity UPLC-MS Single Quad system; column: Acquity UPLC BEH C18 1.7 μm, 50 × 2.1 mm2; eluent A: water (H2O) + 0.2 vol % aqueous (aq) ammonia (NH3, 32%) and eluent B: acetonitrile (MeCN); gradient: 0–1.6 min 1–99% B, 1.6–2.0 min 99% B; flow: 0.8 mL/ min; temperature: 60 °C; diode array detector (DAD) scan: 210–400 nm. Flash chromatography was carried out using a Biotage Isolera One system with a 200–400 nm variable detector, using Biotage SNAP KP-Sil or KP-NH cartridges. Preparative high-performance liquid chromatography (HPLC) was carried out with a Waters AutoPurification MS Single Quad system; column: Waters XBridge C18 5 μm, 100 × 30 mm2; eluent A: H2O + 0.2 vol % aq NH3 (32%) and eluent B: MeCN; gradient: 0–5.5 min 5–100% B; flow: 70 mL/min; temperature: 25 °C; DAD scan: 210–400 nm. Analytical TLC was carried out on aluminum-backed plates coated with Merck Kieselgel 60 F254, with visualization under UV light at 254 nm. All NMR spectra were recorded on Bruker Avance III HD spectrometers. 1H NMR spectra were obtained at 300, 400, 500, or 600 MHz and referenced to the residual solvent signal (2.49 ppm for dimethyl sulfoxide (DMSO)-d6; 7.26 ppm for CDCl3).
13C NMR spectra were obtained at 101 or 151 MHz and also referenced to the residual solvent signal (39.7 ppm for DMSO-d6). All spectra were obtained at ambient temperature (22 ± 1 °C). 1H NMR data are reported as follows: chemical shift (δ) in ppm, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), and integration. Mass spectra were recorded on LC-MS instruments. Analytical LC-MS and UPLC-MS conditions are reported in the Supporting Information.
rac-6-[(4-Fluorophenyl)methyl]-5,5-dimethylcyclohex-2-en-1-one (22a)
A stirred solution of diisopropylamine (1.35 mL, 9.66 mmol) in tetrahydrofuran (4 mL) was cooled to −78 °C and treated with a 2.5 M solution of n-butyllithium in hexanes (3.54 mL, 8.86 mmol). On stirring at this temperature for 30 min, the reaction mixture was allowed to warm to 0 °C. After 10 min of stirring at this temperature, the reaction mixture was cooled to −78 °C whereupon a solution of 5,5-dimethylcyclohex-2-en-1-one (21, 1.0 g, 8.05 mmol) in tetrahydrofuran (2 mL) was added dropwise for 5 min. On warming to −10 °C, 1-(bromomethyl)-4-fluorobenzene (1.1 mL, 8.86 mmol) was added followed by potassium iodide (134 mg, 0.81 mmol) and the mixture was stirred for 4 h while warming to room temperature. On completion, the reaction was diluted with water and extracted in ethyl acetate (EA). The combined organic layers were washed with brine, dried over magnesium sulfate, and the solvents were removed in vacuo. Purification by reverse-phase column chromatography (C18 silica gel, eluting with water–acetonitrile, 9:1–1:9) followed by column chromatography (silica gel, eluting with heptane–tert-butyl methyl ether (TBME), 9:1) gave 190 mg (10% yield, 98% purity) of the title compound as a colorless oil. LC-MS (method C) Rt = 1.36 min, MS (ESIpos): 233 [M + H]+. 1H NMR (500 MHz, CDCl3) δ [ppm]: 1.02 (s, 3H), 1.12 (s, 3H), 2.30 (dd, 2H), 2.41 (dd, 1H), 2.75 (dd, 1H), 2.94 (dd, 1H), 5.97 (dt, 1H), 6.77 (dt, 1H), 6.91–6.95 (t, 2H), 7.13–7.16 (dd, 2H).
rac-2-[(4-Fluorophenyl)methyl]-3,3-dimethylcyclohexan-1-one (23a)
To a stirred solution of rac-6-[(4-fluorophenyl)methyl]-5,5-dimethylcyclohex-2-en-1-one (22a) (190 mg, 0.82 mmol) in toluene (10 mL) was added chloridotris(triphenylphosphine)rhodium(I) (114 mg, 0.12 mmol). The reaction was stirred at room temperature under an atmosphere of hydrogen for 18 h. On completion, the reaction was filtered through celite and the solvents were removed in vacuo. Purification by column chromatography (silica gel, eluting with heptane–tert-butyl methyl ether, 1:0–9:1) gave 172 mg (90%, >99% purity) of the title compound as a cream solid. LC-MS (method C) Rt = 1.43 min, MS (ESIpos): 235 [M + H]+. 1H NMR (500 MHz, CDCl3) δ [ppm]: 0.84 (s, 3H), 1.19 (s, 3H), 1.62 (dtd, 1H), 1.72 (td, 1H), 1.78–1.87 (m, 1H), 1.92 (m, 1H), 2.22 (td, 1H), 2.29–2.36 (m, 1H), 2.42–2.47 (d, 1H), 2.53 (d, 1H), 3.05 (dd, 1H), 6.88–6.94 (m, 2H), 7.12–7.17 (m, 2H).
(1R,2S)-2-[(4-Fluorophenyl)methyl]-3,3-dimethyl-1-(trifluoromethyl)cyclohexan-1-ol (16)
To a stirred solution of rac-2-[(4-fluorophenyl)methyl]-3,3-dimethylcyclohexan-1-one (23a) (172 mg, 0.73 mmol) and trifluoromethyltrimethylsilane (0.54 mL, 3.67 mmol) in tetrahydrofuran (5 mL) at 0 °C was added dropwise a 1 M solution of tetra-n-butylammonium fluoride in tetrahydrofuran (0.88 mL, 0.88 mmol). The cooling bath was removed, and the reaction was stirred at room temperature for 1 h. On completion, concentrated hydrochloric acid (60 μL, 7.34 mmol, 37% solution) in water (2 mL) was added and the mixture was stirred for 30 min before diluting with water and extracting into ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate, and the solvents were removed in vacuo. Purification by column chromatography (silica gel, eluting with heptane–tert-butyl methyl ether, 1:0–9:1) followed by reverse-phase column chromatography (C18 silica gel, eluting with 0.1% formic acid in water–0.1% formic acid in acetonitrile, 9:1–1:9) gave 82 mg (36% yield, 99% purity) of the title compound as a colorless solid. LC-MS (method B) Rt = 4.94 min, MS (ESIpos): no mass ion observed. 1H NMR (500 MHz, CDCl3) δ [ppm]: 0.94 (s, 3H), 1.10 (s, 3H), 1.32 (td, 1H), 1.51–1.63 (m, 3H), 1.76–1.87 (m, 2H), 1.92 (m, 2H), 2.74 (dd, 1H), 2.99 (dd, 1H), 6.91–6.98 (t, 2H), 7.16 (dd, 2H).
rel-(1R,2S)-2-[(4-Fluorophenyl)methyl]-3,3-dimethyl-1-(trifluoromethyl)cyclohexan-1-ol (16a and 16b)
Enantiomers 16a (first eluting) and 16b (second eluting) were obtained after chiral separation via supercritical fluid chromatography (SFC) (N1).
Compounds 24a–h
The following compounds were prepared using the procedures outlined for final compound 16 starting from 5,5-dimethylcyclohex-2-en-1-one and utilizing the commercially available benzyl bromides. Yields and purities quoted are for the reaction stage exemplified for compound 16.
2-[(4-Chlorophenyl)methyl]-3,3-dimethyl-1-(trifluoromethyl)cyclohexan-1-ol (24a)
49% yield, 99% purity. LC-MS (method A) Rt = 4.58 min, MS (ESIPos): no mass ion observed. 1H NMR (500 MHz, CDCl3) δ [ppm]: 0.94 (s, 3H), 1.10 (s, 3H), 1.32 (td, 1H), 1.52–1.63 (m, 3H), 1.76–1.87 (m, 2H), 1.89–1.95 (m, 2H), 2.74 (dd, 1H), 3.00 (dd, 1H), 7.14 (d, 2H), 7.20–7.25 (d, 2H).
4-[[2-Hydroxy-6,6-dimethyl-2-(trifluoromethyl)cyclohexyl]methyl]benzonitrile (24b)
57% yield, >99% purity. LC-MS (method B) Rt = 4.68 min, MS (ESIPos): 353 [M + H + MeCN]+, 312 [M + H]+. 1H NMR (250 MHz, CDCl3) δ [ppm]: 0.95 (s, 3H), 1.11 (s, 3H), 1.23–1.41 (m, 2H), 1.52–1.98 (m, 5H), 2.03 (s, 1H), 2.81 (dd, 1H), 3.12 (dd, 1H), 7.31 (d, 2H), 7.54 (d, 2H).
3,3-Dimethyl-2-[(4-methylphenyl)methyl]-1-(trifluoromethyl)cyclohexan-1-ol (24c)
50% yield, 95% purity. no mass ion observed by LC/MS analysis. 1H NMR (500 MHz, CDCl3) δ [ppm]: 0.94 (s, 3H), 1.11 (s, 3H), 1.33 (td, 1H), 1.51–1.63 (m, 4H), 1.77–1.85 (m, 1H), 1.95 (d, 1H), 2.01 (dd, 1H), 2.32 (s, 3H), 2.75 (dd, 1H), 2.98 (dd, 1H), 7.05–7.15 (m, 4H).
2-[(4-Methoxyphenyl)methyl]-3,3-dimethyl-1-(trifluoromethyl)cyclohexan-1-ol (24d)
39% yield, 95% purity. No mass ion observed by LC/MS analysis. 1H NMR (500 MHz, CDCl3) δ [ppm]: 0.95 (s, 3H), 1.11 (s, 3H), 1.30 (td, 1H), 1.51–1.63 (m, 2H), 1.75–1.86 (m, 2H), 1.90–1.98 (m, 2H), 2.72 (dd, 1H), 2.95 (dd, 1H), 3.79 (s, 3H), 6.77–6.86 (m, 2H), 7.14 (d, 2H).
(rac)-Methyl 4-[(6,6-Dimethyl-2-oxo-cyclohex-3-en-1-yl)methyl]benzoate (22e)
To a solution of 5,5-dimethylcyclohex-2-en-1-one (6 g, 48.32 mmol) in THF (300 mL) was added LDA (2 M in a mixed solution of THF and heptane, 26.6 mL) at −78 °C under a N2 atmosphere. The reaction mixture was stirred at −78 °C for 30 min. Then, methyl 4-(bromomethyl)benzoate (14.4 g, 62.86 mmol) in THF (100 mL) was added. The reaction mixture was warmed to 25 °C and stirred for 3 h. TLC (petroleum ether (PE)/EA = 5:1) indicated that the reaction was completed. Saturated aq NH4Cl (100 mL) was added to the reaction mixture, and the mixture was stirred for 10 min. The reactions were performed for two batches in parallel, and the combined reaction mixture was extracted with EA (200 mL × 3). The combined organic phase was washed with H2O (50 mL × 2) and saturated brine (50 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel chromatography (PE/EA = 50:1–5:1) to give methyl 4-[(6,6-dimethyl-2-oxo-cyclohex-3-en-1-yl)methyl]benzoate (5.6 g, 19% yield, 88% purity) as a yellow oil. LC-MS (method E): Rt = 0.867 min; m/z = 273.2 (M + H)+.
(rac)-Methyl 4-[(2,2-Dimethyl-6-oxo-cyclohexyl)methyl]benzoate (23e)
The reactions were performed for two batches in parallel: to a solution of (rac)-methyl 4-[(6,6-dimethyl-2-oxo-cyclohex-3-en-1-yl)methyl]benzoate (2.8 g, 10.28 mmol) in toluene (50 mL) was added (Ph3P)3RhCl (1.43 g, 1.55 mmol). The reaction mixture was stirred at 25 °C for 24 h under a H2 atmosphere (15 psi). TLC (PE/EA = 3:1) indicated that the reaction was completed. The reactions were performed for two batches in parallel, and the combined reaction mixture was filtered through celite and the filter cake was washed with EA (100 mL). The organic phase was concentrated in vacuo. The residue was purified by silica gel chromatography (PE/EA = 50:1–10:1) to give the crude product, which was further purified by prep-HPLC (0.05% NH3·H2O/CH3CN/H2O) to give methyl 4-[(2,2-dimethyl-6-oxo-cyclohexyl)methyl]benzoate (2.9 g, 50% yield, 98% purity) as a white solid. LC-MS (method E): Rt = 2.380 min; m/z = 275.3 (M + H)+. 1H NMR (400 MHz, CDCl3) δ [ppm]: 7.93 (d, 2H), 7.27 (d, 2H), 3.89 (s, 3H), 3.16 (dd, 1H), 2.62 (d, 1H), 2.52 (d, 1H), 2.32–2.19 (m, 2H), 1.75–1.58 (m, 4H), 1.21 (s, 3H), 0.86 (s, 3H).
N-Ethyl-4-{[2-hydroxy-6,6-dimethyl-2-(trifluoromethyl)cyclohexyl]methyl}benzamide (24e)
Step A: methyl 4-[(2,2-dimethyl-6-oxocyclohexyl)methyl]benzoate (900 mg, 3.28 mmol) was dissolved in THF (10 mL), and trifluoromethyltrimethylsilane (2.4 mL, 16.0 mmol) was added and the mixture was cooled to 0 °C. Afterward, tetra-n-butylammonium fluoride (3.9 mL, 1.0 M, 3.9 mmol) was added and stirring was continued for 10 min. Afterward, the yellow solution was allowed to come to rt and stirred for 1.5 h. The reaction mixture was then poured into an aq HCl solution (2.7 mL, 37% HCl in 40 mL water) and stirred for 10 min. Then, the aq solution was extracted with ethyl acetate (2 × 15 mL) and the combined organic phases were washed with brine (15 mL) and dried over water-repelling filter, and the solvent was removed under reduced pressure. The crude was suspended in diethyl ether and filtered. The precipitate was washed twice with ether and dried to yield 598 mg (96% purity, 51% yield). LC-MS (method F): Rt = 1.42 min; MS (ESIpos): m/z = 345 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 0.92 (s, 3H), 1.05 (s, 3H), 1.30–1.48 (m, 4H), 1.70–1.80 (m, 1H), 1.88–1.92 (m, 3H), 2.74 (d, 1H), 3.10 (dd, 1H), 3.85 (s, 3H), 5.75 (m, 1H), 7.34 (d, 2H), 7.85 (d, 2H).
Step B: methyl 4-{[2-hydroxy-6,6-dimethyl-2-(trifluoromethyl)cyclohexyl]methyl}-benzoate (445 mg, 1.29 mmol) was dissolved in THF/MeOH (5/0.5 mL) and lithium hydroxide monohydrate (542 mg, 12.9 mmol; CAS-RN:[1310-66-3]) in 0.5 mL water was added, and the reaction mixture was stirred at rt overnight. Afterward, another 5 equiv of LiOH was added and stirring was continued for 24 h. Afterward, the reaction mixture was heated at 50 °C for 4 days. After cooling to rt, the solvent was removed under reduced pressure and water was added. The pH was adjusted to pH 4 by the addition of 10% aq citric acid. The aq phase was extracted with ethyl acetate (three times), and the combined organic phases were washed with brine, filtered over water-repelling filter, and the solvent was removed under reduced pressure. The crude (404 mg, 97% purity, 92% yield) was used without further purification in the next step. LC-MS (method F): Rt = 1.19 min; MS (ESIpos): m/z = 331 [M + H]+.
Step C: 4-{[2-hydroxy-6,6-dimethyl-2-(trifluoromethyl)cyclohexyl]-methyl}benzoic acid was prepared according to the general procedures. Afterward, 80.0 mg (235 μmol) was dissolved in 2-Me-THF (3 mL) and HATU (134 mg, 352 μmol), ethylamine hydrogen chloride (57.5 mg, 705 μmol), and triethylamine (98 μL, 700 μmol) were added. The reaction mixture was stirred at rt overnight. Afterward, water was added and the mixture was extracted with ethyl acetate (three times). The combined organic phases were concentrated under reduced pressure. The title compound (47.0 mg, 98% purity, 55% yield) was obtained after HPLC purification. LC-MS (method D) Rt = 1.22 min, MS (ESIPos): 358 [M + H+]+. 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 0.91 (s, 3H), 1.06 (s, 3H), 1.11 (t, 3H), 1.30–1.37 (m, 1H), 1.41–1.49 (m, 3H), 1.69–1.80 (m, 1H), 1.89–1.91 (m, 2H), 2.68–2.72 (m, 1H), 3.07 (dd, 1H), 3.23–3.30 (m, 2H), 5.72 (s, 1H), 7.25 (d, 2H), 7.72 (d, 2H), 8.37 (t, 1H).
2-[(3-Fluorophenyl)methyl]-3,3-dimethyl-1-(trifluoromethyl)cyclohexan-1-ol (24f)
73% yield, 97% purity. LC-MS (method B) Rt = 4.96 min, MS (ESIPos): no mass ion observed. 1H NMR (500 MHz, CDCl3) δ [ppm]: 0.95 (s, 3H), 1.11 (s, 3H), 1.33 (td, 1H), 1.51–1.65 (m, 3H), 1.76–1.87 (m, 2H), 1.89–1.95 (m, 1H), 1.96 (t, 1H), 2.77 (dd, 1H), 3.03 (dd, 1H), 6.85 (td, 1H), 6.92 (d, 1H), 6.99 (d, 1H), 7.21 (td, 1H).
3-[[2-Hydroxy-6,6-dimethyl-2-(trifluoromethyl)cyclohexyl]methyl]benzonitrile (24g)
14% yield, 95% purity. LC-MS (method A) Rt = 3.91 min, MS (ESIPos): 353 [M + H + MeCN]+. 1H NMR (500 MHz, CDCl3) δ [ppm]: 1.00 (s, 3H), 1.13 (s, 3H), 1.36 (td, 1H), 1.56–1.68 (m, 3H), 1.79–1.87 (m, 2H), 1.87–1.96 (m, 2H), 2.81 (d, 1H), 3.09 (dd, 1H), 7.38 (t, 1H), 7.43–7.51 (m, 3H).
2-[(2,4-Difluorophenyl)methyl]-3,3-dimethyl-1-(trifluoromethyl)cyclohexan-1-ol (24h)
93% yield, 94% purity. LC-MS (method B) Rt = 4.99 min, MS (ESIPos): no mass ion observed. 1H NMR (500 MHz, CDCl3) δ [ppm]: 0.90 (s, 3H), 1.12 (s, 3H), 1.32 (td, 1H), 1.49–1.65 (m, 3H), 1.74–1.89 (m, 2H), 1.89–1.97 (m, 1H), 1.99 (t, 1H), 2.69 (dd, 1H), 3.08 (dd, 1H), 6.75 (ddd, 1H), 6.80 (td, 1H), 7.18 (q, 1H).
(rac)-2-[(4-Fluorophenyl)methyl]cyclohexan-1-one (26a)
To a stirred solution of diisopropylamine (2.6 mL, 18.3 mmol) in tetrahydrofuran (25 mL) at −78 °C was added n-butyllithium (6.7 mL, 16.8 mmol, 2.5 M in hexanes). After 10 min, the reaction mixture was warmed to 0 °C, and after 10 min of stirring at this temperature, the reaction mixture was cooled to −78 °C and a mixture of cyclohexanone (1.6 mL, 15.3 mmol) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (2 mL, 16.8 mmol) in tetrahydrofuran (5 mL) was added dropwise. After 30 min of stirring at −78 °C, 1-(bromomethyl)-4-fluorobenzene (2.1 mL, 16.8 mmol) was added. After a further 1 h, the cold bath was removed and the reaction mixture was allowed to slowly warm to room temperature and stirred for a further 16 h. The reaction mixture was quenched by the addition of a saturated aq solution of ammonium chloride and extracted into ethyl acetate (2×). The combined organic phases were washed with brine, dried (MgSO4), filtered, and the filtrate was concentrated in vacuo. The residue was purified by multiple column chromatography using the Biotage Isolera 4 (eluent: 0–10% tert-butyl methyl ether, then 0–5% tert-butyl methyl ether in heptanes) to afford 2.1 g (53% yield, 71% purity) of a colorless oil. LC-MS (method B) Rt = 1.22 min, MS (ESIPos): 207 [M + H]+. 1H NMR (500 MHz, CDCl3) δ [ppm]: 1.34 (qd, 1H), 1.49–1.78 (m, 2H), 1.77–1.90 (m, 1H), 1.91–2.15 (m, 2H), 2.23–2.58 (m, 4H), 3.18 (dd, 1H), 6.89–6.99 (m, 2H), 7.07–7.17 (m, 2H).
(1R,2S or 1S,2R)-2-[(4-Fluorophenyl)methyl]-1-(trifluoromethyl)cyclohexan-1-ol (17)
To a stirred solution of (rac)-2-[(4-fluorophenyl)methyl]cyclohexan-1-one (2.1 g, 10.3 mmol) and trifluoromethyltrimethylsilane (7.6 mL, 51.4 mmol) in THF (60 mL) at 0 °C was added dropwise tetra-n-butylammonium fluoride (15 mL, 15.4 mmol, 1 M solution in THF). The ice bath was then removed, and the reaction mixture was stirred for 1.5 h at rt. The reaction mixture was diluted with 1 M aq HCl (100 mL) and stirred vigorously for 5 min. Solid sodium bicarbonate was then carefully added portion-wise until effervescence could no longer be observed on addition. The aqueous mixture was then extracted into ethyl acetate (three times). The combined organic phases were washed with brine, dried (MgSO4), filtered, and the filtrate was concentrated in vacuo. The residue thus obtained was purified by column chromatography using the Biotage Isolera 4 (eluent: 0–10% tert-butyl methyl ether in heptane) to afford 2.2 g (76% yield), a yellow oil being an approximate 2:5 ratio of racemic trans- and cis products by LC-MS (method B) 27.8% and 69.7%@Rt = 4.73 and 4.80 min (MS (ESIpos), no associated mass ion). Analysis by 1H NMR was in agreement with the LC-MS assessment and suggested an approximate 1:2 ratio of trans-to-cis products.
The crude material (900 mg out of 2.2 g) was submitted for preparative HPLC separations (eluent: 10–100% acetonitrile in water in the presence of 0.1% formic acid) in 3 batches (300 mg × 3). The product-rich fractions were pooled, and the acetonitrile was removed in vacuo. The residual aqueous phase was treated with salt until a saturated brine had been obtained and then extracted into diethyl ether (2×). The combined ethereal phases were washed with brine, dried (MgSO4), filtered, and the filtrate was concentrated in vacuo. The residues thus obtained were transferred to tared vials using the minimum amount of DCM, and the solvent was removed under a stream of nitrogen. The products were then briefly (ca. 10 min) dried under high vacuum (freeze-drier) to afford the racemic mixture of trans product (226 mg). 1H NMR (500 MHz, chloroform-d) δ = 1.27–1.38 (m, 2H), 1.56–1.73 (m, 5H), 1.94 (s, 1H), 1.96–2.05 (m, 2H), 2.43–2.52 (m, 1H), 3.01 (d, J = 13.7, 1H), 6.93–7.01 (m, 2H), 7.09–7.16 (m, 2H).
174 mg of this racemic mixture was then subjected to chiral separation via SFC (conditions: 10% methanol: 90% CO2 with Chiralpak AD-H 25 cm column at 15 mL/min, methanol) to yield the desired enantiomer 17 (44 mg, 99% purity, 25% yield) as a colorless oil. [α]D20 +31,9° (c 1.00, DMSO). LC-MS (method D): Rt = 1.39 min; MS (ESIneg): m/z = 275 [M – H]−. 1H NMR (500 MHz, CDCl3) δ [ppm]: 1.26–1.38 (m, 2H), 1.55–1.72 (m, 5H), 1.94 (s, 1H), 1.95–2.06 (m, 2H), 2.40–2.54 (m, 1H), 3.01 (d, 1H), 6.94–7.01 (m, 2H), 7.09–7.15 (m, 2H).
Compounds 27a–h
The following compounds were prepared using the procedures outlined for final compound 17 starting from cyclohexanone and utilizing the commercially available benzyl bromides. Yields and purities quoted are for the reaction stage exemplified for compound 17. In the case of final targets 27c and 27f, a different approach was used as specified below for the synthesis of their respective intermediates 26.
(1R,2S or 1S,2R)-2-[(5-Chloro-3-thienyl)methyl]-1-(trifluoromethyl)cyclohexanol (27a)
After chiral separation via SFC, 80 mg (14% yield, 100% purity) was obtained. [α]D20 55.0° (c 1.00, DMSO). LC-MS (method D): Rt = 1.45 min, MS (ESIpos): no mass ion observed. 1H NMR (400 MHz, CDCl3) δ [ppm]: 1.33–1.48 (m, 2H), 1.53–1.62 (m, 2H), 1.66–1.71 (m, 1H), 1.73–1.80 (m, 1H), 1.94–2.01 (m, 3H), 2.67 (dd, 1H), 3.09–3.14 (m, 1H), 6.57–6.58 (m, 1H), 6.71–6.72 (m, 1H).
4-[(1R,2S or 1S,2R)-2-[Hydroxy-2-(trifluoromethyl)cyclohexyl]methyl]thiophene-2-carbonitrile (27b)
After chiral separation via SFC, 14 mg (23% yield, 100% purity) was obtained. LC-MS (method D): Rt = 1.27 min; MS (ESIneg): m/z = 288 [M – H]−. 1H NMR (400 MHz, CDCl3) δ [ppm]: 1.33–1.41 (m, 2H), 1.58–1.61 (m, 1H), 1.63–1.70 (m, 2H), 1.72–1.79 (m, 1H), 1.98–2.00 (m, 2H), 2.02–2.08 (m, 1H), 2.81 (dd, 1H), 3.25 (dd, 1H), 6.82–6.83 (m, 1H), 7.47 (d, 1H).
(1R,2S or 1S,2R)-2-[(5-Chlorothiazol-2-yl)methyl]-1-(trifluoromethyl)cyclohexanol (27c)
After chiral separation via SFC (methanol-CO2 15:85 with Chiralpak AD-H 25 cm column at 15 mL/min), 33 mg (34% yield, 100% purity) was obtained. ee: 100% by SFC; LC-MS (method A): Rt = 3.53 min, MS (ESIpos): m/z = 300.0, 302.0 [M + H]+. 1H NMR (250 MHz, CDCl3) δ [ppm]: 1.27–1.83 (m, 7H), 2.03–2.34 (m, 2H), 2.93 (dd, 1H), 3.35 (dd, 1H), 4.71 (s, 1H), 7.44 (s, 1H).
(1R,2S or 1S,2R)-2-[(6-Chloro-3-pyridyl)methyl]-1-(trifluoromethyl)cyclohexanol (27d)
After chiral separation via SCF (method POB), 17 mg (20% yield, 95% purity) was obtained. LC-MS (OA01a01): Rt = 1.21 min, MS (ESIpos): m/z = 294 [M + H]+. 1H NMR (400 MHz, CDCl3) δ [ppm]: 1.24–1.37 (m, 2H), 1.49–1.71 (m, 5H), 1.92–2.05 (m, 3H), 2.50 (t, 1H), 3.02–3.05 (m, 1H), 7.26–7.28 (m, 1H), 7.47 (dd, 1H), 8.20 (d, 1H).
(1R,2S or 1S,2R)-2-[(5-Chloro-3-pyridyl)methyl]-1-(trifluoromethyl)cyclohexanol (27e)
After chiral separation via SFC (ethanol-CO2 18:82 with Chiralpak AD-H 25 cm column at 15 mL/min), 3.4 mg (20% yield, 95% purity) was obtained. ee: 100% by SFC (weak signal); LC-MS (method A): Rt = 3.22 min, MS (ESIpos): m/z = 294.0, 296.0 [M + H]+. 1H NMR (250 MHz, CDCl3) δ [ppm]: 1.21–1.43 (m, 2H), 1.48–1.77 (m, 5H), 1.92–2.09 (m, 2H), 2.18 (s, 1H), 2.50 (t, 1H), 3.07 (d, 1H), 7.50 (s, 1H), 8.31 (s, 1H), 8.44 (d, 1H).
(1R,2S or 1S,2R)-2-(4-Pyridylmethyl)-1-(trifluoromethyl)cyclohexanol (27f)
After chiral separation via SFCB, 38 mg (40% yield, purity 95%) was obtained. [α]D20 31.0° (c 1.00, DMSO); LC-MS (method D): Rt = 1.04 min, MS (ESIpos): m/z = 260.0 [M + H]+. 1H NMR (400 MHz, CDCl3) δ [ppm]: 1.25–1.39 (m, 2H), 1.51–1.73 (m, 5H), 1.97–2.09 (m, 2H), 2.21 (s, 1H), 2.50 (t, 1H), 3.04–3.06 (m, 1H), 7.11–7.12 (m, 2H), 8.52 (s, 2H).
(1R,2S or 1S,2R)-2-[(5-Chloro-2-pyridyl)methyl]-1-(trifluoromethyl)cyclohexanol (27g)
After chiral separation via SFC (5% methanol: 90% CO2 with Chiralpak AD-H 25 cm column at 15 mL/min), 5.2 mg (22% yield, 100% purity) was obtained. LC-MS (method B): Rt = 4.22 min, MS (ESIpos) m/z = 294/296 [M + H]+. 1H NMR (500 MHz, CDCl3) δ [ppm]: 1.28–1.38 (m, 1H), 1.47–1.59 (m, 3H), 1.59–1.74 (m, 3H), 2.13–2.29 (m, 2H), 2.75 (dd, 1H), 3.35 (dd, 1H), 5.87 (s, 1H), 7.14 (d, 1H), 7.61 (dd, 1H), 8.44 (d, 1H).
rac-(1R,2S)-2-(1H-Indol-3-ylmethyl)-1-(trifluoromethyl)cyclohexanol (27h)
After purification by silica flash column chromatography (gradient: hexanes/DCM/EtOH 15/20/1) followed by preparative HPLC (method H), 20 mg (7% yield, 97% purity) was obtained. LC-MS (method D): Rt = 1.29 min, MS (ESIpos): m/z = 298.2 [M + H]+. 1H NMR (400 MHz, CDCl3) δ [ppm]: 1.04–1.14 (m, 1H), 1.31–1.41 (m, 1H), 1.53–1.66 (m, 5H), 1.93–1.97 (m, 1H), 2.03–2.10 (m, 2H), 2.64 (dd, 1H), 3.20–3.24 (m, 1H), 7.02 (d, 1H), 7.11–7.15 (m, 1H), 7.18–7.22 (m, 1H), 7.37 (d, 1H), 7.59 (d, 1H), 7.99 (s, 1H).
(rac)-1-(Trifluoromethyl)cyclohexanol (28)
To a stirred solution of cyclohexanone (1.0 g, 10 mmol) in THF (15 mL) under nitrogen (trifluoromethyl)trimethylsilane (7.2 g, 51 mmol) was added at −40 °C for 10 min. Then, tetrabutylammonium fluoride (10 mL, 1.0 M, 10 mmol, 1.0 equiv) was added slowly over 45 min at max −30 °C. Afterward, the mixture was allowed to warm up to rt for 1 h. The mixture was poured into aq sat. ammonium chloride solution (25 mL) and extracted twice with diethyl ether (15 mL). The combined organic layer was washed with water (10 mL) and brine (10 mL). The organic layer was dried with sodium sulfate, filtered, and concentrated in vacuo at +40 °C, 800–100 mbar, as the product is highly volatile. The crude was dissolved with tetrahydrofuran (5 mL) and hydrochloric acid solution (13 mL, 4.0 M) and stirred for 30 min at rt. The layers were separated, and the aqueous layer was extracted twice with diethyl ether (10 mL). The combined organic layers were washed with aq sat. sodium hydrogen carbonate solution and dried with sodium sulfate, filtered, and concentrated at 40 °C, 800–100 mbar, to yield 1.85 g as a brown oil (108% yield, purity 78%). LC-MS (method D): Rt = 0.53 min; MS (ESIpos): m/z = 167 [M + H]+. 1H NMR (400 MHz, CDCl3) δ [ppm]: 1.15–1.29 (m, 1H), 1.54 (s, 1H), 1.59–1.68 (m, 4H), 1.68–1.72 (m, 1H), 1.72–1.80 (m, 2H), 1.85 (s, 1H), 3.70–3.80 (m, 1H).
1-(Trifluoromethyl)cyclohexene (29)
To a stirred solution of (rac)-1-(trifluoromethyl)-cyclohexanol (1.7 g, 10 mmol, 1.0 equiv) and 4-dimethylaminopyridine (49 mg, 404 μmol, 0.04 equiv) in dichloromethane (10 mL) under nitrogen thionyl chloride (2.2 mL, 30 mmol, 3.0 equiv) was dropped followed by the addition of pyridine (2.5 mL, 30 mmol, 3.0 equiv). Afterward, the reaction mixture was stirred overnight at 45 °C. The mixture was cooled to 0 °C and poured into copper sulfate solution (40 mL). The layers were separated and extracted three times with dichloromethane (25 mL). The combined organic layers were washed with aq sat. sodium hydrogen carbonate solution (50 mL). The organic layer was dried with sodium sulfate, filtered, and stored as a yellow solution (1.52 g, 300 mL, 100% yield, purity 95%).
(rac)-1-(Trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (30)
To a stirred solution of 1-(trifluoromethyl)cyclohexene 29 (1.5 g, 10 mmol, 1.0 equiv) in dichloromethane (100 mL), mCPBA (6.9 g, 40 mmol, 4.0 equiv) in dichloromethane (10 mL) under nitrogen was added. The reaction mixture was stirred for 4 days at rt. The mixture was cooled to 0 °C, and then carefully aq sat. sodium hydrogen carbonate solution (100 mL) was added. The organic layer was extracted four times with sat. sodium hydrogen carbonate solution (100 mL). The layers were separated, and the organic phase was dried with sodium sulfate, filtered, and further reacted as a dichloromethane solution (110 mL, 80% yield).
(1R,2R or 1S,2S)-2-(4-Fluorophenoxy)-1-(trifluoromethyl)cyclohexanol (31)
4-Fluorophenole (613 mg, 5.5 mmol, 1.0 equiv) was dissolved in dry DMF (13 mL), and Cs2CO3 (2.7 g, 8.2 mmol, 1.5 equiv) and epoxide 30 (1.0 g, 6.0 mmol, 1.1 equiv) were added. The brown solution was heated to 85 °C for 1.5 h and then at 60 °C overnight. The reaction mixture was diluted with water (50 mL) and extracted two times with Et2O (50 mL); the combined organic layer was washed once with sat. aq NaCl (100 mL), dried over MgSO4, filtered, and concentrated under reduced pressure to obtain a brown oil. The crude was purified by column chromatography (Grace Reveleris, column: CHROMABOND Flash with 15 g MN silica; mobile phase: cyclohexane (CyH)/TBME) to yield 289 mg of racemic material. Compound 31 was obtained after chiral separation via HPLC (method NP) (80.0 mg, 95% purity, 42% yield). [α]D20 −61.5° (c 1.00, DMSO). LC-MS (method D): Rt = 1.33 min; MS (ESIneg): m/z = 277 [M – H]−. 1H NMR (400 MHz, CDCl3) δ [ppm]: 1.44–1.52 (m, 1H), 1.54–1.60 (m, 1H), 1.60–1.75 (m, 3H), 1.77–1.87 (m, 1H), 1.87–1.96 (m, 1H), 2.04 (s, 1H), 2.16–2.28 (m, 1H), 4.35–4.41 (m, 1H), 6.84–6.91 (m, 2H), 6.93–7.01 (m, 2H).
General Procedure of Aniline Formation (1.1)
Epoxide 30 (1.31 mmol, 1.0 equiv) and the respective anilines (1.98 mmol, 1.5 equiv) were dissolved in toluene (50 equiv) at rt. Scandium triflate (0.66 mmol, 0.5 equiv) was added and stirred at 60 °C for 4–6 h. The reaction mixture was diluted with water and extracted three times with ethyl acetate, washed twice with hydrochloric acid (1.0 M) and once with aq sat. sodium hydrogen carbonate. The layers were separated, and the organic layer was dried and concentrated in vacuo. The crude was purified by Biotage (KP-Sil 25 g, gradient: hexane/ethyl acetate), and the desired enantiomer was obtained subsequently after chiral separation by SFC.
(1R,2R)-2-[(4-Fluorophenyl)amino]-1-(trifluoromethyl)cyclohexanol (18)
Compound 18 was prepared according to general procedure GP1.1 using 1-(trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (5.0 g, 33 mmol) and 4-fluoroaniline (4.7 mL, 50 mmol) and was purified by preparative HPLC (instrument: Agilent HPLC 1260; column: Chiralpak IG 3 μm 100 × 4.6 mm2; eluent A: hexane; eluent B: 2-propanol; isokratic: 95% A + 5% B; flow 1.4 mL/min; temperature: 25 °C; DAD 254 nm) to yield 7.20 g (78% yield, 100% purity) [α]D20 −95.0° (c 1.00, DMSO). LC-MS (method D): Rt = 1.19 min; MS (ESIpos): m/z = 278 [M + H]+. 1H NMR (400 MHz, DMSO-d6): δ [ppm] = 1.31–1.39 (m, 1H), 1.42–1.65 (m, 5H), 1.71–1.81 (m, 1H), 1.94–2.04 (m, 1H), 3.60–3.67 (m, 1H), 5.37 (d, 1H), 5.88 (s, 1H), 6.59–6.65 (m, 2H), 6.85–6.92 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ [ppm]: 18.4, 19.3, 24.9, 40.2, 50.7, 72.8 (q), 113.3, 115.2, 144.2, 153.0, 155.3. Absolute stereochemistry has been confirmed for compound 18, and based on these findings, the absolute stereoconfiguration for the following compounds synthesized according to GP1 is assumed to be also R,R.
(1R,2R)-2-[(4-Fluorophenyl)(methyl)amino]-1-(trifluoromethyl)cyclohexanol (32a)
Following general procedure GP1.1, 1-(trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (200 mg, 1.20 mmol, 1.0 equiv) and 4-fluoro-N-methylaniline (226 mg, 1.8 mmol, 1.5 equiv) were reacted to yield 275 mg (100% purity, 78% yield). After chiral separation by SFC (Instrument: Sepiatec: Prep SFC100; column: Chiralpak IG 5 μm 250 × 30 mm2; eluent A: CO2, eluent B: ethanol; isocratic: 5% B; flow 100.0 mL/min temperature: 40 °C; BPR: 150 bar; MWD@220 nm), 32a was obtained (90.0 mg, 100% purity, 26% yield). [α]D20 −104.4° (c 1.00, DMSO). LC-MS (method E): Rt = 1.36 min; MS (ESIpos): m/z = 291 [M]+. 1H NMR (500 MHz, DMSO-d6) δ [ppm]: 1.37–1.49 (m, 2H), 1.59–1.71 (m, 3H), 1.71–1.77 (m, 1H), 1.78–1.87 (m, 1H), 2.12–2.19 (m, 1H), 2.70 (s, 3H), 3.74 (dd, 1H), 5.95 (s, 1H), 6.91–6.95 (m, 2H), 6.96–7.02 (m, 2H).
(rac)-(1R,2R)-2-[(4-Fluorophenyl)(2-methoxyethyl)amino]-1-(trifluoromethyl)cyclohexanol (32b)
Following general procedure GP1.1, 1-(trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (80 mg, 0.48 mmol, 1.0 equiv) and 4-fluoro-N-(2-methoxyethyl)aniline (123 mg, 0.72 mmol, 1.5 equiv) were reacted to yield 8.80 mg (85% purity, 5% yield) of compound 32b. LC-MS (method D): Rt = 1.37 min; MS (ESIpos): m/z = 336 [M + H]+. 1H NMR (400 MHz, CDCl3): δ [ppm]= 1.24–1.42 (m, 2H), 1.46–1.54 (m, 2H), 1.63–1.69 (m, 1H), 1.72–1.80 (m, 1H), 1.83–1.94 (m, 2H), 2.31–2.36 (m, 1H), 3.31 (s, 3H), 3.32–3.47 (m, 4H), 3.83–3.87 (m, 1H), 6.94–7.00 (m, 2H), 7.04–7.10 (m, 2H).
rac-(1R,2R)-2-Anilino-1-(trifluoromethyl)cyclohexan-1-ol (32c)
Following general procedure GP1.1, 1-(trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (100 mg, 0.6 mmol, 1.0 equiv) and aniline (84.1 mg, 0.9 mmol, 1.5 equiv) were reacted to yield 41.7 mg (90% purity, 24% yield) of compound 32c. LC-MS (method D): Rt = 1.37 min; MS (ESIpos): m/z = 336 [M + H]+. 1H NMR (400 MHz, CDCl3): δ [ppm] = 1.24–1.42 (m, 2H), 1.46–1.54 (m, 2H), 1.63–1.69 (m, 1H), 1.72–1.80 (m, 1H), 1.83–1.94 (m, 2H), 2.31–2.36 (m, 1H), 3.31 (s, 3H), 3.32–3.47 (m, 4H), 3.83–3.87 (m, 1H), 6.94–7.00 (m, 2H), 7.04–7.10 (m, 2H).
(1R,2R)-2-(5-Fluoro-2,3-dihydro-1H-indol-1-yl)-1-(trifluoromethyl)cyclohexanol (32d)
Following general procedure 1.1, 1-(trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (200 mg, 1.20 mmol, 1.0 equiv) and 5-fluoro-2,3-dihydro-1H-indole (248 mg, 1.81 mmol, 1.5 equiv) were reacted to yield 68.0 mg (100% purity, 19% yield) of 2-(5-fluoro-2,3-dihydro-1H-indol-1-yl)-1-(trifluoromethyl)cyclohexanol. After chiral separation by SFC (eluent A: CO2; eluent B: methanol; isokratic: 88% A + 12% B), compound 32d was obtained (6.30 mg, 100% purity, 2% yield, Rt = 7.8–9.0 min). LC-MS (method E): Rt = 1.38 min; MS (ESIpos): m/z = 304 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.45–1.55 (m, 2H), 1.60–1.74 (m, 4H), 1.79–1.90 (m, 1H), 1.92–2.01 (m, 1H), 2.71–2.83 (m, 1H), 2.85–2.95 (m, 1H), 3.42–3.51 (m, 2H), 3.53–3.61 (m, 1H), 3.62–3.67 (m, 1H), 5.83 (s, 1H), 6.34 (dd, 1H), 6.74 (dt, 1H), 6.82–6.87 (m, 1H).
(1R,2R)-4-Fluoro-N-[(1R,2R)-2-hydroxy-2-(trifluoromethyl)cyclohexyl]benzamide (32e)
1-(Trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (200 mg, 1.20 mmol, 1.0 equiv) was dissolved in N,N-dimethylamide (4.0 mL); then, (96.3 mg, 60% purity, 2.41 mmol, 2.0 equiv) was added and 4-fluorobenzamide (335 mg, 2.41 mmol, 2.0 equiv) in N,N-dimethylformamide (1.0 mL) was added and stirred overnight at 60 °C. The reaction mixture was diluted with water (15.0 mL) and extracted three times with dichloroethane (15.0 mL) and washed twice with water (15 mL, 1.0 M) and once with brine (15.0 mL). The layers were separated, and the organic layer was dried with sodium sulfate and concentrated in vacuo. The crude was purified by Biotage (KP-Sil 10 g, gradient: hexane/ethyl acetate 0–60%) to yield 139 mg (95% purity, 36% yield). After chiral separation by SFC (eluent A: CO2; eluent B: 2-propanol; isokratic: 85% A + 15% B), compound 32e was obtained (35.2 mg, 99% purity, 9% yield). [α]D20 −40.0° (c 1.00, DMSO). LC-MS (method D): Rt = 1.02 min; MS (ESIpos): m/z = 306 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.37–1.55 (m, 3H), 1.56–1.67 (m, 3H), 1.85–1.96 (m, 1H), 2.12–2.22 (m, 1H), 4.42–4.49 (m, 1H), 6.05 (s, 1H), 7.26–7.33 (m, 2H), 7.82–7.88 (m, 2H), 8.15 (d, 1H).
(1R,2R)-5-Fluoro-2-[(1R,2R)-2-hydroxy-2-(trifluoromethyl)cyclohexyl]-2,3-dihydro-1H-isoindol-1-one (32f)
To a stirred solution of 5-fluoro-2,3-dihydro-1H-isoindol-1-one (250 mg, 1.65 mmol, 2.0 equiv) and (66.2 mg, 60% purity, 1.65 mmol, 2.0 equiv) in N,N-dimethylformamide (5.0 mL) was added 1-(trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (137 mg, 0.83 mmol, 1.0 equiv) in N,N-dimethylformamide and stirred for 5 h at 60 °C. The reaction mixture was diluted with ethyl acetate, water was added, and the layers were separated. The organic layer was concentrated and purified by Biotage (Kp-Sil 25 g, gradient: hexane/ethyl acetate 0–25%) to yield 105 mg (90% purity, 36% yield) of 5-fluoro-2-[2-hydroxy-2-(trifluoromethyl)cyclohexyl]-2,3-dihydro-1H-isoindol-1-one. After chiral separation by SFC (eluent A: CO2; eluent B: ethanol; isokratic: 90% A + 10% B), compound 32f (44.3 mg, 95% purity, 42% yield) was obtained. [α]D20 −21.6° (c 1.00, DMSO). LC-MS (method D): Rt = 1.15 min; MS (ESIpos): m/z = 318 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.52–1.62 (m, 2H), 1.64–1.82 (m, 4H), 1.99 (s, 1H), 2.02–2.14 (m, 1H), 4.52–4.61 (m, 2H), 4.69–4.76 (m, 1H), 6.25 (s, 1H), 7.33 (td, 1H), 7.46 (dd, 1H), 7.72–7.77 (m, 1H).
4-Fluoro-N-[(1R,2R or 1S,2S)-2-hydroxy-2-(trifluoromethyl)cyclohexyl]-N-methylbenzene-sulfonamide (33)
Step A: commercially available 2-(methylamino)cyclohexanone, hydrochloride (250 mg, 1.5 mmol, 1.0 equiv), and trimethylamine (0.85 mL, 6.1 mmol, 4.0 equiv) were dissolved in DCM (24.6 mL). Then, 4-fluorobenzenesulfonyl chloride (312 mg, 1.6 mmol, 1.05 equiv) was added at 0 °C in portions. After complete addition, the ice bath was removed and stirring was continued for 1 h. The mixture was concentrated and purified by flash chromatography on silica gel (gradient hexane/EtOAc) to yield 4-fluoro-N-methyl-N-[2-oxocyclohexyl]benzenesulfonamide (83% yield, 96% purity). LC-MS (method F): Rt = 1.07 min; MS (ESIpos): m/z = 286 [M + H]+.
Step B: 4-fluoro-N-methyl-N-[2-oxocyclohexyl]benzenesulfonamide (370 mg, 1.30 mmol, 1.0 equiv) and trifluoromethyltrimethylsilane (922 mg, 6.5 mmol, 5.0 equiv) were dissolved in tetrahydrofuran (7.4 mL) and cooled to 0 °C. Afterward, tetra-n-butylammonium fluoride in tetrahydrofuran (1.9 mL, 1.0 M, 1.9 mmol, 1.5 equiv) was added dropwise into the reaction mixture. After complete addition, the ice bath was removed and it was stirred for 1 h at rt. The reaction mixture was diluted with hydrochloric acid (2.6 mL, 1.0 M, 2.6 mmol) and stirred for 10 min. It was diluted with ethyl acetate, the layers were separated, and the aqueous layer was extracted with ethyl acetate twice. The combined organic layers were washed with brine and dried using a water-resistant filter. The clear filtrate was concentrated under reduced pressure. The crude was purified by Biotage (25 g silica ultracolumn, gradient: dichloromethane/ethanol 0–2%) to yield 175 mg (99% purity, 38% yield). After chiral separation, compound 33 (80.0 mg, 100% purity, 47% yield) was obtained. [α]D20 −2.60° (c 1.00, DMSO). LC-MS (method D): Rt = 1.28 min; MS (ESIpos): m/z = 356 [M + H]+. 1H NMR (400 MHz, CDCl3) δ [ppm]: 0.96–1.07 (m, 1H), 1.15–1.30 (m, 1H), 1.33–1.47 (m, 1H), 1.48–1.54 (m, 1H), 1.59–1.69 (m, 1H), 1.70–1.81 (m, 2H), 2.38–2.45 (m, 1H), 2.81 (s, 3H), 3.72–3.79 (m, 1H), 4.02 (s, 1H), 7.24–7.28 (m, 2H), 7.84–7.89 (m, 2H).
(1R,2R)-2-[(4-Chlorophenyl)amino]-1-(trifluoromethyl)cyclohexanol (34a)
Following general procedure GP1.1, 1-(trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (170 μL, 1.2 mmol, 1.0 equiv) and 4-chloroaniline (230 mg, 1.81 mmol, 1.5 equiv) were reacted to yield 230 mg (100% purity, 65% yield). After chiral separation by SFC (eluent A: CO2, eluent B: ethanol; isokratic: 5% B), compound 34a (95 mg, 100% purity, 27% yield) was obtained. [α]D20 −108.3° (c 1.00, DMSO). LC-MS (method E): Rt = 1.27 min; MS (ESIpos): m/z = 294 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.33–1.40 (m, 1H), 1.41–1.65 (m, 5H), 1.74–1.83 (m, 1H), 1.95–2.04 (m, 1H), 3.67 (d, 1H), 5.67 (d, 1H), 5.91 (s, 1H), 6.62–6.66 (m, 2H), 7.04–7.08 (m, 2H).
4-{[(1R,2R)-2-Hydroxy-2-(trifluoromethyl)cyclohexyl]amino}benzonitrile (34b)
Following general procedure GP1.1, 1-(trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (235 mg, 1.41 mmol, 1.0 equiv) and 4-aminobenzonitrile (251 mg, 2.12 mmol, 1.5 equiv) were reacted to yield 120 mg (100% purity, 30% yield). After chiral separation using SFC (eluent A: CO2; eluent B: ethanol; isokratic: 85% A + 15% B), compound 34b was obtained (25.0 mg, 100% purity, 6% yield). [α]D20 −158.4° (c 1.00, DMSO). LC-MS (method E): Rt = 1.05 min; MS (ESIpos): m/z = 284 [M]+. 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.36–1.48 (m, 2H), 1.48–1.67 (m, 4H), 1.80–1.91 (m, 1H), 1.97–2.07 (m, 1H), 3.80 (d, 1H), 6.03 (s, 1H), 6.53 (d, 1H), 6.74 (d, 2H), 7.43 (d, 2H).
(1R,2R)-2-[(3-Fluorophenyl)amino]-1-(trifluoromethyl)cyclohexanol (34c)
Following general procedure GP1.1, 1-(trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (200 mg, 1.20 mmol, 1.0 equiv) and 3-fluoroaniline (201 mg, 1.81 mmol, 1.5 equiv) were reacted to yield 107 mg (98% purity, 32% yield). After chiral separation by SFC (eluent A: CO2; eluent B: methanol; isocratic: 90% A + 10% B), compound 34c (26.0 mg, 99% purity, 8% yield) was obtained. [α]D20 −107.2° (c 1.00, DMSO). LC-MS (method D): Rt = 1.20 min; MS (ESIpos): m/z = 278 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.34–1.41 (m, 1H), 1.42–1.65 (m, 5H), 1.75–1.85 (m, 1H), 1.94–2.03 (m, 1H), 3.69 (d, 1H), 5.82 (d, 1H), 5.93 (s, 1H), 6.22–6.29 (m, 1H), 6.37–6.42 (m, 1H), 6.44–6.49 (m, 1H), 7.04 (q, 1H).
(1R,2R)-2-[(5-Fluoropyridin-2-yl)amino]-1-(trifluoromethyl)cyclohexanol (34d)
Following general procedure GP1.1, 1-(trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (300 mg, 1.81 mmol, 1.0 equiv) and 2-amino-5-fluoropyridine (304 mg, 2.71 mmol, 1.5 equiv) were reacted to yield 127 mg (100% purity, 25% yield). After chiral separation by SFC (eluent A: CO2; eluent B: methanol; isokratic: 85% A + 15% B), compound 34d (33 mg, 97% purity, 7% yield) was obtained. [α]D20 −101.5° (c 1.00, CHCl3). LC-MS (method E): Rt = 0.98 min; MS (ESIpos): m/z = 279 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.35–1.65 (m, 6H), 1.77–1.87 (m, 1H), 1.91–2.01 (m, 1H), 4.40 (d, 1H), 6.06 (s, 1H), 6.55 (d, 1H), 6.62 (dd, 1H), 7.28–7.35 (m, 1H), 7.88 (d, 1H).
(1R,2R)-2-[(4-Chloro-3-fluorophenyl)amino]-1-(trifluoromethyl)cyclohexanol (34e)
Following general procedure GP1.1, 1-(trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (200 mg, 1.20 mmol, 1.0 equiv) and 4-chloro-3-fluoroaniline (263 mg, 1.81 mmol, 1.5 equiv) were reacted to yield 95.0 mg (100% purity, 25% yield). After chiral separation by SFC (eluent A: CO2; eluent B: methanol; isokratic: 92% A + 8% B), compound 34e (24 mg, 99% purity, 6% yield) was obtained. [α]D20 −108.6° (c 1.00, DMSO). LC-MS (method E): Rt = 1.29 min; MS (ESIpos): m/z = 313 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.37–1.65 (m, 6H), 1.75–1.86 (m, 1H), 1.92–2.01 (m, 1H), 3.67 (d, 1H), 5.96 (s, 1H), 6.02 (d, 1H), 6.50 (dd, 1H), 6.59 (dd, 1H), 7.16 (t, 1H).
(1R,2R)-2-[(3-Chloro-4-fluorophenyl)amino]-1-(trifluoromethyl)cyclohexanol (34f)
Following general procedure GP1.1, 1-(trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (200 mg, 1.20 mmol, 1.0 equiv) and 3-chloro-4-fluoroaniline (263 mg, 1.81 mmol, 1.5 equiv) were reacted to yield 202 mg (100% purity, 54% yield). After chiral separation by SFC (eluent A: CO2; eluent B: ethanol; isokratic: 90% A + 10% B), compound 34f (54.9 mg, 100% purity, 15% yield) was obtained. [α]D20 −80.2° (c 1.00, DMSO). LC-MS (method E): Rt = 1.29 min; MS (ESIpos): m/z = 312 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.33–1.65 (m, 6H), 1.74–1.83 (m, 1H), 1.91–1.99 (m, 1H), 3.64 (d, 1H), 5.70 (d, 1H), 5.94 (s, 1H), 6.59 (dt, 1H), 6.75 (dd, 1H), 7.08 (t, 1H).
(1R,2R)-2-[(3,4-Dichloro-phenyl)amino]-1-(trifluoromethyl)cyclohexanol (34g)
Following general procedure GP1.1, 1-(trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (200 mg, 1.20 mmol, 1.0 equiv) and 3,4-dichloroaniline (293 mg, 1.81 mmol, 1.5 equiv) were reacted to yield 125 mg (96% purity, 31% yield). After chiral separation by SFC (instrument: Sepiatec: Prep SFC100; column: Chiralpak IG 5 μm 250 × 30 mm2; eluent A: CO2, eluent B: ethanol; isocratic: 15% B; flow rate 100.0 mL/min; temperature: 40 °C; BPR: 150 bar; MWD@254 nm), compound 34g (36.0 mg, 99% purity, 9% yield) was obtained. [α]D20 −9.5° (c 1.00, DMSO). LC-MS (method G): Rt = 1.32 min; MS (ESIpos): m/z = 328 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.37–1.64 (m, 6H), 1.77–1.86 (m, 1H), 1.93–1.98 (m, 1H), 3.66–3.69 (m, 1H), 5.96 (s, 1H), 5.98–6.01 (m, 1H), 6.61 (dd, 1H), 6.84–6.85 (m, 1H), 7.22–7.24 (m, 1H).
(1R,2R)-2-[(3-Methoxy-4-fluorophenyl)amino]-1-(trifluoromethyl)cyclohexanol (34h)
Following general procedure GP1.1, 1-(trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (200 mg, 1.20 mmol, 1.0 equiv) and 4-fluoro-3-methoxyaniline (255 mg, 1.81 mmol 1.5 equiv) were reacted to yield 129 mg (96% purity, 33% yield). After chiral separation by HPLC (method NP2), compound 34h (35.0 mg, 100% purity, 9% yield) was obtained. [α]D20 −9.5° (c 1.00, DMSO). LC-MS (method D): Rt = 1.18 min; MS (ESIpos): m/z = 308 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.35–1.37 (m, 1H), 1.46–1.63 (m, 5H), 1.73–1.80 (m, 1H), 1.94–2.01 (m, 1H), 3.63–3.66 (m, 1H), 3.74 (s, 3H), 5.39 (d, 1H), 5.88 (s, 1H), 6.08 (dt, 1H), 6.47 (m, 1H), 6.86 (dd, 1H).
(rac)-2-[(3-Methoxy-4-fluorophenyl)amino]-1-(trifluoromethyl)cyclohexanol (34i)
Following general procedure GP1.1, 1-(trifluoromethyl)-7-oxabicyclo[4.1.0]heptane (200 mg, 1.20 mmol, 1.0 equiv) and 4-amino-2-(trifluoromethyl)benzonitrile (280 mg, 1.50 mmol, 1.5 equiv) were reacted to yield 46 mg (49% purity). After separation by HPLC (method NP3), compound 34i (2.8 mg, 100% purity, 1% yield) was obtained. LC-MS (method G): Rt = 1.15 min; MS (ESIpos): m/z = 353 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ [ppm]: 1.44–1.69 (m, 6H), 1.86–2.03 (m, 2H), 3.87–3.90 (m, 1H), 6.11 (s, 1H), 6.92–6.95 (m, 1H), 7.09 (d, 1H), 7.22 (s, 1H), 7.71 (d, 1H).
TRPA1 Calcium Fluorescence Assay
The potency of compounds inhibiting TRPA1-mediated calcium flux was determined on a FLIPR Tetra instrument using the following recombinant cell lines for human, rat, and mouse TRPA1: CHO K1 hTRPA1 GCaMP6, CHO K1 rTRPA1 GCaMP6, and CHO K1 mTRPA1 GCaMP6.
Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco #41965-039), 10% fetal calf serum (FCS), 14.5 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES), 1 mM sodium pyruvate, and 1× nonessential amino acids (Gibco #11140-035) without selection antibiotics and frozen in 90% culture medium/10% DMSO.
For the assay in 384 MTP format, frozen cells were thawed, resuspended in fresh culture medium supplemented with 2 μg/mL poly-d-lysine (Sigma-Aldrich P6407), and seeded into black 384-well microtiter plates with clear bottom (Greiner #781092) at a density of 6000–8000 cells/well in 30 μL/well. The seeded plates were incubated overnight at 37 °C, 5% CO2.
Prior to the measurement, culture medium was removed and cells were incubated for 30 min at 37 °C in 30 μL/well tyrode buffer (2 mM CaCl2, 130 mM NaCl, 5 mM KCl, 20 mM HEPES, 1 mM MgCl2, 5 mM NaHCO3, pH 7.4, 0.01% bovine serum albumin (BSA) and 200 μg/mL brilliant black).
Half-log serial dilutions of the test compounds were prepared in DMSO starting at a concentration of 10 mM. Before the measurement, the compounds were further diluted 100-fold in tyrode buffer and added to the cells (10 μL/well) resulting in final concentrations between 25 μM and 0.8 nM. The plates were incubated with compounds for 10 min at room temperature.
Plates were placed in the FLIPR, and fluorescence was measured for 3s as a baseline read. The agonist super cinnamaldehyde (Sigma-Aldrich S332255) was added at EC80 (typically 1 μM for hTRPA1, 3 μM for rTRPA1, 0.5 μM for mTRPA1), and the change of calcium fluorescence was measured for 2 min. Dose–response data were fitted using a four-parameter logistic function to calculate IC50 values with proprietary analysis software.
For the HTS assay in 1536 MTP format, cells were seeded from culture at a density of 900 cells/well in 6 μL of medium supplemented with 5% FCS. hTRPA1 was activated using 80 μM cinnamaldehyde (CA) or 100 μM ZnCl2, mTRPA1 using 80 μM CA.
TRPA1 Mutants
TRPA1 mutants were tested using transient transfections. A CHO GCaMP6 cell line was transfected with six different genes in pCDNA3 (wild-type hTRPA1, hTRPA1 T874V, hTRPA1 N855S, hTRPA1 F909T, hTRPA1 M911A, hTRPA1 G238K/N249S/K270N) using jetPRIME (Polyplus Transfection).
Cells were seeded at 5 × 105 cells/well (six-well MTP) and incubated for 24 h at 37 °C/5% CO2. 200 μL/well of transfection mix (1600 μL jetPRIME buffer, 16 μg DNA, and 32 μL jetPRIME) was added, and 7 h later cells were seeded at 4–6k cells/well in 384-well MTPs. For one gene, a 6-well plate yielded two 384-well MTPs (one for agonist EC50 and one for compound IC50 determination). Twenty-four hours later, compounds were tested as described above.
Pharmacology
Animals
Adult male Sprague Dawley rats (RjHan:SD; Janvier Laboratories, Saint-Berthevin Cedex, France) were used in these experiments. Animals were obtained and acclimatized for at least 5 days before the start of a study. Rats were housed in a temperature (22 ± 2 °C)- and humidity (55 ± 5%)-controlled room, maintained in a 12 h light/dark cycle with lights on at 06:00 AM. Rats were housed in groups in transparent polycarbonate cages with clean standard bedding and environmental enrichment (paper towels and/or wood sticks), with ad libitum access to food and water. All animal studies were carried out at Evotec SE (Hamburg, Germany) in strict accordance with Evotec SE policies, AAALAC guidelines, and the European Directive 2010/63/EU as well as the German Animal Welfare Act (Tierschutzgesetz, TierSchG). All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) and competent regional Animal Care and Use Committees according to Section 15 TierSchG (Hamburg, Germany).
Cinnamaldehyde-Induced Nocifensive Behaviors in Rats
BAY-390 was tested in the model of intraplantar cinnamaldehyde (CA)-induced nocifensive pain behaviors in male Sprague Dawley rats (175–200 g at delivery). Briefly, 50 μL of CA at 5 mM was injected at the plantar surface of one hind paw. Immediately after injection, the number and the duration of nocifensive behaviors (including licking or flinching) were quantitated by a blinded experimenter for 5 min. BAY-390 or vehicle (10% DMSO, 40% Solutol, 50% water for injection, vol/vol) was dosed via oral route (p.o.) 1 h before CA injection. Data were expressed as the mean number of flinches, the mean duration of licking behaviors, and the mean duration of nocifensive behaviors for each treatment group. Data were analyzed by performing a one-way analysis of variance (ANOVA). Planned comparison of means (each versus vehicle) was performed using Dunnett’s post hoc test, provided that a main effect was detected. For p values less than 0.05, the results were deemed to be statistically significant.
CFA-Induced Mechanical Hyperalgesia in Rats
The compound was tested in the model of intraplantar complete Freund’s adjuvant (CFA)-induced acute (24 h setting) inflammatory pain in male Sprague Dawley rats (175–200 g at delivery). Briefly, 25 μL of CFA at 1 mg/mL was injected into the plantar surface of one hind paw. Mechanical hyperalgesia was measured using the Pressure Application Measurement apparatus (Ugo Basile, Gemonio, Italy). A linearly increasing pressure was applied to an area of approximately 50 mm2 of the plantar side of the hind paw until a behavioral response (paw withdrawal) was observed or until the pressure reached 1000 grams of force (gf). The pressure at which the behavioral response occurred was recorded as the “paw withdrawal threshold” (PWT). Both CFA-injected and contralateral PWTs were determined for each rat, in each treatment group and at each time point of the studies. The measurements were performed blinded. Mechanical hyperalgesia testing was performed before injecting CFA, 24 h after CFA injection (predrug baseline), 2 and 4 h after treatment. Compound or vehicle (10% DMSO, 40% Solutol, 50% water for injection, vol/vol) was dosed via oral route (p.o.) once 24 h after CFA injection. Data were expressed as the mean PWT for each treatment group and at each time point. PWT data were analyzed by performing a two-way ANOVA with repeated measures (time × treatment). Planned comparison of means (each versus vehicle) was performed by using Dunnett’s post hoc test, provided that a main effect was detected. For p values less than 0.05, the results were deemed to be statistically significant.
Spinal Nerve Ligation (SNL) Model
Spinal nerve ligation surgery was performed as described previously.56 Briefly, male Sprague Dawley rats (100–120 g at the time of surgery) were maintained under 2% (vol/vol) isoflurane anesthesia delivered in a mixture with oxygen. A midline incision was made on the back, and the left paraspinal muscles were separated from the spinous processes at the L4–S2 levels. The left L5 nerve was tightly ligated with a 6–0 silk thread. The left L6 spinal nerve, located just caudal and medial to the sacroiliac junction, was tightly ligated with a silk thread. After suturing the left paraspinal muscles, the surrounding skin was then closed. All rats were monitored for normal behaviors (grooming and mobility) and for general health and weight gain post surgery. To quantify mechanical allodynia, the left (ipsilateral) and right (contralateral) hind paw withdrawal thresholds (WTs) were determined 11 days of surgery (baseline) and at different time points during the 10 day treatment period using von Frey filaments (RFM Medical, Rahoforum Medical GmbH, Germany) and the “up-and-down” method.57 All behavioral tests were performed between 9 AM and 2 PM by an examiner blinded to the treatment groups. Compound BAY-390 or vehicle was dosed orally, twice daily (b.i.d.), for 10 consecutive days from day 15 to day 24 post SNL surgery. Compound BAY-390 was dosed at 30 and 90 mg/kg b.i.d., and vehicle (DMSO/Solutol/water (10/40/50, vol/vol)) was dosed at 5 mL/kg b.i.d. Gabapentin (positive control) was dosed at 30 mg/kg once daily (q.d.). VF behavioral testing was performed before (day 11, post SNL baseline) and during chronic treatment (days 15, 18, 21, and 24 post SNL surgery), 2 h post morning dosing. The data were summarized by calculating the mean (±standard error of the mean (s.e.m.)) paw withdrawal thresholds (expressed as a gram of VF force) measured on the left (ipsilateral) and right (contralateral) legs, for each experimental group and at each time point of the study. The contralateral (control, noninjected) and the ipsilateral sides were compared independently. Since the mechanical threshold represents the perceived intensity of a mechanical stimulus proportional to the logarithm of the stimulus (or VF force) strength, statistical comparisons were performed using nonparametric tests. The presence of mechanical allodynia was verified by comparing mean baseline values after SNL surgery (day 11) between the ipsilateral and the contralateral hind paws, using a paired Wilcoxon test. Effects of BAY-390 and gabapentin were compared with those in the control group (vehicle) at each time point independently, using a one-way ANOVA test (Kruskal–Wallis), followed by Dunn’s multiple comparison test, provided that a main effect (difference between group means/medians) was detected. Differences were considered statistically significant if p values are less than 0.05.
Acknowledgments
The work described herein was performed in the context of the Bayer-Evotec strategic alliance, and contributions of the alliance team are gratefully acknowledged. The authors thank Anh-Thu Nguyen, Michaela Messer, Marc Guttzeit, Timo Weber, Daniel May, Catrin Kroeber (medicinal chemistry Berlin), and Jennifer Hannen (Lead Discovery, Wuppertal) for their technical assistance throughout the whole project. Dr. Stephan Gründemann and Bernd Undeutsch are acknowledged for analytical support. In addition, Antje Rottmann (DMPK, Berlin) is acknowledged for the excellent support of compound metabolism studies. Dr. Jens Schamberger is acknowledged for providing the figure of the mutation binding sites.
Glossary
Abbreviations Used
- EPhys
electrophysiology
- CHO
Chinese hamster ovary cells
- cpd
compound
- cryoEM
cryogenic electron microscopy
- CyH
cyclohexane
- DCM
dichloromethane
- DMAP
4-dimethylaminopyridine
- DMF
dimethylformamide
- DMPU
N,N′-dimethylpropyleneurea
- EtOAc/EA
ethyl acetate
- Et2O
diethyl ether
- EtOH
ethanol
- FLIPR
fluorescent imaging plate reader
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uranium
- LDA
lithium diisopropylamide
- mCPBA
meta-chloroperoxybenzoic acid
- MeOH
methanol
- PE
petroleum ether
- PTSA
p-toluene sulfonic acid
- TBAF
tetra-n-butylammonium fluoride
- TBME
tert-butyl methyl ether
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
- SFC
supercritical fluid chromatography
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01830.
Analytical and purification methods used for characterization and isolation of compounds, LC-MS traces and 1H NMR spectra of all final compounds, vibrational circular dichroism measurement of compound 18, off-target selectivity data of compound 18, in vivo exposure data and brain penetration data of compound 18, and analytical data of all intermediates (PDF)
Molecular formula strings (CSV)
Author Present Address
◆ Preclinical Compound Profiling, Nuvisan ICB GmbH, Müllerstr. 178, 13353 Berlin, Germany
The authors declare no competing financial interest.
Supplementary Material
References
- Chen J.; Hackos D. H. TRPA1 as a drug target--promise and challenges. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2015, 388, 451–463. 10.1007/s00210-015-1088-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerratt S. Recent Progress in the Discovery and Development of TRPA1 Modulators. Prog. Med. Chem. 2017, 56, 81–115. 10.1016/bs.pmch.2016.11.003. [DOI] [PubMed] [Google Scholar]
- Jaquemar D.; Schenker T.; Trueb B. An ankyrin-like protein with transmembrane domains is specifically lost after oncogenic transformation of human fibroblasts. J. Biol. Chem. 1999, 274, 7325–7333. 10.1074/jbc.274.11.7325. [DOI] [PubMed] [Google Scholar]
- Viana F. TRPA1 channels: molecular sentinels of cellular stress and tissue damage. J. Physiol. 2016, 594, 4151–4169. 10.1113/JP270935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes E. S.; Fernandes M. A.; Keeble J. E. The functions of TRPA1 and TRPV1: moving away from sensory nerves. Br. J. Pharmacol. 2012, 166, 510–521. 10.1111/j.1476-5381.2012.01851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sághy É.; Sipos É.; Ács P.; et al. TRPA1 deficiency is protective in cuprizone-induced demyelination-A new target against oligodendrocyte apoptosis. Glia 2016, 64, 2166–2180. 10.1002/glia.23051. [DOI] [PubMed] [Google Scholar]
- Shigetomi E.; Jackson-Weaver O.; Huckstepp R. T.; et al. TRPA1 channels are regulators of astrocyte basal calcium levels and long-term potentiation via constitutive D-serine release. J. Neurosci. 2013, 33, 10143–10153. 10.1523/JNEUROSCI.5779-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradpezhouh E.; Choy J. M. C.; Daria V. R.; et al. TRPA1 expression and its functional activation in rodent cortex. Open Biol. 2017, 7, 160314 10.1098/rsob.160314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K.; et al. Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J. Neurosci. 2005, 25, 4052–4061. 10.1523/JNEUROSCI.0013-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nummenmaa E.; Hämäläinen M.; Moilanen L. J.; et al. Transient receptor potential ankyrin 1 (TRPA1) is functionally expressed in primary human osteoarthritic chondrocytes. Arthritis Res. Ther. 2016, 18, 185. 10.1186/s13075-016-1080-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N.; Chen H. Y.; Harris I. S.; et al. Cancer Cells Co-opt the Neuronal Redox-Sensing Channel TRPA1 to Promote Oxidative-Stress Tolerance. Cancer Cell 2018, 33, 985.e7–1003.e7. 10.1016/j.ccell.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade E. L.; Meotti F. C.; Calixto J. B. TRPA1 antagonists as potential analgesic drugs. Pharmacol. Ther. 2012, 133, 189–204. 10.1016/j.pharmthera.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Koivisto A.-P.; Belvisi M. G.; Gaudet R.; et al. Advances in TRP channel drug discovery: from target validation to clinical studies. Nat. Rev. Drug Discovery 2022, 21, 41–59. 10.1038/s41573-021-00268-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivisto A.; Jalava N.; Bratty R.; et al. TRPA1 Antagonists for Pain Relief. Pharmaceuticals 2018, 11, 117 10.3390/ph11040117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza Monteiro de Araujo D.; Nassini R.; Geppetti P.; et al. TRPA1 as a therapeutic target for nociceptive pain. Expert Opin. Ther. Targets 2020, 24, 997–1008. 10.1080/14728222.2020.1815191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremeyer B.; Lopera F.; Cox J. J.; et al. A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron 2010, 66, 671–680. 10.1016/j.neuron.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khairatkar J. N.et al. TRPA1 Antagonist for the Treatment of Pain Associated to Diabetic Neuropathic Pain. WO2016042501 A1, 2016.
- Balestrini A.; Joseph V.; Dourado M.; et al. A TRPA1 inhibitor suppresses neurogenic inflammation and airway contraction for asthma treatment. J. Exp. Med. 2021, 218, e20201637 10.1084/jem.20201637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P.; Ding H. T.; Liederer B. M.; et al. Translational and pharmacokinetic-pharmacodynamic application for the clinical development of GDC-0334, a novel TRPA1 inhibitor. Clin. Transl. Sci. 2021, 14, 1945–1954. 10.1111/cts.13049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippa B. S.et al. Preparation of Purine Derivatives Inhibiting the Transient Receptor Potential A1 Ion Channel. WO2015164643 A1, 2015.
- Kadam S. M.et al. Preparation of Amides of 2-Amino-4-arylthiazole Compounds and Their Salts. WO2013183035 A2, 2013.
- Kumar S.et al. Preparation of Thieno-pyrimidinedione Derivatives as TRPA1 Modulators. WO2010109334 A2, 2010.
- Verma V.; Volgraf M.; Hu B.. Preparation of Sulfonyl Pyridyl Compounds as TRP Inhibitors. WO2018029288 A1, 2018.
- Perner R. J.et al. Method for Treating Disorders or Conditions Ameliorated by Trpa1 Antagonists. US20090176883 A1, 2009.
- Chen J.; Joshi S. K.; DiDomenico S.; et al. Selective blockade of TRPA1 channel attenuates pathological pain without altering noxious cold sensation or body temperature regulation. Pain 2011, 152, 1165–1172. 10.1016/j.pain.2011.01.049. [DOI] [PubMed] [Google Scholar]
- Rooney L.; Vidal A.; D’Souza A. M.; et al. Discovery, optimization, and biological evaluation of 5-(2-(trifluoromethyl)phenyl)indazoles as a novel class of transient receptor potential A1 (TRPA1) antagonists. J. Med. Chem. 2014, 57, 5129–5140. 10.1021/jm401986p. [DOI] [PubMed] [Google Scholar]
- Svensson M.; Weigelt D.. Trpa1 receptor antagonist. US8,859,556 B2, 2014.
- Schenkel L. B.; Olivieri P. R.; Boezio A. A.; et al. Optimization of a Novel Quinazolinone-Based Series of Transient Receptor Potential A1 (TRPA1) Antagonists Demonstrating Potent in Vivo Activity. J. Med. Chem. 2016, 59, 2794–2809. 10.1021/acs.jmedchem.6b00039. [DOI] [PubMed] [Google Scholar]
- Terrett J. A.; Chen H.; Shore D. G.; et al. Tetrahydrofuran-Based Transient Receptor Potential Ankyrin 1 (TRPA1) Antagonists: Ligand-Based Discovery, Activity in a Rodent Asthma Model, and Mechanism-of-Action via Cryogenic Electron Microscopy. J. Med. Chem. 2021, 64, 3843–3869. 10.1021/acs.jmedchem.0c02023. [DOI] [PubMed] [Google Scholar]
- Chen H.et al. Oxadiazoles as Transient Receptor Potential Channel Inhibitors and Their Preparation. WO2018162607 A1, 2018.
- Brotherton-Pleiss C. E.et al. Substituted Sulfonamide Compounds. WO2014049047A1, 2014.
- Arvela R.et al. Preparation of N-Prop-2-ynyl Carboxamide Derivatives and Their Use as TRPA1 Antagonists. WO2014053694 A1, 2014.
- Bilodeau M. T.et al. Novel TRPA1 Antagonists for Treatment of Pain and Other TRPA1-Associated Diseases. WO2011043954 A1, 2011.
- Fruttarolo F.et al. Preparation of Sulfonamidopyridylmethylpropenamide Derivatives and Analogs for Use as TRPA1 Receptor Antagonists. WO2014135617 A1, 2014.
- Klionsky L.; Tamir R.; Gao B.; et al. Species-specific pharmacology of Trichloro(sulfanyl)ethyl benzamides as transient receptor potential ankyrin 1 (TRPA1) antagonists. Mol Pain 2007, 3, 1744. 10.1186/1744-8069-3-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland K. W.; Boezio A. A.; Cheung E.; et al. Development of novel azabenzofuran TRPA1 antagonists as in vivo tools. Bioorg. Med. Chem. Lett. 2014, 24, 3464–3468. 10.1016/j.bmcl.2014.05.069. [DOI] [PubMed] [Google Scholar]
- Chen H.; Terrett J. A. Transient receptor potential ankyrin 1 (TRPA1) antagonists: a patent review (2015-2019). Expert Opin. Ther. Pat. 2020, 30, 643–657. 10.1080/13543776.2020.1797679. [DOI] [PubMed] [Google Scholar]
- Bianchi B. R.; Zhang X. F.; Reilly R. M.; et al. Species comparison and pharmacological characterization of human, monkey, rat, and mouse TRPA1 channels. J. Pharmacol. Exp. Ther. 2012, 341, 360–368. 10.1124/jpet.111.189902. [DOI] [PubMed] [Google Scholar]
- Nyman E.; et al. In vitro pharmacological characterization of a novel TRPA1 antagonist and proof of mechanism in a human dental pulp model. J. Pain Res. 2013, 6, 59–70. 10.2147/JPR.S37567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckmans T.; Aubdool A. A.; Bodkin J. V.; et al. Design and pharmacological evaluation of PF-4840154, a non-electrophilic reference agonist of the TrpA1 channel. Bioorg. Med. Chem. Lett. 2011, 21, 4857–4859. 10.1016/j.bmcl.2011.06.035. [DOI] [PubMed] [Google Scholar]
- Hu H.; Bandell M.; Petrus M. J.; et al. Zinc activates damage-sensing TRPA1 ion channels. Nat. Chem. Biol. 2009, 5, 183–190. 10.1038/nchembio.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R.; Saito S.; Mori Y.; et al. Structural basis of TRPA1 inhibition by HC-030031 utilizing species-specific differences. Sci. Rep. 2016, 6, 37460 10.1038/srep37460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis A.; Carey R. J. S.. Organische Chemie; Wiley-VCH: New York, 1995; Chapter 8. [Google Scholar]
- Moldenhauer H.; Latorre R.; Grandl J. The pore-domain of TRPA1 mediates the inhibitory effect of the antagonist 6-methyl-5-(2-(trifluoromethyl)phenyl)-1H-indazole. PLoS One 2014, 9, e106776 10.1371/journal.pone.0106776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.; Lin King J. V.; Paulsen C. E.; et al. Irritant-evoked activation and calcium modulation of the TRPA1 receptor. Nature 2020, 585, 141–145. 10.1038/s41586-020-2480-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen C. E.; Armache J. P.; Gao Y.; et al. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 2015, 520, 511–517. 10.1038/nature14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuka K.; Gupta R.; Saito S.; et al. Identification of molecular determinants for a potent mammalian TRPA1 antagonist by utilizing species differences. J. Mol. Neurosci. 2013, 51, 754–762. 10.1007/s12031-013-0060-2. [DOI] [PubMed] [Google Scholar]
- Klement G.; Eisele L.; Malinowsky D.; et al. Characterization of a ligand binding site in the human transient receptor potential ankyrin 1 pore. Biophys. J. 2013, 104, 798–806. 10.1016/j.bpj.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng W. C.; Pryde D. C.; Yoger K. E.; et al. TRPA1 ankyrin repeat six interacts with a small molecule inhibitor chemotype. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 12301–12306. 10.1073/pnas.1808142115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suo Y.; Wang Z.; Zubcevic L.; et al. Structural Insights into Electrophile Irritant Sensing by the Human TRPA1 Channel. Neuron 2020, 105, 882.e5–894.e5. 10.1016/j.neuron.2019.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Reese R.; Vu S.; et al. A Non-covalent Ligand Reveals Biased Agonism of the TRPA1 Ion Channel. Neuron 2021, 109, 273.e4–284.e4. 10.1016/j.neuron.2020.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C.; Millan M. J.; Herz A. Unilateral inflammation of the hindpaw in rats as a model of prolonged noxious stimulation: alterations in behavior and nociceptive thresholds. Pharmacol., Biochem. Behav. 1988, 31, 445–451. 10.1016/0091-3057(88)90372-3. [DOI] [PubMed] [Google Scholar]
- Fehrenbacher J. C.; Vasko M. R.; Duarte D. B. Models of inflammation: Carrageenan- or complete Freund’s Adjuvant (CFA)-induced edema and hypersensitivity in the rat. Curr. Protoc. Pharmacol. 2012, 56, 5.4.1. 10.1002/0471141755.ph0504s56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyatake Ondozabal H.et al. TRPA1 Antagonists for the Treatment of Diseases Associated with Pain and Inflammation. WO2021233752 A1, 2021.
- Macpherson L. J.; Dubin A. E.; Evans M. J.; et al. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 2007, 445, 541–545. 10.1038/nature05544. [DOI] [PubMed] [Google Scholar]
- Ho Kim S.; Mo Chung J. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 1992, 50, 355–363. 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Chaplan S. R.; Bach F. W.; Pogrel J. W.; Chung J. M.; Yaksh T. L. Quantitative Assessment of Tactile Allodynia in the Rat Paw. J. Neurosci. Methods 1994, 55–63. 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.