

Abstract

We report [1,2,4]triazolo[3,4-b]benzothiazole (TBT) as a new inhibitor scaffold, which competes with nicotinamide in the binding pocket of human poly- and mono-ADP-ribosylating enzymes. The binding mode was studied through analogues and cocrystal structures with TNKS2, PARP2, PARP14, and PARP15. Based on the substitution pattern, we were able to identify 3-amino derivatives 21 (OUL243) and 27 (OUL232) as inhibitors of mono-ARTs PARP7, PARP10, PARP11, PARP12, PARP14, and PARP15 at nM potencies, with 27 being the most potent PARP10 inhibitor described to date (IC50 of 7.8 nM) and the first PARP12 inhibitor ever reported. On the contrary, hydroxy derivative 16 (OUL245) inhibits poly-ARTs with a selectivity toward PARP2. The scaffold does not possess inherent cell toxicity, and the inhibitors can enter cells and engage with the target protein. This, together with favorable ADME properties, demonstrates the potential of TBT scaffold for future drug development efforts toward selective inhibitors against specific enzymes.
Introduction
ADP-ribosylation is a post-translational modification found in bacteria and eukaryotes, and it is also associated with viral and bacterial infections. The human diphtheria toxin-like ARTD family contains PARP and tankyrase (TNKS) enzymes that can catalyze both mono-ADP-ribosylation (MAR, mono-ARTs) as well as generate elongated and branched chains of poly-ADP-ribose (PAR, poly-ARTs).1 The PARPs and TNKSs form a family of structurally and functionally diverse enzymes, which are involved in the regulation of various key biological and pathological processes such as DNA repair, cell differentiation, gene transcription, and signal transduction pathways.2−4 PARPs and TNKSs use nicotinamide adenine dinucleotide, NAD+, to transfer an ADP-ribose (ADPr) unit onto target proteins or nucleic acids with the release of nicotinamide. The transfer of ADPr in proteins occurs onto amino acid side chains with a nucleophilic oxygen, nitrogen, or sulfur resulting in O-, N-, or S-glycosidic linkage to the ADPr. This can be further extended to PAR by poly-ARTs PARP1–2 and TNKS1–2.5,6 Poly-ARTs contain a triad of amino acids H–Y–E in their active sites. H–Y are important for binding the NAD+, while E stabilizes the oxocarbenium ion transition state and enables the elongation of the ADPr chain by activating the ribose 2′-hydroxyl group.7 However, the H–Y–E motif is not an absolute indicator determining the PARylation activity as there are two enzymes, PARP3 and PARP4, having the H–Y–E motif but appear unable to produce PAR chains.8,9
Over the past decades, PARPs have emerged as drug targets due to their roles in critical cellular processes.10 Especially, the discovery of synthetic lethality of PARP1 inhibition in the context of BRCA-deficient cancers11,12 boosted inhibitor development and led to the first approved drug, olaparib, in 2014. Other PARP1/2 inhibitors, including rucaparib, niraparib, and talazoparib, have also entered clinical applications for the treatment of ovarian and breast cancers deficient in homologous recombination-mediated DNA double-strand break repair.13−15 TNKSs have also emerged as promising drug targets especially due to their role in Wnt/β-catenin signaling,16−18 with two compounds, E7449 and STP1002 (structure undisclosed), having proceeded into clinical studies along with other promising compounds, such as OM-153,19 which is in advanced preclinical testing (Figure 1). The patent literature on poly-ART inhibitors has been expanding and also recently reviewed.20,21
Figure 1.

Examples of the most potent PARP inhibitors.
The majority of mono-ARTs (PARP6–16) have a different active site triad, H–Y−Φ, where Φ represents a hydrophobic amino acid, such as isoleucine, leucine, or tyrosine. Especially the lack of glutamate has been linked to the activity being limited to MARylation. Notably, PARP13 is thought to be inactive22 and PARP9 is modulating ADP-ribosylation activity of the E3 ubiquitin ligase DTX3L.23−25 Although understudied until recently, roles of mono-ARTs in controlling signaling events in cells, along with the recent discovery of their implications in many diseases, make them new possible drug targets. This has resulted in the interest in developing small-molecule inhibitors, precious research tools, which can be used in parallel with biochemical methods to validate the cellular functions of these enzymes.
PARP10 was the first enzyme of the family described as a mono-ART,26 and its inhibitor, OUL35 (Figure 1) described earlier by us,27 could rescue HeLa cells from PARP10-induced cell death and sensitize HeLa cells to DNA damage in agreement with knockdown studies.28,29 Also, patient cells deficient in PARP10 were shown to be sensitized to DNA damage.30 Subsequently, multiple studies have reported additional PARP10 inhibitors,31−35 such as OUL238,35 which is one of the most potent compounds (Figure 1).
In addition to PARP10, inhibitors have also been developed for other mono-ARTs. PARP14 mediates gene transcription through its MARylation activity, and it has been implied as a possible therapeutic target, for example, in lymphoma, myeloma, hepatocellular carcinoma, and in prostate cancer.36−38 At the moment, the most advanced inhibitor for PARP14 is RBN-012759 (Figure 1), disclosed by Ribon Therapeutics, with an IC50 of 3 nM.39 Ribon Therapeutics has also developed RBN-2397, a PARP7 selective inhibitor with <3 nM IC50, demonstrating antitumor effects in xenografts and currently progressed to phase I clinical trials.40 Also, AstraZeneca has contributed to the inhibitor development against mono-ART by describing a potent PARP6 inhibitor, AZ0108 (Figure 1), which prevents ADP-ribosylation of Chk1 that subsequently contributes to antitumor effects in breast cancer mouse models.41 ITK7 has been reported as a selective nM inhibitor of PARP11,42 and recently, the first inhibitors for PARP15 have been described, although the cellular role of this enzyme is not yet well elucidated.43,44 The efforts summarized above were recently reviewed, documenting the high and increasing interest in the development of mono-ART inhibitors.45
Here, we report our contribution in identifying a set of compounds based on a new nicotinamide mimicking chemotype, [1,2,4]triazolo[3,4-b]benzothiazole (TBT), able to inhibit different PARP family enzymes with submicromolar activity depending on the substitution pattern around the central core. The shortlisted compounds were profiled against most of the active human PARP enzymes leading to the most potent inhibitors for PARP10 described to date, with the best compound reaching 7.8 nM IC50 in an enzymatic assay. The binding mode of the TBT scaffold was studied through the synthesis of analogues and their complex crystal structures with PARP2, TNKS2, PARP14, and PARP15. We demonstrate that the compounds enter the cells and engage with the target proteins, with the most potent compound showing 150 nM EC50 value, and the scaffold does not possess inherent cell toxicity. In addition, in vitro ADME studies show that the compounds have good solubility in the 50–150 μM range, are extremely stable in polar solvents and human plasma, and are not susceptible to first pass metabolism by enzymes of the human microsomes. The TBT scaffold therefore forms a basis for drug development efforts toward multiple enzymes of the family.
Results
Biochemical Analysis and Structural Studies of OUL40 (1) and Analogue Design
We previously screened a compound library from the open chemical repository of the National Cancer Institute, which led to the identification of the potent and selective PARP10 inhibitor OUL35.27 From the same screening, a TBT derivative OUL40 (NSC295701) (1) (Figure 2A) emerged, showing an IC50 = 3.2 μM against PARP10. This led us to hypothesize that 1 could be a new nicotinamide mimicking compound with the potential to inhibit multiple PARPs. Indeed, when assayed against a panel of two poly-ARTs, PARP2 and TNKS2, and two additional mono-ARTs, PARP14 and PARP15, IC50 values in the low micromolar range (1.2–5.3 μM) were obtained (Table 1). With X-ray crystallography, we confirmed the binding of 1 into the nicotinamide binding pocket of TNKS2, PARP14, and PARP15 active sites. The inhibitor binding mode is highly similar in all three enzymes, where two hydrogen bonds are generated between N1 and N2 of the triazole ring with glycine and serine residues, respectively. In addition, π–π interactions are present between the inhibitor core and tyrosine residues (Figure 2B–D).
Figure 2.

New nicotinamide mimicking compound. (A) Structural formula of compound 1. (B) TNKS2 (PDB ID 7R3Z), (C) PARP14 (PDB ID 7R3L), and (D) PARP15 (PDB ID 7R3O) crystal structures in complex with 1. TNKS2, PARP14, and PARP15 are colored in blue, brown, and gray, respectively. 1 is presented as a ball-and-stick model and colored in magenta. The hydrogen bonds are indicated with black dashes. The ligand-omitted sigma A weighted Fo – Fc electron density maps are colored in gray and contoured at 3.0 σ.
Table 1. Activity of 1 and the Initial Analoguesa,b.

IC50 (pIC50 ± SEM) values (μM) and PDB IDs are reported.
*Purchased from NCI DTP repository #Purchased from Specs. >denotes less than 50% inhibition in the reported highest concentration. Compound 1 showed IC50 of 1.2 μM against PARP14.
Our preliminary studies on 1 revealed the potency of the compound scaffold by showing reasonable inhibition even in the absence of the typical benzamide moiety. Importantly, no PARP inhibitor based on this tricyclic scaffold has been reported until now. As it offered many options for substitutions, we were encouraged to study this scaffold in more detail.
At first, we were interested to test whether small modifications of the scaffold would lead to any significant changes in selectivity toward mono- or poly-ARTs or to specific inhibition of individual ARTs within the respective subfamily. Iterative medicinal chemistry cycles were performed with a first set of compounds that emerged by working on the benzene ring, where the methyl group of 1 was deleted (2), moved from C-7 to C-6 or C-8 positions (3 and 4), or replaced by bulkier groups as in compounds 5–7. Disubstituted derivatives 8 and 9 were also contemplated (Table 1). Within further derivatives, monomethoxy (10–13), dimethoxy (14 and 15), monohydroxy (16 and 17), and dihydroxy (18) groups decorate the benzene ring (Table 2).
Table 2. Activity of Methoxy- and Hydroxy-Substituted Analoguesa,b.

IC50 (pIC50 ± SEM) values (μM) and PDB IDs are reported.
>Denotes less than 50% inhibition in the reported highest concentration.
Subsequent biochemical and structural analyses described later suggested that the C-3 functionalization of the triazole ring could improve selectivity. Indeed, additional compounds were prepared by placing a heteroatom, oxygen, sulfur, or nitrogen in this position, which was also derivatized while maintaining a 7-methyl (19–25) or a 5,8-dimethoxy (26–31) substitution pattern in the benzene ring (Table 3).
Table 3. Activity of C-3 Substituted Analoguesa,b.

IC50 (pIC50 ± SEM) values (μM) and PDB IDs are reported.
>Denotes less than 50% inhibition in the reported highest concentration, and ≫ denotes no inhibition at the reported concentration; *value limited by the protein concentration used.
Chemistry
All compounds, with the exception of derivatives 1 and 8 that are commercially available, were synthesized as shown in Schemes 1 and 2. In particular, as depicted in Scheme 1, TBT target compounds variously functionalized on the benzene ring were prepared from the key 2-hydrazinobenzothiazole intermediates 33, 64–73, and 80–82, obtained through three different synthetic pathways.
Scheme 1.

Reagents and conditions: (i) hydrazine hydrate, EtOH, 80 °C, overnight (98%); (ii) formic acid, reflux, 7–48 h (10–50%); (iii) NH4SCN, H2O, 12 N HCl, reflux, 6–48 h (15–45%); (iv) Br2, CHCl3, r.t., 2–8 h (30–77%); (v) hydrazine hydrate, CH3COOH, ethylene glycol, 125 °C, 7–48 h (32–88%); (vi) BBr3, dry CH2Cl2, r.t., 3 h (10–22%); (vii) potassium ethyl xanthogenate, dry DMF, 110 °C; 3 h (50–94%).
Scheme 2.
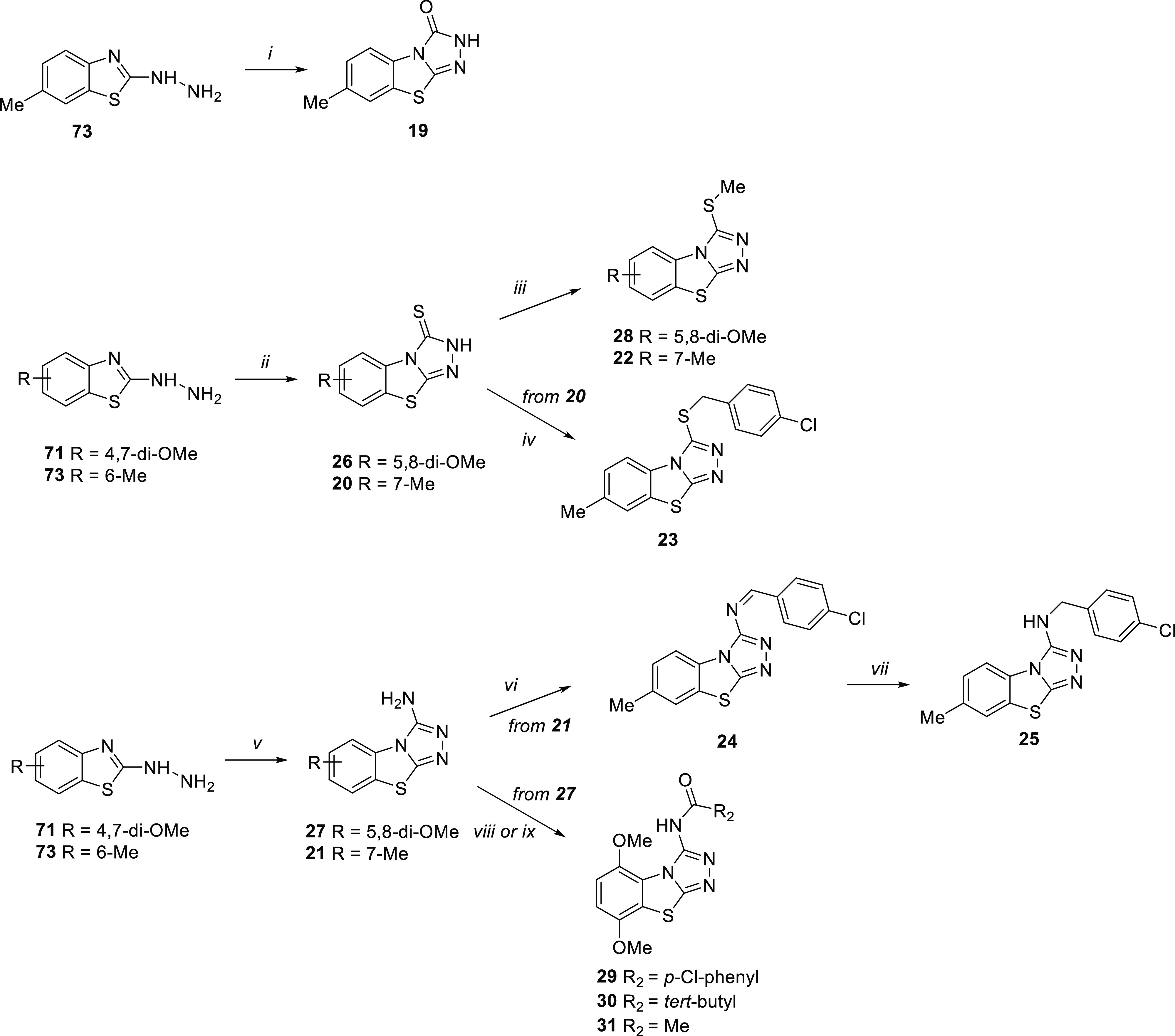
Reagents and conditions: (i) urea, neat, fusion, 3 h (10%); (ii) CS2, KOH, EtOH, reflux, 2 h (22–33%); (iii) MeI, K2CO3, dry DMF, 80 °C, 2 h (15–22%); (iv) p-chlorobenzyl chloride, EtOH, reflux, 4 h (27%); (v) CNBr, MeOH, reflux, 3 h (15–27%); (vi) p-chlorobenzaldehyde, p-TsOH, dry benzene, reflux, 16 h (10%); (vii) NaBH4, absolute EtOH, r.t., overnight (17%); (viii) p-chlorobenzyl chloride, Et3N, dry DMF, 80 °C, 2 h (10%); (ix) trimethylacetyl chloride or acetyl chloride Et3N, dry toluene, 110 °C, 12 h (20–22%).
The unsubstituted hydrazinobenzothiazole 33 was obtained starting from 2-chlorobenzothiazole 32 by reaction with hydrazine hydrate in EtOH with a conversion of 98%. Most of the 2-hydrazinobenzothiazoles (64–73) were instead synthesized starting from the properly substituted anilines 34–43, which were converted into the corresponding arylthiourea derivatives 44–53 by reaction with NH4SCN in acidic solution at reflux. The successive oxidative cyclization of 44–53 using Br2 gave the corresponding 2-aminobenzothiazoles 54–63, which were then treated with hydrazine hydrate to give 64–73. On the other hand, 2-hydrazinobenzothiazoles 80–82 were prepared from 2-mercaptobenzothiazoles 77–79, which in turn was obtained through double nucleophilic substitution of properly substituted 2-fluoroanilines 74–76 with potassium ethyl xanthogenate in dry DMF.
The successive reaction of 2-hydrazinobenzothiazoles with refluxed formic acid in excess led to the synthesis of tricyclic compounds: 2 starting from 33; 3, 5, 6, 9–11, and 13–15 from 64–72; and 4, 7, and 12 from 80–82.
The methoxy derivatives 11, 12, and 14 were further elaborated into the corresponding hydroxyl derivatives 16–18 by using BBr3.
As reported in Scheme 2, TBT variously functionalized at the C-3 position 19–31 were synthesized from 4,7-dimethoxy and 6-methyl hydrazine intermediates 71 and 73. By treating 73 with urea in neat condition and at the fusion temperature (133 °C), benzothiazol-3-one 19 was obtained.
Starting from both 71 and 73 and using CS2 in EtOH, benzothiazole-3-thiones 26 and 20 were obtained, respectively. S-alkylation of 26 and 20 with MeI in the presence of K2CO3 in dry DMF gave the corresponding derivatives 28 and 22. Compound 20 was also S-alkylated by reaction with p-chlorobenzyl chloride in EtOH to give compound 23.
Finally, the reaction of 71 and 73 with CNBr furnished 3-aminobenzothiazole derivatives 27 and 21. 3-Amino-7-methyl derivative 21 was condensed with p-chlorobenzaldehyde to give imine derivative 24, which was then reduced to amine derivative 25 with NaBH4. Amidation of derivative 27 with p-chlorobenzoyl chloride in the presence of Et3N in dry DMF gave compound 29, while the reaction of 27 with trimethylacetyl chloride or acetyl chloride in the presence of Et3N in dry toluene yielded compounds 30 and 31, respectively.
OUL40 (1) Analogues: Biochemical Analysis and Structural Studies
All synthesized TBTs were initially tested against representative members of the PARP family: two poly-ARTs, PARP2 and TNKS2, and two mono-ARTs, PARP10 and PARP15. The latter were selected based on the availability of a robust cell-based readout for PARP10 engagement and for a similarly robust crystal system of PARP15 to study compound binding modes experimentally. In addition, all analogues were also routinely tested for toxicity using a colorimetric WST-1 assay, which only identified isopropyl derivative 6 and dihydroxy derivative 18 as being toxic in a dose-dependent manner (Figure S1).
We first tested the effect of small alkyl groups and halogens on the benzene ring (Table 1). The only commercially available compound 8 had an additional methyl substituent, and it showed improved potency against all tested PARPs. The removal of the methyl group on the other hand reduced the potency of 2, especially against PARP10 and PARP15 (IC50 > 10 μM), indicating that a hydrophobic substituent would be important for potency toward mono-ARTs (Table 1). Shifting of the methyl group to other positions did not have major effects on the potency (3, 4, and 9), and all compounds maintained μM potencies for the tested enzymes. Compound 3 having the methyl in the C-6 position, however, showed higher potency against PARP2 and TNKS2. The TNKS2 crystal structure in complex with 3 revealed that 6-methyl pushed Tyr1050 to a different conformation and provided the additional interaction explaining the poly-ART selectivity (Figure S2A,B). When C-7 methyl found in 1 was extended to a larger alkyl (5 and 6), no improvements in inhibition potency were observed, while the C-7 chlorine derivative 7 maintained a micromolar activity only against PARP2 and PARP15. The minor modifications of the C-6 substituent did not result in significant structural changes as observed from the PARP15 crystal structures in complex with 6, 7, and 8, which showed highly similar binding modes to 1 (Figure S2C–E).
Next, we tested the effects of hydroxy and methoxy groups placed in various positions of the benzene ring of the TBT scaffold (Table 2). Interestingly, the presence of a C-7 hydroxy group made 16 very potent and specific for poly-ARTs with nearly 10-fold selectivity for PARP2 over TNKS2 (IC50 of 44 vs 370 nM). Based on the comparison of complex structures of PARP2 and PARP15 (Figure 3), 16 has a similar binding mode as 1, but the catalytic residue of PARP2 (Glu558), not present in PARP15, interacts with the hydroxyl group of 16 (Figure 3A). The hydroxyl group also interacts with the Met456 backbone amide via a water molecule. In contrast, in PARP15, the hydroxyl group interacts with the carbonyl of Ala583 causing a compound orientation that brings the sulfur atom of 16 in close contact with the side chain of Tyr598, which has therefore changed its conformation compared to the binding mode of 1 (Figures 2D and 3B). In addition, the ligand-omitted Fo – Fc electron density map is not well-defined, indicating flexibility in the binding mode of 16 (Figure 3B).
Figure 3.

Binding mode of 16 between poly-ART and mono-ART showed by the (A) PARP2 (PDB ID 7R59) and (B) PARP15 (PDB ID 7Z1Y) complex crystal structures. The ligand is presented as a ball-and-stick model and colored in yellow. The hydrogen bonds are indicated with black dashes. The ligand-omitted sigma A weighted Fo – Fc electron density maps are colored in gray and contoured at 3.0 σ.
By shifting the hydroxy group from the C-7 to C-8 position, a very different profile was shown by compound 17 that maintained a weak activity only against PARP15. In contrast to the hydroxy of 16, the replacement of the methyl group of 1 with a methoxy gave compound 11 endowed with a similar profile and a similar binding mode (Figure S2F). Dimethoxy derivative 14 showed submicromolar potency against PARP10 (IC50 = 490 nM), and the corresponding di-hydroxy analogue 18 expanded the nanomolar potency also against PARP15 and PARP2. This did not provide us the poly-ART versus mono-ART selectivity, but the presence of an 8-methoxy group made 12 selective against mono-ARTs PARP10 and PARP15. The PARP15 crystal structure in complex with 14 (Figure 4A) revealed plasticity in the compound orientation compared to 1. The small rotation was observed by comparing the crystal structures (Figure 4B), which also revealed conformational changes in the side chains of Leu659 and Tyr598. An even more dramatic change was observed in the complex structure with 5-methoxy derivative 13, which showed a 180° horizontal flip of the compound in comparison to 5,8-dimethoxy 14 (Figure 4A,C).
Figure 4.

Compound rotation in the PARP active site. (A) PARP15 crystal structure in complex with 14 (PDB ID 7Z41). The ligand is presented as a ball-and-stick model and colored in brown. (B) Superimposition of the PARP15 complex structures of 1 (magenta) and 14 (brown) (PDB IDs 7R3O and 7Z41, respectively). Residues having conformational changes are colored with respective colors regarding the ligands. (C) PARP15 crystal structure in complex with 13 (PDB ID 7Z2O) and (D) with 27 (PDB ID 7Z2Q). The ligands are presented as a ball-and-stick model and colored in light blue and purple. The hydrogen bonds are indicated with black dashes. The ligand-omitted sigma A weighted Fo – Fc electron density maps are colored in gray and contoured at 3.0 σ.
We hypothesized that the plasticity would allow compounds such as 13 and 14 to inhibit multiple PARPs as the compound activities were still at a micromolar level against PARP2 and TNKS2 (Table 2). Therefore, we decided to add an anchor point to the C-3 position in order to fix the compound orientation in the binding pocket. A similar strategy was previously successfully used in the development of TNKS inhibitors.46 We tested multiple substituents at the C-3 position while preserving the C-7 methyl of compound 1 or the 5,8-dimethoxy of compound 14. Compounds having an oxygen (19) or small sulfur groups (20 and 22) integrated to the scaffold 1 emerged as selective against PARP10 with micromolar activities. A more interesting compound was achieved when using an amino group as a C-3 substituent that resulted in 21 showing nanomolar activity against PARP10 (IC50 = 180 nM) and PARP15 (IC50 = 300 nM) with a clear selectivity toward the mono-ARTs over the poly-ARTs PARP2 and TNKS2, which were inhibited only with micromolar potencies (IC50 = 1.6 and 5.7 μM, respectively).
To potentially improve the selectivity, we extended the thiol and amino groups with a longer substituent but, independently of the heteroatom, this caused a loss of activity (23–25); however, imine derivative 24 showed some activity against PARP2 (IC50 = 2.9 μM) and PARP15 (IC50 = 7.9 μM) when compared to the more flexible compound 25 (Table 3). Regarding the C-3 substituted dimethoxy analogues, the presence of a thiol group determined a loss of activity for compound 26, while 28 having a thiomethyl group recovered a modest selectivity against PARP2 (IC50 = 8.4 μM). The presence of a 3-amino group emerged as particularly suitable to improve the potency against PARP10 and PARP15, with IC50 ranging from 120 to 300 nM, with dimethoxy derivative 27 that also stood out as selective for MARylating enzymes (Table 3). The PARP15 crystal structure in complex with 27 (Figure 4D) showed a highly similar binding mode to the analogue 14 (Figure 3A). However, the amino group of 27 creates a hydrogen bond with Gly560 which together with the activity profiles (1 vs 21 and 14 vs 27) indicate that the anchor in C-3 is crucial for gaining selectivity against mono-ARTs. To improve the selectivity even more, we extended the anchor this time by preparing amide derivatives 29–31, but in the presence of either longer or shorter substituents, only very weak activity was observed against some enzymes (Table 3).
PARP Profiling and Biological Evaluation
7-Hydroxy derivative 16 (OUL245) and 3-amino derivatives 21 (OUL243) and 27 (OUL232) emerged as the most interesting compounds of the work as they were both potent and showed selectivity toward either mono- or poly-ARTs. We therefore decided to profile them against a large panel of enzymatically active PARPs (Table 4).
Table 4. Profile of the Selected Compounds against the PARP Enzymes, IC50 (pIC50 ± SEM, n = 3), where Mono-ARTs PARP7-PARP16 are Measured Using a Proximity-Enhanced Assay,47 Potency of the Compounds in Rescuing Cells from PARP10 Overexpression along with 95% Confidence Interval for the EC50, and ADME Profiling.
| 16 (OUL245) | 21 (OUL243) | 27 (OUL232) | |
|---|---|---|---|
| Profiling of PARP and TNKS Enzymes | |||
| PARP1 | 570 nM(6.25 ± 0.02) | 1.6 μM(5.79 ± 0.03) | 15 μM |
| PARP2 | 44 nM(7.44 ± 0.12) | 1.6 μM | 10 μM |
| PARP3 | 8.8 μM | 34 μM | 50 μM |
| PARP4 | 6.0 μM | 10 μM | 11 μM |
| TNKS1 | 1.6 μM | 2.1 μM | 5.4 μM |
| TNKS2 | 370 nM(6.43 ± 0.13) | 5.7 μM | 10 μM |
| PARP6 | ≫10 μMa | >10 μMb | 7.7 μM |
| PARP7 | ≫10 μM | 3.8 μM | 83 nM(7.08 ± 0.06) |
| PARP10 | 2.9 μM | 25 nM(7.60 ± 0.03) | 7.8 nM(8.11 ± 0.12) |
| PARP11 | 9.4 μM | 470 nM(6.33 ± 0.13) | 240 nM(6.61 ± 0.07) |
| PARP12 | >10 μM | 4.4 μM | 160 nM(6.80 ± 0.002) |
| PARP14 | 6.7 μM | 650 nM(6.19 ± 0.03) | 300 nM(6.52 ± 0.03) |
| PARP15 | 2.0 μM | 260 nM(6.59 ± 0.11) | 56 nM(7.25 ± 0.01) |
| PARP16 | >10 μM | 5.2 μM | 3.4 μM |
| Activity in Cellular Context | |||
| PARP10 rescue EC50 | inactive | 500 nM(443–750 nM) | 150 nM(103–279 nM) |
| Pharmacokinetic Profile | |||
| water solubility μg/mL (log s) | 24.91 (−3.885) | 37.56 (−3.735) | 12.60 (−4.298) |
| GI Papp × 10–6 cm/s(RM %) | 0.019 (1.4) | 0.281 (1.8) | 0.144 (1.3) |
| BBB Papp × 10–6 cm/s(RM %)c | 0.164 (1.4) | 0.475 (5.3) | 0.143 (3.4) |
| metabolic stablity % | 95.05 (4.95) | 99.14 (0.86) | 99.11 (0.89) |
| stab in human plasma (h) | >24 | >24 | >24 |
| stab. in MeOH (h) | >24 | >24 | >24 |
| stab in PBS pH 7.4 (h) | >24 | >24 | >24 |
≫Denotes no inhibition at the reported concentration.
>Denotes less than 50% inhibition in the reported highest concentration.
Value for olaparib tested in parallel: 0.016 (3.0). RM %: retention membrane percentage.
It should be noted that compounds 21 and 27 had reached the sensitivity limit of the mono-ART assay and that the values reported in Table 3 were artificially high due to the enzyme concentrations needed for a robust conversion of NAD+. We therefore had to improve the assay method and developed a homogeneous proximity enhanced assay for mono-ARTs.47 This new assay was used here to test the selected compounds against PARP7–PARP16 as it allowed us to measure robust IC50 values for the discovered potent inhibitors while using less enzyme (Figure S3).
As emerged from Table 4, both 3-amino derivatives 21 and 27 were potent inhibitors of multiple human mono-ARTs. While 21 still inhibited multiple poly-ARTs at low μM concentration, 27 was overall more selective for mono-ARTs in agreement with our initial assessment. Compound 27 showed the highest potency as it inhibited PARP10 with an IC50 of 7.8 nM making it the best PARP10 inhibitor described to date. Additionally, 27 inhibited PARP7, PARP11, PARP12, PARP14, and PARP15 at low nanomolar potencies. Notably, no inhibitors of PARP12 have been described earlier, and just a few PARP15 inhibitors have been discovered recently.35,44
Compound 16 was confirmed as a weak inhibitor of mono-ARTs while showing potent inhibition of poly-ARTs PARP1–2 and TNKS2. Interestingly, compound 16 shows selectivity toward PARP2 (IC50 = 44 nM) even over highly similar PARP1 (13-fold), and the same behavior was observed when comparing its activity on TNKS2 that was 4-fold higher than on TNKS1. In addition, 16 showed μM IC50 values for the other active site glutamate containing mono-ARTs PARP3–4. This is consistent with the crystal structures where the hydroxyl of 16 forms a hydrogen bond with the glutamate (Figure 3B).
Cell Assays for PARP10 Target Engagement
Taken together, our in vitro experiments identified a potent scaffold that when suitably functionalized gave derivatives that inhibit multiple PARPs. To complement and strengthen our data, we aimed at demonstrating the effectiveness of these compounds in a cell model. We tested 16, 21, and 27 for their capability of rescuing cells from PARP10-induced cell death using a colony formation assay. In line with our results of the WST-1 assay, none of the compounds showed toxicity in this assay using control cells expressing a catalytically inactive PARP10 mutant (PARP10-GW) (Figure 5A).
Figure 5.

Cell assay for PARP10 inhibition. (A) Compounds 21 and 27 effectively rescue the PARP10 overexpressing cells from ADP-ribosylation-dependent cell death, whereas 16 does not show this effect consistent with its lower potency. The compounds are not toxic (-Dox) and do not affect cells expressing catalytically inactive PARP10-GW mutant. Cell colonies were grown for 10–12 days, stained with methylene blue. (B) Quantifications of titration experiments that were measured using ImageJ. Mean and standard deviation for three experiments are shown. Representative titration figures are shown in Figure S4. (C) MARylation of PARP10 is inhibited in cells by 21 and 27 as well as by OUL35. DMSO (D) and inactive PARP10-GW mutant were included for control.
When wild type PARP10 overexpression is induced by doxycyclin (Dox), it leads to cell death observed from the lack of colonies (Figure 5A). At 1 μM, compounds 21 and 27 efficiently rescued cells from PARP10-induced cell death, while 16 did not show any effect at this concentration (Figure 5A). The results are in good agreement with the enzymatic IC50 values (Table 4) and demonstrate the usability of the compounds based on TBT scaffold in inhibiting PARPs in cellular contexts. The titration experiments with 21 and 27 revealed that they are indeed the most potent PARP10 inhibitors described so far also in cell assays (Figure 5B). Especially 27 was effective in rescuing the cells with an EC50 of 150 nM (Table 4), 4-fold more potent than the compounds disclosed earlier,35 and again the ranking is in good agreement with the potencies measured in enzymatic assays.
We then tested that the compounds indeed affect PARP10 through inhibition of the catalytic activity. Both 21 and 27 effectively inhibited auto-MARylation activity of overexpressed PARP10 (Figure 5C). Similarly, we did not observe a MAR-signal for the catalytically inactive PARP10-GW as well as for the previously reported PARP10 inhibitor OUL35.
In Vitro ADME Properties
The colony-forming assay had already shown the ability of the TBT compounds to enter the cells where they engaged the target enzymes, but the successive preclinical studies as well as their use in proof-of-concept in vivo studies require a wider physicochemical characterization. Thus, the in vitro ADME properties were assessed for the best PARP inhibitors, 16, 21, and 27 (Table 4). First, we evaluated the thermodynamic water solubility. The results indicated that compounds 16 and 21 present better solubility of 24.91 and 37.56 μg/mL, respectively, when compared to compound 27. This is likely due to the presence of polar −OH and −NH2 groups. The two −OMe groups instead impart lower solubility to compound 27 (12.60 μg/mL) (Table 4). The solubility of 16 and 21 is still not ideal and should be improved when optimizing the compounds in the future.
Then, a parallel artificial membrane permeability assay (PAMPA) was performed to predict passive permeability through different biological membranes, such as the gastrointestinal tract (GI) and the blood brain barrier (BBB). The BBB permeability is a critical clinically relevant parameter that should be considered when selecting hit compounds for future development potentially targeting brain malignancies. The results shown in Table 4 suggest that all three compounds exhibit suboptimal GI permeability and low membrane retention, reflecting their degree of hydrophilicity already observed in the aqueous solubility tests. The permeability through BBB was better for the smaller compounds 16 (Papp 0.164) and 21 (Papp 0.475) which is 10–30 times higher than that of the clinical PARP inhibitor olaparib tested in parallel (Papp 0.016). Although improved over the control inhibitor, the permeability should be taken into account in the successive optimization steps.48
All compounds showed excellent phase I metabolic stability in human liver microsomes (HLMs), with compounds 21 and 27 showing >99% of unchanged compound, while 16 exhibited a lower although good stability (>95%). The lower steric bulk on the molecule with only a hydroxyl substituent could have enabled the formation of a metabolite likely due to aromatic oxidation (4.95%). Finally, stability tests were performed in MeOH, PBS buffer, and human plasma incubated at 37 °C. All compounds were shown to be stable in these conditions for more than 24 h.
Discussion and Conclusions
Most of the PARP inhibitors, whether inhibiting mono- or poly-ARTs, share a nicotinamide mimic moiety such as a benzamide group or a group rigidified into a cycle (Figure 1).45,49 A few other scaffolds, including triazole as nicotinamide site-binding moiety, have also been explored.50 Unfortunately, sharing the same pharmacophoric requirements, the compounds usually lack the desired selectivity profile.45,51
In this paper, we initially discovered compound 1 based on a new nicotinamide mimic scaffold, TBT, which was able to inhibit multiple PARP enzymes at micromolar potency. TBT is an underexplored scaffold in medicinal chemistry, with few examples of compounds with antifungal,52 anticonvulsant,53 or anti-inflammatory54 properties. Most importantly, no PARP inhibitor based on this scaffold has been reported until now. By making small changes around the tricyclic scaffold of 1, we were able to shift the activity from pan to a selective inhibition of either the mono- or poly-ARTs.
In particular, the benzene ring was decorated with various alkyl substituents, as well as with one or two methoxy or hydroxy groups or with halogen atoms, while the C-3 position of the triazole ring was functionalized with heteroatoms also derivatized with side chains of different lengths. The amino group emerged as the most interesting C-3 substituent that when coupled with a 7-methyl or 5,8-dimethoxy groups on the benzene ring gave compounds 21 and 27, respectively, which had nM potency and selectivity toward mono-ARTs. Of note, compound 27 emerged as the most potent PARP10 inhibitor ever reported to date both in the enzymatic assay (IC50 = 7.8 nM) and in the inhibition of intracellular PARP10 (EC50 = 150 nM). Furthermore, it also potently inhibited PARP15 with IC50 of 56 nM and PARP12 at 160 nM becoming the first potent PARP12 inhibitor. 7-Hydroxy derivative 16 is of special interest as a poly-ART inhibitor since it is both a potent (IC50 = 44 nM) and specific PARP2 inhibitor with 13-fold selectivity over PARP1. Structurally, PARP1 and PARP2 are both able to form the same interactions with the hydroxy moiety (Figure 3A), and therefore, there is no apparent reason for the observed selectivity.
Preliminary ADME analysis indicated good aqueous solubility, not limiting passive permeability in GI and BBB, and excellent stability. The effectiveness of the currently approved PARP1 inhibitors has been shown to be significantly reduced by their poor brain availability due to efflux transporters and restricted delivery across BBB. Comparison studies have shown that niraparib has a greater tumor exposure and sustainability in the brain, while olaparib, rucaparib, and talazoparib have a more limited BBB penetration.55−57 Limited data are available to understand the penetration and residence of PARP inhibitors in a disrupted BBB setting, but pharmacokinetic studies have shown that olaparib, despite the low permeability in the PAMPA model, is able to penetrate recurrent glioblastoma at levels allowing radiosensitization.58
There is still an unmet need to discover PARP inhibitors with the appropriate profile to treat brain metastasis, brain cancers, or neurodegenerative diseases,59 and the TBT scaffold may thus be a potential candidate for further development toward these indications. In summary, the nM potencies measured for the TBT analogues for both mono- and poly-ARTs, experimentally determined binding modes, and favorable ADME properties elucidate possibilities on the development of PARP-specific chemical probes and drug leads based on the compounds disclosed here.
Experimental Section
Chemistry
All starting materials, reagents, and solvents were purchased from common commercial suppliers and were used as such without further purification. Compounds 2,607,6111,5316,5319,6220,5433,6344,6445,6546,6648,6449,6750,6851,6952,7053,6954,7155,7256,7258,7159,7160,7361,7462,7063,7164,7568,7569,7570,7573,7577–79,7680,7781,78 and 82(70) were prepared as described in the literature. Organic solutions were dried over anhydrous Na2SO4 and concentrated with a rotary evaporator at low pressure. All reactions were routinely checked by thin-layer chromatography on silica gel 60F254 (Merck) and visualized by using UV and iodine. Flash chromatography separations were carried out on Merck silica gel 60 (mesh 230–400) or by using automated Buchi Reveleris X2-UV with column FP Ecoflex Si 12 g. Yields were of purified products and were not optimized. 1HNMR spectra were recorded at 400 MHz (Bruker AVANCE DRX-400), while 13C NMR spectra were recorded at 101 MHz (Bruker AVANCE DRX-400). Chemical shifts are given in ppm (δ) relative to TMS. Spectra were acquired at 298 K. Data processing was performed with standard Bruker software XwinNMR, and the spectral data are consistent with the assigned structures. Coupling constant (J) is reported in Hz. The purity of the tested compounds was evaluated by HPLC analysis using a JASCO LC-4000 instrument equipped with a UV–visible diode array JASCO-MD4015 (JASCO Corporation, Tokyo, Japan) and an XTerra MS C18 column, 5 μm × 4.6 mm × 150 mm (Waters Corporation, Massachusetts, USA). Chromatograms were analyzed by ChromNAV2.0 Chromatography Data System software. The purity of the compounds, performed at λ 254 nm, at the λ max of each compound and the absolute maximum of absorbance between 200 and 600 nm was ≥95%. The peak retention time (ret. time) is given in minutes. High-resolution mass detection was performed for some representative compounds, and it was based on electrospray ionization in positive polarity using an Agilent 1290 Infinity system equipped with a MS detector Agilent 6540A Accurate Mass Q-TOF.
4-Fluoro-7-methyl-1,3-benzothiazol-2-amine (57)
A solution of Br2 (0.2 mL, 3.3 mmol) in CHCl3 (5 mL) was slowly added to a suspension of 47 (0.6 g, 3.3 mmol) in CHCl3 (13 mL) at 0 °C. The reaction mixture was stirred at r.t. for 4 h, and then, a solution of 10% Na2SO3 was added to the mixture. CHCl3 was removed under reduced pressure, and NH4OH solution was added until the formation of a precipitate that was filtered, giving compound 57 (0.36 g, 66%). 1H NMR (400 MHz, DMSO-d6) δ: 2.46 (3H, s, CH3), 6.75–6.78 (1H, m, aromatic CH), 6.92–7.00 (1H, m, aromatic CH), 7.67 (2H, br s, NH2).
6-Ethyl-2-hydrazino-1,3-benzothiazole (65)
General Procedure (A) for the Synthesis of Hydrazinobenzothiazoles
Hydrazine hydrate (0.30 mL, 5.88 mmol) and CH3COOH (0.17 mL, 2.94 mmol) were added to a suspension of 55(72) (0.35 g, 1.96 mmol) in ethylene glycol (18 mL), and the reaction mixture was stirred for 12 h at 125 °C. Then, the mixture was then poured into ice/water, and a saturated solution of NaHCO3 was added until pH = 8 to give a precipitate that was filtered yielding 65 (0.158 g, 42%). 1H NMR (400 MHz, DMSO-d6) δ: 1.19 (3H, t, J = 7.4 Hz, CH2CH3), 2.62 (2H, q, J = 7.4 Hz, CH2CH3), 4.96 (2H, br s, NH2), 7.04 (1H, d, J = 7.6 Hz, H4), 7.23 (1H, d, J = 7.9 Hz, H5), 7.51 (1H, s, H7), 8.86 (1H, br s, NH).
6-Isopropyl-2-hydrazino-1,3-benzothiazole (66)
The title compound was prepared according to the general procedure A, starting from 56(72) (30 h) in 52% yield. 1H NMR (400 MHz, DMSO-d6) δ: 1.21 (3H, d, J = 2.1 Hz, CH3), 1.22 (3H, d, J = 2.1 Hz, CH3), 2.87–2.91 (1H, m, CH), 4.97 (2H, br s, NH2), 7.08 (1H, d, J = 8.3 Hz, H4), 7.23 (1H, dd, J = 1.9 and 8.2 Hz, H5), 7.55 (1H, br s, H7), 8.88 (1H, br s, NH).
4-Fluoro-2-hydrazino-7-methyl-1,3-benzothiazole (67)
The title compound was prepared according to the general procedure A, starting from 57 (24 h) in 32% yield. 1H NMR (400 MHz, DMSO-d6) δ: 2.26 (3H, s, CH3), 5.11 (2H, br s, NH2), 6.72–6.76 (1H, m, aromatic H), 6.91–6.96 (1H, m, aromatic H), 9.09 (1H, br s, NH).
2-Hydrazino-4,7-dimethoxy-1,3-benzothiazole (71)
The title compound was prepared according to the general procedure A, starting from 61(74) (26 h) in 20% yield. 1H NMR (400 MHz, DMSO-d6) δ: 3.73 (3H, s, OCH3), 3.77 (3H, s, OCH3), 5.00 (2H, br s, NH2), 6.48 (1H, d, J = 8.6 Hz, H5), 6.70 (1H, d, J = 8.6 Hz, H6), 8.86 (1H, br s, NH).
2-Hydrazino-5,7-dimethoxy-1,3-benzothiazole (72)
The title compound was prepared according to the general procedure A, starting from 62(70) (20 h) in 44% yield. 1H NMR (400 MHz, DMSO-d6) δ: 3.75 (3H, s, OCH3), 3.87 (3H, s, OCH3), 5.00 (2H, br s, NH2), 6.27 (1H, s, H5), 6.56 (1H, s, H6), 8.98 (1H, br s, NH).
6-Methyl[1,2,4]triazolo[3,4-b][1,3]benzothiazole (3)
General Procedure (B) for the Synthesis of [1,2,4]Triazolo[3,4-b][1,3]benzothiazoles
A solution of 64(75) (0.14 g, 0.78 mmol) in formic acid (5 mL) was refluxed for 9 h. The reaction mixture was then poured in ice/water, and the pH was neutralized using a saturated solution of NaHCO3. The reaction mixture was extracted with EtOAc (×3), and the organic layers were washed with brine, dried over Na2SO4, and evaporated to dryness under reduced pressure to give a solid that was purified by crystallization using cyclohexane/EtOAc (2:1) yielding 3 (0.010 g, 10%). 1H NMR (400 MHz, DMSO-d6) δ: 2.41 (3H, s, CH3), 7.28 (1H, d, J = 8.2 Hz, H7), 7.87 (1H, d, J = 8.2 Hz, H8), 7.92 (1H, s, H5), 9.55 (1H, s, H3). 13C NMR (101 MHz, DMSO-d6) δ: 21.34, 115.50, 125.48, 127.99, 128.67, 129.35, 137.00, 137.41, 155.12. HPLC: CH3CN/H2O + 0.1% FA (70:30), ret. time: 2.05 min, peak area: 99.21%.
8-Methyl[1,2,4]triazolo[3,4-b][1,3]benzothiazole (4)
The title compound was prepared according to the general procedure B starting from 80(77) (12 h) in 30% yield as a pink solid, after purification by flash chromatography eluting with CHCl3/MeOH (95:5). 1H NMR (400 MHz, DMSO-d6) δ: 2.42 (3H, s, CH3), 7.27–7.31 (1H, m, H7), 7.48 (1H, t, J = 7.7 Hz, H6), 7.91–7.93 (1H, m, H5), 9.65 (1H, s, H3). 13C NMR (101 MHz, DMSO-d6) δ: 19.70, 112.85, 127.55, 127.61, 129.18, 131.54, 134.71, 137.44, 154.07. HPLC: CH3CN/H2O + 0.1% FA (70:30), ret. time: 2.06 min, peak area: 99.9%.
7-Ethyl[1,2,4]triazolo[3,4-b][1,3]benzothiazole (5)
The title compound was prepared according to the general procedure B starting from 65 (24 h) in 50% yield as a yellow solid, after purification by flash chromatography eluting with CHCl3/MeOH (99:1). 1H NMR (400 MHz, DMSO-d6) δ: 1.23 (3H, t, J = 7.5 Hz, CH2CH3), 2.73 (2H, q, J = 7.6 Hz, CH2CH3), 7.44 (1H, dd, J = 1.0 and 7.3 Hz, H6), 7.89 (1H, s, H8), 8.02 (1H, d, J = 8.3 Hz, H5), 9.60 (1H, s, H3). 13C NMR (101 MHz, DMSO-d6) δ: 16.18, 28.61, 115.13, 124.77, 127.33, 127.62, 132.13, 137.18, 143.33, 154.88. HPLC: CH3CN/H2O (65:35), ret. time: 2.81 min, peak area: 98.49%.
7-Isopropyl[1,2,4]triazolo[3,4-b][1,3]benzothiazole (6)
The title compound was prepared according to the general procedure B starting from 66 (24 h) in 58% yield as a white solid, after purification by flash chromatography eluting with CHCl3/MeOH (99:1) and successive treatment with cyclohexane. 1H NMR (400 MHz, DMSO-d6) δ: 1.26 (6H, d, J = 6.9 Hz, CH3 × 2), 3.1 (1H, sept, J = 7 Hz, CH), 7.47–7.49 (1H, m, H6), 7.95 (1H, s, H8), 8.03 (1H, d, J = 8.3 Hz, H5), 9.60 (1H, s, H3). 13C NMR (101 MHz, DMSO-d6) δ: 24.44, 34.07, 115.15, 123.44, 126.05, 127.70, 132.16, 137.19, 147.99, 154.93. HPLC: CH3CN/H2O (65:35), ret. time: 3.50 min, peak area: 98.13%.
5-Fluoro-8-methyl[1,2,4]triazolo[3,4-b][1,3]benzothiazole (9)
The title compound was prepared according to the general procedure B starting from 67 (10 h) in 13% yield as a yellowish, after purification by flash column chromatography eluting with CH2Cl2/MeOH (98:2). 1H NMR (400 MHz, DMSO-d6) δ: 2.37 (3H, s, CH3), 7.25–7.32 (1H, m, H7), 7.43 (1H, t, J = 8.3 Hz, H6), 9.43 (1H, s, H3). 13C NMR (101 MHz, DMSO-d6) δ: 18.91, 114.06 (d, J = 16.5 Hz), 117.63 (d, J = 16.2 Hz), 127.89 (d, J = 6.5 Hz), 130.21 (d, J = 4.0 Hz), 133.54 (d, J = 2.5 Hz), 138.15, 148.63 (d, J = 245 Hz), 154.06. HPLC: CH3CN/H2O + 0.1% FA (70:30), ret. time: 2.12 min, peak area: 99.9%.
6-Methoxy[1,2,4]triazolo[3,4-b][1,3]benzothiazole (10)
The title compound was prepared according to general procedure B starting from 68(75) (24 h) in 15% yield as a white solid, after purification by flash chromatography eluting with CH2Cl2/MeOH (98:2). 1H NMR (400 MHz, DMSO-d6) δ: 3.93 (3H, s, OCH3), 8.01 (2H, s, aromatic H), 8.30 (1H, s, aromatic H), 9.56 (1H, s, H3). 13C NMR (101 MHz, DMSO-d6) δ: 57.45, 100.22, 108.92, 123.75, 129.21, 129.73, 137.09, 155.21, 155.82. HPLC: CH3CN/H2O + 0.1% FA (70:30), ret. time: 2.19 min, peak area: 99.72%.
8-Methoxy[1,2,4]triazolo[3,4-b][1,3]benzothiazole (12)
The title compound was prepared according to the general procedure B starting from 82(70) (6 h) in 31% yield as a pink solid, after purification by flash chromatography eluting with CH2Cl2/MeOH (98:2). 1H NMR (400 MHz, DMSO-d6) δ: 3.95 (3H, s, OCH3), 7.12 (1H, d, J = 8.2 Hz, H7), 7.49–7.55 (1H, m, H6), 7.67 (1H, d, J = 8.2 Hz, H5), 9.56 (1H, s, H3). 13C NMR (101 MHz, DMSO-d6) δ: 56.87, 107.90, 108.81, 118.83, 129.02, 130.20, 137.42, 154.79. HPLC: CH3CN/H2O + 0.1% FA (70:30), ret. time: 1.70 min, peak area: 98.77%.
5-Methoxy[1,2,4]triazolo[3,4-b][1,3]benzothiazole (13)
The title compound was prepared according to the general procedure B starting from 70(75) (6 h) in 23% yield as a yellow solid, after purification by flash chromatography eluting with CH2Cl2/MeOH (98:2). 1H NMR (400 MHz, DMSO-d6) δ: 4.01 (3H, s, OCH3), 7.22 (1H, d, J = 6.7 Hz, H6), 7.42 (1H, t, J = 8.1 Hz, H7), 7.55 (1H, d, J = 8.2 Hz, H8), 9.34 (1H, s, H3). 13C NMR (101 MHz, DMSO-d6) δ: 56.98, 109.94, 117.20, 119.23, 127.71, 132.88, 138.28, 148.28, 154.57. HPLC: CH3CN/H2O + 0.1% FA (70:30), ret. time: 1.70 min, peak area: 99.47%.
5,8-Dimethoxy[1,2,4]triazolo[3,4-b][1,3]benzothiazole (14)
The title compound was prepared according to the general procedure B starting from 71 (12 h) in 25% yield as a pink solid, after purification by crystallization using cyclohexane/EtOAc (2:1). 1H NMR (400 MHz, DMSO-d6) δ: 3.89 (3H, s, OCH3), 3.95 (3H, s, OCH3), 7.04 (1H, d, J = 9.0 Hz, H6), 7.15 (1H, d, J = 9.0 Hz, H7), 9.29 (1H, s, H3). 13C NMR (101 MHz, DMSO-d6) δ: 56.85, 57.12, 108.40, 110.55, 119.58, 119.97, 138.20, 142.52, 148.15, 154.51. HRMS: m/z calcd for C27H28N3O4S 258.0313 [M + Na+], found 258.0310. HPLC: CH3CN/H2O + 0.1% FA (70:30), ret. time: 2.15 min, peak area: 99.53%.
6,8-Dimethoxy[1,2,4]triazolo[3,4-b][1,3]benzothiazole (15)
The title compound was prepared according to the general procedure B starting from 72 (15 h) in 10% yield as a white solid, after purification by flash chromatography eluting with CH2Cl2/MeOH (98:2). 1H NMR (400 MHz, DMSO-d6) δ: 3.88 (3H, s, OCH3), 3.96 (3H, s, OCH3), 6.76 (1H, s, H7), 7.46 (1H, s, H5), 9.58 (1H, s, H3). 13C NMR (101 MHz, DMSO-d6) δ: 56.72, 57.07, 93.28, 97.86, 110.21, 130.48, 137.27, 155.35, 155.42, 161.11. HPLC: CH3CN/H2O (70:30), ret. time: 1.78 min, peak area: 97.88%.
[1,2,4]Triazolo[3,4-b][1,3]benzothiazol-8-ol (17)
1 M solution of BBr3 in CH2Cl2 (1.22 mL, 1.22 mmol) was added dropwise to a solution of 12 (0.05 g, 0.24 mmol) in dry CH2Cl2 (3 mL) at 0 °C and under a nitrogen atmosphere. The reaction was stirred at r.t. overnight, and then, MeOH was added. The mixture was evaporated under reduced pressure to give a residue that was poured into ice/water, and then 6 N HCl was added until pH 4 and extracted with EtOAc. The organic layers were washed with brine, dried over Na2SO4, and evaporated under reduced pressure, obtaining a solid that was purified by flash chromatography eluting with CH2Cl2/MeOH (95:5), yielding 17 as a brown solid (22%). 1H NMR (400 MHz, DMSO-d6) δ: 6.96 (1H, d, J = 8.2 Hz, H7), 7.43 (1H, t, J = 8.1 Hz, H6), 7.59 (1H, d, J = 8.1 Hz, H5), 9.62 (1H, s, H3), 11.14 (1H, s, OH). 13C NMR (101 MHz, DMSO-d6) δ: 106.30, 112.68, 117.96, 128.84, 130.64, 137.40, 153.73, 154.89. HPLC: CH3CN + 0.1% FA/H2O + 0.1% FA (50:50), ret. time: 2.00 min, peak area: 99.12%.
[1,2,4]Triazolo[3,4-b][1,3]benzothiazol-5,8-diol (18)
The title compound was synthesized following the same procedure as used for the synthesis of compound 17 starting from 14 in 10% yield as a purple solid, after purification with flash column chromatography eluting with CH2Cl2/MeOH (95:5)1H NMR (400 MHz, DMSO-d6) δ: 6.79 (1H, d, J = 8.7 Hz, H7), 6.89 (1H, d, J = 8.8 Hz, H6), 9.24 (1H, s, H3), 10.29 (1H, s, OH), 10.43 (1H, s, OH). 13C NMR (101 MHz, DMSO-d6) δ: 112.72, 114.60, 118.87, 119.17, 138.04, 139.41, 145.59, 154.59. HPLC: CH3CN + 0.1% FA/H2O + 0.1% FA (50:50), ret. time: 1.89 min, peak area: 99.93%.
7-Methyl[1,2,4]triazolo[3,4-b][1,3]benzothiazol-3-amine (21)
CNBr (0.47 g, 4.44 mmol) was added to a solution of 73(75) (0.53 g, 2.96 mmol) in MeOH (10 mL), and the reaction mixture was refluxed for 3.5 h. Then, it was poured in ice/water. A saturated solution of NaHCO3 was added until pH 8, and the obtained precipitate was filtered and purified by flash chromatography eluting with CHCl3/MeOH (95:5), obtaining 21 as a brown solid (0.06 g, 10%). 1H NMR (400 MHz, DMSO-d6) δ: 2.39 (3H, s, CH3), 6.41 (2H, br s, NH2), 7.30 (1H, dd, J = 0.8 and 8.3 Hz, H6), 7.71 (1H, br s, H8), 7.90 (1H, d, J = 8.3 Hz, H5). 13C NMR (101 MHz, DMSO-d6) δ: 21.31, 113.76, 125.43, 127.63, 127.84, 131.43, 135.63, 149.11, 150.90. HRMS: m/z calcd for C9H8N4S 205.0550 [M + H+], found 205.0544. HPLC: CH3CN/H2O (70:30), ret. time: 1.61 min, peak area: 98.97%.
7-Methyl-3-(methylthio)[1,2,4]triazolo[3,4-b][1,3]benzothiazole (22)
K2CO3 (0.21 g, 1.5 mmol) and MeI (0.1 mL, 1 mmol) were added under a nitrogen atmosphere to a solution of 20(54) (0.11 g, 0.5 mmol) in dry DMF (6 mL). The reaction mixture was stirred at 80 °C for 1.5 h and then poured in ice/water. The mixture was acidified with 2 N HCl until pH 5, and the obtained precipitate was filtered and purified by crystallization using cyclohexane/EtOAc (2:1), obtaining 22 as a brown solid (0.03 g, 22%). 1H NMR (400 MHz, DMSO-d6) δ: 2.39 (3H, s, CH3), 2.70 (3H, s, SCH3), 7.37 (1H, d, J = 8.2 Hz, H5), 7.83 (1H, s, H8), 7.86 (1H, d, J = 8.3 Hz, H6). 13C NMR (101 MHz, DMSO-d6) δ: 16.08, 21.26, 113.95, 125.79, 127.62, 128.25, 131.87, 136.63, 144.35, 156.11. HPLC: CH3CN/H2O + 0.1% FA (70:30), ret. time: 2.15 min, peak area: 99.88%.
3-[(4-Chlorobenzyl)thio]-7-methyl[1,2,4]triazolo[3,4-b][1,3]benzothiazole (23)
A suspension of 20(54) (0.12 g, 0.54 mmol) and KOH (0.03 g, 0.54 mmol) in absolute EtOH (8 mL) was refluxed for 30 min under a nitrogen atmosphere. Then, p-chlorobenzyl chloride (0.09 g, 0.54 mmol) was added, and the reaction mixture was stirred at reflux. After 4 h, EtOH was removed under reduced pressure to give a residue that was added to water and extracted with EtOAc (×3). The organic layers were washed with brine, dried over Na2SO4, and evaporated to dryness under a reduced pressure to give a solid that was purified by crystallization using cyclohexane/EtOAc (3:1), to give 23 as a white solid (0.05 g, 27%). 1H NMR (400 MHz, DMSO-d6) δ: 2.36 (1H, s, CH3), 4.39 (2H, s, Bz CH2), 7.22–7.31 (5H, m, aromatic H), 7.80 (1H, s, H8), 7.84 (1H, d, J = 8.3 Hz, H6). 13C NMR (101 MHz, DMSO-d6) δ: 21.25, 37.72, 114.10, 125.61, 127.57, 128.02, 128.73, 131.19, 131.72, 132.53, 136.43, 136.62, 142.34, 156.55. HPLC: CH3CN/H2O + 0.1% FA (70:30), ret. time: 3.32 min, peak area: 99.52%.
N-[(1Z)-(4-Chlorophenyl)methylene]-7-methyl[1,2,4]triazolo[3,4-b][1,3]benzothiazol-3-amine (24)
p-TsOH (10 mg, 10% w/w) and p-chlorobenzaldehyde (0.07 g, 0.5 mmol) were added to a solution of 21 (0.10 g, 0.5 mmol) in dry benzene (20 mL). The reaction mixture was stirred at reflux by using a Dean–Stark apparatus for 16 h and then poured in ice/water. A saturated solution of NaHCO3 was added until pH 8. The mixture was extracted with CH2Cl2 (×3), and the organic layers were washed with brine, dried over Na2SO4, and evaporated to dryness under reduced pressure to give an oil that was purified by flash chromatography eluting with CH2Cl2/MeOH (95:5) and then treated with Et2O to give 24 as a brown solid (0.02 g, 10%). 1H NMR (400 MHz, DMSO-d6) δ: 2.51 (1H, s, CH3), 7.43 (1H, d, J = 8.3 Hz, H5), 7.68 (2H, d, J = 8.5 Hz, H3′ and H5′), 7.85 (1H, s, H8), 8.14 (1H, d, J = 8.2 Hz, H6), 8.21 (2H, d, J = 8.5 Hz, H2′ and H6′), 9.41 (1H, s, CH). 13C NMR (101 MHz, DMSO-d6) δ: 21.55, 115.30, 125.72, 127.98, 128.94, 129.90, 132.03, 132.13, 134.24, 136.83, 138.51, 153.43, 155.14, 163.73. HRMS: m/z calcd for C16H11ClN4S 327.04770 [M + H+], found 327.0472. HPLC: CH3CN/H2O + 0.1% FA (60:40), ret. time: 5.80 min, peak area: 95.64%.
N-(4-Chlorobenzyl)-7-methyl[1,2,4]triazolo[3,4-b][1,3]benzothiazol-3-amine (25)
NaBH4 (0.026 g, 0.69 mmol) was added to a suspension of 24 (0.15 g, 0.46 mmol) in EtOH (10 mL) at 0 °C and under a nitrogen atmosphere. The reaction mixture was stirred overnight at r.t., and then, it was poured in ice/water. A saturated solution of NaHCO3 was added until pH 8 furnishing a precipitate that was filtered and purified by flash chromatography eluting with CH2Cl2/MeOH (97:3) to give 25 as a green solid (0.03 g, 17%). 1H NMR (400 MHz, DMSO-d6) δ: 2.41 (3H, s, CH3), 4.54 (2H, d, J = 5.6 Hz, Bz CH2), 7.23 (1H, br s, NH), 7.34 (1H, d, J = 8.3 Hz, H5), 7.39 (2H, d, J = 8.3 Hz, H3′ and H5′), 7.49 (2H, d, J = 8.3 Hz, H2′ and H6′), 7.72 (1H, s, H8), 7.97 (1H, d, J = 8.3 Hz, H6). 13C NMR (101 MHz, DMSO-d6) δ: 21.13, 46.17, 113.61, 125.33, 127.45, 127.57, 128.47, 129.81, 131.24, 131.70, 135.59, 139.03, 149.76, 150.64. HPLC: CH3CN/H2O + 0.1% FA (60:40), ret. time: 2.64 min, peak area: 99.11%.
5,8-Dimethoxy[1,2,4]triazolo[3,4-b][1,3]benzothiazole-3(2H)-thione (26)
KOH (0.09 g, 1.7 mmol) dissolved in few drops of H2O was added to a suspension of 71 (0.38 g, 1.70 mmol) in EtOH (7 mL), and then, CS2 (0.5 mL, 8.5 mmol) was added. The reaction mixture was refluxed for 2 h, and then, EtOH was removed under reduced pressure, and 2 N HCl was added. The obtained precipitate was filtered, purified by flash chromatography eluting with cyclohexane/EtOAc (80:20), and treated with EtOH to give 26 as a purple solid (0.08 g, 18%). 1H NMR (400 MHz, DMSO-d6) δ: 3.85 (3H, s, OCH3), 3.93 (3H, s, OCH3), 7.13 (1H, d, J = 9.0 Hz, H6), 7.20 (1H, d, J = 9.0 Hz, H7), 13.91 (1H, s, NH). 13C NMR (101 MHz, DMSO-d6) δ: 57.03, 57.82, 109.76, 114.61, 118.81, 122.65, 143.91, 148.03, 151.75, 163.65. HPLC: CH3CN/H2O (65:35), ret. time: 1.95 min, peak area: 96.90%.
5,8-Dimethoxy[1,2,4]triazolo[3,4-b][1,3]benzothiazol-3-amine (27)
The title compound was synthesized following the same procedure as used for the synthesis of compound 21 starting from 71 in 12% yield as a pink solid, after purification by flash chromatography eluting with CHCl3/MeOH 95:5 and successive treatment by EtOH. 1H NMR (400 MHz, DMSO-d6) δ: 3.90 (3H, s, OCH3), 3.99 (3H, s, OCH3), 6.46 (2H, br s, NH2), 7.02 (1H, d, J = 9.0 Hz, H6), 7.16 (1H, d, J = 9.0 Hz, H7). 13C NMR (101 MHz, DMSO-d6) δ: 57.00, 57.78, 108.36, 111.50, 120.03, 120.15, 141.48, 148.45, 148.55, 150.67. HRMS: m/z calcd for C10H10N4O2S 251.0590 [M + H+], found 251.0584. HPLC: CH3CN/H2O (70:30), ret. time: 1.64 min, peak area: 97.78%.
5,8-Dimethoxy-3-(methylthio)-2,3-dihydro[1,2,4]triazolo[3,4-b][1,3]benzothiazole (28)
The title compound was synthesized following the same procedure as used for the synthesis of compound 22 starting from 26 in 15% yield as a pink solid, after purification by flash chromatography eluting with cyclohexane/EtOAc (from 100:0 to 50:50). 1H NMR (400 MHz, DMSO-d6) δ: 2.61 (3H, s, SCH3), 3.87 (3H, s, OCH3), 3.89 (3H, s, OCH3), 7.02 (1H, d, J = 8.9 Hz, H6), 7.11 (1H, d, J = 8.9 Hz, H7). 13C NMR (101 MHz, DMSO-d6) δ: 15.79, 56.95, 57.07, 109.05, 111.58, 120.07, 120.71, 142.13, 146.93, 148.30, 156.33. HPLC: CH3CN/H2O (60:40), ret. time: 2.23 min, peak area: 95.80%.
4-Chloro-N-(5,8-dimethoxy-2,3-dihydro[1,2,4]triazolo[3,4-b][1,3]benzothiazol-3-yl)benzamide (29)
Et3N (0.16 mL, 1.2 mmol) and p-chlorobenzoyl chloride (0.13 mL, 1.04 mmol) were added to a solution of 27 (0.22 g, 0.8 mmol) in dry DMF (6 mL) under a nitrogen atmosphere. The reaction mixture was stirred for 2 h at 80 °C, and then, it was poured in ice/water obtaining a precipitate that was filtered and purified by flash chromatography eluting with CHCl3/MeOH (95:5) to give 29 as a purple solid (0.03 g, 10%). 1H NMR (400 MHz, DMSO-d6) δ: 3.39 (3H, s, OCH3), 3.95 (3H, s, OCH3), 7.09–7.15 (2H, m, H6 and H7), 7.71 (2H, d, J = 6.7 Hz, H3′ and H5′), 8.11 (2H, d, J = 6.7 Hz, H2′ and H6′), 11.42 (1H, s, NH). 13C NMR (101 MHz, DMSO-d6) δ: 57.02, 57.14, 109.15, 111.33, 120.10, 120.38, 129.30, 130.30, 131.38, 137.90, 142.56, 142.92, 148.01, 154.93, 166.58. HRMS: m/z calcd for C17H13ClN4O3S 389.048 [M + H+], found 389.047. HPLC: CH3CN/H2O + 0.1% FA (60:40), ret. time: 2.17 min, peak area: 97.06%.
N-(5,8-Dimethoxy[1,2,4]triazolo[3,4-b][1,3]benzothiazol-3-yl)-2,2-dimethylpropanamide (30)
The title compound was synthesized following the same procedure as used for the synthesis of compound 29 using trimethylacetyl chloride (0.11 mL, 0.88 mmol) and dry toluene as a solvent at 110 °C overnight. After purification by flash chromatography eluting with CHCl3/MeOH (97:3), compound 30 was obtained as a gray solid (0.06 g, 20%). 1H NMR (400 MHz, DMSO-d6) δ: 1.29 (9H, s, CH3) 3.89 (3H, s, OCH3), 3.95 (3H, s, OCH3), 7.13 (1H, d, J = 9.1 Hz, H7), 7.22 (1H, d, J = 9.1 Hz, H6), 10.14 (1H, s, NH). 13C NMR (101 MHz, DMSO-d6) δ: 27.68, 57.18, 57.99, 109.25, 112.11, 120.40, 120.55, 143.45, 143.49, 148.22, 154.51, 156.13, 179.03. HPLC: CH3CN/H2O + 0.1% FA (70:30), ret. time: 1.77 min, peak area: 99.76%.
N-(5,8-Dimethoxy[1,2,4]triazolo[3,4-b][1,3]benzothiazol-3-yl)acetamide (31)
The title compound was synthesized following the same procedure as used for the synthesis of compound 29 using acetyl chloride (0.63 mL, 0.88 mmol) and dry toluene as a solvent at 110 °C overnight. The reaction mixture was extracted with EtOAc, and the organic layers were washed with brine, dried over Na2SO4, and evaporated under reduced pressure, obtaining an oil that was purified by flash chromatography eluting with CHCl3/MeOH (97:3), to give 31 as a pink solid (0.05 g, 20%). 1H NMR (400 MHz, DMSO-d6) δ: 2.19 (3H, s, CH3), 3.87 (3H, s, OCH3), 3.90 (3H, s, OCH3), 6.81 (1H, d, J = 8.7 Hz, H7), 6.92 (1H, d, J = 8.6 Hz, H6), 12.49 (1H, s, NH). 13C NMR (101 MHz, DMSO-d6) δ: 23.22, 56.45, 56.88, 104.96, 108.99, 121.28, 139.95, 140.12, 146.97, 148.23, 157.34, 169.78. HPLC: CH3CN/H2O + 0.1% FA (70:30), ret. time: 1.80 min, peak area: 95.05%.
Protein Production
The proteins used in Tables 1–3 were produced as described earlier. Dockerin constructs to allow enhanced activity assays for mono-ARTs (Table 4) were produced in Escherichia coli with an N-terminal maltose binding protein (MBP) and a C-terminal dockerin domain from Hungateiclostridium thermocellum Cel48s as described recently.47
Human PARP6 (Uniprot #Q2NL67-1) was cloned into a modified pFASTBac1 vector (Addgene #30116) with N-terminal 6× His-MBP tag with a TEV protease site by SLIC cloning. The construct was sequence verified by dideoxy sequencing. Protein was produced as described before.25 Sf21 cells were transfected with bacmids using Fugene6 (Promega #E2693). V0 virus containing media was harvested after 7 days. These viruses were amplified to increase titre (V1). For protein production, Sf21 cells at a density of 1 × 106 cells per mL and a predetermined volume of V1 virus that induces growth arrest at this density were used. Cells were harvested after 72 h of growth arrest and frozen at −20 °C with lysis buffer (50 mM HEPES pH 7.4, 0.5 M NaCl, 10% glycerol, 0.5 mM TCEP, and 10 mM imidazole) until needed.
Pellet was thawed, and 0.2 mM Pefabloc was added to the suspension. The mixture was sonicated and then centrifuged at 16,000 rpm to separate soluble proteins from cellular debris. The supernatant was centrifuged again to remove any carry-over debris. The supernatant was bound to HiTrap IMAC columns (Cytiva) and washed with 4 column volume of lysis buffer and then wash buffer (50 mM HEPES pH 7.4, 0.5 M NaCl, 10% glycerol, 0.5 mM TCEP, and 25 mM imidazole). Proteins were eluted in (50 mM HEPES pH 7.4, 0.5 M NaCl, 10% glycerol, 0.5 mM TCEP, and 350 mM imidazole). Eluted proteins were loaded into MBP-trap columns and washed with size exclusion chromatography (SEC) buffer (30 mM HEPES pH 7.5, 0.35 M NaCl, 10% glycerol, and 0.5 mM TCEP) and eluted in SEC buffer supplemented with 10 mM maltose. TEV protease (1:30) molar ratio was used to cleave tags.79 A reverse IMAC step was used to separate tags and TEV protease from cleaved PARP6. A final SEC was performed, and fractions were pooled, concentrated, and flash frozen. The identity of the purified protein was confirmed using MALDI-TOF analysis.
Activity Assay
Inhibition experiments were performed using a homogenous assay measuring NAD+ consumption.80−82 Reactions were carried out in quadruplicate, and IC50 curves were fitted using sigmoidal dose response curve (four variables) in GraphPad Prism version 8.02. For the compounds showing <1 μM potency, the experiment was repeated three times, and pIC50 ± SEM was calculated. Assay conditions for PARP2, TNKS2, PARP10, and PARP15 (Tables 1–3) were recently reported.44 The conditions for the proximity-enhanced mono-ART assays (Table 4) were also reported recently.47 PARP6 (400 nM) inhibition was measured using the standard buffer (50 mM sodium phosphate pH 7.0) for the enhanced activity assay using 500 nM NAD+ and 18 h incubation in r.t..
Crystallization and Structure Refinement
The crystallizations were carried out using a sitting drop vapor diffusion method at +20 °C for PARP15 and at +4 °C for PARP2 and TNKS2. A hanging drop vapor diffusion method at +20 °C was used for PARP14. The compounds were dissolved in DMSO at 10 mM concentration and were used to obtain protein-inhibitor complex structures either by cocrystallization or soaking, as stated specifically for each protein below. The protein and precipitant solutions were mixed at 2:1–1:2 ratios with the Mosquito crystallization robot (SPT Labtech) resulting in 160–500 nl droplets. The crystallization experiments were monitored using RI54 imagers (Formulatrix) through the IceBear software.83
The inhibitors except 27 were cocrystallized with PARP15 as previously reported.44 A 10 mg/mL of PARP15 was mixed with the compound solution to reach approximately 700 μM concentration. 0.2 M NH4Cl pH 7.5, 16–20% (w/v) PEG 3350 was used as a precipitant solution. The crystals were cryoprotected with a solution containing 0.2 M NH4Cl and 30% (v/v) 2-methyl-2,4-pentanediol (MPD). 27 was soaked to a PARP15 crystal with the cryoprotectant solution containing 1 mM 27 and incubated for 15 min at +20 °C prior to cryo-freezing with liquid nitrogen.
Prior to crystallization, the TNKS2 ART domain (5.3 mg/mL) was mixed with 1:100 chymotrypsin and incubated for 2 h at room temperature. The protein was then mixed with precipitant solution containing 100 mM Tris (pH 8.5), 200 mM lithium sulfate, and 20–24% (w/v) PEG3350. Crystals formed within 2–3 days. TNKS2 crystals were soaked for 8–24 h with 1 or 3 diluted in precipitant solution to a final concentration of approximately 1 mM compound in the crystallization droplets. The crystals were cryoprotected using precipitant solution containing 20% (v/v) glycerol.
15 mg/mL PARP14 was mixed with 1 to reach approximately 860 μM inhibitor concentration. 0.17 M NH4SO4, 15% (v/v) glycerol, and 27% (v/v) PEG 4000 were used as the precipitant solution. Crystals were obtained in 2 days and were cryo-frozen with liquid nitrogen.
Compound 16 complex crystal structure with PARP2 was obtained using dry compound cocrystallization. 20 nL of 10 mM compound 10 were transferred to the crystallization plate and allowed to dry at 37 °C before proceeding. 30 mg/mL PARP2 was then mixed with the precipitant solution containing 100 mM Tris pH 9.5 and 20% PEG 3350. Crystals were cryoprotected with a solution containing 100 mM Tris pH 9.0, 200 mM NaCl, 25% PEG3350, 22% glycerol, and 100 μM compound 16.
All datasets collected from PARP2, TNKS2, PARP14, and PARP15 crystals were processed with XDS.84 Phases were solved by using molecular replacement with the programs MOLREP85 in CCP4i286 or with Phaser.87 The existing models having PDB ids 4TVJ,885OWS,893GOY,51 and 3BLJ(22) were used as search models for PARP2, TNKS2, PARP14, and PARP15, respectively. The models were built by using the Coot program90 and refined with Refmac591 in CCP4i2. Data collections and refinement statistics are shown in Table S1.
Cell Viability Assay
Cell viability was assessed by colorimetric WST-1 (Cellpro-Roche, Sigma-Aldrich) assay following manufacturer’s instructions. Shortly, HEK293T cells were seeded at the density of 2.5 × 104 cells per well in a 96-well plate in 100 μL of Dulbecco’s modified Eagle’s medium (DMEM, Biowest) supplemented with 10% fetal bovine serum (Biowest) and 1% of penicillin and streptomycin. Cells were allowed to grow for 18 h before adding compounds at the indicated concentrations (100, 50, and 10 μM). Also, DMSO and 10 mM hydroxyurea (Sigma) were used as internal controls for induced cell toxicity. Cells were grown for additional 24 h. Thereafter, WST-1 reagent was pipetted followed by 2 h incubation and absorbance measuring by a Tecan Infinite M1000 or a Tecan Spark (Tecan) plate reader. The assay was performed in triplicate and repeated at least three times. Data were normalized to DMSO control.
PARP10 Rescue Assay
HeLa Flp-In T-REx-PARP10 and -PARP10-G888W cells were grown in DMEM medium supplemented with 10% heat-inactivated fetal calf serum at 37 °C in 5% CO2.92 For colony formation assays, 500 HeLa cells were seeded in 6-well culture plates. Once the cells adhered, protein expression was induced by adding 500 ng/mL Dox. Different concentrations of the indicated compounds were added to the cell culture medium as indicated in the figure. The cells were grown for 10–12 days and then stained using methylene blue. The number of colonies was assessed using ImageJ. EC50 curves were fitted using three variables in GraphPad Prism version 8.02.
Cellular MARylation Assay
HeLa-Flp-In TRex PARP10 WT or PARP10-GW (catalytically inactive) cells were cultivated in DMEM and supplemented with 10% heat inactivated FCS at 37 °C and 5% CO2. PARP10 expression was induced by 0.1 μg/μL of Dox, and the cells were treated with DMSO as the vehicle control or 10 μM of the compounds 21, 27, or OUL35 over night for 16 h. Cells were lysed in RIPA lysis buffer [10 mM Tris pH 7.4; 150 mM NaCl; 1% NP-40; 1% desoxycholate; 0,1% SDS; protease inhibitor cocktail (Sigma-Aldrich) and 10 μM olaparib (Selleck Chemicals)], and lysates were analyzed via SDS-PAGE and immunoblotting using specific anti-PARP10 (Eurogentec),93 anti-MAR/PAR (Cell Signaling, E6F6A, anti-poly/Mono-ADP-ribose antibody), and anti-α-tubulin (B 5-1-2, Sigma-Aldrich) antibodies.
In Vitro ADME Studies
All solvents and reagents were from Sigma-Aldrich Srl (Milan, Italy). Dodecane was purchased from Fluka (Milan, Italy). Pooled male donors 20 mg/mL HLM were from Merk-Millipore (Burlington, MA, USA). Milli-Q quality water (Millipore, Milford, MA, USA) was used. Hydrophobic filter plates (MultiScreen-IP, clear plates, 0.45 mm diameter pore size), 96-well microplates, and 96-well UV-transparent microplates were obtained from Merk-Millipore (Burlington, MA, USA).
UV/LC–MS Methods
UV/LC–MS LC analyses for ADME studies were performed by UV/LC–MS with an Agilent 1260 Infinity HPLC-DAD system interfaced with an Agilent MSD 6130 (Agilent Technologies, Palo Alto, CA) system. Chromatographic separation was obtained using a Phenomenex Kinetex C18-100 Å column (150 × 4.6 mm) with 5 μm particle size and gradient elution with a binary solution (eluent A: H2O, eluent B: ACN, both eluents were acidified with formic acid 0.1% v/v) at room temperature. The analysis started with 5% of B (from t = 0 to t = 1 min), then B was increased to 95% (from t = 1 to t = 10 min), then kept at 95% (from t = 10 to t = 19 min), and finally return to 5% of eluent A in 1.0 min. The flow rate was 0.6 mL/min, and injection volumes were 10 μL.
Water Solubility
Each solid compound (1 mg) was added to 1 mL of distilled water. Each sample was mixed at r.t. in a shaker water bath 24 h. The resulting suspension was filtered through a 0.45 μm nylon filter (Acrodisc), and the solubilized compound was quantified in triplicate using UV/LC–MS method reported above by comparison with the appropriate calibration curve that was obtained from samples of the compound dissolved in methanol at different concentrations.94
PAMPA
Each “donor solution” was prepared from a solution of the appropriate compound (DMSO, 1 mM) diluted with phosphate buffer (pH 7.4, 0.025 M) up to a final concentration of 500 μM. Filters were coated with 10 μL of 1% dodecane solution of phosphatidylcholine or 5 μL of brain polar lipid solution (20 mg/mL 16% CHCl3, 84% dodecane) prepared from CHCl3 solution 10% w/v for intestinal permeability and BBB permeability, respectively. Donor solution (150 μL) was added to each well of the filter plate and to each well of the acceptor plate was added 300 μL of solution (50% DMSO in phosphate buffer). The sandwich plate was assembled and incubated for 5 h at r.t. After the incubation time, the plates were separated, and the samples were taken from both the donor and acceptor wells, and the amount of compound was measured by UV/LC–MS. All compounds were tested in three independent experiments. Permeability (Papp) was calculated according to the following equation obtained from the literature95,96 with some modification in order to obtain permeability values in cm/s
Where VA is the volume in the acceptor well, VD is the volume in the donor well (cm3), A is the “effective area” of the membrane (cm2), t is the incubation time (s), and r is the ratio between drug concentration in the acceptor and equilibrium concentration of the drug in the total volume (VD + VA). Drug concentration is estimated by using the peak area integration. Membrane retentions (%) were calculated according to the following equation
where r is the ratio between drug concentration in the acceptor and equilibrium concentration and D, A, and Eq represent drug concentration in the donor, acceptor, and equilibrium solution, respectively.
Metabolic Stability in HLM
Each compound in DMSO solution was incubated at 37 °C for 1 h in phosphate buffer (25 mM pH 7.4), human liver microsomal protein (0.2 mg/mL), and in the presence of an NADPH regenerating system (NADPH 0.2 mM, NADPH+ 1 mM, d-glucose-6-phosphate 4 mM, 4 unit/mL glucose-6-phosphate dehydrogenase) in 48 mM MgCl2 at a final volume of 500 μL. The reaction was stopped by cooling in ice and quenched by adding 1.0 mL of acetonitrile. The reaction mixtures were then centrifuged (4000 rpm for 10 min), and the supernatant was taken, dried under nitrogen flow, and suspended in 100 μL of methanol. The parent drug and metabolites were subsequently determined by UV/LC–MS. The percentage of not metabolized compound was calculated by comparison with reference solutions. For each compound, the determination was performed in three independent experiments.
Stability Tests
For the stability measurements in polar solvents, each compound was dissolved at room temperature in MeOH or PBS (0.025 M, pH 7.4) up to a final concentration of 500 μM. Aliquot samples (20 μL) were taken at fixed time points (0.0, 4.0, 8.0, and 24.0 h) and were analyzed by UV/LC–MS. For each compound, the determination was performed in three independent experiments.
To test the stability in human plasma, the incubation mixture (total volume of 2.0 mL) was constituted of the following components: pooled human plasma (1.0 mL, 55.7 mg protein/mL),97 HEPES buffer (0.9 mL, 25 mM, 140 mM NaCl, pH 7.4), and 0.1 mL of each compound in DMSO (2.0 mM). The solution was mixed in a test tube that was incubated at 37 °C. At set time points (0.0, 0.08, 0.25, 0.50, 1.0, 2.0, 4.0, 8.0, and 24.0 h), samples of 50 μL were taken, mixed with 450 μL of cold acetonitrile, and centrifuged at 5000 rpm for 15 min98 The supernatant was removed and analyzed by UV/LC–MS. For each compound, the determination was performed in three independent experiments.
Acknowledgments
We thank Chiara Bosetti for measuring some dose responses with the TNKS2 enzyme. The use of the facilities and expertise of the Biocenter Oulu Structural Biology core facility (a member of Biocenter Finland, Instruct-ERIC Centre Finland and FINStruct), Proteomics and Protein Analysis core facility (a member of Biocenter Finland), and Biocenter Oulu sequencing center are gratefully acknowledged. We thank the staff members of DLS, ESRF, and MAXIV.
Glossary
Abbreviations
- ADPr
ADP-ribose
- BBB
blood brain barrier
- GI
gastrointestinal
- MAR
mono-ADP-ribosylation
- PAMPA
parallel artificial membrane permeability assay
- PAR
poly-ADP-ribosylation
- TBT
[1,2,4]triazolo[3,4-b]benzothiazole
- TNKS
tankyrase
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01460.
Crystallography data processing and refinement statistic, WST-1 cell toxicity analysis of compounds, binding modes of OUL40 analogues to TNKS2 and PARP15, examples of IC50 measurements, example of HPLC analysis, and 1H NMR and 13C NMR spectra of the target compounds (PDF)
Molecular formula string for all final compounds (CSV)
Author Contributions
⊥ S.M., M.G.N., and M.M.M. authors contributed equally.
The work was supported by the Deutsche Forschungsgemeinschaft funding to BL (Lu 466/16-2), by the Magnus Ehrnrooth Foundation to S.M., and by the Academy of Finland (grant nos. 287063 and 294085) and by Sigrid Jusélius and Jane and Aatos Erkko foundations to L.L.
The authors declare the following competing financial interest(s): S.M., M.G.N., M.M.M., S.M., O.T. and L.L. are inventors listed in a patent application regarding the disclosed inhibitors. HV, PK, BL, and LL are also inventors in granted patents and patent applications for PARP and tankyrase inhibitors. The remaining authors declare no competing interests.
Notes
PDB ID codes: atomic coordinates and structure factors have been deposited to the Protein Data Bank under accession numbers also mentioned in the tables 7R3Z, 7R3L, 7R3O, 7R4A, 7R5X, 7R5D, 7Z1W, 7Z1Y, 7R59, 7Z1V, 7Z41, 7Z2O and 7Z2Q. Raw diffraction images are available at IDA (https://doi.org/10.23729/0b11fe27-a545-48b0-a953-292d1e1e1d38). Authors will release the atomic coordinates and experimental data upon article publication.
Supplementary Material
References
- Lüscher B.; Ahel I.; Altmeyer M.; Ashworth A.; Bai P.; Chang P.; Cohen M.; Corda D.; Dantzer F.; Daugherty M. D.; Dawson T. M.; Dawson V. L.; Deindl S.; Fehr A. R.; Feijs K. L. H.; Filippov D. V.; Gagné J.-P.; Grimaldi G.; Guettler S.; Hoch N. C.; Hottiger M. O.; Korn P.; Kraus W. L.; Ladurner A.; Lehtiö L.; Leung A. K. L.; Lord C. J.; Mangerich A.; Matic I.; Matthews J.; Moldovan G.-L.; Moss J.; Natoli G.; Nielsen M. L.; Niepel M.; Nolte F.; Pascal J.; Paschal B. M.; Pawłowski K.; Poirier G. G.; Smith S.; Timinszky G.; Wang Z.-Q.; Yélamos J.; Yu X.; Zaja R.; Ziegler M. ADP-Ribosyltransferases, an Update on Function and Nomenclature. FEBS J. 2022, 289, 7399–7410. 10.1111/febs.16142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels C. M.; Ong S.-E.; Leung A. K. L. The Promise of Proteomics for the Study of ADP-Ribosylation. Mol. Cell 2015, 58, 911–924. 10.1016/j.molcel.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte R.; Liu Z.; Kraus W. L. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 2017, 31, 101–126. 10.1101/gad.291518.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher B.; Bütepage M.; Eckei L.; Krieg S.; Verheugd P.; Shilton B. H. ADP-Ribosylation, a Multifaceted Posttranslational Modification Involved in the Control of Cell Physiology in Health and Disease. Chem. Rev. 2018, 118, 1092–1136. 10.1021/acs.chemrev.7b00122. [DOI] [PubMed] [Google Scholar]
- Prokhorova E.; Agnew T.; Wondisford A. R.; Tellier M.; Kaminski N.; Beijer D.; Holder J.; Groslambert J.; Suskiewicz M. J.; Zhu K.; Reber J. M.; Krassnig S. C.; Palazzo L.; Murphy S.; Nielsen M. L.; Mangerich A.; Ahel D.; Baets J.; O’Sullivan R. J.; Ahel I. Unrestrained Poly-ADP-Ribosylation Provides Insights into Chromatin Regulation and Human Disease. Mol. Cell 2021, 81, 2640–2655. 10.1016/j.molcel.2021.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa S. T.; Galera-Prat A.; Wazir S.; Alanen H. I.; Maksimainen M. M.; Lehtiö L. A Molecular Toolbox for ADP-Ribosyl Binding Proteins. Cell Rep. Methods 2021, 1, 100121. 10.1016/j.crmeth.2021.100121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruf A.; Rolli V.; de Murcia G.; Schulz G. E. The Mechanism of the Elongation and Branching Reaction of Poly(ADP-Ribose) Polymerase as Derived from Crystal Structures and Mutagenesis. J. Mol. Biol. 1998, 278, 57–65. 10.1006/jmbi.1998.1673. [DOI] [PubMed] [Google Scholar]
- Loseva O.; Jemth A.-S.; Bryant H. E.; Schüler H.; Lehtiö L.; Karlberg T.; Helleday T. PARP-3 Is a Mono-ADP-Ribosylase That Activates PARP-1 in the Absence of DNA. J. Biol. Chem. 2010, 285, 8054–8060. 10.1074/jbc.m109.077834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas S.; Matic I.; Uchima L.; Rood J.; Zaja R.; Hay R. T.; Ahel I.; Chang P. Family-Wide Analysis of Poly(ADP-Ribose) Polymerase Activity. Nat. Commun. 2014, 5, 4426. 10.1038/ncomms5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo J.; Lord C. J.; Serra V.; Tutt A.; Balmaña J.; Castroviejo-Bermejo M.; Cruz C.; Oaknin A.; Kaye S. B.; de Bono J. S. A Decade of Clinical Development of PARP Inhibitors in Perspective. Ann. Oncol. 2019, 30, 1437–1447. 10.1093/annonc/mdz192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant H. E.; Schultz N.; Thomas H. D.; Parker K. M.; Flower D.; Lopez E.; Kyle S.; Meuth M.; Curtin N. J.; Helleday T. Specific Killing of BRCA2-Deficient Tumours with Inhibitors of Poly(ADP-Ribose) Polymerase. Nature 2005, 434, 913–917. 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Farmer H.; McCabe N.; Lord C. J.; Tutt A. N.; Johnson D. A.; Richardson T. B.; Santarosa M.; Dillon K. J.; Hickson I.; Knights C.; Martin N. M.; Jackson S. P.; Smith G. C.; Ashworth A. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Haddad G.; Saadé M. C.; Eid R.; Haddad F. G.; Kourie H. R. PARP Inhibitors: A Tsunami of Indications in Different Malignancies. Pharmacogenomics 2020, 21, 221–230. 10.2217/pgs-2019-0113. [DOI] [PubMed] [Google Scholar]
- Pilié P. G.; Tang C.; Mills G. B.; Yap T. A. State-of-the-Art Strategies for Targeting the DNA Damage Response in Cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. 10.1038/s41571-018-0114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guney Eskiler G. Talazoparib to Treat BRCA-Positive Breast Cancer. Drugs Today 2019, 55, 459–467. 10.1358/dot.2019.55.7.3015642. [DOI] [PubMed] [Google Scholar]
- Riffell J. L.; Lord C. J.; Ashworth A. Tankyrase-Targeted Therapeutics: Expanding Opportunities in the PARP Family. Nat. Rev. Drug Discovery 2012, 11, 923–936. 10.1038/nrd3868. [DOI] [PubMed] [Google Scholar]
- Lehtiö L.; Chi N.-W.; Krauss S. Tankyrases as Drug Targets. FEBS J. 2013, 280, 3576–3593. 10.1111/febs.12320. [DOI] [PubMed] [Google Scholar]
- Haikarainen T.; Krauss S.; Lehtio L. Tankyrases: Structure, Function and Therapeutic Implications in Cancer. Curr. Pharm. Des. 2014, 20, 6472–6488. 10.2174/1381612820666140630101525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenders R. G. G.; Brinch S. A.; Sowa S. T.; Amundsen-Isaksen E.; Galera-Prat A.; Murthy S.; Aertssen S.; Smits J. N.; Nieczypor P.; Damen E.; Wegert A.; Nazaré M.; Lehtiö L.; Waaler J.; Krauss S. Development of a 1,2,4-Triazole-Based Lead Tankyrase Inhibitor: Part II. J. Med. Chem. 2021, 64, 17936–17949. 10.1021/acs.jmedchem.1c01264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta C. C.; Bhatt H. G. Tankyrase Inhibitors as Antitumor Agents: A Patent Update (2013 - 2020). Expert Opin. Ther. Pat. 2021, 31, 645–661. 10.1080/13543776.2021.1888929. [DOI] [PubMed] [Google Scholar]
- Velagapudi U. K.; Patel B. A.; Shao X.; Pathak S. K.; Ferraris D. V.; Talele T. T. Recent Development in the Discovery of PARP Inhibitors as Anticancer Agents: A Patent Update (2016-2020). Expert Opin. Ther. Pat. 2021, 31, 609–623. 10.1080/13543776.2021.1886275. [DOI] [PubMed] [Google Scholar]
- Karlberg T.; Klepsch M.; Thorsell A.-G.; Andersson C. D.; Linusson A.; Schüler H. Structural Basis for Lack of ADP-Ribosyltransferase Activity in Poly(ADP-Ribose) Polymerase-13/Zinc Finger Antiviral Protein. J. Biol. Chem. 2015, 290, 7336–7344. 10.1074/jbc.m114.630160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.-S.; Jividen K.; Spencer A.; Dworak N.; Ni L.; Oostdyk L. T.; Chatterjee M.; Kuśmider B.; Reon B.; Parlak M.; Gorbunova V.; Abbas T.; Jeffery E.; Sherman N. E.; Paschal B. M. Ubiquitin Modification by the E3 Ligase/ADP-Ribosyltransferase Dtx3L/Parp9. Mol. Cell 2017, 66, 503–516. 10.1016/j.molcel.2017.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatrin C.; Gabrielsen M.; Buetow L.; Nakasone M. A.; Ahmed S. F.; Sumpton D.; Sibbet G. J.; Smith B. O.; Huang D. T. Structural Insights into ADP-Ribosylation of Ubiquitin by Deltex Family E3 Ubiquitin Ligases. Sci. Adv. 2020, 6, eabc0418 10.1126/sciadv.abc0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashok Y.; Vela-Rodríguez C.; Yang C.-S.; Alanen H. I.; Liu F.; Paschal B. M.; Lehtiö L. Reconstitution of the DTX3L-PARP9 complex reveals determinants for high-affinity heterodimerization and multimeric assembly. Biochem. J. 2022, 479, 289–304. 10.1042/bcj20210722. [DOI] [PubMed] [Google Scholar]
- Kleine H.; Poreba E.; Lesniewicz K.; Hassa P. O.; Hottiger M. O.; Litchfield D. W.; Shilton B. H.; Lüscher B. Substrate-Assisted Catalysis by PARP10 Limits Its Activity to Mono-ADP-Ribosylation. Mol. Cell 2008, 32, 57–69. 10.1016/j.molcel.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Venkannagari H.; Verheugd P.; Koivunen J.; Haikarainen T.; Obaji E.; Ashok Y.; Narwal M.; Pihlajaniemi T.; Lüscher B.; Lehtiö L. Small-Molecule Chemical Probe Rescues Cells from Mono-ADP-Ribosyltransferase ARTD10/PARP10-Induced Apoptosis and Sensitizes Cancer Cells to DNA Damage. Cell Chem. Biol. 2016, 23, 1251–1260. 10.1016/j.chembiol.2016.08.012. [DOI] [PubMed] [Google Scholar]
- Nicolae C. M.; Aho E. R.; Vlahos A. H. S.; Choe K. N.; De S.; Karras G. I.; Moldovan G.-L. The ADP-Ribosyltransferase PARP10/ARTD10 Interacts with Proliferating Cell Nuclear Antigen (PCNA) and Is Required for DNA Damage Tolerance. J. Biol. Chem. 2014, 289, 13627–13637. 10.1074/jbc.m114.556340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleicher E. M.; Galvan A. M.; Imamura-Kawasawa Y.; Moldovan G.-L.; Nicolae C. M. PARP10 Promotes Cellular Proliferation and Tumorigenesis by Alleviating Replication Stress. Nucleic Acids Res. 2018, 46, 8908–8916. 10.1093/nar/gky658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahrour M. A.; Nicolae C. M.; Edvardson S.; Ashhab M.; Galvan A. M.; Constantin D.; Abu-Libdeh B.; Moldovan G.-L.; Elpeleg O. PARP10 Deficiency Manifests by Severe Developmental Delay and DNA Repair Defect. Neurogenetics 2016, 17, 227–232. 10.1007/s10048-016-0493-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy S.; Desantis J.; Verheugd P.; Maksimainen M. M.; Venkannagari H.; Massari S.; Ashok Y.; Obaji E.; Nkizinkinko Y.; Lüscher B.; Tabarrini O.; Lehtiö L. 4-(Phenoxy) and 4-(Benzyloxy)Benzamides as Potent and Selective Inhibitors of Mono-ADP-Ribosyltransferase PARP10/ARTD10. Eur. J. Med. Chem. 2018, 156, 93–102. 10.1016/j.ejmech.2018.06.047. [DOI] [PubMed] [Google Scholar]
- Korn P.; Classen A.; Murthy S.; Guareschi R.; Maksimainen M. M.; Lippok B. E.; Galera-Prat A.; Sowa S. T.; Voigt C.; Rossetti G.; Lehtiö L.; Bolm C.; Lüscher B. Evaluation of 3- and 4-Phenoxybenzamides as Selective Inhibitors of the Mono-ADP-Ribosyltransferase PARP10. ChemistryOpen 2021, 10, 939–948. 10.1002/open.202100087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan R. K.; Kirby I. T.; Vermehren-Schmaedick A.; Rodriguez K.; Cohen M. S. Rational Design of Cell-Active Inhibitors of PARP10. ACS Med. Chem. Lett. 2019, 10, 74–79. 10.1021/acsmedchemlett.8b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holechek J.; Lease R.; Thorsell A.-G.; Karlberg T.; McCadden C.; Grant R.; Keen A.; Callahan E.; Schüler H.; Ferraris D. Design, Synthesis and Evaluation of Potent and Selective Inhibitors of Mono-(ADP-Ribosyl)Transferases PARP10 and PARP14. Bioorg. Med. Chem. Lett. 2018, 28, 2050–2054. 10.1016/j.bmcl.2018.04.056. [DOI] [PubMed] [Google Scholar]
- Nizi M. G.; Maksimainen M. M.; Murthy S.; Massari S.; Alaviuhkola J.; Lippok B. E.; Sowa S. T.; Galera-Prat A.; Prunskaite-Hyyryläinen R.; Lüscher B.; Korn P.; Lehtiö L.; Tabarrini O. Potent 2,3-Dihydrophthalazine-1,4-Dione Derivatives as Dual Inhibitors for Mono-ADP-Ribosyltransferases PARP10 and PARP15. Eur. J. Med. Chem. 2022, 237, 114362. 10.1016/j.ejmech.2022.114362. [DOI] [PubMed] [Google Scholar]
- Qin W.; Wu H.-J.; Cao L.-Q.; Li H.-J.; He C.-X.; Zhao D.; Xing L.; Li P.-Q.; Jin X.; Cao H.-L. Research Progress on PARP14 as a Drug Target. Front. Pharmacol. 2019, 10, 172. 10.3389/fphar.2019.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra P.; Riley J. P.; Patel R.; Li F.; Voss L.; Goenka S. PARP-14 functions as a transcriptional switch for Stat6-dependent gene activation. J. Biol. Chem. 2011, 286, 1767. 10.1074/jbc.M110.157768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goenka S.; Cho S. H.; Boothby M. Collaborator of Stat6 (CoaSt6)-Associated Poly(ADP-Ribose) Polymerase Activity Modulates Stat6-Dependent Gene Transcription. J. Biol. Chem. 2007, 282, 18732–18739. 10.1074/jbc.m611283200. [DOI] [PubMed] [Google Scholar]
- Schenkel L. B.; Molina J. R.; Swinger K. K.; Abo R.; Blackwell D. J.; Lu A. Z.; Cheung A. E.; Church W. D.; Kunii K.; Kuplast-Barr K. G.; Majer C. R.; Minissale E.; Mo J.-R.; Niepel M.; Reik C.; Ren Y.; Vasbinder M. M.; Wigle T. J.; Richon V. M.; Keilhack H.; Kuntz K. W. A Potent and Selective PARP14 Inhibitor Decreases Protumor Macrophage Gene Expression and Elicits Inflammatory Responses in Tumor Explants. Cell Chem. Biol. 2021, 28, 1158–1168. 10.1016/j.chembiol.2021.02.010. [DOI] [PubMed] [Google Scholar]
- Gozgit J. M.; Vasbinder M. M.; Abo R. P.; Kunii K.; Kuplast-Barr K. G.; Gui B.; Lu A. Z.; Molina J. R.; Minissale E.; Swinger K. K.; Wigle T. J.; Blackwell D. J.; Majer C. R.; Ren Y.; Niepel M.; Varsamis Z. A.; Nayak S. P.; Bamberg E.; Mo J.-R.; Church W. D.; Mady A. S. A.; Song J.; Utley L.; Rao P. E.; Mitchison T. J.; Kuntz K. W.; Richon V. M.; Keilhack H. PARP7 Negatively Regulates the Type I Interferon Response in Cancer Cells and Its Inhibition Triggers Antitumor Immunity. Cancer Cell 2021, 39, 1214. 10.1016/j.ccell.2021.06.018. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Grosskurth S. E.; Cheung T.; Petteruti P.; Zhang J.; Wang X.; Wang W.; Gharahdaghi F.; Wu J.; Su N.; Howard R. T.; Mayo M.; Widzowski D.; Scott D. A.; Johannes J. W.; Lamb M. L.; Lawson D.; Dry J. R.; Lyne P. D.; Tate E. W.; Zinda M.; Mikule K.; Fawell S. E.; Reimer C.; Chen H. Pharmacological Inhibition of PARP6 Triggers Multipolar Spindle Formation and Elicits Therapeutic Effects in Breast Cancer. Cancer Res. 2018, 78, 6691–6702. 10.1158/0008-5472.can-18-1362. [DOI] [PubMed] [Google Scholar]
- Kirby I. T.; Kojic A.; Arnold M. R.; Thorsell A.-G.; Karlberg T.; Vermehren-Schmaedick A.; Sreenivasan R.; Schultz C.; Schüler H.; Cohen M. S. A Potent and Selective PARP11 Inhibitor Suggests Coupling between Cellular Localization and Catalytic Activity. Cell Chem. Biol. 2018, 25, 1547–1553. 10.1016/j.chembiol.2018.09.011. [DOI] [PubMed] [Google Scholar]
- Yuen L. H.; Dana S.; Liu Y.; Bloom S. I.; Thorsell A.-G.; Neri D.; Donato A. J.; Kireev D.; Schüler H.; Franzini R. M. A Focused DNA-Encoded Chemical Library for the Discovery of Inhibitors of NAD+-Dependent Enzymes. J. Am. Chem. Soc. 2019, 141, 5169–5181. 10.1021/jacs.8b08039. [DOI] [PubMed] [Google Scholar]
- Maksimainen M. M.; Murthy S.; Sowa S. T.; Galera-Prat A.; Rolina E.; Heiskanen J. P.; Lehtiö L. Analogs of TIQ-A as Inhibitors of Human Mono-ADP-Ribosylating PARPs. Bioorg. Med. Chem. 2021, 52, 116511. 10.1016/j.bmc.2021.116511. [DOI] [PubMed] [Google Scholar]
- Nizi M. G.; Maksimainen M. M.; Lehtiö L.; Tabarrini O. Medicinal Chemistry Perspective on Targeting Mono-ADP-Ribosylating PARPs with Small Molecules. J. Med. Chem. 2022, 65, 7532–7560. 10.1021/acs.jmedchem.2c00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz M. D.; Majumdar D.; Chin D. N.; Fortin P. D.; Feng Y.; Gould T.; Kirby C. A.; Stams T.; Waters N. J.; Shao W. Structure-Efficiency Relationship of [1,2,4]Triazol-3-Ylamines as Novel Nicotinamide Isosteres That Inhibit Tankyrases. J. Med. Chem. 2013, 56, 7049–7059. 10.1021/jm400826j. [DOI] [PubMed] [Google Scholar]
- Galera-Prat A.; Alaviuhkola J.; Alanen H. I.; Lehtiö L. Protein Engineering Approach to Enhance Activity Assays of Mono-ADP-Ribosyltransferases through Proximity. Protein Eng., Des. Sel. 2022, 35, gzac006. 10.1093/protein/gzac006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L.; Kerns E. H.; Fan K.; McConnell O. J.; Carter G. T. High Throughput Artificial Membrane Permeability Assay for Blood-Brain Barrier. Eur. J. Med. Chem. 2003, 38, 223–232. 10.1016/s0223-5234(03)00012-6. [DOI] [PubMed] [Google Scholar]
- Ferraris D. V. Evolution of Poly(ADP-Ribose) Polymerase-1 (PARP-1) Inhibitors. From Concept to Clinic. J. Med. Chem. 2010, 53, 4561–4584. 10.1021/jm100012m. [DOI] [PubMed] [Google Scholar]
- Yoneyama-Hirozane M.; Matsumoto S.-I.; Toyoda Y.; Saikatendu K. S.; Zama Y.; Yonemori K.; Oonishi M.; Ishii T.; Kawamoto T. Identification of PARP14 Inhibitors Using Novel Methods for Detecting Auto-Ribosylation. Biochem. Biophys. Res. Commun. 2017, 486, 626–631. 10.1016/j.bbrc.2017.03.052. [DOI] [PubMed] [Google Scholar]
- Wahlberg E.; Karlberg T.; Kouznetsova E.; Markova N.; Macchiarulo A.; Thorsell A.-G.; Pol E.; Frostell Å.; Ekblad T.; Öncü D.; Kull B.; Robertson G. M.; Pellicciari R.; Schüler H.; Weigelt J. Family-Wide Chemical Profiling and Structural Analysis of PARP and Tankyrase Inhibitors. Nat. Biotechnol. 2012, 30, 283–288. 10.1038/nbt.2121. [DOI] [PubMed] [Google Scholar]
- Paget C. J.S-Triazolo [5,1-b]benzothiazoles as fungicidal agents. U.S. Patent 3,974,286 A, August 10, 1976.
- Deng X.-Q.; Song M.-X.; Wei C.-X.; Li F.-N.; Quan Z.-S. Synthesis and Anticonvulsant Activity of 7-Alkoxy-Triazolo-[3, 4-b] Benzo[d]Thiazoles. Med. Chem. 2010, 6, 313–320. 10.2174/157340610793358855. [DOI] [PubMed] [Google Scholar]
- Mamatha S. V.; Belagali S. L.; Bhat M. Synthesis and SAR Evaluation of Mercaptotriazolo derivatives as anti-inflammatory agents. Int. J. ChemTech Res. 2020, 13, 187–198. 10.20902/ijctr.2019.130123. [DOI] [Google Scholar]
- Sun K.; Mikule K.; Wang Z.; Poon G.; Vaidyanathan A.; Smith G.; Zhang Z.-Y.; Hanke J.; Ramaswamy S.; Wang J. A Comparative Pharmacokinetic Study of PARP Inhibitors Demonstrates Favorable Properties for Niraparib Efficacy in Preclinical Tumor Models. Oncotarget 2018, 9, 37080–37096. 10.18632/oncotarget.26354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizilbash S. H.; Gupta S. K.; Chang K.; Kawashima R.; Parrish K. E.; Carlson B. L.; Bakken K. K.; Mladek A. C.; Schroeder M. A.; Decker P. A.; Kitange G. J.; Shen Y.; Feng Y.; Protter A. A.; Elmquist W. F.; Sarkaria J. N. Restricted Delivery of Talazoparib Across the Blood-Brain Barrier Limits the Sensitizing Effects of PARP Inhibition on Temozolomide Therapy in Glioblastoma. Mol. Cancer Ther. 2017, 16, 2735–2746. 10.1158/1535-7163.mct-17-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durmus S.; Sparidans R. W.; van Esch A.; Wagenaar E.; Beijnen J. H.; Schinkel A. H. Breast Cancer Resistance Protein (BCRP/ABCG2) and P-Glycoprotein (P-GP/ABCB1) Restrict Oral Availability and Brain Accumulation of the PARP Inhibitor Rucaparib (AG-014699). Pharm. Res. 2015, 32, 37–46. 10.1007/s11095-014-1442-z. [DOI] [PubMed] [Google Scholar]
- Hanna C.; Kurian K. M.; Williams K.; Watts C.; Jackson A.; Carruthers R.; Strathdee K.; Cruickshank G.; Dunn L.; Erridge S.; Godfrey L.; Jefferies S.; McBain C.; Sleigh R.; McCormick A.; Pittman M.; Halford S.; Chalmers A. Pharmacokinetics, safety, and tolerability of olaparib and temozolomide for recurrent glioblastoma: results of the phase I OPARATIC trial. Neuro Oncol. 2020, 22, 1840–1850. 10.1093/neuonc/noaa104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim H.-W.; Galanis E.; Khasraw M. PARP Inhibitors in Glioma: A Review of Therapeutic Opportunities. Cancers 2022, 14, 1003. 10.3390/cancers14041003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gvozdjakova A.; Ivanovičová H. Synthesis and Reactions of Both Tautomers of 2-Hydrazinobenzothiazole. Chem. Pap. 1986, 40, 797–800. [Google Scholar]
- Kuberkar S. V.; Bhosale V. N.; Vartale S. P.; Badne S. G. Synthesis and Some Novel Reactions of 8-Chloro-2H-[1,2,4]Triazino[3,4-b] [1,3]Benzothiazole-3,4-Dione and 6-Chloro-2-Hydrazino-1,3-Benzothiazole. J. Chem. Res. 2005, 2005, 632–635. 10.3184/030823405774663084. [DOI] [Google Scholar]
- Swamy D. K.; Deshmukh M. V. A Facile Conversion of Triazolobenzothiazole-3-Thione to Triazolobenzothiazole-3-Ones by Sulphur Extrusion. J. Res. Pharm. Chem. 2010, 2, 167–170. [Google Scholar]
- Peet N. P.; Sunder S.; Barbuch R. J.; Whalon M. R.; Huffman J. C. Reinvestigation of the Condensation of 2-Hydrazinobenzothiazole with Ethyl Acetoacetate. J. Heterocycl. Chem. 1988, 25, 543–547. 10.1002/jhet.5570250235. [DOI] [Google Scholar]
- Hay M. P.; Turcotte S.; Flanagan J. U.; Bonnet M.; Chan D. A.; Sutphin P. D.; Nguyen P.; Giaccia A. J.; Denny W. A. 4-Pyridylanilinothiazoles That Selectively Target von Hippel–Lindau Deficient Renal Cell Carcinoma Cells by Inducing Autophagic Cell Death. J. Med. Chem. 2010, 53, 787–797. 10.1021/jm901457w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal J.-C.; Pinhas H.; Laure F.; Dumez D.; Poizot A. New antiarrhythmic agents. Piperazine guanidine derivatives. Eur. J. Med. Chem. 1990, 25, 81–85. 10.1016/0223-5234(90)90169-4. [DOI] [Google Scholar]
- Vogt D.; Weber J.; Ihlefeld K.; Brüggerhoff A.; Proschak E.; Stark H. Design, Synthesis and Evaluation of 2-Aminothiazole Derivatives as Sphingosine Kinase Inhibitors. Bioorg. Med. Chem. 2014, 22, 5354–5367. 10.1016/j.bmc.2014.07.044. [DOI] [PubMed] [Google Scholar]
- Gangadhar S. P.; Ramesh D. K.; Mahajan S. K. Synthesis, Characterisation and Anticonvulsant Activity of 3-Substituted 2-Thiohydantoin Derivatives. Int. J. Res. Pharm. Chem. 2013, 3, 793–796. [Google Scholar]
- Feng Y.; Zou M.; Song R.; Shao X.; Li Z.; Qian X. Formation of 1,4,2-Dithiazolidines or 1,3-Thiazetidines from 1,1-Dichloro-2-Nitroethene and Phenylthiourea Derivatives. J. Org. Chem. 2016, 81, 10321–10327. 10.1021/acs.joc.6b01307. [DOI] [PubMed] [Google Scholar]
- Xu H.; Su X.; Liu X.-Q.; Zhang K.-P.; Hou Z.; Guo C. Design, Synthesis and Biological Evaluation of Novel Semicarbazone-Selenochroman-4-Ones Hybrids as Potent Antifungal Agents. Bioorg. Med. Chem. Lett. 2019, 29, 126726. 10.1016/j.bmcl.2019.126726. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang Y.; Chen Z.; Chen L.; Feng T.; Huang R.; Li Q.; Li D.; Sun J.; Xu Y.; Li J.; Li J.; Chen S.. Fgfr Inhibitor and Medical Application Thereof, WO 2019034076A1, February 21, 2019.
- Patel N.; Agravat S. N.; Shaikh F. M. Synthesis and Antimicrobial Activity of New Pyridine Derivatives-I. Med. Chem. Res. 2011, 20, 1033–1041. 10.1007/s00044-010-9440-0. [DOI] [Google Scholar]
- He W.-B.; Gao L.-Q.; Chen X.-J.; Wu Z.-L.; Huang Y.; Cao Z.; Xu X.-H.; He W.-M. Visible-Light-Initiated Malic Acid-Promoted Cascade Coupling/Cyclization of Aromatic Amines and KSCN to 2-Aminobenzothiazoles without Photocatalyst. Chin. Chem. Lett. 2020, 31, 1895–1898. 10.1016/j.cclet.2020.02.011. [DOI] [Google Scholar]
- Wang H.; Hong X.; Zhu X.; Wang P.; LV G.; Fu J.; Luo H.; Zhang J.; Wen M.; Qu C.; Zhu J.; Hu X.. Benzothiazole Derivative and Anti-Tumor Use Thereof. U.S. Patent 20,160,102,066 A1, 2016.
- Hasebe H.; Aoki Y.; Kotani K.; Koiso A.; Nakata H.. Polymerizable Compound, Composition, Polymer, Optically Anisotropic Body, Liquid Crystal Display Device, and Organic El Device. U.S. Patent 20,170,002,123 A1, January 5, 2017.
- Chen G.; Niu C.; Yi J.; Sun L.; Cao H.; Fang Y.; Jin T.; Li Y.; Lou C.; Kang J.; Wei W.; Zhu J. Novel Triapine Derivative Induces Copper-Dependent Cell Death in Hematopoietic Cancers. J. Med. Chem. 2019, 62, 3107–3121. 10.1021/acs.jmedchem.8b01996. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Fan J.; Vu K.; Hong K.; Le Brazidec J.-Y.; Shi J.; Biamonte M.; Busch D. J.; Lough R. E.; Grecko R.; Ran Y.; Sensintaffar J. L.; Kamal A.; Lundgren K.; Burrows F. J.; Mansfield R.; Timony G. A.; Ulm E. H.; Kasibhatla S. R.; Boehm M. F. 7’-Substituted Benzothiazolothio- and Pyridinothiazolothio-Purines as Potent Heat Shock Protein 90 Inhibitors. J. Med. Chem. 2006, 49, 5352–5362. 10.1021/jm051146h. [DOI] [PubMed] [Google Scholar]
- Wang S.-M.; Zha G.-F.; Rakesh K. P.; Darshini N.; Shubhavathi T.; Vivek H. K.; Mallesha N.; Qin H.-L. Synthesis of benzo[d]thiazole-hydrazone analogues: molecular docking and SAR studies of potential H+/K+ATPase inhibitors and anti-inflammatory agents. MedChemComm 2017, 8, 1173–1189. 10.1039/c7md00111h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munirajasekhar D.; Himaja M.; Mali S. V. A Facile and Efficient Synthesis of 2-(5-(4-Substitutedphenyl)-4, 5-Dihydro-3-Phenylpyrazol-1-Yl)-6-Substitutedbenzothiazoles and Their Biological Studies. J. Heterocycl. Chem. 2014, 51, 459–465. 10.1002/jhet.1618. [DOI] [Google Scholar]
- van den Berg S.; Löfdahl P.-A.; Härd T.; Berglund H. Improved Solubility of TEV Protease by Directed Evolution. J. Biotechnol. 2006, 121, 291–298. 10.1016/j.jbiotec.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Narwal M.; Fallarero A.; Vuorela P.; Lehtiö L. Homogeneous Screening Assay for Human Tankyrase. J. Biomol. Screen 2012, 17, 593–604. 10.1177/1087057112436558. [DOI] [PubMed] [Google Scholar]
- Venkannagari H.; Fallarero A.; Feijs K. L.; Lüscher B.; Lehtiö L. Activity-based assay for human mono-ADP-ribosyltransferases ARTD7/PARP15 and ARTD10/PARP10 aimed at screening and profiling inhibitors. Eur. J. Pharm. Sci. 2013, 49, 148–156. 10.1016/j.ejps.2013.02.012. [DOI] [PubMed] [Google Scholar]
- Putt K. S.; Hergenrother P. J. An enzymatic assay for poly(ADP-ribose) polymerase-1 (PARP-1) via the chemical quantitation of NAD+: application to the high-throughput screening of small molecules as potential inhibitors. Anal. Biochem. 2004, 326, 78–86. 10.1016/j.ab.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Daniel E.; Maksimainen M. M.; Smith N.; Ratas V.; Biterova E.; Murthy S. N.; Rahman M. T.; Kiema T. R.; Sridhar S.; Cordara G.; Dalwani S.; Venkatesan R.; Prilusky J.; Dym O.; Lehtiö L.; Koski M. K.; Ashton A. W.; Sussman J. L.; Wierenga R. K. IceBear: An Intuitive and Versatile Web Application for Research-Data Tracking from Crystallization Experiment to PDB Deposition. Acta Crystallogr., Sect. D: Struct. Biol. 2021, 77, 151–163. 10.1107/s2059798320015223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Integration, Scaling, Space-Group Assignment and Post-Refinement. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 133. 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. Molecular replacement withMOLREP. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 22–25. 10.1107/s0907444909042589. [DOI] [PubMed] [Google Scholar]
- Potterton L.; Agirre J.; Ballard C.; Cowtan K.; Dodson E.; Evans P. R.; Jenkins H. T.; Keegan R.; Krissinel E.; Stevenson K.; Lebedev A.; McNicholas S. J.; Nicholls R. A.; Noble M.; Pannu N. S.; Roth C.; Sheldrick G.; Skubak P.; Turkenburg J.; Uski V.; von Delft F.; Waterman D.; Wilson K.; Winn M.; Wojdyr M. CCP4i2: The New Graphical User Interface to the CCP4 Program Suite. Acta Crystallogr., Sect. D: Struct. Biol. 2018, 74, 68–84. 10.1107/s2059798317016035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsell A.-G.; Ekblad T.; Karlberg T.; Löw M.; Pinto A. F.; Trésaugues L.; Moche M.; Cohen M. S.; Schüler H. Structural Basis for Potency and Promiscuity in Poly(ADP-Ribose) Polymerase (PARP) and Tankyrase Inhibitors. J. Med. Chem. 2017, 60, 1262–1271. 10.1021/acs.jmedchem.6b00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkizinkiko Y.; Desantis J.; Koivunen J.; Haikarainen T.; Murthy S.; Sancineto L.; Massari S.; Ianni F.; Obaji E.; Loza M. I.; Pihlajaniemi T.; Brea J.; Tabarrini O.; Lehtiö L. 2-Phenylquinazolinones as Dual-Activity Tankyrase-Kinase Inhibitors. Sci. Rep. 2018, 8, 1680. 10.1038/s41598-018-19872-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/s0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog N.; Hartkamp J. D. H.; Verheugd P.; Treude F.; Forst A. H.; Feijs K. L. H.; Lippok B.; Kremmer E.; Kleine H.; Lüscher B. Caspase-Dependent Cleavage of the Mono-ADP-Ribosyltransferase ARTD10 Interferes with Its pro-Apoptotic Function. FEBS J. 2013, 280, 1330–1343. 10.1111/febs.12124. [DOI] [PubMed] [Google Scholar]
- Forst A.; Karlberg T.; Herzog N.; Thorsell A.-G.; Gross A.; Feijs K. L. H.; Verheugd P.; Kursula P.; Nijmeijer B.; Kremmer E.; Kleine H.; Ladurner A. G.; Schüler H.; Lüscher B. Recognition of Mono-ADP-Ribosylated ARTD10 Substrates by ARTD8 Macrodomains. Structure 2013, 21, 462–475. 10.1016/j.str.2012.12.019. [DOI] [PubMed] [Google Scholar]
- Bard B.; Martel S.; Carrupt P.-A. High Throughput UV Method for the Estimation of Thermodynamic Solubility and the Determination of the Solubility in Biorelevant Media. Eur. J. Pharm. Sci. 2008, 33, 230–240. 10.1016/j.ejps.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Wohnsland F.; Faller B. High-Throughput Permeability PH Profile and High-Throughput Alkane/Water Log P with Artificial Membranes. J. Med. Chem. 2001, 44, 923–930. 10.1021/jm001020e. [DOI] [PubMed] [Google Scholar]
- Sugano K.; Hamada H.; Machida M.; Ushio H. High Throughput Prediction of Oral Absorption: Improvement of the Composition of the Lipid Solution Used in Parallel Artificial Membrane Permeation Assay. J. Biomol. Screen 2001, 6, 189–196. 10.1177/108705710100600309. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. A. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Barthel B. L.; Rudnicki D. L.; Kirby T. P.; Colvin S. M.; Burkhart D. J.; Koch T. H. Synthesis and Biological Characterization of Protease-Activated Prodrugs of Doxazolidine. J. Med. Chem. 2012, 55, 6595–6607. 10.1021/jm300714p. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.