

Abstract
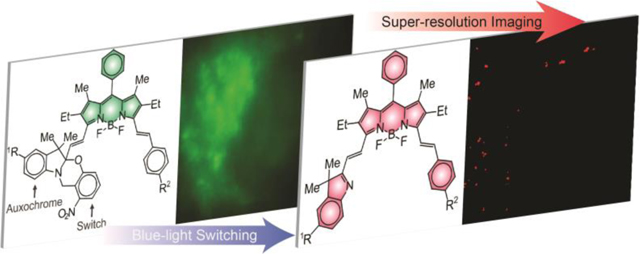
The photoinduced disconnection of an oxazine heterocycle from a borondipyrromethene (BODIPY) chromophore, equipped with a styryl appendage, activates bright far-red fluorescence. The high brightness of the photochemical product together with the lack of any significant autofluorescence in this spectral region allows detection at the single-molecule level even within the intracellular environment of live cells. Indeed, these photoactivatable fluorophores localize in lysosomal compartments and, when functionalized with an active ester, remain covalently immobilized within these organelles. The suppression of any significant diffusion allows the reiterative reconstruction of images with sub-diffraction resolution and the visualization of the labeled intracellular compartments with excellent localization precision in live cells. Thus, the combination of photochemical, photophysical and structural properties designed into these photoactivatable fluorophores translate into the opportunity to visualize live cells with spatial resolution at the nanometer level that is inaccessible to conventional fluorescence imaging schemes.
Graphical Abstract
The diffraction of focused light1 limits the spatial resolution of conventional fluorescence imaging to the micrometer level2 and prevents the visualization of the many nanostructured components present in the intracellular environment.3 These stringent limitations stimulated the development of alternative schemes for the acquisition of images with sub-diffraction resolution.4–6 The ingenious operating principles behind these super-resolution imaging methods are generally based on either patterned illumination with ensemble detection or temporal differentiation of single-molecule events. The former confine the emission of conventional fluorophores within sub-diffraction volumes, relying on the interplay of exciting and depleting laser sources. The latter localizes the emission of immobilized fluorophores sequentially, relying on the stochastic switching of their fluorescence at the single-molecule level. Indeed, a diversity of biological specimens have been imaged already with sub-diffraction resolution on the basis of these protocols and either fluorescent proteins or synthetic dyes.
Most of the synthetic probes developed for super-resolution imaging so far are members of the cyanine and xanthene families of dyes with a few notable exceptions.7–10 In spite of their synthetic versatility and outstanding photophysical properties,11–18 BODIPY chromophores have instead been fairly unexplored in this context, apart from two remarkable examples.19,20 Both are based on photoswitching and detection of BODIPY fluorescence at the single-molecule level. In fact, a number of structural designs to photoactivate the emission of BODIPY fluorophores have actually been reported in the literature already.17–25 Most of them are based on the photoinduced disconnection of a quencher from a BODIPY chromophore with a concomitant fluorescence enhancement. In principle, they could be exploited to reconstruct sub-diffraction images by photoactivated localization microscopy (PALM).26–27 However, the modest contrast ratios inherent to these switching mechanisms complicate detection at the single-molecule level. Additionally, the initial and final states of these systems generally absorb in the same spectral window, preventing the selective photobleaching of the latter in the presence of the former necessary for the reiterative reconstruction of PALM images.
In search of strategies to monitor dynamic events in living organisms with fluorescence measurements, our laboratories devised a photochemical mechanism to shift bathochromically the main absorption of a BODIPY chromophore and allow the selective excitation of the product.28–31 The overall result is fluorescence activation with infinite contrast and the spectral resolution of the absorptions of initial and final states. This behavior suggests that photoactivable BODIPYs with optimal properties for the implementation of PALM schemes might be developed on the basis of these photochemical transformations. This article reports the chemical synthesis and spectroscopic characterization of members of this family of photoactivatable fluorophores specifically designed for PALM and their ability to enable the sub-diffraction visualization of intracellular compartments in live cells.
A boron center locks two conjugated pyrrole heterocycles in a planar arrangement within the BODIPY chromophore.8–15 As a result, BODIPY derivatives absorb electromagnetic radiation with large molar absorption coefficients in the visible region. The two pyrrole rings are generally substituted with alkyl groups to block three of the four available positions on the heterocyclic precursors, direct regioselectively the formation of the methene bridge and facilitate the synthesis of the BODIPY platform. Additionally, the pair of alkyl groups adjacent to the meso-position on the resulting chromophore restrict the conformational freedom of any substituent attached to this particular position, discourage the nonradiative decay of the excited state and ensure high fluorescence quantum yields. Furthermore, the two alkyl groups next to the nitrogen atoms can be condensed with benzaldehydes to extend the π-system over two styryl substituents, shift the BODIPY absorption and emission bands into the far-red region and overcome the characteristic autofluorescence of biological samples.
On the basis of these structural considerations, our laboratory envisaged the possibility of condensing a pre-formed BODIPY chromophore to a photoswitchable auxochrome and a styryl appendage in the shape of 1 and 3 (Figure 1). Both molecules were synthesized in two steps from known precursors (Figure S1) and their structural identity was confirmed by electrospray ionization mass spectrometry (ESIMS) and 1H nuclear magnetic resonance (NMR) spectroscopy. They differ exclusively in the nature of the group (R2 in Figure 1) on position 4 of the styryl substituent, which is a hydrogen atom in 1 and a methoxy group in 3. The corresponding absorption spectra (a in Figures 2 and S3) show the characteristic BODIPY band with a maximum at a wavelength (λAb in Table S1) of 609 nm for 1 and 618 nm for 3 in tetrahydrofuran (THF) and a molar absorption coefficient (ε in Table S1) of 73.0 and 87.6 Mm−1 cm−1 respectively. The emission spectra (c in Figures 2 and S3) also show the characteristic BODIPY band with a maximum at a wavelength (λEm in Table S1) of 624 nm for 1 and 635 nm for 3 and a fluorescence quantum yield (ϕF in Table S1) of 0.85 and 0.80 respectively. These relatively high ε and ϕF values correspond to a brightness (ε × ϕF in Table S1) of 62 mM–1 cm–1 for 1 and 66 mM–1 cm–1 for 3.
Figure 1.

Photoinduced conversion of 1, 3 and 5 into 2, 4 and 6 respectively.
Figure 2.

Normalized absorption and emission spectra of 1 (a and c, λEx = 565 nm) and 2 (b and d, λEx = 620 nm) in THF at 25 °C. Absorption spectra (e) of a THF solution of 1 (10 μM) recorded before and during irradiation (350 nm, 4.2 mW cm–2, 10 min).
Illumination of THF solutions of 1 and 3 at a λAc in the spectral region where their ortho-nitrobenzyl fragment absorbs cleaves irreversibly the 2H,4H-benzo[1,3]oxazine heterocycle to produce 2 and 4 respectively and release 7 (Figure 1). This photoinduced transformation converts the chiral center of the reactant into a sp2 carbon atom in the product and brings the BODIPY chromophore in electronic conjugation with the 3H-indole auxochrome. As a result, the BODIPY absorption shifts bathochromically (b in Figures 2 and S3) to a λAb of 653 nm for 2 and 664 nm for 4 with ε of 72.8 and 86.7 mM–1 cm–1 respectively (Table S1). Indeed, absorption spectra (Figures 3 and S5) of THF solutions of 1 and 3, recorded over the course of the photolytic transformation, show the developing absorption of the photochemical product in both instances. Analysis of the temporal absorbance evolution indicates the quantum yield for the photochemical formation of 2 and 4 to be 0.02 and 0.04 respectively. Furthermore, the pronounced bathochromic shift in absorption associated with the photoinduced conversion of 1 into 3 and of 2 into 4 allows the selective excitation of the photochemical product with concomitant fluorescence. Specifically, 2 emits at a λEm of 666 nm with a ϕF of 0.41 and a ε × ϕF of 30 mM−1 cm−1 and 4 produces fluorescence at a λEm of 679 nm with a ϕF of 0.23 and a ε × ϕF of 20 mM−1 cm−1.
Figure 3.

Emission spectrum (a, λEx = 642 nm), averaged over 8,971 single molecules, of a photoactivated (λAc = 405 nm) PMMA film doped with 1 and spectral evolution (b) of a single molecule after photoactivation. Plots (c and d) of the emission intensity against the spectral centroid and spectral centroid distributions (insets) with Gaussian fitting, showing the spectral fluctuation of a single molecule (c) and the spectral heterogeneity (d) among 8,971 photoactivation events respectively.
The depressive effect of the methoxy group on the ϕF of the photochemical product suggested the structural modification of the unsubstituted system (i.e., 1) only to permit bioconjugation. In particular, an active ester (R1 in Figure 1) was introduced in position 5 of the 2H,3H-indole heterocycle of 1 to generate 5. This molecule was prepared in two synthetic steps from known precursors (Figure S2) and its structural identity was confirmed by ESIMS and 1H NMR spectroscopy. Its absorption and emission spectra (a and c in Figure S3) show the active ester to have negligible influence on the position of the BODIPY bands, in agreement with the fact that the photocleavable auxochrome and fluorescent chromophore are isolated electronically in the ground state. Indeed, λAb, λEm and ε for 5 are essentially the same of those measured for 1 (Table S1). Additionally, the active ester does not seem to affect the excitation dynamics and the ϕF of 5 remains close to that of the parent compound (Table S1).
Photolysis of 5 cleaves the oxazine heterocycle to generate 6 and 7 (Figure 1), as observed for the parent system. Absorption spectra (e in Figure 2), recorded during the course of the photochemical transformation, reveal the developing absorption of 6 at a λAb of 656 nm with a ε of 76.8 mM−1 cm−1 (b in Figure S3 and Table S1). These values differ slightly from those of the parent system (i.e., 2), consistently with the fact that the photoinduced disconnection of the oxazine heterocycle brings the active ester in conjugation with the BODIPY chromophore. Analysis of the temporal absorbance evolution, during photolysis, indicates the quantum yield for the photochemical transformation to be 0.01. Selective excitation of the photochemical product within the developing absorption generates fluorescence (d in Figure S4) at a λEm of 669 nm and a ϕF of 0.40 (Table S1). Once again, these values remain remarkably similar to those of the parent system. Thus, our photoactivatable BODIPY retains its photochemical and photophysical properties almost unaffected even after the attachment of an active ester to its molecular skeleton.
The excellent photophysical properties of our photoactivatable BODIPY‒oxazine dyads permit the characterization of their fluorescence at the single-molecule level. Specifically, the photoactivation of 1 can be monitored within a poly(methyl methacrylate) (PMMA) film using a spectroscopic photon localization microscopy (SPLM) setup, which simultaneously collects the spatial and spectral information of single-molecule emission events.32 Illumination of the doped polymer film at a λEx of 642 nm shows minimal fluorescence because the absorbance of 1 is negligible at this wavelength. After activation at 405 nm, a strong fluorescence signal is detected at the single-molecule level, under the same 642-nm excitation conditions, as a result of photoinduced transformation of 1 into 2. The averaged single-molecule emission spectrum (a in Figure 3) of 8,971 photoactivated species shows a λEm of 670 nm, confirming that the photoactivation process can be replicated in the PMMA matrix at the single-molecule level.
The ultrahigh brightness of 2 permits the observation of the spectral evolution for a single photoactivation process. Upon photoactivation, a single molecule of 2 can be detected unambiguously and its emission spectrum can be monitored until photobleaching (b in Figure 3). The emission intensity and spectrum fluctuate over the entire acquisition time (c in Figure 3). The standard deviation for the spectral centroid distribution (σSc) is 1.74 nm. The relatively high resistance of 2 to photodegradation requires an illumination power of 500 W cm‒2 at 642 nm to ensure photobleaching in tens of milliseconds and enable the high-throughput detection of multiple photoactivation events at the single-molecule level. The emission intensities and spectra of all photoactivated molecules (8,971) reveal larger variations than a single one (c and d in Figure 4). The spectral centroid fluctuates for as many as 60 nm with a σSc of 5.0 nm. Presumably, environmental inhomogeneities across different regions of the substrate are responsible for the relatively large spectral heterogeneity, compared to the spectral fluctuations of a single molecule. Indeed, differences in the local environment around individual emitters are known to affect their spectral output.33
Figure 4.

CLSM images (scale bar = 10 μm) of live COS-7 cells incubated with 5 (a, d and g; λEx = 561 nm, λEm = 575‒650 nm) and lysosomal (b), mitochondrial (e) or nuclear (h) stains (λEx = 488 nm, λEm = 500‒550 nm) together with the corresponding overlays (c, f and i).
Images, acquired with a confocal laser-scanning microscope (CLSM), of COS-7 cells incubated with either 1 (a in Figure S7) or 5 (a, d and g in Figure 4) show intense intracellular fluorescence upon excitation at a λEx of 561 nm. Co-incubation with lysosomal (b in Figures 5 and S7), mitochondrial (e in Figure 4) or nuclear (h in Figure 4) stains reveals exclusive co-localization in the lysosomes (c in Figures 4 and S7) with a Pearson’s coefficient of 0.98. Neither mitochondrial (f in Figure 4) nor nuclear (i in Figure 4) co-localization can be detected.
Figure 5.

PALM image (a, scale bar = 2 μm) of a COS-7 cell, labeled with 5, with magnification (b and c, scale bar = 500 nm) of the regions highlighted in blue and green together with the corresponding epi-fluorescence image (inset in b). Representative single-molecule blinking trajectory (d) recorded within a 7 × 7 pixel area in the same sample after photoactivation over the entire acquisition time. Line measurements (e) with Gaussian fitting of individual lysosomes in b (blue) and c (green). Photon count (f) and localization uncertainty (g) parameters of the single-molecule photoactivation events.
The internalized BODIPY‒oxazine dyads (1 or 5) can be photoactivated in the intracellular environment of live COS-7 cells and to track the bright photochemical products (2 or 6) at the single-molecule level. Interestingly, 2 appears to diffuse for hundreds of nanometers intracellularly (Figure S8), while 6 remains localized at the photoactivated site for prolonged periods of time (Figure S9). Indeed, the active ester of 5 is designed to anchor the internalized photoactivatable fluorophores to the abundant primary amines of the intracellular proteins. In fact, this particular functional group is routinely employed to immobilize synthetic probes intracellularly.34
The suppression of any significant intracellular diffusion and high brightness engineered into our photoactivatable fluorophores provide the opportunity to reconstruct super-resolution images of live cells with optimal localization performance. In particular, the lysosomal localization of the photoactivatable probes, evident from the CLSM images of COS-5 cells incubated with 5 (Figure 4), can be exploited to visualize these particular intracellular compartments with spatial resolution at the nanometer level on the basis of PALM. Furthermore, the photochemical mechanism for fluorescence activation designed into our molecules permits the acquisition of PALM images in the cell-growth medium without the addition of oxygen scavengers or antioxidants.19 Once again, 5 switches to 6, under mild 405-nm illumination, and the latter can be selectively excited with a λEx of 642 nm. Within tens of milliseconds, the photoactivated fluorophores photobleach irreversibly and cannot produce further fluorescence. As a result, the reiterative photoactivation and photobleaching of sub-populations of 5 and 6 respectively enable the reconstruction of sub-diffraction images of the labeled lysosomes with localization precision at the nanometer level. Specifically, the intensity trajectory (a Figure 5) of the photoactivation/photobleaching process from a 7 × 7 pixel area shows distinct intensity spikes over the entire acquisition time. The reconstructed images (b‒d in Figure 5) show individual lysosomes that would otherwise be impossible to resolve with conventional epi-fluorescence microscopy. The line profiles of the green and blue highlighted regions indicate the width of individual lysosomes to be ca. 80 nm (e in Figure 5), consistently with literature values.35 Additionally, the localization precision is ca. 15 nm with a mean photon count of ca. 2,000 (f and g in Figure 5). Thus, these results demonstrate the photochemical and photophysical properties designed into our BODIPY‒oxazine dyads to be appropriate for the acquisition of super-resolution images of intracellular compartments in live cells.
Condensations of a photocleavable oxazine heterocycle at one end of a BODIPY fluorophore and of a styryl appendage at the other enable the photoactivation of bright fluorescence in the far-red region of the electromagnetic spectrum. The additional introduction of an active ester on the molecular skeleton of these photoactivatable fluorophores allows their covalent immobilization within lysosomal compartments of live cells. The lack of any significant diffusion in the intracellular environment, the mild visible illumination sufficient for activation and the bright emission in a spectral window with negligible autofluorescence permit the unprecedented visualization of organelles in live cells with a localization uncertainty smaller than 15 nm. Thus, our structural design for fluorescence activation can evolve into invaluable analytical tools for the single-molecule tracking and localization microscopy of intracellular components in live cells.
Supplementary Material
ACKNOWLEDGMENT
The National Science Foundation (CHE-1505885, CBET-1706642 and EEC-1530734), National Institute of Health (R01EY026078 and R01EY029121) and Northwestern University Innovative Initiative Incubator (I3) Award are acknowledged for financial support.
H.F.Z. and C.S. have financial interests in Opticent Health, which did not fund this work.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: #.
Experimental procedures; spectroscopic data; fluorescence images
All remaining authors declare no competing financial interests.
REFERENCES
- (1).Born M; Wolf E, Principles of Optics. Cambridge University Press: Cambridge: 2002. [Google Scholar]
- (2).Murphy DB, Fundamentals of Light Microscopy and Electronic Imaging. Wiley-Liss: New York: 2001. [Google Scholar]
- (3).Pawley JB (Ed.), Handbook of Biological Confocal Microscopy. Springer: New York: 2006. [Google Scholar]
- (4).Betzig E.Angew. Chem. Int. Ed 2015, 54, 8034–8053. [DOI] [PubMed] [Google Scholar]
- (5).Hell SW Angew. Chem. Int. Ed 2015, 54, 8054–8066. [DOI] [PubMed] [Google Scholar]
- (6).Moerner WE Angew. Chem. Int. Ed 2015, 54, 8067–8093. [DOI] [PubMed] [Google Scholar]
- (7).Raymo FM Phys. Chem. Chem. Phys 2013, 15, 14840–14850. [DOI] [PubMed] [Google Scholar]
- (8).van de Linde S; Sauer M.Chem. Soc. Rev 2014, 43, 1076–1087. [DOI] [PubMed] [Google Scholar]
- (9).Gorka AP; Nani RR; Schnermann MJ Org. Biomol. Chem 2015, 13, 7584–7598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lavis LD Annu. Rev. Biochem 2017, 86, 825–843. [DOI] [PubMed] [Google Scholar]
- (11).Loudet A; Burgess K.Chem. Rev 2007, 107, 4891–4932. [DOI] [PubMed] [Google Scholar]
- (12).Ziessel R; Ulrich G; Harriman A.New J. Chem 2007, 31, 496–501. [Google Scholar]
- (13).Ulrich G; Ziessel R; Harriman A.Angew. Chem. Int. Ed 2008, 47, 1184–1201. [DOI] [PubMed] [Google Scholar]
- (14).Benstead M; Mehl GH; Boyle RW Tetrahedron 2011, 67, 3573–3601. [Google Scholar]
- (15).Boens N; Leen V; Dehaen W.Chem. Soc. Rev 2012, 41, 1130–1172. [DOI] [PubMed] [Google Scholar]
- (16).Kamkaew A; Lim SH; Lee HB; Kiew LV; Chung LY; Burgess K.Chem. Soc. Rev 2013, 42, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lu H; Mack J; Yang Y; Shen Z.Chem. Soc. Rev 2014, 43, 4778–4823. [DOI] [PubMed] [Google Scholar]
- (18).Ni Y; Wu J.Org. Biomol. Chem 2014, 12, 3774–3791. [DOI] [PubMed] [Google Scholar]
- (19).Shim S-H; Xia C; Zhong G; Babcock HP; Vaughan JC; Huang B; Wang X; Xu C; Bi G-Q; Zhuang X.Proc. Natl. Acad. Sci. USA 2012, 109, 13978–13983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wijesooriya CS; Peterson JA; Shrestha P; Gehrmann EJ; Winter AH; Smith E.Angew. Chem. Int. Ed 2018, 57, 10.1002/anie.201805827. [DOI] [PubMed] [Google Scholar]
- (21).Kobayashi T; Komatsu T; Kamiya M; Campos C; González-Gaitán M; Terai T; Hanaoka K; Nagano T; Urano YJ Am. Chem. Soc 2012, 134, 11153–11160. [DOI] [PubMed] [Google Scholar]
- (22).Ragab SS; Swaminathan S; Baker JD; Raymo FM Phys. Chem. Chem. Phys 2013, 15, 14851–14855. [DOI] [PubMed] [Google Scholar]
- (23).Shaban Ragab S; Swaminathan S; Deniz E; Captain B; Raymo FM Org. Lett 2013, 15, 3153–3157. [DOI] [PubMed] [Google Scholar]
- (24).Amamoto T; Hirata T; Takahashi H; Kamiya M; Urano Y; Santa T; Kato MJ Mater. Chem. B 2015, 3, 7427–7433. [DOI] [PubMed] [Google Scholar]
- (25).Goswami PP; Syed A; Beck CL; Albright TR; Mahoney KM; Unash R; Smith EA; Winter AH J. Am. Chem. Soc 2015, 137, 3783–3786. [DOI] [PubMed] [Google Scholar]
- (26).Hess ST; Girirajan TPK; Mason MD Biophys. J 2006, 91, 4258–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Betzig E; Patterson GH; Sougrat R; Lindwasser OW; Olenych S; Bonifacino JS; Davidson MW; Lippincott-Schwartz J; Hess HF Science 2006, 313, 1642–1645. [DOI] [PubMed] [Google Scholar]
- (28).Zhang Y; Swaminathan S; Tang S; Garcia-Amorós J; Boulina M; Captain B; Baker JD; Raymo FM J. Am. Chem. Soc 2015, 137, 4709–4719. [DOI] [PubMed] [Google Scholar]
- (29).Zhang Y; Tang S; Sansalone L; Baker JD; Raymo FM Chem. Eur. J 2016, 22, 15027–15034. [DOI] [PubMed] [Google Scholar]
- (30).Liu X; Zhang Y; Baker JD; Raymo FM J. Mater. Chem. C 2017, 5, 12714–12719. [Google Scholar]
- (31).Sansalone L; Tang S; Garcia-Amoros J; Zhang Y; Nonell S; Baker JD; Captain B; Raymo FM ACS Sens. 2018, 3, 1347–1353. [DOI] [PubMed] [Google Scholar]
- (32).Dong B; Almassalha L; Urban BE; Nguyen T-Q; Khuon S; Chew T-L; Backman V; Sun C; Zhang HF Nat. Commun 2016, 7, 12290–1‒8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lu HP; Xie XS Nature 1997, 385, 143–146. [Google Scholar]
- (34).Johnson ID (Ed), Molecular Probes Handbook: A Guide to Fluorescent Probes and Labeling Technologies. Life Technologies Corporation: Grand Island: 2010. [Google Scholar]
- (35).He H; Ye Z; Zheng Y; Xu X; Guo C; Xiao Y; Yang W; Qian X; Yang Y.Chem. Commun 2018, 54, 2842–2845. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.