

Abstract
The aryl hydrocarbon receptor (AHR) is a ligand-dependent transcription factor regulating adaptive and maladaptive responses toward exogenous and endogenous signals. Research from various biomedical disciplines has provided compelling evidence that the AHR is critically involved in the pathogenesis of a variety of diseases and disorders, including autoimmunity, inflammatory diseases, endocrine disruption, premature aging and cancer. Accordingly, AHR is considered an attractive target for the development of novel preventive and therapeutic measures. However, the ligand-based targeting of AHR is considerably complicated by the fact that the receptor does not always follow the beaten track, i.e. the canonical AHR/ARNT signaling pathway. Instead, AHR might team up with other transcription factors and signaling molecules to shape gene expression patterns and associated physiological or pathophysiological functions in a ligand-, cell- and micromilieu-dependent manner. Herein, we provide an overview about some of the most important non-canonical functions of AHR, including crosstalk with major signaling pathways involved in controlling cell fate and function, immune responses, adaptation to low oxygen levels and oxidative stress, ubiquitination and proteasomal degradation. Further research on these diverse and exciting yet often ambivalent facets of AHR biology is urgently needed in order to exploit the full potential of AHR modulation for disease prevention and treatment.
Keywords: Aryl hydrocarbon receptor, Immune response, Non-canonical signaling, Signal transduction, Transcription factor, Ubiquitination
1. Introduction
Since its initial identification by Alan Poland’s group in 1976, the aryl hydrocarbon receptor (AHR) and its signaling pathways have been and still are a highly relevant research topic in toxicology [1]. Poland had interest in the mechanisms behind the occurrence of industrially acquired acne, called chloracne, in factory workers producing the herbicide 2,4,5-trichlorophenol (2,4,5-T) [2]. As others before, they found unwanted side products like 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) to be the acnegenic factor [3]. These findings ultimately led to the idea to synthesize radiolabeled TCDD, followed by the discovery of its receptor molecule AHR [1]. Of note, several investigations of the laboratory of Daniel Nebert assessing the inducibility of aryl hydroxylase activity by environmental chemicals in genetically different, responsive and non-responsive mouse strains [4–6], preceded this landmark discovery. The critical relevance of AHR for the adverse health effects induced by dioxins is illustrated by several chemical incidents, such as the Seveso disaster in 1976, an explosion of a 2,4,5-T production reactor setting free high amounts of TCDD in a densely populated area nearby [7], the widespread usage of TCDD-contaminated defoliants, in particular Agent Orange, a mixture of 2,4-dichlorophenoxyacetic acid and 2,4,5-T, during Operation Ranch Hand in Vietnam [8] or the dioxin poisoning of the Ukrainian presidential candidate Viktor Yushchenko in 2004 [9].
Since its first description as a cellular signaling molecule that mediates the toxicity of dioxins and related compounds, the mechanistic details and facets of its major signaling route, the so-called canonical AHR signaling pathway, have been elucidated [10–12]. Briefly, in the absence of a ligand, AHR is part of a cytosolic multiprotein complex consisting of AHR-interacting protein (also known as ARA9 or XAP2), a heat shock protein 90 dimer and co-chaperone p23. In addition, an association of the protein tyrosine kinase c-Src with the cytosolic AHR multiprotein complex has been observed in several cell-types [13–15]. Upon ligand-binding, this complex dissociates and AHR translocates into the nucleus where it dimerizes with the AHR nuclear translocator (ARNT). The resulting dimer then binds to xenobiotic-responsive elements (XRE) in the enhancer region of target genes to recruit components of the general transcription machinery and induce their expression [10–12]. The probably best examined target genes of the AHR/ARNT complex encode for cytochrome P450 (CYP) 1A1, CYP1A2 and CYP1B1, xenobiotic-metabolizing enzymes that oxidize the invading (AHR-activating) chemical to enhance its polarity and enable its detoxification via the conjugating enzyme system.
However, CYP1-catalyzed reactions may lead to the formation of reactive metabolites and the generation of oxidative stress which can harm the tissue by damaging the DNA and/or other cellular macromolecules. In fact, AHR-deficient mice were found to be resistant against polycyclic aromatic hydrocarbon (PAH)-initiated tumors, a phenomenon which was attributed to the attenuated expression of CYP1 isoforms in the respective animals [16,17]. Furthermore, gene and protein levels of AHR are frequently elevated in various types of cancer, including brain, breast, lung and pancreatic cancer [18]. Also, it was observed that transgenic mice expressing a constitutively active AHR develop gastric tumors [19]. Yet, the role of AHR in cancer is highly complex since oncogenic as well as tumor-suppressive effects of the receptor have been observed [20]. For example, it was shown that AHR-deficiency in mice had a promoting effect on liver tumors, indicating that AHR acts as a tumor suppressor [21]. Additionally, there is evidence that AHR activation is associated with malignant progression and poor survival in glioblastoma patients [22]. However, this multilateral role of AHR in cancer is also critical to the design of AHR-targeted cancer therapeutics, concerning molecular and pharmacological approaches. Currently, there are first clinical trials in progress for the AHR inhibitors BAY2416964 and IK-175 recruiting participants with uncurable solid cancers to assess their tolerability and toxicity profile [23,24]. However, given the complexity of modulating AHR activity in cancer, it should be considered that under certain circumstances, not only AHR antagonists but also agonists may be useful as cancer therapeutics [25].
Furthermore, the AHR is expressed by a number of immune cells, e.g. T helper 17 (Th17) cells and regulatory T (Tregs) cells [10]. Activation of AHR in Th17 cells leads to the production of cytokine interleukin (IL)-22 [26] which is linked to pro-inflammatory processes such as cutaneous inflammation, psoriasis or Crohn’s disease [27–29]. Since it has been demonstrated that TCDD treatment leads to immunotoxicity in Ahr+/+ but not Ahr−/− mice, as the latter are resistant to TCDD-induced immunosuppression [30], IL-22 production appears to be downstream of AHR [31]. Moreover, activation of AHR by its ligands modulates T cell differentiation and Treg function, contributing to antigen-presenting cell response [32]. A recent study also suggested AHR to be an important regulator for inflammatory diseases, showing that it protects against allergic airway inflammation by controlling cell autophagy [33]. In May 2022, a cream containing 1 % of the AHR agonist tapinarof received its first approval by the US FDA for the topical treatment of the immune-related skin diseases plaque psoriasis in adults [34]. Thus, concomitant with its effects on immune response, AHR may also serve as a target for new therapeutic approaches concerning autoimmune disease [35]. A study from August 2022 for example suggests that AHR might be an interesting target for the therapy of rheumatoid arthritis, as its ubiquitination seems to be involved in an imbalance of Th17/Treg cells [36]. On top of this, AHR is also thought to be involved in COVID-19 hypoxia formation via upregulation of the expression of mucins and hence accumulation of alveolar mucus affecting the blood-gas-barrier [37].
Once considered only a mediator of the toxicity of dioxins, the field of AHR research moved into many different directions. Meanwhile, the multiple tissue-, organ-, and ligand-specific functions and seemingly paradox responses of AHR are still not fully understood. As AHR has recently sparked even more clinical intertest as a target [34,38–40], its complex non-canonical functions should urgently move more sharply into the focus of basic and preclinical research. This review article aims to provide a clear and concise overview of some of the most interesting and potentially important non-canonical AHR signaling events and functions, specifically taking the impact on the development of new pharmacological approaches into account. For that purpose, this review article will first focus on AHR’s involvement in major signaling cascades controlling cell function and fate, including the epidermal growth factor receptor (EGFR), janus kinase (JAK)/signal transducer and activator of transcription (STAT) as well as the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). Further, pathways involved in cellular adaptation towards low oxygen levels and oxidative stress will be addressed with regard to hypoxia-inducible factor 1 alpha (HIF-1α) and nuclear factor erythroid 2-related factor 2 (NRF2). Subsequently, the function of AHR beyond its role as a transcription factor will be discussed with a focus on E3 ubiquitin ligase activity.
2. EGFR signaling pathways
The EGFR is a receptor tyrosine kinase (RTK) of the ErbB family and plays a key role in embryonic development and physiology [41]. As EGFR has an intra- and extracellular domain to bind ligands, activation can occur via both ways. This activation includes receptor trans-autophosphorylation, the hetero- or homodimerization with a member of the ErbB family or another EGFR molecule and the recruitment of signaling proteins or adaptors [41]. Its downstream signaling includes the mitogen-activated protein kinase (MAPK) RAS- RAF- MEK1/2-ERK1/2 and AKT-PI3K-mTOR pathways as well as protein kinase C (PKC), STAT, SRC and NF-κB. Moreover, EGFR is a well-known oncogenic protein and involved in the pathogenesis of several cancers, commonly carrying a mutation that leads to a constitutive activation of the receptor [42,43]. Thus, EGFR plays an important role in several cellular processes such as proliferation, differentiation and apoptosis.
The seemingly first AHR-related interaction with the EGFR was described in 1982 suggesting that benzo[a]pyrene (BaP) and other PAHs may not only activate AHR but also inhibit binding of EGFR by its ligand, the epidermal growth factor (EGF) [44]. Since then, it has been repeatedly reported that an exposure to AHR agonists interferes with the binding of radiolabeled EGF to the plasma membrane [45–47]. The underlying molecular mechanism may involve an AHR ligand-mediated enforcement of EGFR internalization either by stimulating a phosphorylation of the RTK via c-Src or by inducing the release of growth factors that bind to EGFR extracellular domain (ECD). However, in contrast to PAHs that, presumably due to recycling of EGFR, only cause a transient decline [46,47], TCDD reduces the EGF-binding capacity of the plasma membrane for up to 4 days in human keratinocytes [47] and 40 days in rat liver [45]. In 2022, we published a mechanistic study which may serve to better understand the interaction between different AHR ligands and EGFR internalization [48]. Briefly, we found that treatment of human keratinocytes with PAHs, i.e. BaP and benzo[k]fluoranthene, causes a biphasic stimulation of EGFR phosphorylation and downstream MEK/ERK signal transduction. Whereas the early peak, occurring approximately after 15 min after treatment, this seems to be due to a direct c-Src-mediated phosphorylation of EGFR at residue Y845 (Fig. 1), the second temporally delayed peak of EGFR/ERK activation (approximately 2 h after treatment) involves extracellular events, i.e. the release of growth factors. Specifically, this process is driven by the c-Src-dependent sequential activation of PKC and metalloproteinases resulting in the ectodomain shedding of cell surface-bound EGFR ligands, in particular of amphiregulin (AREG) and transforming growth factor (TGF)-α (Fig. 1). These polypeptide growth factors then bind to the ECD of EGFR and initiate dimerization and autophosphorylation of the RTK at various tyrosine residues, including Y1068 and Y1137 [48]. Importantly, bulk RNAseq analyses indicated that this second wave of EGFR-dependent signal transduction seems to be responsible for the major differences in the gene expression profile in BaP- versus polychlorinated biphenyl (PCB) 126-exposed keratinocytes [48]. In fact, we were able to show that binding of dioxin-like compounds, including PCB126 and TCDD, induces similar AHR-dependent and c-Src-driven signaling events culminating in the shedding of EGFR ligands from the plasma membrane. However, dioxin-like compounds stimulate EGFR signal transduction only early (15 min) but not late (2 h) after treatment. In silico as well as further experimental work revealed that dioxin-like compounds bind to the ECD of EGFR directly and thereby inhibit an activation of the RTK by growth factors [48,49].
Fig. 1. Ligand-activated AHR activates EGFR and downstream signaling.
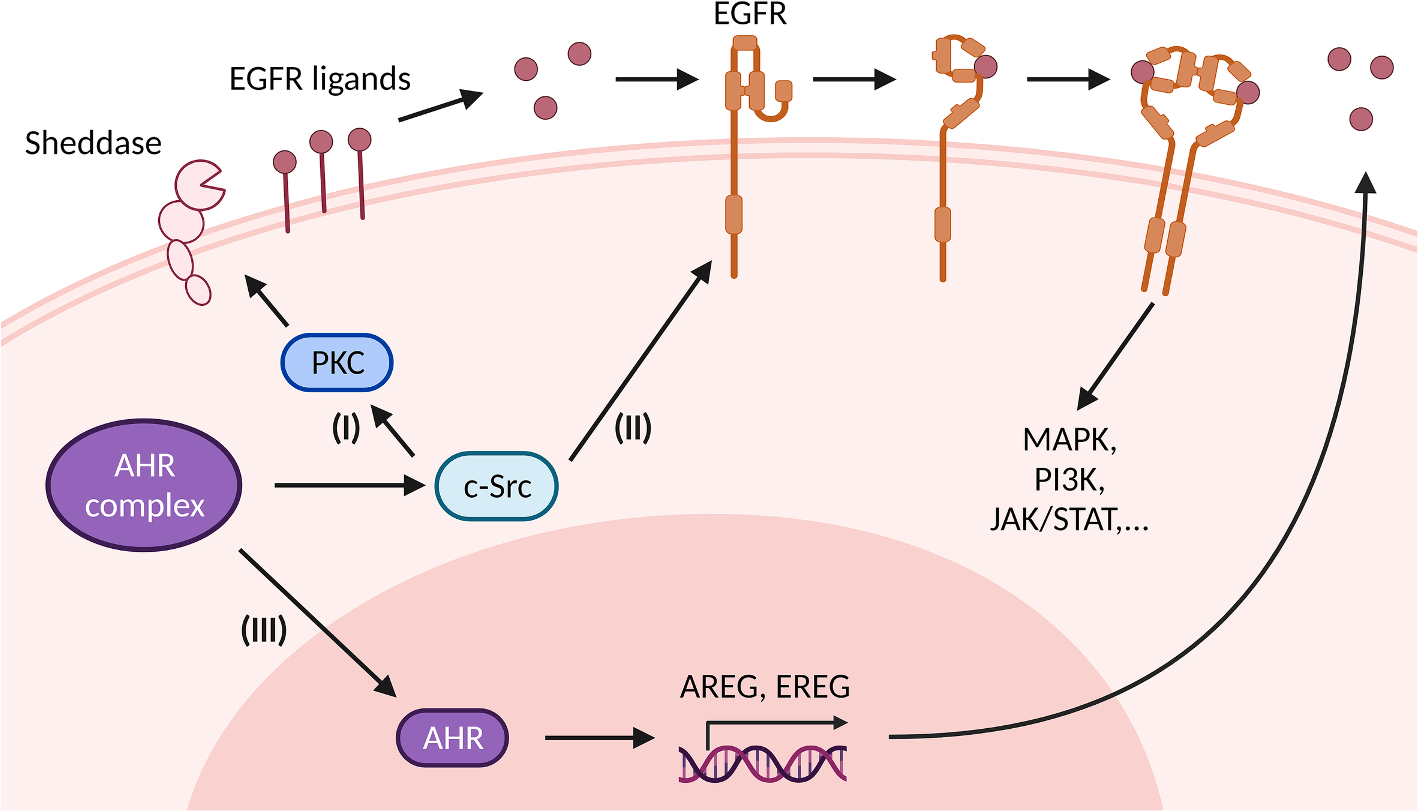
The ligand-driven dissociation of AHR complex leads to the release of c-Src which can (I) directly activate the epidermal growth factor receptor (EGFR) by phosphorylating its intracellular domain, and (II) sequentially activate protein kinase C (PKC) and sheddases resulting in ectodomain shedding of cell surface-bound EGFR ligands. In addition, nuclear AHR transactivates genes encoding EGFR ligands, such as amphiregulin (AREG) and epiregulin (EREG) (III). Independently from its mode of activation, i.e. ligand-binding or intracellular phosphorylation, the EGFR monomer changes its conformation from tethered to untethered and forms a hetero- or homodimer leading to activation of downstream signaling pathways like MAPK, PI3K or JAK/STAT.
Apart from non-canonical signaling events, the ligand-activated AHR has been found to induce the expression of several EGFR ligands (Fig. 1), including epiregulin and AREG, in an XRE-dependent manner [50,51]. Other investigators reported that tobacco smoke induces the expression of AREG in oral epithelial cells via a non-canonical AHR pathway involving the cAMP – protein kinase A signaling axis [52]. However, Lemjabbar et al. identified tobacco smoke to stimulate the proliferation of lung epithelial cells by promoting the metalloprotease-mediated ectodomain shedding of AREG [53]. Hence, depending on ligand, cell type and context, AHR may affect EGFR signal transduction via canonical and non-canonical signaling events or a combination of both.
The laboratory of Thomas Sutter reported an inhibition of the TCDD-induced expression of CYP1A1 and CYP1B1 upon co-treatment of epidermal keratinocytes with EGF. Results from further mechanistic experiments indicated that both pathways compete for the common transcriptional co-activator CBP/p300. Accordingly, the AHR-mediated keratinocyte differentiation was also inhibited by EGF while co-treatment with an EGFR inhibitor promoted differentiation [54]. Hence, these results suggest that EGFR activation might interfere with activation of AHR or at least its canonical signaling pathway via transrepression. This observation is supported by another study, reporting that a pharmacological inhibition of EGFR amplifies the TCDD-induced expression of CYP1 isoforms [55].
Taken together, four different mechanisms of AHR-EGFR-crosstalk have been described so far: (I) AHR-dependent activation of EGFR via release of c-Src, (II) AHR-induced expression of EGFR ligands leading to EGFR activation, (III) AHR-dependent ectodomain shedding of EGFR ligands leading to auto- or paracrine EGFR activation and (IV) the competition for common transcriptional co-activators.
2.1. Pathological consequences of crosstalk between AHR and EGFR
The EGFR is an integral part of multiple signaling pathways controlling tissue homeostasis and disease. Hence, the crosstalk between EGFR and the chemosensor AHR might be a signaling route by which environmental pollutants, drugs or other exogenous chemicals harm cellular functions and tissue integrity. In fact, numerous studies have reported that a modulation of EGFR function by AHR-stimulating factors drives the pathogenesis of diseases, in particular of various types of cancer.
In smokers, the AHR-mediated increased AREG expression results in enhanced cell proliferation and contributes to the procarcinogenic effects of tobacco smoke [52,56]. Moreover, in breast cancer cells, TCDD increased EGFR ligand expression resulting in activation of downstream EGFR signaling and thereby inhibition of apoptosis. Interestingly, this effect could be reversed by addition of the AHR antagonist 3′-methoxy-4′-nitroflavone, suggesting an AHR-driven tumor promoting effect [57].
AHR activation and subsequent non-canonical EGFR signaling are also critically involved in the cellular UVB stress response. UVB radiation leads to the formation of the endogenous AHR ligand 6-formylindolo[3,2-b]carbazole (FICZ) in the skin and further to a c-Src-dependent EGFR internalization and downstream activation of ERK [58]. In contrast to TCDD, which per se activates similar signaling events, FICZ is rapidly metabolized by CYP1 isoforms and, accordingly, causes a transient activation of AHR signaling pathways [59]. However, amongst others, the UVB-induced activation of this AHR signaling cascade leads to an induction of proinflammatory cyclooxygenase-2 (COX-2) [58,60], a modulation of DNA damage-dependent stress responses [61,62], and skin tumor formation [62].
Similar to UVB irradiation [60], exposure to cigarette smoke leads to the induction of the AHR-dependent genes CYP1A1, CYP1B1 and matrix metalloproteinase-1 (MMP-1) [63]. MMP-1 is also induced in the skin of smokers [64] where it leads to collagen breakdown in the dermis, tissue remodeling and promotion of skin aging [65]. In line with that, a knockdown of MMP-1 resulted in inhibition of the progression of colorectal cancer via suppression of the PI3K/AKT signaling pathway downstream of EGFR. This suggests an involvement of AHR-induced MMP-1 in the EGFR-regulated promotion of malignant behavior in colorectal cancer [66]. It has also been shown that EGF induces MMP-1 secretion through EGFR-mediated MAPK signaling, again leading to increased proliferation [67].
A study from 2022 has shown that BaP exposure leads to aggravated TGF-α and MUC5AC expression in an asthma model [68]. MUC5AC is one of the mucins closely related to asthma [69] as it leads to pronounced secretion and accumulation of mucus which blocks the airway and results in breathing difficulties [70]. It was suggested that BaP-induced AHR activation would lead to production of reactive oxygen species (ROS) which activated EGFR and MAPK signaling [68]. The AHR-mediated upregulation of mucins and accumulation of alveolar mucus was also connected to hypoxia and inflammation in COVID-19 patients. Particularly, interferon (IFN)-β and IFN-γ induced mucin expression in an AHR-dependent manner. Concomitant with this, AHR inhibition led to improvement of the lung function in SARS-CoV-2-infected mice [37].
There already are several therapeutics targeting EGFR and its complex signaling pathways, including monoclonal antibodies or inhibitors, which commonly show skin toxicity as a side effect. This often forces the discontinuation of the respective drug [42]. Further, it seems that many EGFR-dependent effects in diseases are also enforced by AHR-driven EGFR ligand expression and/or membrane shedding. Due to the dynamic crosstalk between EGFR and AHR, when developing therapeutics targeting EGFR signaling, AHR should also be taken into consideration as a possible target or co-target – and vice versa. This could possibly lead to reduction of unwanted side effects such as development of drug resistance or other adverse reactions directed at specific tissues or cellular compartments.
3. JAK/STAT pathway
The JAK/STAT pathway transduces signals from the cell surface to the nucleus and, amongst others, is critically involved in the regulation of innate and adaptive immune responses [71]. Accordingly, an aberrant activation of JAK/STAT signaling is associated with the pathogenesis of multiple diseases, including chronic inflammatory diseases, autoimmunity and cancer [72–75]. The molecular mechanisms of JAK/STAT signaling pathways are well characterized. Briefly, members of the JAK family, i.e. JAK1, JAK2, JAK3 and tyrosine kinase 2 in humans, are bound to the intracellular domain of transmembrane cytokine receptors or receptor tyrosine kinases (e.g. EGFR). Upon activation by cytokines or IFNs, the cytokine receptors dimerize or multimerize, leading to a mutual JAK-mediated phosphorylation of specific tyrosine residues in the intracellular domain of the cytokine receptors. These novel phosphotyrosine residues serve as docking sites for the Src-homology 2 (SH2) domain of STAT molecules. After binding to the phosphorylated cytokine receptor, JAKs phosphorylate a C-terminal tyrosine residue in the STAT proteins to form another SH2-binding motif. The latter is reciprocally recognized by two STAT proteins, leading to the formation of a STAT dimer. This dimer translocates to the nucleus and acts as a transcription factor by binding to certain DNA motifs in the enhancer region of target genes. The human genome encodes seven mammalian STAT family members, i.e. STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6.
In 2004, two independent studies were first to report an impact of AHR on STAT signaling. Nukaya et al. showed that a treatment of B6 mice with 3-methylcholanthrene resulted in an AHR-dependent reduction of JAK2 expression in the liver, which was associated by a decreased DNA-binding activity of STAT5 [76]. In the other study, Takanage et al. reported that the AHR agonist β-naphthoflavone attenuates the astrocytic differentiation of glioma cells by inhibiting the expression of IL-6 and downstream STAT3 activation [77]. Hence, AHR seems to control the expression of stimulating cytokines and pathway components to alter JAK/STAT activity and associated cellular consequences. In fact, AHR is known to regulate the expression of various JAK/STAT-stimulating cytokines, including IL-2 [78], IL-10 [79], IL-21 [79], IL-22 [31] and others, via different pathways. For instance, AHR cooperates with the NF-κB subunit RelA/p65 at the IL-6 promoter [80], whereas in lipopolysaccharide (LPS)-treated macrophages AHR sequentially activates c-Src and STAT3 to induce the expression of IL-10 [81]. Moreover, due to parameters such as physicochemical properties of the ligand, cell type, tissue and microenvironment, AHR may either induce or inhibit the expression of these cytokines, as illustrated for instance by IL-6 [82,83] and IL-33 [84,85]. Importantly, the interrelationship between AHR and JAK/STAT signaling pathways is mutual and much more complex than a unilateral regulation of mediators and signaling molecules (Fig. 2).
Fig. 2. Crosstalk of JAK/STAT and AHR.
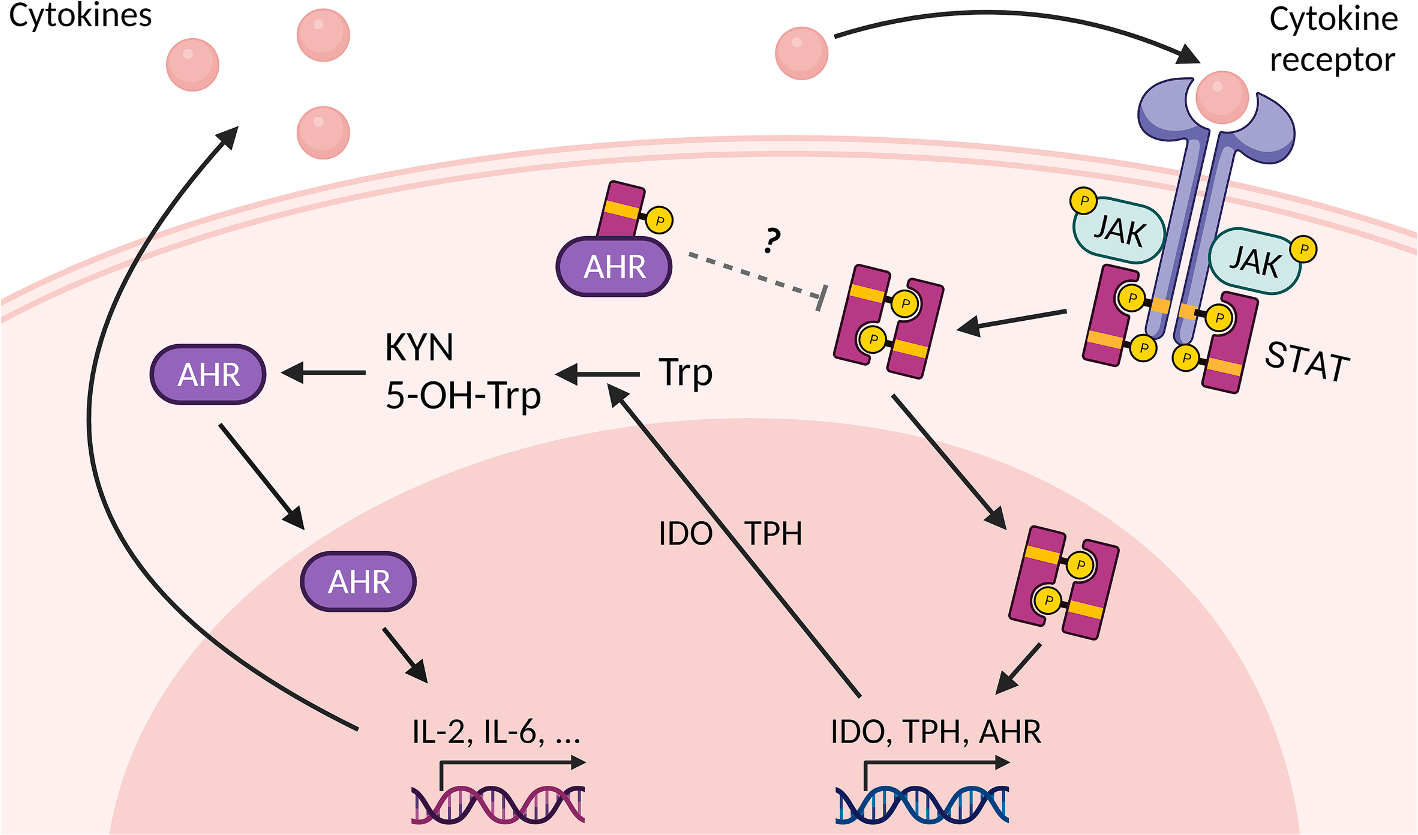
AHR and the signal transducer and activator of transcription (STAT) can induce and repress each other. The phosphorylated STAT forms a dimer which can translocate into the nucleus and induce transcription of target genes including AHR, indoleamine 2,3-dioxygenase (IDO) and tryptophan hydroxylase (TPH). IDO and TPH catalyze the metabolism of tryptophan (Trp) into kynurenine (KYN) and 5-OH-Trp, respectively, which can serve as AHR agonists. AHR activation leads to expression of several cytokines, e.g. IL-2 and IL-6, which bind to cytokine receptors and thereby stimulate the sequential phosphorylation of Janus kinases (JAK) and STATs resulting in the release of STATs from the cell surface receptor. Interestingly, AHR can also form a cytosolic heterodimer with a phosphorylated STAT molecule, i.e. STAT, which is thought to repress the formation and nuclear translocation of the phosphorylated STAT dimer.
In fact, activated STAT1, STAT2 and STAT3 have been reported to bind to the promoter of the AHR gene and induce its transcription (Fig. 2). Upon stimulation of human hepatoma cells with IL-6-type cytokines, STAT3 binds to a STAT-responsive element approximately 2 kilobase pairs 5′-upstream of the transcription initiation site of the AHR gene [86]. This STAT-responsive element is not conserved in the murine Ahr gene, but another functional STAT-binding motif in it has been identified [87]. In murine astrocytes, IFN-β activates JAK1 and tyrosine kinase 2 resulting in the formation of the IFN-stimulated gene factor 2, a trimolecular complex consisting of STAT1, STAT2 and IFN regulatory factor 9, which binds to an IFN-response element in the murine promoter sequence of Ahr to induce its expression [88]. Another study reported an activation of the AHR system in B cells by IL-4 [89]. While the authors found that IL-4 induces AHR expression in a STAT6-dependent manner, the underlying molecular mechanism of the observed nuclear translocation and target gene induction of AHR in response to the cytokine treatment remains less clear.
However, apart from the transcriptional level, STAT proteins facilitate AHR activation by altering tryptophan metabolism (Fig. 2). In human chronic lymphocytic leukemia cells, for instance, IFN-γ induces the expression of indolamine-2,3-dioxygenase (IDO) 1 through the JAK/STAT1 signaling pathway [90]. IDO1 oxidizes tryptophan to N-formyl-kynurenine which is subsequently converted by aryl formamidase to kynurenine (KYN) [91]. KYN and several of its metabolites, including kynurenic acid and xanthurenic acid, serve as low affinity agonists of AHR [82,92,93]. Importantly, AHR activation by KYN was found to resemble the effects of TCDD treatment in mice, i.e. an induction of immunosuppressive Treg cells [92,94,95]. In 2011, Michael Platten and his team identified KYN as an endogenous AHR ligand that is constitutively generated in human brain tumors and responsible for the suppression of antitumor immune responses and the promotion of cancer cell survival and motility [22]. In lung cancer, an autocrine signaling loop consisting of the stimulation of AHR activity by KYN, an AHR-mediated upregulation of IL-6, a stimulation of STAT3 and the associated induction of KYN-producing IDO1 has been described [96]. A similar way of interaction has been identified to be responsible for the exhaustion of CD8+ T cells in the tumor microenvironment. High levels of IL-2 lead to an activation of STAT5, which subsequently induces the expression of tryptophan hydroxylase-1 (TPH). This enzyme converts tryptophan to 5-hydroxytryptophan, another metabolite that serves as an AHR agonist. The resulting activation of AHR signaling leads to an upregulation of inhibitory receptors (e.g. CD39, PD-1) and a downregulation of IFN-γ and tumor necrosis factor (TNF)-α levels and thus to dysfunctional CD8+ T cells [97]. Interestingly, Il-2 is an established target gene of AHR in mice [78], suggesting that there might be an another signaling loop.
Taken together, these studies provide evidence that IL-2-, IL-6- and IFN-triggered JAK/STAT signaling pathways control the expression and activity of AHR (Fig. 2). Vice versa, the AHR system can potentially interfere with JAK/STAT signaling pathways by acting as a transcription factor controlling the expression of cytokines, protein kinases, etc. In addition, AHR can directly interact with STAT molecules to either induce or repress their transcriptional activity (Fig. 2). In peritoneal macrophages, both Ahr- as well as Stat1-deficiency enhances the induction of pro-inflammatory IL-6 in response to LPS treatment [98]. Further mechanistic data, including results from chromatin immunoprecipitation assays, point to the idea that a tripartite complex consisting of AHR, STAT1 and NF-κB subunit p50 is bound to the Il-6 promoter and represses its transactivation. However, it is worth mentioning that meanwhile, additional mechanisms through which AHR may dampen IL-6 expression in LPS-exposed macrophages have been identified [99]. In T cells, AHR was found to facilitate the polarization towards proinflammatory Th17 cells by inhibiting STAT1 phosphorylation which blocks the competing T helper 1 cell differentiation pathway [100]. Remarkably, Th17-polarizing conditions (IL-6 plus TGF-β) induced the AHR-mediated generation of Th17 cells in the absence of exogenous ligands [100]. Given that, later on a critical involvement of STAT3 in the AHR-dependent differentiation of CD4+ T cells into Th17 cells has been reported [101], a STAT3- and IDO1-dependent production of endogenous tryptophan-derived ligands might serve as an explanation for this observation. However, in contrast to the enforced polarization towards Th17 cell differentiation upon AHR-mediated STAT1 inhibition, the same mechanism was reported to impair Th17 differentiation and promote Tregs in a model for idiopathic pneumonia syndrome [102]. In this model, AHR represses the transcription of the activator protein-1 (AP-1) subunit JUND by preventing the binding of STAT1 to the respective gene promoter, resulting in a reduced expression of IL-6 and the associated polarization of CD4+ T cells toward Th17 cells. Accordingly, KYN-mediated activation of AHR induces immunosuppression and thereby attenuates the pneumonia-related symptoms [102]. This discrepancy between the two studies describing the AHR as an inducer of Th17 cells versus an inducer of Tregs has been previously discussed [94,95,103] and may not necessarily solely depend on the structural and biochemical properties of the AHR ligand but also on the applied dose [104]. An inhibitory effect of the IDO1-AHR axis on STAT1 activity has also been observed in a mouse model for melanoma, in which IFN-γ induces dormancy in tumor-repopulating cells by stimulating an iDO-1-driven activation of AHR [105]. This leads to an upregulation of the AHR target and cell cycle inhibitor p27KIP1 [106] which subsequently physically interacts with STAT1, thereby causing a switch from the apoptosis to the dormancy program in the malignant cells [105].
In summary, the existing body of literature provides compelling evidence for a mutual interaction of AHR and STAT family transcription factors on various levels, including transcriptional control, induction of signaling mediators and protein–protein interaction. STAT proteins may critically contribute to the impact of the AHR on multiple immune cells, including macrophages and various T cell populations and thereby determine the downstream pro- or anti-inflammatory outcome. In fact, in numerous types of cancer, including melanoma, oral squamous cell carcinoma, lung cancer and glioblastoma, AHR and JAK/STAT signaling pathways seemingly cooperate to repress anti-tumor immune responses and facilitate tumor growth and progression [96,97,101,107,108]. In contrast to this clear trend observed in malignant diseases, there is also compelling evidence for anti-inflammatory consequences of the AHR-JAK/STAT crosstalk, for instance in the context of central nervous system inflammation and inflammatory bowel disease [88,109]. In conclusion, the currently rather limited amount of available experimental data elucidating the interplay between AHR-dependent and JAK/STAT signaling pathways, emphasizes the urgent need for a deeper functional characterization of this crosstalk. This is necessary to assess whether targeting of the JAK/STAT system supports or counteracts AHR-manipulating therapeutic approaches for certain pathological conditions and vice versa.
4. The NF-κB family signaling pathway and crosstalk with AHR
The NF-κB family of transcription factors regulates the expression of numerous cytokines and immune response genes [110]. NF-κB is critically involved in biological processes, including apoptosis, cell differentiation, immunity and diseases such as autoimmunity or cancer [111]. Considering the physiological and pathological roles of AHR, a significant overlap with the NF-κB signaling pathway becomes evident [112]. When considering AHR as a transcription factor interacting with NF-κB signaling and modulating immune responses, it is crucial to understand its role in diseases based on immune modulatory effects.
The activity of NF-κB is induced by many cytokines and inflammatory signals in a time- and concentration-dependent manner [113]. Early studies have shown that cytokines also affect the activity of AHR-regulated drug-metabolizing enzymes. For instance, treatment with TNF-α clearly suppressed the activity of hepatic CYP-dependent drug metabolism in mice [114]. Subsequent studies noticed suppression of TCDD-induced Cyp1a1 and Cyp1a2 mRNA expression after treatment of isolated rat hepatocytes with IL-1β [115]. In line with this, the acute phase protein IL-6 suppressed the gene expression of CYP1A1, CYP1A2 and CYP3A3 in human hepatoma cells [116]. These early observations strongly indicate that NF-κB activated by inflammatory cytokines interferes with AHR signaling and thereby shapes target gene expression and the associated biological outcome.
The activation of NF-κB changes the expression and activity of AHR and vice versa. One of the first studies reporting a crosstalk between AHR and proteins of the NF-κB family was published by Tian et al. describing the physical interaction of AHR with NF-κB RelA causing a mutual repression of both signaling pathways [117]. In their study, the authors treated mouse hepatoma cells with TNF-α to activate NF-κB and focused on the AHR-mediated induction of CYP1A1 which was significantly repressed after TNF-α treatment. Further, they found that TNF-α-induced NF-κB binding activity was suppressed when Hepa1c1c7 cells were treated with TCDD. Mechanistically, the ligand-dependent activation of AHR induces histone H4 acetylation at the TATA box of the Cyp1a1 promoter region, which was inhibited by TNF-α-induced NF-κB activity [118].
Further studies have shown that AHR interacts with the NF-κB component RelA which supported the TCDD-mediated induction of IL-6, plasminogen activator inhibitor-2, FAS ligand and c-myc [119–122]. Dinatale et al. found that the AHR-mediated amplifying effects on NF-κB signaling requires the AHR/ARNT-heterodimer for the synergistic activation of IL-6 following IL-1β and TCDD treatment. This may involve the dismissal of co-repressors by DNA-bound AHR [119]. The AHR-dependent expression of FasL in thymic stromal cells was found to be regulated through TCDD-mediated activation of the NF-κB subunits p50 and p65 on the Fasl promoter [123]. Another mechanism was described for the expression of C-MYC in breast cancer cells, where RelA and AHR together bind to NF-κB elements and induce the transcription of C-MYC [122].
Conversely, AHR was also identified to suppress NF-κB-mediated gene expression. For instance, the expression of the Ig heavy chain was repressed through the interactions of NF-κB and AHR at a DNA replication-related element and an overlapping κB element [124]. The acute phase protein serum amyloid A was found to be suppressed by the interaction of the activated AHR with RelA, but DNA binding was not involved [125]. Alvaro Puga’s group concluded that TCDD-mediated activation of AHR and induction of CYP1A1 leads to the generation of an oxidative stress signal which enhances NF-κB and AP-1 DNA-binding activity. [126]. Interestingly, the enhanced binding activity of the NF-κB complex was found to be formed by p50/p50 complexes which could be responsible for the inhibitory effect of AHR on NF-κB activity. Another study reported an inhibition of IgM expression by TCDD in LPS-activated B cells, which was associated with an AHR-dependent decreased DNA-binding activity of AP-1 but not of NF-κB [127].
Besides RelA, several studies reported an interaction of AHR with the NF-κB member RelB [128–131]. We identified RelB to physically interact with AHR and bind on a previously unrecognized RelB/AHR-responsive element in the promoter of the IL-8 gene to induce its expression [132]. Further, a ligand-independent activation of AHR via protein kinase A triggers its nuclear translocation and the induction of IL-8. Therefore, this mechanism was described as the non-canonical AHR pathway which involves ligand-independent activation and interaction with proteins other than ARNT, such as RelB.
Similarly, the expression of other chemokines including the B-cell activating factor of the TNF family, B-lymphocyte chemoattractant and chemokine (C–C–motif) ligand (CCL) 1 was also found to involve the binding of RelB/AHR-complexes to the promoter of these genes, suggesting a new regulatory function of AHR [128]. The RelB/AHR-responsive element identified on the promoter of IL-8 is similar to DNA-binding sequences recognized by RelB/p52 dimers in regulatory sequences of the above organogenic chemokine genes [133]. Moreover, AHR signaling can alter the differentiation of mouse bone marrow-derived dendritic cells (BMDC), which involves activation of NF-κB RelB and the AHR-dependent induction of the immune-regulatory enzyme IDO [134,135]. The expression of the cytokines IL-10 and CCL18/DC-CK1 was inhibited by TCDD, but IL-10 was induced in LPS-activated BMDC and in spleen of B6 mice [135,136]. The induction of IDO as well as anti-inflammatory IL-10 in LPS-activated BMDC could critically contribute to the immunosuppressive effect of TCDD. In this context, it is interesting to note that IL-10 was found to protect from colitis [137] and treatment with TCDD may improve colitis in mice associated with an increased number of Tregs [138]. Further, the expression of IL-12A, a key cytokine for Th1 differentiation, was suppressed when TLR4-activated human primary dendritic cells (DCs) were treated with TCDD [139]. This was confirmed in a study using mouse peritoneal macrophages showing that activation of AHR decreased protein levels of pro-inflammatory IL-6 and IL-12 after activation with LPS/IFN-γ [140]. Mechanistically, AHR activation promoted RelA/p65 protein degradation, thus causing the decreased cytokine levels. Besides the interaction of AHR with RelB, studies have demonstrated that ARNT interacts with RelB to regulate expression of the TNF receptor family member CD30 [141]. Since ARNT can dimerize with the AHR repressor (AHRR), the study suggests that AHRR may also affect activity of NF-κB RelB and the expression of cytokines as shown in transgenic mice overexpressing AHRR [142–144]. Interestingly, Ovrevik et al. found that ARNT suppressed the expression of RelB which was associated with a decreased expression of IL-8 in human bronchial epithelial BEAS-2B cells [145].
In summary, these studies indicate that the synergistic or inhibitory effects resulting from the interaction of AHR with NF-κB signaling may involve various mechanisms. This includes physical interaction of members of the AHR pathway with subunits of the NF-κB family and combinatorial effects of AHR and NF-κB elements located on promoters of specific target genes.
4.1. Dysregulation of Toll-like receptor responses through AHR signaling
Ligands of the Toll-like receptor (TLR) family are strong activators of NF-κB signaling. Multiple studies have shown that AHR may interfere with responses triggered by pathogens including the TLR4 ligand LPS [112]. Moreover, AHR-deficiency was found to increase the sensitivity of mice towards LPS toxicity including septic shock [120]. Further studies showed that exposure of AHR-deficient mice to LPS, cigarette smoke or crystalline silica triggers an increased inflammation in the lungs associated with increased NF-κB activity [146,147]. On the other hand, exposure to an AHR antagonist significantly inhibited the LPS shock in mice, including a reduced lethality and inflammatory signaling [148]. This finding is in line with our observation that overexpression of AHRR in mice suppressed LPS-induced toxicity and lethality [143]. Possible mechanisms of AHR’s role in LPS shock were discussed with regard to the immunosuppressive function of an AHR-iDO-mediated pathway, emphasizing the importance of enhanced IDO activity for the establishment of life-threatening conditions in sepsis [149]. The hypothesis of the AHR-iDO-mediated pathway in sepsis is supported by findings showing that AHR activation synergistically increases the LPS-induced expression of IDO1 and IDO2 [139,150].
Studies with AHR−/− mice showed that AHR-deficiency modified the expression of LPS-induced cytokines along with the DNA-binding activity of NF-κB, C/EBPβ and AP-1 in lung and liver [151]. The effects of AHR and TLR ligands in humans were examined in primary DCs derived from human blood monocytes. It was shown that human DCs are very sensitive towards low doses of LPS [139] and that TLR-activated DCs express higher levels of AHR and are more sensitive towards the effects of AHR ligands. Treatment with AHR ligands led to a synergistic or antagonistic effect of the TLR-triggered response in DCs depending on the specific target gene. The effect of AHR ligands in activated DCs involved the expression of RelB and were altered by the specific TLR ligand used to activate DCs. In a study with BMDC from wildtype and RelB-deficient mice, RelB-involvement in ligand-dependent effects on AHR signaling was confirmed [150]. Similar results were received from a study using BEAS-2B cells [145]. The authors found that AHR suppressed activation of RelA stimulated by TNF-α or polyinosinic:polycytidylic acid. However, activation of AHR induced IL-8 expression, but AHR suppressed the expression of IL-8 and CCL5 when cells were activated by the TLR ligand polyinosinic:polycytidylic acid. The authors concluded that AHR may stimulate or suppress NF-κB signaling and that anti-inflammatory effects of AHR possibly involve additional pathways besides the interaction with NF-κB. The dose-dependent induction of IL-8 by TCDD described in human DCs [139] is supported by a study demonstrating increased levels of IL-8 in blood of asthmatic children with high PCB serum levels [152].
A synergistic induction of CCL20 was observed in peritoneal macrophages exposed to LPS and AHR ligands [153]. The authors concluded that this was due to both, LPS-mediated increase of AHR levels and a potent tandem AHR-binding site upstream from the transcriptional start site of CCL20. Recently, we found that both, a XRE consensus site and a RelBAHRE-binding element located on the promoter of the IL-22 gene, are necessary to mediate the synergistic effects of AHR and TLR ligands on the expression of this cytokine [31]. Furthermore, an exposure to particulate matter (PM) collected from traffic-related air pollution and wildfires activated AHR and NF-κB signaling, leading to a robust induction of IL-22. The results are in line with a study investigating the expression of IL-33 after exposure to PM collected from tunnel dust [154]. IL-33 is a critical factor in the development of asthma and was induced in macrophages treated with PM samples derived from the tunnel. The analysis of the PM composition indicated that the concentration of PAHs and endotoxins are important factors in the AHR-/TLR-dependent induction of IL-33.
As described earlier [153], levels of AHR mRNA and protein were enhanced by various TLR ligands in DCs and thymus of B6 mice [155]. A functional NF-κB-binding element was identified upstream of the transcription start site of the AHR gene indicating an important role of RelA/p50 in the transcriptional regulation of AHR [155]. These results demonstrated for the first time that NF-κB RelA is a critical component regulating the expression of AHR and downstream target gene expression. These findings have important implications regarding the effect of inflammatory stimuli and cytokines that activate NF-κB and induce AHR expression during activation and differentiation of immune cells. The results may provide a possible mechanism of the rapid increase of AHR levels after T cell activation described in previous studies [95,103]. This rapid upregulation of AHR, which also occurs upon activation of murine splenocytes, murine B cells [156] and human B cells [157], may contribute to the enhanced sensitivity of leukocytes towards a modulation by dioxin and dioxin-like compounds. Additionally, the type of TLR response activated through the specific TLR agonist may direct the outcome of AHR interaction with NF-κB subunits and the regulatory or immunogenic response. Further, this involves not only a single gene, but a number or a group of genes will be affected by the AHR/NF-κB-crosstalk. The altered innate immune responses and dysregulation of TLR signaling by AHR are likely to be the key steps in triggering chronic inflammatory diseases as well as modulation of antiviral responses during infection (Fig. 3).
Fig. 3. Dysregulation of Toll-like receptor responses through AHR signaling.
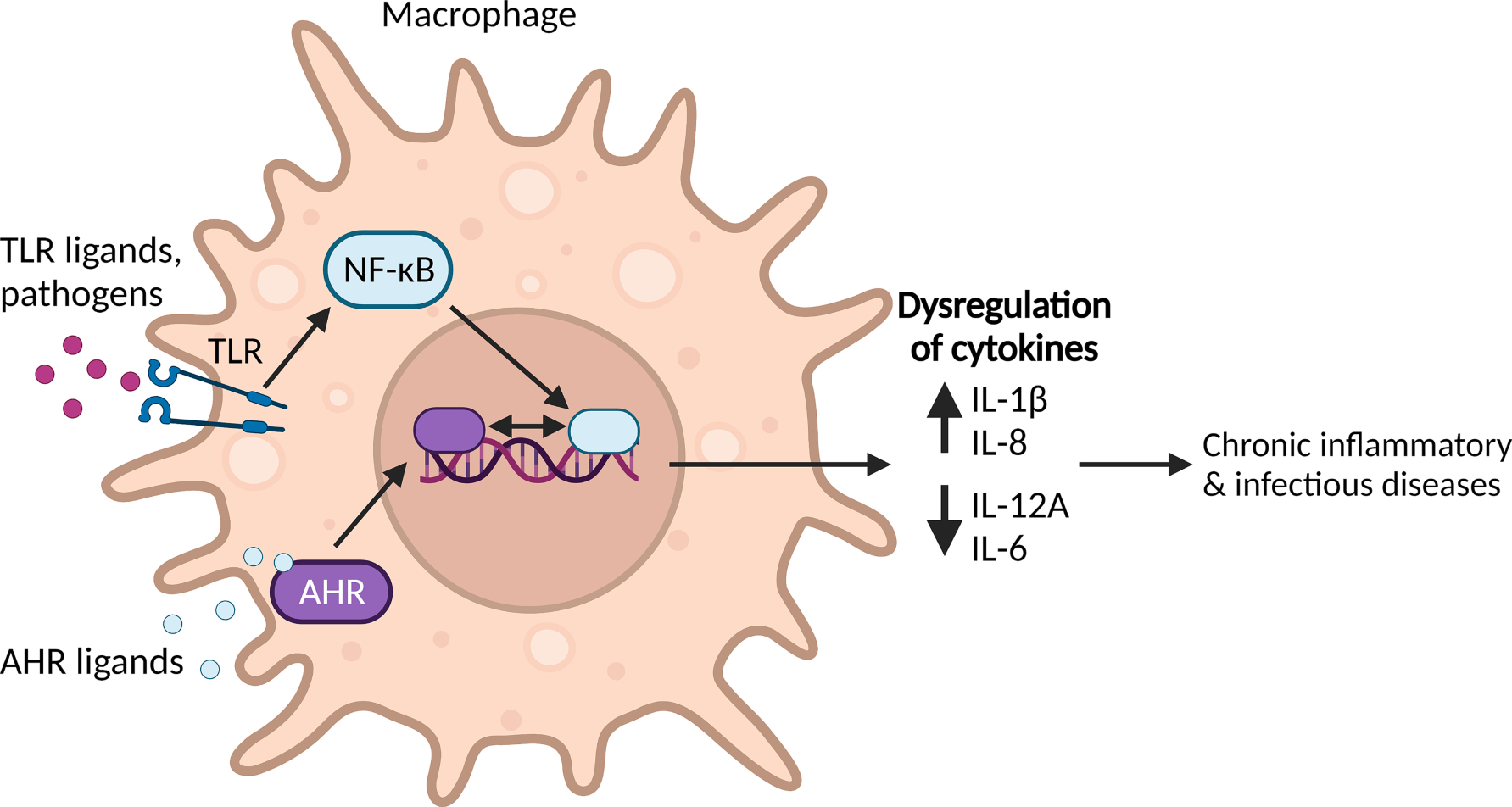
In the macrophage, interaction between AHR and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) can regulate immune response and inflammation via transcriptional inhibition and activation of cytokines and chemokines. If the Toll-like receptor (TLR) is bound by ligands or pathogens it leads to the nuclear translocation of NF-κB, whereas ligand-binding of AHR leads to the nuclear translocation of AHR. When both NF-κB and AHR are activated, a dysregulation of transcription of cytokines can occur, e.g. leading to an increase of interleukin (IL)-1β and -8 and a decrease of IL-12A and -6. This favors occurrence of chronic inflammatory and infectious diseases.
In summary, these findings show that simultaneous activation of AHR and NF-κB signaling pathways leads to synergistic or antagonistic effects on specific target genes by integrating signals of canonical and non-canonical AHR pathways.
4.2. Consequences of AHR and NF-κB interaction in pathology
Many chemotherapeutics are metabolized by AHR-regulated CYP isoforms [158,159]. As described above, inflammatory cytokines can change the expression and activity of CYP enzymes and therefore the metabolism of various drugs. Consequently, changes in the activity of CYP enzymes may ultimately result in clinical endpoints [160]. For instance, altered CYP activities may modify chemotherapeutics exposure and affect the treatment response in cancer patients [161]. In fact, AHR-regulated CYP1 isoforms are primarily responsible for the metabolism of the anti-cancer drugs vemurafenib and imiquimod [162,163]. Alterations in CYP activity also play a critical role in the onset and progression of various diseases including cancer, metabolic disorders and hepatic as well as cardiovascular diseases [160,164–167]. Hence, by attenuating CYP expression and enzyme activity, inflammatory signaling pathways may contribute to adverse health effects.
Inflammatory DCs and macrophages exhibit an increased expression and activity of AHR. Elevated copy numbers of AHR were detected in two disease models, i.e. rheumatoid arthritis and allergic airway inflammation [155]. Further, an increased level of IL-8, an attractant for neutrophils, was found in the blood of asthmatic children which correlated with high PCB serum levels [152]. These data support previous studies showing that AHR activation (via TCDD, PCBs) and activation of NFκB through LPS in vitro or under chronic inflammatory conditions, like asthma, can synergistically increase target gene expression, such as IL-8, in children [168].
The AHR is known to be involved in the pathology of autoimmune diseases, such as rheumatoid arthritis, colitis, and systemic lupus erythematosus (SLE) [169–171]. Interestingly, AHR activation by apoptotic cells was found to depend on interactions between AHR and TLR9 which can inhibit disease progression of SLE in mice [169]. Accordingly, the loss of AHR or deficiency of TLR9 might contribute and accelerate SLE in this mouse model. On the other hand, a synergistic interaction between the shared epitope and the AHR pathway triggered by NFκB signaling has been described to cause a robust increase in arthritis severity in mice [172]. These studies underline the importance of the crosstalk between AHR and NF-κB as a mechanism in the development of autoimmune diseases.
Over the last two decades, numerous reports have shown that the AHR plays an important role in regulating immune responses and that exposure to AHR-activating ligands and toxicants contributes to the promotion of immune system disorders and the development of chronic inflammatory diseases [10,173]. Although AHR ligands and NF-κB stimulation through pathogens or other inflammatory stimuli very specifically activate only one of the two pathways, there is significant crosstalk when both signaling pathways are activated (Fig. 3). The communication between AHR and NF-κB is evidently critical for the immune response, but the type and strength may vary depending upon factors like the length, intensity and timing of the signal, as well as cell-specific receptor distribution. The cross-regulation between these two signaling axes suggests that NF-κB- and AHR-dependent signaling could be viewed within the context of a unique signaling system. It mediates signaling from both pathogen-/inflammatory and ligand-based stimuli that may regulate physiological function and which, if dysregulated, may contribute to pathology of chronic diseases and impact host defenses against infectious diseases. The NF-κB and AHR signaling pathways may run independently in parallel, however, their crosstalk creates multiple opportunities for modulating or fine-tuning of responses to different signals. Such fine-tuning is important as the immune system has to be active enough to fight pathogens or cancer, but should not be hyperactivated and result in chronic inflammatory diseases.
5. HIF-1α and AHR: Interdependence and crosstalk
The Nobel Prize in Physiology or Medicine 2019 was awarded to Gregg Semenza, Sir Peter Ratcliffe and William Kaelin for their joint discovery of hypoxia-inducible factor (HIF)-1α and its functionality under cellular oxygen shortage [174–176]. Of note, the identified co-factor HIF-1β turned out identical to the already known AHR co-factor ARNT. AHR, ARNT, and HIF-1α all belong to the group of basic helix-loop-helix/Per-Arnt-Sim proteins which act as environment-sensing heterodimeric transcription factors [177] governing adaptation to changing external conditions.
HIF-1α function is crucial under physiologic conditions including embryogenesis [178–180] or during anaerobic activity [181,182], but also under pathologic conditions like infection and cancer [180,183,184]. Under normoxic conditions (~21 % O2), the HIF-1α protein is constantly degraded by the proteasome: Oxygen-dependent HIF-prolyl hydroxylases permit binding of the von Hippel–Lindau protein to hydroxylated HIF-1α, leading to ubiquitination and subsequent degradation by the proteasome [174,176,185]. Under hypoxia (~0,1–1 % O2/0 % O2 is termed anoxia), these enzymes are inactive, allowing accumulation of HIF-1α and activation via dimerization with ARNT to induce transcription by binding to hypoxia-responsive elements (HRE) on nuclear DNA (Fig. 4) [175,186].
Fig. 4. Crosstalk of HIF-1α and AHR.
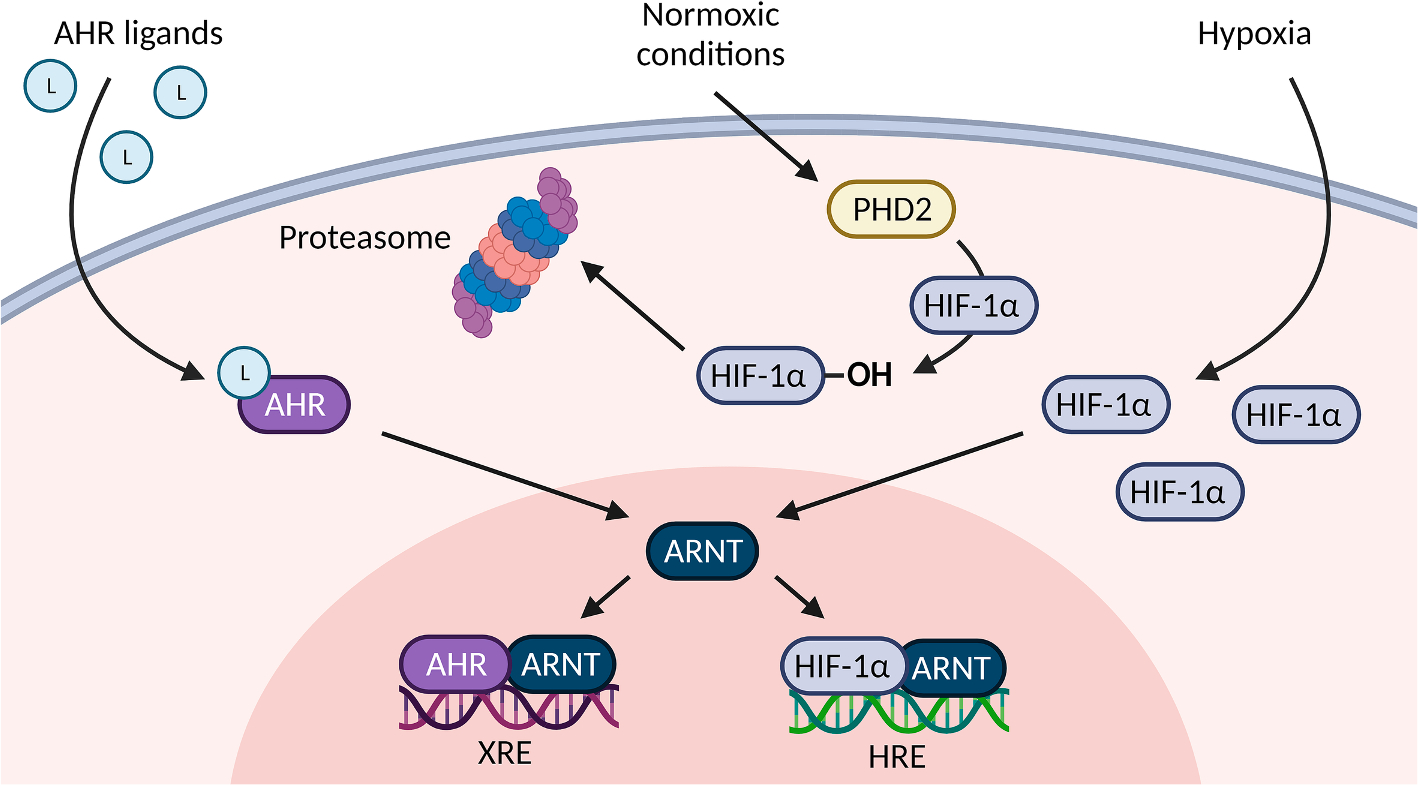
Activated AHR and hypoxia-inducible factor 1 alpha (HIF-1α) both translocate into the nucleus and dimerize with ARNT which results in binding to either xenobiotic response elements (XRE) or hypoxia responsive elements (HRE) in the enhancer/promoter region of respective target genes. Under normoxic conditions, prolyl hydroxylase domain-containing protein 2 (PHD2) executes hydroxylation of HIF-1α resulting in constant proteasomal degradation of HIF-1α. Under hypoxia, HIF-1α can accumulate and then translocate into the nucleus where it then dimerizes with ARNT. Depending on cell-type and tissue, a competition for ARNT may result in the suppression of the AHR pathway when the HIF-1α pathway is activated or vice versa.
There is clear long-standing evidence from independent studies that HIF-1α and AHR activities are interdependent [187–190] in different tissues, indicating a functional crosstalk between their pathways in conditions as diverse as obesity, glioblastoma, prostate cancer, nephrocalcinosis, autoimmune hepatitis and affecting processes like T cell and macrophage differentiation as well as blood–brain barrier function [87,191–199]. However, this issue has rarely been addressed systematically [200]: While a majority of studies suggests downregulation of AHR function through HIF-1α activation and vice versa [201–203], they almost exclusively investigated the function of TCDD, and more physiologic means of AHR activation have been even more scarcely addressed. The modes of interaction can be competitive or counteracting, but could possibly also be additive or compensatory under certain circumstances (Fig. 4). The specific effect may depend on contextual factors ranging from the specific tissue or cell type or pathway trigger to biochemical properties like ARNT availability, binding preference and other parameters. Importantly, it has been reported that ARNT is influenced by both hypoxia and hypoxia mimetics [204,205]. The skin has been a subject to hypoxia research, since especially the avascular epidermis is mildly hypoxic with constant low HIF-1α activity [206–208]. Of note, loss of ARNT in epidermal keratinocytes causes skin barrier failure in neonatal mice [209–211] associated with dysregulation of processes like synthesis of ceramides [212], a group of lipids implicated in oxidative stress balance [213] which in turn is also mediated by ARNT [214]. Since both keratinocyte-specific HIF-1α−/− and AHR−/− mice are viable without an overt phenotype [215–217], these findings underline (I) the importance of cooperative function of HIF-1α and AHR in epidermal keratinocytes and (II) may hint toward the involvement of other ARNT-dependent factors and/or (III) possible compensatory or aberrant pathway activation. Though a majority of studies demonstrates interaction of HIF-1α and AHR via ARNT (Fig. 4), others have argued that cells harbor a relative excess of ARNT not limiting its availability to dimerization partners [218], which also include HIF-2α, AHRR and others [177]. Other molecules proposed in HIF-1α-AHR crosstalk are microRNAs [197] and the proteins NCoA-2 [219] and p23 [203].
Among prototypical targets induced by HIF-1α activity are the vascular endothelial growth factor (VEGF), erythropoietin (EPO) and Glut-1 [220,221] which contribute to metabolic adaptation to hypoxia and improvement of oxygen availability: VEGF induces endothelial vessel growth, EPO is a growth factor for erythrocytes and Glut-1, encoded by the SLC2A1 gene, is an important membrane transport protein enabling the cellular import of glucose and other monosaccharides as well as ascorbic acid. Further, numerous other genes are modulated which can be grouped into inflammation/immune defense, proliferation, tumor promotion/cancer therapy resistance [222]. Taken together, HIF-1α-dependent processes promote cellular survival under stress through oxygen shortage.
The normal oxygen level is tissue-specific and low tissue oxygen availability is not necessarily pathologic: Intermediate O2 levels are termed physoxia (range ~ 1–13 % O2, medium values ~ 5 % O2) [223]. HIF-1α is typically stabilized under hypoxic conditions (below 1 % O2) which arise during high cell proliferation or metabolic activity including embryogenic development, inflammatory processes, physical strain and tumor growth. Further, numerous diseases ranging from respiratory infection over allergic asthma to COPD, besides consuming chronic diseases or placental insufficiency, can increase oxygen demands and/or restrict oxygen availability to the organism. Moreover, ischemia caused by vessel occlusions or tissue damage can induce regional hypoxia.
The HIF-1α pathway can also be upregulated under normoxic (resp. physoxic) conditions [224] through numerous triggers including LPS [225] or pro-inflammatory cytokines, such as IL-6, IL-18 and TNF-α, as well as thrombin [226], glucose [227], angiotensin II [228], cholesterol [229], hyperthermia [230] and UVB radiation [231–233]. A state of normoxic pathologic HIF-1α activation, which occurs in conditions including type II diabetes and cancer, is sometimes referred to as pseudohypoxia. The associated increase in cellular glycolysis is termed the Warburg effect [234,235]. The contextual nature of the HIF-1α pathway trigger will also activate specific other pathways with important implications regarding functional crosstalk.
In this regard, crosstalk of HIF-1α with AHR could be particularly important during UVB-dependent processes like DNA repair, apoptosis and proliferation in epidermal keratinocytes which are essentially dependent on AHR activity [58,61,62]. Intriguingly, HIF-1α may have contrastive effects on non-melanoma skin cancer (arising from keratinocytes) as opposed to melanoma [236–238], which is not fully investigated to date. Moreover, during UVB-induced immunosuppression to contact hypersensitivity, AHR and HIF-1α appear to take opposing roles in keratinocytes [216,239].
Experimentally, hypoxic stabilization of HIF-1α can be achieved by hypoxia mimetics like cobalt chloride or dimethyloxalylglycine [240] which inhibit the HIF-1α-degrading hydroxylases. Also, several HIF inhibitors are available, of which some are specific for HIF-1α [241,242], but others may also target HIF-2α [243]. It should not be neglected that some ligands of AHR vs HIF-1α may have unexpected indirect effects to modulate the other pathway [244].
The HIF-1α target EPO has a long history of therapeutic use for anemia [245], but also in sports doping [246]. Newer drugs include HIF stabilizers like roxadustat and similar HIF-prolyl hydroxylase inhibitors [247,248], which also achieve erythropoiesis besides other hypoxia-mimetic effects. Another presumably hypoxia-mimetic drug class are SGLT2 inhibitors, which palliate diabetic kidney disease and heart failure while improving erythropoiesis [249]. HIF-1α modulation is of particularly high relevance for ophthalmology of both the anterior (cornea) and posterior (retina) eye segments [250,251] and it should be noted that also the AHR is implicated in ocular diseases with some implications for HIF-1α crosstalk, e.g. in the context of cigarette smoking [252]. Blockade of the HIF-1α target VEGF by bevacizumab (Avastin®) is approved for treatment of several advanced solid tumors (lung, colon, breast, kidney, ovary, uterus) [253] and the similar drug ranibizumab (Lucentis®) is in use for ocular diseases involving pathologic angiogenesis [254,255]. Numerous novel approaches of HIF inhibition for advanced or refractory cancers are in clinical development stages [256]. Since AHR activation is implicated in tumor development due to its pro-proliferative impact [20], it would be worthwhile to further assess the role of HIF-1α-AHR interaction not only in cancer pathogenesis but also with regard to therapy schemes. Another field of possible future therapeutic HIF-1α utilization is the promotion of neuronal/axonal regeneration [257,258], in which AHR kinetics are also implicated [259].
An interesting approach to non-pharmacologic application of the HIF-1α pathway is hypoxic preconditioning: While uncontrolled chronic or also episodic hypoxia can be harmful, e.g. due to chronic heart failure resp. obstructive sleep apnea, controlled intermittent hypoxic training has proven beneficial health effects on the cardiovascular system and even confers protection against tissue damage from ischemic events [260,261]. These are similar to the effects of high-altitude training in professional sports. While HIF-1α is activated during ischemic events, reperfusion therapy is accompanied by additional secondary tissue damage involving inflammatory processes and ROS release in the impacted tissue. Novel literature suggests that AHR activation may play a pivotal role in this pathogenesis and suggests that AHR inhibition may pose a therapeutic strategy to mitigate ischemia–reperfusion damage in brain, myocardium and kidney [262–264].
6. NRF2 signaling
Living organisms have a fine-tuned system for maintaining a well-defined low steady-state level of oxidative stress, i.e. ROS production and elimination are well balanced. The term “oxidative stress” was first defined as a technical term by Helmut Sies in 1985 [265]. In this context, the formation of ROS can be initiated by aerobic metabolic processes, endogenous defense mechanisms, exogenous noxious agents and photobiological processes. Thereby, the main sources of ROS are leakage of the mitochondrial respiratory chain as well as CYP-mediated oxidations (phenomenon of “uncoupling”) [11]. Aldo-keto reductases (AKRs), a family of cytosolic NADPH-dependent oxidoreductases, were also suggested to be involved in the metabolic activation of PAHs and an associated generation of ROS. Moreover, several other enzymes were described and summarized in a review by Haarmann-Stemmann and co-workers to stimulate ROS production in an AHR-dependent manner (e.g., NADPH oxidases, COX-2) [11]. Klicken oder tippen Sie hier, um Text einzugeben.Interestingly, already in the beginnings of the 1990s, there was evidence that AHR activation is involved in the generation of ROS as well as the antioxidant stress response [266,267] (Fig. 5). Besides phase I xenobiotic-metabolizing enzymes, also phase II detoxifying enzymes (i.e. glutathione S-transferase, UDP-glucuronosyltransferase, sulfotransferase and N-acetyltransferase) are induced by canonical AHR/ARNT signaling. Moreover, there is evidence that AHR interacts with several regulators of the antioxidant stress response affecting the induction of antioxidants as well as antioxidative enzymes (i.e. catalase, glutathione peroxidase, superoxide dismutase and other peroxidases).
Fig. 5. NRF2-AHR-crosstalk.

Ligand-activation of AHR results in its nuclear translocation where it dimerizes with ARNT and, amongst others, induces the transcription of xenobiotic-responsive element (XRE)-regulated phase I and II detoxifying enzymes, and NRF2. Phase I detoxifying enzymes convert AHR ligands into reactive metabolites which again can lead to the formation of reactive oxygen species (ROS). ROS can trigger the dissociation of the cytosolic NRF2-KEAP1-complex resulting in the nuclear translocation of NRF2 and its subsequent dimerization with small Maf proteins. This results in the expression of antioxidative response elements (ARE)-controlled phase II detoxifying enzymes, as well as AHR. Moreover, the AHR and NRF2 target gene batteries partially overlap, the gene expression of NQO1, GST or UGT1A1, for instance, is under control of both pathways.
These regulators include transcription-factors such as NF-κB [268], AP-1 [269], β-catenin [270], proliferator-activated receptor γ (PPARγ) [271] and the master regulator NRF2, which is a member of the Cap-N-Collar protein family [272]. In addition to several enzymes introduced above, other cytoprotective proteins such as heme oxygenase-1 and NADPH:quinone oxidoreductase-1 (NQO-1) are under the control of the NRF2/Kelch-like ECH-Associated Protein 1 (KEAP1) signaling pathway [273]. Under basal conditions, NRF2 is bound in the cytosol to the adaptor protein KEAP1 [274,275]. Here, KEAP1 serves as an adapter protein linking NRF2 to a cullin-3-mediated ubiquitination and thus proteasomal degradation [276]. In addition, activation of NRF2 occurs predominantly by a conformational change of KEAP1. Thus, it has been shown that the redox-sensitive cysteine residues of KEAP1 are able to react with and oxidize ROS, thereby preventing a cullin-3-mediated ubiquitination and proteasomal degradation of NRF2 [277]. Moreover, ROS have been described to activate specific kinases (PKC, PI3K, MAPK) that phosphorylate NRF2, resulting in an attenuation of NRF2-KEAP1 interaction and inhibition of proteasomal degradation of NRF2 (Fig. 5) [278,279]. Upon activation, NRF2 shuttles into the nucleus and forms a heterodimer with so-called small musculoaponeurotic fibrosarcoma (MAF) proteins. This dimer then binds to the antioxidant response element (ARE) in the promoter region and enhances target gene expression (Fig. 5) [280].
Metabolic activation of procarcinogens initiated by AHR and downstream CYP1 monooxygenases is well known to play a significant role in the human body regarding the development of malignant diseases. Besides this, several enzyme systems involved in the metabolism of xenobiotics initiated by AHR activation lead to the generation of ROS and thus to oxidative stress, activating the NRF2-signaling pathway (Fig. 5).
Apart from the described indirect crosstalk of AHR and NRF2 (Fig. 5), Miao and colleagues showed that the NRF2 gene is a direct target of the canonical AHR signaling pathway containing multiple XREs in its promoter region [281]. Consequently, knockdown of AHR results in a decreased expression of NRF2 and thereby attenuates the induction of the antioxidant stress response. In contrast to this, it was reported that NRF2 expression and activity controls AHR expression [282,283]. Besides this, expression of genes encoding phase II detoxifying enzymes are induced in response to various environmental factors to counteract the deleterious effects of potentially reactive intermediates. Especially the NRF2/KEAP1 signaling pathway is responsible for their induction. However, there appears to be some overlap with target genes of AHR that include NQO1 [284], glutathione S-transferases (GST) [285,286] and some glucuronosyltransferases [287–289]. In addition, Tsuji et al. could prove that treatment of keratinocytes with ketoconazole not only results in an activation of AHR and the corresponding stimulation of CYP1A1 gene expression but also in an increased induction of NRF2 and NQO1 expression. Interestingly, the NRF2 activity stimulated by AHR in ketoconazole-treated primary keratinocytes was independent of ROS [290]. Instead, an involvement of EGFR, a downstream signal transduction of the MEK/ERK signaling pathway and a corresponding phosphorylation of NRF2 and an attenuation of the NRF2/KEAP1 interaction was suggested [272]. In addition, in a recent study we found that PAHs induce AKR1C3 expression in an AHR-EGFR-, MEK/ERK- and NRF2-dependent manner. Specifically, we showed that BaP upregulates AKR1C3 expression via the AHR-EGFR-NRF2 axis, independently of CYP1A1 activity and the associated production of ROS and genotoxic metabolites [48]. But there are also studies showing that it is possible to inhibit AHR activity while stimulating the NRF2/KEAP1 antioxidative system that especially seemed to be important when it comes to metabolic activation of PAHs [291–296]. Interestingly, a treatment with chrysoeriol and curcumin resulted in an inhibition of CYP1A activities and reduced the formation of DNA-adducts induced by BaP metabolites as well as 7,12-dimethylbenz[a]anthracene in MCF-7 breast cancer cells [291,292]. Another study demonstrated that curcumin altered BaP-induced activation and DNA-binding of AHR. In fact, they observed enhanced NRF2 stimulation and an associated increased gene expression of GST and NQO1 with improved detoxification of BaP [293]. There are far more publications pointing to this antithetical activation of AHR and NRF2. In addition, cinnamaldehyde was shown to have an inhibitory function on AHR while having antioxidative capacity, mediated by a stimulation of NRF2 and heme oxygennase-1 expression [297]. This described dual function may point to a suitable modulation of AHR and NRF2 signaling, probably as a good tool to protect tissues, in particular those which are frequently exposed to environmentally or lifestyle-derived AHR ligands (e.g. liver and skin) against the metabolic activation and thus tumor-initiating potency of pro-carcinogenic chemicals. Beyond that, it might also be beneficial for the treatment of disorders related to oxidative stress such as dioxin intoxication, acne and vitiligo as suggested by Furue and co-workers [297]. Beside this, others could show that ROS inhibits the catalytic activity of CYP1A1 and therefore might inhibit the metabolic degradation of AHR ligands. Specifically, it was shown that arsenic-treated mice as well as cells and a corresponding increase of ROS resulted in an accumulation of FICZ and an accompanied upregulation of Cyp1a1 gene expression [298,299]. In addition, increasing ROS levels induced by glutathione deprivation or in the absence of functional NRF2 correlate with an increased expression and activity of AHR in normal and malignant mammary cells. AHR activation thereby fosters the expression of AREG and therefore activation of the EGFR. Subsequent experiments revealed that AHR inhibition sensitized human breast cancer cells to erlotinib and might be an attractive target to change the tumor microenvironment [300]. However, it should be considered that erlotinib itself is a substrate for CYP1A1, so inhibition of AHR/upregulation of CYP1A1 might reinforce an accumulation of erlotinib and would result in an improved impact of erlotinib [301].
Several MAF-proteins, belonging to the family of leucine zipper nuclear transcription factors, were described to have a different impact on transcription of different eukaryotic genes. Thereby, it was demonstrated by Dhakshinamoorthy et al. that overexpression of c-MAF has a negative influence on the gene expression of ARE mediated detoxifying enzymes in HepG2 cells. In particular, it was demonstrated that by forming homodimers or heterodimers with MAFG, c-MAF represses ARE-mediated NQO1 expression [302]. Interestingly, an interaction between AHR and c-MAF could also be reported. There, Apetoh et al. demonstrated that the AHR interacts with c-MAF to promote IL-27-induced differentiation of Type 1 regulatory T cells [79]. For this reason, it might be possible that the activity of NRF2 could be influenced by an interaction of AHR and c-MAF. Thus, it would be conceivable that other transcription factors and/or signaling pathways may have an impact on the interaction of AHR- and NRF2-signaling pathway and not only in terms of the antioxidant stress response.
7. The AHR as a ligand-dependent E3 ubiquitin ligase
While the AHR is infamous for its function as a ligand-activated transcription factor, some studies regarding the toxicity of dioxins and related compounds have shown that the AHR also mediates estrogenic [303,304], anti-estrogenic [305,306] and androgenic effects [307]. The underlying mechanisms were unclear and could not be explained exclusively by CYP1-mediated degradation of steroid hormones or the CYP19A (aromatase)-catalyzed synthesis of the estrogen receptor (ER)-ligand 17β-estradiol. In 2003, Ohtake and co-workers showed that dioxins exert estrogenic effects through the association of ligand-activated AHR/ARNT to unliganded ER [308]. Additionally, Wormke et al. revealed that TCDD induces the proteasome-dependent degradation of endogenous ER-α [309]. Based on this observation, a novel ubiquitin ligase complex was identified. This complex is initiated by ligand activation of the AHR and targets sex steroid receptors [310]. Nevertheless, mechanistic insights into the regulation of both AHR functions remained unclear until 2017, when Luecke-Johansson and co-workers described how the AHR switches between its functions as a transcription factor and an E3 ubiquitin ligase [311].
One of the most important regulators of cellular homeostasis is the ubiquitin proteasome system which specifically degrades targeted proteins [312,313]. In a first step, an activating enzyme (E1) transfers a ubiquitin molecule to a ubiquitin-conjugating enzyme (E2). Ubiquitin protein ligases (E3) recognize specific degradation signals and are therefore responsible for substrate specificity. With the help of an E3 ligase, the E2 enzyme conjugates one or more ubiquitin molecules to the target protein, followed by proteasomal degradation [312,313]. The AHR-associated E3 ligase complex, denoted CUL4BAHR, is composed of the scaffold protein Cullin 4B (CUL4B), damaged-DNA binding protein 1, RING-box protein 1, transducin-β-like protein 3 and the proteasomal 19S particle [310]. Upon ligand-activation of the AHR, it initiates assembly of the CUL4BAHR complex and serves as a substrate-specific adapter protein within the complex (Fig. 6). Then, the availability of ARNT determines the functionality of AHR: when ARNT is available, the AHR functions as a ligand-activated transcription factor and activates the canonical AHR signaling pathway. However, when ARNT is occupied by other proteins, such as the AHRR, the AHR functions as an E3 ligase and induces assembly of the CUL4BAHR complex [311]. Subsequently, substrate proteins are targeted for proteasomal degradation (Fig. 6). Target proteins of the CUL4BAHR complex include ER-α, ER-β and androgen receptor (AR) [310,314]. It was also shown that PPARγ, a transcription factor and important regulator of adipogenesis, is targeted by the CUL4BAHR complex for proteasomal degradation [315]. Moreover, β-catenin, which is a transcription factor downstream from the WNT signaling pathway, has been reported as target protein [316]. Canonical WNT/β-catenin signaling pathway is a key regulator of embryonic development and adult tissue homeostasis [317]. While β-catenin degradation by the CUL4BAHR complex was shown in mouse intestine, other working groups could not find evidence for β-catenin degradation in mouse hepatoma cells despite evidence of physical interaction of AHR and β-catenin. This suggests that AHR/β-catenin interaction, or rather the AHR function as an E3 ligase, may not only be ligand-dependent but also tissue-specific. Interestingly, the AHR was found to induce the ubiquitin-proteasomal and lysosomal degradation of RelA/p65 in mouse peritoneal macrophages [140]. However, whether this process involves AHR’s function as a E3 ubiquitin ligase requires further investigation.
Fig. 6. AHR acts as a ligand-dependent E3 ubiquitin ligase.
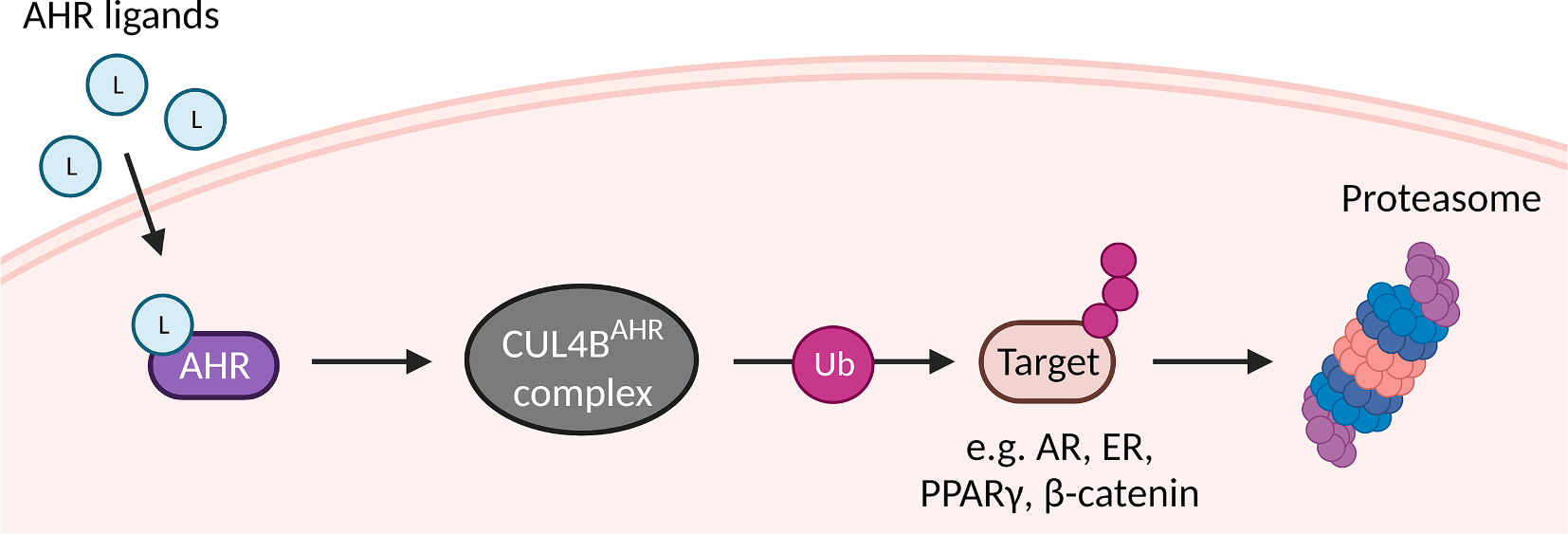
Upon ligand-activation, AHR and the E3 ubiquitin ligase Cullin 4B (CUL4B) can from a cytosolic complex which ubiquitinates and thereby targets various proteins, including androgen receptor (AR), estrogen receptor (ER), peroxisome proliferator-activated receptor γ (PPARγ) and β-catenin, to proteasomal degradation. This process presumably takes place when the AHR binding partner ARNT is absent.
Importantly, upon its ligand-induced nuclear translocation and the subsequent transactivation of target genes, AHR itself becomes degraded [318,319]. Even though this has been known for many years, details about the degradation process only became evident in 2021. Rijo and co-workers demonstrated that the CUL4BAHR complex and TCDD-inducible poly(ADP-ribose) polymerase collaborate in ligand-induced AHR degradation. Knockdown of CUL4B in mouse embryonic fibroblasts partially prevented AHR degradation by TCDD, while additional knockdown of TCDD-inducible poly(ADP-ribose) polymerase completely abolished ligand-induced AHR degradation [320].
Due to the fact that the canonical transcriptional activity and E3-ligase activity of the AHR are in a close relation with each other, it should be considered that molecular targeting of the AHR by classical chemical ligands does also affect other signaling pathways, such as ER, AR, PPARγ and WNT/β-catenin signaling. A dysregulation of these major signaling pathways may contribute to the development of diseases, such as cancer [321–324]. However, the bifunctionality of the AHR gives rise to the opportunity of utilizing the E3 ligase activity of AHR as a new target. Faber et al. summarize the targeted modulation of the AHR in inflammatory gastrointestinal indications and give guidance for drug development in their latest review [39]. Particularly, the Proteolysis targeting chimera (PROTAC) technology is a promising tool that may substitute classical small molecule-based inhibitors [39,325,326]. PROTACs are (hetero-)bifunctional molecules consisting of two domains that are joined by a linker. One domain interacts with an E3 ubiquitin ligase and the other binds the protein of interest (POI), bringing the E3 and POI into spatial proximity, resulting in the targeted degradation of the POI by the proteasome [326,327]. PROTAC technology even allows the targeting of so far undruggable proteins and, in contrast to gene editing approaches, it usually does not cause misinterpretations arising from genetic compensation or spontaneous mutations. Just recently, the first PROTACs, for instance targeting AR in prostate cancer (ARV-110), ER in breast cancer (ARV-471) and Bruton’s tyrosine kinase in B cell malignancies (NX-2127), have entered clinical testing in humans [327]. First PROTACs targeting the AHR [328], but also first attempts to use the AHR as an E3 ligase to degrade target proteins [329], have already been described. As the AHR has been recognized as a pharmacological target by many researchers in recent years, approaches such as PROTACs hold great promise for future research of new treatment strategies for various diseases.
8. Conclusion
While the canonical pathway of AHR is well documented, gaps remain in the bigger picture of AHR-related crosstalk with other signaling proteins. The multiple non-canonical events and functions of AHR are expressed by diverse organ-, tissue- and ligand-specific effects. As described in this review article, AHR is indirectly involved in processes concerning proliferation, differentiation, apoptosis, adaptation to low oxygen levels, oxidative stress, ubiquitination and proteasomal degradation. Even though this already covers a broad spectrum of unquestionably relevant functions, even more AHR crosstalk events have been observed. One of them is the TGF-β/SMAD pathway which seems to negatively regulate canonical AHR signaling [330]. Another interesting non-canonical AHR crosstalk concerns the circadian signaling pathways. Here, ligand-activated AHR can influence the amplitude and phase of rhythms in circadian clock genes, hormones and behavior [331]. Moreover, AHR expression and activity has a critical impact on the self-renewal and maintenance of stem cells and their subsequent differentiation in a tissue-specific manner. Specifically, AHR controls the expression of several pluripotency factors (e.g. OCT4, NANOG) and interacts with multiple signaling pathways (e.g. Wnt/β-catenin, Notch) and epigenetic gene regulatory mechanisms (e.g. DNA methylation) [332–334]. However, deepening the knowledge on non-canonical AHR signaling and further unravelling the potentially even more faceted network following upon AHR activation could contribute to a better understanding of many different physiological and pathological processes. Importantly, this would especially benefit the development of new AHR-targeted therapeutic approaches.
Acknowledgements
NCS and FH are supported by the Jürgen Manchot Foundation. Research of SF is supported by the Deutsche Forschungsgemeinschaft (FA1468/2-1, FA1468/2-2). CFAV is supported by the National Institute of Environmental Health Sciences of the National Institutes of Health (R01ES029126, R01ES032827). Research in the laboratory of THS is supported by the Deutsche Forschungsgemeinschaft (HA 7346/2-2). The scheme shown in the figure was created with BioRender software (www.biorender.com; agreement numbers HM24OFNLD, TQ24OFNH3K, ZU24OFN6I4, VN24OFNB44, ST24PA4ZKF, DF24OFNNQD and HT24OFNJ20).
Abbreviations:
- AHR
aryl hydrocarbon receptor
- AHRR
aryl hydrocarbon receptor repressor
- AKR
aldo-keto reductase
- AP-1
activator protein-1
- AR
androgen receptor
- ARE
antioxidant response element
- AREG
amphiregulin
- ARNT
aryl hydrocarbon receptor nuclear translocator
- BaP
benzo[a]pyrene
- BMDC
bone marrow-derived dendritic cell
- CCL
chemokine (C-C-motif) ligand
- COX-2
cyclooxygenase-2
- CUL4B
cullin 4B
- CYP
cytochrome P450
- DC
dendritic cell
- ECD
extracellular domain
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- EPO
erythropoietin
- ER
estrogen receptor
- FICZ
6-formylindolo[3,2-b]carbazole
- GST
glutathione S-transferase
- HIF-1α
hypoxia-inducible factor 1α
- HRE
hypoxia-responsive element
- IDO
indolamine-2,3-dioxygenase
- IFN
interferon
- IL
interleukin
- JAK
janus-kinase
- KEAP1
Kelch-like ECH-associated protein 1
- KYN
kynurenine
- LPS
lipopolysaccharide
- MAF
musculoaponeurotic fibrosarcoma
- MAPK
mitogen-activated protein kinase
- MMP-1
matrix metalloproteinase-1
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NQO1
NADPH:quinone oxidoreductase-1
- NRF2
nuclear factor erythroid 2-related factor 2
- PAH
polycyclic aromatic hydrocarbon
- PCB
polychlorinated biphenyl
- PHD2
prolyl hydroxylase domain-containing protein 2
- PKC
protein kinase C
- PM
particulate matter
- POI
protein of interest
- PPARγ
peroxisome proliferator-activated receptor γ
- PROTAC
proteolysis targeting chimera
- ROS
reactive oxygen species
- RTK
receptor tyrosine kinase
- SLE
systemic lupus erythematosus
- STAT
signal transducer and activator of transcription
- 2,4,5-T
2,4,5-trichlorophenol
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- TGF
transforming growth factor
- Th17
T helper 17 cells
- TLR
Toll-like receptor
- TNF
tumor necrosis factor
- TPH
tryptophan hydroxylase-1
- Tregs
regulatory T cells
- VEGF
vascular endothelial growth factor
- XRE
xenobiotic-responsive element
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Natalie C. Sondermann: Conceptualization, Writing – original draft, Writing – review & editing, Visualization. Sonja Faßbender: Writing – original draft, Writing – review & editing. Frederick Hartung: Writing – original draft, Writing – review & editing. Anna M. Hätälä: Writing – original draft, Writing – review & editing. Katharina M. Rolfes: Writing – original draft, Writing – review & editing. Christoph F.A. Vogel: Writing – original draft, Writing – review & editing. Thomas Haarmann-Stemmann: Conceptualization, Writing – original draft, Writing – review & editing, Supervision.
References
- [1].Poland A, Glover E, Kende AS, Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol, J. Biol. Chem. 251 (16) (1976) 4936–4946. [PubMed] [Google Scholar]
- [2].Poland AP, Smith D, Metter G, Possick P, A health survey of workers in a 2,4-D and 2,4,5-T plan with special attention to chloracne, porphyria cutanea tarda, and psychologic parameters, Arch. Environ. Health 22 (3) (1971) 316–327. [DOI] [PubMed] [Google Scholar]
- [3].Kimmig J, Schulz KH, Occupational acne (so-called chloracne) due to chlorinated aromatic cyclic ethers, Dermatologica 115 (4) (1957) 540–546. [PubMed] [Google Scholar]
- [4].Nebert DW, Gielen JE, Genetic regulation of aryl hydrocarbon hydroxylase induction in the mouse, Fed. Proc. 31 (4) (1972) 1315–1325. [PubMed] [Google Scholar]
- [5].Gielen JE, Goujon FM, Nebert DW, Genetic regulation of aryl hydrocarbon hydroxylase induction. II. Simple Mendelian expression in mouse tissues in vivo, J. Biol. Chem. 247 (4) (1972) 1125–1137. [PubMed] [Google Scholar]
- [6].Poland AP, Glover E, Robinson JR, Nebert DW, Genetic expression of aryl hydrocarbon hydroxylase activity. Induction of monooxygenase activities and cytochrome P1–450 formation by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice genetically “nonresponsive” to other aromatic hydrocarbons, J. Biol. Chem. 249 (17) (1974) 5599–5606. [PubMed] [Google Scholar]
- [7].Eskenazi B, Warner M, Brambilla P, Signorini S, Ames J, Mocarelli P, The Seveso accident: A look at 40 years of health research and beyond, Environ. Int. 121 (2018) 139–71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Patterson AT, Kaffenberger BH, Keller RA, Elston DM, Skin diseases associated with Agent Orange and other organochlorine exposures, J. Am. Acad. Dermatol. 74 (1) (2016) 143–170. [DOI] [PubMed] [Google Scholar]
- [9].Sorg O, Zennegg M, Schmid P, Fedosyuk R, Valikhnovskyi R, Gaide O, Kniazevych V, Saurat JH, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) poisoning in Victor Yushchenko: identification and measurement of TCDD metabolites, Lancet 374 (9696) (2009) 1179–1185. [DOI] [PubMed] [Google Scholar]
- [10].Rothhammer V, Quintana FJ, The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease, Nat. Rev. Immunol. 19 (3) (2019) 184–197. [DOI] [PubMed] [Google Scholar]
- [11].Vogel CFA, Van Winkle LS, Esser C, Haarmann-Stemmann T, The aryl hydrocarbon receptor as a target of environmental stressors - Implications for pollution mediated stress and inflammatory responses, Redox Biol. 34 (2020), 101530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Avilla MN, Malecki KMC, Hahn ME, Wilson RH, Bradfield CA, The Ah Receptor: Adaptive Metabolism, Ligand Diversity, and the Xenokine Model, Chem. Res. Toxicol. 33 (4) (2020) 860–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jia Y, Tao Y, Lv C, Xia Y, Wei Z, Dai Y, Tetrandrine enhances the ubiquitination and degradation of Syk through an AhR-c-src-c-Cbl pathway and consequently inhibits osteoclastogenesis and bone destruction in arthritis, Cell Death Dis. 10 (2) (2019) 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Manni G, Mondanelli G, Scalisi G, Pallotta MT, Nardi D, Padiglioni E, Romani R, Talesa VN, Puccetti P, Fallarino F, Gargaro M, Pharmacologic Induction of Endotoxin Tolerance in Dendritic Cells by L-Kynurenine, Front. Immunol. 11 (2020) 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Paris A, Tardif N, Baietti FM, Berra C, Leclair HM, Leucci E, Galibert MD, Corre S, The AhR-SRC axis as a therapeutic vulnerability in BRAFi-resistant melanoma, EMBO Mol. Med. (2022) e15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shimizu Y, Nakatsuru Y, Ichinose M, Takahashi Y, Kume H, Mimura J, Fujii-Kuriyama Y, Ishikawa T, Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor, PNAS 97 (2) (2000) 779–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nakatsuru Y, Wakabayashi K, Fujii-Kuriyama Y, Ishikawa T, Kusama K, Ide F, Dibenzo[A, L]pyrene-induced genotoxic and carcinogenic responses are dramatically suppressed in aryl hydrocarbon receptor-deficient mice, Int. J. Cancer 112 (2) (2004) 179–183. [DOI] [PubMed] [Google Scholar]
- [18].Paris A, Tardif N, Galibert MD, Corre S, AhR and Cancer: From Gene Profiling to Targeted Therapy, Int. J. Mol. Sci. 22 (2) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Andersson P, McGuire J, Rubio C, Gradin K, Whitelaw ML, Pettersson S, Hanberg A, Poellinger L, A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors, PNAS 99 (15) (2002) 9990–9995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Murray IA, Patterson AD, Perdew GH, Aryl hydrocarbon receptor ligands in cancer: Friend and foe, Nat. Rev. Cancer 14 (12) (2014) 801–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fan Y, Boivin GP, Knudsen ES, Nebert DW, Xia Y, Puga A, The aryl hydrocarbon receptor functions as a tumor suppressor of liver carcinogenesis, Cancer Res. 70 (1) (2010) 212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, Jugold M, Guillemin GJ, Miller CL, Lutz C, Radlwimmer B, Lehmann I, von Deimling A, Wick W, Platten M, An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor, Nature 478 (7368) (2011) 197–203. [DOI] [PubMed] [Google Scholar]
- [23].Bayer A First-in-Humans Dose Finding Study for an Aryl Hydrocarbon Receptor Inhibitor (AhRi) in Patients With Advanced Cancer. ClinicalTrials.gov identifier: NCT04069026. Updated August 4. 2022. Accessed August 15, 2022. https://clinicaltrials.gov/ct2/show/NCT04069026. [Google Scholar]
- [24].Ikena Oncology, A Phase 1a/b Study of IK-175 as a Single Agent and in Combination With Nivolumab in Patients With Locally Advanced or Metastatic Solid Tumors and Urothelial Carcinoma. ClinicalTrials.gov Identifier: NCT04200963. Updated February 4, 2022. Accessed August 15, 2022. https://clinicaltrials.gov/ct2/show/NCT04200963. [Google Scholar]
- [25].Narasimhan S, Stanford Zulick E, Novikov O, Parks AJ, Schlezinger JJ, Wang Z, Laroche F, Feng H, Mulas F, Monti S, Sherr DH, Towards Resolving the Pro- and Anti-Tumor Effects of the Aryl Hydrocarbon Receptor, Int. J. Mol. Sci. 19 (5) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dumoutier L, Van Roost E, Ameye G, Michaux L, Renauld JC, IL-TIF/IL-22: genomic organization and mapping of the human and mouse genes, Genes Immun. 1 (8) (2000) 488–494. [DOI] [PubMed] [Google Scholar]
- [27].Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, Kolls JK, IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia, Nat. Med. 14 (3) (2008) 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wolk K, Witte E, Wallace E, Döcke WD, Kunz S, Asadullah K, Volk HD, Sterry W, Sabat R, IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis, Eur. J. Immunol. 36 (5) (2006) 1309–1323. [DOI] [PubMed] [Google Scholar]
- [29].Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R, IL-22 increases the innate immunity of tissues, Immunity 21 (2) (2004) 241–254. [DOI] [PubMed] [Google Scholar]
- [30].Vorderstrasse BA, Steppan LB, Silverstone AE, Kerkvliet NI, Aryl hydrocarbon receptor-deficient mice generate normal immune responses to model antigens and are resistant to TCDD-induced immune suppression, Toxicol. Appl. Pharmacol. 171 (3) (2001) 157–164. [DOI] [PubMed] [Google Scholar]
- [31].Ishihara Y, Kado SY, Bein KJ, He Y, Pouraryan AA, Urban A, Haarmann-Stemmann T, Sweeney C, Vogel CFA, Aryl Hydrocarbon Receptor Signaling Synergizes with TLR/NF-kappaB-Signaling for Induction of IL-22 Through Canonical and Non-Canonical AhR Pathways, Front Toxicol 3 (2021), 787360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].O’Driscoll CA, Mezrich JD, The Aryl Hydrocarbon Receptor as an Immune-Modulator of Atmospheric Particulate Matter-Mediated Autoimmunity, Front. Immunol. 9 (2018) 2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang J, Zhao Y, Zhang X, Tu W, Wan R, Shen Y, Zhang Y, Trivedi R, Gao P, Type II alveolar epithelial cell aryl hydrocarbon receptor protects against allergic airway inflammation through controlling cell autophagy, Front. Immunol. 13 (2022), 964575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Keam SJ, Tapinarof Cream 1%: First Approval, Drugs (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Esser C, Rannug A, Stockinger B, The aryl hydrocarbon receptor in immunity, Trends Immunol. 30 (9) (2009) 447–454. [DOI] [PubMed] [Google Scholar]
- [36].Wang W, Xiang T, Yang Y, Wang Z, Xie J, E3 Ubiquitin Ligases STUB1 Contributes to the Th17/Treg Imbalance via the Ubiquitination of Aryl Hydrocarbon Receptor in Rheumatoid Arthritis, Clin. Exp. Immunol. (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liu Y, Lv J, Liu J, Li M, Xie J, Lv Q, Deng W, Zhou N, Zhou Y, Song J, Wang P, Qin C, Tong WM, Huang B, Mucus production stimulated by IFN-AhR signaling triggers hypoxia of COVID-19, Cell Res. 30 (12) (2020) 1078–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Guarnieri T, Hypothesis: Emerging Roles for Aryl Hydrocarbon Receptor in Orchestrating CoV-2-Related Inflammation, Cells 11 (4) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Faber SC, Lahoti TS, Taylor ER, Lewis L, Sapiro JM, Toledo Sales V, Dragan YP, Jeffy BD, Current Therapeutic Landscape and Safety Roadmap for Targeting the Aryl Hydrocarbon Receptor in Inflammatory Gastrointestinal Indications, Cells 11 (10) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Grishanova AY, Perepechaeva ML, Aryl Hydrocarbon Receptor in Oxidative Stress as a Double Agent and Its Biological and Therapeutic Significance, Int. J. Mol. Sci. 23 (12) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Oda K, Matsuoka Y, Funahashi A, Kitano H, A comprehensive pathway map of epidermal growth factor receptor signaling, Mol. Syst. Biol. 1 (2005) (2005) 0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wee P, Wang Z, Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways, Cancers 9 (5) (2017) 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Shostak K, Chariot A, EGFR and NF-kappaB: partners in cancer, Trends Mol. Med. 21 (6) (2015) 385–393. [DOI] [PubMed] [Google Scholar]
- [44].Ivanovic V, Weinstein IB, Benzo[a]pyrene and other inducers of cytochrome p 1–450 inhibit binding of epidermal growth factor to cell surface receptors, Carcinogenesis 3 (5) (1982) 505–510. [DOI] [PubMed] [Google Scholar]
- [45].Madhukar BV, Brewster DW, Matsumura F, Effects of in vivo-administered 2,3,7,8-tetrachlorodibenzo-p-dioxin on receptor binding of epidermal growth factor in the hepatic plasma membrane of rat, guinea pig, mouse, and hamster, PNAS 81 (23) (1984) 7407–7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kärenlampi SO, Eisen HJ, Hankinson O, Nebert DW, Effects of cytochrome P1–450 inducers on the cell-surface receptors for epidermal growth factor, phorbol 12,13-dibutyrate, or insulin of cultured mouse hepatoma cells, J. Biol. Chem. 258 (17) (1983) 10378–10383. [PubMed] [Google Scholar]
- [47].Hudson LG, Toscano WA Jr., Greenlee WF, Regulation of epidermal growth factor binding in a human keratinocyte cell line by 2,3,7,8-tetrachlorodibenzo-p-dioxin, Toxicol. Appl. Pharmacol. 77 (2) (1985) 251–259. [DOI] [PubMed] [Google Scholar]
- [48].Vogeley C, Sondermann NC, Woeste S, Momin AA, Gilardino V, Hartung F, Heinen M, Maaß SK, Mescher M, Pollet M, Rolfes KM, Vogel CFA, Rossi A, Lang D, Arold ST, Nakamura M, Haarmann-Stemmann T, Unraveling the differential impact of PAHs and dioxin-like compounds on AKR1C3 reveals the EGFR extracellular domain as a critical determinant of the AHR response, Environ. Int. 158 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hardesty JE, Al-Eryani L, Wahlang B, Falkner KC, Shi H, Jin J, Vivace BJ, Ceresa BP, Prough RA, Cave MC, Epidermal Growth Factor Receptor Signaling Disruption by Endocrine and Metabolic Disrupting Chemicals, Toxicol. Sci. 162 (2) (2018) 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Patel RD, Kim DJ, Peters JM, Perdew GH, The aryl hydrocarbon receptor directly regulates expression of the potent mitogen epiregulin, Toxicol. Sci. 89 (1) (2006) 75–82. [DOI] [PubMed] [Google Scholar]
- [51].John K, Lahoti TS, Wagner K, Hughes JM, Perdew GH, The Ah receptor regulates growth factor expression in head and neck squamous cell carcinoma cell lines, Mol. Carcinog. 53 (10) (2014) 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Du B, Altorki NK, Kopelovich L, Subbaramaiah K, Dannenberg AJ, Tobacco smoke stimulates the transcription of amphiregulin in human oral epithelial cells: evidence of a cyclic AMP-responsive element binding protein-dependent mechanism, Cancer Res. 65 (13) (2005) 5982–5988. [DOI] [PubMed] [Google Scholar]
- [53].Lemjabbar H, Li D, Gallup M, Sidhu S, Drori E, Basbaum C, Tobacco smoke-induced lung cell proliferation mediated by tumor necrosis factor alpha-converting enzyme and amphiregulin, J. Biol. Chem. 278 (28) (2003) 26202–26207. [DOI] [PubMed] [Google Scholar]
- [54].Sutter CH, Yin H, Li Y, Mammen JS, Bodreddigari S, Stevens G, Cole JA, Sutter TR, EGF receptor signaling blocks aryl hydrocarbon receptor-mediated transcription and cell differentiation in human epidermal keratinocytes, Proc. Natl. Acad. Sci. 106(11) (2009) 4266–4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Joiakim A, Mathieu PA, Shelp C, Boerner J, Reiners JJ Jr., Epidermal Growth Factor Receptor Kinase Inhibitors Synergize with TCDD to Induce CYP1A1/1A2 in Human Breast Epithelial MCF10A Cells, Drug Metab. Dispos. 44 (5) (2016) 665–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shao J, Lee SB, Guo H, Evers BM, Sheng H, Prostaglandin E2 stimulates the growth of colon cancer cells via induction of amphiregulin, Cancer Res. 63 (17) (2003) 5218–5223. [PubMed] [Google Scholar]
- [57].Davis JW Jr., Burdick AD, Lauer FT, Burchiel SW, The aryl hydrocarbon receptor antagonist, 3’methoxy-4’nitroflavone, attenuates 2,3,7,8-tetrachlorodibenzo-p-dioxin-dependent regulation of growth factor signaling and apoptosis in the MCF-10A cell line, Toxicol. Appl. Pharmacol. 188 (1) (2003) 42–49. [DOI] [PubMed] [Google Scholar]
- [58].Fritsche E, Schäfer C, Calles C, Bernsmann T, Bernshausen T, Wurm M, Hübenthal U, Cline JE, Hajimiragha H, Schroeder P, Klotz LO, Rannug A, Fürst P, Hanenberg H, Abel J, Krutmann J, Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation, PNAS 104 (21) (2007) 8851–8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bergander L, Wincent E, Rannug A, Foroozesh M, Alworth W, Rannug U, Metabolic fate of the Ah receptor ligand 6-formylindolo[3,2-b]carbazole, Chem. Biol. Interact. 149 (2–3) (2004) 151–164. [DOI] [PubMed] [Google Scholar]
- [60].Tigges J, Haarmann-Stemmann T, Vogel CFA, Grindel A, Hubenthal U, Brenden H, Grether-Beck S, Vielhaber G, Johncock W, Krutmann J, Fritsche E, The new aryl hydrocarbon receptor antagonist E/Z-2-benzylindene-5,6-dimethoxy-3,3-dimethylindan-1-one protects against UVB-induced signal transduction, J, Invest. Dermatol. 134 (2) (2014) 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Frauenstein K, Sydlik U, Tigges J, Majora M, Wiek C, Hanenberg H, Abel J, Esser C, Fritsche E, Krutmann J, Haarmann-Stemmann T, Evidence for a novel anti-apoptotic pathway in human keratinocytes involving the aryl hydrocarbon receptor, E2F1, and checkpoint kinase 1, Cell Death Differ. 20 (10) (2013) 1425–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Pollet M, Shaik S, Mescher M, Frauenstein K, Tigges J, Braun SA, Sondenheimer K, Kaveh M, Bruhs A, Meller S, Homey B, Schwarz A, Esser C, Douki T, Vogel CFA, Krutmann J, Haarmann-Stemmann T, The AHR represses nucleotide excision repair and apoptosis and contributes to UV-induced skin carcinogenesis, Cell Death Differ. 25 (10) (2018) 1823–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ono Y, Torii K, Fritsche E, Shintani Y, Nishida E, Nakamura M, Shirakata Y, Haarmann-Stemmann T, Abel J, Krutmann J, Morita A, Role of the aryl hydrocarbon receptor in tobacco smoke extract-induced matrix metalloproteinase-1 expression, Exp. Dermatol. 22 (5) (2013) 349–353. [DOI] [PubMed] [Google Scholar]
- [64].Lahmann C, Bergemann J, Harrison G, Young AR, Matrix metalloproteinase-1 and skin ageing in smokers, Lancet 357 (9260) (2001) 935–936. [DOI] [PubMed] [Google Scholar]
- [65].Tanaka H, Ono Y, Nakata S, Shintani Y, Sakakibara N, Morita A, Tobacco smoke extract induces premature skin aging in mouse, J. Dermatol. Sci. 46 (1) (2007) 69–71. [DOI] [PubMed] [Google Scholar]
- [66].Wang K, Zheng J, Yu J, Wu Y, Guo J, Xu Z, Sun X, Knockdown of MMP-1 inhibits the progression of colorectal cancer by suppressing the PI3K/Akt/c-myc signaling pathway and EMT, Oncol. Rep. 43 (4) (2020) 1103–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ancha HR, Kurella RR, Stewart CA, Damera G, Ceresa BP, Harty RF, Histamine stimulation of MMP-1(collagenase-1) secretion and gene expression in gastric epithelial cells: role of EGFR transactivation and the MAP kinase pathway, Int. J. Biochem. Cell Biol. 39 (11) (2007) 2143–2152. [DOI] [PubMed] [Google Scholar]
- [68].Sun Y, Miao X, Zhu L, Liu J, Lin Y, Xiang G, Wu X, Wang X, Ni Z, Li S, Autocrine TGF-alpha is associated with Benzo(a)pyrene-induced mucus production and MUC5AC expression during allergic asthma, Ecotoxicol. Environ. Saf. 241 (2022), 113833. [DOI] [PubMed] [Google Scholar]
- [69].Fahy JV, Dickey BF, Airway mucus function and dysfunction, N. Engl. J. Med. 363 (23) (2010) 2233–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bonser LR, Zlock L, Finkbeiner W, Erle DJ, Epithelial tethering of MUC5AC-rich mucus impairs mucociliary transport in asthma, J. Clin. Invest. 126 (6) (2016) 2367–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Villarino AV, Kanno Y, O’Shea JJ, Mechanisms and consequences of Jak-STAT signaling in the immune system, Nat. Immunol. 18 (4) (2017) 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Salas A, Hernandez-Rocha C, Duijvestein M, Faubion W, McGovern D, Vermeire S, Vetrano S, Vande Casteele N, JAK-STAT pathway targeting for the treatment of inflammatory bowel disease, Nat. Rev. Gastroenterol. Hepatol. 17 (6) (2020) 323–337. [DOI] [PubMed] [Google Scholar]
- [73].Welsch K, Holstein J, Laurence A, Ghoreschi K, Targeting JAK/STAT signalling in inflammatory skin diseases with small molecule inhibitors, Eur. J. Immunol. 47 (7) (2017) 1096–1107. [DOI] [PubMed] [Google Scholar]
- [74].Schwartz DM, Bonelli M, Gadina M, O’Shea JJ, Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases, Nat. Rev. Rheumatol. 12 (1) (2016) 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tzeng HT, Chyuan IT, Lai JH, Targeting the JAK-STAT pathway in autoimmune diseases and cancers: A focus on molecular mechanisms and therapeutic potential, Biochem. Pharmacol. 193 (2021), 114760. [DOI] [PubMed] [Google Scholar]
- [76].Nukaya M, Takahashi Y, Gonzalez FJ, Kamataki T, Aryl hydrocarbon receptor-mediated suppression of GH receptor and Janus kinase 2 expression in mice, FEBS Lett. 558 (1–3) (2004) 96–100. [DOI] [PubMed] [Google Scholar]
- [77].Takanaga H, Yoshitake T, Yatabe E, Hara S, Kunimoto M, Beta-naphthoflavone disturbs astrocytic differentiation of C6 glioma cells by inhibiting autocrine interleukin-6, J. Neurochem. 90 (3) (2004) 750–757. [DOI] [PubMed] [Google Scholar]
- [78].Jeon MS, Esser C, The murine IL-2 promoter contains distal regulatory elements responsive to the Ah receptor, a member of the evolutionarily conserved bHLH-PAS transcription factor family, J. Immunol. 165 (12) (2000) 6975–6983. [DOI] [PubMed] [Google Scholar]
- [79].Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, Burns EJ, Sherr DH, Weiner HL, Kuchroo VK, The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27, Nat. Immunol. 11 (9) (2010) 854–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hollingshead BD, Beischlag TV, Dinatale BC, Ramadoss P, Perdew GH, Inflammatory signaling and aryl hydrocarbon receptor mediate synergistic induction of interleukin 6 in MCF-7 cells, Cancer Res. 68 (10) (2008) 3609–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Zhu J, Luo L, Tian L, Yin S, Ma X, Cheng S, Tang W, Yu J, Ma W, Zhou X, Fan X, Yang X, Yan J, Xu X, Lv C, Liang H, Aryl Hydrocarbon Receptor Promotes IL-10 Expression in Inflammatory Macrophages Through Src-STAT3 Signaling Pathway, Front. Immunol. 9 (2018) 2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].DiNatale BC, Murray IA, Schroeder JC, Flaveny CA, Lahoti TS, Laurenzana EM, Omiecinski CJ, Perdew GH, Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling, Toxicol. Sci. 115 (1) (2010) 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Jensen BA, Leeman RJ, Schlezinger JJ, Sherr DH, Aryl hydrocarbon receptor (AhR) agonists suppress interleukin-6 expression by bone marrow stromal cells: an immunotoxicology study, Environ. Health 2 (1) (2003) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ishihara Y, Haarmann-Stemmann T, Kado NY, Vogel CFA, Interleukin 33 Expression Induced by Aryl Hydrocarbon Receptor in Macrophages, Toxicol. Sci. 170 (2) (2019) 404–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Tsuji G, Hashimoto-Hachiya A, Yen VH, Miake S, Takemura M, Mitamura Y, Ito T, Murata M, Furue M, Nakahara T, Aryl Hydrocarbon Receptor Activation Downregulates IL-33 Expression in Keratinocytes via Ovo-Like 1, J. Clin. Med. 9 (3) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Stobbe-Maicherski N, Wolff S, Wolff C, Abel J, Sydlik U, Frauenstein K, Haarmann-Stemmann T, The interleukin-6-type cytokine oncostatin M induces aryl hydrocarbon receptor expression in a STAT3-dependent manner in human HepG2 hepatoma cells, FEBS J. 280 (24) (2013) 6681–6690. [DOI] [PubMed] [Google Scholar]
- [87].Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, Siddiqui S, Basso AS, Otterbein LE, Pardoll DM, Pan F, Priel A, Clish CB, Robson SC, Quintana FJ, Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha, Nat. Med. 21 (6) (2015) 638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, Chao CC, Patel B, Yan R, Blain M, Alvarez JI, Kebir H, Anandasabapathy N, Izquierdo G, Jung S, Obholzer N, Pochet N, Clish CB, Prinz M, Prat A, Antel J, Quintana FJ, Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor, Nat. Med. 22 (6) (2016) 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Tanaka G, Kanaji S, Hirano A, Arima K, Shinagawa A, Goda C, Yasunaga S, Ikizawa K, Yanagihara Y, Kubo M, Kuriyama-Fujii Y, Sugita Y, Inokuchi A, Izuhara K, Induction and activation of the aryl hydrocarbon receptor by IL-4 in B cells, Int. Immunol. 17 (6) (2005) 797–805. [DOI] [PubMed] [Google Scholar]
- [90].Atene CG, Fiorcari S, Mesini N, Alboni S, Martinelli S, Maccaferri M, Leonardi G, Potenza L, Luppi M, Maffei R, Marasca R, Indoleamine 2, 3-Dioxygenase 1 Mediates Survival Signals in Chronic Lymphocytic Leukemia via Kynurenine/Aryl Hydrocarbon Receptor-Mediated MCL1 Modulation, Front. Immunol. 13 (2022), 832263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Platten M, Nollen EAA, Rohrig UF, Fallarino F, Opitz CA, Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond, Nat. Rev. Drug Discov. 18 (5) (2019) 379–401. [DOI] [PubMed] [Google Scholar]
- [92].Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA, An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells, J. Immunol. 185 (6) (2010) 3190–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Novikov O, Wang Z, Stanford EA, Parks AJ, Ramirez-Cardenas A, Landesman E, Laklouk I, Sarita-Reyes C, Gusenleitner D, Li A, Monti S, Manteiga S, Lee K, Sherr DH, An Aryl Hydrocarbon Receptor-Mediated Amplification Loop That Enforces Cell Migration in ER-/PR-/Her2- Human Breast Cancer Cells, Mol. Pharmacol. 90 (5) (2016) 674–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Funatake CJ, Marshall NB, Steppan LB, Mourich DV, Kerkvliet NI, Cutting edge: activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin generates a population of CD4+ CD25+ cells with characteristics of regulatory T cells, J. Immunol. 175 (7) (2005) 4184–4188. [DOI] [PubMed] [Google Scholar]
- [95].Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL, Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor, Nature 453 (7191) (2008) 65–71. [DOI] [PubMed] [Google Scholar]
- [96].Litzenburger UM, Opitz CA, Sahm F, Rauschenbach KJ, Trump S, Winter M, Ott M, Ochs K, Lutz C, Liu X, Anastasov N, Lehmann I, Hofer T, von Deimling A, Wick W, Platten M, Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR, Oncotarget 5 (4) (2014) 1038–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Liu Y, Zhou N, Zhou L, Wang J, Zhou Y, Zhang T, Fang Y, Deng J, Gao Y, Liang X, Lv J, Wang Z, Xie J, Xue Y, Zhang H, Ma J, Tang K, Fang Y, Cheng F, Zhang C, Dong B, Zhao Y, Yuan P, Gao Q, Zhang H, Xiao-Feng Qin F, Huang B, IL-2 regulates tumor-reactive CD8(+) T cell exhaustion by activating the aryl hydrocarbon receptor, Nat. Immunol. 22 (3) (2021) 358–369. [DOI] [PubMed] [Google Scholar]
- [98].Kimura A, Naka T, Nakahama T, Chinen I, Masuda K, Nohara K, Fujii-Kuriyama Y, Kishimoto T, Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses, J. Exp. Med. 206 (9) (2009) 2027–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Masuda K, Kimura A, Hanieh H, Nguyen NT, Nakahama T, Chinen I, Otoyo Y, Murotani T, Yamatodani A, Kishimoto T, Aryl hydrocarbon receptor negatively regulates LPS-induced IL-6 production through suppression of histamine production in macrophages, Int. Immunol. 23 (10) (2011) 637–645. [DOI] [PubMed] [Google Scholar]
- [100].Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T, Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells, PNAS 105 (28) (2008) 9721–9726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Liu X, Hu H, Fan H, Zuo D, Shou Z, Liao Y, Nan Z, Tang Q, The role of STAT3 and AhR in the differentiation of CD4+ T cells into Th17 and Treg cells, Medicine (Baltimore) 96 (17) (2017) e6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Lee SM, Kim CE, Park HY, Yoon EH, Won HJ, Ahn JM, Nguyen NZN, Kim M, Jang WH, Lee WS, Kang MS, Jeong M, Yun H, Park S, Wu S, Kim DH, Kwon B, Seo SK, Aryl hydrocarbon receptor-targeted therapy for CD4+ T cell-mediated idiopathic pneumonia syndrome in mice, Blood 139 (22) (2022) 3325–3339. [DOI] [PubMed] [Google Scholar]
- [103].Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B, The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins, Nature 453 (7191) (2008) 106–109. [DOI] [PubMed] [Google Scholar]
- [104].Ehrlich AK, Pennington JM, Bisson WH, Kolluri SK, Kerkvliet NI, TCDD, FICZ, and Other High Affinity AhR Ligands Dose-Dependently Determine the Fate of CD4+ T Cell Differentiation, Toxicol. Sci. 161 (2) (2018) 310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Liu Y, Liang X, Yin X, Lv J, Tang K, Ma J, Ji T, Zhang H, Dong W, Jin X, Chen D, Li Y, Zhang S, Xie HQ, Zhao B, Zhao T, Lu J, Hu ZW, Cao X, Qin FX, Huang B, Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-gamma-induced immunologic dormancy of tumor-repopulating cells, Nat. Commun. 8 (2017) 15207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Kolluri SK, Weiss C, Koff A, Gottlicher M, p27(Kip1) induction and inhibition of proliferation by the intracellular Ah receptor in developing thymus and hepatoma cells, Genes Dev. 13 (13) (1999) 1742–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Kenison JE, Wang Z, Yang K, Snyder M, Quintana FJ, Sherr DH, The aryl hydrocarbon receptor suppresses immunity to oral squamous cell carcinoma through immune checkpoint regulation, PNAS 118 (19) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao CC, Gutierrez-Vazquez C, Kenison J, Tjon EC, Barroso A, Vandeventer T, de Lima KA, Rothweiler S, Mayo L, Ghannam S, Zandee S, Healy L, Sherr D, Farez MF, Prat A, Antel J, Reardon DA, Zhang H, Robson SC, Getz G, Weiner HL, Quintana FJ, Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39, Nat. Neurosci. 22 (5) (2019) 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Nakajima K, Maekawa Y, Kataoka K, Ishifune C, Nishida J, Arimochi H, Kitamura A, Yoshimoto T, Tomita S, Nagahiro S, Yasutomo K, The ARNT-STAT3 axis regulates the differentiation of intestinal intraepithelial TCRalphabeta (+)CD8alphaalpha(+) cells, Nat. Commun. 4 (2013) 2112. [DOI] [PubMed] [Google Scholar]
- [110].Mitchell S, Vargas J, Hoffmann A, Signaling via the NFkappaB system, Wiley Interdiscip. Rev. Syst. Biol. Med. 8 (3) (2016) 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Taniguchi K, Karin M, NF-kappaB, inflammation, immunity and cancer: coming of age, Nat. Rev. Immunol. 18 (5) (2018) 309–324. [DOI] [PubMed] [Google Scholar]
- [112].Matsumura F, Vogel CF, Evidence supporting the hypothesis that one of the main functions of the aryl hydrocarbon receptor is mediation of cell stress responses, Biol. Chem. 387 (9) (2006) 1189–1194. [DOI] [PubMed] [Google Scholar]
- [113].Son M, Wang AG, Tu HL, Metzig MO, Patel P, Husain K, Lin J, Murugan A, Hoffmann A, Tay S, NF-kappaB responds to absolute differences in cytokine concentrations, Sci. Signal. 14 (666) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Ghezzi P, Saccardo B, Bianchi M, Recombinant tumor necrosis factor depresses cytochrome P450-dependent microsomal drug metabolism in mice, Biochem. Biophys. Res. Commun. 136 (1) (1986) 316–321. [DOI] [PubMed] [Google Scholar]
- [115].Barker CW, Fagan JB, Pasco DS, Interleukin-1 beta suppresses the induction of P4501A1 and P4501A2 mRNAs in isolated hepatocytes, J. Biol. Chem. 267 (12) (1992) 8050–8055. [PubMed] [Google Scholar]
- [116].Fukuda Y, Ishida N, Noguchi T, Kappas A, Sassa S, Interleukin-6 down regulates the expression of transcripts encoding cytochrome P450 IA1, IA2 and IIIA3 in human hepatoma cells, Biochem. Biophys. Res. Commun. 184 (2) (1992) 960–965. [DOI] [PubMed] [Google Scholar]
- [117].Tian Y, Ke S, Denison MS, Rabson AB, Gallo MA, Ah receptor and NF-kappaB interactions, a potential mechanism for dioxin toxicity, J. Biol. Chem. 274 (1) (1999) 510–515. [DOI] [PubMed] [Google Scholar]
- [118].Ke S, Rabson AB, Germino JF, Gallo MA, Tian Y, Mechanism of suppression of cytochrome P-450 1A1 expression by tumor necrosis factor-alpha and lipopolysaccharide, J. Biol. Chem. 276 (43) (2001) 39638–39644. [DOI] [PubMed] [Google Scholar]
- [119].DiNatale BC, Schroeder JC, Francey LJ, Kusnadi A, Perdew GH, Mechanistic insights into the events that lead to synergistic induction of interleukin 6 transcription upon activation of the aryl hydrocarbon receptor and inflammatory signaling, J. Biol. Chem. 285 (32) (2010) 24388–24397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Sekine H, Mimura J, Oshima M, Okawa H, Kanno J, Igarashi K, Gonzalez FJ, Ikuta T, Kawajiri K, Fujii-Kuriyama Y, Hypersensitivity of aryl hydrocarbon receptor-deficient mice to lipopolysaccharide-induced septic shock, Mol. Cell Biol. 29 (24) (2009) 6391–6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Singh NP, Nagarkatti M, Nagarkatti PS, Role of dioxin response element and nuclear factor-kappaB motifs in 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated regulation of Fas and Fas ligand expression, Mol. Pharmacol. 71 (1) (2007) 145–157. [DOI] [PubMed] [Google Scholar]
- [122].Kim DW, Gazourian L, Quadri SA, Romieu-Mourez R, Sherr DH, Sonenshein GE, The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells, Oncogene 19 (48) (2000) 5498–5506. [DOI] [PubMed] [Google Scholar]
- [123].Camacho IA, Singh N, Hegde VL, Nagarkatti M, Nagarkatti PS, Treatment of mice with 2,3,7,8-tetrachlorodibenzo-p-dioxin leads to aryl hydrocarbon receptor-dependent nuclear translocation of NF-kappaB and expression of Fas ligand in thymic stromal cells and consequent apoptosis in T cells, J. Immunol. 175 (1) (2005) 90–103. [DOI] [PubMed] [Google Scholar]
- [124].Sulentic CE, Kang JS, Na YJ, Kaminski NE, Interactions at a dioxin responsive element (DRE) and an overlapping kappaB site within the hs4 domain of the 3’alpha immunoglobulin heavy chain enhancer, Toxicology 200 (2–3) (2004) 235–246. [DOI] [PubMed] [Google Scholar]
- [125].Patel RD, Murray IA, Flaveny CA, Kusnadi A, Perdew GH, Ah receptor represses acute-phase response gene expression without binding to its cognate response element, Lab. Invest. 89 (6) (2009) 695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Puga A, Barnes SJ, Chang C, Zhu H, Nephew KP, Khan SA, Shertzer HG, Activation of transcription factors activator protein-1 and nuclear factor-kappaB by 2,3,7,8-tetrachlorodibenzo-p-dioxin, Biochem. Pharmacol. 59 (8) (2000) 997–1005. [DOI] [PubMed] [Google Scholar]
- [127].Suh J, Jeon YJ, Kim HM, Kang JS, Kaminski NE, Yang KH, Aryl hydrocarbon receptor-dependent inhibition of AP-1 activity by 2,3,7,8-tetrachlorodibenzo-p-dioxin in activated B cells, Toxicol. Appl. Pharmacol. 181 (2) (2002) 116–123. [DOI] [PubMed] [Google Scholar]
- [128].Vogel CF, Sciullo E, Matsumura F, Involvement of RelB in aryl hydrocarbon receptor-mediated induction of chemokines, Biochem. Biophys. Res. Commun. 363 (3) (2007) 722–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Bankoti J, Rase B, Simones T, Shepherd DM, Functional and phenotypic effects of AhR activation in inflammatory dendritic cells, Toxicol. Appl. Pharmacol. 246 (1–2) (2010) 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Memari B, Bouttier M, Dimitrov V, Ouellette M, Behr MA, Fritz JH, White JH, Engagement of the Aryl Hydrocarbon Receptor in Mycobacterium tuberculosis-Infected Macrophages Has Pleiotropic Effects on Innate Immune Signaling, J. Immunol. 195 (9) (2015) 4479–4491. [DOI] [PubMed] [Google Scholar]
- [131].Salisbury RL, Sulentic CE, The AhR and NF-kappaB/Rel Proteins Mediate the Inhibitory Effect of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin on the 3’ Immunoglobulin Heavy Chain Regulatory Region, Toxicol. Sci. 148 (2) (2015) 443–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Vogel CF, Sciullo E, Li W, Wong P, Lazennec G, Matsumura F, RelB, a new partner of aryl hydrocarbon receptor-mediated transcription, Mol. Endocrinol. 21 (12) (2007) 2941–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, Jegga AG, Aronow BJ, Ghosh G, Rickert RC, Karin M, Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers, EMBO J. 23 (21) (2004) 4202–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Vogel CF, Goth SR, Dong B, Pessah IN, Matsumura F, Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase, Biochem. Biophys. Res. Commun. 375 (3) (2008) 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Vogel CF, Wu D, Goth SR, Baek J, Lollies A, Domhardt R, Grindel A, Pessah IN, Aryl hydrocarbon receptor signaling regulates NF-kappaB RelB activation during dendritic-cell differentiation, Immunol. Cell Biol. 91 (9) (2013) 568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Vogel CF, Chang WL, Kado S, McCulloh K, Vogel H, Wu D, Haarmann-Stemmann T, Yang G, Leung PS, Matsumura F, Gershwin ME, Transgenic Overexpression of Aryl Hydrocarbon Receptor Repressor (AhRR) and AhR-Mediated Induction of CYP1A1, Cytokines, and Acute Toxicity, Environ Health Perspect 124 (7) (2016) 1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Ranatunga DC, Ramakrishnan A, Uprety P, Wang F, Zhang H, Margolick JB, Brayton C, Bream JH, A protective role for human IL-10-expressing CD4+ T cells in colitis, J. Immunol. 189 (3) (2012) 1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Benson JM, Shepherd DM, Aryl hydrocarbon receptor activation by TCDD reduces inflammation associated with Crohn’s disease, Toxicol. Sci. 120 (1) (2011) 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Kado S, Chang WLW, Chi AN, Wolny M, Shepherd DM, Vogel CFA, Aryl hydrocarbon receptor signaling modifies Toll-like receptor-regulated responses in human dendritic cells, Arch. Toxicol. 91 (5) (2017) 2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Dominguez-Acosta O, Vega L, Estrada-Muniz E, Rodriguez MS, Gonzalez FJ, Elizondo G, Activation of aryl hydrocarbon receptor regulates the LPS/IFNgamma-induced inflammatory response by inducing ubiquitin-proteosomal and lysosomal degradation of RelA/p65, Biochem. Pharmacol. 155 (2018) 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Wright CW, Duckett CS, The aryl hydrocarbon nuclear translocator alters CD30-mediated NF-kappaB-dependent transcription, Science 323 (5911) (2009) 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Vogel CFA, Haarmann-Stemmann T, The aryl hydrocarbon receptor repressor - More than a simple feedback inhibitor of AhR signaling: Clues for its role in inflammation and cancer, Curr Opin Toxicol 2 (2017) 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Vogel CFA, Ishihara Y, Campbell CE, Kado SY, Nguyen-Chi A, Sweeney C, Pollet M, Haarmann-Stemmann T, Tuscano JM, A Protective Role of Aryl Hydrocarbon Receptor Repressor in Inflammation and Tumor Growth, Cancers (Basel) 11 (5) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Vogel CFA, Lazennec G, Kado SY, Dahlem C, He Y, Castaneda A, Ishihara Y, Vogeley C, Rossi A, Haarmann-Stemmann T, Jugan J, Mori H, Borowsky AD, La Merrill MA, Sweeney C, Targeting the Aryl Hydrocarbon Receptor Signaling Pathway in Breast Cancer Development, Front. Immunol. 12 (2021), 625346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Ovrevik J, Lag M, Lecureur V, Gilot D, Lagadic-Gossmann D, Refsnes M, Schwarze PE, Skuland T, Becher R, Holme JA, AhR and Arnt differentially regulate NF-kappaB signaling and chemokine responses in human bronchial epithelial cells, Cell Commun. Signal 12 (2014) 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Beamer CA, Seaver BP, Shepherd DM, Aryl hydrocarbon receptor (AhR) regulates silica-induced inflammation but not fibrosis, Toxicol. Sci. 126 (2) (2012) 554–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Thatcher TH, Maggirwar SB, Baglole CJ, Lakatos HF, Gasiewicz TA, Phipps RP, Sime PJ, Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB, Am. J. Pathol. 170 (3) (2007) 855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Muku GE, Lahoti TS, Murray IA, Podolsky MA, Smith KJ, Hubbard TD, Kuzu G, Gowda K, Amin SG, Perdew GH, Ligand-mediated cytoplasmic retention of the Ah receptor inhibits macrophage-mediated acute inflammatory responses, Lab. Invest. 97 (12) (2017) 1471–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Wirthgen E, Hoeflich A, Endotoxin-Induced Tryptophan Degradation along the Kynurenine Pathway: The Role of Indolamine 2,3-Dioxygenase and Aryl Hydrocarbon Receptor-Mediated Immunosuppressive Effects in Endotoxin Tolerance and Cancer and Its Implications for Immunoparalysis, J. Amino Acids 2015 (2015), 973548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Ishihara Y, Kado SY, Hoeper C, Harel S, Vogel CFA, Role of NF-kB RelB in Aryl Hydrocarbon Receptor-Mediated Ligand Specific Effects, Int. J. Mol. Sci. 20 (11) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Wu D, Li W, Lok P, Matsumura F, Vogel CF, AhR deficiency impairs expression of LPS-induced inflammatory genes in mice, Biochem. Biophys. Res. Commun. 410 (2) (2011) 358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Tsuji M, Vogel CF, Koriyama C, Akiba S, Katoh T, Kawamoto T, Matsumura F, Association of serum levels of polychlorinated biphenyls with IL-8 mRNA expression in blood samples from asthmatic and non-asthmatic Japanese children, Chemosphere 87 (11) (2012) 1228–1234. [DOI] [PubMed] [Google Scholar]
- [153].Lahoti TS, Boyer JA, Kusnadi A, Muku GE, Murray IA, Perdew GH, Aryl Hydrocarbon Receptor Activation Synergistically Induces Lipopolysaccharide-Mediated Expression of Proinflammatory Chemokine (c-c motif) Ligand 20, Toxicol. Sci. 148 (1) (2015) 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Ishihara N, Okuda T, Hagino H, Oguro A, Tani Y, Okochi H, Tokoro C, Fujii-Kuriyama Y, Itoh K, Vogel CFA, Ishihara Y, Involvement of polycyclic aromatic hydrocarbons and endotoxin in macrophage expression of interleukin-33 induced by exposure to particulate matter, J. Toxicol. Sci. 47 (5) (2022) 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Vogel CF, Khan EM, Leung PS, Gershwin ME, Chang WL, Wu D, Haarmann-Stemmann T, Hoffmann A, Denison MS, Cross-talk between aryl hydrocarbon receptor and the inflammatory response: a role for nuclear factor-kappaB, J. Biol. Chem. 289 (3) (2014) 1866–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Marcus RS, Holsapple MP, Kaminski NE, Lipopolysaccharide activation of murine splenocytes and splenic B cells increased the expression of aryl hydrocarbon receptor and aryl hydrocarbon receptor nuclear translocator, J. Pharmacol. Exp. Ther. 287 (3) (1998) 1113–1118. [PubMed] [Google Scholar]
- [157].Allan LL, Sherr DH, Constitutive activation and environmental chemical induction of the aryl hydrocarbon receptor/transcription factor in activated human B lymphocytes, Mol. Pharmacol. 67 (5) (2005) 1740–1750. [DOI] [PubMed] [Google Scholar]
- [158].Zanger UM, Schwab M, Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation, Pharmacol. Ther. 138 (1) (2013) 103–141. [DOI] [PubMed] [Google Scholar]
- [159].Mescher M, Haarmann-Stemmann T, Modulation of CYP1A1 metabolism: From adverse health effects to chemoprevention and therapeutic options, Pharmacol. Ther. 187 (2018) 71–87. [DOI] [PubMed] [Google Scholar]
- [160].Nebert DW, Wikvall K, Miller WL, Human cytochromes P450 in health and disease, Philos. Trans. R. Soc. Lond. B Biol. Sci. 368 (1612) (2013) 20120431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Crake RLI, Strother MR, Phillips E, Doogue MP, Zhang M, Frampton CMA, Robinson BA, Currie MJ, Influence of serum inflammatory cytokines on cytochrome P450 drug metabolising activity during breast cancer chemotherapy: a patient feasibility study, Sci. Rep. 11 (1) (2021) 5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Mescher M, Tigges J, Rolfes KM, Shen AL, Yee JS, Vogeley C, Krutmann J, Bradfield CA, Lang D, Haarmann-Stemmann T, The Toll-like receptor agonist imiquimod is metabolized by aryl hydrocarbon receptor-regulated cytochrome P450 enzymes in human keratinocytes and mouse liver, Arch. Toxicol. 93 (7) (2019) 1917–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Rolfes KM, Sondermann NC, Vogeley C, Dairou J, Gilardino V, Wirth R, Meller S, Homey B, Krutmann J, Lang D, Nakamura M, Haarmann-Stemmann T, Inhibition of 6-formylindolo[3,2-b]carbazole metabolism sensitizes keratinocytes to UVA-induced apoptosis: Implications for vemurafenib-induced phototoxicity, Redox Biol. 46 (2021), 102110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Nebert DW, Dalton TP, The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis, Nat. Rev. Cancer 6 (12) (2006) 947–960. [DOI] [PubMed] [Google Scholar]
- [165].Li F, Zhu W, Gonzalez FJ, Potential role of CYP1B1 in the development and treatment of metabolic diseases, Pharmacol. Ther. 178 (2017) 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Yi T, Wang J, Zhu K, Tang Y, Huang S, Shui X, Ding Y, Chen C, Lei W, Aryl Hydrocarbon Receptor: A New Player of Pathogenesis and Therapy in Cardiovascular Diseases, Biomed Res. Int. 2018 (2018) 6058784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Elbekai RH, El-Kadi AO, Cytochrome P450 enzymes: central players in cardiovascular health and disease, Pharmacol. Ther. 112 (2) (2006) 564–587. [DOI] [PubMed] [Google Scholar]
- [168].Charrad R, Kaabachi W, Rafrafi A, Berraies A, Hamzaoui K, Hamzaoui A, IL-8 Gene Variants and Expression in Childhood Asthma, Lung 195 (6) (2017) 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Shinde R, Hezaveh K, Halaby MJ, Kloetgen A, Chakravarthy A, da Silva Medina T, Deol R, Manion KP, Baglaenko Y, Eldh M, Lamorte S, Wallace D, Chodisetti SB, Ravishankar B, Liu H, Chaudhary K, Munn DH, Tsirigos A, Madaio M, Gabrielsson S, Touma Z, Wither J, De Carvalho DD, McGaha TL, Apoptotic cell-induced AhR activity is required for immunological tolerance and suppression of systemic lupus erythematosus in mice and humans, Nat. Immunol. 19 (6) (2018) 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Monteleone I, Pallone F, Monteleone G, Aryl hydrocarbon receptor and colitis, Semin. Immunopathol. 35 (6) (2013) 671–675. [DOI] [PubMed] [Google Scholar]
- [171].Nguyen NT, Nakahama T, Kishimoto T, Aryl hydrocarbon receptor and experimental autoimmune arthritis, Semin. Immunopathol. 35 (6) (2013) 637–644. [DOI] [PubMed] [Google Scholar]
- [172].Fu J, Nogueira SV, Drongelen VV, Coit P, Ling S, Rosloniec EF, Sawalha AH, Holoshitz J, Shared epitope-aryl hydrocarbon receptor crosstalk underlies the mechanism of gene-environment interaction in autoimmune arthritis, PNAS 115 (18) (2018) 4755–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Marshall NB, Kerkvliet NI, Dioxin and immune regulation: emerging role of aryl hydrocarbon receptor in the generation of regulatory T cells, Ann. N. Y. Acad. Sci. 1183 (2010) 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Wang GL, Semenza GL, General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia, PNAS 90 (9) (1993) 4304–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Wood SM, Gleadle JM, Pugh CW, Hankinson O, Ratcliffe PJ, The role of the aryl hydrocarbon receptor nuclear translocator (ARNT) in hypoxic induction of gene expression, Studies in ARNT-deficient cells, J Biol Chem 271 (25) (1996) 15117–15123. [DOI] [PubMed] [Google Scholar]
- [176].Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG, Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein, Nat. Cell Biol. 2 (7) (2000) 423–427. [DOI] [PubMed] [Google Scholar]
- [177].McIntosh BE, Hogenesch JB, Bradfield CA, Mammalian Per-Arnt-Sim proteins in environmental adaptation, Annu. Rev. Physiol. 72 (2010) 625–645. [DOI] [PubMed] [Google Scholar]
- [178].Dunwoodie SL, The role of hypoxia in development of the Mammalian embryo, Dev. Cell 17 (6) (2009) 755–773. [DOI] [PubMed] [Google Scholar]
- [179].Kotch LE, Iyer NV, Laughner E, Semenza GL, Defective vascularization of HIF-1alpha-null embryos is not associated with VEGF deficiency but with mesenchymal cell death, Dev. Biol. 209 (2) (1999) 254–267. [DOI] [PubMed] [Google Scholar]
- [180].Ryan HE, Lo J, Johnson RS, HIF-1 alpha is required for solid tumor formation and embryonic vascularization, EMBO J. 17 (11) (1998) 3005–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Lindholm ME, Rundqvist H, Skeletal muscle hypoxia-inducible factor-1 and exercise, Exp. Physiol. 101 (1) (2016) 28–32. [DOI] [PubMed] [Google Scholar]
- [182].Abe T, Kitaoka Y, Kikuchi DM, Takeda K, Numata O, Takemasa T, High-intensity interval training-induced metabolic adaptation coupled with an increase in Hif-1alpha and glycolytic protein expression, J. Appl. Physiol. (1985) 119(11) (2015) 1297–302. [DOI] [PubMed] [Google Scholar]
- [183].Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M, NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha, Nature 453 (7196) (2008) 807–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS, HIF-1alpha is essential for myeloid cell-mediated inflammation, Cell 112 (5) (2003) 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Bruick RK, McKnight SL, A conserved family of prolyl-4-hydroxylases that modify HIF, Science 294 (5545) (2001) 1337–1340. [DOI] [PubMed] [Google Scholar]
- [186].Slemc L, Kunej T, Transcription factor HIF1A: downstream targets, associated pathways, polymorphic hypoxia response element (HRE) sites, and initiative for standardization of reporting in scientific literature, Tumour Biol. 37 (11) (2016) 14851–14861. [DOI] [PubMed] [Google Scholar]
- [187].Kim JE, Sheen YY, Inhibition of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-stimulated Cyp1a1 promoter activity by hypoxic agents, Biochem. Pharmacol. 59 (12) (2000) 1549–1556. [DOI] [PubMed] [Google Scholar]
- [188].Gradin K, McGuire J, Wenger RH, Kvietikova I, Fhitelaw ML, Toftgard R, Tora L, Gassmann M, Poellinger L, Functional interference between hypoxia and dioxin signal transduction pathways: competition for recruitment of the Arnt transcription factor, Mol. Cell Biol. 16 (10) (1996) 5221–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Gassmann M, Kvietikova I, Rolfs A, Wenger RH, Oxygen- and dioxin-regulated gene expression in mouse hepatoma cells, Kidney Int. 51 (2) (1997) 567–574. [DOI] [PubMed] [Google Scholar]
- [190].Chan WK, Yao G, Gu YZ, Bradfield CA, Cross-talk between the aryl hydrocarbon receptor and hypoxia inducible factor signaling pathways. Demonstration of competition and compensation, J. Biol. Chem. 274 (17) (1999) 12115–12123. [DOI] [PubMed] [Google Scholar]
- [191].Myre M, Imbeault P, Persistent organic pollutants meet adipose tissue hypoxia: does cross-talk contribute to inflammation during obesity? Obes. Rev. 15 (1) (2014) 19–28. [DOI] [PubMed] [Google Scholar]
- [192].Asai H, Hirata J, Hirano A, Hirai K, Seki S, Watanabe-Akanuma M, Activation of aryl hydrocarbon receptor mediates suppression of hypoxia-inducible factor-dependent erythropoietin expression by indoxyl sulfate, Am. J. Physiol. Cell Physiol. 310 (2) (2016) C142–C150. [DOI] [PubMed] [Google Scholar]
- [193].Vuerich M, Harshe R, Frank LA, Mukherjee S, Gromova B, Csizmadia E, Nasser IAM, Ma Y, Bonder A, Patwardhan V, Robson SC, Longhi MS, Altered aryl-hydrocarbon-receptor signalling affects regulatory and effector cell immunity in autoimmune hepatitis, J. Hepatol. 74 (1) (2021) 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Gabriely G, Wheeler MA, Takenaka MC, Quintana FJ, Role of AHR and HIF-1alpha in Glioblastoma Metabolism, Trends Endocrinol Metab 28 (6) (2017) 428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Button EL, Bersten DC, Whitelaw ML, HIF has Biff - Crosstalk between HIF1a and the family of bHLH/PAS proteins, Exp. Cell Res. 356 (2) (2017) 141–145. [DOI] [PubMed] [Google Scholar]
- [196].Fritz WA, Lin TM, Peterson RE, The aryl hydrocarbon receptor (AhR) inhibits vanadate-induced vascular endothelial growth factor (VEGF) production in TRAMP prostates, Carcinogenesis 29 (5) (2008) 1077–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Yang X, Liu H, Ye T, Duan C, Lv P, Wu X, Liu J, Jiang K, Lu H, Yang H, Xia D, Peng E, Chen Z, Tang K, Ye Z, AhR activation attenuates calcium oxalate nephrocalcinosis by diminishing M1 macrophage polarization and promoting M2 macrophage polarization, Theranostics 10 (26) (2020) 12011–12025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Zhang M, Hu Y, Yang F, Zhang J, Zhang J, Yu W, Wang M, Lv X, Li J, Bai T, Chang F, Interaction between AhR and HIF-1 signaling pathways mediated by ARNT/HIF-1beta, BMC Pharmacol. Toxicol. 23 (1) (2022) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Jacob A, Potin S, Saubamea B, Crete D, Scherrmann JM, Curis E, Peyssonnaux C, Decleves X, Hypoxia interferes with aryl hydrocarbon receptor pathway in hCMEC/D3 human cerebral microvascular endothelial cells, J. Neurochem. 132 (4) (2015) 373–383. [DOI] [PubMed] [Google Scholar]
- [200].Vorrink SU, Domann FE, Regulatory crosstalk and interference between the xenobiotic and hypoxia sensing pathways at the AhR-ARNT-HIF1alpha signaling node, Chem. Biol. Interact. 218 (2014) 82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Vorrink SU, Sarsour EH, Olivier AK, Robertson LW, Goswami PC, Domann FE, PCB 126 perturbs hypoxia-induced HIF-1alpha activity and glucose consumption in human HepG2 cells, Exp. Toxicol. Pathol. 66 (8) (2014) 377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Vorrink SU, Severson PL, Kulak MV, Futscher BW, Domann FE, Hypoxia perturbs aryl hydrocarbon receptor signaling and CYP1A1 expression induced by PCB 126 in human skin and liver-derived cell lines, Toxicol. Appl. Pharmacol. 274 (3) (2014) 408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Salminen A, Mutual antagonism between aryl hydrocarbon receptor and hypoxia-inducible factor-1alpha (AhR/HIF-1alpha) signaling: Impact on the aging process, Cell. Signal. 99 (2022), 110445. [DOI] [PubMed] [Google Scholar]
- [204].Wolff M, Jelkmann W, Dunst J, Depping R, The Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT/HIF-1beta) is influenced by hypoxia and hypoxia-mimetics, Cell. Physiol. Biochem. 32 (4) (2013) 849–858. [DOI] [PubMed] [Google Scholar]
- [205].Mandl M, Depping R, Hypoxia-inducible aryl hydrocarbon receptor nuclear translocator (ARNT) (HIF-1beta): is it a rare exception? Mol. Med. 20 (2014) 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Bedogni B, Welford SM, Cassarino DS, Nickoloff BJ, Giaccia AJ, Powell MB, The hypoxic microenvironment of the skin contributes to Akt-mediated melanocyte transformation, Cancer Cell 8 (6) (2005) 443–454. [DOI] [PubMed] [Google Scholar]
- [207].Boutin AT, Weidemann A, Fu Z, Mesropian L, Gradin K, Jamora C, Wiesener M, Eckardt KU, Koch CJ, Ellies LG, Haddad G, Haase VH, Simon MC, Poellinger L, Powell FL, Johnson RS, Epidermal sensing of oxygen is essential for systemic hypoxic response, Cell 133 (2) (2008) 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Stucker M, Struk A, Altmeyer P, Herde M, Baumgartl H, Lubbers DW, The cutaneous uptake of atmospheric oxygen contributes significantly to the oxygen supply of human dermis and epidermis, J. Physiol. 538 (Pt 3) (2002) 985–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Geng S, Mezentsev A, Kalachikov S, Raith K, Roop DR, Panteleyev AA, Targeted ablation of Arnt in mouse epidermis results in profound defects in desquamation and epidermal barrier function, J. Cell Sci. 119 (Pt 23) (2006) 4901–4912. [DOI] [PubMed] [Google Scholar]
- [210].Wondimu A, Weir L, Robertson D, Mezentsev A, Kalachikov S, Panteleyev AA, Loss of Arnt (Hif1beta) in mouse epidermis triggers dermal angiogenesis, blood vessel dilation and clotting defects, Lab. Invest. 92 (1) (2012) 110–124. [DOI] [PubMed] [Google Scholar]
- [211].Robertson ED, Weir L, Romanowska M, Leigh IM, Panteleyev AA, ARNT controls the expression of epidermal differentiation genes through HDAC- and EGFR-dependent pathways, J. Cell Sci. 125 (Pt 14) (2012) 3320–3332. [DOI] [PubMed] [Google Scholar]
- [212].Takagi S, Tojo H, Tomita S, Sano S, Itami S, Hara M, Inoue S, Horie K, Kondoh G, Hosokawa K, Gonzalez FJ, Takeda J, Alteration of the 4-sphingenine scaffolds of ceramides in keratinocyte-specific Arnt-deficient mice affects skin barrier function, J. Clin. Invest. 112 (9) (2003) 1372–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Garcia-Ruiz C, Colell A, Mari M, Morales A, Fernandez-Checa JC, Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione, J. Biol. Chem. 272 (17) (1997) 11369–11377. [DOI] [PubMed] [Google Scholar]
- [214].Gu C, Gonzalez J, Zhang T, Kamel-Reid S, Wells RA, The aryl hydrocarbon receptor nuclear translocator (ARNT) modulates the antioxidant response in AML cells, Leuk. Res. 37 (12) (2013) 1750–1756. [DOI] [PubMed] [Google Scholar]
- [215].Haas K, Weighardt H, Deenen R, Kohrer K, Clausen B, Zahner S, Boukamp P, Bloch W, Krutmann J, Esser C, Aryl hydrocarbon receptor in keratinocytes is essential for murine skin barrier integrity, J. Invest. Dermatol. (2016). [DOI] [PubMed] [Google Scholar]
- [216].Fassbender S, Sondenheimer K, Majora M, Schindler J, Opitz FV, Pollet M, Haarmann-Stemmann T, Krutmann J, Weighardt H, Keratinocytes Counteract UVB-Induced Immunosuppression in Mice Via HIF-1a Signaling, J, Invest. Dermatol. (2021). [DOI] [PubMed] [Google Scholar]
- [217].Peyssonnaux C, Boutin AT, Zinkernagel AS, Datta V, Nizet V, Johnson RS, Critical role of HIF-1alpha in keratinocyte defense against bacterial infection, J, Invest. Dermatol. 128 (8) (2008) 1964–1968. [DOI] [PubMed] [Google Scholar]
- [218].Pollenz RS, Davarinos NA, Shearer TP, Analysis of aryl hydrocarbon receptor-mediated signaling during physiological hypoxia reveals lack of competition for the aryl hydrocarbon nuclear translocator transcription factor, Mol. Pharmacol. 56 (6) (1999) 1127–1137. [DOI] [PubMed] [Google Scholar]
- [219].Tsai CH, Li CH, Liao PL, Cheng YW, Lin CH, Huang SH, Kang JJ, NcoA2-Dependent Inhibition of HIF-1alpha Activation Is Regulated via AhR, Toxicol. Sci. 148 (2) (2015) 517–530. [DOI] [PubMed] [Google Scholar]
- [220].Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ, An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia, Nucleic Acids Res. 37 (14) (2009) 4587–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Tirpe AA, Gulei D, Ciortea SM, Crivii C, Berindan-Neagoe I, Hypoxia: Overview on Hypoxia-Mediated Mechanisms with a Focus on the Role of HIF Genes, Int. J. Mol. Sci. 20 (24) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Ghosh R, Samanta P, Sarkar R, Biswas S, Saha P, Hajra S, Bhowmik A, Targeting HIF-1alpha by Natural and Synthetic Compounds: A Promising Approach for Anti-Cancer Therapeutics Development, Molecules 27 (16) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Soni S, Padwad YS, HIF-1 in cancer therapy: two decade long story of a transcription factor, Acta Oncol. 56 (4) (2017) 503–515. [DOI] [PubMed] [Google Scholar]
- [224].Cimmino F, Avitabile M, Lasorsa VA, Montella A, Pezone L, Cantalupo S, Visconte F, Corrias MV, Iolascon A, Capasso M, HIF-1 transcription activity: HIF1A driven response in normoxia and in hypoxia, BMC Med. Genet. 20 (1) (2019) 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Li JP, Li FY, Xu A, Cheng B, Tsao SW, Fung ML, Leung WK, Lipopolysaccharide and hypoxia-induced HIF-1 activation in human gingival fibroblasts, J. Periodontol. 83 (6) (2012) 816–824. [DOI] [PubMed] [Google Scholar]
- [226].Kuschel A, Simon P, Tug S, Functional regulation of HIF-1alpha under normoxia–is there more than post-translational regulation? J. Cell. Physiol. 227 (2) (2012) 514–524. [DOI] [PubMed] [Google Scholar]
- [227].Isoe T, Makino Y, Mizumoto K, Sakagami H, Fujita Y, Honjo J, Takiyama Y, Itoh H, Haneda M, High glucose activates HIF-1-mediated signal transduction in glomerular mesangial cells through a carbohydrate response element binding protein, Kidney Int. 78 (1) (2010) 48–59. [DOI] [PubMed] [Google Scholar]
- [228].Page EL, Robitaille GA, Pouyssegur J, Richard DE, Induction of hypoxia-inducible factor-1alpha by transcriptional and translational mechanisms, J. Biol. Chem. 277 (50) (2002) 48403–48409. [DOI] [PubMed] [Google Scholar]
- [229].Anavi S, Hahn-Obercyger M, Madar Z, Tirosh O, Mechanism for HIF-1 activation by cholesterol under normoxia: a redox signaling pathway for liver damage, Free Radic. Biol. Med. 71 (2014) 61–69. [DOI] [PubMed] [Google Scholar]
- [230].Moon EJ, Sonveaux P, Porporato PE, Danhier P, Gallez B, Batinic-Haberle I, Nien YC, Schroeder T, Dewhirst MW, NADPH oxidase-mediated reactive oxygen species production activates hypoxia-inducible factor-1 (HIF-1) via the ERK pathway after hyperthermia treatment, PNAS 107 (47) (2010) 20477–20482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Rezvani HR, Dedieu S, North S, Belloc F, Rossignol R, Letellier T, de Verneuil H, Taieb A, Mazurier F, Hypoxia-inducible factor-1alpha, a key factor in the keratinocyte response to UVB exposure, J. Biol. Chem. 282 (22) (2007) 16413–16422. [DOI] [PubMed] [Google Scholar]
- [232].Wunderlich L, Paragh G, Wikonkal NM, Banhegyi G, Karpati S, Mandl J, UVB induces a biphasic response of HIF-1alpha in cultured human keratinocytes, Exp. Dermatol. 17 (4) (2008) 335–342. [DOI] [PubMed] [Google Scholar]
- [233].Cho YS, Kim CH, Park JW, Involvement of HIF-1alpha in UVB-induced epidermal hyperplasia, Mol. Cells 28 (6) (2009) 537–543. [DOI] [PubMed] [Google Scholar]
- [234].Hayashi Y, Yokota A, Harada H, Huang G, Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1alpha in cancer, Cancer Sci. 110 (5) (2019) 1510–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Song J, Yang X, Yan LJ, Role of pseudohypoxia in the pathogenesis of type 2 diabetes, Hypoxia (Auckl) 7 (2019) 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Nys K, Maes H, Dudek AM, Agostinis P, Uncovering the role of hypoxia inducible factor-1alpha in skin carcinogenesis, Biochim. Biophys. Acta 1816 (1) (2011) 1–12. [DOI] [PubMed] [Google Scholar]
- [237].Seleit I, Bakry OA, Al-Sharaky DR, Ragab RAA, Al-Shiemy SA, Evaluation of Hypoxia Inducible Factor-1alpha and Glucose Transporter-1 Expression in Non Melanoma Skin Cancer: An Immunohistochemical Study, J. Clin. Diagn. Res. 11 (6) (2017) EC09–EC16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Malekan M, Ebrahimzadeh MA, Sheida F, The role of Hypoxia-Inducible Factor-1alpha and its signaling in melanoma, Biomed. Pharmacother. 141 (2021), 111873. [DOI] [PubMed] [Google Scholar]
- [239].Navid F, Bruhs A, Schuller W, Fritsche E, Krutmann J, Schwarz T, Schwarz A, The Aryl hydrocarbon receptor is involved in UVR-induced immunosuppression, J, Invest. Dermatol. 133 (12) (2013) 2763–2770. [DOI] [PubMed] [Google Scholar]
- [240].Munoz-Sanchez J, Chanez-Cardenas ME, The use of cobalt chloride as a chemical hypoxia model, J. Appl. Toxicol. 39 (4) (2019) 556–570. [DOI] [PubMed] [Google Scholar]
- [241].Miranda E, Nordgren IK, Male AL, Lawrence CE, Hoakwie F, Cuda F, Court W, Fox KR, Townsend PA, Packham GK, Eccles SA, Tavassoli A, A cyclic peptide inhibitor of HIF-1 heterodimerization that inhibits hypoxia signaling in cancer cells, J. Am. Chem. Soc. 135 (28) (2013) 10418–10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Mylonis I, Chachami G, Simos G, Specific Inhibition of HIF Activity: Can Peptides Lead the Way? Cancers (Basel) 13 (3) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Yu T, Tang B, Sun X, Development of Inhibitors Targeting Hypoxia-Inducible Factor 1 and 2 for Cancer Therapy, Yonsei Med. J. 58 (3) (2017) 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Terzuoli E, Puppo M, Rapisarda A, Uranchimeg B, Cao L, Burger AM, Ziche M, Melillo G, Aminoflavone, a ligand of the aryl hydrocarbon receptor, inhibits HIF-1alpha expression in an AhR-independent fashion, Cancer Res. 70 (17) (2010) 6837–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Goodnough LT, Monk TG, Andriole GL, Erythropoietin therapy, N. Engl. J. Med. 336 (13) (1997) 933–938. [DOI] [PubMed] [Google Scholar]
- [246].Reichel C, Gmeiner G, Erythropoietin and analogs, Handb. Exp. Pharmacol. 195 (2010) 251–294. [DOI] [PubMed] [Google Scholar]
- [247].Dhillon S, Roxadustat: First Global Approval, Drugs 79 (5) (2019) 563–572. [DOI] [PubMed] [Google Scholar]
- [248].Hirota K, HIF-alpha Prolyl Hydroxylase Inhibitors and Their Implications for Biomedicine: A Comprehensive Review, Biomedicines 9 (5) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Vallon V, Verma S, Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function, Annu. Rev. Physiol. 83 (2021) 503–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Cheng L, Yu H, Yan N, Lai K, Xiang M, Hypoxia-Inducible Factor-1alpha Target Genes Contribute to Retinal Neuroprotection, Front. Cell. Neurosci. 11 (2017) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Nicholas MP, Mysore N, Corneal neovascularization, Exp. Eye Res. 202 (2021), 108363. [DOI] [PubMed] [Google Scholar]
- [252].Hammond CL, Roztocil E, Gupta V, Feldon SE, Woeller CF, More than Meets the Eye: The Aryl Hydrocarbon Receptor is an Environmental Sensor, Physiological Regulator and a Therapeutic Target in Ocular Disease, Front. Toxicol. 4 (2022), 791082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Garcia J, Hurwitz HI, Sandler AB, Miles D, Coleman RL, Deurloo R, Chinot OL, Bevacizumab (Avastin(R)) in cancer treatment: A review of 15 years of clinical experience and future outlook, Cancer Treat. Rev. 86 (2020), 102017. [DOI] [PubMed] [Google Scholar]
- [254].Dedania VS, Bakri SJ, Current perspectives on ranibizumab, Clin. Ophthalmol. 9 (2015) 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Allen E, Keir A, Ranibizumab-The jury is still out, Acta Paediatr. 111 (3) (2022) 698–699. [DOI] [PubMed] [Google Scholar]
- [256].Fallah J, Rini BI, Inhibitors HIF, Status of Current Clinical Development, Curr. Oncol. Rep. 21 (1) (2019) 6. [DOI] [PubMed] [Google Scholar]
- [257].An S, Zhou M, Li Z, Feng M, Cao G, Lu S, Liu L, Administration of CoCl2 Improves Functional Recovery in a Rat Model of Sciatic Nerve Transection Injury, Int. J. Med. Sci. 15 (13) (2018) 1423–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Cho Y, Shin JE, Ewan EE, Oh YM, Pita-Thomas W, Cavalli V, Activating Injury-Responsive Genes with Hypoxia Enhances Axon Regeneration through Neuronal HIF-1alpha, Neuron 88 (4) (2015) 720–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Di Giaimo R, Durovic T, Barquin P, Kociaj A, Lepko T, Aschenbroich S, Breunig CT, Irmler M, Cernilogar FM, Schotta G, Barbosa JS, Trumbach D, Baumgart EV, Neuner AM, Beckers J, Wurst W, Stricker SH, Ninkovic J, The Aryl Hydrocarbon Receptor Pathway Defines the Time Frame for Restorative Neurogenesis, Cell Rep 25(12) (2018) 3241–3251 e5. [DOI] [PubMed] [Google Scholar]
- [260].Navarrete-Opazo A, Mitchell GS, Therapeutic potential of intermittent hypoxia: a matter of dose, Am. J. Physiol. Regul. Integr. Comp. Physiol. 307 (10) (2014) R1181–R1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Sharp FR, Ran R, Lu A, Tang Y, Strauss KI, Glass T, Ardizzone T, Bernaudin M, Hypoxic preconditioning protects against ischemic brain injury, NeuroRx 1 (1) (2004) 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Kwon JI, Heo H, Ham SJ, Chae YJ, Lee DW, Kim ST, Min J, Sung YS, Kim KW, Choi Y, Woo DC, Woo CW, Aryl hydrocarbon receptor antagonism before reperfusion attenuates cerebral ischaemia/reperfusion injury in rats, Sci. Rep. 10 (1) (2020) 14906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Wang JX, Wang BB, Yuan SZ, Xue K, Zhang JS, Xu AJ, Blocking the Aryl Hydrocarbon Receptor Alleviates Myocardial Ischemia/Reperfusion Injury in Rats, Curr, Med. Sci. (2022). [DOI] [PubMed] [Google Scholar]
- [264].Eleftheriadis T, Pissas G, Filippidis G, Liakopoulos V, Stefanidis I, Reoxygenation induces reactive oxygen species production and ferroptosis in renal tubular epithelial cells by activating aryl hydrocarbon receptor, Mol. Med. Rep. 23 (1) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Sies H, Oxidative Stress: Introductory Remarks, in: Sies H (Ed.), Oxidative Stress, Academic Press, London, 1985, pp. 1–8. [Google Scholar]
- [266].Prochaska HJ, Talalay P, Regulatory mechanisms of monofunctional and bifunctional anticarcinogenic enzyme inducers in murine liver, Cancer Res. 48 (17) (1988) 4776–4782. [PubMed] [Google Scholar]
- [267].Stohs SJ, Oxidative stress induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), Free Radic. Biol. Med. 9 (1) (1990) 79–90. [DOI] [PubMed] [Google Scholar]
- [268].Shi H, Liu J, Gao H, Benzo(alpha)pyrene induces oxidative stress and inflammation in human vascular endothelial cells through AhR and NF-kappaB pathways, Microvasc. Res. 137 (2021), 104179. [DOI] [PubMed] [Google Scholar]
- [269].Dietrich C, Antioxidant Functions of the Aryl Hydrocarbon Receptor, Stem Cells Int. 2016 (2016) 7943495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].Braeuning A, Kohle C, Buchmann A, Schwarz M, Coordinate regulation of cytochrome P450 1a1 expression in mouse liver by the aryl hydrocarbon receptor and the beta-catenin pathway, Toxicol. Sci. 122 (1) (2011) 16–25. [DOI] [PubMed] [Google Scholar]
- [271].Borland MG, Krishnan P, Lee C, Albrecht PP, Shan W, Bility MT, Marcus CB, Lin JM, Amin S, Gonzalez FJ, Perdew GH, Peters JM, Modulation of aryl hydrocarbon receptor (AHR)-dependent signaling by peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) in keratinocytes, Carcinogenesis 35 (7) (2014) 1602–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Haarmann-Stemmann T, Abel J, Fritsche E, Krutmann J, The AhR-Nrf2 pathway in keratinocytes: on the road to chemoprevention? J, Invest. Dermatol. 132 (1) (2012) 7–9. [DOI] [PubMed] [Google Scholar]
- [273].Schafer M, Werner S, Nrf2–A regulator of keratinocyte redox signaling, Free Radic. Biol. Med. 88 (Pt B) (2015) 243–252. [DOI] [PubMed] [Google Scholar]
- [274].Moi P, Chan K, Asunis I, Cao A, Kan YW, Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region, PNAS 91 (21) (1994) 9926–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M, Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain, Genes Dev. 13 (1) (1999) 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M, Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2, Mol. Cell Biol. 24 (16) (2004) 7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Yamamoto T, Suzuki T, Kobayashi A, Wakabayashi J, Maher J, Motohashi H, Yamamoto M, Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity, Mol. Cell Biol. 28 (8) (2008) 2758–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Nakaso K, Yano H, Fukuhara Y, Takeshima T, Wada-Isoe K, Nakashima K, PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells, FEBS Lett. 546 (2–3) (2003) 181–184. [DOI] [PubMed] [Google Scholar]
- [279].Huang HC, Nguyen T, Pickett CB, Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription, J. Biol. Chem. 277 (45) (2002) 42769–42774. [DOI] [PubMed] [Google Scholar]
- [280].Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y, An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements, Biochem. Biophys. Res. Commun. 236 (2) (1997) 313–322. [DOI] [PubMed] [Google Scholar]
- [281].Miao W, Hu L, Scrivens PJ, Batist G, Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: direct cross-talk between phase I and II drug-metabolizing enzymes, J. Biol. Chem. 280 (21) (2005) 20340–20348. [DOI] [PubMed] [Google Scholar]
- [282].Shin S, Wakabayashi N, Misra V, Biswal S, Lee GH, Agoston ES, Yamamoto M, Kensler TW, NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis, Mol. Cell Biol. 27 (20) (2007) 7188–7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Anwar-Mohamed A, Degenhardt OS, El Gendy MA, Seubert JM, Kleeberger SR, El-Kadi AO, The effect of Nrf2 knockout on the constitutive expression of drug metabolizing enzymes and transporters in C57Bl/6 mice livers, Toxicol. In Vitro 25 (4) (2011) 785–795. [DOI] [PubMed] [Google Scholar]
- [284].Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD, Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H: quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence, Biochem. J 374 (Pt 2) (2003) 337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Hayes JD, Flanagan JU, Jowsey IR, Glutathione transferases, Annu. Rev. Pharmacol. Toxicol. 45 (2005) 51–88. [DOI] [PubMed] [Google Scholar]
- [286].Rushmore TH, Pickett CB, Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants, J. Biol. Chem. 265 (24) (1990) 14648–14653. [PubMed] [Google Scholar]
- [287].Yueh MF, Huang YH, Hiller A, Chen S, Nguyen N, Tukey RH, Involvement of the xenobiotic response element (XRE) in Ah receptor-mediated induction of human UDP-glucuronosyltransferase 1A1, J. Biol. Chem. 278 (17) (2003) 15001–15006. [DOI] [PubMed] [Google Scholar]
- [288].Kalthoff S, Ehmer U, Freiberg N, Manns MP, Strassburg CP, Coffee induces expression of glucuronosyltransferases by the aryl hydrocarbon receptor and Nrf2 in liver and stomach, Gastroenterology 139(5) (2010) 1699–710, 1710 e1–2. [DOI] [PubMed] [Google Scholar]
- [289].Munzel PA, Schmohl S, Buckler F, Jaehrling J, Raschko FT, Kohle C, Bock KW, Contribution of the Ah receptor to the phenolic antioxidant-mediated expression of human and rat UDP-glucuronosyltransferase UGT1A6 in Caco-2 and rat hepatoma 5L cells, Biochem. Pharmacol. 66 (5) (2003) 841–847. [DOI] [PubMed] [Google Scholar]
- [290].Tsuji G, Takahara M, Uchi H, Matsuda T, Chiba T, Takeuchi S, Yasukawa F, Moroi Y, Furue M, Identification of ketoconazole as an AhR-Nrf2 activator in cultured human keratinocytes: the basis of its anti-inflammatory effect, J, Invest. Dermatol. 132 (1) (2012) 59–68. [DOI] [PubMed] [Google Scholar]
- [291].Takemura H, Nagayoshi H, Matsuda T, Sakakibara H, Morita M, Matsui A, Ohura T, Shimoi K, Inhibitory effects of chrysoeriol on DNA adduct formation with benzo[a]pyrene in MCF-7 breast cancer cells, Toxicology 274 (1–3) (2010) 42–48. [DOI] [PubMed] [Google Scholar]
- [292].Ciolino HP, Daschner PJ, Wang TT, Yeh GC, Effect of curcumin on the aryl hydrocarbon receptor and cytochrome P450 1A1 in MCF-7 human breast carcinoma cells, Biochem. Pharmacol. 56 (2) (1998) 197–206. [DOI] [PubMed] [Google Scholar]
- [293].Garg R, Gupta S, Maru GB, Dietary curcumin modulates transcriptional regulators of phase I and phase II enzymes in benzo[a]pyrene-treated mice: mechanism of its anti-initiating action, Carcinogenesis 29 (5) (2008) 1022–1032. [DOI] [PubMed] [Google Scholar]
- [294].Kalpana Deepa Priya D, Gayathri R, Sakthisekaran D, Role of sulforaphane in the anti-initiating mechanism of lung carcinogenesis in vivo by modulating the metabolic activation and detoxification of benzo(a)pyrene, Biomed. Pharmacother. 65(1) (2011) 9–16. [DOI] [PubMed] [Google Scholar]
- [295].Zhang Y, Kensler TW, Cho CG, Posner GH, Talalay P, Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates, PNAS 91 (8) (1994) 3147–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [296].Hwang YP, Han EH, Choi JH, Kim HG, Lee KJ, Jeong TC, Lee ES, Jeong HG, Chemopreventive effects of Furan-2-yl-3-pyridin-2-yl-propenone against 7,12-dimethylbenz[a]anthracene-inducible genotoxicity, Toxicol. Appl. Pharmacol. 228 (3) (2008) 343–350. [DOI] [PubMed] [Google Scholar]
- [297].Uchi H, Yasumatsu M, Morino-Koga S, Mitoma C, Furue M, Inhibition of aryl hydrocarbon receptor signaling and induction of NRF2-mediated antioxidant activity by cinnamaldehyde in human keratinocytes, J. Dermatol. Sci. 85 (1) (2017) 36–43. [DOI] [PubMed] [Google Scholar]
- [298].Mohammadi-Bardbori A, Vikstrom Bergander L, Rannug U, Rannug A, NADPH Oxidase-Dependent Mechanism Explains How Arsenic and Other Oxidants Can Activate Aryl Hydrocarbon Receptor Signaling, Chem. Res. Toxicol. 28 (12) (2015) 2278–2286. [DOI] [PubMed] [Google Scholar]
- [299].Wincent E, Bengtsson J, Mohammadi Bardbori A, Alsberg T, Luecke S, Rannug U, Rannug A, Inhibition of cytochrome P4501-dependent clearance of the endogenous agonist FICZ as a mechanism for activation of the aryl hydrocarbon receptor, PNAS 109 (12) (2012) 4479–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Kubli SP, Bassi C, Roux C, Wakeham A, Göbl C, Zhou W, Jafari SM, Snow B, Jones L, Palomero L, Thu KL, Cassetta L, Soong D, Berger T, Ramachandran P, Baniasadi SP, Duncan G, Lindzen M, Yarden Y, Herranz C, Lazaro C, Chu MF, Haight J, Tinto P, Silvester J, Cescon DW, Petit A, Pettersson S, Pollard JW, Mak TW, Pujana MA, Cappello P, Gorrini C, AhR controls redox homeostasis and shapes the tumor microenvironment in BRCA1-associated breast cancer, Proc. Natl. Acad. Sci. 116(9) (2019) 3604–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Bart AG, Scott EE, Structures of human cytochrome P450 1A1 with bergamottin and erlotinib reveal active-site modifications for binding of diverse ligands, J. Biol. Chem. 293 (50) (2018) 19201–19210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Dhakshinamoorthy S, Jaiswal AK, c-Maf negatively regulates ARE-mediated detoxifying enzyme genes expression and anti-oxidant induction, Oncogene 21 (34) (2002) 5301–5312. [DOI] [PubMed] [Google Scholar]
- [303].Brauze D, Crow JS, Malejka-Giganti D, Modulation by β-naphthoflavone of ovarian hormone dependent responses in rat uterus and liver in vivo, Can. J. Physiol. Pharmacol. 75 (8) (1997) 1022–1029. [DOI] [PubMed] [Google Scholar]
- [304].Boverhof DR, Kwekel JC, Humes DG, Burgoon LD, Zacharewski TR, Dioxin induces an estrogen-like, estrogen receptor-dependent gene expression response in the murine uterus, Mol. Pharmacol. 69 (5) (2006) 1599–1606. [DOI] [PubMed] [Google Scholar]
- [305].Astroff B, Eldridge B, Safe S, Inhibition of the 17β-estradiol-induced and constitutive expression of the cellular protooncogene c-fos by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the female rat uterus, Toxicol. Lett. 56 (3) (1991) 305–315. [DOI] [PubMed] [Google Scholar]
- [306].Boverhof DR, Burgoon LD, Williams KJ, Zacharewski TR, Inhibition of estrogen-mediated uterine gene expression responses by dioxin, Mol. Pharmacol. 73 (1) (2008) 82–93. [DOI] [PubMed] [Google Scholar]
- [307].Lin TM, Ko K, Moore RW, Simanainen U, Oberley TD, Peterson RE, Effects of aryl hydrocarbon receptor null mutation and in utero and lactational 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure on prostate and seminal vesicle development in C57BL/6 mice, Toxicol. Sci. 68 (2) (2002) 479–487. [DOI] [PubMed] [Google Scholar]
- [308].Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, Tohyama C, Krust A, Mimura J, Chambon P, Yanagisawa J, Fujii-Kuriyama Y, Kato S, Modulation of oestrogen receptor signalling by association with the activated dioxin receptor, Nature 423 (6939) (2003) 545–550. [DOI] [PubMed] [Google Scholar]
- [309].Wormke M, Stoner M, Saville B, Walker K, Abdelrahim M, Burghardt R, Safe S, The aryl hydrocarbon receptor mediates degradation of estrogen receptor alpha through activation of proteasomes, Mol. Cell Biol. 23 (6) (2003) 1843–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H, Takahashi S, Kouzmenko A, Nohara K, Chiba T, Fujii-Kuriyama Y, Kato S, Dioxin receptor is a ligand-dependent E3 ubiquitin ligase, Nature 446 (7135) (2007) 562–566. [DOI] [PubMed] [Google Scholar]
- [311].Luecke-Johansson S, Gralla M, Rundqvist H, Ho JC, Johnson RS, Gradin K, Poellinger L, A Molecular Mechanism To Switch the Aryl Hydrocarbon Receptor from a Transcription Factor to an E3 Ubiquitin Ligase, Mol. Cell Biol. 37 (13) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [312].Hershko A, Ciechanover A, The ubiquitin system, Annu. Rev. Biochem 67 (1998) 425–479. [DOI] [PubMed] [Google Scholar]
- [313].Nandi D, Tahiliani P, Kumar A, Chandu D, The ubiquitin-proteasome system, J. Biosci. 31 (1) (2006) 137–155. [DOI] [PubMed] [Google Scholar]
- [314].Ohtake F, Baba A, Fujii-Kuriyama Y, Kato S, Intrinsic AhR function underlies cross-talk of dioxins with sex hormone signalings, Biochem. Biophys. Res. Commun. 370 (4) (2008) 541–546. [DOI] [PubMed] [Google Scholar]
- [315].Dou H, Duan Y, Zhang X, Yu Q, Di Q, Song Y, Li P, Gong Y, Aryl hydrocarbon receptor (AhR) regulates adipocyte differentiation by assembling CRL4B ubiquitin ligase to target PPARgamma for proteasomal degradation, J. Biol. Chem. 294 (48) (2019) 18504–18515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Kawajiri K, Kobayashi Y, Ohtake F, Ikuta T, Matsushima Y, Mimura J, Pettersson S, Pollenz RS, Sakaki T, Hirokawa T, Akiyama T, Kurosumi M, Poellinger L, Kato S, Fujii-Kuriyama Y, Aryl hydrocarbon receptor suppresses intestinal carcinogenesis in ApcMin/+ mice with natural ligands, PNAS 106 (32) (2009) 13481–13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [317].Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang X, Zhou Z, Shu G, Yin G, Wnt/beta-catenin signalling: function, biological mechanisms, and therapeutic opportunities, Signal Transduct. Target. Ther. 7 (1) (2022) 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [318].Roberts BJ, Whitelaw ML, Degradation of the basic helix-loop-helix/Per-ARNT-Sim homology domain dioxin receptor via the ubiquitin/proteasome pathway, J. Biol. Chem. 274 (51) (1999) 36351–36356. [DOI] [PubMed] [Google Scholar]
- [319].Ma Q, Baldwin KT, 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway. Role of the transcription activaton and DNA binding of AhR, J. Biol. Chem. 275 (12) (2000) 8432–8438. [DOI] [PubMed] [Google Scholar]
- [320].Rijo MP, Diani-Moore S, Yang C, Zhou P, Rifkind AB, Roles of the ubiquitin ligase CUL4B and ADP-ribosyltransferase TiPARP in TCDD-induced nuclear export and proteasomal degradation of the transcription factor AHR, J. Biol. Chem. 297 (2) (2021), 100886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [321].Clevers H, Nusse R, Wnt/beta-catenin signaling and disease, Cell 149 (6) (2012) 1192–1205. [DOI] [PubMed] [Google Scholar]
- [322].Culig Z, Santer FR, Androgen receptor signaling in prostate cancer, Cancer Metastasis Rev. 33 (2–3) (2014) 413–427. [DOI] [PubMed] [Google Scholar]
- [323].Jia M, Dahlman-Wright K, Gustafsson JA, Estrogen receptor alpha and beta in health and disease, Best Pract. Res. Clin. Endocrinol. Metab. 29 (4) (2015) 557–568. [DOI] [PubMed] [Google Scholar]
- [324].Kono M, Fujii T, Lim B, Karuturi MS, Tripathy D, Ueno NT, Androgen Receptor Function and Androgen Receptor-Targeted Therapies in Breast Cancer: A Review, JAMA Oncol. 3 (9) (2017) 1266–1273. [DOI] [PubMed] [Google Scholar]
- [325].Kannt A, Dikic I, Expanding the arsenal of E3 ubiquitin ligases for proximity-induced protein degradation, Cell Chem. Biol. 28 (7) (2021) 1014–1031. [DOI] [PubMed] [Google Scholar]
- [326].Crews CM, Inducing Protein Degradation as a Therapeutic Strategy, J. Med. Chem. 61 (2) (2018) 403–404. [DOI] [PubMed] [Google Scholar]
- [327].Bekes M, Langley DR, Crews CM, PROTAC targeted protein degraders: the past is prologue, Nat. Rev. Drug Discov. 21 (3) (2022) 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [328].Puppala D, Lee H, Kim KB, Swanson HI, Development of an aryl hydrocarbon receptor antagonist using the proteolysis-targeting chimeric molecules approach: a potential tool for chemoprevention, Mol. Pharmacol. 73 (4) (2008) 1064–1071. [DOI] [PubMed] [Google Scholar]
- [329].Ohoka N, Tsuji G, Shoda T, Fujisato T, Kurihara M, Demizu Y, Naito M, Development of Small Molecule Chimeras That Recruit AhR E3 Ligase to Target Proteins, ACS Chem. Biol. 14 (12) (2019) 2822–2832. [DOI] [PubMed] [Google Scholar]
- [330].Wolff S, Harper PA, Wong JM, Mostert V, Wang Y, Abel J, Cell-specific regulation of human aryl hydrocarbon receptor expression by transforming growth factor-beta(1), Mol. Pharmacol. 59 (4) (2001) 716–724. [DOI] [PubMed] [Google Scholar]
- [331].Tischkau SA, Mechanisms of circadian clock interactions with aryl hydrocarbon receptor signalling, Eur. J. Neurosci. 51 (1) (2020) 379–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [332].Zablon HA, Ko CI, Puga A, Converging Roles of the Aryl Hydrocarbon Receptor in Early Embryonic Development, Maintenance of Stemness, and Tissue Repair, Toxicol. Sci. 182 (1) (2021) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [333].Rejano-Gordillo C, Ordiales-Talavero A, Nacarino-Palma A, Merino JM, Gonzalez-Rico FJ, Fernandez-Salguero PM, Aryl Hydrocarbon Receptor: From Homeostasis to Tumor Progression, Front. Cell Dev. Biol. 10 (2022), 884004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [334].Akhtar S, Hourani S, Therachiyil L, Al-Dhfyan A, Agouni A, Zeidan A, Uddin S, Korashy HM, Epigenetic Regulation of Cancer Stem Cells by the Aryl Hydrocarbon Receptor Pathway, Semin. Cancer Biol. 83 (2022) 177–196. [DOI] [PubMed] [Google Scholar]